

Abstract
Rapidly changing features in an intact biological sample are challenging to efficiently trap and image by conventional electron microscopy (EM). For example, the model organism C. elegans is widely used to study embryonic development and differentiation, yet the fast kinetics of cell division makes the targeting of specific developmental stages for ultrastructural study difficult. We set out to image the condensed metaphase chromosomes of an early embryo in the intact worm in 3-D. To achieve this, one must capture this transient structure, then locate and subsequently image the corresponding volume by EM in the appropriate context of the organism, all while minimizing a variety of artifacts. In this methodological advance, we report on the high-pressure freezing of spatially constrained whole C. elegans hermaphrodites in a combination of cryoprotectants to identify embryonic cells in metaphase by in situ cryo-fluorescence microscopy. The screened worms were then freeze substituted, resin embedded and further prepared such that the targeted cells were successfully located and imaged by focused ion beam scanning electron microscopy (FIB-SEM). We reconstructed the targeted metaphase structure and also correlated an intriguing punctate fluorescence signal to a H2B-enriched putative polar body autophagosome in an adjacent cell undergoing telophase. By enabling cryo-fluorescence microscopy of thick samples, our workflow can thus be used to trap and image transient structures in C. elegans or similar organisms in a near-native state, and then reconstruct their corresponding cellular architectures at high resolution and in 3-D by correlative volume EM.
1. Introduction
Numerous advances have recently been made in individual imaging technologies in cell biology. In parallel, there are also increasingly powerful ways in which these, as well as more traditional microscopic approaches, may be combined. Most prominently, correlative light and electron microscopy (CLEM) has been applied to a variety of experimental systems. Conventional protocols involve chemical fixation during sample preparation for either light microscopy (LM) or electron microscopy (EM) or both; the accompanying correlation may be for spatial identification of the region of interest (ROI) by LM and EM, or for colocalization of specific probes. For an in-depth CLEM primer, the reader is directed to a previous volume of this publication and the references therein (Muller-Reichert & Verkade, 2012). Reliable cryogenic fixation and technological innovations have significantly advanced cryo-imaging of thin cellular specimens. For instance, leading edges of adherent cells can be directly imaged by cryo-EM, and slightly thicker samples can be thinned down or lifted out by cryo-FIB to render them so amenable (Hsieh, Schmelzer, Kishchenko, Wagenknecht, & Marko, 2014; Rigort & Plitzko, 2015; Schaffer et al., 2019). Since cryo-fluorescence microscopy of these samples eliminates distortions and movements inherent to chemical fixation protocols, cryo-CLEM has been used to accurately correlate features of interest at high resolutions and near-native conditions in such thin samples (Gorelick et al., 2019; Jun et al., 2011; Kukulski et al., 2011; Schorb et al., 2017).
On the other hand, correlative imaging of microns-thick samples in 3-D is challenging; while still amenable to fluorescence techniques, these are too thick to be imaged whole by transmission EM (TEM). Traditional serial-section TEM protocols are being supplanted by a raft of so-called volume EM approaches (Briggman & Bock, 2012; Peddie & Collinson, 2014; Titze & Genoud, 2016), which can now automatically section and image vast volumes of biological specimens at nanoscale resolutions in 3-D; indeed, volume EM of chemically fixed neuronal samples is now a mainstay of connectomics studies (Bock et al., 2011; Helmstaedter et al., 2013; Kasthuri et al., 2015; Takemura et al., 2017).
Microns-thick volumes are typically too large to be vitrified by plunge freezing, so high-pressure freezing (HPF) protocols have been developed to minimize chemical fixation-induced artifacts, and these samples are then freeze substituted to incorporate heavy metal staining for subsequent room temperature volume EM (Czymmek et al., 2020; McDonald et al., 2010; Tsang et al., 2018). Some FIB- SEM imaging under cryogenic conditions has also been demonstrated, as FIB milling can be executed with relative ease at low temperatures (Akiva et al., 2019; Vidavsky et al., 2016). Several groups have combined volume EM with fluorescence readouts in a variety of ways, typically with fluorescence imaging executed at room temperature, either with live, chemically fixed or high-pressure frozen, or indeed resin-embedded cells (Giepmans, Adams, Ellisman, & Tsien, 2006; Narayan et al., 2014; Ou et al., 2017; Peddie et al., 2017; Scotuzzi et al., 2017). In a recent report, Hoffmann et al. combined cryo-fluorescence microscopy with FIB-SEM imaging to visualize protein-ultrastructure relationships in 3D in single mammalian cells at ∼8 nm isotropic resolutions (Hoffman et al., 2020).
Even with these advances, targeted EM imaging of fleeting events or structures in a thick sample or an intact model organism is challenging. For example, capturing embryos at specific developmental stages within the C. elegans worm, with adults measuring >1 mm long and >100 μm thick (Brenner, 1973), presents a significant technical problem. Embryos develop rapidly from the one-cell stage embryo to a hatched L1 larvae in 9 h at 22 °C (Sulston, Schierenberg, White, & Thomson, 1983). Following fertilization, the embryos are encased in a chitinous layer, a physical barrier that prevents chemical fixatives from penetrating and arresting fast mitotic events in a timely manner. These issues obviate traditional aldehyde-based sample preparation for ultrastructure analysis, and high-pressure freezing is the preferred approach for EM studies of C. elegans (Li et al., 2017; Muller-Reichert, Kiewisz, & Redemann, 2018). In a fertilized zygote, the parental chromosomes can be located at the metaphase plate for about 90 s during late prometaphase through metaphase, before anaphase is initiated (Oegema & Hyman, 2006). Therefore, the capture of architectural changes associated with metaphase plate formation and initiation of anaphase necessitates snap freezing by HPF. Most such studies are performed ex vivo, where the worms are dissected, embryos tracked individually, and HPF executed at an appropriate time (Muller-Reichert, Srayko, Hyman, O’Toole, & McDonald, 2007; Rahman et al., 2020). Tracking these alterations in cellular structures during mitosis within an intact worm is thus daunting; not only do the same limitations apply as before, but an intermediate screening step is also required after HPF, i.e., under cryogenic conditions. Without a way to select for worms of interest, “brute force” volume EM of these vast volumes at unknown development stages would be prohibitively time-and resource-consuming. Here we describe a straightforward methodology to identify and selectively image embryos trapped at the desired developmental or cell cycle stage within intact C. elegans worms. We subject whole worms expressing specific fluorescent markers (here, signal peptidase (SP12)::GFP and histone2B (H2B)::mCherry) to HPF fixation and then screen embryos in the intact worm for instances of metaphase or other stages of cell division cycle by super-resolution (Airyscan) cryo-fluorescence microscopy. We follow this by freeze substituting the sample in situ, locating the resin-embedded region of interest, and imaging the desired embryo by FIB-SEM to reconstruct in 3-D the targeted mitotic structures at nanoscale resolutions (Fig. 1).
FIG. 1.

Schematic for cryo-LM/FIB-SEM workflow. Left panel: C. elegans worms are enclosed in cellulose capillaries and high-pressure frozen in planchettes (top). Dividing embryos in the intact worm are imaged in situ by cryo-fluorescence microscopy, revealing targeted and unexpected or rare events (bottom, green, SP12::GFP, red, H2B::mCherry). Right panel: Sectioning of a freeze-substituted sample reveals a cross section of the worm (top), which is used to position the FIB-SEM trench and image acquisition. The resulting nanoscale EM image volume is examined, and correlatively identified features of interest are reconstructed in 3-D (bottom).
2. A note on selecting the right cryoprotectant
Cryoprotectants are crucial for the ultrastructural preservation of high-pressure frozen samples. However, fluorescence studies of developing C. elegans embryos are almost always conducted at room temperature and often ex vivo, so basic optical properties of cryoprotectants such as opacity and autofluorescence are rarely, if ever, considered. However, during cryo-fluorescence microscopy, in order to image and screen for embryos trapped at the desired developmental stage, it is necessary to locate and enumerate embryonic cells within an intact C. elegans worm that is high-pressure-frozen in a significant volume of cryoprotectant. 20% BSA is widely used for cryoprotection of C. elegans worms and embryos (Muller-Reichert et al., 2007), and has consistently generated well-preserved samples for FIB-SEM studies in our hands (Rahman et al., 2020). In preliminary experiments, we high-pressure froze single transgenic OCF5 (SP12::GFP and H2B::mCherry) worms enclosed individually inside cellulose capillary tubes in planchette sandwiches, as outlined in Section 4.1; the worms were cryoprotected by a solution of 20% BSA both inside the tubes as well as in the planchette recess and were examined under a cryo-fluorescence microscope. The intense autofluorescence of BSA in the GFP channel (λem 495–535 nm) greatly reduced visibility of the worm and, combined with the punctate nature of the H2B::mCherry label, almost completely masked the signal from individual embryos (data not shown). Therefore, we worked to identify alternative cryoprotectants that would allow us to clearly visualize details within embryos in intact worms, while also minimizing damage during the HPF process. Below, we detail experiments that led to our choice of the combination of 15% dextran + 5% BSA inside the cellulose capillary and perfluorodecalin inside the planchette sandwich but outside the capillary; the reader can skip ahead to the main methods section 4.1 if they so wish.
2.1. Evaluation of toxicity, ease of handling, and degree of cryoprotection
A search of the literature yielded yeast paste, bacteria (Manning & Richmond, 2015; McDonald et al., 2010; Weimer, 2006), 10% (w/v) polyvinylpyrrolidone (Fabig, Schwarz, Striese, Laue, & Muller-Reichert, 2019), and 10% glycerol (Fuest et al., 2019) as alternatives for cryo-preservation of C. elegans worms and/or embryos. We also included 0.15 M sucrose, previously used to cryo-fix Hela cells in suspension (Schlegel, Giddings, Ladinsky, & Kirkegaard, 1996), 15% dextran and 5% BSA, previously used for electron tomography of Tetrahymena thermophila (Giddings, Meehl, Pearson, & Winey, 2010), and 1-hexadecene, a well-known, chemically inert filler for HPF preparation of both biological and non-biological specimens (Deirieh, Chang, Casey, Joester, & Germaine, 2019; McDonald, 1999). Although not applied for cryogenic experiments, perfluorodecalin used as a mounting medium for plant samples yielded improved clarity and consistency of images of mesophyll from multiple microscopy techniques (Littlejohn et al., 2014). The cryoprotectants that we evaluated, including 20% BSA, are listed in Table 1.
Table 1.
Characterization of cryoprotectants for high-pressure freezing of whole C. elegans worms.
| Cryoprotectant | Adverse effects | Ease of handling | Degree of cryoprotection | Autofluorescence (λex 488 nm, a.u.) |
|---|---|---|---|---|
| 20% BSA | None | +b | +++ | 80.68 |
| 10% glycerol | +a | +++ | +++ | 3.18 |
| 0.15 M sucrose | None/+ | +++ | +++ | 5.55 |
| 20% polyvinyl pyrrolidone (PP) | None | +++ | +++ | 25.35 |
| 15% dextran +5% BSA | None | +++ | ++/+++ | 38.97 |
| Baker’s yeast paste | N/A | +b | +++ | 236.88 |
| Perfluorodecalin | +++c | +++ | ++/+++ | 0.82 |
| 1-Hexadecene | +++c | +++ | +++ | 0.85 |
Candidates were evaluated in terms of their macroscopic adverse effects and toxicity on C. elegans worms, ease of handling during sample preparation, degree of cryoprotection ascertained by microscopic examination, and autofluorescence. The scores are ranked from none to +++ or for each category based on empirical evidence and observations for the first three parameters, made in our laboratory. Autofluorescence scores are semi-quantitative and represent fluorescence values from up to three images acquired with the same parameters and averaged after masking out areas of contamination.
Worms appeared swollen.
High viscosity of medium.
Worms lost structural integrity and became soft and pasty.
One obvious requirement of the alternative cryoprotectant is the absence of adverse effects or toxicity on C. elegans worms during sample preparation. We evaluated macroscopic physical changes in the worms under a dissection microscope during the roughly 3 min it was exposed to the liquid cryoprotectant, i.e., starting from the transfer of muscle relaxant-treated worms from an agar plate to a droplet of cryoprotectant, up until HPF of the worm in the assembled planchette (see Section 4.2). We found that exposure of the worms to 1-hexadecene and perfluorodecalin quickly reduced movement of the worms; they became soft and pasty and could not be drawn into a cellulose capillary by pipetting. This observation extends previous findings of 1-hexadecene toxicity (Hohenberg, Mannweiler, & Muller, 1994; Littlejohn et al., 2014) and suggests that perfluorodecalin is similarly harmful to C. elegans. Even during the short times required for sample preparation, worms prepared in 0.15 M sucrose and 10% glycerol exhibited signs of shrinkage and swelling, respectively. It is possible that at some optimal concentration, these compounds offer perfect osmotic balance and cryoprotection, but this was not tested at length. On the other hand, no adverse effects were observed in worms immersed in the 20% BSA, 15% dextran+ 5% BSA, and 20% polyvinyl pyrrolidine solutions. Since yeast paste was too thick to draw into a cellulose tube, we considered it only as a candidate for the cryoprotectant inside the planchette sandwich but outside the cellulose tubing.
We also evaluated “ease of handling,” a seemingly innocuous qualitative measure of cryoprotectants (Table 1), but an important factor given the manual nature of these steps. For example, the high viscosity of the 20% BSA solution sometimes caused cellulose capillaries to stick to tweezers during transfer and capillary crimping, resulting in delays and drying or loss of samples. Similarly, filling the shallow 100 μm-deep cavity or recess of a planchette sandwich with yeast paste was cumbersome due to the thickness of this cryoprotectant. Yeast paste also dried quickly, so sample preparation before HPF must be executed rapidly—this is possible but chal lenging for a large number of samples. The remaining cryoprotectants on the list pipetted readily into capillary tubes and planchettes, but we note that speed is still of the essence during these steps.
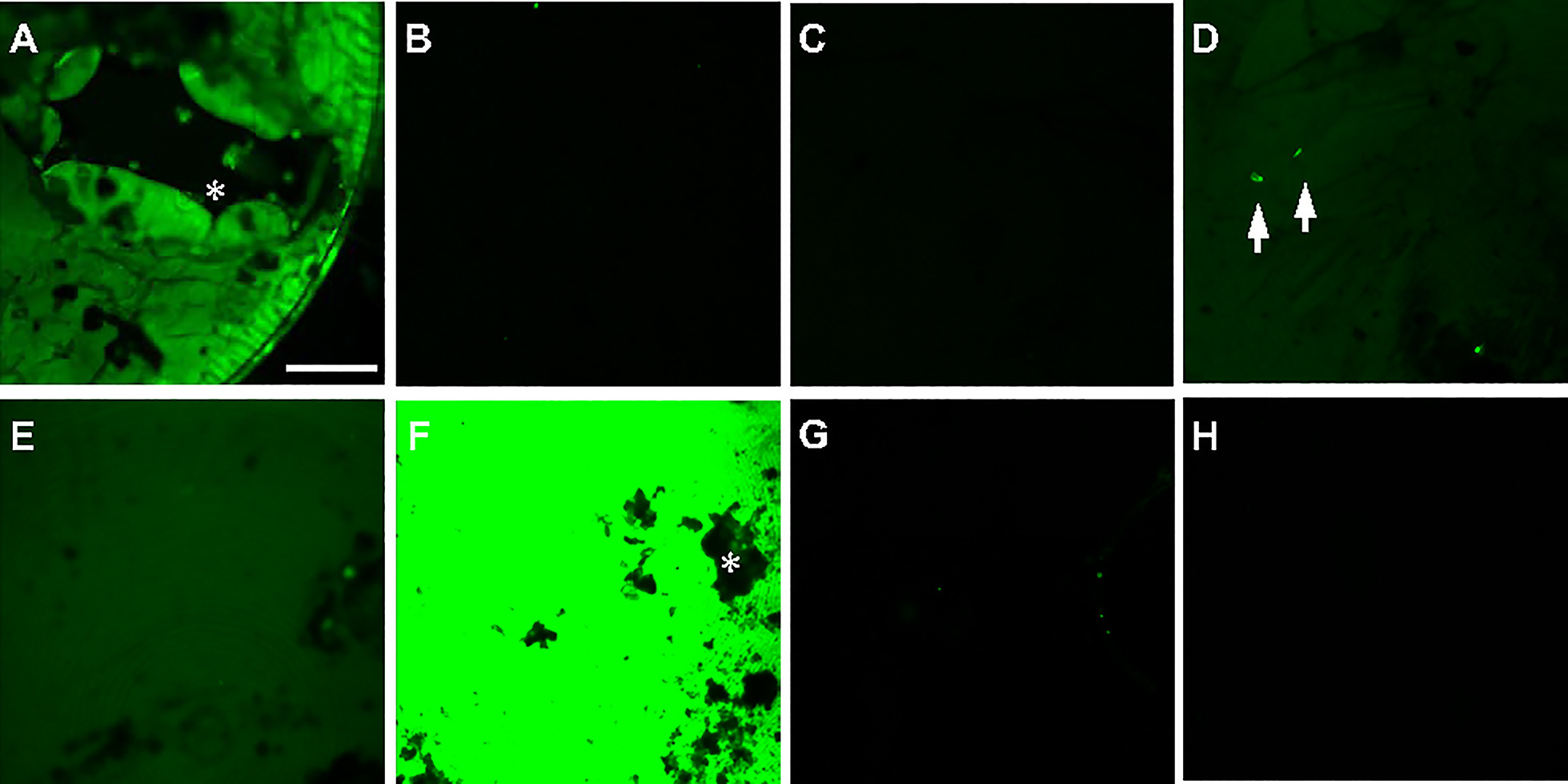
The evaluation of a cryoprotectant’s efficacy by simple fluorescence microscopy is an admittedly coarse approach, however, it is a helpful step to triage samples damaged during or after the freezing process, whether due to poor freezing or devitrification during handling; indeed this evaluation happens by default during the screening of worm samples (Section 4.2.2). Here, we pipetted 0.8 μL of a cryoprotectant into the 100 μm-deep cavity of a type A+flat side type B planchette sandwich and performed HPF on the assembly. The frozen sample sandwiches either opened up in the collection dewar or were manually separated under liquid nitrogen. The type A planchettes were loaded into a planchette cassette, and their 100 μm cavities were inspected by cryo-fluorescence microscopy with a 10 × long working distance objective (see Section 4.2). Samples typically showed one of three characteristics: in a perfect scenario, the entire field of view was clear, suggesting good vitrification and little “frost” contamination. Commonly, the clear field of view was obscured in a few areas by small dark patches—these are possibly small regions of contamination accrued during sample handling and transfer under cryogenic conditions. In the worst case (accidental exposure of samples to elevated temperatures, HPF instrument breakdown), there was obvious ice crystallization, resulting in elevated background signal and appearance of visible cracks (data not shown). Visual inspection outside of a fluorescence microscope reveals an icy mass. We observed occasional small patches of slightly opaque ice in a few of the planchettes containing perfluorodecalin and 15% dextran + 5% BSA, possibly from small variations in samples and handling. No obvious ice damage was observed in samples frozen with the other cryoprotectants tested.
2.2. Measurement of autofluorescence
For the successful execution of our workflow, it was further critical to identify cryoprotectants with least amount of autofluorescence. Previous cryo-correlative studies (Kukulski et al., 2011; Schwartz, Sarbash, Ataullakhanov, McIntosh, & Nicastro, 2007) have typically used cryo-TEM of plunge-frozen samples, meaning that the volume of cryoprotectant is minimal, resulting in a negligible contribution toward background fluorescence signal. In contrast, the cryo-fluorescence microscopy experiments described here require the light path to penetrate into and fluorescence signal to emanate from a minimum of 100 μm; the bulk of this volume occupied by the cryoprotectant results in a non-negligible (and in some cases overwhelming) background fluorescence signal.
In order to exclude cryoprotectants with such undesirable properties, images were acquired from samples high-pressure frozen in the presence of the compounds listed above (Supplementary Fig. S1 in the online version at https://doi.org/10.1016/bs.mcb.2020.09.009). This work was associated with a larger biological study of C. elegans where a variety of proteins were genetically tagged with green and sometimes red fluorophores, hence we focused on background fluorescence in the GFP (λex 488 nm) channel. We note that the autofluorescence of many candidate cryoprotectants is lower at longer wavelengths (data not shown), so using fluorophores with longer λex and λem may allow a wider choice of cryoprotectant. High-pressure frozen cryoprotectant-only samples were placed in a cryostage and imaged under cryogenic conditions with a Zeiss LSM 710 microscope equipped a 10 × and a 100 × long working distance objective (Fig. 2) and an AiryScan detector. While this detector is utilized for super-resolution imaging studies, we clarify that here the images themselves do not break resolution barriers. However, since the resolution, quality and signal-to-noise ratios of images recorded at the AiryScan detector were superior to those acquired at the regular photomultiplier tube, for these and following experiments we recorded images using this detector, with calibrations performed before independent acquisition runs according to the manufacturer’s suggestions. For each sample, 1–3 images were taken at separate locations across the frozen planchette. We avoided or masked out surface contaminants such as frost and ice crystals that appeared as dark or bright spots on the sample surface (Supplementary Fig. S1 in the online version at https://doi.org/10.1016/bs.mcb.2020.09.009A, D, F). Sometimes a loss in homogeneity of cryoprotectant, likely from drying out, appeared as dark holes in the image (Supplementary Fig. S1 in the online version at https://doi.org/10.1016/bs.mcb.2020.09.009, *). Images were acquired with the 10 × and 100 × objectives, at a fixed laser power and gain. Typical acquisition parameters were: pinhole 600.5, bit depth 8, image size 1024 × 1024 pixels, scan bi-directional, speed 5, averaging 4, zoom 1. These were based on observations from a variety of worm strains with endogenous GFP and mCherry labels. After acquisition, the histogram function in the microscope software (ZEN Black, Carl Zeiss Inc) was applied and mean fluorescence intensity plotted.
FIG. 2.

Experimental setup for cryo-fluorescence microscopy with HPF planchettes. (A, B) An upright Zeiss LSM710 equipped with an Airyscan detector and fitted with a Linkam cryostage (boxed) with controller and dewar to the left (A, at room temperature; B, at cryogenic imaging conditions). (C) A round notched “puck” with a cassette in its slot; the cassette contains three inserted planchettes. The magnetic tool is also captured in this image. (D) Cryostage, with metal bridge, imaging window and well in the middle, and LCD display on right. The LN2 dewar is to the left just out of frame. The two round notched slots in the blue well (*) accommodate pucks. (E) Transfer of the loaded cassette from puck to the imaging window with the magnetic tool now attached. (F) Metal bridge with loaded cassette and three planchettes ready for imaging. Images (C)–(F) were taken with empty planchettes and at room temperature and (C, E) taken outside the cryostage well for clarity.
Yeast paste clearly exhibited the highest mean background fluorescence (236.88 a.u.), followed by 20% BSA (80.68 a.u.) and 15% dextran and 5% BSA (38.97 a.u.). 10% glycerol (3.18 a.u.) and 0.15 M sucrose (5.55 a.u.) had low levels and 1-hexadecene (0.85 a.u.) and perfluorodecalin (0.82 a.u.) the lowest levels of autofluorescence. This trend persisted with the 100 × objective, albeit with lower overall signals, as would be expected (data not shown). We eventually settled on the combination of 15% dextran + 5% BSA inside the cellulose capillary, with perfluorodecalin inside the planchette sandwich but outside the capillary. With this combination, the dextran+BSA provided good cryoprotection to large volumes of the worm trapped in the cellulose capillary, while the perfluorodecalin, which was not proximal to the worm but occupied the large void volume within the planchette sandwich, reduced background fluorescence to the point that small features such as individual nuclei deep in the bulk of the worm could be imaged at reasonable signal-to-noise ratios.
3. Rationale
FIB-SEM imaging of distinct but transient or rare cellular events, especially in large biological samples, is extremely inefficient unless executed in a spatially and temporally targeted manner. However, it is technically challenging to trap and record certain developmental or cell cycle stages in intact organisms such as C. elegans in such a way that the same features can be later located and imaged by FIB- SEM with minimal artifacts. Here we outline a workflow that includes high-pressure freezing (HPF) of intact worms, in situ cryo-fluorescence microscopic screening for specific embryonic intermediates, and then correlative room temperature FIB-SEM imaging of the corresponding volumes (Fig. 1). This workflow to trap (HPF), capture (cryo-LM) and image (FIB-SEM) may thus be used to obtain nanoscale volume EM reconstructions of fleeting intermediates in large biological samples.
4. Methods
In this section we provide step-by-step instructions and include comments and/or observations where pertinent.
4.1. High-pressure freezing of C. elegans hermaphrodites
Maintain C. elegans strain OCF5 (Golden, Liu, & Cohen-Fix, 2009) on E. coli OP50 agar plates at 20 °C using standard methods (Brenner, 1974). Move the worm plates to a station adjacent to the high-pressure freezer (EM-ICE, Leica microsystems Inc) and pick young adults with a few eggs.
Transfer selected worms to a large drop of 5% BSA+ 15% dextran and 5 mM levamisole in M9 medium for approximately 2 min. Levamisole is a muscle relaxant, which straightens the worms; the exclusion of this step complicates straightforward imaging and identification of cellular features (see below).
Draw individual worms into a cellulose capillary (Muller-Reichert et al., 2007; Rahman et al., 2020), and crimp and seal the section of capillary containing the suspended worm using a blunted scalpel, forming a 2- or 3-mm pouch.
Cut the pouch with the trapped worm from the rest of the tube using the sharp edge of the scalpel and quickly transfer to the planchette filled with a cryoprotectant filler (perfluorodecalin) in the loading slot of the HPF instrument. In our hands, preliminary experiments suggested that the combination of a diffuse signal from SP12::GFP (labeling ER) and a localized H2B::mCherry signal (labeling DNA) permitted easier identification of embryos inside a worm as well as an approximate enumeration of cells inside the embryos.
Freeze worm samples between type-A and type-B 3 mm gold-coated planchettes. Use a black marker to mark a spot on the 200 μm recess of a type-A planchette. This facilitates subsequent identification of the sample carrier under LN2 during sample loading into a planchette cassette.
Coat a type-B planchette (300 μm recess/flat) with lecithin for ease of removal by immersing it in a 2% lecithin solution (dissolved in chloroform) for 90 s, blotting off the excess solution with filter paper, and drying it in a glass petri dish.
Using tweezers, transfer the worm in the sealed cellulose capillary pouch to the 100 μm-cavity of the type-A planchette. The recess should be pre-filled with ∼0.8 μL of perfluorodecalin, which exhibits a low background fluorescence (see Section 2.2) and will later ease visualization of the worm during cryofluorescence microscopy (see Section 4.2.2).
Place the type-B planchette on top of this planchette, with its flat side facing downward, forming a sandwich. This planchette orientation reduces sample volume and improved HPF performance as well as cryo-LM images in our hands. Quickly place this sandwich in the HPF cartridge set pre-assembled on a Leica EM-ICE according to the manufacturer’s instructions, and actuate a high-pressure freeze cycle.
In the EM-ICE, frozen samples will be automatically deposited in a transfer dewar in the HPF instrument at the end of each HPF cycle, and at the end of a run of no more than eight cycles, quickly transfer these samples to a sample unloading station filled with LN2.
Punch out the planchettes from the middle plate of the HPF cartridge setup with a 3 mm punch into the LN2 bath. Typically, the two halves of the sandwich should fall apart, if not, they can be teased apart manually with pre-cooled tweezers before imaging. The black spot marking the 200 μm recessed side of a type-A planchette should be easily visible under bright lights. The opposite side 100 μm recess contains the pouch with the worm.
Move the type-A planchettes into pre-labeled cryovials under LN2. Keep worms from different strains or experimental conditions separate! Up to three vials can be transferred to a 50 mL conical tube under LN2, which itself can be moved to a large sample storage LN2 dewar until use. Both the cryovials and 50 mL conical tubes should have holes drilled in the side walls and lids to allow complete filling of the containers with LN2 and prevent catastrophic pressure buildup.
4.2. Cryo-fluorescence microscopy
4.2.1. Preparation of the cryostage and sample loading
Cryo-fluorescence microscopy was performed using a Zeiss LSM710 with a Zeiss Airyscan detector, with the standard stage exchanged for a Linkam cryostage and support (Fig. 2A and B). In our hands, it was preferable to mount the stage support, but not the stage itself, at this time.
Power up the cryostage for an hour in advance of an imaging session to warm up the stage and remove moisture. Similarly, pre-bake accessories such as tweezers and an external LN2 dewar on a hot plate set at 40 °C or thoroughly dry with a hairdryer. Use fresh dry LN2 for all experiments.
Retrieve high-pressure frozen samples in cryovials and transport them to a Leica HPF unloading station the microscope using a portable LN2 dewar. At the unloading station discard planchettes without the marked black dot if not done previously already.
Cool down the cryostage by slowly pouring LN2 into the built-in LN2 dewar, and close the stage lid. When the “Fill LN2” icon appears on the cryostage display panel (Fig. 2B–D) it may be activated to enable filling of the sample chamber with LN2 from the built-in dewar either manually or automatically.
Cool down the pre-warmed planchette “puck” holding the planchette “cassette” in its slot in the cryostage, and quickly decant sample planchettes into the cryostage well. Unlock the puck and insert one to three 3 mm-planchettes into the cassette using the provided magnetic tool (magnet on one end of a slender metal rod, screwdriver on the other; Fig. 2C–E). We typically filled each cassette with duplicates or triplicates to increase chances of success and speed of screening.
Again, using the magnetic tool, lock the puck—this clamps the planchettes into the cassette—and transfer and magnetically secure the cassette to the “bridge” on the cryostage. The bridge is a flat metallic plate with a cut-out in the optical path that can be moved laterally via electronic controls to allow imaging of up to three planchettes in a single imaging session (Fig. 2F). The LN2 level in the well of the cryostage is maintained with an electronic sensor such that the bridge is in contact with but not flooded by the cryogen.
Close the cryostage well with its heated lid and plug to reduce the risk of condensation on the exposed planchette surfaces. At this point the cryostage may be mounted on the stage support, and the external LN2 dewar and the sample stage position controller connected to the cryostage (Fig. 2B). A lowering of LN2 levels in the well will automatically trigger a refill from the external dewar; in our hands, this took a variable amount of time, approximately 10–15 min. The injection of fresh LN2 may cause a perturbation in the field of view, so we suggest pre-emptively executing a “Fill LN2” and allowing few seconds for settling before acquiring high resolution images.
4.2.2. Imaging of samples
A significant difference between cryo-correlative imaging of thin samples on a TEM grid and these experiments is that high-pressure frozen samples in planchettes completely occlude the transmitted light path. Hence low-resolution imaging for initial identification of regions of interest (ROIs) in the 3 mm-field of view (FOV) relies on low level background fluorescence signals especially from the cellulose capillary at λex 561 nm (Fig. 3A–D).
FIG. 3.

Airyscan cryo-fluorescence images of OCF5 C. elegans worms. (A) Airyscan cryo-fluorescence image of young OCF5 strain adult worm (H2B::mCherry marking DNA, red; SP12::GFP, marking ER, green) enclosed in cellulose tubing (inside dotted lines) and high-pressure frozen between planchettes. Cryoprotectants inside and outside of the tubing were 15% dextran + 5% BSA and perfluorodecalin respectively. (B) Higher magnification image of dotted area in A showing embryonic cells near the vulva (arrowhead) trapped in metaphase (*) and telophase (#), with a bright H2B signal (arrow) proximal to the latter. (C) Example of late-stage embryos (red) and spermatheca (arrow) captured in a separate dataset. (D) Worm imaged in 10% glycerol, showing signs of loss of structural integrity of the cuticle (inset) possibly due to osmotic imbalances. (E) Worm imaged without muscle relaxant and affixed on a flat side of a planchette with sticky carbon tape, showing significant ice contamination and debris (double arrowheads). Scale bars: A, D, E: 100 μm, B: 10 μm, C: 50 μm.
Move the worms trapped inside capillaries in planchettes into the field of view, identify them using a 10 × or 100 × long working distance (LD) objectives, and acquire overview images of the entire young adult worms with the 10 × objective (Fig. 3A–D). The SP12::GFP signal will mark the bulk of the worms, with significantly lower expression in the developing embryos. Paired with the characteristic staining pattern of nuclei (DNA) by H2B::mCherry, regions containing the embryos at different stages of cell division cycle should be readily recognized (Fig. 3B and C). Typical acquisition parameters we used were: laser power = 3–4 (λex 488 nm) and 4–5 (λex 561 nm); gain = 650–800 (GFP) and 700–900 (mCherry), bit depth = 8 bit; image direction = double; image speed 5; line average = 4; size = 1024 × 1024 pixels.
Next, use the 100 × objective to examine specific embryos within the worm. In this study, we examined the embryos proximal to the vulva, a structure readily visualized in these organisms (Fig. 3B) and useful for downstream identification of ROIs. We suggest the reader images embryonic cells briefly during the location step, even though in our hands subsequent experiments suggest that there was little damage from local heating effects in the imaged volume even with slow and deliberate fluorescence imaging, at least at the resolutions afforded by FIB-SEM imaging (see below).
Load an Airyscan imaging template into ZEN Black, and align the Airyscan detector in the “Continuous” scan mode in the GFP channel on an adjacent area with abundant structural features. A “sacrificial” first planchette with a worm may be used for this step, as well as for evaluating freezing and transfer.
Move the ROI into the field of view, and acquire images with the 100 × objective; the following parameters were typically used: laser power = 5.5–7 (λex 488 nm) and 6.5–7.5 (λex 561 nm); gain 700–900 (GFP) and 800–1050 (mCherry); bit depth 8 bit; image direction double; image speed = 5; line average = 4; image size 1024 × 1024 pixels.
Compared to most adult tissues in this worm strain, the embryos exhibited very weak GFP fluorescence; if this is the case, saturate the GPF signal to enable discernment of the individual embryos. Different stages of the cell division cycle in a multi-cell embryo can be then distinguished by the position and compaction of H2B::mCherry signal (Fig. 3B and C).
Here we show one such cell, where metaphase was easily detected by a characteristic “linear” fluorescence pattern from the chromosomes, condensed and aligned at the metaphase plate (Fig. 3B); immediately adjacent to this cell with the embryo was an intriguing H2B::mCherry staining pattern of three adjacent puncta. As our overall study focused on cell division intermediates that captured nuclear envelope break-down events, this area was imaged and selected for correlative FIB-SEM imaging later. In other samples, late-stage embryos and spermatheca could be easily identified with this approach (Fig. 3C). We note that if there are features of interest present in reasonably well-separated regions even in a single worm, this workflow is amenable to multiple FIB-SEM image acquisition runs as long as the direction of milling and imaging is chosen carefully. We include an example of morphological perturbations induced by the cryoprotectant (Fig. 3D), where HPF of the worm in the presence of 10% glycerol resulted in a “ribbed” appearance, possibly due to shrinkage of the cuticle (Fig. 3D, inset). In initial experiments, we had not included a muscle relaxant, causing the worm to twist and leading to poorer results. Similarly, we attempted to circumvent the challenges arising from autofluorescence by sticking the cellulose tubing to the flat side of a planchette sandwich using carbon tape, and this in fact worsened the degree of contamination observed (Fig. 3E). On rare occasions, problems with either the freezing or with the cryostage caused catastrophic warming and refreezing, destroying the sample in the process (data not shown).
4.1.1. Recovery of samples
After cryo-fluorescence microscopy imaging, unmount the cryostage from the stage support, remove the lid, and using the magnetic rod, return the planchette cassette to the cassette holder puck in the chamber well.
Set the puck to the “open” position in order to remove the sample planchettes. Fill an open cryovial with LN2, place it horizontally in the sample chamber, and transfer the sample planchettes into the cryovial with cooled insulated tweezers. Work fast to avoid frost falling on the samples during this manual step, and only planchettes with useful imaged features need be transferred for subsequent processing.
Cap the tube, place in a 50 mL conical tube and store in LN2 until freeze substitution can be executed.
4.3. Freeze substitution and resin embedding
High-pressure frozen samples were freeze substituted using the quick free substitution (QFS) method (McDonald & Webb, 2011) with a few modifications that allow completion in about 5 h.
Dissolve the contents of three ampules, each with 0.1 g of OsO4 granules, in 13.5 mL of acetone in a glass flask using a magnetic stirrer.
Add 0.75 mL of 2% uranyl acetate in methanol and 0.75 mL of double deionized (DD) water to the OsO4 solution, and after thorough mixing, pipette the QFS cocktail into 8–9 cryovials, about 1 mL per cryovial.
Place a metal heating block with 13 mm holes in an ice bucket, and cool it down by filling the bucket with LN2. Insert the tubes containing the QFS cocktail into the holes of the metal block to freeze the cocktail.
Place individual planchettes of imaged HPF C. elegans worms in each cryovial to avoid mixing of samples. Decant liquid nitrogen from the ice bucket and quickly transfer the metal block to a second ice bucket containing about 5 cm of dry ice, with the metal block rotated 90 degrees so that the cryovials lie horizontally.
Fill this ice bucket with dry ice so the metal block is completely submerged, cover the bucket with a lid, and place it on a rotary shaker (60 cycles/min) for 3 h inside a chemical hood.
At the end of this period, remove the dry ice and continue shaking for another hour before removing the lid. Continue shaking until the metal block reaches room temperature, typically about 1 h more, or as determined by an infrared thermometer (McDonald & Webb, 2011; Muller-Reichert, Hohenberg, O’Toole, & McDonald, 2003).
Afterwards, wash sample planchettes carefully, thrice for 10 min with 100% acetone at room temperature inside the hood so as to not dislodge the samples, and then subject the samples to one-hour graded resin infiltration steps with 1:2, 1:1, and 2:1 (v/v) resin: acetone ratios. We used Polybed 812 resin, medium hardness: 14.6 g Polybed, 8.4 g DDSA, 7.0 g NMA and 0.42 mL DMP-30.
Transfer planchettes into 100% resin and incubate overnight at room temperature. The cellulose capillary pouches containing worms may become dislodged during high-pressure freezing, specimen transfers or washing steps; most times they can be located and recovered. We attempted placing a small cut-out of double-sided sticky carbon tape inside the planchettes, however, this did not significantly increase the success rate of the experiments (Fig. 3E). Further, the black tape obscured the worm during LM inspection (below), so this approach was abandoned.
Following the overnight resin infiltration, make fresh Polybed 812 resin and exchange the samples into the new resin and incubate for 5–6 h.
Embed the planchettes in Beem capsules. Cut off the bottom of a capsule using a razor blade and attach a planchette to the inner side of the flattened lid, sample side up, with a small drop of resin. Close the lid, and gently fill the upturned Beem capsule (cut open bottom now facing up) with resin.
Cure the samples in an oven at 75 °C for about 2 days.
4.4. Light microscopy of resin-embedded worms
Punch out the resin blocks from the Beem capsules and remove excess resin around the planchettes using a jeweler saw, razor blade and scalpel.
Remove the planchette by repeated immersion into LN2 followed by heating with a heat gun, revealing the resin-embedded sample underneath, and then trim the sample block, which should be shaped like a round mesa, into a rectangle and the surface smoothened using a microtome.
-
To align with previously acquired cryo-fluorescence image (Fig. 4A, rotated for ease of visualization), acquire a low magnification overview (Fig. 4B); this should roughly correspond to the cryo-LM image.
Unlike mechanical serial sectioning approaches, FIB-SEM milling is orthogonal to the sample surface and cannot mill beyond tens of μm depth without accruing significant artifacts (Kizilyaprak, Longo, Daraspe, & Humbel, 2015; Xu et al., 2017). Further, milling a trench “blind” with the ion beam risks milling into the features of interest. Therefore, for this correlative approach, it was important to section the block until the cellulose tubing and enclosed worm were exposed at a glancing angle. The use of the muscle relaxant levamisole and cellulose capillary together are thus critical in limiting the degrees of freedom of the worm within the resin block.
Using a microtome, section away material in 1 μm increments (cutting speed: 0.2 mm/s), and for every 5 or 10 μm advancement collect two 200 nm sections on a glass slide and stain them with toluidine blue to check for presence of biological material. Repeat until the embryos are just exposed on the surface of the sample block.
Inspect the corresponding toluidine blue-stained sections under a dissecting microscope to assess the preservation quality of the sample. Obviously damaged samples can be detected and discarded at this point, saving time and resources downstream. A worm prepared using 15% dextran +5% BSA as the inner cryoprotectant and perfluorodecalin as the outer cryoprotectant with well-preserved internal structures at the LM level is shown in Fig. 4C.
FIG. 4.

ROI identification by correlation of features in cryo-LM and SEM images. (A) cryo-LM image of OCF5 worm, rotated to align with (B, C), below. The vulva is easily identified (A, arrowhead, also Fig.3B, top right). (B) Snapshot of freeze-substituted and resin-embedded worm.(C) Transmitted light microscopic image of the final semi-thin section of the worm stained with toluidine blue before SEM and FIB-SEM imaging. (D) SEM image of top surface of remaining resin block, revealing exposed worm and feature corresponding to vulva (arrow).(E) Higher magnification of worm, boxed area and thick arrow indicate area of protective Pt pad and FIB-SEM acquisition. (F) Polished “cliff face” of protected ROI. The appearance of a void (arrowhead) indicates a soon-to-be-revealed embryo. (G) FIB-SEM image section through targeted metaphase (*) and telophase nuclei (#). The plane of the images (F) and (G) is orthogonal to (A)–(E). Scale bars: (A, D): 100 μm, (E): 50 μm, (F, G): 10 μm.
4.5. FIB-SEM sample preparation and imaging
4.5.1. Sample preparation
Cut down the sample block to a roughly 2 mm height using a jeweler saw. Place the trimmed block on a large piece of filter paper, wash 3 times with water followed by three times with 95% ethanol, and then transfer to a 50 mL conical tube with 5 mL of double-distilled (DD) water and ultrasonicate for 4 min.
Replace double-distilled water and repeat this process thrice, followed by a 2 min rinse in 95% ethanol and a subsequent final wash in DD water. Place the sample block on filter paper, dry with compressed air, and examine under a dissection microscope to verify cleanliness of the sample surface.
Mount the sample on a SEM stub using super glue and allow to dry for 30 min. Paint the stub and sample with colloidal silver to cover the entire block except the top face of the raised mesa containing the embryo, and then dry overnight in a fume hood.
Prior to imaging, coat the sample with 10 nm of carbon in a Leica ACE600 carbon coater.
4.5.2. FIB-SEM data acquisition
Transfer the carbon coated sample, with the worm exposed in cross section, to a Zeiss Crossbeam 550 FIB-SEM to obtain a “reference overview” SEM image. The heavy metal-stained cellular structures within the worm will give a strong mass-to-charge (m/z) signal compared to the low atomic number carbon coating and surrounding resin. An image of the entire worm can therefore be recorded at the back-scatter electron detector; we used the ATLAS 5 (Fibics Inc) module in the Zeiss microscope control software.
Acquire a single large ROI (e.g., 16,000 × 16,000 pixel) image at a reasonable pixel sampling, say 50 nm, with the SEM beam operated at approximately 2.3 kV and 1.2 nA (Fig. 4D and E). In an ideal experiment, the worm should be perfectly straight and flat against the surface plane; in the case we show here, the worm is slightly canted, meaning that the SEM image aligned well but incompletely with the low magnification image of the embedded worm.
Nevertheless, enough biological features (here, the characteristic bulge in the cuticle indicative of the vulva, Fig. 4D and E) should be identified such that ROIs for subsequent FIB-SEM imaging can be ascertained with some confidence. It is still possible to screen worms for ultrastructural preservation at this stage in case freezing artifacts are discovered. As these workflows and hardware components become more robust, we expect that there will be fewer to no areas of poor freezing within the physical limits of HPF.
At this point, several ROIs may be chosen for FIB-SEM image acquisition (see below). Tilt the sample to 54° and switch control to ATLAS 3D (Fibics Inc) module for the rest of the sample preparation steps, as previously detailed (Narayan et al., 2014).
Deposit a layer of platinum 1.5 μm thick over this site with the FIB operated at 30 kV and 1.5 nA current, and mill 3D tracking/auto focus lines into the pad surface using the 50 pA aperture followed by a final 1 μm deposition of carbon over the whole pad at 1 nA.
Mill a coarse trench (FIB operated at 30 nA) followed by fine trench (3 nA) with depth set to 40 μm; in our case here, the ultrastructure on the sample surface indicates that the worm was sectioned approximately half-way through the ∼80 μm diameter of a young adult animal, so this depth was appropriate. Continue milling the fine trench until the chitinous layer of the targeted embryo is revealed on the cross section face (Fig. 4F).
At this point initiate image acquisition, with the FIB operated at 1.5 nA and SEM operated at 1.5 kV and 1 nA. Pixel sampling may be chosen as 5 nm in the imaging plane and 15 nm in the axial (FIB step size) plane. At a 3 μs dwell time and ∼ 10 nm/min FIB advancement rate, there should be minimal “wash” artifacts, namely, cyclical and cascading contrast variations in the imaging plane in an image stack.
Collect image datasets corresponding to ROIs identified from the cryo-fluorescence images, with dimensions approximately 50 × 30 × 25 μm. During image acquisition, you should be able to observe satisfactory structural preservation in the worm throughout the multi-cell embryo; here we show an early-stage embryo also captured at high resolutions (Fig. 4G; Supplementary Movie M1).
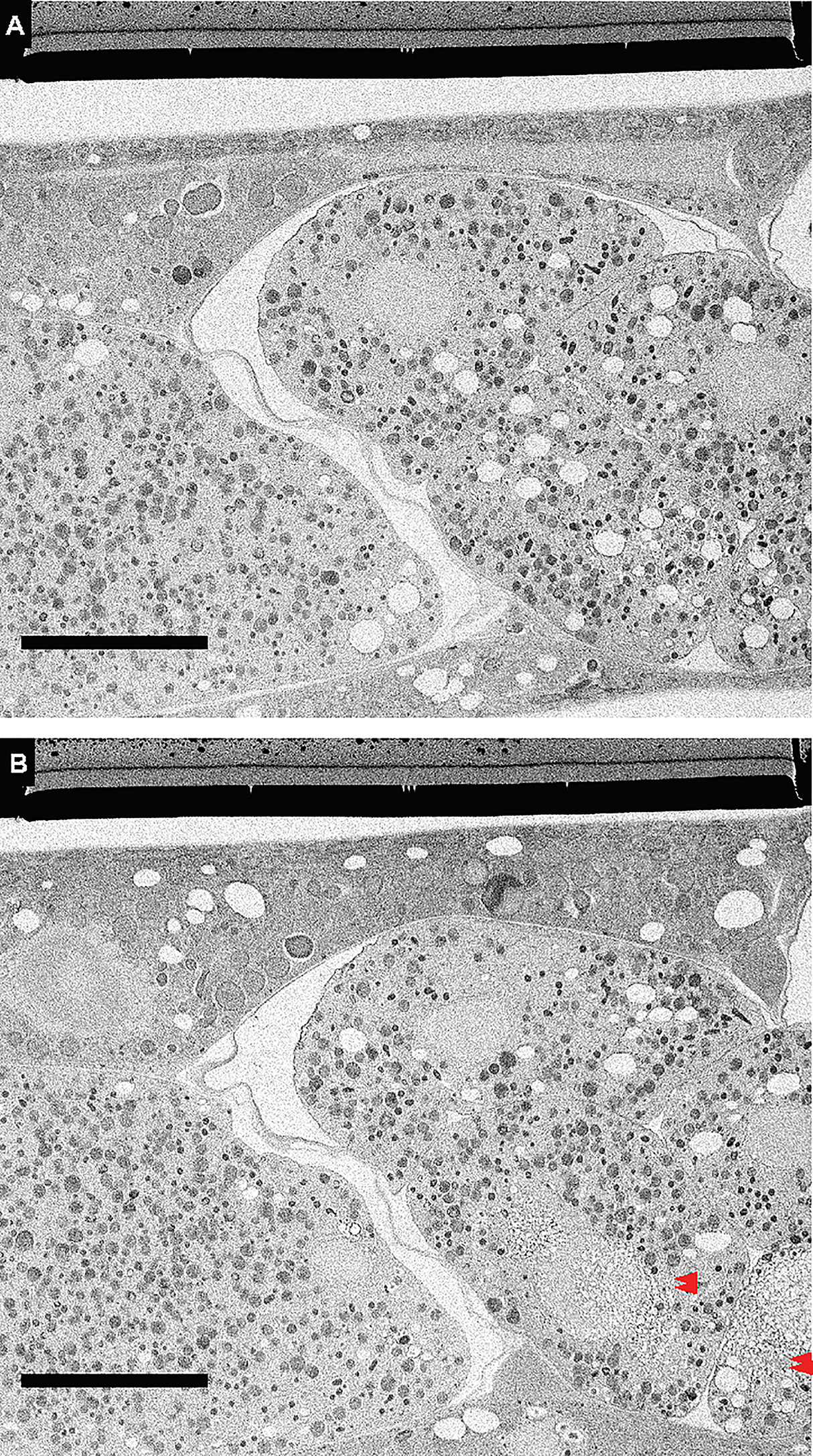
Importantly, thorough cryo-fluorescence imaging of these regions with reasonably high-powered lasers did not appear to cause structural perturbations, at least at these imaging resolutions. However, in a different run, some areas but not others in early-stage embryo far away from the planchette metal surface showed some possible damage (Supplementary Fig. S2 in the online version at https://doi.org/10.1016/bs.mcb.2020.09.009). Interestingly, immediately adjacent non-embryonic cells at the same depth that were also imaged by cryo-LM did not show the artifact, suggesting a cumulative and negative impact of worm thickness, distance from planchette metal surface and chitinous layer on good freezing. Certainly, improper handling and deviations from cryogenic temperatures could also be an issue; a newer design of the cryo holder promises to be more robust, but testing this was outside the scope of this paper.
4.6. Image processing and analysis
Process the .tiff image stack acquired using the ATLAS3D acquisition module as required; we typically crop, export, register, contrast invert and bin (Narayan et al., 2014) the images to yield an isotropic 15 nm voxel .mrc image volume (Fig. 5A).
Render this information-rich image volume in color to reveal a detailed picture of the targeted embryo (Fig. 5B, Supplementary Movie M2), further rendering, segmentation and visualization may be done according to the question asked, using available open-source or proprietary software.
FIG. 5.

FIB-SEM imaging and reconstruction of correlated mitotic intermediates in the C. elegans embryo. (A) Isotropic FIB-SEM image volume and (B) volume rendering of the region in Fig.4E, encompassing nearly the entire targeted embryo. (C, D) Parallel slices from the image volume, resliced and positioned to match the thicker “xy” cryo-LM imaging plane (E); two nuclei in telophase (#), a neighboring cell in metaphase (*) and a putative autophagosome enriched in fluorescently tagged histone protein H2B (arrow) are revealed. Also shown, L to R, xy, xz and yz cross sections through autophagosome. (F) Segmentation and 3-D reconstruction of these features, showing an identical pattern to the fluorescence signal in (E). Scale bars: (B)–(F): 10 μm, (D) bottom panel: 1 μm.
4.7. Observations and results
Here, we detail our process and describe the cellular discoveries made possible by this methodological advance. The reconstructed image volume was visually examined in IMOD; the putative metaphase fluorescence signal was easily identified as a bona fide cell trapped in metaphase at the moment of HPF (Fig. 5C and D, *). Immediately to the left of this area, corresponding to the punctate H2B::mCherry signal, was a neighboring cell in telophase (Fig. 5C and D, #). This accounted for two of the intriguing three spots captured by cryo-LM. The image volume was resliced in a plane orthogonal to FIB-SEM image acquisition, i.e., parallel to cryo-LM image acquisition. The axial resolution of the FIB-SEM image volume was significantly greater than that of the cryo-LM image, especially given the wide pinhole, so the volume corresponding to the LM “image slab” was examined and a spherical and dark staining compartment ∼1 μm in diameter and consistent with an autophagosome was observed (Fig. 5D, arrow and panel). The colocalization with the bright H2B::mCherry signal suggest that this autophagosome could contain the remnants of the second polar body in a developing embryo; indeed, these data provide direct ultrastructural evidence of the clearance of the second polar body at an embryonic stage similar to that reported in a recent fluorescence microscopy study (Fazeli, Stetter, Lisack, & Wehman, 2018). The physical gap between the image planes in Fig. 5C and D is 600 nm, within the z resolution of the cryo-LM set up, and a simple overlay of the three features corresponding to the metaphase and telophase chromosomes leaves only this structure as a possibility (Fig. 5C–F). Amira 6.5.0 (ThermoFisher Inc) was used segment cellular features of interest from the .mrc file. We used a general scheme for segmentation that we used previously to segment nuclear structures in C. elegans embryos imaged ex vivo (Rahman et al., 2020). Briefly, we utilized a threshold-based selection of lipid membranes or chromosomes. Label maps were reanalyzed by slice-by-slice visual inspection of the xy, yz and xz image planes separately; sporadic gaps added to the primary segmented volume and occasional irrelevant cellular were removed manually. For the purposes of this methods paper, and to prove the successful correlation of the features of interest, we rendered in 3-D the chromosomes and autophagosome corresponding to the bright fluorescent structures captured by cryo-fluorescence microscopy (Fig. 5F, Supplementary Movie M3) in the online version at https://doi.org/10.1016/bs.mcb.2020.09.009).
5. Instrumentation and materials
5.1. High-pressure freezing of C. elegans worms
5.1.1. Cryoprotectants and worm strains
Instrumentation:
Stereo dissection microscope with transmitted light; worm pick; EM-ICE (Leica, Austria); Zeiss Examiner.Z1 LSM710 microscope (Zeiss, Germany) with 10 × objective (EC Plan-NEOFLUAR 10 × /0.3, ∞/–; #440330–9901) and 100 × objective (LD EC Plan-NEOFLUAR 100 × /0.75 DIC, ∞/0; #422492–9900). Zen Black microscope software (Zeiss, Germany); Microsoft Excel software.
Materials:
Transgenic labeled C. elegans strain OCF5 (SP12::GFP; histone2B:: mCherry); glass slides; 10 μL and 200 μL pipettes; 1–10 μL and 20–200 μL pipette tips; cellulose tube (#16706869; Leica, Austria); crimping tool (blunted scalpel Bard-Parker #371620, Thermo-Fisher Scientific, USA); sharp-point tweezers (#72919-OA, Electron Microscopy Sciences, Hatfield, PA); type-A gold-coated copper planchette (#16770152; Leica, Austria); type-B gold-coated copper plan- chette (#16770153; Leica, Austria); Nalgene cryo tubes (#5000–1012; Thermo Scientific, USA).
Reagents:
BSA, albumin fraction V (#A3294, Sigma, USA); M9 buffer (#11006–517; IPM Scientific, USA); 1-hexadecene (#H2131–100ML; Sigma, USA); perfluorodecalin (#P9900–25G; Sigma, USA); sucrose (#84097–250G; Sigma, USA); glycerol (#G9012–100ML; Sigma, USA); dextran (Ave. MW 9000–11,000 kDa, #D9260–10G; Sigma, USA); polyvinylpyrrolidone (#PVP10–100G; Sigma, USA); Baker’s yeast; dry liquid nitrogen (LN2).
5.1.2. Preparation and freezing of worms
Instrumentation:
Stereo dissection microscope with transmitted light (Nikon SM2765); worm pick (#59-AWP, Genesee Scientific, El Cajon, CA); High-pressure freezer EM-ICE (Leica, Austria); cartridge assembly (#16771833 and #16771849, Leica, Austria); unloading station with 3 mm punch (Leica, Austria); portable LN2 dewar (#5028944, Thomas Scientific, NJ); sample storage LN2 dewar (#HC34; Taylor-Wharton, USA).
Materials:
Transgenic C. elegans strain OCF5 (see Section 4.1); NGM-agar plates (6 cm in diameter) seeded with E. coli strain OP50 (ATCC, Manassas, VA); petri dish 60 mm (Falcon #353002, Corning, NC); superfrost 1 mm thick glass slides (Daigger Scientific Inc., Hamilton, NJ); cellulose tube (#16706869; Leica, USA); crimping tool blunted scalpel (Bard-Parker #37160, Thermo-Fisher Scientific, USA); sharp-point tweezers (#72919-OA, Electron Microscopy Sciences, Hatfield, PA); type-A gold-coated copper planchette (#16770152; Leica, Austria); type-B gold-coated copper planchette (#16770153; Leica, Austria); Nalgene cryo-vial (#5000–1012; Thermo Scientific, USA); ice pack.
Reagents:
Levamisole (#L-025, Sigma, USA); M9 buffer (#11006–517; IPM Scientific, USA); BSA, albumin fraction V (#A3294, Sigma, USA); perfluorodecalin (#P9900–25G; Sigma, USA); sucrose (#84097–250G; Sigma, USA); glycerol (#G9012–100ML; Sigma, USA); dextran (Ave. MW = 9,000–11,000 kDa, #D9260–10G; Sigma, USA); dry LN2.
5.2. Cryo super-resolution fluorescence microscopy
5.2.1. Preparation of cryostage and sample loading
Instrumentation:
Zeiss Examiner.Z1 LSM 710 with 10× and 100× objectives (see Section 5.1; Zeiss, Germany); Linkam CMS 196 cryo stage and associated accessories (Linkam, UK); cryo stage support (Linkam, UK); planchette cassette for 3 mm planchettes (Linkam, UK); planchette cassette holder (Linkam, UK); magnetic rod and plug (part of cryo stage accessories; Linkam, UK); external LN2 dewar (Linkam, UK); slide warmer (#XH-2002; Premiere, USA); hairdryer; unloading station with 3 mm punch (Leica, Austria).
Materials:
Forceps insulated with PVC (#16701955; Leica, Austria); portable LN2 dewar (#5028944, Thomas Scientific, NJ).
Reagents:
Dry liquid nitrogen.
5.2.2. Imaging of samples
Instrumentation:
Zeiss Examiner.Z1 LSM710 with 10× and 100× objectives (see Section 5.1; Zeiss, Germany); Zeiss Airyscan module (Zeiss, Germany).
5.2.3. Recovery of samples
Materials:
Forceps insulated with PVC (#16701955; Leica, Austria); Nalgene cryo tubes (#5000–1012; Thermo Scientific, USA); 50 mL conical tubes.
Reagents:
Dry liquid nitrogen.
5.3. Freeze substitution and resin embedding
Instrumentation:
Stirring plate (SP195025, Thermo-Fisher Scientific, USA); standard metal heating block (VWR Scientific, USA); rotary shaker (#3500 VWR Scientific, USA); infrared thermometer (IRT207; General Tools & Instruments, USA); isotemp oven (#637F, Thermo-Fisher Scientific, USA).
Materials:
Glass flasks, 250 mL (Corning, USA); cryovials (#34506; EMS, USA); tweezers (#78080-CP, EMS, USA); insulated tweezers (#78460–702A, EMS, USA) (use with LN2; Leica, Austria); Wheaton type I glass vials for preparing diluted resin solutions (Wheaton, San Diego, CA); Permanox petri dishes 6 cm (Nalge Nunc International, USA); plastic cups for weighing resin (#13915–089, VWR, USA); desiccator connected to mechanical pump; razor blades (#55411–050, VWR, USA); Beem capsules (#70000-B, VWR, USA); and Beem capsule holder (#69916–01, EMS, USA).
Reagents:
OsO4 granules (#19134; EMS, USA); acetone (#9011; Thermo Scientific, USA); uranyl acetate (#22400; EMS, USA); methanol (#3016; Mallinckrodt, USA); double deionized water; dry LN2; dry ice; Poly/Bed 812 embedding kit/DMP-30 (#08792–1; Polysciences, USA).
5.4. Light microscopy of resin-embedded worms
Instrumentation:
Beem capsule sample punch; jeweler saw; razor blades; pointed tip scalpel #11 (Bard-Parker #371611, Thermo-Fisher Scientific, USA); styrofoam box for LN2; heat gun (#HG 201; Master Appliance Corp., USA); Leica Ultracut ultramicrotome (Leica, Austria); loop to pick up sample sections; hot plate (#HP-A1915B; Thermolyne, USA); Laxco stereomicroscope (Laxco, USA) with SeBa-Cam5C digital camera (Laxco, USA).
Materials:
Superfrost 1 mm thick glass slides (Daigger Scientific Inc., Hamilton, NJ).
Reagents:
Dry liquid nitrogen; toluidine blue (#T-161; Fisher Scientific, USA); double deionized water and nitrogen gas (both in-house supply).
5.5. FIB-SEM sample preparation and imaging
5.5.1. Sample preparation
Instrumentation:
Stationary sample clamp; jeweler saw; Rapidograph ultrasonic cleaner (#3069 USC2; Laboratory Supplies Company, USA); dissection microscope (Nikon SM2765); chemical hood; Leica ACE600 coater (Leica, Austria).
Materials:
Whatman filter paper (#1001–240; GE Healthcare, USA); compressed air cans (#75780–350; VWR, USA); laboratory tweezers (#82027–386 VWR, USA); conical tube 50 mL (Falcon 352098, VWR, USA); super glue; SEM stub (#16111–9; Ted Pella, USA).
Reagents:
Double deionized water (in-house supply); 95% ethanol (#111000190; Pharmco, USA); colloidal silver (#12630; EMS, USA).
5.5.2. FIB-SEM data acquisition
Instrumentation:
450 Gemini 2 scanning electron microscope (Carl Zeiss, Germany); 550 FIB-SEM (Carl Zeiss, Germany); ACE600 coater (Leica, Austria); Atlas 5 software package (Fibics, Canada).
5.6. Image analysis
Instrumentation/Software:
Amira 6.5.0 (release 2018-03-07; Thermo-Fisher Scientific, USA) software with XImagePAQ, XMesh, and XSkeleton extension packages, 3dmod version 4.9.13 (University of Colorado) and ImageJ version 1.52q (National Institutes of Health, USA).
6. Discussion
Here we have described a workflow that enables the trapping, imaging and screening of desired events in an intact high-pressure frozen model organism by cryo-fluorescence microscopy, followed by targeted FIB-SEM imaging and reconstruction of their corresponding 3-D architectures. With this approach, we show that it is possible to target transient features for volume EM (here, the condensed and aligned chromosomes in metaphase), but also pursue unexpected events for further elucidation. We imaged three bright H2B puncta by cryo-LM, of which two were revealed to be condensed chromosomes in telophase. Based on these data, it appears that the third localized signal can be ascribed to excess H2B is packaged for destruction via inclusion into an autophagic vacuole. H2B monoubiquitination has been linked with upregulation of autophagy (Chen et al., 2017). Our reconstructions are consistent with the autophagy of the second polar body in the developing embryo, provide compelling ultrastructural evidence for observations made in a recent study using light microscopy (Fazeli et al., 2018).
From a methodological perspective, while cryo-CLEM with cryo-FIB to thin down samples has been demonstrated recently (Gorelick et al., 2019), imaging thick samples in 3-D presents unique challenges. In our hands, a combination of prioritizing good cryoprotection in the immediate vicinity of the sample but low auto-fluorescence in the rest of the volume strikes a balance, allowing for adequate screening of fluorescently labeled targets. We acknowledge and hope that there may be as-yet-unproven compounds that provide both cryoprotection and low auto- fluorescence and may be used by themselves, such as polyvinylpyrrolidone (Fabig et al., 2019). Alternatively, fluorophores with longer wavelength profiles may be used. We note that there may be limits to how much this workflow can be extrapolated to other organisms. For example, there are inherent limitations to volumes that can be adequately preserved by HPF. In our C. elegans samples, there were patches outside of the ROI and farthest away from the planchette surface during HPF that were not well cryopreserved (Supplementary Fig. S2 in the online version at https://doi.org/10.1016/bs.mcb.2020.09.009). We suspect that there will always be some unpredictability in freezing quality, therefore multiple repeats are advised. In addition, we were able to modestly constrain the orientation of intact worms in cellulose capillaries, but other organisms may be less conducive to such manipulation. This is a small but central factor for the success of a given experiment; not only does this reduce movements during sample preparation steps, allowing correct correlation later, but the orientation (“flatness” against the resin surface) of the sample is essential in ensuring tangential sectioning of the worm. This is turn allows for a precise definition of the FIB-SEM target volume and ensures that the ROIs are close to the surface, a requirement for artifact-free FIB-SEM imaging (Narayan & Subramaniam, 2015; Xu et al., 2017).
There is always some degree of error involved, and there are times when we milled through or partially missed our intended regions of interest (data not shown). Of course, our workflow can be adapted for other volume EM approaches such as SBF-SEM and array tomography (Mulcahy et al., 2018) which section much larger areas of the resin-embedded sample, in which case this is no longer a concern. A more painstaking but higher resolution approach would be to execute a targeted FIB lift-out followed by TEM tomography, which could certainly be done in special cases. Irrespective, here we show that it is possible to reconstruct and visualize in 3-D at nanoscale resolutions, targeted as well as unexpected structures captured in the intact worm by cryo-fluorescence microscopy and subsequently imaged by FIB- SEM. This opens new and exciting avenues for research in cell biology, where scientists can now interrogate any variety of events in vivo in small model organisms, as long as they can be fluorescently marked, and then generate nanoscale 3-D reconstructions of their corresponding architectures.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
We thank Alexandra Elli (Carl Zeiss Microscopy GmbH) and Kirk Czymmek (currently at Donald Danforth Plant Science Center) for collaborative support on instrumentation. We thank Alexandre Laquerre and Michael Phaneuf (Fibics Inc) for help with executing targeted FIB-SEM imaging. This project was funded in part with Federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E. Part of the project, including provision of hardware was funded via a CRADA executed between Carl Zeiss Microscopy GmbH and the National Cancer Institute. MMR and OCF were supported by and intramural grant from NIDDK # DK069012. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
References
- Akiva A, Nelkenbaum O, Schertel A, Yaniv K, Weiner S, & Addadi L (2019). Intercellular pathways from the vasculature to the forming bone in the zebrafish larval caudal fin: Possible role in bone formation. Journal of Structural Biology, 206(2), 139–148. 10.1016/j.jsb.2019.02.011. [DOI] [PubMed] [Google Scholar]
- Bock DD, Lee WC, Kerlin AM, Andermann ML, Hood G, Wetzel AW, et al. (2011). Network anatomy and in vivo physiology of visual cortical neurons. Nature, 471(7337), 177–182. 10.1038/nature09802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S (1973). The genetics of behaviour. British Medical Bulletin, 29(3), 269–271. 10.1093/oxfordjournals.bmb.a071019. [DOI] [PubMed] [Google Scholar]
- Brenner S (1974). The genetics of Caenorhabditis elegans. Genetics, 77(1), 71–94. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/4366476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggman KL, & Bock DD (2012). Volume electron microscopy for neuronal circuit reconstruction. Current Opinion in Neurobiology, 22(1), 154–161. 10.1016/j.conb.2011.10.022. [DOI] [PubMed] [Google Scholar]
- Chen S, Jing Y, Kang X, Yang L, Wang DL, Zhang W, et al. (2017). Histone H2B monoubiquitination is a critical epigenetic switch for the regulation of autophagy. Nucleic Acids Research, 45(3), 1144–1158. 10.1093/nar/gkw1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czymmek K, Sawant A, Goodman K, Pennington J, Pedersen P, Hoon M, et al. (2020). Imaging plant cells by high-pressure freezing and serial block-face scanning electron microscopy. Methods in Molecular Biology, 2177, 69–81. 10.1007/978-1-0716-0767-1_7. [DOI] [PubMed] [Google Scholar]
- Deirieh A, Chang IY, Casey B, Joester D, & Germaine JT (2019). Impact of drying and effective stresses on the pore space and microstructure of mudrocks. Journal of Geophysical Research: Solid Earth, 124(5), 4290–4304. [Google Scholar]
- Fabig G, Schwarz A, Striese C, Laue M, & Muller-Reichert T (2019). In situ analysis of male meiosis in C. elegans. Methods in Cell Biology, 152, 119–134. 10.1016/bs.mcb.2019.03.013. [DOI] [PubMed] [Google Scholar]
- Fazeli G, Stetter M, Lisack JN, & Wehman AM (2018). C. elegans Blastomeres clear the corpse of the second polar body by LC3-associated phagocytosis. Cell Reports, 23(7), 2070–2082. 10.1016/j.celrep.2018.04.043. [DOI] [PubMed] [Google Scholar]
- Fuest M, Schaffer M, Nocera GM, Galilea-Kleinsteuber RI, Messling J-E, Heymann M, et al. (2019). In situ microfluidic cryofixation for cryo focused ion beam milling and cryo electron tomography. Nature, 9, 19133–19142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giddings TH Jr., Meehl JB, Pearson CG, & Winey M (2010). Electron tomography and immuno-labeling of Tetrahymena thermophila basal bodies. Methods in Cell Biology, 96, 117–141. 10.1016/S0091-679X(10)96006-8. [DOI] [PubMed] [Google Scholar]
- Giepmans BN, Adams SR, Ellisman MH, & Tsien RY (2006). The fluorescent tool-box for assessing protein location and function. Science, 312(5771), 217–224. 10.1126/science.1124618. [DOI] [PubMed] [Google Scholar]
- Golden A, Liu J, & Cohen-Fix O (2009). Inactivation of the C. elegans lipin homolog leads to ER disorganization and to defects in the breakdown and reassembly of the nuclear envelope. Journal of Cell Science, 122(Pt. 12), 1970–1978. 10.1242/jcs.044743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelick S, Buckley G, Gervinskas G, Johnson TK, Handley A, Caggiano MP, et al. (2019). PIE-scope, integrated cryo-correlative light and FIB/SEM microscopy. eLife, 8, e45919. 10.7554/eLife.45919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmstaedter M, Briggman KL, Turaga SC, Jain V, Seung HS, & Denk W (2013). Connectomic reconstruction of the inner plexiform layer in the mouse retina. Nature, 500(7461), 168–174. 10.1038/nature12346. [DOI] [PubMed] [Google Scholar]
- Hoffman DP, Shtengel G, Xu CS, Campbell KR, Freeman M, Wang L, et al. (2020). Correlative three-dimensional super-resolution and block-face electron microscopy of whole vitreously frozen cells. Science, 367(6475). 10.1126/science.aaz5357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohenberg H, Mannweiler K, & Muller M (1994). High-pressure freezing of cell suspen- sions in cellulose capillary tubes. Journal of Microscopy, 175, 34–43. [DOI] [PubMed] [Google Scholar]
- Hsieh C, Schmelzer T, Kishchenko G, Wagenknecht T, & Marko M (2014). Practical workflow for cryo focused-ion-beam milling of tissues and cells for cryo-TEM tomo- graphy. Journal of Structural Biology, 185(1), 32–41. 10.1016/j.jsb.2013.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun S, Ke D, Debiec K, Zhao G, Meng X, Ambrose Z, et al. (2011). Direct visualization of HIV-1 with correlative live-cell microscopy and cryo-electron tomography. Structure, 19(11), 1573–1581. 10.1016/j.str.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasthuri N, Hayworth KJ, Berger DR, Schalek RL, Conchello JA, Knowles- Barley S, et al. (2015). Saturated reconstruction of a volume of neocortex. Cell, 162(3), 648–661. 10.1016/j.cell.2015.06.054. [DOI] [PubMed] [Google Scholar]
- Kizilyaprak C, Longo G, Daraspe J, & Humbel BM (2015). Investigation of resins suitable for the preparation of biological sample for 3-D electron microscopy. Journal of Structural Biology, 189(2), 135–146. 10.1016/j.jsb.2014.10.009. [DOI] [PubMed] [Google Scholar]
- Kukulski W, Schorb M, Welsch S, Picco A, Kaksonen M, & Briggs JA (2011). Correlated fluorescence and 3D electron microscopy with high sensitivity and spatial precision. The Journal of Cell Biology, 192(1), 111–119. 10.1083/jcb.201009037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Ji G, Chen X, Ding W, Sun L, Xu W, et al. (2017). Large scale three-dimensional reconstruction of an entire Caenorhabditis elegans larva using AutoCUTS-SEM. Journal of Structural Biology, 200(2), 87–96. 10.1016/j.jsb.2017.09.010. [DOI] [PubMed] [Google Scholar]
- Littlejohn GR, Mansfield JC, Christmas JT, Witterick E, Fricker MD, Grant MR, et al. (2014). An update: Improvements in imaging perfluorocarbon-mounted plant leaves with implications for studies of plant pathology, physiology, development and cell biol- ogy. Frontiers in Plant Science, 5, 140. 10.3389/fpls.2014.00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning L, & Richmond J (2015). High-pressure freeze and freeze substitution electron microscopy in C. elegans. Methods in Molecular Biology, 1327, 121–140. 10.1007/978-1-4939-2842-2_10. [DOI] [PubMed] [Google Scholar]
- McDonald K (1999). High-pressure freezing for preservation of high resolution fine structure and antigenicity for Immunolabeling. In Hajibagheri N (Ed.) Vol. 117, Methods in molecular biology: Electron microscopy methods and protocols (pp. 77–97). Totowa, NJ: Humana Press Inc. [DOI] [PubMed] [Google Scholar]
- McDonald K, Schwarz H, Muller-Reichert T, Webb R, Buser C, & Morphew M (2010). “Tips and tricks” for high-pressure freezing of model systems. Methods in Cell Biology, 96, 671–693. 10.1016/S0091-679X(10)96028-7. [DOI] [PubMed] [Google Scholar]
- McDonald KL, & Webb RI (2011). Freeze substitution in 3 hours or less. Journal of Microscopy, 243(3), 227–233. 10.1111/j.1365-2818.2011.03526.x. [DOI] [PubMed] [Google Scholar]
- Mulcahy B, Witvliet D, Holmyard D, Mitchell J, Chisholm AD, Meirovitch Y, et al. (2018). A pipeline for volume electron microscopy of the Caenorhabditis elegans nervous system. Front Neural Circuits, 12, 94. 10.3389/fncir.2018.00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller-Reichert T, Hohenberg H, O’Toole ET, & McDonald K (2003). Cryoimmobilization and three-dimensional visualization of C. elegans ultrastructure. Journal of Microscopy, 212(Pt. 1), 71–80. 10.1046/j.1365-2818.2003.01250.x. [DOI] [PubMed] [Google Scholar]
- Muller-Reichert T, Kiewisz R, & Redemann S (2018). Mitotic spindles revisited—new insights from 3D electron microscopy. Journal of Cell Science, 131(3). 10.1242/jcs.211383. [DOI] [PubMed] [Google Scholar]
- Muller-Reichert T, Srayko M, Hyman A, O’Toole ET, & McDonald K (2007). Correlative light and electron microscopy of early Caenorhabditis elegans embryos in mitosis. Methods in Cell Biology, 79, 101–119. 10.1016/S0091-679X(06)79004-5. [DOI] [PubMed] [Google Scholar]
- Muller-Reichert T, & Verkade P (2012). Introduction to correlative light and electron microscopy. Methods in Cell Biology, 111, xvii–xix. 10.1016/B978-0-12-416026-2.03001-6. [DOI] [PubMed] [Google Scholar]
- Narayan K, Danielson CM, Lagarec K, Lowekamp BC, Coffman P, Laquerre A, et al. (2014). Multi-resolution correlative focused ion beam scanning electron microscopy: Applications to cell biology. Journal of Structural Biology, 185(3), 278–284. 10.1016/j.jsb.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayan K, & Subramaniam S (2015). Focused ion beams in biology. Nature Methods, 12(11), 1021–1031. 10.1038/nmeth.3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oegema K, & Hyman AA (2006). Cell division. WormBook, 1–40. 10.1895/wormbook.1.72.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou HD, Phan S, Deerinck TJ, Thor A, Ellisman MH, & O’Shea CC (2017). ChromEMT: Visualizing 3D chromatin structure and compaction in interphase and mitotic cells. Science, 357(6349). 10.1126/science.aag0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peddie CJ, & Collinson LM (2014). Exploring the third dimension: Volume electron microscopy comes of age. Micron, 61, 9–19. 10.1016/j.micron.2014.01.009. [DOI] [PubMed] [Google Scholar]
- Peddie CJ, Domart MC, Snetkov X, O’Toole P, Larijani B, Way M, et al. (2017). Correlative super-resolution fluorescence and electron microscopy using conventional fluorescent proteins in vacuo. Journal of Structural Biology, 199(2), 120–131. 10.1016/j.jsb.2017.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman M, Chang IY, Harned A, Maheshwari R, Amoateng K, Narayan K, et al. (2020). C. elegans pronuclei fuse after fertilization through a novel membrane structure. The Journal of Cell Biology, 219(2). 10.1083/jcb.201909137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigort A, & Plitzko JM (2015). Cryo-focused-ion-beam applications in structural biology. Archives of Biochemistry and Biophysics, 581, 122–130. 10.1016/j.abb.2015.02.009. [DOI] [PubMed] [Google Scholar]
- Schaffer M, Pfeffer S, Mahamid J, Kleindiek S, Laugks T, Albert S, et al. (2019). A cryo-FIB lift-out technique enables molecular-resolution cryo-ET within native Caenor-habditis elegans tissue. Nature Methods, 16(8), 757–762. 10.1038/s41592-019-0497-5. [DOI] [PubMed] [Google Scholar]
- Schlegel A, Giddings TH Jr., Ladinsky MS, & Kirkegaard K (1996). Cellular origin and ultrastructure of membranes induced during poliovirus infection. Journal of Virology, 70(10), 6576–6588. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/8794292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorb M, Gaechter L, Avinoam O, Sieckmann F, Clarke M, Bebeacua C, et al. (2017). New hardware and workflows for semi-automated correlative cryo-fluorescence and cryo-electron microscopy/tomography. Journal of Structural Biology, 197(2), 83–93. 10.1016/j.jsb.2016.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz CL, Sarbash VI, Ataullakhanov FI, McIntosh JR, & Nicastro D (2007). Cryo-fluorescence microscopy facilitates correlations between light and cryo-electron microscopy and reduces the rate of photobleaching. Journal of Microscopy, 227(Pt. 2), 98–109. 10.1111/j.1365-2818.2007.01794.x. [DOI] [PubMed] [Google Scholar]
- Scotuzzi M, Kuipers J, Wensveen DI, de Boer P, Hagen KC, Hoogenboom JP, et al. (2017). Multi-color electron microscopy by element-guided identification of cells, organelles and molecules. Scientific Reports, 7, 45970. 10.1038/srep45970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulston JE, Schierenberg E, White JG, & Thomson JN (1983). The embryonic cell lineage of the nematode Caenorhabditis elegans. Developmental Biology, 100(1), 64–119. 10.1016/0012-1606(83)90201-4. [DOI] [PubMed] [Google Scholar]
- Takemura SY, Aso Y, Hige T, Wong A, Lu Z, Xu CS, et al. (2017). A connectome of a learning and memory center in the adult Drosophila brain. eLife, 6. 10.7554/eLife.26975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titze B, & Genoud C (2016). Volume scanning electron microscopy for imaging biological ultrastructure. Biology of the Cell, 108(11), 307–323. 10.1111/boc.201600024. [DOI] [PubMed] [Google Scholar]
- Tsang TK, Bushong EA, Boassa D, Hu J, Romoli B, Phan S, et al. (2018). High-quality ultrastructural preservation using cryofixation for 3D electron microscopy of genetically labeled tissues. eLife, 7. 10.7554/eLife.35524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidavsky N, Akiva A, Kaplan-Ashiri I, Rechav K, Addadi L, Weiner S, et al. (2016). Cryo-FIB-SEM serial milling and block face imaging: Large volume structural analysis of biological tissues preserved close to their native state. Journal of Structural Biology, 196(3), 487–495. 10.1016/j.jsb.2016.09.016. [DOI] [PubMed] [Google Scholar]
- Weimer RM (2006). Preservation of C. elegans tissue via high-pressure freezing and freeze-substitution for ultrastructural analysis and immunocytochemistry. Methods in Molecular Biology, 351, 203–221. 10.1385/1-59745-151-7:203. [DOI] [PubMed] [Google Scholar]
- Xu CS, Hayworth KJ, Lu Z, Grob P, Hassan AM, Garcia-Cerdan JG, et al. (2017). Enhanced FIB-SEM systems for large-volume 3D imaging. eLife, 6. 10.7554/eLife.25916. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.