

Abstract
Background:
Preclinical studies suggest that sphingosine-1-phophate (S1P) influences blood pressure regulation primarily through nitric-oxide (NO)-induced vasodilation. Since microvascular tone significantly contributes to mean arterial pressure, the mechanism of S1P on human resistance arterioles was investigated. We hypothesized that S1P induces NO-mediated vasodilation in human arterioles from adults without coronary artery disease (nonCAD) through activation of two receptors, S1PR1 and S1PR3. Further, we tested whether this mechanism is altered in vessels from patients diagnosed with coronary artery disease (CAD).
Methods:
Human arterioles (50-200 μm in luminal diameter) were dissected from otherwise discarded surgical adipose tissue, cannulated, and pressurized. Following equilibration, resistance vessels were pre-constricted with endothelin-1 (ET-1) and changes in internal diameter to increasing concentrations of S1P (10−12 to 10−7 M) in the presence or absence of various inhibitors were measured.
Results:
S1P resulted in significant dilation that was abolished in vessels treated with S1PR1 and S1PR3 inhibitors and in vessels with reduced expression of each receptor. Dilation to S1P was significantly reduced in the presence of the NOS inhibitor L-NAME and the NO scavenger cPTIO. Interestingly, dilation was also significantly impaired in the presence of PEG-catalase, apocynin, and specific inhibitors of NADPH oxidases (NOX) 2 and 4. Dilation in vessels from patients diagnosed with coronary artery disease (CAD) was dependent on H2O2 alone which was only dependent on S1PR3 activation.
Conclusions:
These translational studies highlight the inter-species variation observed in vascular signaling and provide insight into the mechanism by which S1P regulates microvascular resistance and ultimately blood pressure in humans.
Keywords: Endothelium, Microvascular, Vasodilation, Nitric Oxide, sphingosine-1-phosphate (S1P), microvascular tone, CAD, translational, human
Graphical Abstract
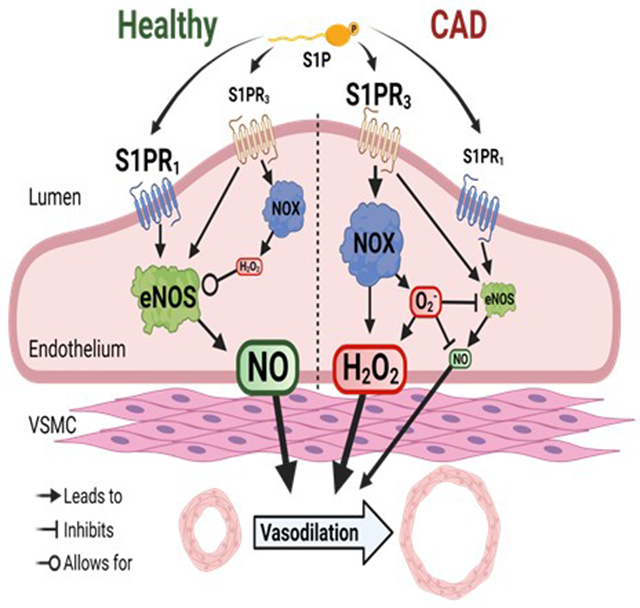
INTRODUCTION
Hypertension remains the dominating risk factor for the development of cardiovascular disease, the number one cause of mortality both in the United States and globally. Maintaining normal blood pressure requires tight control of arterial tone predominantly in the resistance arterioles, a subtype of blood vessels that regulate peripheral vascular resistance. Resistance within arterioles, or microvascular tone, is modulated by various stimuli including shear-induced vasoactive mediators such as nitric oxide (NO)1, hydrogen sulfide (H2S)2, and reactive oxygen species (ROS) such as H2O23, to name a few. While many vasoactive compounds exist that freely diffuse out of the endothelium to relax surrounding smooth muscle, NO production via endothelial nitric oxide synthase (eNOS or NOS3) is preferred due to its anti-inflammatory properties on surrounding tissues as opposed to the damaging effects of H2O24.
Circulating sphingolipids such as sphingosine-1-phosphate (S1P) have been shown to activate eNOS/NOS3 to cause NO-mediated dilation and reduce blood pressure in animals5. Within endothelial cells, S1P is formed by the phosphorylation of sphingosine by one of two sphingosine kinase isoforms (SpK 1 and 2). S1P then exits the cell exclusively through transporter Spinster-2 6–8 and participates in “inside-out signaling” where it can activate its receptors within the endothelial cell membrane. The importance of S1P and its role in blood pressure homeostasis is highlighted by rodent in vivo studies showing that inhibition of S1P formation results in endothelial dysfunction and elevated mean arterial pressure9. S1P-induced activation of eNOS/NOS3 has been shown to be via endothelial membrane-bound G-protein coupled receptors, namely S1P receptors 1 and 3 (S1PR1&3). S1PR1 is the dominant receptor in the endothelium10, whereas S1PR3 is present in both the endothelial and smooth muscle layer of arteries11. Activation of S1PR3 along with S1PR2 in vascular smooth muscle cells has been shown to induce constriction12 which is in sharp contrast to the NO-mediated vasodilatory effects observed by S1PR3 in the endothelium. While the majority of studies suggest a strong link between S1PR1 and NO formation, the role of S1PR3 is more elusive with prior work demonstrating that it is capable of increasing ROS13.
Although the beneficial effects of S1P in rodents is clear, the diversity in mechanisms of vasodilation between species is dramatic14 and no studies to-date have examined S1P signaling in the human vasculature. The effects of S1P on microvascular tone during the disease state (e.g. cardiovascular disease) are also not well established. Here, we investigated the effect of S1P on human microvascular tone and sought to elucidate the mechanism(s) by which S1P elicits dilation in peripheral arterioles from subjects without coronary artery disease (nonCAD) as well as those diagnosed with coronary artery disease (CAD).
METHODS
The authors declare that all supporting data are available within the article (and its Supplemental Material).
Human Specimen Acquisition
Surgical discard human adipose tissue (pericardial, subcutaneous, visceral) was collected for the isolation of arterioles. Patients were included if they were under the age of 80 and had 0-1 risk factor(s) for CAD (e.g., hypertension, hyperlipidemia, diabetes mellitus type 1 or 2, congestive heart failure), or had a confirmed formal diagnosis of CAD via cardiac catheterization. For more detailed information regarding the collection of human specimens for this study please refer to the Supplemental Material. The research protocols for tissue acquisition and processing were approved by the Institutional Review Board of the Medical College of Wisconsin.
Microvascular Reactivity Studies
Human resistance arterioles with luminal diameters of 50-200μm (113 ± 17μm, mean±SD) were dissected from adipose tissue within 36 hrs of specimen arrival. Microvessels were cannulated on glass micropipettes of matched resistance in an organ chamber and pressurized at 30 mmHg and 60 mmHg for 30 min each in warm Krebs buffer while bubbled with a 74%/21%/5% mixture of N2, O2, and CO2, respectively as previously reported15, 16. Following equilibration, arterioles were pre-constricted with endothelin-1 (ET-1) to 30-70% of their passive diameters. Vessel wall diameter changes in response to agents were then measured by visualizing the internal wall and using a digital caliper videomicroscopy system. Measurements were made both pre- and post-S1P administration at 1 min intervals.
During equilibration at 60 mmHg, vessels were exposed for 30 min to one of several different compounds: nitric oxide synthase (NOS) inhibitor Nω-nitro-L-arginine methyl ester (L-NAME) (100 μM), the NO scavenger 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (cPTIO) (100 μM), polyethylene glycol-catalase (PEG-Catalase) 500 U/mL, S1PR1 inhibitor W146 (10 μM), S1PR3 inhibitor CAY10444 (10 μM), NADPH-oxidase (NOX) inhibitor apocynin (300 μM), NOX4 inhibitor GKT137831 (1 μM) or NOX2 inhibitor GSK2795039 (1 μM). All interventions were administered into the organ bath and constituted ≤1% of the total bath volume and all concentrations represent the final concentrations in the organ bath. Increasing concentrations of S1P were administered (10−12 M to 10−6 M) at 1-minute intervals. For endothelial viability assessment, acetylcholine was administered in increasing concentrations (10−9 M to 10−4 M) at 2-minute intervals. Up to two studies were performed in each vessel, separated by a wash (organ chamber bath replaced x3). At the end of the study, arterioles were exposed to the endothelial-independent dilator papaverine (100 μM) to determine the vessel’s maximal diameter. Microvessels that did not achieve a stable constriction with ET-1 for 10 min or dilated less than 75% of maximal resting diameter to papaverine were not included in the data analysis.
siRNA in Human Arterioles
Decreased expression of S1PR1 and S1PR3 was achieved as previously described17. Arterioles were cannulated on glass micropipettes at one end and transfused with either the negative control siRNA or target protein siRNA (50nM), both with Lipofectamine RNAiMAX (Invitrogen). Both ends of the arterioles were tied off and vessels were incubated in fresh EBM-2 medium overnight (16-20 hrs). Both ends of the vessels were cut following intraluminal exposure then cannulated for functional studies. Confirmation of decreased expression in arterioles was performed using immunohistochemistry.
Detection of NO in Microvessels
Fluorescent detection of NO in cannulated vessels was achieved using the Enzo Life Sciences NO detection Kit (Enzo Life Sciences, Enzo Biochem, Farmingdale, New York). Cannulated vessels were intraluminally exposed to the NO fluorescent probe (1:400 in HEPES buffer) for 2 hrs then pre-constricted with ET-1 to 50% of its passive diameter. Images were taken using an Olympus IX73 microscope following exposure to S1P (10−7) and maximal flow (ΔP 100 cmH2O) every min up to 5 min. The NOS inhibitor L-NAME was used to confirm probe specificity. Relative intensity was measured at excitation and emission wavelengths of 650/670 using ImageJ software. The total corrected fluorescence (TCF) = integrated density – (vessel area x mean background intensity) was calculated for each vessel. Data was normalized and reported as fold change from the pre-S1P or pre-flow baseline value.
Immunohistochemistry
Confirmation of S1PR1 and S1PR3 expression in human arterioles was accomplished using immunohistochemistry. Microvessels collected from nonCAD adult patients (0-1 risk factors for CAD) or those diagnosed with CAD were fixed in 10% zinc formalin buffer for 24-72 hrs. Samples were then embedded in paraffin and cut into 4μm slices using a HMS355 microtome. S1P receptor expression was assessed by immunolabeling with a rabbit polyclonal IgG antibody against human S1PR1 (1:200, 1 hr incubation) and rabbit polyclonal IgG against human S1PR3 (1:100, overnight incubation). Sections were then incubated with secondary antibody or normal serum IgG (negative control). Immunohistochemical staining proceeded with biotinylated goat anti-rabbit IgG followed by alkaline phosphatase streptavidin and alkaline phosphatase chromogen substrate. A Leica Bond MAX immunostainer was used. Following staining, slides underwent heat-induced epitope retrieval (HIER) for 10 min. Slides were scanned with a NanoZoomer HT slide scanner (Hamamatsu, Japan).
Cell Culture
Human umbilical vein endothelial cells (HUVEC; ATCC, Manassas, VA, USA) were cultured in endothelial cell growth medium (EGM-2; Lonza, Basel, Switzerland), supplemented with 5% fetal bovine serum, growth factors, heparin, hydrocortisone, and ascorbic acid (EGM-2 SingleQuot; Lonza, Basel, Switzerland). Cells were used at passage 4-5 at 70-90% confluency. Treatments included 1% FBS with S1P vehicle (methanol), NOX4 inhibitor, NOX4 inhibitor vehicle (ethanol), and PEG-Catalase for 30 min. Following the 30 min incubation, cells were exposed to S1P (10−7) for 10 min.
Western Blot
Treated HUVECs were scraped and submerged in cold lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1% deoxycholic acid, 0.1% SDS, 0.5% NP40), which was previously mixed with a protease and phosphatase inhibitor cocktail (Roche, Basel, Switzerland). The cell lysates were sonicated and kept at −80°C until further use. Protein concentration was determined using the BCA protein assay (Thermo Scientific, Waltham, MA, USA). Samples were then subjected to electrophoresis using 4-20% SDS-PAGE gel and transferred to nitrocellulose membranes. Primary antibodies for proteins of interest were then mixed in 1% BSA and added for 1 hr (eNOS, 1:1000, rabbit, monoclonal, Cell Signaling, and phosphorylated-eNOS Ser1177, 1:1000, rabbit, monoclonal, Cell Signaling). Expression of GAPDH (1:1000, rabbit, monoclonal, Cell signaling, #D16H11) was used as a loading control. Peroxidase conjugated secondary antibodies and chemiluminescent substrate (Bio-Rad, Hercules, CA, USA) were used for detection.
Materials
1% bovine serum albumin (BSA) solution was used to dissolve endothelin-1 (Millipore Sigma, St. Louis, MO, USA). Methanol (MeOH) and 0.3M NaOH solution were used to reconstitute S1P (Millipore Sigma, St. Louis, MO, USA and Cayman Chemical, Ann Arbor, MI, USA) according to the manufacturer’s recommendations. W146 (Cayman Chemical & Tocris Bioscience, Minneapolis, MN, USA) was reconstituted in MeOH and 0.03M NaOH. CAY10444 and c-PTIO (Cayman Chemical) were dissolved in dimethylformamide and water, respectively. GKT137831 (Cayman Chemical) and GSK2795039 (Millipore Sigma) were both dissolved in ethanol. Apocynin and PEG-Catalase (Millipore Sigma) were dissolved in MeOH and water, respectively. Acetylcholine (Millipore Sigma) was diluted in water. S1P receptor antibodies were purchased from Invitrogen by Thermo Fisher (Thermo Fisher Scientific, Waltham, MA, USA). ROS-ID® NO Detection kit (Enzo Life Sciences, Enzo Biochem, Farmingdale, New York) was dissolved in HEPES buffer at the time of the experiment.
Statistical Analysis
For functional vascular studies, data is presented as mean ± SEM. Dilation is expressed as a percentage of maximal dilation with 100% dilation representing the maximal dilation achieved with the endothelium-independent dilator papaverine. % maximal dilation = (vessel diameter – stable diameter after ET-1 constriction) / (maximal diameter – stable diameter after ET-1 constriction). To compare the dose-response relationship, a two-way ANOVA was used with dose-response to S1P and treatment as parameters with a Geisser-Greenhouse correction. When a significant difference was observed between curves (p < 0.05), results were compared using a Dunnett’s multiple comparisons test. For NO detection studies, all data was normalized and reported as fold-change from baseline. A student’s t-test was performed between intervention (S1P dose or time at maximal flow) and intervention + L-NAME. Western blot data were plotted as ratios of interventions compared against S1P analyzed using a ratio t-test; interventions were compared against S1P. All analyses were performed using GraphPad Prism 9.1.0 (GraphPad Software, San Diego, CA, USA) with statistical significance defined at p < 0.05.
RESULTS
Demographic data for patients is included in Table 1. nonCAD individuals are defined as those having 0-1 risk factor(s) for CAD while the CAD group consisted of patients with a confirmed clinical diagnosis of significant (obstructive) coronary atherosclerosis. Risk factors for CAD included hypertension, hyperlipidemia, active smoker, diabetes mellitus (type 1 and 2), and congestive heart failure. A total of 69 surgical discard specimens were collected from adults aged 22-79. 47 of the 69 patients had 0 risk factors for CAD, while others had only one risk factor. 15 patients had a diagnosis of CAD and could have more than 1 risk factor for the disease. The average age was 42.6±10.8 yrs (mean ± SD) and 67.5±10.2 for the nonCAD and CAD groups, respectively. BMI was similar between the nonCAD and CAD groups. The initial diameters at cannulation and after administration of papaverine (mean ± SD), were 113 ± 17μm and 164 ± 22μm, respectively.
Table 1. Sample demographics.
A total of 69 discarded human samples (54 nonCAD and 15 CAD) were utilized for this study. A subject was categorized as nonCAD if they carried no more than one risk factor for CAD (e.g. hypertension, hyperlipidemia, diabetes mellitus, or congestive heart failure). Subjects categorized as CAD had to have angiographic evidence of obstructive CAD and could have more than 1 risk factor for disease.
| Patient Demographics: Total Samples (n = 69) |
nonCAD (n=54) | CAD (n=15) |
|---|---|---|
| Characteristics | ||
| Sex, M/F/unknown | 2/51/1 | 10/5/0 |
| Age, years (average±SD) | 42.6±10.8 | 67.5±10.2 |
| BMI (average±SD) | 29.2±6.8 | 30.0±6.6 |
| Underlying disease/risk factor(s) | ||
| Hypertension | 4 | 8 |
| Hyperlipidemia | 2 | 11 |
| Diabetes Mellitus | 1 | 3 |
| Congestive Heart Failure | 0 | 3 |
| None of the Above | 47 | 0 |
M = male, F = female, nonCAD = non-coronary artery disease, CAD = coronary artery disease, BMI = body mass index
S1PR1 and S1PR3 contribute to S1P-induced dilation in arterioles from nonCAD adults
To confirm that both S1PR1 and S1PR3 are present in human resistance arterioles, immunohistochemistry was performed. As shown in Figure 1A, both S1PR1 and S1PR3 are present in the endothelium of microvessels from nonCAD adults as indicated by the brown staining which is absent in the negative controls (Fig S1A).
Figure 1. S1P elicits dilation in human arterioles from nonCAD patients via activation of S1PR1 or S1PR3.
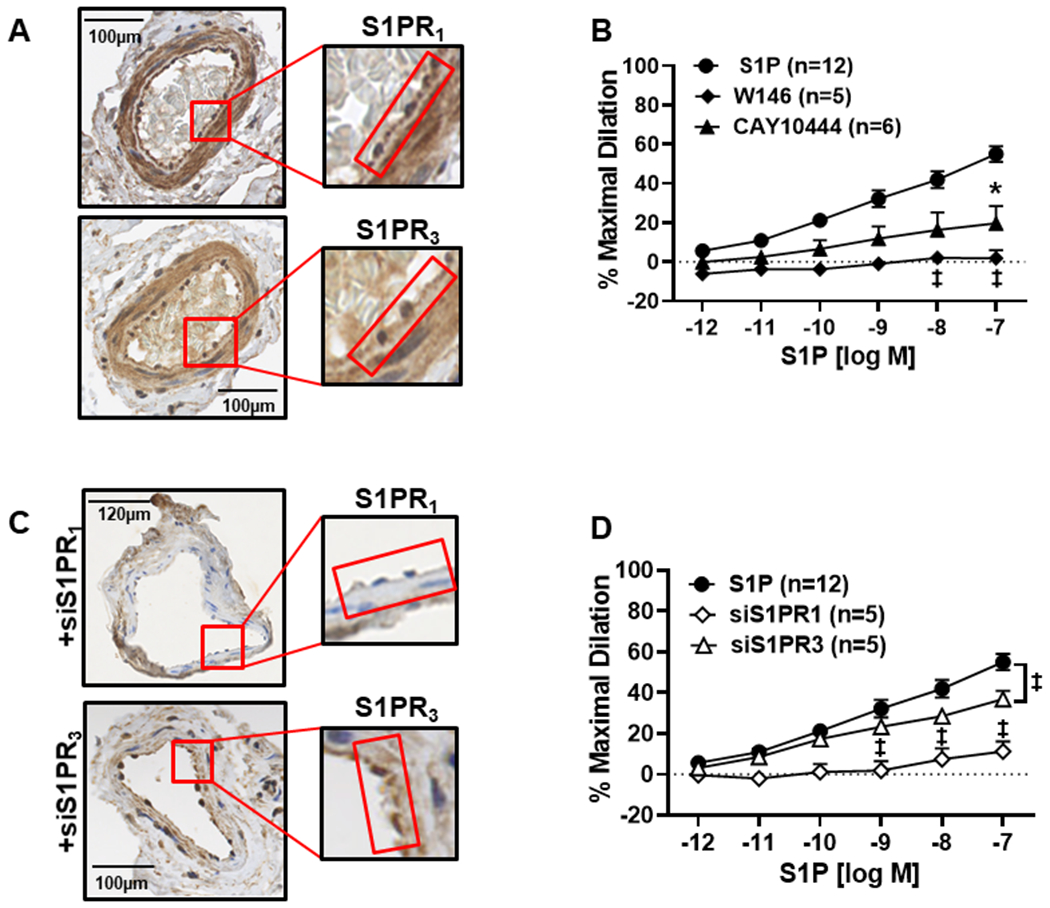
A) Immunohistochemistry (IHC) staining (representative images of n=5) shows that S1P receptors 1 and 3 are expressed in the human microvasculature from nonCAD adults (left). The endothelium is highlighted by the red box. B) S1P elicits a dose-dependent dilation (10−12 to 10−7, n=12) which is inhibited by the S1PR1 inhibitor W146 (10μM, n=5) and CAY10444 (10μM), an S1PR3 inhibitor (n=5) compared to S1P alone (n=12, ‡p<0.01, *p<0.05) C) Confirmation of siRNA-mediated knock-down of S1PR1 and S1PR3 in human arterioles with endothelium outlined in red (representative images from n=3). D) Dilation to S1P is significantly decreased in microvessels treated with either siS1PR1 (n=5) or siS1PR3 (n=5, ‡p<0.01 and *p<0.05) siRNA treatment versus S1P. Data are expressed as mean % maximal dilation±SEM.
To determine whether exogenous S1P is capable of eliciting dilation in human arterioles, increasing doses of exogenous S1P (10−12 M to 10−7 M) were administered to pressurized microvessels pre-constricted with ET-1. A linear, dilatory dose response curve was observed (Figure 1B) with maximal dilation occurring at 10−7 (% maximal dilation [MD] mean ± SEM, 55 ± 4.1, n=12) which was significantly increased compared to vehicle control (%MD 1.9 ± 1.0, n=5 ‡p<0.01, *p<0.05) (Figure S1B). Interestingly, administration of 10−6 S1P elicited constriction and reduced the diameters on average to 22.7 ± 5.8, n=12 (Figure S3A), suggesting higher doses of S1P cause constriction, a phenomenon previously reported in animal studies18. To determine whether the response to S1P requires activation of S1PR1 and/or S1PR3, a dose response curve (DRC) to S1P was performed both in the presence of the S1PR1 antagonist W146 and the S1PR3 antagonist CAY10444. As shown in Figure 1B, maximal dilation to S1P was significantly reduced with inhibition of either S1PR1 or S1PR3 compared to the administration of S1P alone (%MD 1.7 ± 4, n=5 and 19.7 ± 8.7, n=6, respectively, versus 55 ± 4.1 for S1P alone, ‡p<0.01, *p<0.05), suggesting that both receptors are critical for S1P-induced dilation in humans. To demonstrate the viability of the endothelium in the presence of these inhibitors, dose response curves to the endothelial-dependent vasodilator acetylcholine (Ach) were performed following incubation with either W146 or CAY10444. Despite the presence of these inhibitors, vessels maintained the ability to dilate to Ach (80% ± 7.6, n=4 and 89.1% ± 2.7, n=4, for W146 and CAY10444, Figure S2B).
To confirm the role of S1PR1 and S1PR3 in this mechanism of dilation, siRNA against both receptors was utilized to decrease expression in nonCAD human arterioles. As shown in Figure 1C, a decreased amount of brown staining is observed in the endothelium (denoted by red box) in microvessels intraluminally exposed to siRNA for S1PR1 and S1PR3. Dilation to S1P was significantly decreased in vessels treated with siRNA against S1PR1 (%MD 11.2 ± 4.9, n=5) compared to S1P alone (%MD 55 ± 4.1, n=12, ‡p<0.01). Likewise, a significant reduction in S1P-induced maximal dilation was seen in arterioles treated with siRNA against S1PR3 (%MD 36.8 ± 4.1, n=5) compared to untreated vessels (‡p<0.01) (Figure 1D). S1P was capable of eliciting dilation in arterioles intraluminally treated with negative control siRNA (Figure S1C).
S1P-induced dilation in human microvessels requires formation of NO
To determine the role of NO in dilation, we performed S1P DRCs in the presence of the NOS inhibitor L-NAME and cPTIO, a potassium salt capable of scavenging NO. As shown in Figure 2, nonCAD vessels exposed to either L-NAME or cPTIO exhibited signficantly reduced dilation to S1P (%MD 15.8 ± 6.5, n=5, and 9.5 ± 3.1, n=5 for L-NAME and cPTIO, respectively) compared to S1P alone (%MD 55.0 ± 4.1, n=12, ‡p<0.01).
Figure 2. Role of NO in S1P-induced dilation of the human microvasculature.
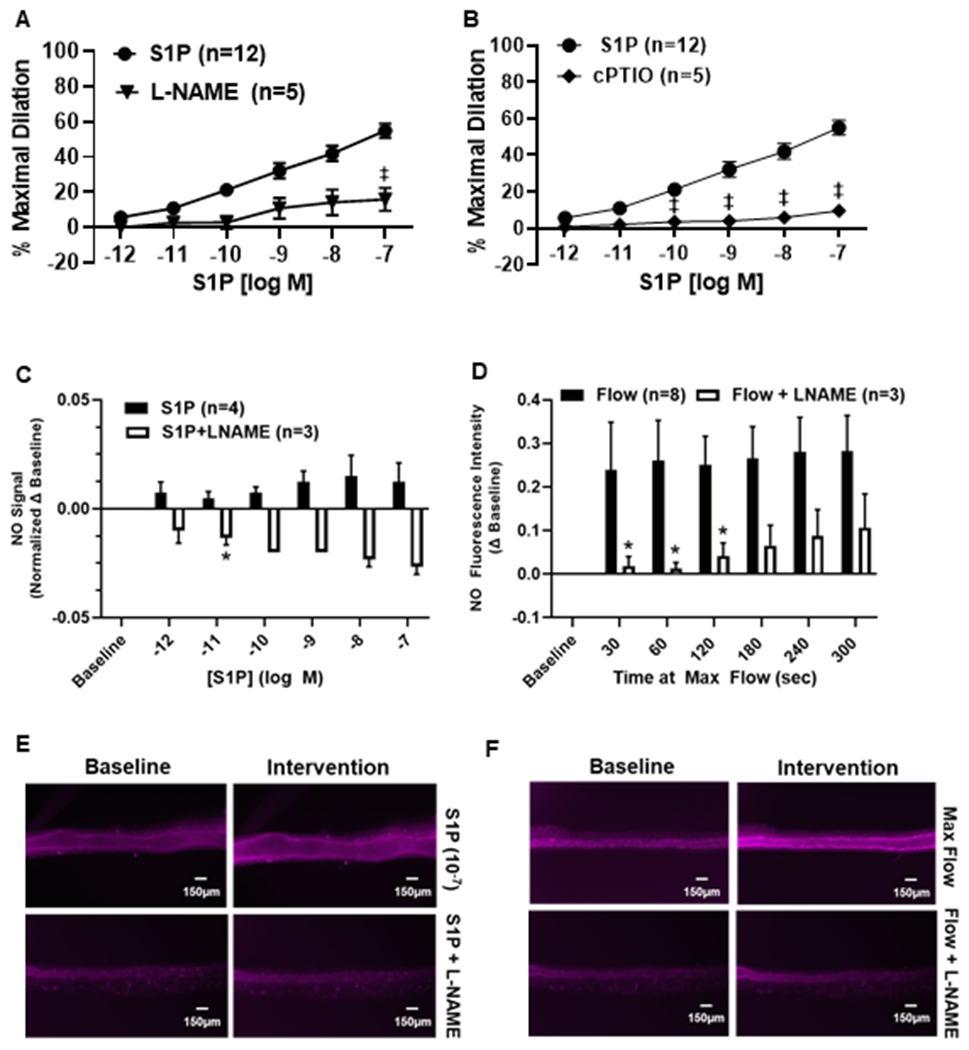
A) S1P-induced dilation in arterioles from nonCAD patients is reduced in the presence of the nitric oxide synthase (NOS) inhibitor L-NAME (100μM, n=5, ‡p<0.01) L-NAME versus S1P alone, as well as during exposure to the NO scavenger cPTIO (100μM, n=5, ‡p<0.01) (B). C) NO increases from baseline following exposure to increasing doses of S1P (n=4) which is quenched in the presence of L-NAME (100μM, n=3). D) NO formation in response to maximal flow (20μL/min, n=8) which is significantly reduced with L-NAME (100μM, n=3, *p<0.05). E) Representative images of NO production in vessels exposed to S1P +/− L-NAME (100μM). F) Representative images of NO formation in arterioles exposed to maximal flow (20μL/min) +/− L-NAME (100μM). Data are expressed as the mean±SEM.
To confirm that S1P induces formation of NO in microvessels, NO was detected with a fluorescent probe and measured using microscopy. After intraluminal incubation with the probe, vessels were exposed to increasing concentrations of S1P (10−12 M to 10−7 M). Figure 2C illustrates that administration resulted in a slight increase in signal intensity which was quenched by the addition of L-NAME at 10−11 M of S1P (n=3, *p<0.05). Since shear stress is a robust stimulus for NO production, changes in fluorescence intensity were also measured in vessels exposed to maximal flow (pressure gradient of 100cm H2O) to serve as a comparison. NO formation rose dramatically within 30 s of being exposed to maximal flow (Figure 2D) as indicated by an increase in fluorescence intensity which peaked at 300 s (5 min) of flow (n=8). The signal was significantly reduced in the presence of L-NAME at 30, 60, and 120 s of flow (n=3, *p<0.05). Figures 2E and 2F are representative images for NO detection from S1P and flow, respectively.
H2O2 is necessary for NO-mediated S1P-induced dilation
It has been shown that H2O2 sensitizes S1P receptors in endothelial cells and results in a higher degree of eNOS activation compared to S1P alone19. To understand the role of H2O2 in S1P-mediated vasodilation, S1P DRCs were performed in nonCAD human microvessels during exposure to PEG-Catalase which resulted in impaired dilation compared to S1P (%MD 8.4 ± 3.1, n=5, ‡p<0.01, Figure 3A). Since sphingolipids are known to acutely activate NADPH oxidases (NOXs), we then tested whether inhibition of NOX would cause a similar impairment in dilation. Microvessels treated with the non-specific NOX inhibitor apocynin exhibited a significant decrease in S1P-induced dilation (%MD 10.1 ± 2.9, n=5) compared to S1P alone (%MD 55.0 ± 4.1, n=12, ‡p<0.01, Figure 3B). To determine which specific NOX is required for dilation, inhibitors for the two major NOX isoforms in endothelial cells, NOX2 and NOX4, were administered during exposure to S1P. The use of either inhibitor significantly decreased S1P-induced dilation (%MD 22.9 ± 6.6, n=5 and 11.2 ± 8.3, n=5, for NOX2 and NOX4, respectively) compared to S1P (‡p<0.01, Figure 3C–D). Finally, to add mechanistic insight into the role of NOX in S1P vasodilation, western blots were performed in human umbilical vascular endothelial cells (HUVECs) to determine if NOX4, an enzyme that has been shown to be necessary for eNOS signaling20, is required for S1P-induced increases in phosporylated eNOS21. Figure 4 shows that the Ser1177 phosphorylated:total eNOS ratio is decreased in endothelial cells treated with both S1P and the NOX4 inhibitor compared to S1P alone (n=4 for all groups, *p<0.05). Of note, constriction to S1P was observed at the higher dose (10−6) in nonCAD arterioles treated with the various inhibitors as well as siRNA (Figure S3).
Figure 3. H2O2 production via NOX is necessary for S1P-induced dilation in microvessels from nonCAD subjects.
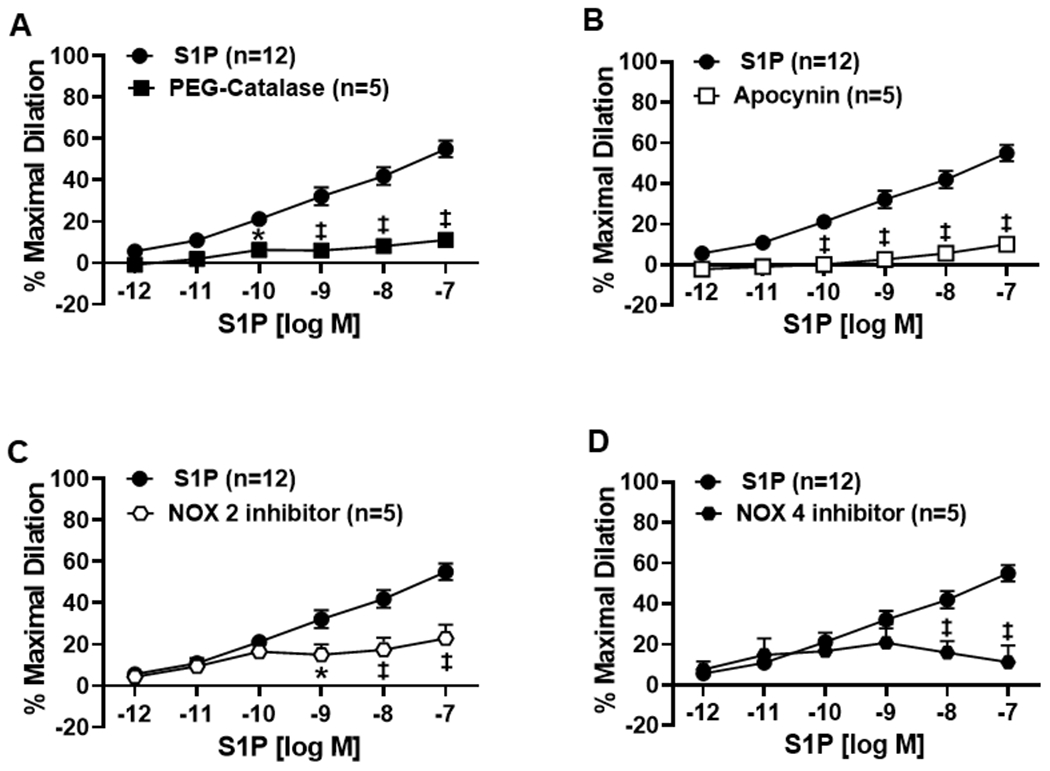
A) Dilation elicited by S1P is significantly reduced in the presence of PEG-Catalase (500U, n=5), the non-specific NOX inhibitor apocynin (300μM, n=5) (B), the specific NOX2 inhibitor GSK2795039 (1μM, n=5) (C), and the NOX4 inhibitor GKT137831 (1μM, n=5) compared to S1P alone (n=12, ‡p<0.01, *p<0.05) (D).
Figure 4. Role of H2O2 in phosphorylated eNOS from S1P.
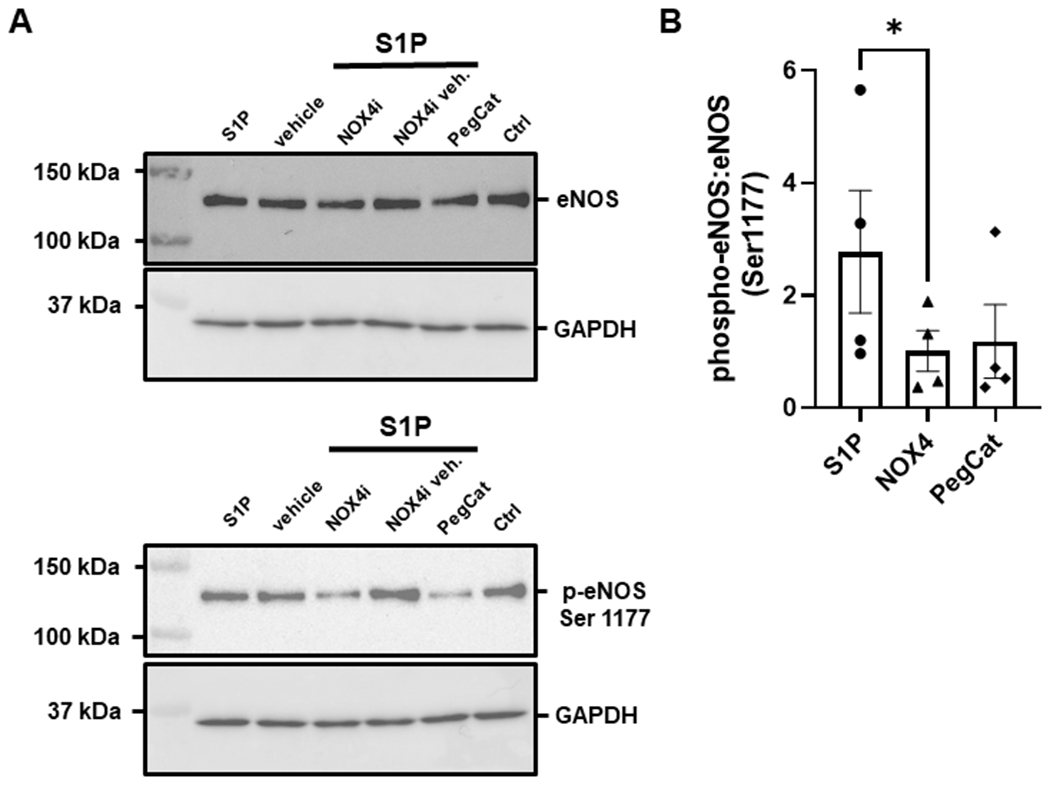
S1P-induced Ser1177 phosphorylation of eNOS in HUVECs (n=4) is impaired in the presence of PEG-Catalase (500U, n=4) and significantly reduced during inhibition of NOX4 with GKT137831 (1μM, n=4). Representative images of western blots on the left (A) and phospho-eNOS:eNOS ratio comparison on the right (B). Data are expressed as the mean ratio of phosphorylated eNOS to total eNOS±SEM. *p<0.05 compared to S1P alone.
Dilation to S1P during disease relies primarily on activation of S1PR3 and formation of H2O2
Similar to arterioles from nonCAD individuals, both S1PR1 and S1PR3 were identified in microvessels collected from patients with CAD (Figure 5A). Although S1P elicited dilation in vessels from individuals with disease (%MD 56.3 ± 12.6, n=5), this effect was not signficantly reduced during inhibition of S1PR1 (%MD 30.8 ± 13.4, n=5, Figure 5B) but rather was only impaired during exposure to the S1PR3 antagonist (%MD 8.1 ± 8.4, n=4, *p<0.05) as opposed to S1P alone (%MD 56.3 ± 12.6, n=5) (Figure 5C). To establish the mediator of S1P-induced dilation in human vessels during disease, we exposed the vessels to L-NAME and PEG-Catalase. Exposure of the vessels to L-NAME did not significantly impact the dilation caused by S1P (%MD 27 ± 6, n=5, Figure 5D), however was significantly impaired with PEG-Catalase (%MD 12.4 ± 8.8, n=5, *p<0.05) compared to S1P alone (%MD 56.3 ± 12.6, n=5, Figure 5E). Ach-induced dilation did not differ in arterioles from patients with CAD versus nonCAD (Figure S2A). A relative constricion was also observed following administration of a higher dose of S1P (10−6) in presence of all inhibitors (Figure S4).
Figure 5. The S1P signaling pathway during disease (CAD).
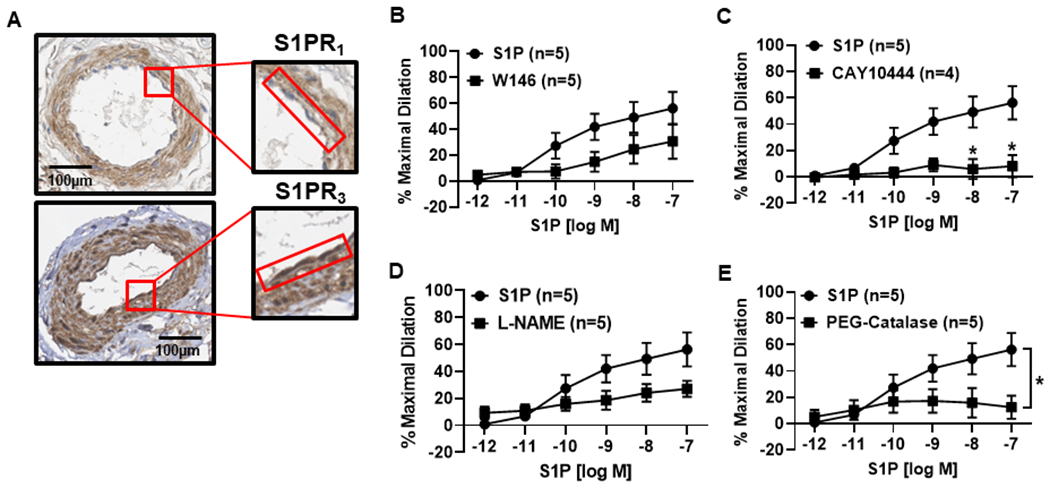
A) Representative immunohistochemistry (IHC) images showing staining (representative images of n=5) of S1P receptors 1 and 3 in the microvascular endothelium from patients with CAD. The endothelium is highlighted by the red box. B) S1P induced a dose-dependent dilation (10−12 to 10−7) was not significantly reduced in the presence of the S1PR1 inhibitor W146 (10μM, n=5) in arterioles collected from patients diagnosed with CAD. (C) Dilation to S1P was significantly impaired during exposure to the S1PR3 inhibitor CAY10444 (10μM, n=5) compared to S1P alone (n=5). D) In vessels from patients with CAD, dilation was not significantly impaired with the addition of L-NAME (100μM, n=5), but was significantly reduced in the presence of PEG-Catalase (500U, n=5) (E). Data are expressed as mean % maximal dilation±SEM., *p<0.05 compared to S1P treatment alone.
DISCUSSION
To our knowledge this is the first study to examine the mechanism by which S1P elicits vasodilation in humans during both health and disease. There are several major findings here, the first of which is that S1P promotes dilation in the human microvasculature in physiological concentrations, suggesting a possible major role in blood pressure homeostasis. We hypothesized and confirmed that S1P receptors 1 and 3 are present in the endothelium of resistance arterioles. Both pharmacological inhibition and genetic knock-down of S1PR1 and S1PR3 supported our hypothesis that both receptors are critical for S1P-induced dilation in microvessels from nonCAD individuals. Our fourth major and unexpected finding was that the S1P pathway requires both NO and H2O2 to elicit dilation, and activation of NADPH oxidases (NOX 2 and 4) is a key contributor to this mechanism. However, unlike the robust formation of NO during shear, exogenous exposure to S1P results in a modest increase in NO. Finally, although both receptors are present in the microvascular endothelium from patients with disease (CAD), dilation from S1P relies primarily on activation of S1PR3 as opposed to S1PR1. These translational findings suggest that S1P signaling in the human endothelium regulates vascular tone however the mechanism differs between nonCAD adult individuals versus those with cardiovascular disease.
S1P signals through three G-protein coupled receptors (GPCRs), S1PR1-3. Expression of these receptors in vascular tissue is heterogenous, with S1PR1 being the dominant receptor in the endothelium10, S1PR2 being more highly expressed in the medial layer, and S1PR3 being expressed in both the endothelium and smooth muscle cells11. The downstream signaling of each receptor has also proven unique as S1PR1 signals exclusively through the Gαi subunit22, S1PR3 signals through Gi, Gq, and G12/13, and S1PR2 relies primarily on G12/13 10, 23–27. Several studies done in human cell lines 28–30 and rodents 31 have shown that increases in NO upon administration of S1P are dependent on activation of S1PR132 and S1PR333, 34, which lead to eNOS activation through PI3K and Akt that is initiated by the shared Gi subunit. Our data suggests that inhibition or decreased expression of either receptor results in decreased dilation suggesting a necessary communication between the two downstream signaling pathways. With the exception of pharmacological inhibition of S1PR1, residual dilatory capacity to S1P was observed in vessels with reduced activity or expression of either receptor. The residual dilation may be the result of incomplete inhibition of the targeted receptor, activity of the non-targeted receptor, or dilation through an unknown mechanism.
Generation of mouse models in which S1PR1 is deleted in endothelial cells by tamoxifen-activated Cre recombinase (S1PR1flox/flox or S1PR1 ECKO) have been crucial to understand the role of S1P in blood pressure regulation in animals. Using this mouse model, Cantalupo et al. elegantly demonstrated that S1PR1, as opposed to S1PR3, is primarily responsible for blood pressure homeostasis 35. Others have provided compelling evidence that S1PR3 is equally important in controlling arterial pressure. Nofer et al. have shown that high-density lipoprotein (HDL) exerts its dilatory effects through bound S1P and activation of S1PR333. Forrest et al. also proposed that S1PR3 is essential for blood pressure homeostasis as arterial pressure was significantly increased in S1PR3−/− mice 36. It is important to note that these preclinical studies were not only done in animals, but also in varying sized vessels from different vascular beds. Although these studies have added insight to the importance of S1PR signaling, the abundance of S1PR1 versus S1PR3 in large (conduit vessels) versus small (arterioles) is unknown as well as how receptor expression changes from health to disease. It is clear from our data and others that both receptors have an important role in vascular tone and blood pressure regulation. Perhaps the more interesting question is whether expression of one receptor over the other is more beneficial for long-term vascular health and if receptor expression or signaling is altered in the early stages of hypertension.
While the majority of studies have focused on how S1P reduces vascular tone through formation of NO, only a few have suggested an important role for H2O2. Dantas and colleagues have reported that dilation from S1P relies exclusively on eNOS-derived NO, as opposed to other endothelium-derived hyperpolarizing factors (EDHFs)31. Our results suggest that NO formed during exposure to S1P is modest compared to NO generated during increased flow, although still critical as S1P-induced dilation is inhibitable by both L-NAME and cPTIO. One possible explanation for this finding could be that the level of NO produced from S1P is below the threshold of detection for the fluorescent probe. Another possibility is the fact that S1P also promotes ROS formation, therefore NOX-produced superoxide could potentially quench S1P-induced NO thus falling below the threshold for detection. Finally, the majority of S1P in human plasma is bound to HDL37, however for this study, as common in other studies 30, 31, 38, S1P was administered directly, unbound to lipoproteins. It is possible that S1P triggers a higher level of NO formation if bound to HDL.
Based on our findings, the human vasodilatory response to S1P requires production of both NO and H2O2, presumably through nitric oxide synthase (NOS) and NOX, respectively. Although the deleterious effects of H2O2 on the cardiovascular system are well-documented in the literature39, formation of this ROS during health has also proven critical. Generation of H2O2 has been shown to be a necessary component in the activation of eNOS during laminar shear stress40. Some speculate the source of H2O2 is the key factor in the determination of whether its downstream result is beneficial or detrimental to the vascular wall. For instance, emerging evidence suggests that NOX4-derived H2O2 is vasculoprotective 41–43. Furthermore, Tian et al. have demonstrated that H2O2 promotes eNOS localization to the plasma membrane to enhance NO production during shear44. More specifically, studies performed in BAECs have shown that activation of NOX4 with subsequent activation of eNOS is critical in NO-formation during flow45. Our data add to this growing body of evidence as we found that NOX 2 and 4 are needed for dilation from S1P. Further, we provide data suggesting that S1P-induced increases in phosphorylated eNOS rely on activation of NOX4. While studies have suggested that NO is the primary mediator responsible for S1P dilation, our study underscores the importance of ROS in the mechanism by which S1P may regulate blood pressure in humans. The H2O2 formed from S1P may itself trigger dilation through various proposed mechanisms including activation of transient receptor potential vanilloid 4 (TRPV4) channels46, or act as a component of eNOS activation47.
Although animal studies have been instrumental in deciphering the S1P signaling pathway, the use of discarded surgical tissue allowed for interrogation of S1P signaling specific to humans and also shed light on how this mechanism changes during times of disease, a model which is difficult to replicate in animals. Interestingly, although both S1PR1 and S1PR3 were both identified and found to be functional in arterioles from patients with CAD, dilation to S1P was observed to be predominantly due to activation of S1PR3. Of note, since collection of tissue from diseased patients is more scarce, only pharmacological studies were conducted. A key hallmark of cardiovascular disease is decreased NO bioavailability due to increased cellular oxidative stress48. Our data here suggests that dilation due to acute S1P exposure relies primarily on activation of S1PR3 and formation of reactive oxygen species (H2O2). Although S1P may regulate tone by promoting vasorelaxation, the mechanism by which this is accomplished may cause further oxidative damage. We have previously shown that chronic exposure (16-20 hrs) to S1P is capable of restoring flow-induced NO-mediated dilation in vessels from CAD patients49. This, combined with the fact that plasma S1P levels are reduced in patients with CAD, suggests that during times of low S1P exposure or a high oxidative stress environment, the S1P signaling pathway is pro-oxidant but can be manipulated over time to reestablish a healthy endothelium.
Study limitations
As common in other studies 30, 31, 38 including this study, S1P was administered freely dissolved in methanol. This may not accurately represent the physiological effects of S1P in human blood, as most of the lipid is bound to either albumin or HDL 37. Another issue is that to enhance clarity during video microscopy, arterioles were stripped of perivascular adipose tissue (PVAT) prior to cannulation. As such, possible interactions between S1P and PVAT remain unknown, and it is possible that S1P also elicits effects on PVAT which ultimately influence tone on the vessel it surrounds. Finally, a major limitation of this study is the amount of heterogeneity that exists among the patients from which tissue was collected. Please refer to the Supplemental Material for a complete description of limitations associated with the use of human tissue for translational research.
Perspectives
Preclinical studies have revealed an important role for S1P in the regulation of blood pressure. Although S1P is effective at reducing microvascular tone, the mechanism of dilation in humans is more complex and favors formation of oxidative stress in individuals with cardiovascular disease. The question remains whether targeting of S1PR1 is a safe and effective strategy to reduce blood pressure in this patient population.
Supplementary Material
Novelty and Relevance.
What is New?
Physiologic levels of S1P elicit dilation in human resistance arterioles through activation of S1PR1 and S1PR3.
Both S1PR1 and S1PR3 are expressed in microvessels from nonCAD adult patients as well as individuals diagnosed with CAD.
S1P signaling in arterioles from nonCAD adults requires formation of both NO and H2O2, as opposed to predominantly H2O2 in vessels from subjects with CAD.
What is Relevant?
S1P signaling in nonCAD humans requires formation of both NO and reactive oxygen species.
The mechanism of S1P-induced dilation is altered during disease and favors formation of H2O2 via activation of S1PR3.
Clinical/Pathological Implications.
S1P signaling has emerged as a potential therapeutic target in the regulation of blood pressure. The mechanism by which S1P causes dilation in human resistance arterioles is altered during disease and may amplify production of intracellular oxidative stress.
Acknowledgments
The authors wish to thank the surgeons and nurses at Froedtert Memorial Lutheran Hospital and the Ascension Healthcare Group for assistance in providing human tissue.
Sources of Funding
This work is supported by a National Institutes of Health (NHLBI) K08 HL141562 (JKF).
Abbreviations and Acronyms
- BAEC
bovine aortic endothelial cells
- CAD
coronary artery disease
- cPTIO
2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide
- EDHF
endothelial-derived hyperpolarizing factor
- eNOS
endothelial nitric oxide synthase
- FID
flow-induced dilation
- H2O2
hydrogen peroxide
- HUVEC
human umbilical vein endothelial cells
- L-NAME
Nω-nitro-L-arginine methyl ester
- MD
maximal dilation
- NADPH
nicotinamide adenine dinucleotide phosphate
- NO
nitric oxide
- NOX
nicotinamide adenine dinucleotide phosphate oxidase
- PEG-Catalase
polyethylene glycol - catalase
- ROS
reactive oxygen species
- S1P
sphingosine-1-phosphate
- S1PR
sphingosine-1-phosphate receptor
Footnotes
Previous Presentations: This work was presented as a poster and oral presentation at the Experimental Biology Annual meeting in 2021.
Disclosures
The authors have no financial conflicts of interest to disclose.
References
- 1.Dosunmu-Ogunbi AAM, Galley JC, Yuan S, Schmidt HM, Wood KC and Straub AC. Redox Switches Controlling Nitric Oxide Signaling in the Resistance Vasculature and Implications for Blood Pressure Regulation: Mid-Career Award for Research Excellence 2020. Hypertension. 2021;78:912–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Szijarto IA, Marko L, Filipovic MR, Miljkovic JL, Tabeling C, Tsvetkov D, Wang N, Rabelo LA, Witzenrath M, Diedrich A, Tank J, Akahoshi N, Kamata S, Ishii I and Gollasch M. Cystathionine gamma-Lyase-Produced Hydrogen Sulfide Controls Endothelial NO Bioavailability and Blood Pressure. Hypertension. 2018;71:1210–1217. [DOI] [PubMed] [Google Scholar]
- 3.Kadlec AO, Chabowski DS, Ait-Aissa K, Hockenberry JC, Otterson MF, Durand MJ, Freed JK, Beyer AM and Gutterman DD. PGC-1alpha (Peroxisome Proliferator-Activated Receptor gamma Coactivator 1-alpha) Overexpression in Coronary Artery Disease Recruits NO and Hydrogen Peroxide During Flow-Mediated Dilation and Protects Against Increased Intraluminal Pressure. Hypertension. 2017;70:166–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gutterman DD, Chabowski DS, Kadlec AO, Durand MJ, Freed JK, Ait-Aissa K and Beyer AM. The Human Microcirculation: Regulation of Flow and Beyond. Circ Res. 2016;118:157–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cantalupo A, Gargiulo A, Dautaj E, Liu C, Zhang Y, Hla T and Di Lorenzo A. S1PR1 (Sphingosine-1-Phosphate Receptor 1) Signaling Regulates Blood Flow and Pressure. Hypertension. 2017;70:426–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fukuhara S, Simmons S, Kawamura S, Inoue A, Orba Y, Tokudome T, Sunden Y, Arai Y, Moriwaki K and Ishida J. The sphingosine-1-phosphate transporter Spns2 expressed on endothelial cells regulates lymphocyte trafficking in mice. The Journal of clinical investigation. 2012;122:1416–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hisano Y, Kobayashi N, Yamaguchi A and Nishi T. Mouse SPNS2 functions as a sphingosine-1-phosphate transporter in vascular endothelial cells. PloS one. 2012;7:e38941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kawahara A, Nishi T, Hisano Y, Fukui H, Yamaguchi A and Mochizuki N. The sphingolipid transporter spns2 functions in migration of zebrafish myocardial precursors. Science. 2009;323:524–527. [DOI] [PubMed] [Google Scholar]
- 9.Cantalupo A, Zhang Y, Kothiya M, Galvani S, Obinata H, Bucci M, Giordano FJ, Jiang XC, Hla T and Di Lorenzo A. Nogo-B regulates endothelial sphingolipid homeostasis to control vascular function and blood pressure. Nat Med. 2015;21:1028–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee MJ, Thangada S, Claffey KP, Ancellin N, Liu CH, Kluk M, Volpi M, Sha’afi RI and Hla T. Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell. 1999;99:301–12. [DOI] [PubMed] [Google Scholar]
- 11.Means CK and Brown JH. Sphingosine-1-phosphate receptor signalling in the heart. Cardiovascular research. 2009;82:193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cantalupo A and Di Lorenzo A. S1P Signaling and De Novo Biosynthesis in Blood Pressure Homeostasis. J Pharmacol Exp Ther. 2016;358:359–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hou J, Chen Q, Wu X, Zhao D, Reuveni H, Licht T, Xu M, Hu H, Hoeft A, Ben-Sasson SA, Shu Q and Fang X. S1PR3 Signaling Drives Bacterial Killing and Is Required for Survival in Bacterial Sepsis. Am J Respir Crit Care Med. 2017;196:1559–1570. [DOI] [PubMed] [Google Scholar]
- 14.Durand MJ and Gutterman DD. Diversity in mechanisms of endothelium-dependent vasodilation in health and disease. Microcirculation. 2013;20:239–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nishijima Y, Cao S, Chabowski DS, Korishettar A, Ge A, Zheng X, Sparapani R, Gutterman DD and Zhang DX. Contribution of KV1.5 Channel to Hydrogen Peroxide-Induced Human Arteriolar Dilation and Its Modulation by Coronary Artery Disease. Circ Res. 2017;120:658–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beyer AM, Zinkevich N, Miller B, Liu Y, Wittenburg AL, Mitchell M, Galdieri R, Sorokin A and Gutterman DD. Transition in the mechanism of flow-mediated dilation with aging and development of coronary artery disease. Basic Res Cardiol. 2017;112:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kadlec AO, Chabowski DS, Ait-Aissa K, Hockenberry JC, Otterson MF, Durand MJ, Freed JK, Beyer AM and Gutterman DD. PGC-1α (Peroxisome Proliferator–Activated Receptor γ Coactivator 1-α) Overexpression in Coronary Artery Disease Recruits NO and Hydrogen Peroxide During Flow-Mediated Dilation and Protects Against Increased Intraluminal Pressure. Hypertension. 2017;70:166–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Igarashi J and Michel T. Sphingosine-1-phosphate and modulation of vascular tone. Cardiovascular research. 2009;82:212–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Igarashi J, Miyoshi M, Hashimoto T, Kubota Y and Kosaka H. Hydrogen peroxide induces S1P1 receptors and sensitizes vascular endothelial cells to sphingosine 1-phosphate, a platelet-derived lipid mediator. American Journal of Physiology-Cell Physiology. 2007;292:C740–C748. [DOI] [PubMed] [Google Scholar]
- 20.Craige SM, Chen K, Pei Y, Li C, Huang X, Chen C, Shibata R, Sato K, Walsh K and Keaney JF Jr. NADPH oxidase 4 promotes endothelial angiogenesis through endothelial nitric oxide synthase activation. Circulation. 2011;124:731–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Igarashi J, Bernier SG and Michel T. Sphingosine 1-phosphate and activation of endothelial nitric-oxide synthase. differential regulation of Akt and MAP kinase pathways by EDG and bradykinin receptors in vascular endothelial cells. J Biol Chem. 2001;276:12420–6. [DOI] [PubMed] [Google Scholar]
- 22.Reinhard NR, Mastop M, Yin T, Wu Y, Bosma EK, Theodorus WJ, Gadella J, Goedhart J and Hordijk PL. The balance between Gαi-Cdc42/Rac and Gα12/13-RhoA pathways determines endothelial barrier regulation by sphingosine-1-phosphate. Molecular Biology of the Cell. 2017;28:3371–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee M-J, Van Brocklyn JR, Thangada S, Liu CH, Hand AR, Menzeleev R, Spiegel S and Hla T. Sphingosine-1-phosphate as a ligand for the G protein-coupled receptor EDG-1. Science. 1998;279:1552–1555. [DOI] [PubMed] [Google Scholar]
- 24.ZONDAG CG, POSTMA RF, ETTEN IV, VERLAAN I and MOOLENAAR HW. Sphingosine 1-phosphate signalling through the G-protein-coupled receptor Edg-1. Biochemical Journal. 1998;330:605–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Okamoto H, Takuwa N, Yatomi Y, Gonda K, Shigematsu H and Takuwa Y. EDG3 is a functional receptor specific for sphingosine 1-phosphate and sphingosylphosphorylcholine with signaling characteristics distinct from EDG1 and AGR16. Biochemical and biophysical research communications. 1999;260:203–208. [DOI] [PubMed] [Google Scholar]
- 26.Windh RT, Lee M-J, Hla T, An S, Barr AJ and Manning DR. Differential coupling of the sphingosine 1-phosphate receptors Edg-1, Edg-3, and H218/Edg-5 to the Gi, Gq, and G12 families of heterotrimeric G proteins. Journal of Biological Chemistry. 1999;274:27351–27358. [DOI] [PubMed] [Google Scholar]
- 27.Kon J, Sato K, Watanabe T, Tomura H, Kuwabara A, Kimura T, Tamama K-i, Ishizuka T, Murata N and Kanda T. Comparison of intrinsic activities of the putative sphingosine 1-phosphate receptor subtypes to regulate several signaling pathways in their cDNA-transfected Chinese hamster ovary cells. Journal of Biological Chemistry. 1999;274:23940–23947. [DOI] [PubMed] [Google Scholar]
- 28.Igarashi J and Michel T. Agonist-modulated targeting of the EDG-1 receptor to plasmalemmal caveolae: eNOS activation by sphingosine 1-phosphate and the role of caveolin-1 in sphingolipid signal transduction. Journal of Biological Chemistry. 2000;275:32363–32370. [DOI] [PubMed] [Google Scholar]
- 29.Kwon Y-G, Min J-K, Kim K-M, Lee D-J, Billiar TR and Kim Y-M. Sphingosine 1-phosphate protects human umbilical vein endothelial cells from serum-deprived apoptosis by nitric oxide production. Journal of Biological Chemistry. 2001;276:10627–10633. [DOI] [PubMed] [Google Scholar]
- 30.Igarashi J, Bernier SG and Michel T. Sphingosine 1-phosphate and activation of endothelial nitric-oxide synthase differential regulation of Akt and MAP kinase pathways by EDG and bradykinin receptors in vascular endothelial cells. Journal of Biological Chemistry. 2001;276:12420–12426. [DOI] [PubMed] [Google Scholar]
- 31.Dantas APV, Igarashi J and Michel T. Sphingosine 1-phosphate and control of vascular tone. American Journal of Physiology-Heart and Circulatory Physiology. 2003;284:H2045–H2052. [DOI] [PubMed] [Google Scholar]
- 32.Igarashi J, Miyoshi M, Hashimoto T, Kubota Y and Kosaka H. Statins induce S1P1 receptors and enhance endothelial nitric oxide production in response to high-density lipoproteins. British journal of pharmacology. 2007;150:470–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nofer J-R, Van Der Giet M, Tölle M, Wolinska I, von Wnuck Lipinski K, Baba HA, Tietge UJ, Gödecke A, Ishii I and Kleuser B. HDL induces NO-dependent vasorelaxation via the lysophospholipid receptor S1P 3. The Journal of clinical investigation. 2004;113:569–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Theilmeier G, Schmidt C, Herrmann Jr, Keul P, Schäfers M, Herrgott I, Mersmann J, Larmann J, Hermann S and Stypmann Jr. High-density lipoproteins and their constituent, sphingosine-1-phosphate, directly protect the heart against ischemia/reperfusion injury in vivo via the S1P3 lysophospholipid receptor. Circulation. 2006;114:1403–1409. [DOI] [PubMed] [Google Scholar]
- 35.Cantalupo A, Gargiulo A, Dautaj E, Liu C, Zhang Y, Hla T and Lorenzo AD. S1PR1 (Sphingosine-1-Phosphate Receptor 1) Signaling Regulates Blood Flow and Pressure. Hypertension. 2017;70:426–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Forrest M, Sun S-Y, Hajdu R, Bergstrom J, Card D, Doherty G, Hale J, Keohane C, Meyers C and Milligan J. Immune cell regulation and cardiovascular effects of sphingosine 1-phosphate receptor agonists in rodents are mediated via distinct receptor subtypes. Journal of Pharmacology and Experimental Therapeutics. 2004;309:758–768. [DOI] [PubMed] [Google Scholar]
- 37.Takuwa Y, Okamoto Y, Yoshioka K and Takuwa N. Sphingosine-1-phosphate signaling in physiology and diseases. BioFactors (Oxford, England). 2012;38:329–37. [DOI] [PubMed] [Google Scholar]
- 38.Cantalupo A, Zhang Y, Kothiya M, Galvani S, Obinata H, Bucci M, Giordano FJ, Jiang X-C, Hla T and Di Lorenzo A. Nogo-B regulates endothelial sphingolipid homeostasis to control vascular function and blood pressure. Nature Medicine. 2015;21:1028–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ballinger SW, Patterson C, Yan CN, Doan R, Burow DL, Young CG, Yakes FM, Van Houten B, Ballinger CA, Freeman BA and Runge MS. Hydrogen peroxide- and peroxynitrite-induced mitochondrial DNA damage and dysfunction in vascular endothelial and smooth muscle cells. Circ Res. 2000;86:960–6. [DOI] [PubMed] [Google Scholar]
- 40.Breton-Romero R, Gonzalez de Orduna C, Romero N, Sanchez-Gomez FJ, de Alvaro C, Porras A, Rodriguez-Pascual F, Laranjinha J, Radi R and Lamas S. Critical role of hydrogen peroxide signaling in the sequential activation of p38 MAPK and eNOS in laminar shear stress. Free Radic Biol Med. 2012;52:1093–100. [DOI] [PubMed] [Google Scholar]
- 41.Datla SR, Peshavariya H, Dusting GJ, Mahadev K, Goldstein BJ and Jiang F. Important role of Nox4 type NADPH oxidase in angiogenic responses in human microvascular endothelial cells in vitro. Arteriosclerosis, thrombosis, and vascular biology. 2007;27:2319–2324. [DOI] [PubMed] [Google Scholar]
- 42.Craige SM, Kant S, Reif M, Chen K, Pei Y, Angoff R, Sugamura K, Fitzgibbons T and Keaney JF Jr. Endothelial NADPH oxidase 4 protects ApoE−/−mice from atherosclerotic lesions. Free Radical Biology and Medicine. 2015;89:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gray SP, Di Marco E, Kennedy K, Chew P, Okabe J, El-Osta A, Calkin AC, Biessen EA, Touyz RM and Cooper ME. Reactive oxygen species can provide atheroprotection via NOX4-dependent inhibition of inflammation and vascular remodeling. Arteriosclerosis, thrombosis, and vascular biology. 2016;36:295–307. [DOI] [PubMed] [Google Scholar]
- 44.Tian J, Hou Y, Lu Q, Wiseman DA, Vasconcelos Fonsesca F, Elms S, Fulton DJ and Black SM. A novel role for caveolin-1 in regulating endothelial nitric oxide synthase activation in response to H2O2 and shear stress. Free Radic Biol Med. 2010;49:159–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sanchez-Gomez FJ, Calvo E, Breton-Romero R, Fierro-Fernandez M, Anilkumar N, Shah AM, Schroder K, Brandes RP, Vazquez J and Lamas S. NOX4-dependent Hydrogen peroxide promotes shear stress-induced SHP2 sulfenylation and eNOS activation. Free Radic Biol Med. 2015;89:419–30. [DOI] [PubMed] [Google Scholar]
- 46.Bubolz AH, Mendoza SA, Zheng X, Zinkevich NS, Li R, Gutterman DD and Zhang DX. Activation of endothelial TRPV4 channels mediates flow-induced dilation in human coronary arterioles: role of Ca2+ entry and mitochondrial ROS signaling. Am J Physiol Heart Circ Physiol. 2012;302:H634–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Drummond GR, Cai H, Davis ME, Ramasamy S and Harrison DG. Transcriptional and posttranscriptional regulation of endothelial nitric oxide synthase expression by hydrogen peroxide. Circ Res. 2000;86:347–54. [DOI] [PubMed] [Google Scholar]
- 48.Miura H, Bosnjak JJ, Ning G, Saito T, Miura M and Gutterman DD. Role for Hydrogen Peroxide in Flow-Induced Dilation of Human Coronary Arterioles. Circulation Research. 2003;92:e31–e40. [DOI] [PubMed] [Google Scholar]
- 49.Schulz ME, Katunaric B, Hockenberry JC, Gutterman DD and Freed JK. Manipulation of the Sphingolipid Rheostat Influences the Mediator of Flow-Induced Dilation in the Human Microvasculature. Journal of the American Heart Association. 2019;8:e013153. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.