

Abstract
Herein, we developed a special strategy for the fast sensitization of red emitting copper nanoclusters with the assistance of green emitting copper nanoclusters. Compared to most previous methods based on AIE, which do not maintain the water solubility or tiny size of nanoclusters, the charming features of the copper nanoclusters were retained after the fabrication. Furthermore, the product was employed for the detection of sulphide, which revealed its ratiometric sensing ability in water since the ratio of the intensity change for green and red emission was related to the sulphide concentration. In addition, after the addition of Zn2+, the green and red emission was either enhanced or quenched via the corresponding mechanism. This enables the facile fabrication of promising white light-harvesting materials since the species of the emitting color can be simply tuned.
Herein, we developed a special strategy for the fast sensitization of red emitting copper nanoclusters with the assistance of green emitting copper nanoclusters.
1. Introduction
Noble metal nanoclusters (NCs) have been proven as promising materials for several applications due to their molecule-like behaviors, including fluorescence properties, biocompatibility and ultra-small size.1 For the fabrication of NCs with excellent water solubility and tiny size, the use of small thiol-containing molecules to protect NCs was effective, but it was quite difficult to obtain bright fluorescence.2 Among them, the fluorescence brightness of the red-emitting copper nanoclusters (R-CuNCs) was especially weak though they tended to be favorable for optical applications. Aggregation-induced emission (AIE) resulted in fluorescence enhancement, where the vibration and rotation of NCs was inhibited and the corresponding nonradiative process was reduced.3 The fluorescence quantum yield (QY) of dispersed NCs could be boosted using various AIE strategies, such as placing NCs in a confined environment.4,5 Fluorescence enhancement was also obtained by fabricating NCs as large-size nanomaterials.6 For instance, Zhao found that the dissociation of ligands on NCs could induce the formation of aggregates and enhance the fluorescence.7 Additionally, there other fluorescence enhancement methods have been reported, such as encapsulating NCs in a polymer matrix or metal–organic frameworks (MOF).8–12 However, it should be noted that all these protocols actually changed the application potential of the original NCs.13 This is because were no longer well distributed in water as ultra-small size materials.14 In summary, these methods could enhance the fluorescence, but failed to reserve the special molecule-like properties of the original NCs.15–18
NCs were found to demonstrate remarkable catalytic activity for oxidization/reduction.19–22 Among them, copper nanoclusters (CuNCs) were employed to boost the rate for several types of reactions.23–25 An increase in activity was realized by the introduction of different CuNCs.26 However, to the best of our knowledge, there are no reports on the use of CuNCs for the formation of other NCs. Herein, we designed a facile protocol for the synthesis of R-CuNCs via the assistance of green emitting CuNCs (G-CuNCs). In the absence of G-CuNCs, R-CuNCs were not formed until a long reaction time using dihydrolipoic acid (DHLA) as a protecting ligand. On the other hand, in the presence of G-CuNCs, R-CuNCs were fabricated using bis[tetrakis(hydroxymethyl)phosphonium] sulfate (THPS) as a reducing reagent quite fast, and their red emission was enhanced significantly via combination with appropriate amounts of Cu2+. Since this process retained the tiny size and water solubility of the NCs, it has great advantages for further applications.18 The reaction mechanism is demonstrated in Fig. 1. Initially, R-CuNCs were formed fast due to the assistance of G-CuNCs. Simultaneously, the distance between G-CuNCs and R-CuNCs was long due to the dilute solution. As R-CuNCs became concentrated via the reduction of more Cu2+, they moved closer to G-CuNCs until the chance for their electrostatic interaction was considerable. Unlike the big nanoparticles with a relatively large repulsive force, the distance between each group of NCs was extremely small since it was a “big crowd” and the electrostatic interaction as “soft aggregates” was simpler. Thereafter, a FRET system may have been constructed between the CuNCs with different emissions when their proximity was close enough.
Fig. 1. Scheme for the fast formation of R-CuNCs and enhancement of their fluorescence through the assistance of G-CuNCs.

Since the as obtained product (G-R-CuNCs) were constructed using two types of emitters, they could show different responses to analytes under certain conditions, and thus they may be applied ratiometrically either by inducing a change in the red or green emission.27 Ratiometric sensors based on NCs have been confirmed to be capable of demonstrating anti-interference effects.28 However, most NCs are combined with additional organic fluorescence dyes or quantum dots to fabricate sensors, which involves toxic chemicals and complicated procedures.29,30 Multi-color emission was found for DNA-protected NCs and the as-prepared products facilitated the fabrication of ratiometric sensors without the introduction of other types of fluorophores. In this case, different emissions had to be excited at different wavelengths.31 Besides, DNA templates are much more expensive than common reagents, which limit their application. Herein, ratiometric sensing materials were totally constructed using cost-effective CuNCs. Subsequently, their ratiometric sensing behavior for a typical analyte (S2−) confirmed this assumption. In addition, it was found that the red emission of the product could be enhanced, quenched or changed ratiometrically with the green emission upon the addition of Zn2+. Furthermore, was interesting to find the generation of white light during this process. These phenomena suggest that all-copper NC products can be developed as promising white light-harvesting materials because this protocol is very convenient and no dyes or other protocols were required.32
2. Results and discussions
2.1. Preparation of G-CuNCs
Initially, G-CuNCs were obtained via a facile synthetic strategy involving the modification of PVP–CuNCs with DHLA. The typical product was denoted as G-CuNCs-1, which was characterized and described in Fig. 2. As shown in Fig. 2a, its UV-Vis absorbance spectrum demonstrated an insignificant peak that does not support the surface plasma resonance (SPR) of larger copper nanoparticles (CuNPs) (normally around 560 nm). This was similar to the previously reported water-soluble CuNCs with tiny sizes.33 Meanwhile, it was demonstrated in Fig. 2a (inset) that the as-prepared G-CuNCs-1 exhibited green emission under UV light irradiation upon excitation at 365 nm. As can be observed from Fig. 2b, G-CuNCs-1 demonstrated a fluorescence emission at 491 nm with the maximum excitation at 398 nm. The QY was calculated to be about 2.46% using FITC (QY = 92% in pH 8 solution) as a reference. Furthermore, its morphology and size distribution were studied by TEM, as shown in Fig. 2c and d, respectively. It can be seen that it was approximately 0.5–2.0 nm (n = 100) in diameter, which exhibited typical NC-level features.34,35
Fig. 2. (a) UV-Vis absorption spectrum of G-CuNCs-1 (inset, photograph of the sample under a 365 nm UV lamp), (b) fluorescence excitation (EX) and emission (EM) spectra of G-CuNCs-1 with the slit widths of 10 nm for both excitation and emission, (c) typical TEM image and (d) statistic size distribution of G-CuNCs-1.

2.2. Facile formation of R-CuNCs
It was found in previous work that R-CuNCs could be prepared using DHLA as a protecting ligand, but the reaction takes a long time.33 In the current work, we found that R-CuNCs could be obtained quite fast with the assistance of G-CuNCs. Firstly, for the evaluation of the formation tendency of R-CuNCs, different amounts of G-CuNCs were used as the assistant, as shown in Fig. 3. Initially, the fluorescence emission spectra were monitored for the synthesis of R-CuNCs without G-CuNCs. It can be seen from Fig. 3a a quite weak fluorescence with a maximum emission intensity at ca. 600 nm was only obtained a relatively high concentration of Cu2+ was combined with DHLA, THPS and Cu2+. Also, various methods were used for mixing the precursors including DHLA, THPS and Cu2+; however, still no R-CuNCs with satisfactory red emitting response could be observed within a short time. This revealed that it was difficult to obtain water-soluble R-CuNCs with excellent fluorescence behaviors quite fast using the one-pot synthetic strategy without additional assistance. However, it can be seen from Fig. 3b that remarkable red emission was obtained within two minutes using the same synthetic protocol in the presence of G-CuNCs. In addition, specific fluorescence emission spectra for R-CuNCs were even exhibited with the combination of a relatively low concentration of Cu2+. On the other hand, no red emission was observed using such low amounts of Cu2+ in the absence of G-CuNCs. Apparently, R-CuNCs were formed much more easily due to the additional assistance. Thus, to deeply understand what assisted the formation of R-CuNCs (the entire G-CuNCs or the pre-synthesis materials for G-CuNCs), it was worth investigating whether R-CuNCs could form quite fast with the assistance of other compounds. Subsequently, Cu2+ was combined with a mixture treated with the same protocol as that for the preparation of G-CuNCs without the involvement of the copper precursor (see Fig. 3b, green and yellow lines). It should be noted that PVP, l-ascorbic acid and DHLA were present in the mixture (G-pre). However, no fluorescence response corresponding to the generation of R-CuNCs was observed within a short time using a same strategy. This revealed that the fast formation of R-CuNCs could not occur until G-CuNCs were used.
Fig. 3. (a) Fluorescence emission spectra of the mixtures with the combination of the protecting ligands (DHLA), reducing reagent (THPS), and various amounts of Cu2+. (b) Fluorescence response for the fabrication of R-CuNCs in the absence (green and yellow lines) and presence (red and blue lines) of G-CuNCs (100 μL) using 40 μL of 0.079% THPS after the addition of 0 (red and yellow lines) and 100 μM (green and blue line) of Cu2+. G-pre + THPS indicates the medium underwent the same treatments for the preparation of G-CuNCs in the absence of Cu2+.

This phenomenon was further evaluated with different amounts of G-CuNCs, as shown in Fig. 4. For the investigations, 500 μL of 0.079% THPS was used as the reducing agent. The mixture was prepared in the presence of different amounts of G-CuNCs with a fixed volume. After the combination with different amounts of Cu2+, the fluorescence emission spectra were recorded. Meanwhile, a product (R-CuNCs-0) in the absence of G-CuNCs was prepared with a similarly long reaction time. It was found that the most intense fluorescence intensity was not generated until overnight reaction. After the synthesis, the product was characterized and described in Fig. S1.† It can be seen from Fig. S1† that the fluorescence was quite weak. However, when 25–50 μL of G-CuNCs was employed, the fluorescence was obtained within two minutes, as shown in Fig. 4a and b. Since the fluorescence emission spectra were demonstrated using the same measurement conditions as that in Fig. S1,† it could be concluded that the as-formed G-R-CuNCs exhibited the brightest fluorescence in Fig. 4a and b. At this stage, the enhanced fluorescence was possibly caused by the fast reaction, which facilitated the formation of brighter NCs due the acceleration of the nucleation of NCs and vice versa.36 Since ab increase in the content of G-CuNCs did not enhance the fluorescence emission intensity of R-CuNCs dramatically, it seemed the presence of G-CuNCs was only capable of effectively boosting the formation rate of R-CuNCs.33 However, it was further found that the fluorescence emission intensity at 610 nm could be significantly enhanced while that at 490 nm decreased if 100 μL-1000 μL of G-CuNCs was applied, as shown in Fig. 4c–f. This revealed that the presence of G-CuNCs at higher concentrations played a more important role than just assistants for accelerating the formation rate of R-CuNCs. The enhancement and quenching of the fluorescence at 610 nm and 490 nm, respectively, agreed with the typical FRET phenomenon, where both donor and acceptor existed.37 However, for the occurrence of FRET, the distance between the donor and acceptor is important. By using low amounts of G-CuNCs, the NCs were relatively isolated from each other in the solution. This meant that the chance for the approximation or interaction from G-CuNCs to R-CuNCs was low. At the same time, it can be seen from Fig. 4a and b that the emission intensity at 490 nm for G-CuNCs was quite weak, which could hardly play a role as a donor. On the other hand, when enough G-CuNCs were present in the system, the intensity of the green emission was stronger. Meanwhile, the occupation of more space due to the high concentrations facilitated G-CuNCs approaching R-CuNCs. Then, the opportunities for proximity between the promising donor and acceptor would be greatly increased. Therefore, the problem for realizing the FRET process could possibly be overcome.
Fig. 4. Fluorescence emission spectra of the generation of G-R-CuNCs as a function of the amount of G-CuNCs, (a) 25 μL, (b) 50 μL, (c) 100 μL, (d) 150 μL, (e) 250 μL, and (f) 1000 μL with the slit widths of 10 nm for both excitation and emission. (d1) (150 μL), (e1) (250 μL), and (f1) (1000 μL) were with the slit widths of 10 nm for excitation and 5 nm for emission.

According to the results from Fig. 4d–f, all the emission spectra for G-R-CuNCs were significantly enhanced after the combination with enough Cu2+. The typical fluorescence change as a function of Cu2+ in detail was also described in Fig. S2.† It can be seen that the emission intensity at 610 nm did not increase linearly as a function of Cu2+ from low concentration to high concentration. On the contrary, the fluorescence exhibited little change initially and dramatically jumped at a certain concentration. This was not surprising since the R-CuNCs formed at first possibly did not influence the fluorescence behavior of G-CuNCs. Also, the fluorescence emission demonstrated overlapping behavior. A scheme is presented in Fig. S3† to demonstrate the increasing approximation of G-CuNCs and R-CuNCs during the latter process. Similar to the requirement of relatively large amounts of G-CuNCs, the opportunity for approximation was not realized until relatively large amounts of R-CuNCs were formed. It should be noted from Fig. 4d1–f1 that the highest emission intensity for R-CuNCs was almost same when the amounts of G-CuNCs were different. This was quite different from the AIE phenomenon that the enhancement of fluorescence increases as a function of the induced materials when their content is not high.38
Meanwhile, it was found that much larger amounts of Cu2+ were required for the generation of the lowest response of red emission when larger amounts of G-CuNCs were applied. This was caused by the complexing effect of DHLA. If the ratio of Cu2+ was not considerable, it would complex with DHLA rather than transfer to Cu0 by reduction with THPS. Thereafter, no R-CuNCs could be formed to generate red emission until DHLA was complexed with enough Cu2+. Besides this, the increasing requirement of higher amounts of Cu2+ to reach both the strongest brightness and lowest response for red emission was found as a function of THPS within a certain range, as shown in Fig. S4 and Table S1.† It was observed that only after enough Cu2+ was combined, additional Cu2+ would sensitize the fluorescence of R-CuNCs. This indicated both THPS and DHLA play the role of complexing reagents because previous work also found that the reducing reagent functioned as a complexing reagent as well.39 Additionally, in the absence of THPS, the fluorescence enhancement at 610 nm was observed as a function of Cu2+ (Fig. S4a†). This indicated that the adsorbed LAA on the surface of G-CuNCs had the ability to reduce Cu2+ to Cu0, but the as obtained product exhibited very weak fluorescence behaviors. Thus, the application of THPS as an additional reducing reagent is necessary to obtain G-R-CuNCs with excellent features.
G-CuNCs could significantly enhance the formation rate for the generation of R-CuNCs, which appears to be similar to a catalytic process according to the above results. However, G-CuNCs had to be reserved after they assisted the formation of R-CuNCs. Based on the phenomena in Fig. 4f, the feature of green emission for G-CuNCs was not observed anymore after the addition of high amounts of Cu2+, which indicated G-CuNCs were probably used up since their green emission was almost quenched. However, the formation of R-CuNCs enabled the quenching of the green fluorescence if FRET occurred. In that case, we could not observe the fluorescence of G-CuNCs even though they were still present in the colloid. On the other hand, if the FRET was truly the reason that we could not observe the fluorescence of G-CuNCs after the generation of R-CuNCs, the green emission should be recovered to a certain extent when red emission was destroyed or masked.
To investigate the phenomena and eliminate the possible interferences for the enhancement/quenching behaviors, G-CuNCs without the protection of PVP were prepared (G-CuNCs-2). Subsequently, they were used as assistants for the fabrication of R-CuNCs (R-CuNCs-2). The typical process for the generation and inhibition of red emission is described in Fig. S5.† Initially, it could be observed the fluorescence of G-CuNCs-2 was quenched as a function of Cu2+ due to the formation of R-CuNCs-2 (Fig. S5a†). This was similar to the fluorescence evolution behavior in Fig. 4f. Subsequently, we tried to select a reagent that would mask the fluorescence of R-CuNCs, but would not be strong enough to inhibit the fluorescence of G-CuNCs. Since thio-containing molecules would easily react with both G-CuNCs and R-CuNCs at high concentrations, the amino acid l-histidine was explored. It was reported R-CuNCs could be quenched after the combination of a relatively high concentration of l-histidine through the formation of a more stable His/Cu2+ complex due to the participation of the imidazole ring of His.40 Herein, the masking of R-CuNCs was demonstrated since the red emission was quenched, as shown in Fig. S5b.† Meanwhile, it was interesting to observe that the green emission of G-CuNCs-2 was recovered. However, the emission peak in Fig. S5b† became broader and its relative intensity was not exactly the same as that in Fig. S5a.† This indicated the possible occurrence of a recovery process, but it was not completely reversible. Notwithstanding, since the green emission could not be generated from the other applied materials, but only CuNCs, it can be concluded that the presence of G-CuNCs was significant. This indicated G-CuNCs may still exist after the formation of R-CuNCs although their feature were not observed. Therefore, the enhanced formation rate for R-CuNCs was possibly caused by the involvement of G-CuNCs as a catalyst. However, since the generation and enhancement of the red emission were apparently controlled by more complicated processes, G-CuNCs were considered as an ‘assistant’ rather than a catalyst. Although G-CuNCs-2 could also be used for assisting the formation of R-CuNCs, it was not as stable as the PVP-protected G-CuNCs. Besides, it was found that large amounts of G-CuNCs-2 were required for realizing a similar process, which is possibly attributed to the different surface states, sizes, and other conditions.41 As a result, they were only employed for understanding the reaction mechanism. For further applications, the PVP-protected G-CuNCs were employed.
For fabrication of the red-emitting product, a typical sample (G-R-CuNCs-1) was obtained using 200 μL of G-CuNCs as the assistant and 500 μL of 0.079% THPS as a reducing reagent. After the combination with enough Cu2+, the product with the highest fluorescence enhancement was characterized and described in Fig. 5. As shown in Fig. 5a, the UV-Vis absorption spectrum of G-R-CuNCs-1 exhibited an insignificant peak, which indicated that the molecule-like behavior of CuNCs was not destroyed.19 A strong red emission was observed with its maximum peak at 610 nm in the fluorescence spectrum when the sample was irradiated with 424 nm. It also exhibited a bright red emission under UV light at 365 nm (inset of Fig. 5a). The QY was calculated to be 4.83% using acridine yellow (47% in ethanol) as a reference. As shown in Fig. 5c and d, the G-R-CuNCs-1 were approximately 0.5–3.0 nm (n = 100), and were dispersed as uniformly as the G-CuNCs in Fig. 2c. Table S2† presents a comparison of the synthetic conditions for the fabrication of the water-soluble R-CuNCs. It can be seen that our work show great advantages for the development of water-soluble products for CuNCs with fast reaction time.
Fig. 5. (a) UV-Vis absorption spectrum of G-R-CuNCs-1, (b) fluorescence excitation (EX) and emission (EM) spectra of G-R-CuNCs-1 with the slit widths of 10 nm for excitation and 5 nm for emission, (c) typical TEM images (d) and statistic size distributions of G-R-CuNCs-1.

2.4. Comparison of the properties of the CuNCs
For further understanding the relationship between the modified product of G-R-CuNCs-1, original G-CuNCs-1 and the slowly formed R-CuNCs-0, other characterizations including DLS, FT-IR and XPS were performed, as shown in Fig. S7–S9,† respectively. It can be seen from Fig. S7† that the DLS study for the CuNCs demonstrated small sizes, which were similar to the TEM study. The zeta potentials of G-CuNCs-1, G-R-CuNCs-1 and R-CuNCs-0 were determined to be ca. −29.6 mV, −38.1 mV, and −25.2 mV respectively, revealing that the surface of all these CuNCs were negatively charged. This indicated the CuNCs could maintain excellent stability due to the electrostatic repulsion effects. The surface groups of the synthesized CuNCs were further investigated using FT-IR, as shown in Fig. S8.† The typical S–H stretching of DHLA in the range of ca. 2500–2600 cm−1 was not observed for all the products of CuNCs. This indicated that the surface of the CuNCs were capped by the S–H group from the ligand.42 Furthermore, XPS analysis was employed to investigate the oxidation state of Cu, as shown in Fig. S9.† The peaks at 932.4 eV and 952.1 eV can be ascribed to the binding energy of the 2p3/2 and 2p1/2 electrons of the Cu atoms (Fig. S9a†), respectively.12,19 Meanwhile, the XPS spectrum shows a more positive peak for G-R-CuNCs-1 at 952.3 eV, which can be assigned to the binding energy of the Cu 2p1/2 electrons of metallic copper for both Cu(0) and Cu(i) (Fig. S9c†).24,43,44 These results suggest that more electrons were lost for Cu in G-R-CuNCs-1 compared to G-CuNCs-1. Furthermore, it could be observed that Cu 2p for R-CuNCs-0 showed even more positive peaks for both Cu 2p3/2 and Cu 2p1/2 at 932.7 eV and 952.6 eV, respectively (Fig. S9e†). These results revealed that R-CuNCs-0 tended to exhibit more positive values than G-CuNCs-1.45 On the other hand, the XPS survey spectra in Fig. S9b, d and f† for the all the products described the presence of C, S and O, which belonged to DHLA. This agreed with the FT-IR investigation that CuNCs were well protected by the DHLA ligand.
2.5. Mechanism analysis
To understand the possible mechanisms for the fluorescence change in G-CuNCs due to the generation of R-CuNCs, we proposed that FRET may control the process. The efficiency for FRET (EFRET) was initially calculated according to Förster's theory:![]() where, R0 is the critical distance at which the energy transfer efficiency is 50% and r0 is the distance between the donor and the acceptor. Meanwhile, FDA and FD represent the fluorescence intensity of the donor in the presence and absence of the acceptor, respectively. For instance, the fluorescence intensity of a typical process at the peak (490 nm) after the formation of G-R-CuNCs for FD and FDA was 937.8 and 2262.4 (as shown in Fig. S2†), respectively. Then, based on the above formula, EFRET could be calculated as 58.5%. In theory, the distance between the two CuNCs could be as short as the size of the two DHLA molecules between them ca. 2.6 nm, see Fig. S10.† Then R0 could be calculated as 2.7 nm according to the above formula. In this close proximity, G-CuNCs could emit a virtual photon, which would then be absorbed by R-CuNCs. Then, after excitation, we observed a significant fluorescence enhancement for the red emission and quenching for the green emission.
where, R0 is the critical distance at which the energy transfer efficiency is 50% and r0 is the distance between the donor and the acceptor. Meanwhile, FDA and FD represent the fluorescence intensity of the donor in the presence and absence of the acceptor, respectively. For instance, the fluorescence intensity of a typical process at the peak (490 nm) after the formation of G-R-CuNCs for FD and FDA was 937.8 and 2262.4 (as shown in Fig. S2†), respectively. Then, based on the above formula, EFRET could be calculated as 58.5%. In theory, the distance between the two CuNCs could be as short as the size of the two DHLA molecules between them ca. 2.6 nm, see Fig. S10.† Then R0 could be calculated as 2.7 nm according to the above formula. In this close proximity, G-CuNCs could emit a virtual photon, which would then be absorbed by R-CuNCs. Then, after excitation, we observed a significant fluorescence enhancement for the red emission and quenching for the green emission.
To obtain more information on this process, the fluorescence decay curves for the typical transformation of G-CuNCs to G-R-CuNCs were measured, as shown in Fig. S11.† Herein, the lifetimes of the donor and the as-obtained products were investigated. The curves could all be fitted by a three-exponential function, with three components. The triple exponential decay curve for the lifetime study can be described as follows: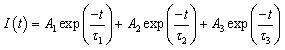
E
FRET is obtained from the donor exciton lifetime upon interaction with the acceptor, where τDA (G-CuNCs-b) is the fluorescence lifetime of the donor in the presence of the acceptor and τD (G-CuNCs-a) is the fluorescence lifetime of the donor alone.![]()
The lifetime results were described in Table S3† and the EFRET was calculated to be 47.2%. Herein, the as obtained EFRET was not optimized. It could be tuned via the appropriate combination of G-CuNCs, reducing reagent (THPS), and Cu2+. Thus, it is necessary for the details of this process to be investigated in the future.
Unlike AuNCs, the absorption peak for the water-soluble CuNCs was not quite obvious without theoretical calculation.46,47 Normally, when some materials were excited to a higher state, they tended to end up in the lowest excited state and then emitted radiation. In this case, the excitation peak agreed well with the absorption peak. For determination of the typical band gap, the absorption was analyzed using the excitation peak as a reference. Then, the absorption edge was estimated as 390 nm (excitation, 398 nm) and 412 nm (excitation, 424 nm), respectively according to Fig. S12.† Furthermore, based on the following equation, it was concluded that G-CuNCs and R-CuNCs exhibited a band gap of ca. 3.2 eV (Fig. S12a†) and ca. 3.0 eV (Fig. S12b†), respectively.![]()
Meanwhile, the overlap of the emission spectra of G-CuNCs and the excitation spectra of R-CuNCs was analyzed, as shown in Fig. S12c.† Using the Origin software, the overlapping potion was calculated to be more than 42% for both CuNCs. For quantum dots with similar structures, it was found that energy transfer could occur if the acceptors had a bandgap smaller than that of the donors of 55 meV.48 Additionally, for quantum dots emitting in the NIR, the energy difference should be bigger than ca. 59 meV.49 The CuNCs exhibited quite similar fluorescence properties to quantum dots. Thus, it was reasonable to assume that more than 200 meV should be enough for the energy transfer between the two NCs.
Additionally, a cyclic voltammetry study was conducted to estimate the valence and conduction band energy levels of the CuNCs (see Fig. S13†). The CuNCs were not as stable as AuNCs in water. With an increase or decrease in potential, the dissociation of the protecting ligands may interfere with the observation. Thus, to avoid unexpected errors, we calculated the valence and conduction band energy levels from the onset oxidation (Eox) and reduction (Ered) potentials using a previously reported paper as a reference. As the structures of the protecting ligands for l-cysteine and DHLA were similar, we assumed our CuNCs would exhibit similar electrochemical behaviour to their CuNCs.50 Thereafter, according to Fig. S13a,† the onset Eox and Ered were calculated to be 0.364 V and −1.0 V and the estimated VB and CB energy levels were −5.16 eV and −3.8 eV for G-CuNCs, respectively. Meanwhile, based on Fig. S13b,† the estimated VB and CB energy levels were calculated to be −5.12 V and −3.82 V, respectively. This agreed well with the VB and CB energy levels of the l-cysteine-protected CuNCs, which were −5.11 eV and −3.7 eV, respectively. The gap between G-CuNCs and R-CuNCs suggested that the electrons could transfer from a higher to the lower energy level.
To obtain more information on these CuNCs, ESI-MS was employed for the characterization of the unknown molecular species. Negative-ion mode ESI was used since it has been employed to appropriately characterize other CuNCs.51 A representative ESI-MS investigation of the CuNCs is shown in Fig. S14.† It can be seen from Fig. S14a† that the dominant ions in the mass spectra correspond to different combinations of copper and DHLA. There were several types of CuNCs with different atomic numbers. For example, Cu2, Cu3, Cu4, and Cu(DHLA)x complexes were observed for G-CuNCs. On the other hand, other species of NCs containing Cu6 and Cu7 were present for G-R-CuNCs-1. It should be noted that the Cu(DHLA)x complex disappeared in G-CuNCs-1 (Fig. S14b†). This indicated that the green emission was possibly quenched partly due to the transformation of the Cu(DHLA)x complex to Cu6 and Cu7. Meanwhile, the energy transfer from Cu2, Cu3 and Cu4 to Cu6 and Cu7 should play an important role for this fluorescence enhancement process. Interestingly, it was observed that Cu6 was not present in the slowly formed R-CuNCs-0 (Fig. S14c†). This revealed that more complicated processes could be involved in the generation of G-R-CuNCs-1. For instance, Cu6 may transform into Cu4 and Cu2, while Cu7 could transform into Cu3 and Cu4 in the ESI mode. Based on the ESI-MS, the CuNCs should be smaller than 1 nm according to the structure analysis in Fig. S15.† However, larger particles were observed in both Fig. 2c and 5c. This was caused by the aggregation associated with the organic ligand-capped small particles in the presence of the 200 keV strong electron beam.52
Besides this, we investigated many theoretical modes to predict the accurate structures of the CuNCs. However, we are not sure that the simulated structure is the optimized one since the system is a mixture, which is quite complicated. The credible way to simulate the structure of a sample would be based on the formation of a single crystal. However, the fabrication of crystals for CuNCs capped by flexible ligands is extremely difficult.53 Thus, further effort is required to obtain more pure materials in combination with the calculation.
2.6. Ratiometric sensing applications
Combined systems with two types of fluorophores are normally excellent candidates for sensing because each emission can be employed to exhibit a response to analytes. For instance, S2− could sensitively quench R-CuNCs and a turn-off sensor was developed.14 On the other hand, we found a much higher concentration of S2− was required to quench G-CuNCs. This indicated a facile ratiometric probe could be fabricated based on the current system since its response would be different from a turn-off sensor. It was demonstrated that the product with the most significant enhancement was not obtained until large amounts of THPS and Cu2+ were employed. Normally, employing a lower concentration of fluorescence sensor will result in high sensitivity because fewer analytes are required to react or function with it. Meanwhile, the change in the fluorescence intensity will be more significant in the presence of same amount of analyte. It was proven in Fig. S4† that the requirement of Cu2+ increased as a function of THPS by employing the same amount of G-CuNCs. Thus, the tendency for the required copper in the product will be the same as that of THPS. Then, the detection of S2− using the sensor obtained with different amounts of THPS was investigated and demonstrated in Fig. S16.† It can be clearly observed that the fluorescence intensity at 610 nm changed less significantly by using greater amounts of THPS. This revealed that high sensitivity could be obtained by using low amounts of Cu2+. However, the weak brightness of a sensor suffers from high signal-noise interferences. Thus, for balancing both the sensitivity and interferences, 150 μL of G-CuNCs and 20 μL of 0.079% THPS were chosen to fabricate the sensor. As a result, a sensing product (G-R-CuNCs-3) was obtained after combination with Cu2+. Herein, the amount of applied THPS was relatively low, but the fluorescence of the product was strong enough for analysis, which exhibited both excellent sensitivity and insignificant signal-noise interferences.
To evaluate the sensitivity and selectivity of the sensor for the determination of S2−, the linear relationship and frequently encountered interferences were studied, as shown in Fig. 6. Fig. 6a depicts the titration of G-R-CuNCs-3 with respect to different concentrations of S2−. It can be observed that the fluorescence intensity at 610 nm decreased with an increase in S2−, revealing that S2− effectively quenched the red emitting species (R-CuNCs-3). To understand the change in NCs in the presence of S2−, the TEM image was obtained, as shown in Fig. S17.† It can be seen that large aggregates were present. This is attributed to the exchange reaction between the surface ligands (DHLA) of R-CuNCs-3 and S2−, which enabled the formation of large Cu2S precipitates. On the other hand, it can be observed that the intensity at 490 nm changed much more gently as a function of S2− than that at 610 nm because G-CuNCs were normally more stable. The ratio of the different responses between the intensity at 610 nm and 490 nm (I610/I490) revealed a good linear relationship with log(C/μM) of S2−. This enabled the signal of the sensor to change ratiometrically as a function of S2− at both low and high concentrations. To further explore the feasibility to use the G-R-CuNCs-3 as an S2− probe, its calibration plot was investigated. The Y axis was established using the intensity ratio for the emission intensity at 610 nm (I610) and 490 nm (I490). Meanwhile, the logarithm of the concentration (log(C/μM)) was employed as the X axis. The regression equation was Y = −2.285X + 0.7266, with the correlation coefficient of 0.994 (Fig. 6b). Additionally, the limits of detection were measured to be much smaller than 100 nM based on the smallest fluorescence response upon the combination of S2−. For the interference analysis, 10 μM of common amino acids and glutathione (GSH), which usually exist in biological media, were investigated. From the results presented in Fig. 6c and d, it can be concluded that the fluorescence response for other interferences was not comparable to that of S2− at the same level in the current system. However, the mercapto amino acid GSH influenced the red emission at higher concentrations for CuNCs and the other amino acids quenched R-CuNCs at even higher concentrations. Nevertheless, their effects cannot compare with the quenching ability of S2− at trace levels. Besides, other common cations and biomolecules with comparable concentrations did not show significant quenching behaviors for the sensing of S2− using the current medium, as shown in Fig. S18.†
Fig. 6. (a) Fluorescence emission spectra of the sensing system in the presence of different amounts of S2−, (b) ratio of the fluorescence emission intensity at 610 nm to 490 nm (excitation, 400 nm) as a function of log S2− concentration, (c) fluorescence emission spectra of the sensing system in the presence of 10 μM of amino acid and S2− and (d) selectivity of the sensor for S2− over common amino acids (GSH was also used).

The proposed sensor was applied to detect S2− in chicken blood serum samples and the analytical results are presented in Table S4.† It was found that the recovery ratio of S2− reached 95.3–113% with a relative standard deviation (RSD) of 0.94–1.18%, which demonstrated the excellent analytical performance of the ratiometric sensor for the detection of S2− in aqueous samples. The results were also compared to that of previously reported ratiometric sensors based on NCs, as shown in Table S5.† It can be observed that the as-fabricated sensor required more convenient protocols and lower cost, but its sensing performance was as excellent as the other sensors. The comparison indicated the as-obtained CuNC system is a promising sensing material.
2.7. White light harvesting system
Since different turn-off behaviors were exhibited for S2− in the system for G-CuNCs and R-CuNCs, it was worth investigating whether the green emission or red emission could be induced differently by some other factors. Firstly, the requirement of low amounts of Cu2+ was designed for the fabrication of the materials based on our strategy. Then, some products were synthesized using 500 μL of G-CuNCs as the catalyst and 50 μL of 0.079% THPS as the reducing reagent. After adding 100 μL of 0.01 M Cu2+, the product (G-R-CuNCs-4) was formed. Furthermore, the fluorescence emission spectra of G-R-CuNCs-4 was demonstrated as a function of Zn2+ in Fig. 7. It was interesting to find that the emission peak located at 490 nm significantly increase with a slight blue shift upon the addition of Zn2+, while the emission peak at 610 nm slightly decreased, as shown in Fig. 7a. The CIE1931 chromaticity coordinate calculation was performed to illustrate the details of the light tuning in the system. As displayed in Fig. 7b and S19† (the corresponding picture), it can be seen that white light emission was obtained after 200 μL of Zn2+ (0.1 M) was added. The CIE co-ordinate for the system also indicated the light could be tuned from natural white to cold white light. For instance, the CIE co-ordinate for the product obtained by the combination of 200 μL of Zn2+ at X = 0.354 and Y = 0.3121 did not demonstrate a significant difference from that of natural white light (X = 0.33, Y = 0.33). Meanwhile, the CIE co-ordinate obtained from the addition of 250 μL of Zn2+ at X = 0.3134 and Y = 0.2803 was typical cold white light. Since the product and fluorescence spectra of G-R-CuNCs could be modified by tuning the amounts of G-CuNCs and other reagents, it can be concluded that other types of white light can also be obtained. Different from most white light-harvesting methods, this system does not require the introduction of other types of materials.32 Thus, this system provides a facile strategy for the generation of white light.
Fig. 7. (a) Fluorescence emission spectra of the as-prepared G-R-CuNCs-4 by the addition of different amounts of Zn2+ (with the slit widths of 10 nm for excitation and 5 nm for emission). Synthetic conditions: 500 μL of G-CuNCs, 50 μL of 0.079% THPS and 100 μL of 0.01 M Cu2+. (b) Image of the CIE1931 chromaticity coordinate calculation for the corresponding samples.

Besides, G-CuNCs and another product fabricated by the addition of a relatively high concentration of Cu2+ (G-R-CuNCs-5) were investigated, as shown in Fig. S20.† It can be seen from Fig. S20a† that the fluorescence emission intensity was significantly enhanced for G-CuNCs. This was similar to that in Fig. 7a. However, different from G-R-CuNCs-4, the red emission of G-R-CuNCs-5 was enhanced rather than quenched by the titration of Zn2+ (Fig. S20b†). The comparison of G-R-CuNCs-4 and G-R-CuNCs-5 indicated that the red emission of the as-prepared CuNCs could be either enhanced or quenched based on the synthetic conditions. When the product was fabricated with low amounts of Cu2+, the concentration of R-CuNCs was low. The red emission was mainly contributed by the FRET phenomenon. Also, the addition of Zn2+ inhibited R-CuNCs from getting close to G-RuNCs, which would decrease the energy transfer. Thus, a reversible phenomenon of FRET was observed (Fig. 7a). On the other hand, after the combination with a relatively high concentration of Cu2+, additional R-CuNCs would be formed. The red emission was contributed both by FRET and the residual R-CuNCs that did not participate in the fluorescence enhancement process. It was demonstrated in previous report that Zn2+ could light up the red emission of GSH-CuNCs based on AIE.54 Meanwhile, the red emission for our product was enhanced due to AIE since the complex of Zn2+ with the carboxylate groups of the ligands rigidifies the Cu(i) thiolates, which restricted the intramolecular rotation-vibrational motion. In summary, it can be concluded the fluorescence of the products can be controlled appropriately such as quenched, enhanced, or ratiometrically changed according to the corresponding applications and these processes can be well applied in different fields.
3. Experimental
3.1. Chemical and materials
Deionized water was used as the aqueous solution for dissolving all reagents. Copper(ii) chloride (CuCl2, 99%), l-ascorbic acid (LAA, ≥99%), (±)-α-lipoic acid (≥99%), polyvinylpyrrolidone (PVP, mol wt. 40 000), sodium hydroxide (NaOH, ≥98%), bis[tetrakis(hydroxymethyl)phosphonium], sulfate solution (THPS, 70–75% in water) and sodium sulfide nonahydrate (Na2S, ≥98%) were purchased from Sigma Aldrich. Other reagents were all of analytical grade and obtained from Aladdin Company. All glassware was cleaned with aqua regia and rinsed with water prior to use.
3.2. Synthesis of PVP–CuNCs
PVP-protected CuNCs (PVP–CuNCs) were synthesized according to a previously reported paper with modifications.55 PVP (0.5 g) was added to a 20 mL water solution. After the complete dissolution, 2 mL of 0.1 M CuCl2 was injected. After the combination of 0.1 M LAA, the mixer was shaken evenly. The reactants were transferred to an 80 °C water bath and left for 3–5 hours until a light yellow color supernatant was observed.
3.3. Synthesis of G-CuNCs
37.8 mg of (±)-α-lipoic acid was added to a vial with 7.5 mL of water. 250 μL of 1 M NaOH was added to the solution to dissolve (±)-α-lipoic acid to form DHLA. Further, 1 mL of the as-obtained PVP–CuNCs was added. Herein, the excess DHLA ligand in the colloid was further used as the protecting ligand for R-CuNCs. Thus, the as-formed colloid without separation was stored at 4 °C in a refrigerator for further use. For material characterization, G-CuNCs were purified by ultrafiltration with a 20 kDa Millipore membrane. Besides this, G-CuNCs-2 were synthesized following the same procedure as that for G-CuNCs in the absence of PVP.
3.4. Formation of G-R-CuNCs
In a typical synthetic process, a certain amount of G-CuNCs were used. 100 μL of 1 M NaOH was added to the solution. The total volume of the mixture was fixed as 2.5 mL. Subsequently, 500 μL of 0.079% THPS was added to the mixture. Finally, different amounts of 0.01 M CuCl2 solution was added to the above mixture. For the sample requiring a large amount of Cu2+, 0.1 M of CuCl2 was employed. In order to obtain the brightest red emission, F97 was employed to monitor the fluorescence emission spectra upon the addition of Cu2+ at 2 minutes intervals until the highest fluorescence emission intensity was reached. Then, a mixture of fluorescence-enhanced CuNCs (G-R-CuNCs) was obtained. After vacuum freeze drying, the as-obtained powder was preserved for long-term use. Besides, CuNCs with red emission was obtained with the assistance of G-CuNCs-2 (R-CuNCs-2) and the corresponding fluorescence-enhanced product (G-R-CuNCs-2) was fabricated.
3.5. Synthesis of R-CuNCs-0
37.8 mg of (±)-α-lipoic acid was added to a vial with 7.5 mL of water. 250 μL of 1 M NaOH was dropped in the solution to dissolve (±)-α-lipoic acid to form DHLA. Next, 200 μL of 0.1 M CuCl2 and 500 μL of 0.079% THPS was added to the above mixture. The mixture was stirred overnight until a light-yellow colloid with the strongest emission intensity was obtained. The sample were purified by ultrafiltration with a 20 kDa Millipore membrane for characterization.
3.6. Characterization
All ultraviolet-visible (UV-Vis) absorption spectra were recorded using a UV-7504 UV-Vis spectrophotometer (Shanghai Xinmao). Fluorescence spectra were obtained on a fluorescence spectrophotometer (F97, Shanghai Lingguang). X-ray photoelectron spectroscopy (XPS) was performed on an ESCALAB 250 XI electron spectrometer (Thermo). The fluorescence lifetime was determined on a FL920 time-correlated single-photon-counting fluorescence lifetime spectrometer (Edinburgh Analytical Instruments, Edinburgh, U.K.). Transmission electron microscopy (TEM) images were obtained on a FEI Tecnai G2 F20 microscope. Dynamic light scattering (DLS) and zeta potential measurements were performed with a Malvern Zetasizer. FT-IR spectra were measured using an FT-IR spectrometer equipped with a DGTS detector (Nicolet is5/is10).
3.7. Ratiometric sensing of S2−
For S2− analysis, 100 μL 1 M NaOH was added to 2 mL of PBS buffer (pH 8). After that, 150 μL colloid of G-CuNCs was injected. Then, 20 μL of 0.079% THPS was injected to the mixture. Next, 40 μL of 0.01 M Cu2+ was added to the above solution and the pH value was tuned to 8 using HCl. After mixing with 100 μL of EDTA (50 mM), the sensor (G-CuNCs-3) in the absence of S2− was used as the blank. Next, different concentrations S2− were combined with the system for analysis of the calibration curve. Besides, other solutions containing amnion acids, GSH and cations were studied in the place of S2− by measuring their fluorescence emission spectra for the selectivity study, which were recorded at an excitation wavelength of 400 nm. All measurements were performed at room temperature.
3.8. S2− sensing in real sample
For the analysis of serum samples, chicken blood samples were collected from a local market. All the blood samples were centrifuged at 12 000 rpm for 15 min at 4 °C. Then, the supernatant was subjected to 10-fold dilution with PBS (10 mmol L−1, pH 8), and different concentrations of S2− were added to the serum supernatant to prepare the spiked samples. Besides, all the samples were mixed with 100 μL of EDTA (50 mM). After incubation at room temperature for 2 min, the resulting solution was diluted to the same volumes and investigated similarly to the standard samples.
3.9. Fabrication of white light-emitting colloid
100 μL of 1 M NaOH was added to 2 mL of water. Subsequently, 500 μL colloid of G-CuNCs was injected. Then, 50 μL of 0.079% THPS was injected to the mixture. Next, 100 μL of 0.01 M Cu2+ was combined with the above solution and the product was obtained (G-CuNCs-4). Finally, 0.1 M of ZnSO4 solution with different volumes was added to the system when the total volume was fixed. F97 was used to measure the fluorescence emission spectra and CIE1931 was employed to analyze the CIE of the light.
4. Conclusions
The strategy of using green emitting copper nanoclusters as assistants for the fabrication of red emitting copper nanoclusters was demonstrated. This method for the fabrication of the product not only provides a facile protocol for the fast generation of nanoclusters with much brighter fluorescence with assistance from the nanoclusters themselves, but also can provide avenues for future ratio-metric analysis with all-copper nanoclusters. Additionally, it has potential for the development of light-harvesting materials via a simple process. The as-obtained products of all-copper nanoclusters are excellent materials for application in different fields.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
We are grateful for the financial support from the National Natural Science Foundation of China (Grant No. 81702105 and No. 81671742) and Liaoning (Grant No. 201602288).
Electronic supplementary information (ESI) available: Details of the characterization of the materials; factors influencing the catalysis and FRET effect. See DOI: 10.1039/c8na00224j
Notes and references
- Xu S. Nie Y. Jiang L. Wang J. Xu G. Wang W. Luo X. Anal. Chem. 2018;90:4039–4045. doi: 10.1021/acs.analchem.7b05253. [DOI] [PubMed] [Google Scholar]
- Chen L. Y. Wang C. W. Yuan Z. Chang H. T. Anal. Chem. 2015;87:216–229. doi: 10.1021/ac503636j. [DOI] [PubMed] [Google Scholar]
- Goswami N. Yao Q. Luo Z. Li J. Chen T. Xie J. J. Phys. Chem. Lett. 2016;7:962–975. doi: 10.1021/acs.jpclett.5b02765. [DOI] [PubMed] [Google Scholar]
- Tian R. Zhang S. Li M. Zhou Y. Lu B. Yan D. Wei M. Evans D. G. Duan X. Adv. Funct. Mater. 2015;25:5006–5015. doi: 10.1002/adfm.201501433. [DOI] [Google Scholar]
- Goswami N. Lin F. Liu Y. Leong D. T. Xie J. Chem. Mater. 2016;28:4009–4016. doi: 10.1021/acs.chemmater.6b01431. [DOI] [Google Scholar]
- Wu Z. Liu H. Li T. Liu J. Yin J. Mohammed O. F. Bakr O. M. Liu Y. Yang B. Zhang H. J. Am. Chem. Soc. 2017;139:4318–4321. doi: 10.1021/jacs.7b00773. [DOI] [PubMed] [Google Scholar]
- Zhao M. Qian Z. Zhong M. Chen Z. Ao H. Feng H. ACS Appl. Mater. Interfaces. 2017;9:32887–32895. doi: 10.1021/acsami.7b09659. [DOI] [PubMed] [Google Scholar]
- Hembury M. T. Beztsinna N. Asadi H. van den Dikkenberg J. Meeldijk J. D. Hennink W. E. Vermonden T. Biomacromolecules. 2018;19(7):2841–2848. doi: 10.1021/acs.biomac.8b00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z. Liu J. Gao Y. Liu H. Li T. Zou H. Wang Z. Zhang K. Wang Y. Zhang H. Yang B. J. Am. Chem. Soc. 2015;137:12906–12913. doi: 10.1021/jacs.5b06550. [DOI] [PubMed] [Google Scholar]
- Pyo K. Thanthirige V. D. Kwak K. Pandurangan P. Ramakrishna G. Lee D. J. Am. Chem. Soc. 2015;137:8244–8250. doi: 10.1021/jacs.5b04210. [DOI] [PubMed] [Google Scholar]
- Cao F. Ju E. Liu C. Li W. Zhang Y. Dong K. Liu Z. Ren J. Qu X. Nanoscale. 2017;9:4128–4134. doi: 10.1039/C7NR00073A. [DOI] [PubMed] [Google Scholar]
- Su X. Liu J. ACS Appl. Mater. Interfaces. 2017;9:3902–3910. doi: 10.1021/acsami.6b13914. [DOI] [PubMed] [Google Scholar]
- Zhou T. Y. Lin L. P. Rong M. C. Jiang Y. Q. Chen X. Anal. Chem. 2013;85:9839–9844. doi: 10.1021/ac4023764. [DOI] [PubMed] [Google Scholar]
- Chen P. C. Li Y. C. Ma J. Y. Huang J. Y. Chen C. F. Chang H. T. Sci. Rep. 2016;6:24882. doi: 10.1038/srep24882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia X. Li J. Wang E. Small. 2013;9:3873–3879. doi: 10.1002/smll.201300896. [DOI] [PubMed] [Google Scholar]
- Tao Chen S. Y. Chai J. Song Y. Fan J. Rao B. Sheng H. Yu H. Sci. Adv. 2017;3:e1700956. doi: 10.1126/sciadv.1700956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y. Feng H. Liu W. Zhang S. Tang C. Chen J. Qian Z. J. Mater. Chem. B. 2017;5:5120–5127. doi: 10.1039/C7TB00901A. [DOI] [PubMed] [Google Scholar]
- Li D. Chen Z. Mei X. Adv. Colloid Interface Sci. 2017;250:25–39. doi: 10.1016/j.cis.2017.11.001. [DOI] [PubMed] [Google Scholar]
- Lu Y. Wei W. Chen W. Chin. Sci. Bull. 2012;57:41–47. doi: 10.1007/s11434-011-4896-y. [DOI] [Google Scholar]
- Zhao J. Jin R. Nano Today. 2018;18:86–102. doi: 10.1016/j.nantod.2017.12.009. [DOI] [Google Scholar]
- Yang X. Sun J.-K. Kitta M. Pang H. Xu Q. Nat. Catal. 2018;1:214–220. doi: 10.1038/s41929-018-0030-8. [DOI] [Google Scholar]
- Liu L. Corma A. Chem. Rev. 2018;118(10):4981–5079. doi: 10.1021/acs.chemrev.7b00776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z. Li Y. Liu J. Lu Z. Zhang H. Yang B. Angew. Chem., Int. Ed. 2014;53:12196–12200. doi: 10.1002/anie.201407390. [DOI] [PubMed] [Google Scholar]
- Masaki Takahashi H. K. Chun W.-J. Kori M. Imaoka T. Yamamoto K. Sci. Adv. 2017;3:e1700101. doi: 10.1126/sciadv.1700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C. Wang C. Xu L. Cheng H. Lin Q. Zhang C. Nanoscale. 2014;6:1775–1781. doi: 10.1039/C3NR04835G. [DOI] [PubMed] [Google Scholar]
- Vilar-Vidal N. Rivas J. López-Quintela M. A. ACS Catal. 2012;2:1693–1697. doi: 10.1021/cs300355n. [DOI] [Google Scholar]
- Wang L.-L. Qiao J. Liu H.-H. Hao J. Qi L. Zhou X.-P. Li D. Nie Z.-X. Mao L.-Q. Anal. Chem. 2014;86:9758–9764. doi: 10.1021/ac5023293. [DOI] [PubMed] [Google Scholar]
- Del Bonis-O'Donnell J. T. Thakrar A. Hirschberg J. W. Vong D. Queenan B. N. Fygenson D. K. Pennathur S. ACS Chem. Neurosci. 2018;9:849–857. doi: 10.1021/acschemneuro.7b00444. [DOI] [PubMed] [Google Scholar]
- Yu Q. Gao P. Zhang K. Y. Tong X. Yang H. Liu S. Du J. Zhao Q. Huang W. Light: Sci. Appl. 2017;6:e17107. doi: 10.1038/lsa.2017.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y. Yu H. Zhang K. Sun M. Zhang Y. Wang X. Wang S. Nano Res. 2016;9:2088–2096. doi: 10.1007/s12274-016-1099-5. [DOI] [Google Scholar]
- Shahad J. G. C. Alsaiari K. Hammami M. Omar H. W. Neelakanda P. Adv. Healthcare Mater. 2017;6:1601135. doi: 10.1002/adhm.201601135. [DOI] [PubMed] [Google Scholar]
- Barman M. K. Paramanik B. Bain D. Patra A. Chemistry. 2016;22:11699–11705. doi: 10.1002/chem.201601849. [DOI] [PubMed] [Google Scholar]
- Li D. Chen Z. Wan Z. Yang T. Wang H. Mei X. RSC Adv. 2016;6:34090–34095. doi: 10.1039/C6RA01499B. [DOI] [Google Scholar]
- Upashi Goswam A. D. Raza A. Kandimalla R. Kalita S. Ghosh S. Chattopadhyay A. ACS Appl. Mater. Interfaces. 2018;10:3282–3294. doi: 10.1021/acsami.7b15165. [DOI] [PubMed] [Google Scholar]
- Raju Y. Krishnamurthi P. Paulose P. L. Manoharan P. T. Nanoscale. 2017;9:17963–17974. doi: 10.1039/C7NR07136A. [DOI] [PubMed] [Google Scholar]
- Shang L. Yang L. Stockmar F. Popescu R. Trouillet V. Bruns M. Gerthsen D. Nienhaus G. U. Nanoscale. 2012;4:4155–4160. doi: 10.1039/C2NR30219E. [DOI] [PubMed] [Google Scholar]
- Otto J. P. Wang L. Pochorovski I. Blau S. M. Aspuru-Guzik A. Bao Z. Engel G. S. Chiu M. Chem. Sci. 2018;9:3694–3703. doi: 10.1039/C8SC00818C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z. Yuan X. Yu Y. Zhang Q. Leong D. T. Lee J. Y. Xie J. J. Am. Chem. Soc. 2012;134:16662–16670. doi: 10.1021/ja306199p. [DOI] [PubMed] [Google Scholar]
- Li D. Chen Z. Yang T. Wang H. Lu N. Mei X. RSC Adv. 2016;6:19182–19189. doi: 10.1039/C6RA00912C. [DOI] [Google Scholar]
- Liu Y. Ding D. Zhen Y. Guo R. Biosens. Bioelectron. 2017;92:140–146. doi: 10.1016/j.bios.2017.01.036. [DOI] [PubMed] [Google Scholar]
- Wang Z. Susha A. S. Chen B. Reckmeier C. Tomanec O. Zboril R. Zhong H. Rogach A. L. Nanoscale. 2016;8:7197–7202. doi: 10.1039/C6NR00806B. [DOI] [PubMed] [Google Scholar]
- Jiang J. Conroy C. V. Kvetny M. M. Lake G. J. Padelford J. W. Ahuja T. Wang G. J. Phys. Chem. C. 2014;118:20680–20687. doi: 10.1021/jp5060217. [DOI] [Google Scholar]
- Porta M. Nguyen M. T. Ishida Y. Yonezawa T. RSC Adv. 2016;6:105030–105034. doi: 10.1039/C6RA17291A. [DOI] [Google Scholar]
- Huang C. Liu Q. Fan W. Qiu X. Sci. Rep. 2015;5:16736. doi: 10.1038/srep16736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. Biradar A. V. Wang G. Sharma K. K. Duncan C. T. Rangan S. Asefa T. Chemistry. 2010;16:10735–10743. doi: 10.1002/chem.201000354. [DOI] [PubMed] [Google Scholar]
- Das N. K. Ghosh S. Priya A. Datta S. Mukherjee S. J. Phys. Chem. C. 2015;119:24657–24664. doi: 10.1021/acs.jpcc.5b08123. [DOI] [Google Scholar]
- Baral A. Basu K. Ghosh S. Bhattacharyya K. Roy S. Datta A. Banerjee A. Nanoscale. 2017;9:4419–4429. doi: 10.1039/C7NR00353F. [DOI] [PubMed] [Google Scholar]
- Crooker S. A. Hollingsworth J. A. Tretiak S. Klimov V. I. Phys. Rev. Lett. 2002;89:186802. doi: 10.1103/PhysRevLett.89.186802. [DOI] [PubMed] [Google Scholar]
- Ranojoy Bose J. F. M. Gao J. Rickey K. M. Chen C. J. Talapin D. V. Murray C. B. Won A. C. W. Nano Lett. 2008;8:2006–2011. doi: 10.1021/nl8011243. [DOI] [PubMed] [Google Scholar]
- Maity S. Bain D. Bhattacharyya K. Das S. Bera R. Jana B. Paramanik B. Datta A. Patra A. J. Phys. Chem. C. 2017;122:13354–13362. doi: 10.1021/acs.jpcc.7b09959. [DOI] [Google Scholar]
- Jeremy Essner X. C. Wood T. Baker G. Analyst. 2018;143:1036–1041. doi: 10.1039/C7AN01867C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu K. Gayen K. Mitra T. Baral A. Roy S. S. Banerjee A. ChemNanoMat. 2017;3:808–814. doi: 10.1002/cnma.201700162. [DOI] [Google Scholar]
- Kang X. Chong H. Zhu M. Nanoscale. 2018;10:10758–10834. doi: 10.1039/C8NR02973C. [DOI] [PubMed] [Google Scholar]
- Lin L. Hu Y. Zhang L. Huang Y. Zhao S. Biosens. Bioelectron. 2017;94:523–529. doi: 10.1016/j.bios.2017.03.038. [DOI] [PubMed] [Google Scholar]
- Shi Y. E. Luo S. Ji X. Liu F. Chen X. Huang Y. Dong L. Wang L. Dalton Trans. 2017;46:14251–14255. doi: 10.1039/C7DT02193C. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.