

Abstract
Treatment with immune checkpoint inhibitors targeting programmed cell death protein 1 (PD-1) or its ligand (PD-L1) can generate durable responses in various cancer types, but only in a subset of patients. The use of predictive biomarkers for response to PD-1/PD-L1 inhibitors is critical for patient selection. Expression of PD-L1 has demonstrated utility in patient selection. Tumour mutational burden (TMB) is an emerging biomarker for response to PD-1/PD-L1 inhibitors. The evaluation of this biomarker is based on the hypothesis that a high number of mutations in somatic exonic regions will lead to an increase in neoantigen production, which could then be recognised by CD8+ T cells, resulting in improved immune responses. In this review, we will discuss rationale and implementation of TMB usage in patients, development of different methods to assess it, current limitations and technical issues to use this biomarker as a diagnostic test and propose future perspectives beyond TMB.
Keywords: Biomarker, Tumour mutational burden, Immunotherapy
1. Introduction
Immune checkpoint inhibitors (ICIs) have demonstrated durable antitumor effects in treatment of multiple cancer types. Since the initial approval of ipilimumab in metastatic melanoma in 2011, multiple ICIs have been developed for a variety of cancer types. Phase III clinical trials have demonstrated the efficacy of anti-PD-l/PD-L1 alone or in combination with other agents in many cancer types such as small and non-small cell lung cancer (NSCLC), urothelial, head and neck, clear renal cell, hepatocellular carcinoma and triple negative breast cancer [1–10]. Furthermore, evidence of ICI efficacy in tumours with mismatch repair deficiency led to the first tumour-agnostic FDA approval, based on molecular alteration rather than tumour location.
Despite the impressive results obtained with ICIs, only a subset of patients have a durable clinical benefit from such therapies. It is estimated that only 12% of all cancer patients will benefit from ICIs used as monotherapy [11]. In addition, ICIs have substantial costs, carry significant risks of immune-related toxicities and may even cause tumour hyperprogression and worsen tumour prognosis in a subset of patients [12]. To improve patient selection, there is an urgent need of biomarkers to identify the subgroup of patients with a durable survival upon ICI treatment.
A. Immunotherapy in solid cancer: Development of predictive biomarkers is crucial
Optimisation of ICI treatment requires predictive biomarkers to identify patients, who are likely to benefit from this treatment. Biomarkers for ICI effectiveness have been found; nevertheless, their predictive role remains unsatisfying. Currently, only PD-L1 is validated to predict response to ICIs. Indeed, pembrolizumab as a monotherapy is approved for the first-line of NSCLC in patients selected with PD-L1 > 50% [2]. In breast cancer, atezolizumab is approved in combination with nab paclitaxel in metastatic triple negative breast cancer with PD-L1 > 1%, evaluated using SP142 antibody [9].
However, PD-L1 is an imperfect biomarker due to tumour intrinsic PD-L1 heterogeneity and its lability during cancer history [13]. Despite association between PD-L1 expression and response rate to immunotherapy in a pan cancer analysis, many patients with PD-L1 expression are resistant to ICIs and some patients without PD-L1 expression gain benefit from the therapy. Hence, this marker is not optimal to stratify patients who will have a clinical benefit from ICI treatment. Therefore, other approaches are tested such as transcriptomic signatures or tumour infiltrate lymphocytes (TILs) presence. For example, we have shown that in NSCLC patients treated with anti-PD-1, addition of CD8+ T lymphocytes infiltration analysis improves PD-L1 ability to predict progression-free survival (PFS) [14]. Furthermore, CD8 and PD-L1 combination seems to be more effective as a biomarker when assessed by transcriptomic analysis rather than IHC. When compared to other transcriptomic signatures like IFN or EIG [15], these two gene signatures provide similar results.
Genomic analysis is another promising way to find biomarkers of response to ICIs. Cancer is inherently a somatic genetic disease. Tumour cells are characterised by abnormal functions (proliferation, apoptosis resistance, etc.), which are consequences of genomic alterations. Some of these mutations arc considered ‘driver mutations’, leading the oncogenic process, while other mutations are considered ‘passenger mutations’ and are less essential for cancer cell survival. Yet, the majority of tumour DNA alterations are defined as passenger mutations, with few or little implicated in carcinogenesis or tumour growth. However, all driver or passenger mutations code for mutant proteins. These mutant proteins, like all other cell proteins, are recycled through the proteasome pathway. After degradation into 8–9 amino acid length peptides, these molecules are processed in endoplasmic reticulum and loaded in HLA class I molecules that will be presented on cancer cell surface. When peptides originated from mutant proteins have higher affinity for HLA molecules than normal peptides, they will be presented by HLA molecules on cancer cells surface, allowing CD8+ T cells to recognise and kill cancer cells. Consequently, genetic mutations in cancer cells could be considered as the first event driving antitumor immune response. Therefore, tumours that contain a high level of mutations will have an increased level of tumour neoantigens and will present a higher immunogenicity (Figure 1). The hypothesis put forward implies that a high number of mutation in somatic exonic region will lead to an increased neoantigen production, which then could be recognised by CD8+ T cells, explaining the improved immune response [16]. In this context, a new promising biomarker emerged recently: the tumour mutational burden (TMB) which is classically defined as the number of non-synonymous exonic mutations per megabase (Mb).
Fig. 1.
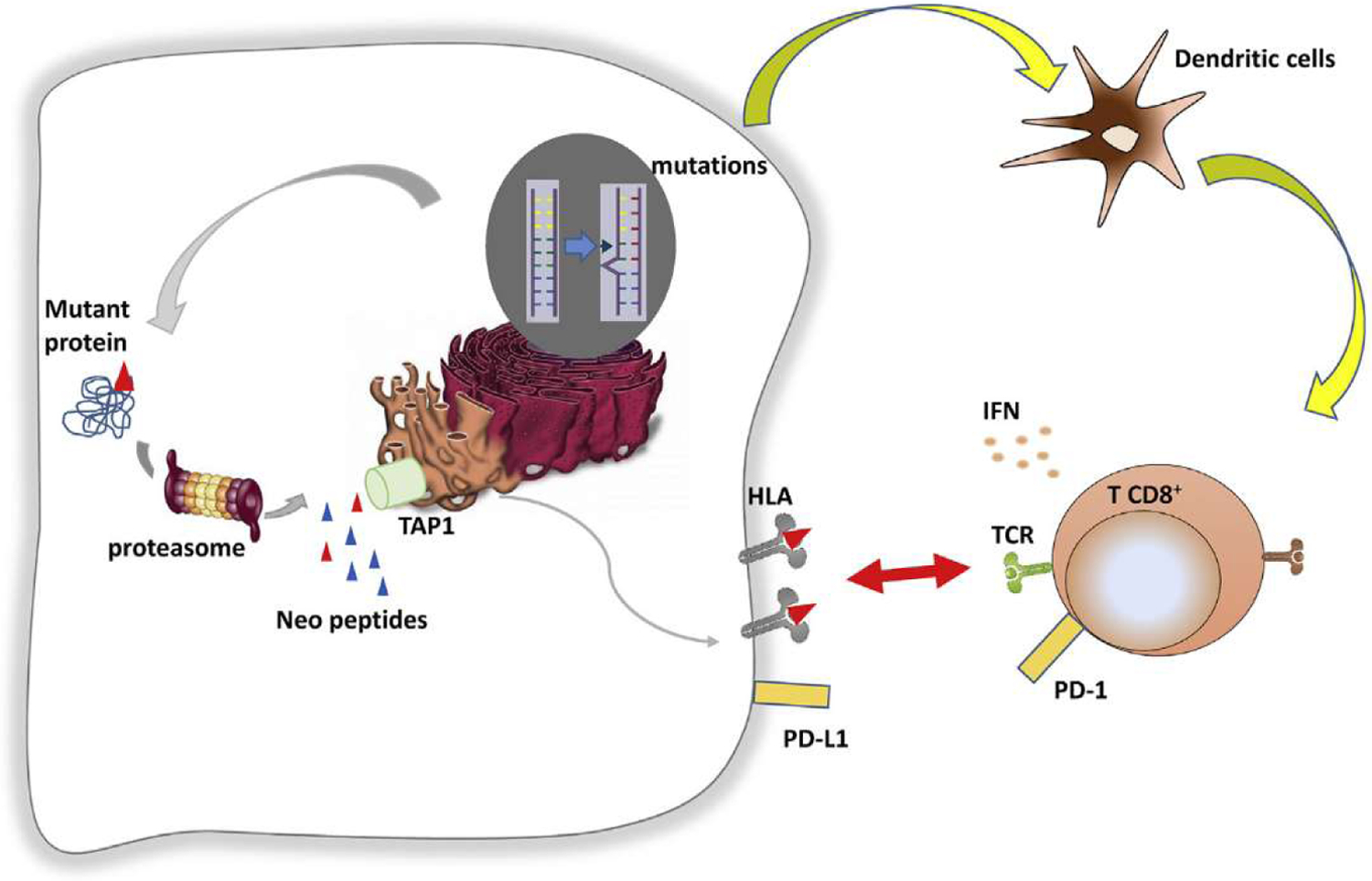
Mechanisms of Antigen (Ag) presentation.
A novel biomarker should have an analytic and clinical utility, economic feasibility and a biologic rationale [17]. In this review, these parameters will be discussed. In addition, we will specially highlight the current clinical results concerning TMB, development of different methods to assess it, current limitations and technical issues to use biomarker as a diagnostic test and discuss future perspectives beyond TMB.
B. TMB as a potential biomarker of ICI response
Analytic and clinical utility of TMB as a biomarker was initially tested and validated in several retrospective studies showing the potential capacity of a high number of mutations or high number of neoantigens to predict ICI efficacy. The first studies supporting this hypothesis were published by Snyder et al. [18] and Rizvi et al. [19]. These studies showed that higher numbers of exonic non-synonymous mutations were associated with higher response rates in patients with melanoma treated with anti-CTLA-4 and NSCLC treated with anti-PD-1, respectively. Similarly, our group also observed in 77 patients with NSCLC treated in second-line with nivolumab that higher exonic non-synonymous mutations and neoantigens are associated with a better outcome [20]. More recently, in pan tumour analysis published in 2017, TMB has been evaluated in TCGA cohort and median TMB value per tumour type shows a correlation with the rate of objective response in clinical trials testing ICIs [21]. Such data suggest that TMB is a pan-cancer genomic biomarker related to checkpoint inhibitors efficacy.
Overall, in several retrospective studies performed in different types of cancer, the hypothesis of correlation between high TMB and better response to ICIs seems to be validated. TMB appears as a promising predictive biomarker of ICI efficacy, whatever the ICI and in numerous cancer types [22]. Furthermore, in mismatch repair deficient tumours, TMB provides additional information although mismatch repair status seems to be the main parameter [23].
However, the relationship between TMB and ICI response is imperfect both across tumour types and within tumour types. For example, Merkel cell carcinoma (MCC), renal cell cancers (RCC) and mesothelioma all have higher response rates to ICIs than would be anticipated from their TMBs [24]. This may be related to the higher quality of antigens in these tumour types, resulting from viral antigens (in MCC), a high number of indel mutations (in RCC) and complex chromosomal rearrangements (in mesothelioma). TMB also has poor predictive value within some tumour types such as gliomas and renal cell cancers [25]. Concerning clear cell renal cancer, Swanton’s group showed that TMB is not a biomarker of efficacy of ICI therapy. However, clear cell renal cancers are characterised by high number of insertions/deletions in the genome (indel). Neoantigens derived from indel mutations were nine times enriched for mutant-specific binding, as compared with non-synonymous single nuclear variant-derived neoantigens, which classically constitute TMB. In contrast to TMB, indel or mutant-specific neoantigens were associated with T-cell recruitment. Finally, frameshift indel count is significantly associated with ICI therapy efficacy in both renal cancer and melanoma [26].
C. TMB as a biomarker in clinical trials
In addition, with these retrospective data, TMB was analysed retrospectively in prospective trials and then prospectively to determine its ability to be a predictive biomarker of ICI’s response.
TMB was analysed in the prospective clinical trial KEYNOTE 158 to assess its ability to be a validated clinical biomarker in ten different tumour types (anal, biliary, cervical, endometrial, salivary, thyroid, vulvar carcinoma, mesothelioma, neuroendocrine tumour and small cell lung cancer) treated by pembrolizumab [27]. In this trial, high TMB was defined as more that 10 mutations per Mb using FoundationOne assay. High TMB, using the selected cutoff, concerned 13% of patients. ORR and PFS were improved in patients with high TMB. No difference in terms of OS was detected. Accordingly, such data suggest that TMB may be predictive of the efficacy of pembrolizumab monotherapy in this multi-organ trial.
Moreover, in several trials using different combinations of anti-PD-l/PD-Ll and anti-CTLA-4, the higher the TMB, the higher the response rate to ICIs. Nevertheless, it should be noted that some contradictory results were observed [21].
In NSCLC, results of the Checkmate 026 and 227 trials suggest that TMB determined using either exome sequencing or FoundationOne assay might become a new diagnostic biomarker to predict response to ICI, either alone or in combinatorial regimens [28,29]. Indeed, in these two studies, ICI alone or in combination has shown a benefit against chemotherapy only in a TMBhigh subgroup (defined as > 243 mutations in exonic sequence or > 10 mutation per Mb using FoundationOne assay). Studies with pembrolizumab and atezolizumab in NSCLC used different cutoffs, but seem to give the same conclusions. Similar results were also obtained in small cell lung cancer and melanoma treated with combotherapy nivolumab plus ipilimumab (summary in Table 1) [18,19,24,28–35,36–45].
Table 1.
Evidence of TMB as a biomarker in clinical trial.
| Tumour type | Testing drug | TMB method | TMB cutoff | Type of benefit | |
|---|---|---|---|---|---|
| NSCLC | Checkmate 026 | Nivolumab | WES | >243 mut/exome | ORR, PFS |
| Rozenblum et al, 2017 | Nivolumab or pembrolizumab | FoundationOne | >9,6 mut/Mb | ORR | |
| KEYNOTE 001 | Pembrolizumab | WES | >178 mut/exome | ORR, PFS | |
| POPLAR/OAK | Atezolizumab | Foundation bTMB | >10 mut/Mb | PFS, OS | |
| POPLAR/FIR/BIRCH | Atezolizumab | FoundationOne | 1st line: >13,5 mut/Mb 2nd line: >17,1 mut/Mb |
ORR, PFS, OS | |
| BFAST and BF1RST | Atezolizumab | Foundation bTMB | Unknown | ORR, PFS, OS | |
| MSKCC, Rizvi et al, 2018 | Multiagent (alone or in combination) | WES | >8,5 mut/Mb | ORR, PFS | |
| Checkmate 227 | Ipilimumab + nivolumab | FoundationOne | >10 mut/Mb | ORR, PFS | |
| Checkmate 568 | Ipilimumab + nivolumab | FoundationOne | >10 mut/Mb | ORR, PFS | |
| Checkmate 012 | Ipilimumab + nivolumab | WES | >158 mut/exome | ORR, PFS and clinical benefit | |
| Melanoma | Checkmate 064 | Nivolumab | WES | Unknown | ORR, OS |
| Van Allen et al 2015 | Ipilimumab | WES | 197 mut/exome | Clinical benefit | |
| Snyder et al 2014 | Ipilimumab | WES | >100 mut/exome | OS | |
| Johnson et al, 2016 | Multiagent (alone or in combination) | FoundationOne | >23,1 mut/Mb | ORR, PFS, OS | |
| Hugo et al, 2016 | Multiagent (alone or combination) | WES | >495 mut/exome | OS | |
| Checkmate 038 | Nivolumab | WES | >100 mut/exome | OS | |
| Bladder | Checkmate 275 | Nivolumab | WES | >167 mut/exome | ORR, PFS, OS |
| Imvigor 210 | Atezolizumab | FoundationOne | >16 mut/Mb | ORR, OS | |
| Imvigor 211 | Atezolizumab | FoundationOne | > median | OS | |
| Snyder et al, 2017 | Atezolizumab | WES | > median | PFS | |
| SCLC | Hellman et al, 2018 | Nivolumab + ipilimumab | WES | >248 mut/exome | ORR, PFS, OS |
| CRC | KEYNOTE 012 - 028 | Pembrolizumab | WES | Unknown | ORR, PFS |
| Multiple solid tumours | Goodman et al, 2017 | Multiagent (alone or combination) | FoundationOne | >20 mut/Mb | ORR, PFS, OS |
| Yarchoan et al, 2017 | Multiagent (alone or combination) | Various | Various | ORR |
In contrast, in other tumour types, TMB is not associated with ICI therapy efficacy. In particular, it should be noted that in germ cell tumours, which are known to be less genetic unstable, TMB median is low and non-predictive of ICI’s response. In the recent ABACUS trial, testing atezolizumab as a neoadjuvant monotherapy for localised urothelial cancer, TMB did not predict response rate or survival [46].
As shown in Table 1, the selected cutoff values change between clinical studies suggesting that it could be difficult to find a pan tumour cutoff value. In the recent large MSKCC-IMPACT trial, association between TMB and ICI efficacy was tested in multiple cancer types. Higher TMB is associated with better outcome in most cancer types but the optimal cutoff point may vary according to the tumour type, thus raising the hypothesis that it would be suboptimal to propose a general cutoff common to all cancers. In addition, TMB is a continuous Gaussian variable. It is important to determine if the effect of this biomarker on survival is continuous or an ON/OFF effect, thus enforcing the rational to find an optimal cutoff. In this study, top 10% TMB seem to have better outcome than top 20%, suggesting a continuous dose effect of TMB. Optimal cutoff analysis suggests a continuous effect of TMB with low cutoff values and then an ON/OFF effect after 20Mt/base [47]. Despite these data, 10 mut/Mb cutoff seems to become widely used. Based on Yarchoan meta-analysis [24], we could confirm in the TCGA cohort that the percentage of patients with more than 10 mut/Mb is strongly associated with response rate to immunotherapy (Figure 2A).
Fig. 2.
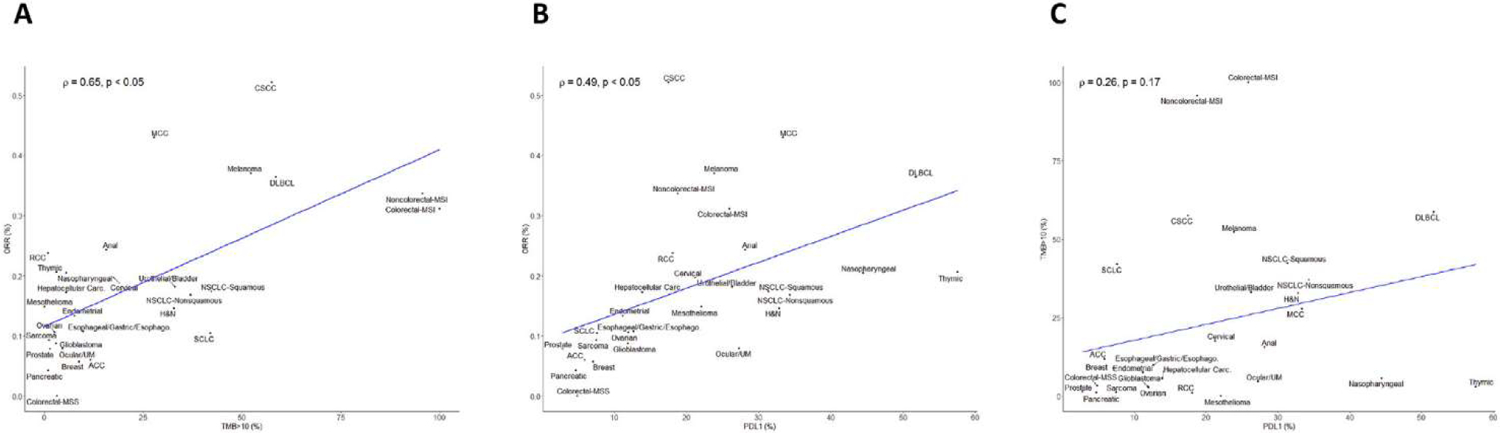
Correlation between TMB, PDL1 expression and response rate to ICIs. A: correlation between TMB and overall response rate. B: correlation between PD-L1 and overall response rate. C: correlation between TMB and PD-L1.
In most clinical studies, PD-L1 expression is associated with a high response rate to ICI. Pan tumour analysis shows a strong correlation between the percentage of tumour tissue expressing PD-L1 and response rate to immunotherapy (Figure 2B). Surprisingly TMB and PD-L1 expression seem to be independent, poorly correlated predictive biomarkers both within most tumour types and across tumour types (Figure 2C). These results suggest that these two markers are probably independent and their combination may improve prediction of patient’s prognosis.
Surprisingly, results from two different exploratory analyses of KEYNOTE trials recently presented at the ESMO 2019 congress showed that TMB failed to prove effective as a biomarker in response to chemotherapy plus ICI or chemotherapy alone as first-line treatment in NSCLC [48,49]. In both analyses, researchers observed no significant association among TMB, used as a continuous variable, and response rates, progression-free survival or overall survival. Similar to previous studies, they also found no association between TMB and PD-L1 expression. It has been suspected that prior chemotherapy, which could generate additional somatic mutations, was not associated with a statistical increase of TMB [50]. Additional data are needed before generating definitive conclusion.
D. Technical issue with TMB usage
For initial studies, TMB was determined by whole exome sequencing (WES) carried out on tumour DNA and matching normal DNA. Germline DNA sequencing is used to remove constitutional variation [51]. Using such type of analysis, TMB is estimated as the number of somatic non-synonymous mutations and reported in numbers per Mb. Recent reports also demonstrated the feasibility to estimate TMB by targeted panel Next Generation Sequencing (NGS). Such strategy may simplify the determination of TMB and could extend its availability, because WES could be difficult to upscale to a large population due to its high costs, substantial turnaround time for analysis and sequencing. Concerning panel strategy, two competitive approaches are available. Some tests are only based on somatic sequencing. To eliminate germline variants, a bioinformatics pipeline is developed to filter out potential germline variants according to published databases of known germline polymorphisms, like dbSNP and ExAC [52,53]. Current leader in such technology is Roche Diagnostics with the Foundation Medicine (FM) NGS approach (FICDx), which uses a panel of 0.8 Mb. The main problem of technologies using databases for germline subtraction is a trend for race-dependent increases in TMB scores [54]. The other technology available uses panel analysis of somatic and germline DNA, similarly to exome sequencing. The MSKCC NGS approach (MSK-IMPACT) is the most developed panel and sequences 468 genes covering 1.22 Mb. Exome and panels tested (FICDx and MSK-IMPACT) have demonstrated their ability to predict ICI response. Both FICDx and MSK-IMPACT have been approved by FDA. Comparison of WES results and estimation of TMB using different panels based on TCGA data show a strong correlation in tumours where ICIs are clinically developed in monotherapy (Figure 3).
Fig. 3.
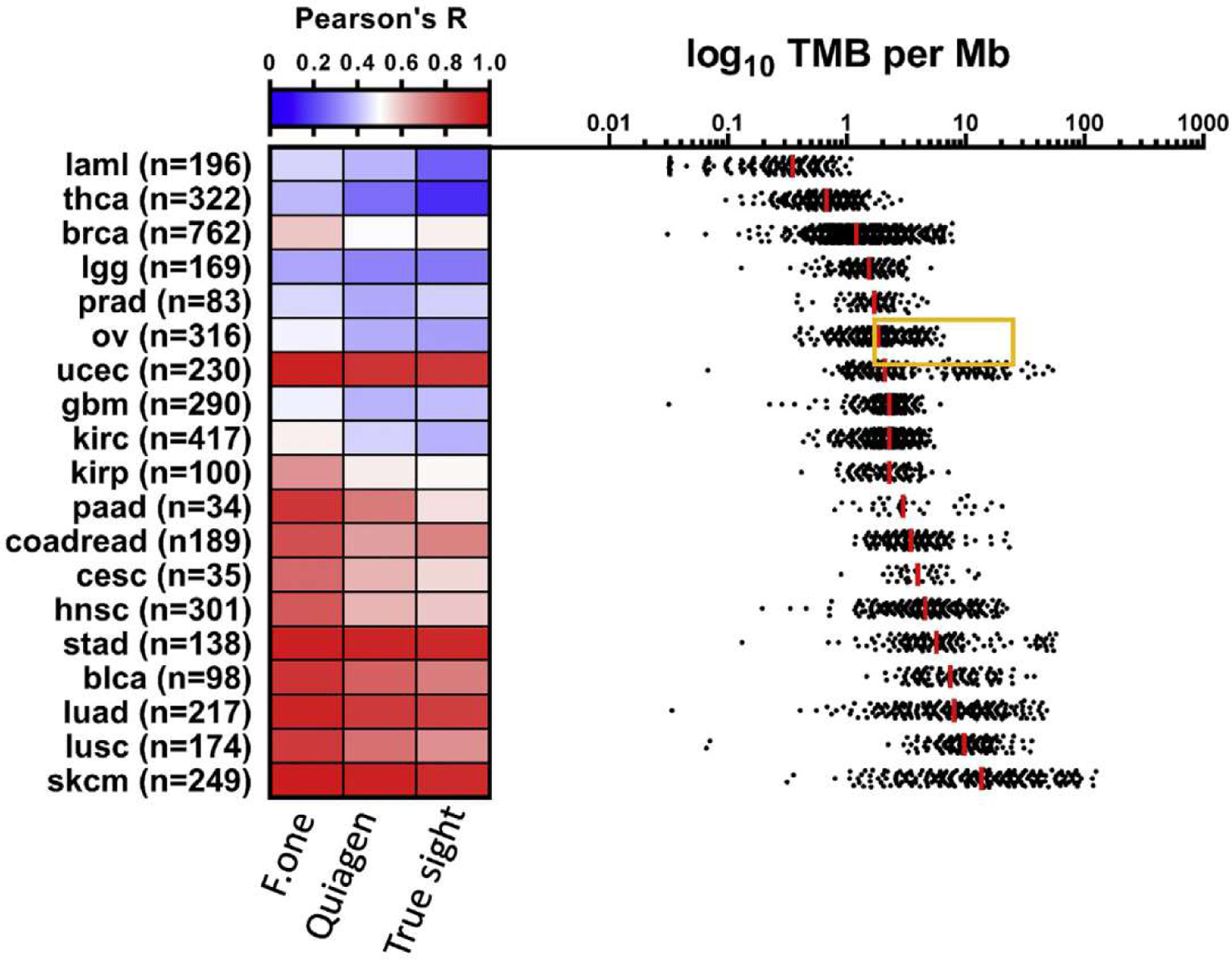
Correlation between NGS panels and WES data to predict TMB.
Nevertheless, some technological issues must be addressed to correctly use such technologies. Firstly, TMB calculation varies between tests. Thus, Exome and MSK-IMPACT count only non-synonymous exonic mutations, while FICDx uses the number of synonymous and non-synonymous mutations and includes insertions, deletions and splicing mutations. From an immunological point of view, such a TMB sequencing method is not very relevant since it considers mutations that would not be translated into neoantigens, which are responsible for driving tumour immunity. This strategy is a technical palliation to artificially enhance the number of detected mutations. In practice, panel size influences the sensibility and specificity of the test. A cutoff of 10 mut/Mbp corresponds to the detection of 306 non-synonymous mutations in the whole exome, 13 mutations with MSK-IMPACT and 8 using F1CDx. Statistical analysis could be performed to estimate precision of TMB estimation with panels. For a TMB of 10 mutations/MB, 95% confidence intervals (CIs) ranged between 3.0–21.8 and 5.5–14.6, for panel sizes of 0.533 and 1.942. Such data are important as for a panel (<1 Mb) such as F1CDx, we have observed large CIs for TMBs, between 0 and 30 mutations per Mb. These data suggest that TMB estimation using small gene panels can be highly imprecise and thus clinically suboptimal when used for patient stratification and response prediction. Such problem is particularly important in tumours like NSCLC where TMB value is a continuous variable. For some other diseases, such as colorectal cancer where TMB has a bimodal distribution, this problem is less relevant (Figure 3). Consequently, Stenzinger’s group recently proposed a mathematical method to estimate accuracy limitations inherent to panel TMB and define a new variable called coefficient of variant (CV) to estimate panel TMB accuracy and proposed to include a grey zone where panel could not accurately predict TMB [55]. Table 2 provides the major metrics related to main TMB estimation techniques. TMB calculation included both synonymous and non-synonymous mutations requiring a formula to scale results to the number of missense mutations determined by WES (scale factor = sum (all mutations)/sum (missense mutations). Scaling factors range between 1.44 and 1.66. Such difference between tumours may artificially elevate TMB. Some tumours present mutations not related to neoantigens and may impede TMB to predict checkpoint efficacy.
Table 2.
Main types of TMB assays.
| Type | Number of genes | Size (MB) | Germline analysis | TMB definition | Number of mutations detected for 10 m/MB in the genome | Cl 95% for a TMB evaluated at 10 m/MB | CV |
|---|---|---|---|---|---|---|---|
| WES | 22 000 | 30 | Yes | No. of somatic, missense mutations | 306 | Gold standard | |
| MSK-IMPACT | 468 | 1.5 MB | Yes | No. of somatic, missense mutations | 14 | [5.1; 15.7] | 25 |
| FoundationOne CDx | 384 | 0.8 | No | Synonymous and non-synonymous, short indel | 8 | [4.3; 19.7] | 35 |
| True Sight | 500 | 2 | No | Synonymous and non-synonymous, short indel | 18 | [5.3; 14.2] | 22 |
| Oncomine | 409 | 1.7 | No | Synonymous and non-synonymous, short indel | 15 | [4.9; 14.5] | 24 |
| QIAGEN Comprehensive Cancer Panel | 160 | 0.7 | No | Synonymous and non-synonymous, short indel | 7 | [4.0; 20.6] | 37 |
Bioinformatics algorithms, which could differ between tests, might also have a major impact when comparing reproducibility based on different tests. Friends of Cancer Research established a working group to create a universal reference and harmonise these methods [56]. The most frequently used variation caller is MuTect for somatic variant detection. For somatic and constitutional sequencing, the most widely used pipeline involves the Genome Analysis Toolkit supplementing with GATK-Mutect 2, which is based on MuTect and GATK-HaplotypeCaller. Applications applied for InDel detection present a wide variety and application reliability is difficult to determine [57]. Moreover, filtering algorithms for putative germline variants, variant allele frequency and FFPE-induced deamination artefacts may vary between assays and can strongly affect TMB values. Lastly, subtracting algorithms used for tumour only analysis are not adapted for Asian and African people. A trend for race-dependent TMB scores increase is observed [54].
Several pre-analytic variables could affect TMB determination and should be standardised across platforms: depth of sequencing, length of sequencing reads. Type of fixative agents and fixation time influence the degree of formaldehyde fixed-paraffin embedded (FFPE)-induced deamination artefacts, which impacts analysis of TMB. In addition, low tumour purity, resulting from sampling errors may lead to reduced TMB assay sensitivity. A particular difference between panel sequencing and WES is the depth of sequencing, around 100X for exome and 2000X for panels. Such difference may enhance the capacity of panels to detect sub-clonal mutations and artificially increase TMB number with sub-clonal mutations. Although such mutations are known to be unrelated to ICI efficacy, this issue may limit the clinical efficacy of panel analysis [58].
Altogether, such data support that presently WES should remain the gold standard for TMB assessment. Large panels of more that 2–5 MB using somatic and germline sequencing could be a cost-effective alternative solution [59]. However, to improve the usage of TMB, standardisation of pre-analytic procedures, harmonisation of bioinformatics pipeline and comparison between tests in large patient cohorts, determining scaling factors to be able to compare tests, are necessary.
E. Exome analysis beyond TMB
TMB is a good surrogate marker of the number of neo-peptides, but looking at additional genetic variables could be interesting to predict response to ICI. Recently an editor raised questions concerning the quality rather than quantity of neoantigens [60]. Authors based their argumentation on recent fundamental studies, which underlined that many additional parameters could influence the ability of neoantigens to stimulate effective immune responses. Expression of mRNA and protein may influence the quantity of antigens presented by HLA molecules. Mutation clonality is also important as subclonality and intratumoural heterogeneity are frequently associated with the lack of ICI efficacy despite high TMB [58]. The quality of neoantigens and in particular their hydrophobicity, affinity to HLA molecules and resemblance to foreign peptides could influence immunogenicity. Another important point is the requirement of peptide presentation by HLA molecules. Mutations in the machinery required for optimal peptide presentation to HLA class I molecules, such as TAP protein or B2m mutations, could easily explain lack of efficacy of ICI therapy despite high TMB. Functional diversity of the HLA-I genes could also be involved in ICI efficacy. Recent reports underline that loss of HLA locus heterozygosity is associated with lack of ICI efficacy [61]. In addition, an HLA-I genotype with two alleles with divergent sequences enables presentation of an increased diversity neoantigens, suggesting that HLA type may also influence ICI efficacy. This divergence could be estimated using a metric called HLA-I evolutionary divergence. This metric was recently evaluated in patients treated with ICI and was a strong determinant of survival [62]. Data connecting divergent HLA allele advantage to immunotherapy efficacy might enhance prediction of TMB efficacy. In agreement with this hypothesis, we have shown that complex genomic analysis of WES involving neoantigens, clonality and HLA status could outperform TMB analysis [20]. In addition to an elevated TMB, whole exome analysis could analyse other parameters allowing a better response prediction. In this publication, mutations in DNA repair, TCR clonal restriction and HLA A 01 phenotype were associated with ICI clinical efficacy. Additionally, prospective studies are required to confirm these results and to benchmark these composite biomarkers with TMB alone, but we can logically expect that combination of TMB plus HLA typing and addition of genomic variables will improve the predictive power of genetic testing.
An alternative strategy is to combine TMB with other biomarkers such as RNA signature, PD-L1 or histological parameters. A meta-analysis published in JAMA oncology showed that each individual biomarker shows similar predictive capacities concerning response to ICI but a combination of multimodal biomarkers improves prediction of response in comparison to a unique biomarker [63]. Considering these data, validating the presence of many robust biomarkers, some publications confirmed the interest of multimodality of biomarkers to improve prediction. The association of TMB and T cell-inflamed gene expression profile (GEP) has shown interesting results in tissue-agnostic to independently predict response to anti-PD-1 therapy in a large cohort of patients treated in prospective clinical trials. Improved responses were noted in patients with high TMB and high T cell-inflamed GEP signature. TMB and GEP signatures are not correlated and bring benefit. Furthermore, GEP signature is correlated with PD-L1 expression and high TMB and high PD-L1 could also be combined to improve prediction of response to ICI [45]. These data suggest that using biomarkers of inflammation such as PD-L1 or T cell-inflamed GEP in combination with TMB may improve the identification of patients who will better respond to ICI. In melanoma, a multimodal biomarker with high TMB, >50% decrease of cell free DNA and undetectable ctDNA is the best combo to predict overall survival under anti-PDLl + anti-CTLA4 [64]. A recent publication has shown that the combination of TMB and copy number alterations can be used to stratify different types of metastatic tumours into groups with different prognosis and different clinical responses to ICI treatment. Patients with high TMB and low copy number alteration cancer can be an optimal subgroup for ICI therapy [65], Figure 4 summarises the different strategies to improve ICI efficacy combining TMB and other different biomarkers.
Fig. 4.
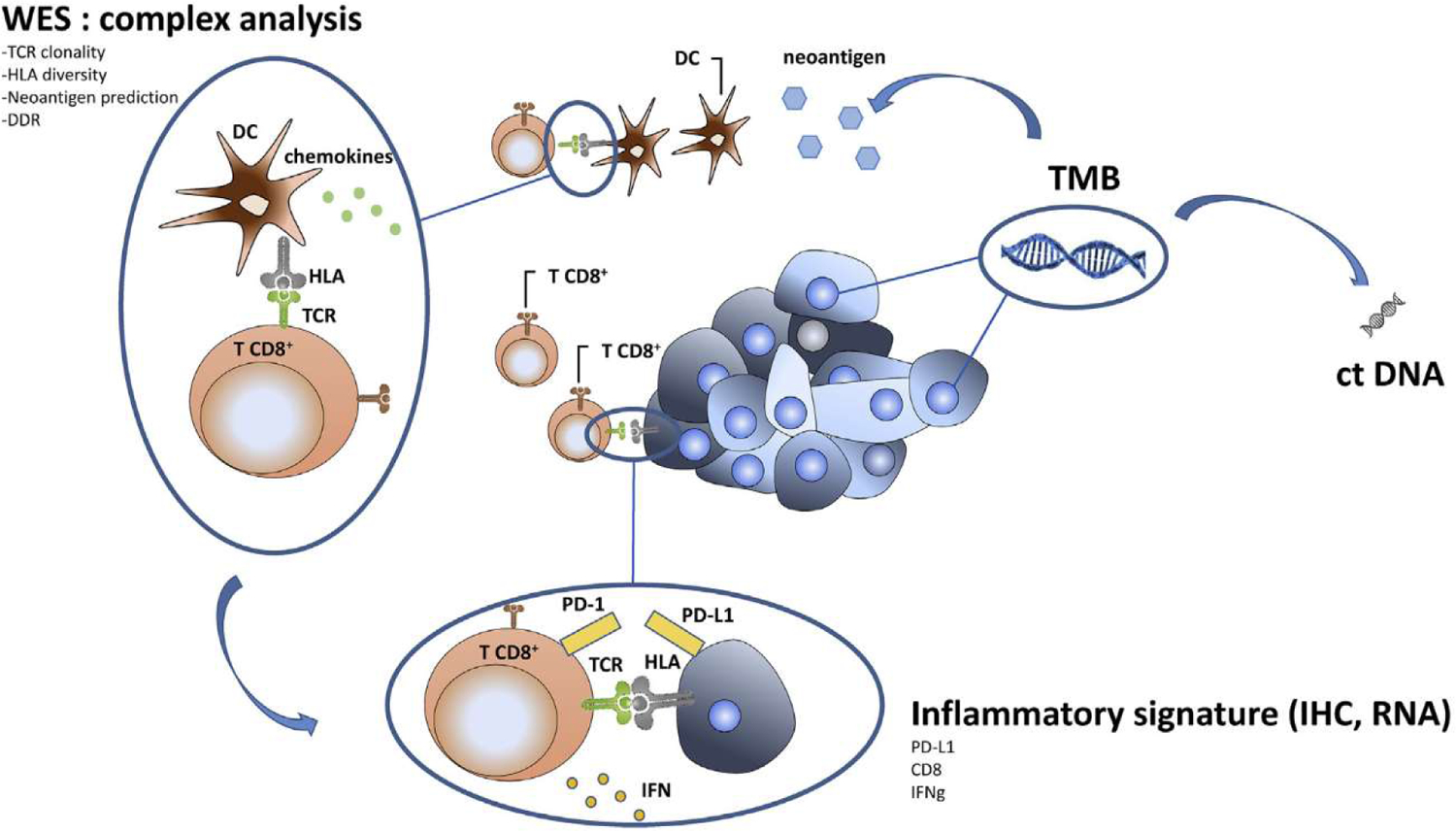
Different biomarkers that could be combined to predict ICI efficacy.
2. Conclusion
In this review, we provide an overview of the current evidence supporting the use of TMB as a biomarker to predict ICI outcomes. High TMB is correlated with an increased number of neoantigens in most cancers and could provide predictors for the efficacy of ICI treatment. Overall, available data suggest that TMB is a good biomarker to identify patients that could be treated by ICI alone or in combination, but not for patients treated with chemotherapy plus ICI. However, additional data are required before definitive validation of this conclusion. Similarly to PD-L1 expression, TMB is also an imperfect biomarker since there are different assays, platforms and cutoffs to characterise TMB. Exploratory studies mostly used whole exome sequencing, which is not widely used in clinical practice. Indeed, for many practical reasons (high cost, substantial turnaround times, limited availability of fresh unfixed tissue, …), gene panel sequencing became a standard to approximately determine TMB in routine diagnostics. However, data on parameters influencing panel-based TMB estimation are limited. Calibration and estimation of available tests precision are urgently needed for clinical development. Along with this line, the recent report published by Buchhalter et al [59] has shown that ‘size does matter’ with an optimal panel size being reached between 1.5 and 3 Mbp considering the benefit–cost ratio. Probably, TMB estimation by panel will gain in precision by defining a grey zone with undetermined predictive role. In the same way, using both somatic and constitutional analyses reinforces the precision of TMB estimation. Additionally, defining an optimal cutoff remains a challenge. Indeed, the optimal cutoff for prediction seems to vary between cancer types [47]. In the future, we may expect that an algorithm using TMB as a continuous variable and other factors such as tumour type, some clinical conditions and PD-L1 testing could improve TMB usage. Although pembrolizumab was approved in a microsatellite instable tumor (MSI) agnostic tumour, for the moment it is not clear if it will be possible to approve ICIs in TMB high agnostic-tumours. Whether TMB performs sufficiently well to be used as a predictive biomarker for ICI response across multiple tumour types may be answered by CheckMate 848, on on-going study of nivolumab alone or in combination with ipilimumab in patients with solid tumours with a high TMB as determined by the FoundationOne assay. Future perspectives will probably be based on the implementation of other WES informative variables such as clonality or HLA divergence and combination of TMB with transcriptomics and histological parameters such PD-L1 or immune infiltrates.
In conclusion, TMB appears to provide clinically valuable and non-redundant information when compared with PD-L1 expression. A lack of standardisation and paucity of randomised clinical trial data have restricted its clinical application. The concurrent use of multiple different approaches and technologies may be the most effective way to enrich for patient response to ICI therapy, as shown in studies combining WES and RNA or WES and IHC to predict patient responses to therapy.
Acknowledgements
Authors thank Isabel Gregoire, Medical Writer, Centre Georges Francois Leclerc for carefully reading the manuscript, Corentin Richard for generation of Figure 3 data and Elise Ballot for generation of Figure 2.
Footnotes
Conflict of interest statement
All authors declare no conflict of interest.
References
- [1].Gandhi L, Rodríguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, et al. Pembrolizumab plus chemotherapy in metastatic non–small-cell lung cancer. N Engl J Med 31 mai 2018;378(22):2078–92. [DOI] [PubMed] [Google Scholar]
- [2].Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, et al. Pembrolizumab versus chemotherapy for PD-L1–positive non–small-cell lung cancer. N Engl J Med 10 nov 2016;375(19):1823–33. [DOI] [PubMed] [Google Scholar]
- [3].Bellmunt J, de Wit R, Vaughn DJ, Fradet Y, Lee J-L, Fong L, et al. Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N Engl J Med 16 mars 2017;376(11):1015–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med 5 nov 2015;373(19):1803–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Motzer RJ, Tannir NM, McDermott DF, Arén Frontera O, Melichar B, Choueiri TK, et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N Engl J Med 5 avr 2018;378(14):1277–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus docetaxel in advanced nonsquamous non–small-cell lung cancer. N Engl J Med 22 oct 2015;373(17):1627–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WEE, Poddubskaya E, et al. Nivolumab versus docetaxel in advanced squamous-cell non–small-cell lung cancer. New England Journal of Medicine 9 juill 2015;373(2):123–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Larkin J, Chiarion-Sileni V, Gonzalez R, Grob J-J, Rutkowski P, Lao CD, et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med 17 oct 2019;381(16):1535–46. [DOI] [PubMed] [Google Scholar]
- [9].Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med 29 nov 2018;379(22):2108–21. [DOI] [PubMed] [Google Scholar]
- [10].Horn L, Mansfield AS, Szczęsna A, Havel L, Krzakowski M, Hochmair MJ, et al. First-line atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N Engl J Med 6 déc 2018;379(23):2220–9. [DOI] [PubMed] [Google Scholar]
- [11].Haslam A, Prasad V. Estimation of the percentage of US patients with cancer who are eligible for and respond to checkpoint inhibitor immunotherapy drugs. JAMA Netw Open 3 mai 2019;2(5):e192535–e192535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Champiat S, Dercle L, Ammari S, Massard C, Hollebecque A, Postel-Vinay S, et al. Hyperprogressive disease (HPD) is a new pattern of progression in cancer patients treated by anti-PD-1/PD-L1. Clinical Cancer Research;2016. Clincanres–1741. [DOI] [PubMed] [Google Scholar]
- [13].Kim H, Chung J-H. PD-L1 testing in non-small cell lung cancer: past, present, and future. J Pathol Transl Med juill 2019;53(4):199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Fumet J-D, Richard C, Ledys F, Klopfenstein Q, Joubert P, Routy B, et al. Prognostic and predictive role of CD8 and PD-L1 determination in lung tumor tissue of patients under anti-PD-1 therapy. Br J Cane oct 2018;119(8):950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest 26 juin 2017;127(8):2930–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Yarchoan M, Johnson BA, Lutz ER, Laheru DA, Jaffee EM. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer, avr 2017;17(4):209–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hayes DF. Biomarker validation and testing. Mol Oncol mai 2015;9(5):960–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 4 déc 2014;371(23):2189–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 3 avr 2015;348(6230):124–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Richard C, Fumet J-D, Chevrier S, Derangère V, Ledys F, Lagrange A, et al. Exome analysis reveals genomic markers associated with better efficacy of nivolumab in lung cancer patients. Clin Cane Res 2019. Feb 1;25(3):957–66. clincanres. 1940.2018. [DOI] [PubMed] [Google Scholar]
- [21].Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther 1 janv 2017. [Internet] [cité 3 déc 2019]; Disponible sur: https://mct.aacrjournals.org/content/early/2017/08/23/1535-7163.MCT-17-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wu Y, Xu J, Du C, Wu Y, Xia D, Lv W, et al. The predictive value of tumor mutation burden on efficacy of immune checkpoint inhibitors in cancers: a systematic review and meta-analysis [Internet] Front Oncol 2019. [cité 3 déc 2019];9. Disponible sur: https://www.frontiersin.org/articles/10.3389/fonc.2019.01161/full. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Schrock AB, Ouyang C, Sandhu J, Sokol E, Jin D, Ross JS, et al. Tumor mutational burden is predictive of response to immune checkpoint inhibitors in MSI-high metastatic colorectal cancer. Ann Oncol 1 juill 2019;30(7):1096–103. [DOI] [PubMed] [Google Scholar]
- [24].Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med 21 déc 2017;377(25):2500–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Labriola M, Zhu J, Gupta R, McCall S, Jackson J, White JR, et al. Characterization of tumor mutational burden (TMB), PD-L1, and DNA repair genes to assess correlation with immune checkpoint inhibitors (ICIs) response in metastatic renal cell carcinoma (mRCC). JCO 26 févr 2019;37(7_suppl). 589–589. [Google Scholar]
- [26].Turajlic S, Litchfield K, Xu H, Rosenthal R, McGranahan N, Reading JL, et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. The Lancet Oncology 1 août 2017;18(8):1009–21. [DOI] [PubMed] [Google Scholar]
- [27].Study of Pembrolizumab (Mk-3475) in Participants With Advanced Solid Tumors. (MK-3475–158/KEYNOTE-158) - Full Text View - ClinicalTrials.gov [Internet], [cité 3 déc 2019]. Disponible sur: https://clinicaltrials.gov/ct2/show/NCT02628067.
- [28].Hellmann MD, Ciuleanu T-E, Pluzanski A, Lee JS, Otterson GA, Audigier-Valette C, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med 31 mai 2018;378(22):2093–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Carbone DP, Reck M, Paz-Ares L, Creelan B, Horn L, Steins M, et al. First-line nivolumab in stage IV or recurrent Non?Small-cell lung cancer. N Engl J Med 22 juin 2017;376(25):2415–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Rozenblum AB, Ilouze M, Dudnik E, Dvir A, Soussan-Gutman L, Geva S, et al. Clinical impact of hybrid capture–based next-generation sequencing on changes in treatment decisions in lung cancer. J Thorac Oncol 1 févr 2017;12(2):258–68. [DOI] [PubMed] [Google Scholar]
- [31].Legrand FA, Gandara DR, Mariathasan S, Powles T, He X, Zhang W, et al. Association of high tissue TMB and atezolizumab efficacy across multiple tumor types. JCO 20 mai 2018;36(15_suppl):12000. [Google Scholar]
- [32].Mok TSK, Gadgeel S, Kim ES, Velcheti V, Hu S, Riehl T, et al. 1383TiPBlood first line ready screening trial (B-F1RST) and blood first assay screening trial (BFAST) enable clinical development of novel blood-based biomarker assays for tumor mutational burden (TMB) and somatic mutations in 1L advanced or metastatic NSCLC [Internet]. 1 sept Ann Oncol 2017. [cité 12 déc 2019];28(suppl_5). Disponible sur: https://academic.oup.com/annonc/article/28/suppl_5/mdx380.084/4109440. [Google Scholar]
- [33].Rizvi H, Sanchez-Vega F, La K, Chatila W, Jonsson P, Halpenny D, et al. Molecular determinants of response to anti–programmed cell death (PD)-l and anti–programmed death-ligand 1 (PD-L1) blockade in patients with non–small-cell lung cancer profiled with targeted next-generation sequencing. J Clin Oncol 1 mars 2018;36(7):633–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ready N, Hellmann MD, Awad MM, Otterson GA, Gutierrez M, Gainor JF, et al. First-line nivolumab plus ipilimumab in advanced non–small-cell lung cancer (CheckMate 568): outcomes by programmed death ligand 1 and tumor mutational burden as biomarkers. JCO 20 févr 2019;37(12):992–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hellmann MD, Rizvi NA, Goldman JW, Gettinger SN, Borghaei H, Brahmer JR, et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): results of an open-label, phase 1, multicohort study. Lancet Oncol 2017;18(1):31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Weber JS, Gibney G, Sullivan RJ, Sosman JA, Slingluff CL, Lawrence DP, et al. Sequential administration of nivolumab and ipilimumab with a planned switch in patients with advanced melanoma (CheckMate 064): an open-label, randomised, phase 2 trial. The Lancet Oncology 1 juill 2016;17(7):943–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 9 oct 2015;350(6257):207–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Johnson DB, Frampton GM, Rioth MJ, Yusko E, Xu Y, Guo X, et al. Targeted next generation sequencing identifies markers of response to PD-1 blockade. Cancer Immunol Res nov 2016;4(11):959–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell mars 2016;165(1):35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell 2 nov 2017;171(4):934–49. e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Gaisky MD, Saci A, Szabo PM, Azrilevich A, Horak C, Lambert A, et al. 848PDImpact of zumor mutation burden on nivolumab efficacy in second-line urothelial carcinoma patients: exploratory analysis of the phase ii checkmate 275 study [Internet] Ann Oncol 1 sept 2017. [cité 12 déc 2019];28(suppl_5). Disponible sur: https://academic.oup.com/annonc/article/28/suppl_5/mdx371.003/4108934. [Google Scholar]
- [42].Balar AV, Gaisky MD, Rosenberg JE, Powles T, Petrylak DP, Bellmunt J, et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: a single-arm, multicentre, phase 2 trial. The Lancet 7 janv 2017;389(10064):67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Powles T, Loriot Y, Ravaud A, Vogelzang NJ, Duran I, Retz M, et al. Atezolizumab (atezo) vs. chemotherapy (chemo) in platinum-treated locally advanced or metastatic urothelial carcinoma (mUC): immune biomarkers, tumor mutational burden (TMB), and clinical outcomes from the phase III IMvigor211 study. J Clin Orthod 20 févr 2018;36(6_suppl):409. [Google Scholar]
- [44].Snyder A, Nathanson T, Funt SA, Ahuja A, Novik JB, Hellmann MD, et al. Contribution of systemic and somatic factors to clinical response and resistance to PD-L1 blockade in urothelial cancer: an exploratory multi-omic analysis. PLoS Med 26 mai 2017;14(5):e1002309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade–based immunotherapy [Internet] Science 2018. Oct 12;362(6411). pii: eaar3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Powles T, Kockx M, Rodriguez-Vida A, Duran I, Crabb SJ, Heijden MSVD, et al. Clinical efficacy and biomarker analysis of neoadjuvant atezolizumab in operable urothelial carcinoma in the ABACUS trial. Nat Med nov 2019;25(11):1706–14. [DOI] [PubMed] [Google Scholar]
- [47].Samstein RM, Lee C-H, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet févr 2019;51(2):202–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Garassino M, Rodriguez-Abreu D, Gadgeel S, Esteban E, Felip E, Speranza G, et al. OA04.06 evaluation of TMB in KEYNOTE-189: pembrolizumab plus chemotherapy vs placebo plus chemotherapy for nonsquamous NSCLC. J Thorac Oncol 1 oct 2019;14(10):S216–7. [Google Scholar]
- [49].Langer C, Gadgeel S, Borghaei H, Patnaik A, Powell S, Gentzler R, et al. OA04.05 KEYNOTE-021: TMB and outcomes for carboplatin and pemetrexed with or without pembrolizumab for nonsquamous NSCLC. J Thorac Oncol 1 oct 2019;14(10):S216. [Google Scholar]
- [50].Jonna S, Vanderwalde AM, Nieva JJ, Poorman KA, Saul M, von Buttlar X, et al. Impact of prior chemotherapy or radiation therapy on tumor mutation burden in NSCLC. JCO 20 mai 2019;37(15_suppl):2627. [Google Scholar]
- [51].Chan TA, Yarchoan M, Jaffee E, Swanton C, Quezada SA, Stenzinger A, et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann Oncol janv 2019;30(1):44–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Sherry ST, Ward M-H, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 1 janv 2001;29(1):308–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Karczewski KJ, Weisburd B, Thomas B, Solomonson M, Ruderfer DM, Kavanagh D, et al. The ExAC browser: displaying reference data information from over 60 000 exomes. Nucleic Acids Res 4 janv 2017;45:D840–5. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Chang H, Sasson A, Srinivasan S, Golhar R, Greenawalt DM, Geese WJ, et al. Bioinformatic methods and bridging of assay results for reliable tumor mutational burden assessment in non-small-cell lung cancer. Mol Diagn Ther 2019;23(4):507–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Budczies J, Allgäuer M, Litchfield K, Rempel E, Christopoulos P, Kazdal D, et al. Optimizing panel-based tumor mutational burden (TMB) measurement. Ann Oncol 1 sept 2019;30(9):1496–506. [DOI] [PubMed] [Google Scholar]
- [56].Friends of Cancer Research Launches Tumor Mutational Burden Harmonization Project - The ASCO Post [Internet], [cité 9 déc 2019], Disponible sur: https://www.ascopost.com/issues/october-25-2018/friends-of-cancer-research-launches-tumor-mutational-burden-harmonization-project/.
- [57].Bartha Á, Győrffy B. Comprehensive outline of whole exome sequencing data analysis tools available in clinical oncology. Cancers nov 2019;11(11):1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].McGranahan N, Furness AJS, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 25 mars 2016;351(6280):1463–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Buchhalter I, Rempel E, Endris V, Allgäuer M, Neumann O, Volckmar A-L, et al. Size matters: dissecting key parameters for panel-based tumor mutational burden analysis. Int J Canc 2019;144(4):848–58. [DOI] [PubMed] [Google Scholar]
- [60].McGranahan N, Swanton C. Neoantigen quality, not quantity [Internet] Sci Transl Med 2019. Aug 21;11(506). pii: eaax7918. [DOI] [PubMed] [Google Scholar]
- [61].Chowell D, Morris LGT, Grigg CM, Weber JK, Samstein RM, Makarov V, et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science 2 févr 2018;359(6375):582–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Chowell D, Krishna C, Pierini F, Makarov V, Rizvi NA, Kuo F, et al. Evolutionary divergence of HLA class I genotype impacts efficacy of cancer immunotherapy. Nat Med nov 2019;25(11):1715–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Lu S, Stein JE, Rimm DL, Wang DW, Bell JM, Johnson DB, et al. Comparison of biomarker modalities for predicting response to PD-1/PD-L1 checkpoint blockade: a systematic review and meta-analysis. JAMA Oncol 1 août 2019;5(8):1195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Forschner A, Battke F, Hadaschik D, Schulze M, Weißgraeber S, Han C-T, et al. Tumor mutation burden and circulating tumor DNA in combined CTLA-4 and PD-1 antibody therapy in metastatic melanoma – results of a prospective biomarker study. J Immuno Ther Cane 12 juill 2019;7(1):180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Liu L, Bai X, Wang J, Tang X-R, Wu D-H, Du S, et al. Combination of TMB and CNA stratifies prognostic and predictive responses to immunotherapy across metastatic cancer [Internet], 1 janv. Clin Cancer Res;2019. [cité 4 déc 2019]; Disponible sur: https://clincancerres.aacrjournals.org/content/early/2019/09/12/1078-0432.CCR-19-0558. [DOI] [PubMed] [Google Scholar]