

Abstract
Glioblastoma (GBM) remains the most frequently diagnosed primary malignant brain cancer in adults. Despite recent progress in understanding the biology of GBM, the clinical outcome for patients remains poor, with a median survival of approximately one year after diagnosis. One factor contributing to failure in clinical trials is the fact that traditional models used in GBM drug discovery poorly recapitulate patient tumors. Previous studies have shown that monensin (MON) analogs, namely esters and amides on C-26 were potent towards various types of cancer cell lines. In the present study we have investigated the activity of these molecules in GBM organoids, as well as in a host:tumor organoid model. Using a mini-ring cell viability assay we have identified seven analogs (IC50 = 91.5 ± 54.4–291.7 ± 68.8 nM) more potent than parent MON (IC50 = 612.6 ± 184.4 nM). Five of these compounds induced substantial DNA fragmentation in GBM organoids, suggestive of apoptotic cell death. The most active analog, compound 1, significantly reduced GBM cell migration, induced PARP degradation, diminished phosphorylation of STAT3, Akt and GSK3β, increased ɣH2AX signaling and upregulated expression of the autophagy associated marker LC3-II. To investigate the activity of MON and compound 1 in a tumor microenvironment, we developed human cerebral organoids (COs) from human induced pluripotent stem cells (iPSCs). The COs showed features of early developing brain such as multiple neural rosettes with a proliferative zone of neural stem cells (Nestin+), neurons (TUJ1 +), primitive ventricular system (SOX2 +/Ki67 +), intermediate zone (TBR2 +) and cortical plate (MAP2 +). In order to generate host:tumor organoids, we co-cultured RFP-labeled U87MG cells with fully formed COs. Compound 1 and MON reduced U87MG tumor size in the COs after four days of treatment and induced a significant reduction of PARP expression. These findings highlight the therapeutic potential of MON analogs towards GBM and support the application of organoid models in anti-cancer drug discovery.
Keywords: Glioblastoma, Monensin, Analogs, Organoid
1. Introduction
Glioblastoma multiforme (GBM) continues to be the most common primary brain tumor in adults [1]. Despite numerous clinical trials, the 5-year median survival rate of less than 5% remains unchanged [2]. Among various factors contributing to high resistance to standard therapy are the influence of blood-brain barrier, vascularization, molecular heterogeneity among tumors and within tumors, tumor stem cell-like characteristics, activation of alternative signaling pathways, and the expression level of O6-methylguanine-DNA methyltransferase [3–6]. Therefore, there is a need for development of new treatment approaches, as well as improved ex vivo GBM models which better recapitulate tumor heterogeneity for timely empirical testing of personalized therapeutic strategies [7].
One of the main barriers for the development of novel treatment strategies is the challenge of translating scientific discoveries from bench to bedside [8]. This is mostly related to the fact that traditional models of cancer poorly recapitulate patient tumors [9], and consequently many drugs which perform well in pre-clinical models eventually fail in clinical trials [8]. Traditional in vitro culture models have contributed tremendously to our understanding of biological mechanisms underlying GBM pathogenesis but possess several limitations, such as significant adaptation and selection to two-dimensional culture conditions, which results in the loss of genetic heterogeneity of original tumor, lack of a stromal compartment and a non-cancerous tissue control [7,8]. Recent advances in the development of organoid models that more accurately recapitulate three-dimensional tumor architecture open new possibilities in GBM-targeted drug discovery [10,11].
Monensin A (MON) is a compound of natural origin, which was first isolated from Streptomyces cinnamonensis in 1967 and belongs to the well-known group of polyether ionophore antibiotics. It exhibits a wide spectrum of biological activities such as antimicrobial, antiproliferative, anti-parasitic and anti-viral [12]. The biological properties of MON arise from its ability to bind ions and transport them across lipid membranes into the cellular environment. As a consequence, the Na+ /K+ gradient concentration and intercellular pH are disturbed, which leads to programmed cell death induced by cell swelling, vacuolization and mitochondrial injury. Polyether ionophore antibiotics differ in respect to their preferences for monovalent ions, with MON showing at least a 10-fold preference for Na+ over K+ [13]. Currently, MON is commonly used in veterinary medicine as an anti-coccidial drug for poultry and cattle [14]. Most recently, MON has been recognized in various studies for its anti-cancer activity [15]. MON has been shown to be active against human melanoma cells in vitro and in vivo [16], human gastric stem-like cells [17], acute myeloid leukemia and adenoid cystic carcinoma cells [18], and prostate cancer cells [19]. In a study conducted by Yoon et al. in glioma cells, MON enhanced cytotoxicity by TRAIL (tumor necrosis factor-related apoptosis-induced ligand) [20]. In a recent study, MON has been shown to effectively target tumor-associated endothelial cells, resulting in decreased GBM angiogenesis and growth inhibition [21]. These findings are of particular importance in the context of potential drug repurposing, which has become a new focus in cancer treatment [16]. New drug development involves a long research period, a large risk of failure and biosafety issues [22]. Since safety, pharmacological properties and potential toxicity are established for approved drugs, repurposing allows many time-consuming and expensive steps in the drug discovery pipeline to be circumvented [23].
Progress in the development of novel three-dimensional cancer organoid models and discoveries on the anti-GBM potential of MON prompted us to employ these models to study the activity of MON analogs as possibly more potent anti-GBM agents. MON analogs obtained by modification at the C-26 position have been previously synthesized and preliminary studies showed anti-cancer activity against human uterine sarcoma and colon adenocarcinoma cell lines as well as their doxorubicin resistant sublines [24,25,26]. In the present work, we investigated the activity of a series of MON esters and urethanes at the C-26 position against GBM organoids as well as infiltrative GBM in cerebral organoid (CO) models.
2. Materials and methods
2.1. Compounds
Monensin (MON) and its analogs were synthesized according to the procedure shown in Fig. 1 and characterized by Huczyński and co-workers as previously reported.
Fig. 1.
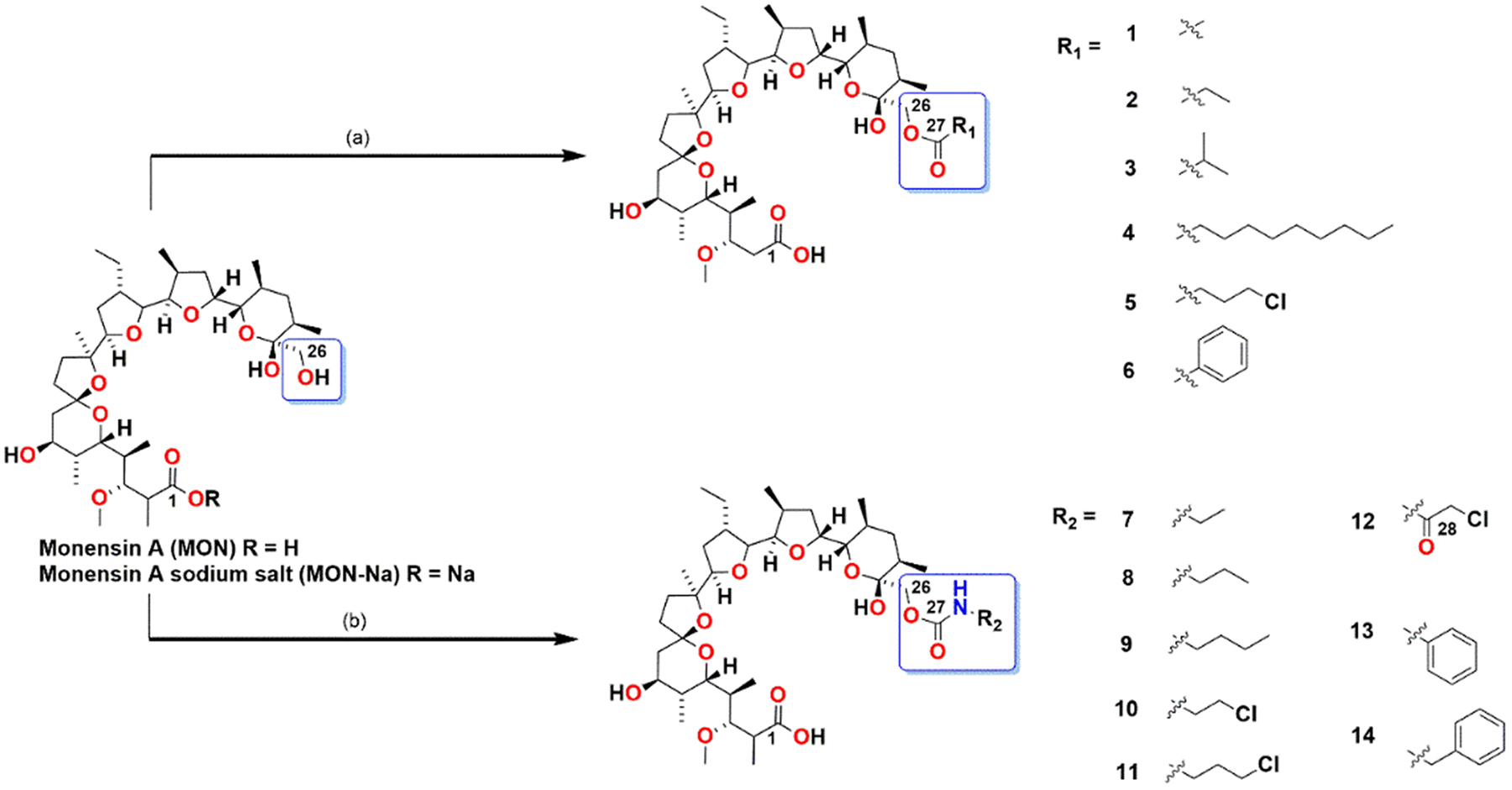
Schematic representation of the synthesis of MON analogs. Reactions and conditions: (a) MON-Na (1 eq.), DMAP (catalytic amount), acyl chloride (3 eq.), pyridine, rt, 48 h; (b) MON-H (1 eq.), respective isocyanate (0.95 eq.), anhydrous toluene, rt, 14 days.
2.1.1. General procedure
All reagents and all solvents were obtained from Merck or Trimen Chemicals S.A. (Poland), and were used as received without further purification. CD2Cl2 and CD3CN spectral grade solvents were stored over 3 Å molecular sieves for several days. Reaction mixtures were stirred using Teflon-coated magnetic stir bars and were monitored by thin layer chromatography (TLC) using aluminum-backed plates (Merck 60 F254). TLC plates were visualized by UV-light (254 nm), followed by treatment with phosphomolybdic acid (PMA, 5% in absolute EtOH) and gentle heating. Products of the reactions were purified using CombiFlash®Rf+ Lumen Flash Chromatography System (Teledyne Isco) with integrated ELS and UV detectors. All solvents used in flash chromatography were of HPLC grade (Merck) and were used as received. Solvents were removed using a rotary evaporator.
NMR spectra were recorded on a Varian 400 (1H NMR at 403 MHz, 13C NMR at 101 MHz) magnetic resonance spectrometer. 1H NMR spectra are reported in chemical shifts downfield from TMS using the respective residual solvent peak as internal standard (CD2Cl2 δ 5.32 ppm and CD3CN δ 2.04 ppm). 1H NMR spectra are reported as follows: chemical shift (δ, ppm), multiplicity (s = singlet, d = doublet, q = quartet, dd = doublet of doublets, dt = doublet of triplets, dq = doublet of quartets, ddd = doublet of doublet of doublets, ddt = doublet of doublet of triplets, dddd = doublet of doublet of doublet of doublets, m = multiplet), coupling constant(s) in Hz, and integration. Significant peaks are reported within the overlapping ~2.00–0.50 ppm region of the 1H NMR spectra. The 13C NMR spectra are reported in chemical shifts downfield from TMS using the respective residual solvent peak as internal standard (CD2Cl2 δ 53.84 ppm and CD3CN δ 118.69 ppm and 1.39 ppm). Line broadening parameters were 0.5 or 1.0 Hz, while the error of chemical shift value was 0.1 ppm.
Infrared spectra in the mid infrared region were recorded in KBr tablets on an IFS 113 v FT-IR spectrophotometer (Bruker) equipped with a DTGS detector, and are reported as follows: wavenumbers (cm−1), description (w = weak, m = medium, s = strong, br = broad). The spectra were taken on resolution 2 cm−1, NSS = 64. The Happ-Genzel apodization function was used.
The electrospray ionization (ESI) mass spectra were recorded on a Waters/Micromass ZQ mass spectrometer (Waters Alliance) equipped with a Harvard syringe pump. The samples were prepared in dry acetonitrile, and were infused into the ESI source using a Harvard pump at a flow rate of 20 ml min−1. The ESI source potentials were: capillary 3 kV, lens 0.5 kV, and extractor 4 V. The standard ESI mass spectra were recorded at the cone voltages of 10 and 30 V. The source temperature was 120 °C and the desolvation temperature was 300 °C. Nitrogen was used as the nebulizing and desolvation gas at flow-rates of 100 dm3h−1. Mass spectra were acquired in the positive ion detection mode with unit mass resolution at a step of 1 m/z unit. The mass range for ESI experiments was from m/z = 300 to m/z = 1100.
Monensin salt was isolated from commercially available veterinary feed additive – Coxidin®. The sodium salt of monensin was dissolved in CH2Cl2 and extracted with H2SO4 solution (pH=1) followed by washing with water to obtain MON. The isolation was conducted according to the procedure described by Huczyński in 2006 [27,28].
2.1.2. General procedure for the synthesis of 1–6
To a solution of MON-Na (1 eq.) in pyridine, the respective acyl chloride (3 eq.) and DMAP (catalytic amount) were added. The reaction mixture was stirred for 3 days, then was concentrated under reduced pressure to dryness. The residue was dissolved in CH2Cl2 and extracted three times with the solution of H2SO4 (pH=1) and once with water. Then the organic layer was evaporated under reduced pressure with silica gel. The residue was purified by column flash chromatography using CombiFlash®Rf+ (dry loading on silica gel, hexane/ethyl acetate, increasing concentration gradient) with an integrated Evaporative Light Scattering Detector (ELSD) and UV detector, and Electrospray Ionization Mass Spectrometry (ESI-MS). The structures and purity of derivatives were determined by HR-MS, FT-IR, 1H, and 13C NMR methods. The spectral characterization of obtained analogs is described in detail in our previous paper, where we investigated their antibacterial and anti-trypanosomal activity [29].
2.1.3. General procedure for the synthesis of 7–14
To a solution of MON (1 eq.) in anhydrous toluene, the respective isocyanate (0.95 eq.) was added. The mixture was stirred for 14 days and then was concentrated under reduced pressure to dryness. The course of the synthesis was monitored by thin-layer chromatography, the reaction was completed when the formation of larger amounts of product ceased. The residue was purified by column flash chromatography using CombiFlash®Rf+ (hexane/ethyl acetate, increasing concentration gradient) with an integrated Evaporative Light Scattering Detector (ELSD) and UV detector, and Electrospray Ionization Mass Spectrometry (ESI-MS). The structures and purity of derivatives were determined by HR-MS, FT-IR, 1H, and 13C NMR methods. The spectral characterization of obtained analogs is described in detail in our previous paper, where we investigated their antibacterial and anti-trypanosomal activity [29].
2.2. Cell culturing conditions
The human glioblastoma U-118 MG cell line was cultured in Minimum Essential Medium (MEM) (cat. no. 11095–080, Gibco) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS) (cat. no. FP-0500-A, Atlas Biologicals, Fort Collins, CO, USA). The human glioblastoma cell line U-87 MG stably expressing red fluorescent protein (RFP) under the control of the CMV promoter was purchased from Cellomics Technology (cat. no. SC-1031) and cultured in Dulbecco’s Modified Eagle Medium (DMEM) (cat. no. 10–013-CV, Corning) supplemented with 10% (v/v) heat-inactivated FBS. All cell lines were maintained at 37°C with 5% CO2. Cells were reported negative for mycoplasma contamination in March 2021 (U-118 MG) and in August 2021 (U-87/RFP). Cell lines were validated as authentic, giving a 100% match when compared to the known reference profile through short tandem repeat profiling in March 2021 (U-118 MG) and August 2021 (U-87 MG/RFP) performed by Genetica DNA Laboratories (Burlinton, NC, USA) [30].
2.3. Human induced pluripotent stem cells culturing conditions
Mono-allelic mEGFP-tagged ACTB WTC human induced pluripotent stem cells (hiPSCs) were purchased from Coriell Institute (cat. no. AICS-0016) and cultured in complete mTeSR Plus (cat. no. 05825, StemCell Technologies). hiPSCs were maintained at 37°C with 5% CO2. Cells were reported to be negative for mycoplasma contamination in March 2021 by Genetica DNA Laboratories (Burlinton, NC, USA).
2.4. Mini-ring cell viability assay
10 μl/well of single-cell suspensions of U-118 MG cells (10^4/well) in 4:3 ratio of Matrigel (cat. no. 354277, Corning) and complete NeuroCult (cat. no. 05751, StemCell Technologies) were plated in a ring shape around the rim in 96-well plates (cat. no. 92096, TPP) (Fig. 2) according to the previously published protocols by Phan et al. and Nguyen and Soragni [10,31]. Cells suspended in Matrigel:NeuroCult mix were kept on ice at all times, and continuously agitated while plating the rings. After plating of every eight wells the pipette tip was changed and vortexing of the mix performed. 100 μl of unsupplemented medium was added to all surrounding wells, which didn’t contain rings. After 15 min of incubation at 37 °C with 5% CO2 to allow rings to solidify, 100 μl of complete NeuroCult was added to each working well. Plates were incubated for 48 h, followed up by the complete removal of medium, and replacement with 100 μl of fresh complete NeuroCult containing 0.1% DMSO (control), or increasing concentrations (1 nM – 10 μM) of MON and its analogs. Medium with DMSO or compounds was changed every 24 h. After 72 h of treatment, medium was removed and wells were washed with 100 μl of pre-warmed DPBS (cat. no. 21–031-CV, Corning). Cells were then released by incubating with 50 μl/well of 5 mg/ml dispase (cat. no. 17105–041, Gibco) for 40 min in 37 °C. In the next step 10 μl of MTT reagent (5 mg/ml) (M2128, Sigma) was added to each well, and the plate was incubated for 24 h at 37°C with 5% CO2. 100 μl of 10% SDS in 0.01 M HCl was added to each well and the plate was incubated at 37°C for additional 24 h. Absorbance was recorded at 540 nm employing a BioTek Plate Reader. GraphPad Prism 9 for Windows (GraphPad Software) was used to determine IC50 values by non-linear regression analysis.
Fig. 2.
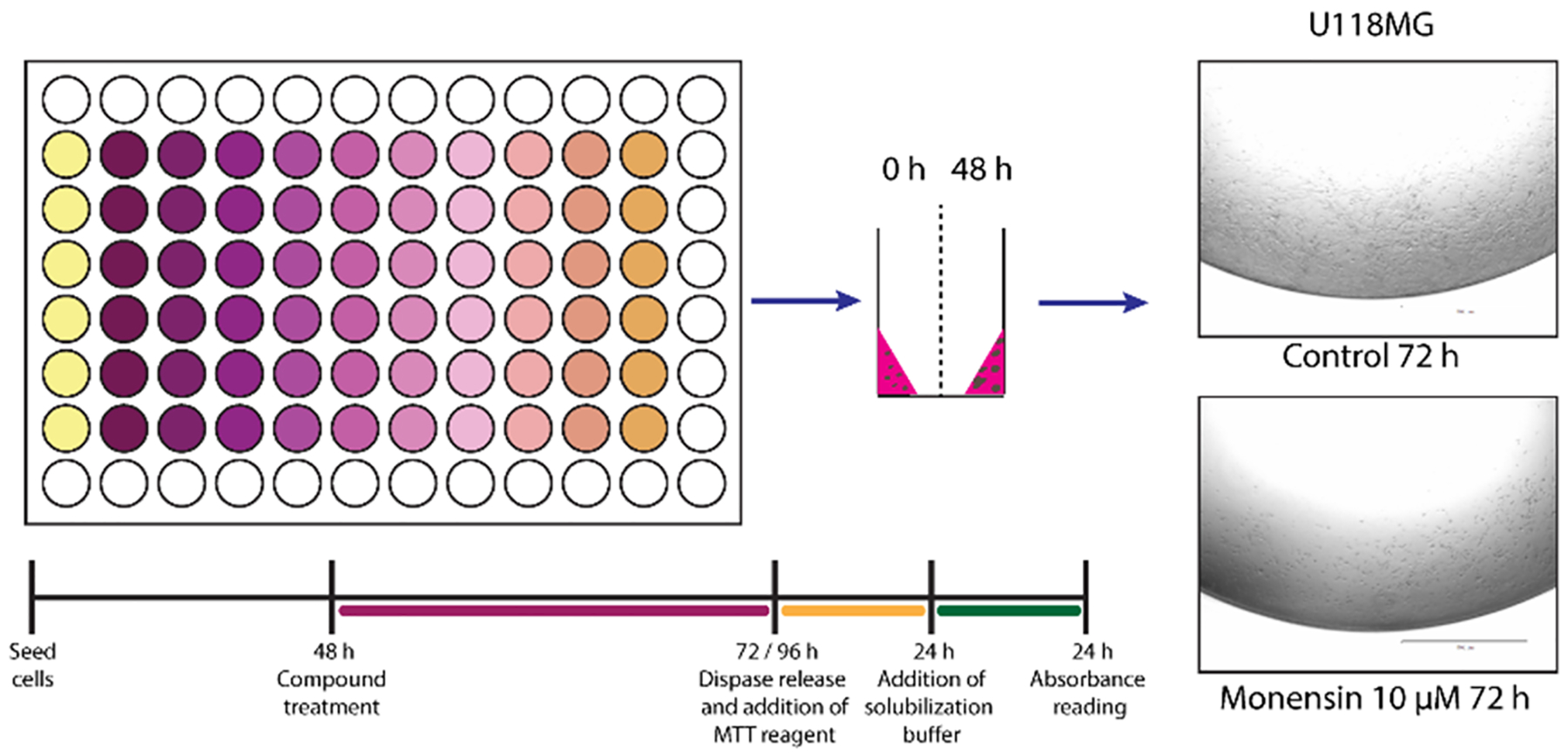
Treatment scheme for assessing compound response using the 3D mini-ring cell viability assay. Representative images of U-118MG cells seeded in mini-ring format were taken with EVOS FL Auto Cell Imaging System (Thermo Fisher Scientific) after 72 h of treatment with 0.1% DMSO (control) or 10 μM MON.
2.5. GBM organoids acquisition and culturing
Cancer organoids were generated as shown in Fig. 3A according to our previously published protocol [32]. Briefly, droplets of a complete NeuroCult and Matrigel in 1:4 ratio, containing 8 × 10^3 U-118 MG cells/droplet were generated by pipetting 20 μl of the cold mixture (stored on ice) onto a base mold of Parafilm. Droplets were subsequently allowed to solidify in 37 °C, 5% CO2 humidified incubator for 1 h, and gently scooped into 24 well plate containing 1 ml/well of complete NeuroCult (2 droplets/well). Droplets were grown for 96 h at 37°C with 5% CO2 without agitation. 0.5 ml of fresh NeuroCult was added to each well every other day. After 96 h, the plate was placed on the shaking platform at 70 rpm (Orbi-Shaker Jr, Benchmark Scientific, USA). After 48 h approximately 3 mm diameter organoids were cut into 0.5–1 mm diameter pieces using Excelta scissors (cat. no. 17–467–493, 17–456–004, 17–467–497, Fisher Scientific) in order to prevent the formation of a necrotic core [7].
Fig. 3.
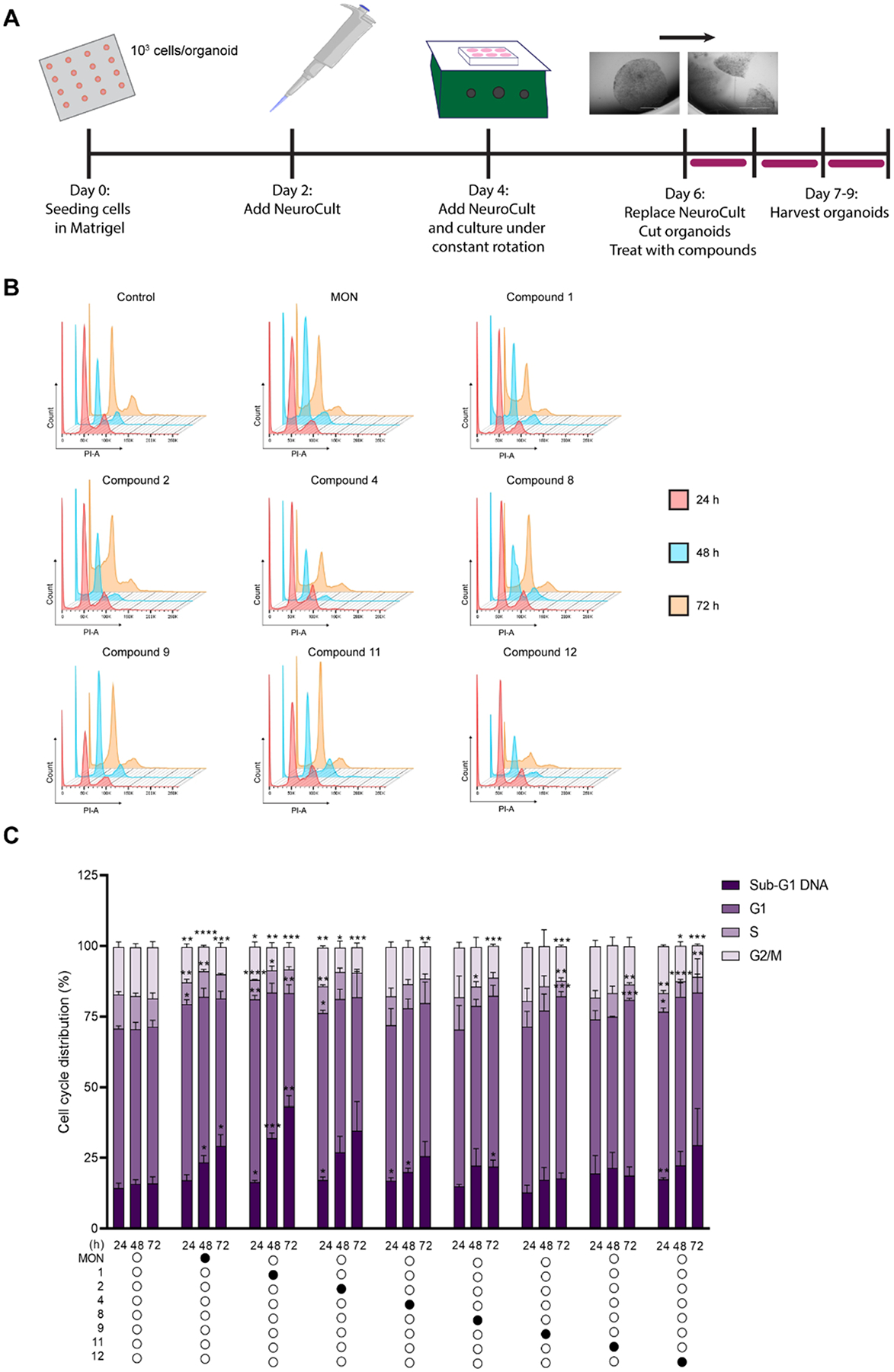
MON analogs induced DNA fragmentation in U-118MG organoids. U-118MG organoids were treated with 0.1% DMSO (control), parent MON or its select analogs at concentrations equal to 5 × IC 50 values for 24, 48 or 72 h and subsequently subjected to propidium iodide staining and flow cytometry. A. Schematic representation of U-118MG organoids generation and treatment; B. Representative cytograms; C. Distribution of cells in different phases of cell cycle or with Sub-G1 DNA determined by PI staining. Data represent mean ± SD (n = 6 for control, and n = 3 for compound treatment) *p ≤ 0.05, **p ≤ 0.01, *** p ≤ 0.001, ****p ≤ 0.0001.
2.6. Cell cycle analysis of GBM organoids by flow cytometry
Cancer organoids were generated as described in GBM organoids acquisition and culturing. Two organoids/well were cultured in a 24 well plate in 1 ml of complete NeuroCult in the presence of 0.1% DMSO (control) or MON, or compounds 1, 2, 4, 8, 9, 11, 12 at concentrations equal to 5 × IC50 values (Table 1) for 24, 48, or 72 h. Organoids were then harvested with gentle cell dissociation reagent (cat. no. 07174, StemCell Technologies) containing 0.2% anti-clumping agent, 2-naphtol-6,8-disulfonic acid dipotassium salt (NDA) (cat. no. 439013, Frontier Scientific) and dispersed by gentle pipetting with 1250 μl tip. Cells were washed with DPBS, fixed with 1 ml of 70% ice-cold ethanol and stored in 20 °C until processing. Cells were centrifuged, stained with 300 μl propidium iodide/RNase Staining buffer (cat. no. 550825, BD Biosciences, San Jose, CA, USA) and stored for 1 h in the dark at room temperature (rt). DNA content was measured with FacsAria Ilu Flow Cytometer (BD Biosciences, San Jose, CA, USA) and data were analyzed employing FlowJo software.
Table 1.
Antiproliferative activity (IC50) values of monensin (MON) and its analogs.
| Compound | IC50 [nm] |
|---|---|
| MON | 612.6 ± 184.4& |
| 1 | 291.7 ± 68.8 ** |
| 2 | 189.8 ± 79.3 ** |
| 3 | 604.9 ± 149.6 |
| 4 | 168.7 ± 82.2** |
| 5 | 487.6 ± 161.2 |
| 6 | 622.7 ± 203.8 |
| 7 | 994.9 ± 330.3 |
| 8 | 257.3 ± 135.6 * |
| 9 | 100.0 ± 92.0 *** |
| 10 | 808.3 ± 103.4 |
| 11 | 139.0 ± 105.4 ** |
| 12 | 91.5 ± 54.4 *** |
| 13 | 424.0 ± 288.6 |
| 14 | 337.0 ± 217.1 |
IC50 value is defined as the concentration of the compound which induces 50% growth inhibition.
Values are given as mean ± SD (n = 4 each).
The value is an average from two independent experiments (n = 4 each).
p ≤ 0.05,
p ≤ 0.01,
p ≤ 0.001.
2.7. Wound healing assay
U-118 MG cells were seeded in 60 mm petri dishes (cat. no. 430166, Corning) (0.15 ×10^6)/dish) and grown in complete MEM medium for nine days to obtain a confluent cell monolayer. The medium has been aspirated and replaced with MEM without FBS. After 48 h the medium has been removed, and cross shaped scratches have been made with 1250 μl pipette tip in cell monolayer. Cells were gently washed twice with DPBS and incubated in medium containing 0.1% DMSO, or MON (306 nM) or compound 1 (146 nM) at concentrations equal to respective ½ of IC50 values (Table 1). The wound surface was photographed with the inverted phase contrast microscope EVOS FL Auto Cell Imaging System (Thermo Fisher Scientific) at the beginning of the experiment (0 h), as well as after 4, 28, 48, and 74 h. Three biological replicates were performed, and seven measurements for each replicate, at each time point were taken, and mean ± SD was calculated. The wound surface at each time point was divided by the surface at 0 h, as a measure of wound closure. The wound surface was defined as 100% at 0 h for each condition.
2.8. Immunoblot analysis of GBM organoids
U-118 MG organoids were generated as described above in GBM organoids acquisition and culturing. Two organoids/well were cultured in 24 well plate in 1 ml of complete NeuroCult/well for 24 or 48 h in the presence of 0.1% DMSO, or MON (3 μM) or compound 1 (1.5 μM) at the concentrations equal to 5 × IC50 values (Table 1). Organoids were than washed with Dulbecco′s Phosphate Buffered Saline (DPBS), snap frozen in liquid nitrogen and stored in −80 °C until processing. Organoids were further thawed and lysed with lysis buffer (25 mM HEPES, pH 7.5, 300 mM NaCl, 0.1% w/v Triton X-100, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, EDTA-free complete protease inhibitor tablets (Roche), 20 μg/ml aprotinin, 50 μg/ml leupeptin, 10 μM pepstatin, 1 mM phenylmethylsulfonyl fluoride, 20 mM β-glycerophosphate, 1 mM Na3VO4, and 1 μM okadaic acid). Protein content was evaluated by Bradford assay and equal amounts (20 μg) were separated by electrophoresis using 12% (w/v) acrylamide Mini-PROTEAN® precast gels (Bio-Rad). Proteins were transferred electrophoretically onto a PVDF membrane (Immobilon-FL, cat. no. IPFL00010, Merck Millipore) and next stained with Ponceau S to assess transfer efficiency and verify equal loading. The membrane was blocked with 5% (w/v) non-fat milk (cat. no. M17200, Research Products International) in Tris-buffered saline containing 0.1% (w/v) TWEEN-20 (TBS-T) for 1 h at rt and incubated overnight at 4°C with primary antibodies against PARP (cat. no. 9532, Cell Signaling Technology) (1:1000 dilution), STAT3 (cat. no. 9139, Cell Signaling Technology) (1:1000 dilution), phospho-STAT3 (Tyr705) (cat. no. 9145, Cell Signaling Technology) (1:2000 dilution), phospho-Akt (Ser473) (cat. no. 4060, Cell Signaling Technology) (1:2000 dilution), phospho-GSK-3β (Ser9) (cat. no. 9323, Cell Signaling Technology) (1:1000 dilution), ɣH2AX (cat. no. ab81299, abcam) (1:5000 dilution), LC3B (cat. no. 2775, Cell Signaling Technology) (1:1000 dilution), GAPDH (cat. no. 2118, Cell Signaling Technology) (1:10000 dilution). After washing with 1X Tris-Buffered Saline, 0.1% Tween® 20 Detergent (TBS-T) for 5 × 5 min the membrane was incubated with secondary HRP-conjugated goat anti-rabbit IgG (H+L) antibody (1:5000 dilution) (cat. no. 170–6515, Bio-Rad) or goat anti-mouse (cat. no. 1706516, Bio-Rad) for 1 h at rt. After washing in TBS-T the membrane was exposed to ClarityTM Western ECL Substrate luminol enhancer solution and peroxide solution (Bio-Rad) for 5 min and visualized and quantified using Image J software. For uncropped immunoblots see Figs. S2 and S3 in Supplementary material.
2.9. Co-treatment of GBM organoids with autophagy inhibitor
U-118 MG organoids were acquisitioned as described in GBM organoids acquisition and culturing. Three organoids/well were cultured in a 24 well plate in 1 ml of complete NeuroCult/well and treated with MON (3 μM) or compound 1 (1.5 μM) or 0.1% DMSO control in the presence or absence of bafilomycin A1 (cat. no. A8627, ApexBio Houston, TX) (3 nM) for 24 or 48 h. After the treatment organoids were harvested, subjected to PI staining and analyzed by flow cytometry as described in Cell cycle analysis of GBM organoids by flow cytometry.
2.10. Cerebral organoids acquisition and culturing
Cerebral organoids (COs) were generated using STEMdiff Cerebral Organoid Kit (cat. no. 08570, StemCell Technologies) according to the manufacturer’s instructions (Fig. 7A). Briefly, once large, compact hiPSCs colonies showing less than 10% differentiation reached 70–80% confluency they were individually plated in ultra-low attachment 96-well round-bottom plates (cat. no. 7007, Corning) (9000 cells/well). Embryoid bodies (EBs) were supplemented every other day with 100 μl/well of EB Formation Medium. On day five EBs were transferred to 24-well ultra-low attachment plates (cat. no. 3473, Corning) containing 0.5 ml of Induction Medium / well. On day seven, EBs were embedded in cold Matrigel droplets (cat. no. 354277, Corning), and further cultured in 6-well ultra-low adherent plates (12–16 droplets per well) in Expansion Medium (3 ml/well). On day ten the medium was replaced with Maturation Medium (3 ml/well), and plate with organoids has been placed on the orbital shaker (Orbi-Shaker Jr, Benchmark Scientific, USA) at 70 rpm at 37°C with 5% CO2. Medium has been replaced every three to four days. At day 51, two representative COs were harvested, fixed with 4% paraformaldehyde for 45 min in RT, followed by three ice-cold DPBS washes. Fixed COs were further incubated for 1 h with 15% sucrose in RT, the solution was replaced with 30% sucrose, and additional overnight incubation in 4 °C was performed. The solution was removed, and COs were embedded in Optimal Cutting Temperature Compound (OCT) (cat. no. 4585, Fisher HealthCare), snap frozen in liquid nitrogen and stored at – 20 °C until processing.
Fig. 7.
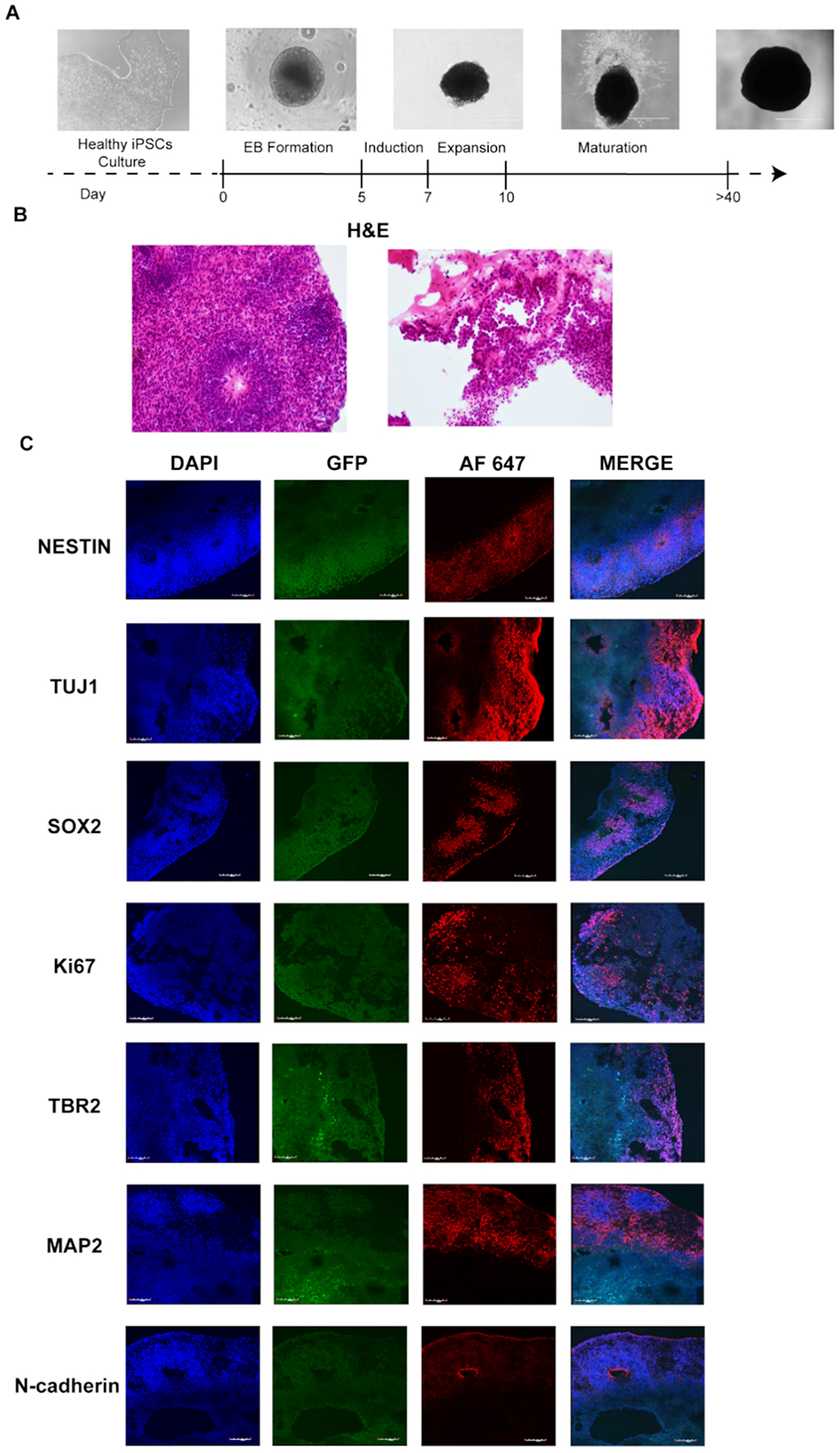
Cerebral organoids acquisition and validation. A. Schematic representation of cerebral organoids generation, scale bar 1000 μm; B. H&E staining of normal cerebral organoids at 51 days of development, 20X magnification; C. Immunofluorescence staining of DAPI, GFP and AF 647 – Alexa Fluor 647, a secondary antibody used for Nestin, Tuj1, Sox2, Ki67, TBR2, MAP2, N-cadherin, 20X magnification.
2.11. Co-culture of COs and GBM cells
In order to establish GLICO tumors, by co-culturing COs with cancer cells, individual COs were transferred to 24-well plate (1 CO/well). 2 ml of fresh, complete Maturation Medium containing 10^5 U-87MG/RFP cells has been added to each well. COs were incubated for 24 h at 37°C with 5% CO2 with no agitation. COs were further transferred with wide-bore pipette tips to the clean wells in 24-well plates (1 CO/well) and washed with DPBS (1 ml/well). Tumor-bearing COs (GLICO tumors) were cultured in 2 ml/well of fresh, complete Maturation Medium on 70 rpm orbital shaker at 37°C with 5% CO2 for seven days. Images of each individual GLICO were taken every 24 h using EVOS FL Auto Cell Imaging System (Thermo Fisher Scientific). After seven days complete Maturation Medium has been replaced (2 ml/well), and GLICO tumors (tumor-bearing COs) were cultured for additional four days in the presence of 0.1% DMSO (control), or compounds of interest: MON (1226 nM), or compound 1 (584 nM) at concentrations equal to the respective 2 × IC50 values (Table 1). Each treatment condition has been performed on 4–5 individual GLICOs. Representative images of each GLICO tumors were taken every 24 h as described above. After four days of treatment GLICO tumors were harvested and fixed as described in Cerebral organoids acquisition and culturing.
2.12. Histopathological evaluation and immunofluorescence
Immunohistochemical staining was performed by UAMS Experimental Pathology Core. For histopathological examination, OCT embedded COs were sectioned to 25 μm thick sections and stained with hematoxylin and eosin (H&E) (cat. no. 7231, Richard-Allan Scientific, Thermo Fisher Scientific, Waltham, MA, USA).
For immunofluorescence frozen blocks were cryosectioned to 25 μm thick sections. Sections were permeabilized with 0.02% Triton-X in 1X DPBS for 20 min in RT, followed by blocking with 10% goat serum in DPBS for 1 h in RT. Staining with the following primary antibodies was performed for 2 h in RT: Nestin (cat. no. 33475, Cell Signaling Technology, 1:500), Tubulin β3/TUJ1 (cat. no. 801201, Bio Legend, 1:100), TBR2 (cat. no. 23345, abcam, 1:100), N-cadherin (cat. no. 13116, Cell Signaling Technology, 1:200), SOX2 (cat. no. 4900, Cell Signaling Technology, 1:200), Ki-67 (cat. no. 16667, abcam, 1:100), MAP2 (cat. no. 4542, Cell Signaling Technology, 1:50), PARP (cat. no. 9532, Cell Signaling Technology, 1:100), ɣH2AX (cat. no. ab81299, abcam, 1:100). Sections were washed three times in DPBS for 5 min in RT, followed by the incubation with secondary antibodies: (cat. no. A21244 (anti-rabbit) or A32728 (anti-mouse), Invitrogen, 1:100) for 1 h in RT. Sections were mounted to cover slips using ProLong® Gold antifade reagent with DAPI (cat. no. P36935, Molecular Probes). Images were taken with Zeiss LSM8800 Confocal System, 20x objective, 405 nm, 488 nm, and 633 nm lasers, and analyzed with Zeiss Zen 2.3 software. At least 139 cells were scored per condition and blind scored via ImageJ and Cell Profiler.
2.13. Statistical analysis
Unpaired t-test with Welch’s correction was employed and p values of < 0.05 were considered significant. For the analysis of large data sets with multiple replicates (immunofluorescence in GLICO model) one-way ANOVA was performed. Results are presented as a mean ± SD. GraphPad Prism 9 for Windows (GraphPad Software) was used for statistical analysis.
3. Results and discussion
3.1. Analog design and synthesis
Chemical modifications of MON in the C-26 position are well described in the literature. In 2018, a series of MON esters was prepared by modification of the carboxyl group at the C-1 position and the hydroxyl group at the C-26 position. All compounds were tested against four human cancer cell lines [33] and among these six C-26 esters (1–6) (Fig. 1), which exhibited the highest antiproliferative activity, were selected for further evaluation of biological activity. The ester derivatives (1–6) (Fig. 1) were synthesized according to Gaboyard’s method in the reaction of MON-Na with the respective acyl chlorides in the presence of 4-(dimethylamino)pyridine (DMAP) in pyridine [34].
The preliminary in vitro studies of fourteen MON urethanes, obtained by Westley’s group, showed certain derivatives exhibited a ten-fold higher activity against Gram-positive microorganisms, compared to unmodified MON. The highest activities were exhibited by the phenyl urethanes, with MIC values lower than 0.1 μg/ml [24]. Whereas in 2011, Huczyński et al. reassessed the structure of the phenyl urethane of MON sodium salt and proved that the oxygen from the carbonyl urethane group is not involved in the coordination of sodium cation, but the presence of the urethane moiety improves antibacterial activity. The differences between the biological activity of MON and its urethane are connected to the stability of the complex and the chemical and biological nature and size of the urethane substituent. Huczyński’s group provided evidence that the sodium salt of the phenylurethane derivative exhibits higher antibacterial activity against human pathogenic bacteria S. aureus and S. epidermidis, including resistant strains, than the parent compound [35]. This result inspired us to synthesize eight MON urethanes and examine their anticancer activities. The series of urethanes (7–14) (Fig. 1) was obtained in the reaction between a respective isocyanate and MON in anhydrous toluene [24].
3.2. Evaluation of the anti-proliferative activity of MON and its analogs in three-dimensional mini-ring cell viability screening
MON, and its 14 analogs (Fig. 1) were evaluated for their antiproliferative activity towards the human glioblastoma cell line U-118MG via three-dimensional mini-ring cell viability assays. The mini-ring system [10,31] was optimized and adapted here for an MTT endpoint assay. In this model, cells are seeded in a three-dimensional format around the rim of the well (mini-ring) (Fig. 2).
Such a system more reliably mimics cell-cell interactions, tumor microenvironment and response to the compounds when compared with conventional cell monolayer assays [10]. The results presented as IC50 ± SD are summarized in Table 1, with corresponding cell viability curves shown in Fig. S1 in Supplementary material.
We identified three esters (1, 2, and 4 IC50 = 168.7 ± 82.2–291.7 ± 68.8 nM) and four urethanes (8, 9, 11, and 12 IC50 = 91.5 ± 54.4–257.3 ± 135.6 nM) of MON that were significantly more active than the parental MON compound (IC50 = 612.6 ± 184.4 nM) (as indicated by lower IC50 values). Of note, the IC50 value for MON towards GBM organoids appears to be lower than previously reported for colon cancer (2.5 μM) [36], myeloma (1 μM) [37], and Mel-624 melanoma (0.7 μM) [16] cells. This suggests a higher sensitivity of GBM to MON, especially since our model is three-dimensional to mimic tissue architecture and drug permeability may be reduced. MON was also tested by Wan et al. towards the panel of GBM cell lines, however, the authors did not provide IC50 values, which precludes an accurate comparison with our results [21].
Based on the analysis of cell viability curves of the most potent compounds, MON, 1, 2, 4 and 12 appeared to be cytotoxic (indicated by the dose response curve reaching near complete inhibition of cell viability) versus compounds 8, 9 and 11, which showed a curve characteristic for cytostatic compounds (Fig. S1 in Supplementary material). The most active analogs, as well as parental MON were selected for further study.
3.3. MON and its analogs induce DNA fragmentation in GBM organoids
The two-dimensional cell monolayer model commonly used in drug discovery poorly reflects a native tumor, and this may be at least partially responsible for the high failure rate of new drug candidates in clinical trials [8,9,38]. To overcome these limitations, we employed three-dimensional tumor organoids as described in Materials and methods, and shown in Fig. 3A. Cancer organoids better recapitulate the three-dimensional architecture of cancer, and constitute a superior model to identify and test novel anticancer agents [8].
To further elucidate the mechanism underlying the promising activity of MON and its analogs towards GBM organoids, we employed flow cytometry to evaluate DNA content and its fragmentation. Briefly, DNA content was measured by propidium iodide (PI) staining, and cells with sub-G1 DNA (<2 N) were assessed as dying or dead. GBM organoids were treated for 24, 48 or 72 h with the parental MON, or the most potent esters and urethanes 1, 2, 4, 8, 9, 11 and 12 each at concentrations equal to 5 × respective IC50 values (Table 1). Treatment with 0.1% DMSO (control) at equal time points was employed as a negative control. The full set of representative cytograms is shown in Fig. 3B. The summary of cells in different phases of cell cycle are summarized from the mean of three replicates and presented in Fig. 3C. Statistically significant increases in sub-G1 DNA content when compared to the DMSO control were observed as early as 24 h after of treatment with compounds 1, 2, 4 and 12. Importantly, since the concentrations of the compounds were adjusted to the respective 5 × IC50 values, those analogs were able to induce DNA fragmentation earlier, and in lower doses than unmodified MON. After 48 h, significant sub-G1 DNA content was recorded for the treatment with MON, and further increased for the treatments with 1 and 4. After 72 h MON and compound 1 induced an additional increase of sub-G1 DNA and this was also seen after treatment with compound 8. Based on the cytotoxic profile of compound 1 (Fig. S1 in Supplementary material), as well as the above observation that this molecule was able induce DNA fragmentation rapidly and in lower dose than parent MON, compound 1 was prioritized for further studies.
3.4. MON and compound 1 attenuate U-118MG cell migration
Since one of the major issues contributing to the failure of GBM therapeutics is its diffuse infiltrative growth, novel drug candidates should inhibit not only cell proliferation, but also their migration [39]. We therefore investigated the ability of MON, and compound 1 to inhibit GBM cell migration in a wound healing assay. The data are presented in Fig. 4. A significant reduction of the migration of U-118MG cells was seen with compound 1 and MON and this was observed as early as 28 h after treatment (82% and 74% vs. 55% of wound surface area treatments vs. control respectively) (Fig. 4B). Significant inhibition of cell migration was maintained after 48 h and 74 h of incubation with compound 1 and the parental MON (86% and 74% vs. 32% of wound surface area treatments vs. control respectively for the 48 h time point) (90% and 78% vs. 9% of wound surface area treatments vs. control respectively for the 74 h time point). Importantly, due to the differences in IC50 values (Table 1) compound 1 was used in this assay in approximately twice lower concentration than parent MON. Of note, the small increase of wound surface area for MON and compound 1 treatments over time might be related to the cytotoxic activity of those molecules, although concentrations equal to ½ of IC50 values (Table 1) were employed in the present experiment to minimize such an effect. Based on these results compound 1 showed superior anti-migratory effect over the parental MON.
Fig. 4.
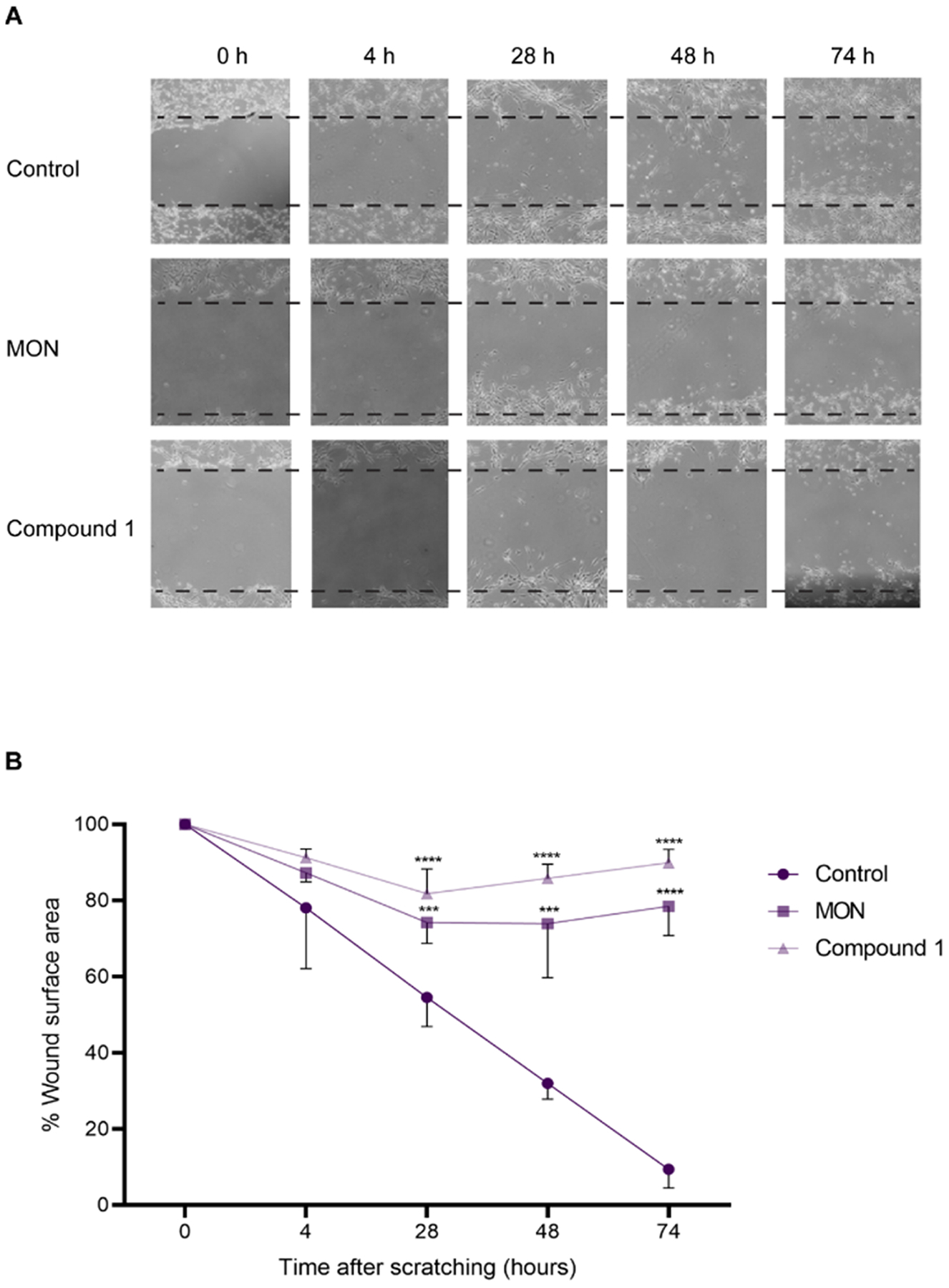
Wound healing assay of U-118MG cells after treatment with 0.1% DMSO (control), 306 nM MON, or 146 nM compound 1 (concentrations equal to respective ½ of IC50 values) for 74 h. Wounds were created on a confluent cell monolayer and images were taken at 0, 4, 28, 48, and 74 h after scratching. Three biological replicates were made. A. Representative images of wound healing; B. Quantification of wound healing. Data represent mean ± SD (n = 3), *** p ≤ 0.001, ****p ≤ 0.0001.
3.5. MON and compound 1 induce features of apoptotic cell death, DNA damage response and downregulate p-STAT3, p-Akt and p-GSK-3β in GBM organoids
In order to determine whether MON and its analog induce apoptosis, GBM organoids were treated with MON, compound 1 or DMSO control for 24 or 48 h and changes in the levels of poly (ADP-ribose) polymerase (PARP) were measured via immunoblotting as described in Materials and methods. The molecular weight of intact PARP protein is 116 kDa, and during the apoptosis it is cleaved by caspases to produce a major fragment of 89 kDa [40].
As shown in Fig. 5A and B, treatment with MON for 48 h induced significant loss of PARP expression with its characteristic degradation product detected. Loss of the 116 kDa PARP was more dramatic after treatment with compound 1 for 48 h (Fig. 5A and B). An initial slight increase in the expression of PARP after 24 h treatment with compound 1 may suggest the induction of defense mechanisms protecting cells from the toxic effects of the compound (Fig. 5B).
Fig. 5.
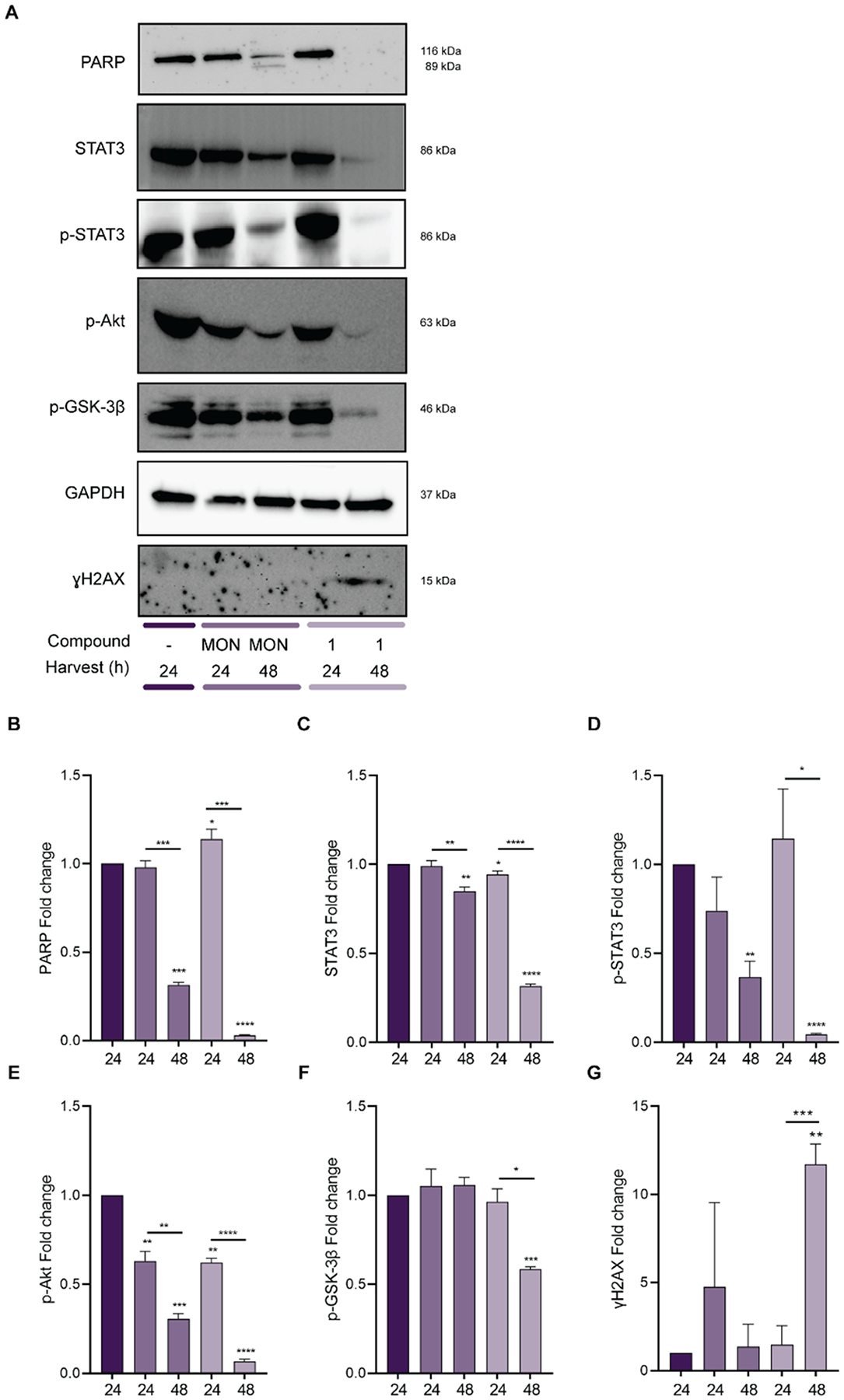
Effect of the treatment with MON or its analog 1 on PARP, STAT3, p-STAT3, p-AKT, p-GSK-3β, and ɣH2AX expression. U-118MG organoids were treated with 0.1% DMSO (control), or 3 μM MON, 1.5 μM compound 1 for 24 or 48 h as indicated, and extracts were prepared as described in the Materials and methods and subjected to immunoblotting with anti-sera indicated. GAPDH was used as a loading control. A. Representative immunoblots; B-G. Bar diagrams representing the fold changes of proteins normalized to GAPDH. Immunoblots were quantified with ImageJ software by measuring the band densitometry. Data represent mean ± SD (n = 3), *p ≤ 0.05, **p ≤ 0.01, *** p ≤ 0.001, ****p ≤ 0.0001.
Persistent activation of signal transducer and activator of transcription 3 (STAT3) has been shown to be a driver of GBM cell proliferation, anti-apoptosis, glioma stem cell maintenance, tumor invasion, angiogenesis, and immune evasion [41]. Therefore STAT3 is an attractive target for anti-GBM drug discovery as well as an indicator of GBM prognosis [41]. The application of MON as a STAT3 specific inhibitor has been previously patented (patent no. CN102552300A). For this reason, it was of interest to examine the effect of MON or compound 1 on the status of STAT3 activation in GBM organoids. As evident in Fig. 5A, C-D the levels of total STAT3 and phosphorylated STAT3 were downregulated upon 48 h treatment with MON or compound 1.
The PI3K/Akt/GSK-3β pathway is known to promote cancer survival, and its inhibition is associated with apoptosis and/or autophagy [22]. Both Akt and GSK-3β play a role in numerous cellular processes such as proliferation, cell cycle, growth and mortality [42]. Deregulated activity of one of these molecules or their targets is often seen in GBM, and contributes to tumor progression [42]. Therefore, Akt and GSK-3β signaling pathways are attractive targets for anti-GBM drug discovery [43]. MON has been previously shown to reduce phosphorylation of Akt in U373 glioma cells, EGF-treated head and neck squamous cell carcinoma SCC9 and GBM [21,44,45]. Therefore, it was of our interest to investigate the effect of treatment with MON or compound 1 on phosphorylated Akt (p-Akt) in GBM organoids by immunoblotting. The results shown in Fig. 5A and E indicate that after 24 h treatment MON or compound 1 significantly reduced phosphorylation of Akt, with further significant decrease in the phosphorylation of this protein after 48 h. Moreover, both MON and compound 1 induced significant downregulation of p-GSK-3β in GBM organoids in a time dependent manner as compared to control treatment (Fig. 5A and F). Since phosphorylation of GSK-3β may be mediated by several kinases [46], its decrease upon treatment with MON and compound 1 may not be a direct consequence of the downregulated phosphorylation of Akt. The above data suggest that decreased phosphorylation of Akt and GSK-3β may contribute to decreased viability of GBM organoids, however, since the activity of Akt is tightly regulated at different levels in a specific context dependent manner [47], further study is warranted to precisely the explain mechanism underlying inhibition of phosphorylation of these proteins by compound 1.
The mode of action of the majority of current anti-cancer drugs is induction of double-strand DNA breaks, which are the most severe of all mammalian DNA lesions [48]. ɣH2AX, a histone H2AX variant, phosphorylated at the Ser-139 residue has become known as a highly specific and sensitive molecular marker for determination of double stranded DNA damage [49–52]. To investigate whether the cytotoxicity induced by MON or compound 1 in GBM organoids involves DNA damage, organoids were treated with for 24 or 48 h in the presence of control or compound of interest and immunoblotting was performed with antibody to ɣH2AX. Our data show that treatment of GBM organoids for 48 h with compound 1 induced double strand DNA damage (Fig. 5A and G). Interestingly such an effect was not observed after treatment with parent MON, suggesting potential differences in the mechanisms of action of these compounds in GBM organoids.
3.6. MON and compound 1 induce autophagy in GBM organoids
Autophagy is a self-degradable process during which proteins and organelles are sequestered into autophagic vesicles named autophagosomes, which further merge with lysosomal acidic cell components to form autophagolysosomes [53]. Since MON has been previously reported to induce autophagy in UOK146 renal carcinoma cell line [54] it was of interest to investigate whether MON or compound 1 would show similar effects in GBM organoids. At the molecular level, the light chain 3 (LC3-I) protein associated with microtubules is cleaved and bound to phosphatidylethanolamine to form LC3-II, which is further recruited to the membranes of the autophagosomes [55]. Western blot analysis (Fig. 6A and B) suggests that MON and compound 1 induced a significant upregulation in LC3II expression (14 kDa) in GBM organoids after 24 and 48 h of treatment. Autophagy can function as a pro-death or pro-survival pathway depending on the stimulus [44,56]. In order to determine which role autophagy played in this context, GBM organoids were treated with MON and compound 1 in the absence or presence of bafilomycin A1, and DNA fragmentation was measured by flow cytometry. Bafilomycin A1 is a chemical inhibitor of late stage autophagy, which targets the vacuolar-type H+-ATPase responsible for acidifying lysosomes [57]. Since bafilomycin A1 is inherently toxic, we first performed MTT cell viability assay on U-118MG mini-rings in order to identify its sub-lethal concentration towards these cells (Fig. 6C). Based on the results from this experiment, the concentration of 3 nM was employed as this is below the IC50 value. As determined in Fig. 6D the amount of sub-G1 DNA significantly increased in GBM organoids treated with MON or compound 1 in the presence of bafilomycin A1 versus treatment with ionophores alone. Importantly, there was no statistically significant difference in the amount of fragmented DNA induced by control and bafilomycin A1 alone, confirming that the increase induced by co-treatment was not associated with additive effect of the inhibitor and tested compounds. The data suggest that similarly to the majority of anti-cancer agents [58,59] autophagy may function as a salvage pathway to protect GBM organoids from cell death induced by MON and compound 1. These results indicate that autophagy inhibitors may enhance the cytotoxic effect of MON and its analogs. This finding encourages more detailed studies involving combination of autophagy inhibitors and other polyether ionophore antibiotics.
Fig. 6.
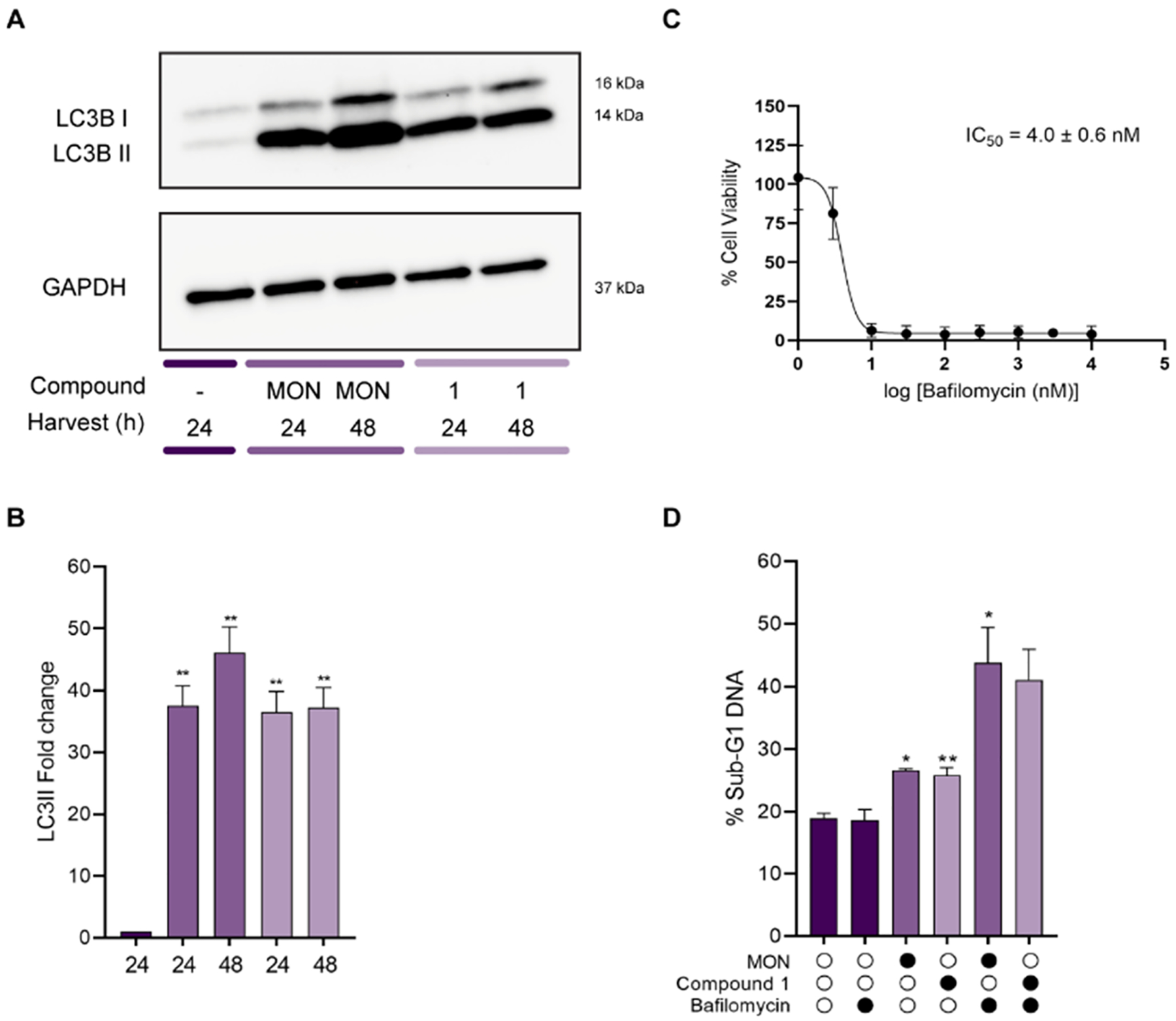
Effect of MON and compound 1 on autophagy induction in U-118MG organoids. Organoids were treated with 0.1% DMSO (control), 3 μM MON, or 1.5 μM compound 1 (concentrations equal to respective 5 × IC50 values) for times indicated. A. Extracts were prepared and subjected to immunoblotting for LC3 (16 and 14 kDa). GAPDH was employed as a loading control; B. Bar diagram showing changes of proteins normalized to GAPDH. Images were quantified by measuring the band intensity using ImageJ software. Data are represented as mean ± SD (n = 3), **p ≤ 0.01; C. Cell viability of Bafilomycin A1 as determined by MTT mini-ring assay. Results are given as mean ± SD (n = 4). Mini-rings were treated with vehicle (100% viability) or increasing concentrations of Bafilomycin A1 (see Materials and methods for details); D. Effect of the autophagy inhibitor Bafilomycin A1 on MON and compound 1 induced DNA fragmentation. U-118MG organoids were treated with 0.1% DMSO (control), 3 μM MON, 1.5 μM compound 1, or 3 nM Bafilomycin A1, alone or in combination for 48 h, and Sub-G1 DNA was determined by PI staining and flow cytometry as described in the Materials and methods. Results are given as mean ± SD (n = 3 and n = 2 for compound 1 and Bafilomycin A1 treatment), *p ≤ 0.05, **p ≤ 0.01.
3.7. MON and compound 1 show anti-GBM activity in a 3D host:tumor organoid model by inducing apoptotic cell death and DNA damage
The in vivo cancer microenvironment can have an acute effect on the cancer cells response to cellular stress, including genotoxic damage from radio- and chemotherapy [11]. Therefore, in order to investigate the activity of MON and compound 1 in a surrogate normal host tissue: tumor hybrid microenvironment we developed hiPSCs-derived cerebral organoids (COs) co-cultured with GBM cells (in a modification of the previously described GLICO (cerebral organoid glioma) model) [11]. Here, we generated human COs from hiPSCs constitutively expressing the green fluorescent protein (GFP) under control of the ACTB promoter according to a four stage protocol, where CO formation is initiated through intermediate embryoid bodies followed by expansion of neuroepithelia (Fig. 7A). COs are three-dimensional culture models characterized by structural organization and cellular composition characteristic of the developing human brain [11,60]. After a period of maturation, we sectioned COs and performed H&E as well as immunofluorescence staining. On routine H&E staining, COs recapitulated embryonal central nervous system structures, such as multiple neural rosettes / neural tubes (Fig. 7B, left panel) as well as papillary structures lined by cuboidal epithelium recapitulating ependyma and choroid plexus (Fig. 7B, right panel), with a proliferative zone of neural stem cells (Nestin+) (Fig. 7C), neurons (TUJ1 +), primitive ventricular system (SOX2 +/Ki67 +), intermediate zone (TBR2 +) and cortical plate (MAP2 +) (Fig. 7C). Positive staining for the apical tight junction protein N-cadherin provided additional evidence of regulated development suggesting the same type of polarity found within the neural tube of the embryonic neural plate (Fig. 7C). These morphological and immunofluorescence findings suggested the level of differentiation and development of generated COs as early stage human fetal brain.
To model human GBM infiltration we co-cultured individual, fully formed COs with varying numbers of red fluorescent protein (RFP)-labeled U-87MG cells for 24 h (Fig. 8A). Subsequently, tumor growth rate was monitored daily by immunofluorescence microscopy to track tumor formation. As shown in Fig. 8A, RFP expression at day 5 was substantially higher for tumors originating from 100,000 cells compared with GLICOs from 10,000 or 50,000 cells. Consequently, we chose 100,000 RFP-U-87MG cells for all subsequent assays. Fig. 8B demonstrated increasing RFP expression at days 1 and 6 for GLICO cultures, indicating that the RFP-U-87MG tumor cells are proliferating over time. Upon reaching considerable tumor formation after six days of co-culture COs co-treated with U-87MG/RFP cells were subjected to the treatment with 0.1% DMSO (control), MON or compound 1 (Fig. 8B). Compound 1 reduced the tumor size after four days of treatment as compared with day one (Fig. 8B).
Fig. 8.

GLICO tumors show differential response to treatment with MON and compound 1. A. 10,000, 50,000 or 100,000 RFP expressing U-87MG cells were co-cultured with COs as described in Materials and methods. Shown are representative images of RFP expression in GLICO tumors at day 5 post co-culture; B. Representative fluorescent microscopic images of GFP (left) and RFP (right) in GLICO tumors at day 1 of co-culture with U-87MG RFP expressing cells, day 6 (first day of 0.1% DMSO (control) or 1226 nM MON or 584 nM compound 1 treatment) and day 9 of the treatment, n = 4–5 COs per group. C. GLICO tumors at day 6 post co-culture with RFP expressing U87MG cells were treated with 0.1% DMSO (control) or 1226 nM MON or 584 nM compound 1. Shown are representative images of immunofluorescence staining for PARP, 20X magnification; D. Quantification of PARP intensity from 139 to 1171 cells after four days of treatment, *p ≤ 0.05, ****p ≤ 0.0001; E. GLICO tumors were treated as described above. Representative images of immunofluorescence staining for ɣH2AX are shown, 20X magnification; F. Quantification of ɣH2AX intensity from 185 to 532 cells after four days of treatment, *p ≤ 0.05.
In order to assess whether GBM’s sensitivity to MON and compound 1 is similar or different in the GLICO model compared with cancer organoids (Figs. 2, 3, 5 and 6), GLICOs were treated with MON or compound 1 as described in Materials and methods and analyzed for PARP and ɣ H2AX (Fig. 8C–F). In order to ensure that the compound’s effect was measured only in the tumor area instead of the entire CO, all markers were analyzed in areas of co-localization with RFP expressed by U-87MG cells (Fig. 8C, E and G second panel). Consistent with our previous findings (Fig. 5A and B) both MON and compound 1 induced significant decrease of total PARP (Fig. 8C and D). Importantly, since the doses were adjusted to the respective 2 × IC50 values compound 1 was able to induce significant loss of PARP expression in approximately 2 times lower concentration than parental MON. Both MON and compound 1 induced the upregulation of the expression of DNA damage marker ɣH2AX, however only the effect of MON was statistically significant (Fig. 8E and F). This is contrary to our observations in the GBM organoid model, where only compound 1 induced double strand DNA damage (Fig. 5A and G) and highlights the utility of GLICO model to reveal mechanistic differences in possible drug behavior in more complex host:tumor surrogate 3D culture systems.
The ability to cross blood-brain barrier (BBB) is of special concern when it comes to development of glioblastoma targeting agents [42]. Molecules of less than 400–500 Da forming less than 8–10 hydrogen bonds with water are believed to be able to cross the BBB [61]. With a molecular mass of approximately 670–713 Da, MON and compound 1 seem not to meet the first criteria, and further studies would be needed to evaluate whether the second requirement is fulfilled. If unable to cross the BBB in their current compositions, the potential treatment of GBM with MON or its analogs could be achieved through localized delivery via an Ommaya reservoir, which is a soft, plastic dome-shaped device placed under the scalp. Ommaya reservoirs are used to deliver medications such as chemotherapy, monoclonal antibodies, or antibiotics directly into cerebrospinal fluid, as well as to collect it [62]. Another option to circumvent any BBB limitation could come from drug-loaded nanocarriers. Such nanocarriers can improve drug delivery to gliomas by chemical stabilization of the drug in the bloodstream, enhanced permeability and retention, active targeting through carrier- and receptor-mediated transporters, P-gp inhibition or cell-mediated targeting [63]. Summing up, although MON and its analog 1 may not pass BBB, which presents a limitation of their utility for the treatment of GBM, there exist several options such as an Ommaya reservoir for localized delivery or use of nanocarriers to improve delivery and retention across the BBB.
4. Conclusions
In the present study we identified seven analogs of MON characterized by significantly lower IC50 values in mini-ring cell viability screen than the parental MON. Five of these molecules induced significant DNA fragmentation in GBM organoids, suggestive of apoptotic cell death. The most potent compound 1 was selected for further studies where it showed enhanced anti-migratory properties in GBM cells over MON. Furthermore, compound 1 induced PARP degradation, diminished phosphorylation of STAT3, Akt and GSK-3β, increased ɣH2AX signaling and upregulated expression of the autophagy associated marker LCII in GBM organoid model in two times lower doses than parent MON. In order to study the activity of compound 1 and MON in a surrogate host:tumor microenvironment, we developed human COs from GFP expressing hiPSCs, which were co-cultured with U-87MG-RFP expressing cells to generate a modified GLICO model. Compound 1 and MON decreased RFP-U-87MG cell counts in the GLICO model, as well as a decrease in PARP expression. However, the exact molecular targets of MON scaffold-based compounds in GBM require further validation. A discrepancy between a novel compound’s activity in vitro and in vivo is a common phenomenon [42], and so there remains a question regarding the in vivo activity and efficacy of these molecules which will require further testing in animal models. We conclude that MON and its analogs constitute promising scaffolds for anti-GBM drug discovery, with further study warranted towards pre-clinical development of these compounds.
Supplementary Material
Acknowledgments
This work was supported by Winthrop P. Rockefeller Cancer Institute Team Science Award (to AMM, RLE and AR), a Barton Pilot Grant from UAMS College of Medicine (to AU), Equipment Award Program from UAMS College of Medicine (to AU), and grants from the National Institutes of Health: R01HD093461 (to AMM), P20GM121293 (to AJT), R24GM137786 (to AJT), and R01CA236209 (to AJT). This publication was also supported by Grant Number P30 GM103450 from the National Institute of General Medical Sciences of the National Institutes of Health (NIH). We thank Ms. Andrea Harris from the UAMS Flow Cytometry Core, Ms. Jennifer James from the UAMS Experimental Pathology Core and Mr. Jeff Kamykowski from the UAMS Digital Microscopy Core Laboratory. We would like to thank Drs. Eric Norris and HoWon Kim from StemCell Technologies for valuable discussions concerning iPSC culture and COs generation. We also thank Dr. Amanda Linkous for consultations and helpful suggestions for the generation of the GLICO model. Some elements in the figures were created using BioRender (BioRender.com).
Footnotes
CRediT authorship contribution statement
Alicja Urbaniak: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Validation, Visualization, Funding acquisition, Writing – original draft. Megan R. Reed: Data curation, Formal analysis, Investigation, Methodology, Visualization, Writing – review & editing. Billie Heflin: Data curation, Investigation, Visualization. John Gaydos: Data curation, Investigation, Visualization. Sergio Piña-Oviedo: Data curation, Formal analysis, Investigation, Methodology, Visualization, Writing – review & editing. Marta Jędrzejczyk: Data curation, Formal analysis, Resources, Visualization, Writing – review & editing. Greta Klejborowska: Data curation, Resources. Natalia Stępczyńska: Data curation, Resources. Timothy C. Chambers: Resources, Writing – review & editing. Alan J. Tackett: Funding acquisition, Resources. Analiz Rodriguez: Conceptualization, Funding acquisition, Methodology, Resources. Adam Huczyński: Conceptualization, Data curation, Project administration, Resources, Supervision, Writing – review & editing. Robert L. Eoff: Conceptualization, Funding acquisition, Project administration, Resources, Supervision. Angus M. MacNicol: Conceptualization, Formal analysis, Funding acquisition, Methodology, Project administration, Resources, Supervision, Writing – review & editing.
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Appendix A. Supplementary material
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.biopha.2022.113440.
References
- [1].Ostrom QT, Gittleman H, Truitt G, Boscia A, Kruchko C, Barnholtz-Sloan JS, Duncan DL, CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2011–2015 introduction, Neuro-Oncology 20 (2018) 1–86, 10.1093/neuonc/noy131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Stupp R, Taillibert S, Kanner AA, Kesari S, Steinberg DM, Toms SA, Taylor LP, Lieberman F, Silvani A, Fink KL, Barnett GH, Zhu J-J, Henson JW, Engelhard HH, Chen TC, Tran DD, Sroubek J, Tran ND, Hottinger AF, Landolfi J, Desai R, Caroli M, Kew Y, Honnorat J, Idbaih A, Kirson ED, Weinberg U, Palti Y, Hegi ME, Ram Z, Maintenance therapy with tumor-treating fields plus temozolomide vs temozolomide alone for glioblastoma a randomized clinical trial preliminary communication, TRIAL Regist. Clin. Identifier NCT00916409, JAMA 314 (2015) 2535–2543, 10.1001/jama.2015.16669. [DOI] [PubMed] [Google Scholar]
- [3].Brennan CW, Verhaak RGW, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, Beroukhim R, Bernard B, Wu CJ, Genovese G, Shmulevich I, Barnholtz-Sloan J, Zou L, Vegesna R, Shukla SA, Ciriello G, Yung WK, Zhang W, Sougnez C, Mikkelsen T, Aldape K, Bigner DD, Van Meir EG, Prados M, Sloan A, Black KL, Eschbacher J, Finocchiaro G, Friedman W, Andrews DW, Guha A, Iacocca M, O’Neill BP, Foltz G, Myers J, Weisenberger DJ, Penny R, Kucherlapati R, Perou CM, Hayes DN, Gibbs R, Marra M, Mills GB, Lander E, Spellman P, Wilson R, Sander C, Weinstein J, Meyerson M, Gabriel S, Laird PW, Haussler D, Getz G, Chin L, The somatic genomic landscape of glioblastoma, Cell 155 (2) (2013) 462–477, 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Darmanis S, Sloan SA, Croote D, Mignardi M, Chernikova S, Samghababi P, Zhang Y, Neff N, Kowarsky M, Caneda C, Li G, Chang SD, Connolly ID, Li Y, Barres BA, Gephart MH, Quake SR, Single-cell RNA-Seq analysis of infiltrating neoplestic cells at the migrating front of human glioblastoma, Cell Rep. 21 (5) (2017) 1399–1410, 10.1016/j.celrep.2017.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, Richman AR, Silverbush D, Shaw ML, Hebert CM, Dewitt J, Gritsch S, Perez EM, Gonzalez Castro LN, Lan X, Druck N, Rodman C, Dionne D, Kaplan A, Bertalan MS, Small J, Pelton K, Becker S, Bonal D, Nguyen QD, Servis RL, Fung JM, Mylvaganam R, Mayr L, Gojo J, Haberler C, Geyeregger R, Czech T, Slavc I, Nahed BV, Curry WT, Carter BS, Wakimoto H, Brastianos PK, Batchelor TT, Stemmer-Rachamimov A, Martinez-Lage M, Frosch MP, Stamenkovic I, Riggi N, Rheinbay E, Monje M, Rozenblatt-Rosen O, Cahill DP, Patel AP, Hunter T, Verma IM, Ligon KL, Louis DN, Regev A, Bernstein BE, Tirosh I, Suvà ML, An integrative model of cellllular states, plasticity, and genetics for glioblastoma, Cell 178 (4) (2019) 835–849, 10.1016/j.cell.2019.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Noch EK, Ramakrishna R, Magge R, Challenges in the treatment of glioblastoma: multisystem mechanisms of therapeutic resistance, World Neurosurg. 116 (2018) 505–517, 10.1016/J.WNEU.2018.04.022. [DOI] [PubMed] [Google Scholar]
- [7].Jacob F, Salinas RD, Zhang DY, Nguyen PTT, Schnoll JG, Wong SZH, Thokala R, Sheikh S, Saxena D, Prokop S, ao Liu D, Qian X, Petrov D, Lucas T, Chen HI, Dorsey JF, Christian KM, Binder ZA, Nasrallah M, Brem S, O’Rourke DM, li Ming G, Song H, A patient-derived glioblastoma organoid model and biobank recapitulates inter- and intra-tumoral heterogeneity, e22, Cell 180 (2020) 188–204, 10.1016/J.CELL.2019.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Drost J, Clevers H, Organoids in cancer research, Nat. Rev. Cancer 18 (7) (2018) 407–418, 10.1038/s41568-018-0007-6. [DOI] [PubMed] [Google Scholar]
- [9].Kamb A, What’s wrong with our cancer models, Nat. Rev. Drug Discov (2005) 161–165, 10.1038/nrd1635. [DOI] [PubMed] [Google Scholar]
- [10].Phan N, Hong JJ, Tofig B, Mapua M, Elashoff D, Moatamed NA, Huang J, Memarzadeh S, Damoiseaux R, Soragni A, A simple high-throughput approach identifies actionable drug sensitivities in patient-derived tumor organoids, Commun. Biol 2 (78) (2019), 10.1038/s42003-019-0305-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Linkous A, Balamatsias D, Snuderl M, Edwards L, Miyaguchi K, Milner T, Reich B, Cohen-Gould L, Storaska A, Nakayama Y, Schenkein E, Singhania R, Cirigliano S, Magdeldin T, Lin Y, Nanjangud G, Chadalavada K, Pisapia D, Liston C, Fine HA, Modeling patient-derived glioblastoma with cerebral organoids, e5, Cell Rep. 26 (2019) 3203–3211, 10.1016/J.CELREP.2019.02.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Nachliel E, Finkelstein Y, Gutman M, The mechanism of monensin-mediated cation exchange based on real time measurements, Biochim. Biophys. Acta Biomembr 1285 (1996) 131–145, 10.1016/S0005-2736(96)00149-6. [DOI] [PubMed] [Google Scholar]
- [13].Pressman BC, Fahim M, Pharmacology and toxicology of the monovalent carboxylic ionophores, Annu. Rev. Pharmacol. Toxicol 22 (1982) 465–490, 10.1146/ANNUREV.PA.22.040182.002341. [DOI] [PubMed] [Google Scholar]
- [14].Chapman HD, Jeffers TK, Williams RB, Forty years of monensin for the control of coccidiosis in poultry, Poult. Sci 89 (2010) 1788–1801, 10.3382/ps.2010-00931. [DOI] [PubMed] [Google Scholar]
- [15].Markowska A, Kaysiewicz J, Markowska J, Huczyński A, Doxycycline, salinomycin, monensin and ivermectin repositioned as cancer drugs, Bioorg. Med. Chem. Lett 29 (2019) 1549–1554, 10.1016/J.BMCL.2019.04.045. [DOI] [PubMed] [Google Scholar]
- [16].Zhang Z, Xin H, Li J, Zhang H, Li Y, Zeng S, Wang Z, Deng F, Monensin may inhibit melanoma by regulating the selection between differentiation and stemness of melanoma stem cells, Peer J. 25 (7) (2019), 10.7717/peerj.7354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Pádua D, Barros R, Amaral AL, Mesquita P, Freire AF, Sousa M, Maia AF, Caiado I, Fernandes H, Pombinho A, Pereira CF, Almeida R, A SOX2 reporter system identifies gastric cancer stem-like cells sensitive to monensin, Cancers 12 (2) (2020) 495, 10.3390/cancers12020495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Yusenko MV, Trentmann A, Andersson MK, Ghani LA, Jakobs A, Arteaga Paz MF, Mikesch JH, Peter von Kries J, Stenman G, Klempnauer KH, Monensin, a novel potent MYB inhibitor, suppresses proliferation of acute myeloid leukemia and adenoid cystic carcinoma cells, Cancer Lett. 479 (2020) 61–70, 10.1016/J.CANLET.2020.01.039. [DOI] [PubMed] [Google Scholar]
- [19].Vanneste M, Huang Q, Li M, Moose D, Zhao L, Stamnes MA, Schultz M, Wu M, Henry MD, Carver LA, High content screening identifies monensin as an eMt-selective cytotoxic compound, Sci. Rep 9 (1200) (2019), 10.1038/s41598-018-38019-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Yoon MJ, Kang J, Kim IY, Kim EH, Lee JA, Lim H, Kwon TK, Choi KS, Monensin, a polyether ionophore antibiotic, overcomes TRAIL resistance in glioma cells via endoplasmic reticulum stress, DR5 upregulation and c-FLIP downregulation, Carcinogenesis 34 (2013) 1918–1928, 10.1093/carcin/bgt137. [DOI] [PubMed] [Google Scholar]
- [21].Wan W, Zhang X, Huang C, Chen L, Yang X, Bao K, Peng T, Monensin inhibits glioblastoma angiogenesis via targeting multiple growth factor receptor signaling, Biochem. Biophys. Res. Commun 530 (2020) 479–484, 10.1016/J.BBRC.2020.05.057. [DOI] [PubMed] [Google Scholar]
- [22].Omoruyi SI, Ekpo OE, Semenya DM, Jardine A, Prince S, Exploitation of a novel phenothiazine derivative for its anti-cancer activities in malignant glioblastoma, apoptosis, Apoptosis 25 (2020) 261–274, 10.1007/s10495-020-01594-5. [DOI] [PubMed] [Google Scholar]
- [23].Ashburn TT, Thor KB, Drug repositioning: identifying and developing new uses for existing drugs, Nat. Rev. Drug Discov 3 (2004) 3, 10.1038/nrd1468. [DOI] [PubMed] [Google Scholar]
- [24].Westley JW, Liu CM, Evans RH, Sello LH, Troupe N, Hermann T, Preparation, properties and biological activity of natural and semisynthetic urethanes of monensin, J. Antibiot 36 (1983) 1195–1200, 10.7164/antibiotics.36.1195. [DOI] [PubMed] [Google Scholar]
- [25].Huczyński A, Ratajczak-Sitarz M, Stefańska J, Katrusiak A, Brzezinski B, Bartl F, Reinvestigation of the structure of monensin A phenylurethane sodium salt based on X-ray crystallographic and spectroscopic studies, and its activity against hospital strains of methicillin-resistant S. epidermidis and S. aureus, J. Antibiot 64 (2011) 249–256, 10.1038/ja.2010.167. [DOI] [PubMed] [Google Scholar]
- [26].Klejborowska G, Jędrzejczyk M, Stępczyńska N, Maj E, Wietrzyk J, Huczyński A, Antiproliferative activity of ester derivatives of monensin A at the C-1 and C-26 positions, Chem. Biol. Drug Des 94 (2019) 1859–1864, 10.1111/cbdd.13581. [DOI] [PubMed] [Google Scholar]
- [27].Huczyński A, Przybylski P, Brzezinski B, Complexes of monensin A methyl ester with Mg2+, Ca2+, Sr2+, Ba2+ cations studied by electrospray ionization mass spectrometry and PM5 semiempirical method, J. Mol. Struct 788 (2006) 176–183, 10.1016/J.MOLSTRUC.2005.11.032. [DOI] [Google Scholar]
- [28].Huczyński A, Michalak D, Przybylski P, Brzezinski B, Bartl F, Monensin A benzyl ester and its complexes with monovalent metal cations studied by spectroscopic, mass spectrometry and semiempirical methods, J. Mol. Struct 797 (2006) 99–110, 10.1016/J.MOLSTRUC.2006.03.014. [DOI] [Google Scholar]
- [29].Jędrzejczyk M, Stępczyńska N, Klejborowska G, Podsiad M, Stefańska J, Steverding D, Huczyński A, Synthesis and evaluation of antibacterial and trypanocidal activity of derivatives of monensin A, Bioorg. Med. Chem. Lett 58 (2022), 128521, 10.1016/J.BMCL.2021.128521. [DOI] [PubMed] [Google Scholar]
- [30].Robin T, Capes-Davis A, Bairoch A, CLASTR: the cellosaurus STR similarity search tool - a precious help for cell line authentication, Int. J. Cancer (2020), 10.1002/ijc.32639. [DOI] [PubMed] [Google Scholar]
- [31].Nguyen HTL, Soragni A, Patient-derived tumor organoid rings for histologic characterization and high-throughput screening, STAR Protoc. 1 (2020), 10.1016/J.XPRO.2020.100056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Urbaniak A, Reed MR, Fil D, Moorjani A, Heflin S, Antoszczak M, Sulik M, Huczyński A, Kupsik M, Eoff RL, MacNicol MC, Chambers TC, MacNicol AM, Single and double modified salinomycin analogs target stem-like cells in 2D and 3D breast cancer models, Biomed. Pharmacother 141 (2021), 111815, 10.1016/J.BIOPHA.2021.111815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Klejborowska G, Jędrzejczyk M, Stępczyńska N, Maj E, Wietrzyk J, Huczyński A, Antiproliferative activity of ester derivatives of monensin A at the C-1 and C-26 positions, Chem. Biol. Drug Des 94 (2019) 1859–1864, 10.1111/CBDD.13581. [DOI] [PubMed] [Google Scholar]
- [34].Gaboyard C, Dauphin G, Vaufrey F, Jeminet G, Analogs of monensin with a modified C25–C26 moiety: Na+/K+ selectivity and biological activity, Agric. Biol. Chem 54 (1990) 1149–1155, 10.1080/00021369.1990.10870088. [DOI] [Google Scholar]
- [35].Huczyński A, Ratajczak-Sitarz M, Stefańska J, Katrusiak A, Brzezinski B, Bartl F, Reinvestigation of the structure of monensin A phenylurethane sodium salt based on X-ray crystallographic and spectroscopic studies, and its activity against hospital strains of methicillin-resistant S. epidermidis and S. aureus, J. Antibiot 646 (64) (2011) 249–256, 10.1038/ja.2010.167. [DOI] [PubMed] [Google Scholar]
- [36].Park WH, Kim ES, Jung CW, Kim BK, Lee YY, Monensin-mediated growth inhibition of SNU-C1 colon cancer cells via cell cycle arrest and apoptosis, Int. J. Oncol 22 (2003) 377–382, 10.3892/IJO.22.2.377/HTML. [DOI] [PubMed] [Google Scholar]
- [37].Park WH, Kim ES, Kim BK, Lee YY, Monensin-mediated growth inhibition in NCI-H929 myeloma cells via cell cycle arrest and apoptosis, Int. J. Oncol 23 (2003) 197–204, 10.3892/IJO.23.1.197/HTML. [DOI] [PubMed] [Google Scholar]
- [38].Caponigro G, Sellers WR, Advances in the preclinical testing of cancer therapeutic hypotheses, Nat. Rev. Drug Discov 103 (10) (2011) 179–187, 10.1038/nrd3385. [DOI] [PubMed] [Google Scholar]
- [39].Lefranc F, Le Rhun E, Kiss R, Weller M, Glioblastoma quo vadis: Will migration and invasiveness reemerge as therapeutic targets? Cancer Treat. Rev 68 (2018) 145–154, 10.1016/J.CTRV.2018.06.017. [DOI] [PubMed] [Google Scholar]
- [40].Simbulan-Rosenthal CM, Rosenthal DS, Iyer S, Boulares AH, Smulson ME, Transient poly(ADP-ribosyl)ation of nuclear proteins and role of poly(ADP-ribose) polymerase in the early stages of apoptosis, J. Biol. Chem 273 (1998) 13703–13712, 10.1074/JBC.273.22.13703. [DOI] [PubMed] [Google Scholar]
- [41].Chang N, Ahn SH, Kong DS, Lee HW, Nam DH, The role of STAT3 in glioblastoma progression through dual influences on tumor cells and the immune microenvironment, Mol. Cell. Endocrinol 451 (2017) 53–65, 10.1016/J.MCE.2017.01.004. [DOI] [PubMed] [Google Scholar]
- [42].Szeliga M, Karpińska M, Rola R, Niewiadomy A, Design, synthesis and biological evaluation of novel 1,3,4-thiadiazole derivatives as anti-glioblastoma agents targeting the AKT pathway, Bioorg. Chem 105 (2020), 10.1016/J.BIOORG.2020.104362. [DOI] [PubMed] [Google Scholar]
- [43].Majewska E, Szeliga M, AKT/GSK3β signaling in glioblastoma, Neurochem. Res 42 (2017) 918–924, 10.1007/s11064-016-2044-x/Published. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Fan Q-W, Cheng C, Hackett C, Feldman M, Houseman BT, Nicolaides T, Haas-Kogan D, David James C, Oakes SA, Debnath J, Shokat KM, Weiss WA, Akt and autophagy cooperate to promote survival of drug-resistant glioma, Sci. Signal 3 (147) (2010), 10.1126/scisignal.2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Dayekh K, Johnson-Obaseki S, Corsten M, Villeneuve PJ, Sekhon HS, Weberpals JI, Dimitroulakos J, Monensin inhibits epidermal growth factor receptor trafficking and activation: synergistic cytotoxicity in combination with EGFR inhibitors, Mol. Cancer Ther 13 (2014) 2559–2571, 10.1158/1535-7163.MCT-13-1086/85531/AM/MONENSIN-INHIBITS-EPIDERMAL-GROWTH-FACTOR-RECEPTOR. [DOI] [PubMed] [Google Scholar]
- [46].Beurel E, Grieco SF, Jope RS, Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases, Pharmacol. Ther 148 (2015) 114–131, 10.1016/J.PHARMTHERA.2014.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Hart JR, Vogt PK, Phosphorylation of AKT: a mutational analysis, Oncotarget 2 (6) (2011) 467–476, 10.18632/oncotarget.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Annovazzi L, Mellai M, Schiffer D, Chemotherapeutic drugs: DNA damage and repair in glioblastoma, Cancers 9 (6) (2017) 57, 10.3390/cancers9060057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Wu C-H, Bai L-Y, Tsai M-H, Chu P-C, Chiu C-F, Chen MY, Chiu S-J, Chiang JH, Weng J-R, Pharmacological exploitation of the phenothiazine antipsychotics to develop novel antitumor agents-A drug repurposing strategy, Sci. Rep 6 (2016), 10.1038/srep27540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Gangopadhyay S, Karmakar P, Dasgupta U, Chakraborty A, Trifluoperazine stimulates ionizing radiation induced cell killing through inhibition of DNA repair, Mutat. Res. Genet. Toxicol. Environ. Mutagen 633 (2007) 117–125, 10.1016/J.MRGENTOX.2007.05.011. [DOI] [PubMed] [Google Scholar]
- [51].Zong D, Hååg P, Yakymovych I, Lewensohn R, Viktorsson K, Chemosensitization by phenothiazines in human lung cancer cells: impaired resolution of gamma H2AX and increased oxidative stress elicit apoptosis associated with lysosomal expansion and intense vacuolation, Cell Death Dis. 2 (2011), 10.1038/cddis.2011.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM, DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139, J. Biol. Chem 273 (1998) 5858–5868, 10.1074/JBC.273.10.5858. [DOI] [PubMed] [Google Scholar]
- [53].Mathew R, Karantza-Wadsworth V, White E, Role of autophagy in cancer, Nat. Rev. Cancer 7 (2007) 961–967, 10.1038/nrc2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Prakash Verma S, Das P, Monensin induces cell death by autophagy and inhibits matrix metalloproteinase 7 (MMP7) in UOK146 renal cell carcinoma cell line,In Vitro Cell, Dev. Biol. Anim 54 (2018) 736–742, 10.1007/s11626-018-0298-7. [DOI] [PubMed] [Google Scholar]
- [55].Tanida I, Ueno T, Kominami E, LC3 conjugation system in mammalian autophagy, Int. J. Biochem. Cell Biol 36 (2004) 2503–2518, 10.1016/J.BIOCEL.2004.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Galluzzi L, Vitale I, Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018, Cell Death Differ. 25 (2018) 486–541, 10.1038/s41418-017-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Mauvezin C, Neufeld TP, Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/ SERCA-dependent autophagosome-lysosome fusion, Autophagy 11 (2015) 1437–1438, 10.1080/15548627.2015.1066957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].White E, Deconvoluting the context-dependent role for autophagy in cancer, Nat. Rev. Cancer 12 (2012) 401–410, 10.1038/nrc3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Amaravadi R, Kimmelman AC, White E, Recent insights into the function of autophagy in cancer, Genes Dev. 30 (17) (2016) 1913–1930, 10.1101/gad.287524.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, Knoblich JA, Cerebral organoids model human brain development and microcephaly, Nature 501 (2013) 373–379, 10.1038/NATURE12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Pardridge WM, The blood-brain barrier: bottleneck in brain drug development, NeuroRx 2 (1) (2005) 3–14, 10.1602/neurorx.2.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Boiardi A, Antonio AE, Ae S, Ae ME, Ae EL, Salmaggi A, Paola AE, Ae G, Ae AF, Botturi A, Chiara AE, Ae F, Solari A, Graziella AE, Ae F, Di F, Ae M, Broggi G, Treatment of recurrent glioblastoma: can local delivery of mitoxantrone improve survival? J. Neurooncol 88 (1) (2008) 105–113, 10.1007/s11060-008-9540-6. [DOI] [PubMed] [Google Scholar]
- [63].Ferraris C, Cavalli R, Panciani PP, Battaglia L, Overcoming the blood-brain barrier: successes and challenges in developing nanoparticle-mediated drug delivery systems for the treatment of brain tumours, Int. J. Nanomed 15 (2020) 2999–3022, 10.2147/IJN.S231479. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.