

Abstract
Deubiquitinating enzymes (DUBs) are a class of isopeptidases that regulate ubiquitin dynamics through catalytic cleavage of ubiquitin from protein substrates and ubiquitin precursors. Despite growing interest in DUB biological function and potential as therapeutic targets, few selective small molecule inhibitors and no approved drugs currently exist. To identify chemical scaffolds targeting specific DUBs and establish a broader framework for future inhibitor development across the gene-family, we performed high-throughput screening of a chemically diverse small molecule library against eight different DUBs, spanning three well-characterized DUB families. Promising hit compounds were validated in a series of counter-screens and orthogonal assays, as well as further assessed for selectivity across expanded panels of DUBs. Through these efforts, we have identified multiple highly selective DUB inhibitors and developed a roadmap for rapidly identifying and validating selective inhibitors of related enzymes.
Keywords: Deubiquitinase, high-throughput screening, small molecule inhibitor
eTOC blurb
Deubiquitinases (DUBs) have emerged as therapeutically interesting targets due to their involvement in protein turnover in cells. Varca et. al. describe a multi-DUB high throughput screen and orthogonal validation campaign to identify new tool compounds for elucidating DUB function and provide a roadmap for future DUB inhibitor screens.
Graphical Abstract
INTRODUCTION
Since its discovery over 40 years ago, the ubiquitin-proteasome system (UPS) has been the subject of extensive research interest. The UPS mediates ubiquitination, a process whereby ubiquitin, a small 76 amino acid protein, is added to substrate proteins by the concerted action of ubiquitin activating (E1), conjugating (E2), and ligating (E3) enzymes and removed by deubiquitinases (DUBs) (Kerscher et al., 2006). The most common functional outcome of polyubiquitination from UPS mediated attachment of multiple ubiquitin molecules to a single substrate is proteasomal degradation of the substrate protein (Kleiger and Mayor, 2014, Ciechanover, 2005). This process is critical for regulating protein turn-over and homeostasis. Therefore, processes such as oncogenic transformation affect the UPS and render molecular players involved in the UPS potential drug targets of interest (Ding et al., 2009, Senft et al., 2018). The approval of bortezomib (Richardson et al., 2003), a proteasome inhibitor, as a cancer therapeutic as well as the recent discovery that immunomodulatory drugs (IMiDs) target E3 ligases to promote targeted degradation of specific neo-substrates (Petzold et al., 2016, Matyskiela et al., 2016, Winter et al., 2015, Sakamoto et al., 2001) have heightened the interest in UPS drug discovery and development. In this context, DUBs have also emerged as an enzyme family of growing interest for therapeutic targeting.
DUBs are isopeptidases that catalyze the removal of ubiquitin from their target proteins. There are currently around 100 reported mammalian DUBs divided broadly into two classes based on their catalytic mechanism: cysteine proteases and zinc metalloproteases. Additionally, the approximately 90 cysteine protease DUBs can be further subdivided into six families based on sequence homology (Mevissen and Komander, 2017, Abdul Rehman et al., 2016, Kwasna et al., 2018, Haahr et al., 2018, Hermanns et al., 2018, Hewings et al., 2018, Gopinath et al., 2016). DUBs play significant roles in physiology, including growth factor signaling, genome stability, redox regulation, cell fate, apoptosis, and others (Turcu et al., 2009, Amerik and Hochstrasser, 2004, Cotto-Rios et al., 2012, Ramakrishna et al., 2011). A myriad of studies have nominated DUBs as an emergent class of drug targets to multiple human pathologies such as cancer (Fraile et al., 2012, D’Arcy et al., 2015, Pinto-Fernandez and Kessler, 2016), neurodegeneration (Das et al., 2020, Lim et al., 2020, Leroy et al., 1998), and inflammation/immune response (Hu and Sun, 2016, Zinngrebe et al., 2014, Wertz et al., 2004). In some instances, genomic alteration renders DUBs as primary drivers of pathogenesis (Oliveira et al., 2004, Reincke et al., 2015, Chen et al., 2018). In other contexts, DUBs can be therapeutically targeted to mediate degradation of protein drivers of a disease state. In a growing number of examples, DUBs were shown to stabilize “undruggable” proteins, including transcription factors such as Myc, Np63 and HIF1α (Prieto-Garcia et al., 2020, Popov et al., 2007, Bingol et al., 2014, Flugel et al., 2012, Boselli et al., 2017). The ability to promote the selective degradation of these proteins through DUB inhibition highlights the immense potential of DUBs as therapeutic targets. Despite this broad array of function and therapeutic potential, there is a significant lack of well-validated probe compounds for a majority of DUBs hindering the ability to further study the biological outcomes of DUB inhibition.
Recent studies focused on one of the best studied DUBs, USP7, demonstrated that developing potent and selective DUB inhibitors is achievable, and that pharmacological DUB inhibition can promote degradation of disease relevant proteins for therapeutic benefit (Lamberto et al., 2017, Turnbull et al., 2017, Kategaya et al., 2017, Gavory et al., 2018, Stolte et al., 2018, Schauer et al., 2020a, Leger et al., 2020). However, selective compounds have been identified for only a very limited number of DUBs beyond USP7 (Ndubaku and Tsui, 2015, Schauer et al., 2020b, Ritorto et al., 2014). A majority of the reported DUB inhibitors exhibit weak inhibitory activity (double digit micromolar range), contain undesirable chemical features, and/or are now known to possess poor selectivity across DUB enzyme family (Ritorto et al., 2014, Schauer et al., 2020b, Ndubaku and Tsui, 2015, Farshi et al., 2015). Therefore, despite the accumulating evidence that DUBs represent promising therapeutic targets, the number of high-quality chemical probes and lead compounds with appropriate potency and selectivity continues to be limited.
Here, we describe generation of a dense dataset of DUB-ligand interactions that enabled rapid generation of small molecule inhibitors and chemical probes for multiple DUBs as well as a wealth of data to facilitate future inhibitor development studies. To achieve this, we employed high-throughput, parallel screening of eight DUBs against the same library of 47.48K small molecules, comprised of well-curated diversity compounds as well as natural products. Unlike previous efforts in this area that prioritized screening hits based on potency, we placed a strong emphasis on selectivity as a strategy to identify and prioritize hits. Validation studies confirmed this approach facilitated rapid confirmation of hits as bona fide selective DUB inhibitors. Indeed, we credentialed best-in-class probes of USP28 as well as identified selective starting points for medicinal chemistry optimization to achieve chemical probe or clinical compound development for other DUBs.
RESULTS
Assay Development and Optimization Provides Robust Screening Assay
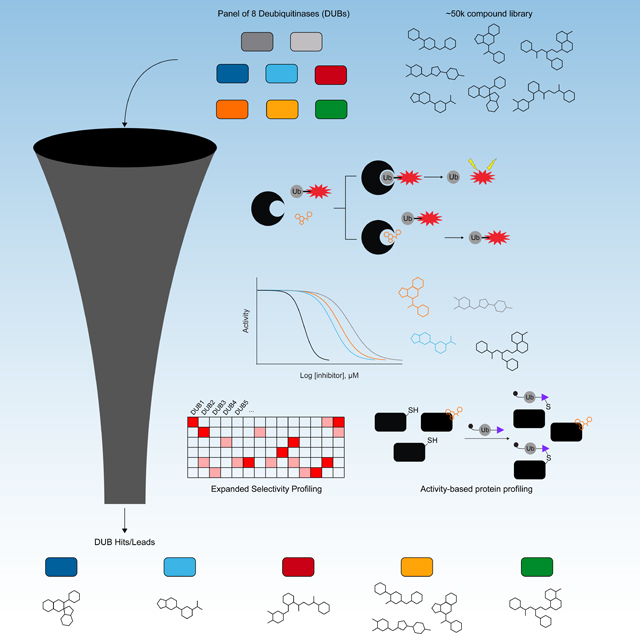
This effort was conducted in collaboration with the Novartis Institute for Biomedical Research’s (NIBR) FAST Lab program which enables academic labs to access a nonproprietary compound collection assembled at NIBR and work side-by-side with NIBR scientists to perform screens. For our screening campaign, we employed recombinant DUB enzymes and a fluorogenic DUB substrate, ubiquitin-rhodamine110 (Ub-Rho) (Figure 1A). This is a robust assay that is adaptable to most DUBs and proved amenable to high-throughput screening. The eight DUBs screened were USP7, USP8, USP10, USP17, USP28, USP30, UCHL1 and OTUD3 (Figure 1B). Selection of these DUB enzymes was based on multiple considerations. First was ease of access to ensure enough quantity of high purity enzyme (Figure S1A). After taking this into account, we sought the inclusion of multiple members of the largest DUB family (the USP subfamily) in addition to representatives from the two other most well-studied cysteine protease DUB families (UCHL and OTU). In addition, we selected DUBs that were associated with interesting biology and/or were lacking in chemical probes to further explore their biological function. By using this assortment of DUBs, we were able to assess targetability and cross-reactivity within and across DUB families from hits from the initial screen. The approximately 50K compound library was comprised of a curated small molecule diversity set as well as a smaller natural product collection. Both compound sets demonstrate good drug-like properties with reasonable lipophilicity, polar surface area, and molecular weight(Waring, 2010, Lipinski et al., 2001, Navia and Chaturvedi, 1996, Leeson and Springthorpe, 2007) with the natural product set showing a wider range of properties as shown in Figure 1C.
Figure 1. Overview of DUB HTS campaign.

A) Schematic of Ub-Rho screening assay. Uninhibited DUB cleaves rhodamine from ubiquitin (top) resulting in a fluorescent signal. In the presence of an inhibitor (bottom), the DUB cannot cleave the substrate, and fluorescence is unchanged.
B) DUBs included in the screen along with constructs used (cat denotes catalytic domain, FL denotes full length protein, cat + UBA denotes the catalytic domain plus UBA domain). All proteins are human isoforms except USP28, which is from mouse (mus musculus).
C) Summary of druglike properties partition coefficient (LogP) to polar surface area (PSA) and molecular weight (MW) among library members.
D) Diagram of screening cascade workflow.
Our screening and compound triage cascade is summarized in Figure 1D. For the primary assay, all compounds were tested against each DUB at a concentration of 20 μM, 25 μM, or 50 μM. Selected actives were subsequently tested in dose-response against a minimum of two DUBs to confirm activity and selectivity. To assess the ability of parallel DUB screening to accelerate identification of high-quality DUB hit and lead compounds, we applied filters to the dose-response dataset to identify potent and selective scaffolds for each DUB. The selectivity and potency filters, discussed in more detail later on, enabled us to identify actives with selectivity for a single DUB amongst the entire library as well as rank these compounds by potency against the target DUB. These top hits were selected for resynthesis and confirmation via the Ub-Rho screening assay along with other purified enzyme and cell-based orthogonal assays.
We initiated our screening campaign by performing comprehensive assay development for USP7 and UCHL1, two well-studied DUBs with well-characterized positive control compounds. A Design of Experiment (DOE) that investigated buffer, pH, salt, BSA, EDTA, detergent and reducing agent was performed (Figure S1B) and allowed us to select a single buffer that produced robust signal for both DUBs. Notably, the conditions allowed us to reduce enzyme and substrate concentration up to over 200-fold compared to starting conditions (Figure S1C); reagent consumption was an important consideration for this study given the large number of enzymes and expense of the substrate. We next performed a time-course titration of both enzyme and substrate to identify reagent concentrations and a single timepoint for assay readout in which substrate turnover was in the linear range with a signal to noise ratio of at least 5x (Figure S2A). Using the optimized assay conditions, we confirmed that known inhibitors of each DUB displayed the expected inhibitory activity (Figure S2B).
As an initial assessment of assay performance and potential for identifying selective DUB inhibitors from the compound collection, we screened the entire compound collection at a concentration of 20 μM against USP7 and UCHL1 followed by dose-response analysis of all actives against both DUBs. The primary assays performed well with Z’ values of 0.89 and 0.92 for USP7 and UCHL1, respectively (Table S1). Using > 20% inhibition as criteria for calling actives, we observed 289 hits for USP7 and 175 hits for UCHL1. Hit compounds exhibiting inhibition based on our cutoff for each enzyme were tested in dose-response against both DUBs in triplicate starting at 25 μM for UCHL1 and 50 μM for USP7. Based on compound availability, 128 of the 289 USP7 hits and 101 of the 175 UCHL1 hits were retested at dose. As was the case for the primary assay, the confirmatory assays performed well with Z’ values greater than 0.8 for each DUB. We classified any compounds that exhibited ≥ 20% inhibition at the highest tested dose in the dose-response testing as active. Using these criteria, the confirmation rate was 83% for USP7 (106 of 128 compounds) and 57% for UCHL1 (58 of 101 compounds). Notably, we found 34 compounds that demonstrated inhibition of USP7 (IC50 < 50 μM and ≥ 20% inhibition) that were inactive against UCHL1 (IC50 >25 μM) and 12 compounds inhibiting UCHL1 (IC50 <25 μM and ≥ 20% inhibition) that were inactive against USP7 (IC50 >50 μM) (Figure S3). Given the excellent assay performance and initial indication of selective inhibitors, we proceeded to screening the other six DUBs. Prior to screening the additional DUBs, we confirmed that each DUB was active in the same buffer utilized for USP7 and UCHL1 and performed titration and time-course experiments to select screening conditions (Figure S4). The use of the optimized buffer for all DUBs further aided comparison of the datasets by preventing any buffer-specific artifacts from complicating the analysis.
Primary Screen Reveals Multiple Hits Across All DUBs
The same set of compounds screened against USP7 and UCHL1 were screened against the 6 additional DUBs at a concentration of 20 μM (USP8 and USP10), 25 μM (USP28, USP30, and OTUD3), or 50 μM (USP17). All screens performed well with Z’ values between 0.49 and 0.92 (Table S1). We classified compounds exhibiting ≥ 30% inhibition of target as active. Hit rates were variable, with 0.4% for UCHL1 on the low end and 6.5% for USP17 on the high end. Among the USP family, USP7 had the lowest hit rate of 0.6%. (Table S1). The heatmap in Figure 2A depicts all actives from the primary, single point screen across all eight DUBs. Each DUB is indicated along the y-axis and hits, defined as ≥ 30% inhibition of target, are highlighted and clustered by structural similarity as determined by Tanimoto score. Visual inspection of the heatmap indicates several qualitative conclusions: 1) selective inhibitors for each DUB 2) multi-targeted inhibitors, and 3) examples of multiple structurally related hits. To further demonstrate our findings of selective and multi-targeted compounds we plotted the hits in an UpSet plot (Figure 2B) (Conway et al., 2017). Gratifyingly, the analysis revealed selective hits for each DUB. The number of selective compounds for each DUB was quite varied, ranging from 12 for UCHL1 and USP7 to 1403 for USP17. In general, the USP family seemed to have more hits compared to UCHL1 (12 selective hits) and OTUD3 (34 selective hits) although there were notable exceptions like USP7 (12 selective hits) and USP10 (70 selective hits). Encouragingly, the selective hits for USP28 (671 selective hits), USP8 (968 selective hits), USP30 (1198 selective hits), and USP17 (1403 selective hits) represented the largest clusters of compounds observed in the UpSet plot. As anticipated, we also observed multi-targeted inhibitors. In the diversity set, we observed 127 multi-targeted inhibitors, defined as hitting 5 of the 8 DUBs included in the screen. In the natural product set we observed a higher percentage of multi-targeted inhibitors (Figure S5A). Examination of these multi-targeted compounds revealed structures or substructures that have been annotated in the literature as potential PAINS compounds (Baell and Holloway, 2010, Daina et al., 2017); however, some of the compounds contained structures that do not contain obvious flags (PAINS/electrophiles) underscoring the importance of selectivity profiling up-front. Some of these compounds from the diversity set are depicted in Figure S5B as a resource for others conducting DUB HTS, but we note that we have not performed additional experiments to confirm the compounds as pan-inhibitors or investigated whether they are real multi-targeted binders or false positives. Overall, the data from the primary screen indicated potential selective hit compounds for all the DUBs screened and supported proceeding to dose-response confirmation of the screening hits.
Figure 2. Primary Screening Data.

A) Heatmap of primary screen hits. Maximal inhibition of each compound is plotted for each DUB. Compounds are clustered on the x-axis by structural similarity (Tanimoto score). Patches of blue in each of the rows show compound clusters that demonstrate inhibition of the corresponding DUB.
B) UpSet plot summarizing primary screening data. Compounds with activity against at least one DUB, as defined by maximal inhibition ≥ 30%, are clustered by activity against each DUB in the panel. The bar chart represents the number of compounds in a particular cluster, the dot underneath the bar chart correlates that cluster with one or more DUBs (the presence of a single dot correlates to a specific DUB on the left, the presence of multiple dots means those compounds are hits against those particular DUBs). Single DUB hits are colored in red to highlight potentially selective hits in the primary screen for each DUB. The bar chart on the lower left represents the total number of hits for each DUB regardless of selectivity. The top 40 clusters of compounds are shown.
Dose-response Screen Leads to Identification of Potent and Selective Hits
For the dose-response confirmation assay with the additional six DUBs, we screened USP8 and USP10 together followed by USP17, OTUD3, USP28, and USP30 as these primary screens were completed in different timeframes. All hits against USP8 and USP10 were tested in 8-point dose-response in triplicate starting at 40 μM for USP8 and 50 μM for USP10 against both DUBs to confirm inhibitory activity against the target and provide further evidence of selectivity over other DUBs. All USP17, USP28, USP30 and OTUD3 active compounds were evaluated starting at 50 μM in 4-point dose-response against all four DUBs. All assays performed well with Z’ values ranging from 0.58 to 0.75 and confirmation rates ranged from 33 to 91% (Table S1), using ≥ 30% inhibition at the highest concentration tested for calling a compound active.
Next, we analyzed the dose-response data of the active compounds through the application of a series of selectivity and potency filters. These different filters were implemented individually and in combination to allow us to assess the impact of each parameter in triaging compounds and narrow in on the most promising actives for each DUB. Section I in Figure 3A depicts the number of active compounds for each DUB in the dose-response dataset using the initial criteria of maximum inhibition ≥ 30% and an IC50 below 40 μM for the target DUB (or below 25 μM for UCHL1 as this was the highest dose for UCHL1). With these criteria, we observed a disparate number of confirmed hits amongst the DUBs that was in line with what was observed for the primary screen. DUBs like UCHL1 and USP7 were on the low end with 54 and 53, respectively, whereas DUBs on the other end of the spectrum had >500 hits confirm in the dose-response (USP17, USP28, and USP30). To focus in on the top compounds for each DUB we applied a series of filters to the data. First, we applied a selectivity filter which removed compounds with an IC50 at or below 40 μM (25 μM for UCHL1) against any other DUB evaluated (Figure 3A section II). Filtering the dataset in this manner produced a marked reduction in the number of hits for most DUBs including eliminating >90% of hits for some DUBs. To this set of compounds, we then applied a potency filter where we only included compounds with an IC50 below 10 μM with maximum inhibition ≥ 30% and this produced a reduction in the number of hits down to the tens to hundreds range for most DUBs (Figure 3A section III). In some cases, in fact, no qualifying hits remained while for other DUBs there were anywhere from 1 to 160 hits that qualified. We applied one last filter of increasing the maximum inhibition to ≥ 80% to help us further focus in on the most promising compounds (Figure 3A section IV). This ultimately narrowed the number of hits down to around 80 overall, but they were not split evenly among DUBs. There were 74 hits for USP28 while there were none for USP8, USP17, UCHL1 or OTUD3 and only 1 each for USP7 and USP30 and 2 for USP10. Visual inspection of all the hits revealed the vast majority contained synthetically tractable drug-like structures. Additionally, examination of the datasets for each DUB, particularly in the case of USP28, revealed multiple analogs of the same core structure that scored as hits leading to increased confidence that these compounds represented real inhibitors. Interestingly, the IC50 values of the most potent compound for each DUB was quite varied, with some in the low nanomolar range (Figure 3B).
Figure 3. Dose-response screening data with selectivity and potency filters.

A) Number of compounds that qualify as hits for each DUB based on filter criteria listed in the table below. The arrow denotes increasing potency and/or selectivity of hit compounds for each DUB. The * denotes that for UCHL1, 25 μM was the highest tested dose in the dose-response and thus this value was used for calling hits as active against UCHL1.
B) The violin plot shows the distribution of IC50s for DUBs that had qualifying hits in section IV in Fig 3A.
Expansion Library Leads to Identification of Additional USP7 and USP8 Hit Compounds
We noted that one of the hits for USP7, AV-9606–27, contained a core motif that was present in a significant number of members of a larger 250K screening deck, so we proceeded to screen an expansion set containing compounds with structural similarity to this hit (Figure 4A). The key structural element of this collection was the presence of a spiro indolinone group with various fused ring cores and substituents. Upon examination of the USP8 hits in the selective group (Figure 3A section II), we observed a hit that shared structural similarity with some of the compounds in the USP7 spiro indolinone expansion set. While the USP8 hit did not contain a spiro indolinone group, it did contain a similar fused ring system that appeared on numerous derivatives in the expansion set. We thus screened the expansion set against USP8 to potentially identify additional USP8 hits while also providing a counter screen for USP7. The compounds were assessed in 8-point dose-response in triplicate against both USP7 and USP8. Gratifyingly, we identified a different spiro indolinone scaffold active against USP7 and inactive against USP8 (AV-9606–41, Figure 4A). In addition, we identified compounds with a spiro indolinone group sharing the same fused ring as the initial USP8 hit with increased activity over the screening hit that were inactive against USP7. AD-10942–8 represents the best hit from among those compounds (Figure 4A).
Figure 4. Conformation of screening hits.

A) Structures of USP7 and USP8 screening hits prompted further screening of a spiro indolinone expansion set leading to the identification of new USP7 and USP8 hits for validation studies.
B) Structures of screening hits for each corresponding DUB that were selected for validation studies.
C) Dose-response data for screening hits in Fig 4A and 4B. Compounds were screened in a 12-point dose-response using a kinetic Ub-Rho biochemical assay. Data are represented as mean ± SEM, n ≥ 2.
Resynthesis and Biochemical Assays Confirm Activity of Selected Hits.
We focused on the most stringently triaged group of compounds for each DUB for validation studies and to assess the effectiveness of our approach in yielding highly selective DUB inhibitors. We initially selected two of the USP7 hits (AV-9606–27 – PubChem CID: 2998850 and AV-9606–41 – PubChem CID: 3624054) as our lab has significant experience with this DUB (Lamberto et al., 2017, Schauer et al., 2020a), as well as one USP10 hit (Z56965384 – PubChem CID: 3908773), one USP8 hit (AD-10942–8), four USP28 hits (AV-9606–99 – PubChem CID: 3487576, AV-9606–129 – PubChem CID: 3985183, AV-11324–75 – PubChem CID: 136087907, and bin-01–07-07 – PubChem CID: 2019582) and the single USP30 hit (AV-11324–5, PubChem CID: 4383255) (Figure 4B). The lone USP17 hit was not included in validation studies as it contained a core scaffold similar to one with reported redox activities (Lor et al., 2007).
Fresh aliquots of each compound were obtained by chemical synthesis or from commercial vendors and tested in 12-point dose-response in the Ub-Rho assay. For USP28, as we had employed the mouse isoform of the protein in the screen, the catalytic domain of the human protein was utilized to confirm activity against the human isoform. All the compounds confirmed upon retest with IC50s against the respective DUB target ranging from 50 nM to 30 μM (Table 1), which was in good agreement with the screening IC50 values. The USP30 compound and several of the USP28 inhibitors were the most potent with low nanomolar IC50s. The USP30 hit, AV-11324–5, was found to have an IC50 value of 125 nM (Figure 4C, Table 1). The four USP28 inhibitors we selected inhibited the DUB with IC50 values from the low nanomolar to low micromolar: AV-9606–99 (4.33 μM), AV-9606–129 (402 nM), AV-11324–75 (176 nM) and bin-01–07-07 (46 nM) (Figure 4C, Table 1). The USP7 (AV-9606–27 and AV-9606–41), USP8 (AD-10942–8), and USP10 (Z56965384) hits all exhibited micromolar inhibitory activity in the dose-response confirmation with IC50s of 6.77, 3.92, 9.28, and 26.6 μM respectively (Figure 4C, Table 1).
Table 1.
IC50 data for selected screening hits
| DUB | Compound | IC50 ± SEM μM (n) |
|---|---|---|
| USP7 | AV-9606–27 | 4.68 ± 1.05 (3) |
| AV-9606–41 | 3.83 ± 1.60 (3) | |
| USP8 | AD-10942–8 | 6.01 ± 0.18 (4) |
| USP10 | Z56965384 | 26.6 ± 4.69 (14) |
| USP28 | AV-9606–99 | 4.33 ± 1.08 (8) |
| AV-9606–129 | 0.402 ± 0.197 (6) | |
| bin-01–07-07 | 0.046 ± 0.017 (3) | |
| AV-11324–75 | 0.176 ± 0.073 (5) | |
| USP30 | AV-11324–5 | 0.275 ± 0.070 (6) |
IC50 values for selected screening hits. Data are represented as mean ± SEM with the number of replicates for each compound (n).
As DUBs are susceptible to inactivation by oxidation of the catalytic cysteine residue by redox active compounds, we analyzed all confirmed actives using a resazurin based redox assay in dose-response(Lor et al., 2007). Most of the compounds exhibited little to no redox activity with those that did only exhibiting redox behavior at concentrations well above their IC50s (Figure S6). The exception to this was the USP8 hit, AD-10942–8, which generated significant signal at lower concentrations. Based on this finding, AD-10942–8 may inhibit USP8 through oxidation of the catalytic cysteine residue rather than inhibiting the enzyme through a direct binding interaction.
Selectivity of Hit Compounds Confirmed Against an Expanded DUB Panel
While our screening results indicated that these compounds are selective toward a single DUB over those included in the screen, we next sought to confirm this result by profiling the compounds against a comprehensive library of DUBs. The USP7, USP8, USP10, USP28, and USP30 hits were assessed for inhibitory activity against a panel of 41 purified DUBs using Ubiquigent’s DUBprofiler™ platform. This assay measured cleavage of Ub-Rho using an end point assay. In addition to the activity assay, compounds are also evaluated in two separate control experiments for autofluorescence and for modulation of the product signal to determine potential compound interference with assay readout. Five of the eight compounds tested were confirmed to inhibit their intended target with excellent selectivity. Figure 5 shows the profiling data for these compounds with each DUB in the panel shown on the x-axis while percent activity of the DUB with compound treatment is plotted on the y-axis. Compounds that inhibit the DUB will show a reduced percent activity remaining. The USP30 inhibitor and all four USP28 compounds exhibited a high degree of selectivity across the entire panel for the target DUBs identified from the screen. The USP30 inhibitor AV-11324–5 showed remarkable selectivity for USP30 over all other DUBs in the panel. All four of the USP28-targeting compounds significantly inhibited two DUBs, USP28 and USP25, with no other DUBs inhibited. USP25 and USP28 share high sequence homology within the DUB domain as well as identical domain structure compared to all other USP family members(Valero et al., 2001, Liu et al., 2018, Gersch et al., 2019, Sauer et al., 2019). To independently confirm USP25 inhibition, we expressed and purified USP25 and tested the hit compounds in dose-response in the Ub-Rho assay and confirmed the compounds demonstrated comparable activity against USP25 and USP28 (Figure S7A). The USP7 inhibitor, AV-9606–41, did not confirm to inhibit USP7, or any other DUBs in the panel (Figure S7B). One possibility this result raised was that AV-9606–41 inhibits the catalytic domain construct of USP7 used in our studies but not full-length enzyme, although the compound exhibited autofluorescence in the control experiment at Ubiquigent rendering it difficult to confidently draw a conclusion. We evaluated the inhibitory activity of AV-9606–41 against full-length USP7 in dose-response using a kinetic Ub-Rho assay, conditions less sensitive to assay interference, and observed no inhibition of full-length protein (Figure S7D). Based on this finding we dropped AV-9606–41 from further validation experiments, although the compound may be a useful tool for studying conformation differences between catalytic domain and full-length USP7. The USP8 compound, AD-10942–8, inhibited multiple DUBs in the panel, however, the compound also exhibited autofluorescence in the control experiment (Figure S7B). These results, coupled with the apparent redox activity of the compound observed previously, led to us excluding the USP8 hit from further validation studies. The USP10 compound, Z56965384, also showed inhibition of multiple DUBs within the panel (Figure S7B). However, the compound exhibited assay interference consistent with quenching fluorescence based on a control experiment in the absence of enzyme. We confirmed inhibition of an ‘off-target’ DUB, USP8, using our kinetic assay (Figure S7C). We also tested the compound against both USP8 and USP10 in dose-response using Ubiquitin-AMC, a different substrate containing a fluorophore that emits at a different wavelength. In this experiment we observed a 3–10 fold increase in IC50 value indicating the compound is affecting rhodamine fluorescence (Supplemental Figure S7C). These results exclude Z56965384 as a potent or selective starting point for USP10 inhibitor development.
Figure 5. Selectivity Profiling.

Compounds were assessed for selectivity against an expanded panel of purified recombinant DUBs using DUBProfiler (Ubiquigent). Compounds were tested at 100 μM, except bin-01–07-07 which was tested at 10 μM, and the percent activity of each DUB remaining following compound treatment is reported for each compound.
Orthogonal Assays Validate USP28 and USP30 Hits
We focused further validation studies on the UPS28 and USP30 hits given these DUBs had sub-micromolar hits with good selectivity. To assess target binding to native enzyme and selectivity in a cellular environment, we performed competitive activity-based protein profiling (ABPP) coupled with quantitative mass spectrometry(Hemelaar et al., 2004, Pinto-Fernandez et al., 2019, Borodovsky et al., 2002) (Figure 6A). Briefly, HEK293T lysates were treated with inhibitor followed by co-incubation with a 1:1 mixture of DUB activity-based probes, biotin-ubiquitin-propargylamine (Ub-PA) and biotin-ubiquitin-vinyl methyl ester (Ub-VME). The probes will irreversibly label the active site cysteine of DUBs not already bound by inhibitor and contain a biotin moiety for streptavidin enrichment. Following affinity purification, the samples are subjected to tryptic digest and peptide sequencing by LC-MS/MS enabled identification of DUBs that are bound and pulled down by the probe. Based on comparison to a DMSO control sample, we can identify which DUB(s) the compounds are binding to and thus preventing labeling with the covalent probe. Figure 6B shows the results of the target engagement experiments. For the USP30 compound we see robust inhibition of USP30 with little to no off-target activity further highlighting the selectivity of this compound. For the dual USP25/USP28, we observed strong binding to native USP25 with all four compounds but only the two most potent compounds bin-01–07-7 and AV-9606–180 (a close derivative of screening hit AV-11324–75 with similar potency in the Ub-Rho dose-response assay, Figure S7E) exhibited strong inhibition of USP28. To further confirm the ABPP results we performed a dose-response treatment of the most potent compounds (AV-9606–180, AV-11324–75 and bin-01–07-07) in cellular lysate followed by addition of the covalent activity-based probes and readout by western blot for USP28 (Figure 6C). Compound binding prevents probe labeling resulting in the presence of the native USP28 band. Probe binding results in a higher molecular weight adduct (DUB + probe) that is resolvable by western blot as a band above the native protein. Results demonstrate good dose-dependent inhibition and prevention of probe labeling in line with expected compound activity.
Figure 6. Validation of USP28 and USP30 hits by Quantitative Activity Based Protein Profiling.

A) Schematic for quantitative activity-based protein profiling.
B) Heatmap showing results of screening compounds profiled in ABPP assay. Compounds demonstrate blockage of probe labeling of target DUBs compared to all other DUBs detected in cell lysate. AZ1 is a literature reported USP25/28 inhibitor (Wrigley et al., 2017). XL-188 is a USP7 inhibitor included as a positive control(Lamberto et al., 2017). See also Supplemental Table 2.
C) Western blots show blockage of USP28 labeling by covalent probe (Ub-PA/Ub-VME) in cellular lysate after 1 hour pre-treatment with indicated compounds in dose-response.
A dual USP25/USP28 inhibitor AZ1 has been reported in literature so we sought to benchmark our inhibitors against this compound(Wrigley et al., 2017). We observed biochemical inhibition of USP25 and USP28 by AZ1 with IC50s of 3.47 ± 0.73 μM (n = 4) and 0.98 ± 0.02 μM (n = 2) respectively, in line with reported values for the compound. In the ABPP assay, AZ1 strongly blocked labeling of USP25 by DUB ABP, however, it had little impact on USP28. The compound, as previously reported, did not exhibit activity toward any other DUBs detected in the experiment. These results nominate bin-01–07-07 and AV-9606–180 as best-in-class probes of USP28. Both compounds inhibit the DUB with IC50 values around or below 100 nM, approximately 10-fold more potent that AZ1, and exhibit substantially stronger cellular target engagement (Wrigley et al., 2017). The combination of two structurally distinct scaffolds in studies that probe USP28 function will be highly valuable.
Overall, the generated dataset also provides a number of scaffolds that may retain activity and selectivity against single DUBs but were not selected for follow up studies based on our initial focus on the more potent hits. These weaker compounds may provide starting points for medicinal chemistry campaigns to generate more potent analogs that retain selectivity against a single DUB.
DISCUSSION
DUBs control diverse processes within the cell and have been implicated in many different diseases including cancer, neurodegeneration, and inflammation/immune response (Harrigan et al., 2018). Studies have demonstrated that pharmacologic inhibition of specific DUBs can lead to degradation of their substrate proteins, and result in a beneficial therapeutic effect (Kapuria et al., 2010, Schauer et al., 2020a, Boselli et al., 2017). While there is great interest in DUBs as a way to provide a high-precision mechanism to target oncogenic ‘undruggable’ proteins for degradation, the number of selective small molecule probes remains limited (Ndubaku and Tsui, 2015, Schauer et al., 2020b). One of the main challenges in developing inhibitors selective for a given DUB is the similarity between DUB family members, especially USPs, which ultimately leads to compounds with poor selectivity profiles and limits their utility in elucidating DUB function(Ritorto et al., 2014). Recent reports in the literature validate the plausibility of developing selective inhibitors of single DUBs (Kluge et al., 2018, Lee et al., 2010, Schlierf et al., 2016, Kategaya et al., 2017, Lamberto et al., 2017, Turnbull et al., 2017). These results encouraged us to perform HTS with the focus on using selectivity, rather than potency, as a way to identify and prioritize hits. We screened eight DUBs (USP7, USP8, USP10, USP17, USP28, USP30, UCHL1, and OTUD3) spanning three of the DUB families against a 50,000-member compound library to identify selective inhibitors for each of the DUBs at the earliest stages of inhibitor development.
We screened 47.26K compounds against each DUB that produced variable hit rates in the primary screen ranging from 0.4–6.9%. These data may also indicate a difference in ‘druggability’ amongst the different DUB families as we had much lower hit rates for UCHL1 and OTUD3 compared to the USP family on average. Follow up hit validation studies that comprised of profiling dose-response against target DUBs and one to three of the other DUBs included in the screen resulted in confirmation of inhibitors for five of the eight DUBs screened providing starting points for further medicinal chemistry optimization for probe development. In this study, USP28 was a clear outlier with 73 hits satisfying the most stringent criteria. Inspection of the structures of these hits showed multiple compounds with the same core scaffold. This accounts for the larger number of hits observed as well as gave us confidence in these compounds as inhibitors of USP28 and provided some initial SAR. In the case of USP30, the lone hit that was discovered was reported as a potent and selective USP30 inhibitor during the course of our screening by another group validating the ability of our approach to identify selective DUB inhibitors (Kluge et al., 2018).
To further map the selectivity profiles of our hit compounds, we employed a larger DUB panel, which disqualified some of the hits based on their activity against other DUBs not included in the screen. In the case of the USP28 inhibitors, all of the compounds displayed activity against USP25 in addition to USP28 which is not unsurprising given the high sequence homology shared by both DUBs (Liu et al., 2018, Valero et al., 2001, Sauer et al., 2019, Gersch et al., 2019). Indeed, this is borne out by the literature given that reported USP28 inhibitors, AZ1 and vismodegib, have equipotent activity against USP25 (Wrigley et al., 2017, Wang et al., 2021). Some of the compounds discovered through our screening efforts demonstrate roughly 10-fold better potency than AZ1 and offer multiple different scaffolds that can be utilized as probe compounds for USP25/28 biology or as starting points for medicinal chemistry efforts to tune selectivity towards one of the two DUBs. Our finding with the USP7 inhibitor, AV-9606–41, where the hit displayed potent and selective activity in the screen done with catalytic domain but no activity against a full-length protein, provides a cautionary note to anyone using isolated domains for screening campaigns. Our results with the USP8 inhibitor, where the hit displayed significant redox activity, demonstrates the importance of performing this type of analysis for large screens in general and for DUB inhibitors specifically. USP8 has been a difficult DUB to target as evidenced by the lack of well-validated, selective inhibitors in the literature. These results may indicate that some DUBs, like USP8, may be more susceptible to redox inactivation(Kulathu et al., 2013, Lee et al., 2013, Cotto-Rios et al., 2012). The Ubiquigent profiling results for the USP7, USP8, and USP10 hits, where we saw compound interference with assay readout by autofluorescence or fluorescence quenching, emphasize the importance of these types of analysis as part of an effort to validate compound activity. Taken together, our results illustrate the power of taking selectivity into account at a very early stage in the screening process, when conducting work on a large protein family. Keeping selectivity in mind, we were able to identify potential probes along with many viable starting points for probe development for a multitude of DUBs from a library of approximately 50,000 compounds which is modest compared to some of the larger screening collections deployed for HTS. Indeed, it is entirely possible and even likely that this compound collection is too small of a sample to identify viable hits for some of the more recalcitrant DUBs as we observed in our screen. While our focus has been on the identification of selective inhibitors, our method for screening has produced a robust dataset that can be mined for other compounds of interest. For example, although we have identified number of potential pan-DUB inhibitors, the pan-DUB activity of these compounds remains to be formally validated. Additionally, while our focus was on the most potent and selective inhibitors, the data generated by these experiments includes many selective but weaker inhibitors that could serve as starting points for medicinal chemistry campaigns. Collectively, the work described here represents the most comprehensive DUB inhibitor screening and profiling effort to date. As such, it offers number of future opportunities for development of pan- or selective DUB inhibitors as chemical tool compounds and/or drug leads to both further explore the biological functions of these DUBs and examine their value as therapeutic targets.
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Sara Buhrlage (saraj_buhrlage@dfci.harvard.edu).
Materials Availability
The materials generated in this study will be distributed upon request to the lead contact. There are restrictions to availability due to a Material Transfer Agreement (MTA).
Data and Code Availability
The primary and dose-response screening datasets generated during this study are available at PubChem (PubChem AID: 1645869). https://pubchem.ncbi.nlm.nih.gov/bioassay/1645869
Raw mass spectrometry data files for TMT 6-Plex activity-based protein profiling have been deposited in MassIVE (Mass spectrometry Interactive Virtual Environment, https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp). Dataset Identifier: MSV000087089
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Microbe strains
Recombinant proteins were expressed in E. coli BL21(DE3) bacteria, which were grown in Luria Broth (LB) at 37 °C with shaking at 200 rpm and then induced at 16 °C with shaking at 200 rpm.
Cell lines
HEK293T (female human origin) cell lines were used in this study. HEK293T cells (ATCC, Cat# CRL-3216) are a clonal isolate of HEK293 cells transformed with the SV40 large T antigen. For growth, HEK293T cells were maintained in DMEM containing 4 mM L-glutamine (Gibco) supplemented with 10% (v/v) 10% heat inactivated fetal bovine serum (FBS) (Gibco), and penicillin/streptomycin (100 U/mL penicillin and 0.1 mg/mL streptomycin) (Gibco). Cells were grown at 37°C with 5% CO2 in a water-saturated incubator. For passaging, cells were incubated with trypsin/EDTA at 37°C to detach cells.
METHOD DETAILS
Recombinant Protein Expression and Purification
The constructs encoding USP7 catalytic domain (AA 208–560), USP7 full-length (AA 1–1102), USP8 catalytic domain (AA 743–1110), USP10 catalytic domain (AA 376–798), USP30 catalytic domain (AA 65–517), USP28 catalytic domain (AA 140–697 for mouse, AA 149–703 for human), and USP25 (AA 157–714) were cloned into pET28 expression vectors with an N-terminal 6xHis tag in NEB® 10-beta Competent E. coli (High Efficiency) cells. The construct encoding UCHL1 full length (AA 1–223) was cloned into a pGEX6P1 expression vector with an N-terminal GST tag in NEB® 10-beta Competent E. coli (High Efficiency) cells. Plasmids containing desired constructs were then isolated, transfected into E. coli BL21 (DE3) cells and overexpressed. Cells were grown at 37 °C to an OD of 0.9, cooled to 16 °C, induced with 500 μM isopropyl-1-thio-D-galactopyranoside (IPTG), incubated overnight at 16 °C, collected by centrifugation, and stored at −80 °C. Cell pellets were sonicated in lysis buffer (25 mM Tris pH 8, 1 M NaCl, and 10 mM BME) supplemented with 10 μg/ml phenylmethanesulfonylfluoride (PMSF) and the resulting lysate was centrifuged at 30,000 ×g for 40 min. Ni-NTA beads (Qiagen) were mixed with lysate supernatant for 2 hours, and washed with lysis buffer supplemented with 25 mM imidazole. The bound protein was eluted with lysis buffer supplemented with 300 mM imidazole. The sample was then concentrated to 1 ml (30 kDa concentrator; Amicon Ultra, Millipore), and run on a Superdex 200 (GE healthcare) in buffer containing 25 mM HEPES pH 7.5, 200 mM NaCl, and 1 mM DTT. Fractions were pooled, concentrated, and frozen at −80 °C. A plasmid expressing USP17 (1–530) was synthesized by BioBasic in a pET28 expression vector, and expressed and purified as described previously (Hjortland and Mesecar, 2016). A plasmid expressing OTUD3 (52–275) was purchased from Addgene (Cat# 61411) and was expressed and purified as described(Mevissen et al., 2013). As judged by gel all proteins were at or above 90% purity, except for USP17 and USP30 which were approximately 70% pure. Yield ranged from 0.5–2 mg/L.
Design of Experiments (DOE) operation.
To test for optimal buffer conditions a design of experiments (DOE) approach was used. This involves first deciding on relevant buffer components to vary, taking from literature sources or baseline buffer conditions. These components are then varied at different concentrations and typically to cover a full permutation of all possible combinations yields several thousand experiments. Therefore, once all the factors are chosen the experimental design is created in JMP statistical software (developed by SAS Institute) which provides the optimal combination of factors to achieve ~ 2% coverage of all conditions allowing adequate modeling of each component’s effect on the assay. We used the output of the JMP design to create a dispense list to deliver the buffer components to the assay plate using a Formulatrix Tempest liquid handler that is capable of dispensing up to 12 reagents in varying volumes and combinations. Once the data is collected the results are imported into JMP to make a model of the assay response to all the factors and the quality of the model is considered good when the actual vs predicted plot of the data achieves a r2 ~ 0.9. Profiles of the buffer effects on the assay signal can be used to make decisions on buffer composition and the top buffers are also ranked based on overall signal so that the top scoring buffers can be chosen for confirmation testing. In the DUB assay DOE, HEPES and Tris buffers were prepared at three pH values (7, 7.5, and 8) and NaCl was varied a 0, 25, and 50mM. Bovine serum albumin was varied at 0 and 1% and the detergents CHAPS, Tween-20, PF127, NP-40, and Triton-X, were varied at 0, 0.25, and 0.5 of their CMC values. As well, the reducing reagents DTT and TCEP were varied at 0 and 1mM and EDTA was varied at 0 and 1mM. For this DOE, full permutation of all conditions would test 4,320 different buffers and the experimental design employed tested 8.6% of these. Following the JMP modeling the top buffers were prepared and tested in the assay to confirm performance (Supplemental Figure 1).
Ubiquitin rhodamine 110 endpoint assay (for screening USP7 and UCHL1)
2.5 μL of a solution of DUB in assay buffer (50 mM Tris pH 8, 50 mM NaCl, 0.002% Tween-20, 5 mM DTT) at 2x the final concentration in the assay was added to columns 1–46 on a Greiner medium binding 1536 well plate (catalog number: 782076). Compounds were acoustically transferred to columns 1–46 (columns 45–46 contained only DMSO as a neutral control). 2.5 μL of assay buffer was dispensed to columns 47–48. Plates were then incubated for 1 hour. 2.5 μL of Ub-Rho110 at 2x the final concentration in the assay was then added to all wells on the plate. Plates were incubated at room temperature for 10 minutes and the fluorescence was recorded using a PheraStar fluorescence plate reader at excitation and emission wavelengths of 485 nm and 535 nm respectively.
Ubiquitin rhodamine 110 endpoint assay (for screening other DUBs)
2.5 μL of a solution of DUB in assay buffer (50 mM Tris pH 8, 50 mM NaCl, 0.002% Tween-20, 5 mM DTT) at 2x the final concentration in the assay was added to columns 1–46 on a Greiner medium binding 1536-well plate (catalog number: 782076). Compounds were acoustically transferred to columns 1–46 (columns 45–46 contained only DMSO as a neutral control). 2.5 μL of assay buffer was dispensed to columns 47–48. Plates were then incubated for 10 minutes. 2.5 μL of Ub-Rho110 at 2x the final concentration in the assay was then added to all wells on the plate. Plates were incubated at room temperature for 30 minutes – 3 hours depending on the DUB. After incubation, reactions were quenched with the addition of 2.5 μL of stop buffer (assay buffer + 0.2% trifluoroacetic acid) and fluorescence was recorded using a PheraStar fluorescence plate reader at excitation and emission wavelengths of 485 nm and 535 nm respectively.
Analysis of screening data.
Normalization of raw data to positive and negative control wells, systematic pattern models, and curve fitting were applied to the data using Helios, a high-throughput screening data analysis program developed at Novartis.(Gubler et al., 2018) For measuring compound potency, a four-parameter sigmoid Hill curve model was fitted to the data (Gubler et al., 2018) and the absolute IC50 was used for potency measurements, that is the concentration of compound where the data showed 50% inhibition. If no data points achieved 50% inhibition, then the absolute IC50 was reported as >X μM (where X is the highest tested concentration).
Ubiquitin rhodamine 110 kinetic assay
DUBs from the high-throughput screen were tested for their respective activity in the Ub-Rho110 assay in the presence or absence of inhibitors using Nunc™ 384-well plates (Thermo Scientific™ catalog number: 2262260). Individual DUBs were pre-incubated with different concentrations of inhibitors in assay buffer (50 mM Tris pH 8, 50 mM NaCl, 0.002% Tween-20, 5 mM DTT). Compounds and protein were incubated for 30 minutes at room temperature prior to the addition of Ub-Rho110 (Boston Biochem) substrate. The initial rate of the reaction was measured by collecting fluorescence data at one-minute intervals over 30-minute period using a Clariostar fluorescence plate reader at excitation and emission wavelengths of 485 nm and 535 nm respectively. The calculated initial rate values were normalized to no compound control wells and were plotted against inhibitor concentrations to determine IC50s. All the experimental data were plotted using GraphPad Prism (log(inhibitor) vs. normalized response - Variable slope, least squares fit). All assays were performed at least twice for each compound and error bars are reported as SEM.
Ubiquitin-AMC kinetic assay
The same assay protocol as the Ubiquitin rhodamine 110 kinetic assay was followed, substituting Ub-AMC for the Ub-Rho110 and reading at the specified wavelengths for AMC (excitation and emission wavelengths of 345 nm and 445 nm respectively).
Redox Assay
50 μL of redox assay buffer (50 mM Tris pH 7.5, 50 mM NaCl, 5 mM DTT, and 25 μM resazurin) was added to column 1 of an Axygen® 96-well Polypropylene PCR Microplate (product number: PCR-96-FS-C). 25 μL of redox assay buffer was added to remaining wells in columns 2–11. Column 12 received 25 μL of redox assay buffer with 0.5 μL DMSO. 0.5 μL of 10 mM compound was then added to each row in column 1 for an initial concentration of 100 μM. A serial two-fold dilution of each compound was then performed down each row, omitting column 12. The plate was then incubated for 30 minutes in darkness, transferred to a Nunc™ 384-well plate (Thermo Scientific™ catalog number: 2262260) and then read on a Clariostar fluorescence plate reader at excitation and emission wavelengths of 545 nm and 600 nm respectively. The baseline signal based on the averaged DMSO control wells was then subtracted from all data points. Fluorescence values for each compound in dose were plotted as a function of compound dose using GraphPad Prism (log(agonist) vs. response - Variable slope (four parameters), least squares fit). All assays were performed at least twice for each compound.
In Vitro DUB Selectivity Profiling
Compounds were screened using the Ubiquigent DUBProfiler™ SPT system (www.ubiquigent.com/drug-discovery-services/dubprofiler/). Each of 41 purified DUBs was incubated with compound for 15 minutes, then Ubiquitin rhodamine 110 (Ub-Rho110) was added and percent inhibition was determined based on fluorescence relative to a DMSO control. The autofluorescence control experiment determines the fluorescence signal generated by the compound at screening concentration in absence of enzyme relative to the enzyme and no enzyme control for each of the DUBs in the panel. The product signal modulation (PSM) control experiment determines the fluorescence signal generated by the compound (at screening concentration) in the presence of synthetic rhodamine 110 glycine (the product of enzymatic cleavage of the DUBprofiler Ub-Rho substrate) and DUBprofiler substrate (at standard assay concentration) expressed as a percentage relative to the fluorescence signal of synthetic rhodamine 110 glycine and DUBprofiler substrate (at standard assay concentration). Autofluorescence and PSM data can indicate autofluorescence (positive %) or quenching (negative %) of background signal and/or assay signal.
Quantitative activity-based protein profiling
DUB activity based protein profiling was performed using conditions modified from those in Schauer et al.(Schauer et al., 2020a), based on work by Lawson et al.(Lawson et al., 2017). HEK 293T cells were lysed (50 mM Tris pH 8.0, 150 mM NaCl, 5 mM MgCl2, 0.5 mM EDTA, 0.5% NP-40, 10% glycerol, 1 mM TCEP, protease and phosphatase inhibitors) and the lysate was clarified by centrifugation, then diluted to 10 mg/mL. 200 μL aliquots were incubated with 250 μM compound or DMSO for 30 minutes at RT. Selective USP7 inhibitor XL188 was included in each run as a positive control.(Lamberto et al., 2017) Afterwards, the treated lysates were incubated with 1 μM each of Biotin-Ub-PA and BiotinUb-VME for 15 minutes at RT. 125 μL magnetic streptavidin sepharose slurry was added to each sample, followed by incubation at RT for 30 minutes with end-to-end rotation. After immobilizing the beads using a magnetic rack, the beads were washed (3× 0.2% SDS, 3x PBS, 2x ddH2O). After the final wash, supernatant was removed, and the resin was flash frozen and stored at −80° C.
Streptavidin beads were resuspended in 95 μL 100 mM Tris pH 8.0. Each sample was denatured with 0.1% rapigest, reduced (10 mM dithiothreitol), alkylated (22.5 mM iodoacetamide), and digested with trypsin at 37 °C overnight. The next day, beads were captured using a magnetic rack, and supernatants were acidified with 10% TFA, incubated at 37o C for 30 minutes, and centrifuged at 14,000 rpm for 15 minutes at 4o C to remove rapigest. Peptides were then desalted by C18 and dried by vacuum centrifugation.
Dried peptides were reconstituted in 40μL 50mM pH 8.0 TEAB, and 1/4 unit of TMT reagent was added, and reactions incubated at RT for 1 hour. TMT reactions were pooled and treated with hydroxylamine according to the manufacturer’s instructions. Peptide mixtures were then dried, reconstituted in 100 mM ammonium bicarbonate and desalted by SP3(Hughes et al., 2014). Eluted TMT labeled peptides were dissolved in 5% acetonitrile with 100 mM ammonium formate, pH 10, and analyzed by our DEEP-SEQ multidimension peptide fractionation platform (Zhou et al., 2013, Ficarro et al., 2011, Zhou et al., 2011) built around a NanoAcquity UPLC system (Waters, Milford, MA) and coupled directly to a timsTOF Pro ion mobility mass spectrometer (Bruker, Billerica, MA) (Meier et al., 2015, Meier et al., 2018). As described (Fu et al., 2020), each of 11 peptide fractions was eluted from the first dimension reversed phase column (5 cm × 150 μm packed with 5 μm XBridge C18, Waters, Milford, MA) and second dimension anion exchange column (5 cm × 150 μm I.D. fused silica packed with 10 μm POROS 10HQ Applied Biosystems, Foster City, CA) using injections of acetonitrile or ammonium formate at pH 10.0. Peptide fractions were diluted on-line with solvent A (0.1% formic acid in water) to reduce pH and organic concentration and facilitate retention on the third dimension precolumn (5 cm × 100 μm I.D. fused silica packed with 7 μm Symmetry C18, Waters). Peptides were eluted with an HPLC gradient (6.5–35% B in 120 minutes, A = 0.1% formic acid in water, B = 0.1% formic acid in acetonitrile), resolved on an analytical column (30 μm I.D. × 50 cm Monitor C18, Orochem, Naperville, IL) and introduced to the mass spectrometer by electrospray ionization using a captive spray ion source (spray voltage = 1.8 kV). The mass spectrometer acquired ion mobility MS spectra over a mass range of m/z 100–1700 and 1/k0 of 0.6 to 1.6, and then performed 10 cycles of PASEF MS/MS with a target intensity of 20k, a threshold of 2500 and a quadrupole isolation width of 1 Da. Active exclusion was enabled with a release time of 0.4 minutes. For TMT reporter ion detection, TIMS stepping function (Ogata and Ishihama, 2020) was used. Briefly, the collision energy was ramped stepwise against the ion mobility: 39.88 eV for 1/k0 0.6–0.83, 46.25 eV for 1/k0 0.83–1.0, 52.50 eV for 1/k0 1.0–1.18, 58.75 eV for 1/k0 1.18–1.35, and 64.63 eV for 1/k0 1.35–1.6. The collision energy for the second step is decreased by 20%. Other settings corresponding to the two-step collision energy include collision RF of 500 and 1500, transfer time of 25 μs and 60 μs, and pre pulse storage time of 8 μs and 12 μs, respectively.
Raw data files were converted to mgf using multiplierz scripts (Alexander et al., 2017), and searched using Mascot 2.6.1 against a forward and reversed human refseq database (NCBI). Search parameters specified a precursor mass tolerance of 20 ppm, a product ion tolerance of 50 mmu, fixed carbamidomethylation of cysteine, TMT 6-plex labeling of lysine and N-termini, and variable oxidation of methionine. For TMT quantitation, identified peptides were filtered to a peptide score no less than 13 and a false discovery rate of 1%. We used Pep2gene (Askenazi et al., 2010) to reduce the set of peptides to those which mapped to unique gene identifiers, and then aggregated reporter ions to derive a protein-level quantification value (Supplemental Table 2).
Competitive Activity Based Protein Profiling
HEK293T cells were pelleted, washed with PBS, lysed on ice with lysis buffer (20 mM Tris pH 8, 150 mM NaCl, 1% NP-40, 10% glycerol, 1 mM TCEP, phosphatase inhibitor cocktails (Sigma P5726 and Calbiochem 524624), and protease inhibitors (pepstatin, leupeptin, PMSF, and aprotinin), and clarified by centrifugation. Protein content was quantified by BCA and diluted to 2 mg/mL in lysis buffer. 40 μL samples of lysate were incubated with compounds or DMSO at desired concentrations and incubated at room temperature for 1 hour. Samples were then supplemented with 1:1 Ub-Pa/Ub-VME probe at a final concentration in the sample of 2 μM and incubated at room temperature with shaking for 30 minutes. Reactions were quenched with 4x LDS sample buffer (Thermo Fisher B0007) supplemented with 10% BME, vortexed vigorously, and heated to 95°C for 5 minutes. Samples were resolved by SDS-PAGE and analyzed by Western blot with the indicated antibodies (Primary antibodies used: USP28 rabbit monoclonal antibody from Abcam, catalog number: ab126604. GAPDH rabbit monoclonal antibody from Cell Signaling, catalog number: 2118S. Secondary antibody used: IRDye® 800CW Goat anti-Rabbit IgG Secondary Antibody from LI-COR, catalog number: 926–32211)
UpSet plot
UpSet plot was generated using UpSetR code from GitHub
Quantification and Statistical Analysis
All biochemical curves and associated statistical analyses were produced using GraphPad Prism Software. Details of replicates and data analysis for specific experiments can be found in figure legends or the methods section.
Chemical Synthesis
List of abbreviations: DCM (dichloromethane); DMSO (dimethyl sulfoxide); Et2O (diethyl ether); EtOAc (ethyl acetate); EtOH (ethanol); Et3N (triethylamine); g (gram); HCl (hydrochloric acid); LC/MS (liquid chromatography-mass spectrometry); MeCN (acetonitrile); MeOH (methanol); MHz (megahertz); mg (milligram); mL (milliliter); mmol (millimole); NaHCO3 (sodium bicarbonate); Na2SO4 (sodium sulfate); NH4Cl (ammonium chloride); NMR (nuclear magnetic resonance); THF (tetrahydrofuran); μL (microliter).
General Methods and Materials
All commercially available starting materials were purchased from Sigma Aldrich, Fisher Scientific, Oakwood Chemical, and Combi Block. All reagents were used as received without further purification. Anhydrous solvents, such as tetrahydrofuran (THF), dichloromethane (DCM), diethyl ether (Et2O), dimethyl formamide (DMF), dimethylsulfoxide (DMSO), 1,4-dioxane, and toluene were purchased from Fisher Scientific, and used as received. If necessary, air or moisture sensitive reactions were carried out under an inert atmosphere of nitrogen. Removal of solvents was accomplished on a Büchi R-300 rotary evaporator and further concentration was done under a Welch 1400B-01 vacuum line, and Labconco FreeZone 6 plus system. Purification of compounds was performed by normal phase column chromatography using Teledyne CombiFlash chromatography system, and/or reversed phase chromatography on Waters Micromass ZQ preparative system with SunFire® Prep C18 OBD™ 5μM column. The purity was analyzed on Waters Acquity UPLC system. Analytical thin layer chromatography (TLC) plates were purchased from Fisher Scientific (EMD Millipore TLC Silica Gel60 F254). Visualization was accomplished by irradiation under UV light (254 nm).
All 1H-NMR spectra were recorded at 298K on a Bruker ARX 500 (500 MHz) spectrometer. Samples were dissolved in CDCl3, DMSO-d6, or CD3OD obtained from Cambridge Isotope Laboratories. The spectra were referenced to the residual solvent peak (chloroform-d: 7.26 ppm for 1H-NMR and 77.16 ppm for 13C-NMR; DMSO-d6: 2.50 ppm for 1H-NMR and 39.25 ppm for 13C-NMR, CD3OD: 3.31 ppm for 1H NMR and 49.00 ppm for 13C NMR or tetramethylsilane (TMS) as the internal standard. Chemical shift, multiplicity (s=singlet, d=doublet, t=triplet, q=quartet, m=multiplet, br=broad peak), coupling constants (Hz), and number of protons. Mass spectrometry (LCMS) data were obtained on Waters Acquity UPLC system in positive ESI mode.
Z56965384 was purchased directly from Enamine at 85% purity. The compound was purified via HPLC to afford the desired product. LC/MS (ESI) m/z 486.57 [M+H]+; calcd for C22H19N2O9S+: 487.08. 1H NMR (500 MHz, DMSO-d6) δ 10.26 (s, 1H), 8.27 (d, J = 2.4 Hz, 1H), 8.01 – 7.98 (m, 2H), 7.97 (dd, J = 7.9, 1.6 Hz, 1H), 7.85 (dd, J = 8.9, 2.4 Hz, 1H), 7.65 (ddd, J = 8.9, 7.4, 1.6 Hz, 1H), 7.52 (dd, J = 8.3, 1.1 Hz, 1H), 7.28 (td, J = 7.6, 1.2 Hz, 1H), 7.23 (d, J = 8.9 Hz, 1H), 7.16 – 7.08 (m, 2H), 5.69 (s, 2H), 3.88 (s, 3H).
Final compounds submitted for testing were prepared and stored as 10 mM stock solutions in DMSO. Stocks were stored at −20°C and aliquoted as needed for specific experiments to reduce the number of freeze thaw cycles. We observed no apparent solubility issues with the compounds either in DMSO or aqueous buffer conditions at the concentrations used.
Compound synthesis
3-methyl-1-phenyl-1H-spiro[benzo[4,5]thiazolo[3,2-a]pyrazolo[3,4-d]pyrimidine-4,3’-indolin]-2’-one (AV-9606–27)

Step 1: N-(3-methyl-1-phenyl-1H-pyrazol-5-yl)benzo[d]thiazol-2-amine (AV-9606–26)
2-chlorobenzo[d]thiazole (173.9 mg, 1.0 mmol) was suspended in THF (5 mL). To the reaction was added sodium hydride (37.9 mg, 1.5 mmol), and the reaction was stirred at room temperature for 30 minutes under nitrogen. 3-methyl-1-phenyl-1H-pyrazol-5-amine (130.2 uL, 1.0 mmol) was then added to the reaction and the reaction was heated to 60°C for 18 hours. The reaction mixture was diluted with EtOAc and washed with water and brine. The organics were combined, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting crude was purified via silica gel chromatography (0–100% EtOAc: hexanes) to afford the desired product (25.9 mg, 8.5%). LC/MS (ESI) m/z 306.87 [M+H]+; calcd for C17H15N4S+: 307.10.

Step 2: 3-methyl-1-phenyl-1H-spiro[benzo[4,5]thiazolo[3,2-a]pyrazolo[3,4-d]pyrimidine-4,3’-indolin]-2’-one (AV-9606–27)
AV-9606–26 (25.9 mg, 0.08 mmol) and isatin (14.2 mg, 0.10 mmol) were combined and suspended in DMF (2 mL). To the reaction was then added TMS-Cl (32.3 uL, 0.24 mmol). The reaction was heated at 100°C for 16 hours. The reaction was diluted with water and placed in a sonication bath for 90 minutes. The reaction was then filtered to isolate the resulting precipitate. The precipitate was then purified via silica gel chromatography (0–100% EtOAc: hexanes) to afford the desired product (2.1 mg, 6.0%). LC/MS (ESI) m/z 435.87 [M+H]+; calcd for C25H18N5OS+: 436.12. 1H NMR (500 MHz, Methanol-d4) δ 7.86 – 7.77 (m, 1H), 7.59 (d, J = 7.7 Hz, 1H), 7.48 (q, J = 7.6 Hz, 2H), 7.38 – 7.27 (m, 2H), 7.17 (dd, J = 12.5, 7.8 Hz, 2H), 7.09 (s, 1H), 6.55 (d, J = 8.3 Hz, 1H), 1.70 (s, 3H).
6’-amino-3’-isopropyl-2-oxo-2’H-spiro[indoline-3,4’-pyrano[2,3-c]pyrazole]-5’-carbonitrile (AV-9606–41)

Methyl isobutyrylacetate (96.7 μL, 0.69 mmol) and hydrazine hydrate (21.2 μL, 0.68 mmol) were combined and diluted in EtOH (3 mL). The reaction was heated to 65°C for 15 min at which point isatin (102.0 mg, 0.69 mmol), DMAP (8.6 mg, 0.07 mmol), and malononitrile (48.4 mg, 0.73 mmol) were added. The reaction was stirred at 65°C for 1 hour. The reaction mixture was concentrated under reduced pressure and resuspended in EtOAc then washed with water and brine. The organics were combined and dried over anhydrous Na2SO4 then filtered and concentrated under reduced pressure. The crude residue was then purified via silica gel chromatography (0–100% EtOAc: hexanes) to afford the desired product (43.9 mg, 20.1%). LC/MS (ESI) m/z 322.07 [M+H]+; calcd for C17H16N5O2+: 322.13. 1H NMR (500 MHz, DMSO-d6) δ 12.30 (s, 1H), 10.60 (s, 1H), 7.23 (t, J = 7.6 Hz, 1H), 7.17 (s, 2H), 7.04 (d, J = 7.2 Hz, 1H), 6.98 (t, J = 7.5 Hz, 1H), 6.89 (d, J = 7.7 Hz, 1H), 4.10 (q, J = 5.1 Hz, 1H), 3.17 (d, J = 4.8 Hz, 2H), 2.14 – 2.01 (m, 1H), 0.95 (d, J = 7.1 Hz, 3H), 0.66 (d, J = 7.0 Hz, 3H).
(2’R,3R,3’S)-2’-(4-fluorobenzoyl)-3’-(thiophene-2-carbonyl)-2’,3’,8’,8a’-tetrahydro-7’H-spiro[indoline-3,1’-indolizin]-2-one (AD-10942–8)

Step 1: (Z)-3-(2-(4-fluorophenyl)-2-oxoethylidene)indolin-2-one (AV-9606–80)
Isatin (149.0 mg, 1.0 mmol) was suspended in EtOH (1 mL) and 4’-fluoroacetophenone (121.4 μL, 1.0 mmol) was added. The reaction was stirred at room temperature for 6 hours. The reaction was filtered to isolate the resulting precipitate in the reaction mixture. The precipitate was resuspended in EtOH (5 mL) and concentrated HCl (0.5 mL) was added, and the reaction was stirred at 60°C for 2 hours. The reaction was filtered again to afford the desired product (139.1 mg, 52.0%). LC/MS (ESI) m/z 268.07 [M+H]+; calcd for C16H11FNO2+: 268.08. 1H NMR (500 MHz, Chloroform-d) δ 8.30 (d, J = 7.8 Hz, 1H), 8.15 (ddd, J = 10.9, 6.6, 3.8 Hz, 3H), 7.82 (s, 1H), 7.33 (td, J = 7.7, 1.2 Hz, 1H), 7.21 (t, J = 8.6 Hz, 2H), 7.02 (td, J = 7.7, 1.0 Hz, 1H), 6.88 (d, J = 7.8 Hz, 1H).

Step 2: 2-bromo-1-(thiophen-2-yl)ethan-1-one (AV-9606–87)
Copper(II) Bromide (338.1 mg, 1.5 mmol) was suspended in EtOH (5 mL). To the reaction was then added 2-acetylthiophene (108.1 μL, 1.0 mmol) and the reaction was heated to 60°C for 24 hours. The reaction was concentrated under reduced pressure and the crude residue (AV-9606–87) was used in the subsequent reaction without further purification. LC/MS (ESI) m/z 204.98 [M+H]+; calcd for C6H6BrOS+: 204.93.
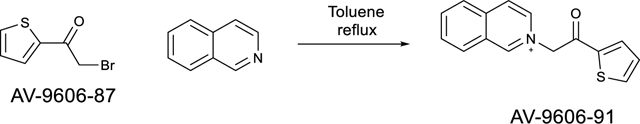
Step 3: 2-(2-oxo-2-(thiophen-2-yl)ethyl)isoquinolin-2-ium bromide (AV-9606–91)
AV-9606–87 (102.0 mg, 0.5 mmol) was dissolved in toluene (2 mL). To the reaction was then added isoquinoline (58.7 μL, 0.5 mmol). The reaction was then heated to reflux for 30 minutes. The reaction was cooled to room temperature and filtered then washed with hexanes. The oily residue on the bottom of the reaction vessel was dissolved in DCM and concentrated. The crude residue was suspended in 1,4-dioxane and sonicated to precipitate out the desired product from solution. The reaction was filtered to afford the desired product (92.4 mg, 55.3%). The crude material was used in the next step without further purification. LC/MS (ESI) m/z 253.87 [M]+; calcd for C15H12NOS+: 254.06.

Step 4: (2’R,3R,3’S)-2’-(4-fluorobenzoyl)-3’-(thiophene-2-carbonyl)-2’,3’,8’,8a’-tetrahydro-7’H-spiro[indoline-3,1’-indolizin]-2-one (AD-10942–8)
AV-9606–91 (92.4 mg, 0.28 mmol) and AV-9606–80 (74.8 mg, 0.28 mmol) were combined and suspended in EtOH (2.25 mL). Et3N (46.8 μL, 0.34 mmol) was then added and the reaction was stirred at room temperature for 4.5 hours. The reaction was filtered, and the precipitate was washed with EtOH. The precipitate was recrystallized from EtOH/DCM to afford the desired product (52.2 mg, 43.6%). LC/MS (ESI) m/z 521.18 [M+H]+; calcd for C31H22FN2O3S+: 521.13. 1H NMR (500 MHz, DMSO-d6) δ 10.52 (s, 1H), 8.10 (dd, J = 4.9, 1.2 Hz, 1H), 8.03 (dd, J = 3.9, 1.2 Hz, 1H), 7.42 – 7.34 (m, 2H), 7.28 (dd, J = 4.9, 3.9 Hz, 1H), 7.18 (dd, J = 7.6, 1.2 Hz, 1H), 7.13 (t, J = 8.8 Hz, 2H), 6.88 (dd, J = 7.4, 1.3 Hz, 1H), 6.82 (dt, J = 9.0, 7.0 Hz, 2H), 6.70 (ddt, J = 21.5, 7.6, 6.4 Hz, 3H), 6.53 (d, J = 7.6 Hz, 1H), 6.27 (d, J = 7.7 Hz, 1H), 6.14 (d, J = 5.3 Hz, 1H), 5.34 (s, 1H), 5.14 (d, J = 7.5 Hz, 1H), 4.73 (d, J = 5.3 Hz, 1H).
5-bromo-3’-phenyl-1’H-spiro[indoline-3,2’-quinazoline]-2,4’(3’H)-dione (AV-9606–99)

5-bromoisatin (112.4 mg, 0.50 mmol) and isatoic anhydride (122.6 mg, 0.75 mmol) were combined and suspended in acetic acid (2 mL). Aniline (68.0 μL, 0.75 mmol) was added and the reaction mixture was stirred at room temperature for 15 minutes. The reaction mixture was then transferred to a microwave vial, sealed and heated to 160°C for 30 min. The reaction mixture was then diluted with EtOAc and washed with water and brine. The organics were collected, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The resulting crude was purified via silica gel chromatography (0-100% EtOAc: hexanes) to afford the desired product (65.2 mg, 31.0%). LCMS (ESI) m/z = 420.07 [M+H]+; calcd for C21H15BrN3O2+: 420.03. 1H NMR (500 MHz, DMSO-d6) δ 10.56 (s, 1H), 7.75 (d, J = 2.0 Hz, 1H), 7.67 (d, J = 6.2 Hz, 2H), 7.33 (td, J = 8.5, 1.9 Hz, 2H), 7.26 (t, J = 7.6 Hz, 2H), 7.19 (t, J = 7.3 Hz, 1H), 7.03 (d, J = 8.1 Hz, 2H), 6.78 (t, J = 7.5 Hz, 1H), 6.70 (d, J = 8.1 Hz, 1H), 6.61 (d, J = 8.3 Hz, 1H).
2-(3,4-dichlorophenyl)-N-(5-(N,N-dimethylsulfamoyl)-2-methylphenyl)acetamide (AV-9606–129)

3,4-dichlorophenylacetic acid (100.4 mg, 0.49 mmol) was combined with HATU (226.4 mg, 0.60 mmol) and suspended in DMF (3 mL). Et3N (204.9 μL, 1.47 mmol) was added and the reaction was stirred at room temperature for about 15 minutes. To the reaction was then added 3-amino-N,N,4-trimethylbenzene-1-sulfonamide (108.8 mg, 0.51 mmol). Stir at room temperature for 42 hours. The reaction was diluted with EtOAc and washed with water and brine. The organic layer was collected, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude was purified via silica gel chromatography (0–100% EtOAc:hexanes) to afford the pure compound as a light brown powder (41.7 mg, 21%). LC/MS (ESI) m/z 400.87 [M+H]+; calcd for C17H19Cl2N2O3S+: 401.05. 1H NMR (500 MHz, Methanol-d4) δ 7.86 (d, J = 1.9 Hz, 1H), 7.57 (d, J = 2.1 Hz, 1H), 7.55 – 7.45 (m, 3H), 7.32 (dd, J = 8.3, 2.1 Hz, 1H), 3.78 (s, 2H), 2.67 (s, 6H), 2.31 (s, 3H).
5-(5-(3-chlorobenzyl)-1,2,4-oxadiazol-3-yl)pyridin-2(1H)-one (AV-11324–75)

Step 1: 6-chloro-N-hydroxynicotinimidamide (AV-11324–22)
2-chloro-5-cyanopyridine (1542.1 mg, 11.1 mmol) and hydroxylamine HCl (897.0 mg, 12.9 mmol) were combined and suspended in EtOH (25 mL). Et3N (7735.6 uL, 55.5 mmol) was then added and the reaction was heated to 65°C for 6 hours. White precipitate was observed in the reaction. Filtration afforded the desired product as a white powder (1378.1 mg, 72%). LC/MS (ESI) m/z 171.99 [M+H]+; calcd for C6H7ClN3O+: 172.03.
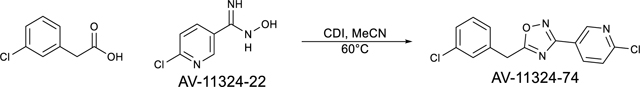
Step 2: 5-(3-chlorobenzyl)-3-(6-chloropyridin-3-yl)-1,2,4-oxadiazole (AV-11324–74)
3-chlorophenylacetic acid (173.2 mg, 1.02 mmol) and CDI (255.5 mg, 1.68 mmol) were combined and suspended in MeCN (3 mL). The reaction was stirred at room temperature for 2 hours. The reaction was then filtered and 6-chloro-N-hydroxynicotinimidamide (197.0 mg, 1.15 mmol) was then added and the reaction was stirred at room temperature for 30 minutes. DBU (305.1 uL, 2.04 mmol) was then added and the reaction was heated to 60°C for 1 hour. The reaction was concentrated under reduced pressure and resuspended in EtOAc and washed with water and brine. The organic layer was collected, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude was purified via silica gel chromatography (0–100% EtOAc:hexanes) to afford the pure compound (194.5 mg, 62%). LC/MS (ESI) m/z 305.77 [M+H]+; C14H10Cl2N3O+: 306.02.

Step 3: 5-(5-(3-chlorobenzyl)-1,2,4-oxadiazol-3-yl)pyridin-2(1H)-one (AV-11324–75)
5-(3-chlorobenzyl)-3-(6-chloropyridin-3-yl)-1,2,4-oxadiazole (192.1 mg, 0.63 mmol) was dissolved in concentrated HCl (3 mL) and heated to reflux for 2h. The reaction was removed from heat and quenched with water and neutralized with saturated NaHCO3. The aqueous solution was extracted with DCM. The organic layer was collected and washed with brine. The organic layer was then isolated, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude was then purified via silica gel chromatography (0–10% MeOH:DCM) to afford the desired product as a white powder (5.7 mg, 2%). LC/MS (ESI) m/z 287.87 [M+H]+; calcd for C14H11ClN3O2+: 288.05. 1H NMR (500 MHz, DMSO-d6) δ 7.97 (d, J = 2.6 Hz, 1H), 7.87 (dd, J = 9.6, 2.7 Hz, 1H), 7.50 (d, J = 2.1 Hz, 1H), 7.45 – 7.33 (m, 2H), 6.48 (d, J = 9.6 Hz, 1H), 4.43 (s, 2H).
N-(3,4-dimethoxyphenethyl)-4-propyl-2,3-dihydro-1H-cyclopenta[4’,5’]pyrido[3’,2’:4,5]thieno[3,2-d]pyrimidin-7-amine (bin-01–07-07)
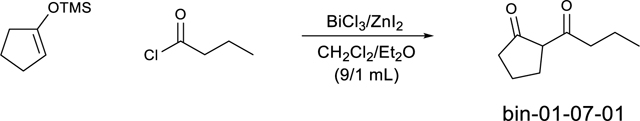
Step 1: 2-butyrylcyclopentan-1-one (bin-01–07-01)
To a dried flask charged with DCM/Et2O (9/1; 25 mL) was added bismuth(III) chloride (0.5 g, 1.6 mmol, 0.05 eq) and zinc iodide (0.77 g, 2.4 mmol, 0.075 eq) rapidly (under dryer). This is followed by the addition of butyryl chloride (3.66 mL, 35.2 mmol, 1.1 eq) and the suspension was stirred for 5 minutes at room temperature. After that, the 1-(trimethylsiloxy)cyclopentane was added at once under rapid stirring. After 40 minutes at room temperature, the reaction was quenched by saturated NaHCO3. After the organic layers were separated, the aqueous layer was washed by DCM (3 × 30 mL). The organic layers were combined, dried over Na2SO4, and concentrated to give the crude product bin-01–07-01 which was used directly without further purification. LC/MS (ESI) m/z 155.10 [M+H]+; calcd for C9H15O2+: 155.11.

Step 2: 3-chloro-1-propyl-6,7-dihydro-5H-cyclopenta[c]pyridine-4-carbonitrile (bin-01–07-03)
The reaction mixture of bin-01–07-01 (32 mmol, crude reaction mixture obtained directly from last step), cyano acetamide (2.5 g, 30 mmol), diethyl amine (3.1 mL, 30 mmol) in EtOH (50.0 mL) was stirred at room temperature for 24 hours. The reaction mixture was then concentrated and washed with cooled EtOH to give pure product bin-01–07-02 that was used in the next step directly. The reaction mixture of bin-01–07-02 (2.0 g, 10 mmol) in anhydrous 1,4-dioxane (10.0 mL) and POCl3 (5.0 mL) was heated at 90°C overnight. The reaction mixture was then poured into ice water and adjusted to pH = 7. The resulting precipitate was then filtered, dried to give pure product as white solid bin-01–07-03, (30% yield over three steps). LC/MS (ESI) m/z 221.28 [M+H]+; calcd for C12H14ClN2+: 221.08.

Step 3: ethyl 1-amino-5-propyl-7,8-dihydro-6H-cyclopenta[d]thieno[2,3-b]pyridine-2-carboxylate (bin-01–07-04)
To a solution of bin-01–07-03 (220 mg, 1 mmol, 1.0 eq) in EtOH (5.0 mL) was added sodium ethoxide (75 mg, 1.1 mmol, 1.1 eq) and ethyl thioglycolate (120 μL, 1.1 mmol, 1.1 eq.). The reaction mixture was stirred under reflux for 5 hours. The reaction mixture was then cooled down to room temperature and concentrated under reduced pressure. The resulting yellow brown solid was then dissolved with water and EtOAc (combined 100 mL). The organic layer was then washed with saturated brine and concentrated. The crude material was purified through silica gel chromatography (0–15% EtOAc:hexanes) to give pure product bin-01–07-04 as white solid, (85% yield). LC/MS (ESI) m/z 305.07 [M+H]+; calcd for C16H21N2O2S+: 305.13.

Step 4: 7-chloro-4-propyl-2,3-dihydro-1H-cyclopenta[4’,5’]pyrido[3’,2’:4,5]thieno[3,2-d]pyrimidine (bin-01–07-06)
A mixture of bin-01–07-04 (220 mg, 1 mmol, 1.0 eq) in formamide (5 mL) was refluxed for 4 hours. After that, the reaction mixture was poured into water and extracted with EtOAc. The combined organic phase was then concentrated and purified through silica gel chromatography (0–10% MeOH:DCM) to give pure product bin-01–07-05. LC/MS (ESI) m/z 286.07 [M+H]+; calcd for C15H16N3OS+: 286.10. To a solution of bin-01–07-05 (1 mmol) in 1,4-dioxane (1.0 mL) was added POCl3 (1.0 mL). The reaction was heated at 95°C overnight. The reaction was poured into icy water and adjusted to pH around 7. The resulting precipitate was isolated via filtration to give the desired product as a pure white solid. The filtrate was extracted with EtOAc, concentrated, and purified via silica gel chromatography (0–10% EtOAc:hexanes) to afford additional product. The product from both conditions were combined to afford bin-01–07-06 (60% yield over two steps).
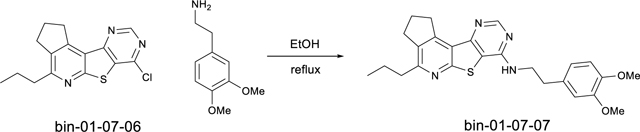
Step 5: N-(3,4-dimethoxyphenethyl)-4-propyl-2,3-dihydro-1H-cyclopenta[4’,5’]pyrido[3’,2’:4,5] thieno[3,2-d]pyrimidin-7-amine (bin-01–07-07)
A solution of bin-01–07-05 (28.5 mg, 0.1 mmol, 1.0 eq) and 3,4-dimeoxyphenethylamine (36 mg, 0.2 mmol) in EtOH (5 mL) was heated to reflux overnight. The reaction mixture was then concentrated and the crude material was purified via silica gel chromatography (0–60% EtOAc:hexanes) to give pure product bin-01–07-07 (85% yield). LC/MS (ESI) m/z 449.18 [M+H]+; calcd for C25H29N4O2S+: 449.20. 1H NMR (500 MHz, DMSO-d6) δ 8.61 (s, 1H), 7.89 (t, J = 5.5 Hz, 1H), 6.87 ‒ 6.84 (m, 2H), 6.76 (dd, J = 8.2, 1.9 Hz, 1H), 3.75 ‒ 3.72 (m, 2H), 3.71 (s, 3H), 3.70 (s, 3H), 3.47 (t, J = 7.5 Hz, 2H), 2.98 (t, J = 7.5 Hz, 2H), 2.91 ‒ 2.86 (m, 2H), 2.84 ‒ 2.78 (m, 2H), 2.18 (Pent, J = 7.5 Hz, 2H), 1.75 (Sextet, J = 7.5 Hz, 2H), 0.96 (t, J = 7.5 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 159.26, 158.61, 156.59, 155.10, 153.51, 150.57, 148.59, 147.23, 135.73, 131.93, 122.81, 120.52, 112.79, 112.59, 111.92, 55.50, 55.34, 42.10, 37.44, 34.34, 32.04, 29.57, 24.24, 21.21, 13.92.
(S)-N-(1-((5-(N-isopropylsulfamoyl)naphthalen-1-yl)amino)-1-oxo-3-phenylpropan-2-yl)benzamide (AV-11324–5)
Step 1: sodium 5-acetamidonaphthalene-1-sulfonate
5-Aminonaphthalene-1-sulfonic acid (1.12 g, 5.0 mmol) was dissolved in 5 mL pyridine, and acetic anhydride (0.76 g, 7.5 mmol) was added dropwise. The mixture was stirred at room temperature for 2 hours and then concentrated under reduced pressure. The crude was then dissolved in 2N NaOH solution and stirred at room temperature for 2 hours. The reaction mixture was then poured into 100% EtOH. Solid precipitate was collected by filtration and washed with additional cold EtOH, and dried under high vacuum to afford the desired product (0.74 g, 52%). LC/MS (ESI) m/z 265.97 (sulfonic acid form +H) [M+H]+; calcd for C12H11NNaO4S+: 288.03.
Step 2: 5-acetamidonaphthalene-1-sulfonyl chloride
A solution of sodium 5-acetamidonaphthalene-1-sulfonate (0.61 g, 2.1 mmol) in chlorosulfuric acid (10 mL) was stirred at 65°C for 1 hour. Then the reaction mixture was carefully poured onto ice, and the crude product was precipitated and collected by filtration. The solid material was further washed with ice-cold water and dried under vacuum to afford crude product which was used directly without further purification.

Step 3: N-(5-(N-isopropylsulfamoyl)naphthalen-1-yl)acetamide
To the solution of 5-acetamidonaphthalene-1-sulfonyl chloride (0.28 g, 1.0 mmol) in 5 mL DCM was added isopropylamine (0.08 g, 1.2 mmol) and Et3N (0.42 mL, 3.0 mmol). The reaction mixture was stirred at room temperature overnight, then concentrated under reduced pressure. The crude was purified by silica gel chromatography (0–100% EtOAc:hexanes) to afford the desired product (0.08 g, 26%). LC/MS (ESI) m/z 306.87 [M+H]+; calcd for C15H19N2O3S+: 307.11.

Step 4: 5-amino-N-isopropylnaphthalene-1-sulfonamide (XL-9872–143)
N-(5-(N-isopropylsulfamoyl)naphthalen-1-yl)acetamide (0.08 g, 0.27 mmol) was dissolved in 5N NaOH aqueous solution (15.0 eq) and 3 mL MeOH. The mixture was stirred at 65°C then 80°C for 48 hours. Then the mixture was diluted with DCM and washed with saturated NH4Cl and brine. The organic phase was concentrated under reduced pressure and purified by silica gel chromatography using (0–100% EtOAc:hexanes) to afford desired product (0.04 g, 56%). LC/MS (ESI) m/z 264.87 [M+H]+; calcd for C13H17N2O2S+: 265.10.

Step 5: tert-butyl benzoyl-L-phenylalaninate (AV-9606–205)
Benzoic acid (205.4 mg, 1.68 mmol) and HATU (857.0 mg, 2.25 mmol) were combined and suspended in DMF (4 mL). To the reaction was then added L-phenylalanine tert-Butyl ester hydrochloride (332.6 mg, 1.5 mmol) followed by Et3N (1045.4 μL, 7.5 mmol). The reaction was stirred at room temperature 1 hour. The reaction was then heated to 100°C for an additional 30 minutes at which point the reaction became fully soluble. The reaction was then diluted with EtOAc and washed with water and brine. The organics were collected and dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude material was purified via silica gel chromatography (0–100% EtOAc:hexanes) to afford the desired product (385.9 mg, 79.1%). LC/MS (ESI) m/z 269.87 [M-tert-Butyl+H]+; calcd for C16H16NO3+: 270.11.

Step 6: benzoyl-L-phenylalanine
AV-9606–205-1 (385.9 mg, 1.19 mmol) was then suspended in 4N HCl in 1,4-dioxane (5 mL) and stirred at room temperature for 24 hours. The reaction was filtered to isolate the precipitate observed in the reaction as the desired product (287.2 mg, 89.6%). LC/MS (ESI) m/z 269.97 [M+H]+; calcd for C16H16NO3 +: 270.11. The resulting material was used in the next step without further purification.

Step 7: (S)-N-(1-((5-(N-isopropylsulfamoyl)naphthalen-1-yl)amino)-1-oxo-3-phenylpropan-2-yl)benzamide (AV-11324–5)
AV-9606–205-2 (41.4 mg, 0.15 mmol) was suspended in DMF (1 mL) and placed in an ice bath. To the reaction was then added EDC hydrochloride (30.7 mg, 0.16 mmol) followed by HOBt (17.6 mg, 0.11 mmol). The reaction was stirred at room temperature for 1 hour. To the reaction was then added XL-9872–143 (19.9 mg, 0.075 mmol) and the reaction was stirred at room temperature for 72 hours. The reaction was then purified directly via silica gel column chromatography to afford the desired product (13.3 mg, 34%). LC/MS (ESI) m/z 515.39 [M+H]+; calcd for C29H30N3O4S+: 516.20. 1H NMR (500 MHz, DMSO-d6) δ 10.32 (s, 1H), 8.83 (d, J = 7.7 Hz, 1H), 8.53 (d, J = 8.5 Hz, 1H), 8.22 – 8.11 (m, 2H), 7.94 – 7.84 (m, 3H), 7.72 – 7.60 (m, 3H), 7.57 – 7.51 (m, 1H), 7.50 – 7.43 (m, 4H), 7.33 (t, J = 7.5 Hz, 2H), 7.24 (t, J = 7.4 Hz, 1H), 5.03 (ddd, J = 9.6, 7.6, 5.6 Hz, 1H), 3.33 – 3.17 (m, 4H), 0.87 (dd, J = 8.1, 6.5 Hz, 6H).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-GAPDH | Cell Signaling Technology | Cat#2118S; RRID: AB_1031003 |
| Rabbit monoclonal anti-USP28 | Abcam | Cat#ab126604; RRID: AB_11127442 |
| IRDye® 800CW Goat anti-Rabbit IgG Secondary Antibody | LI-COR Bioscience | Cat#926–32211; RRID: AB_621843 |
| Bacterial and Virus Strains | ||
| NEB® 10-beta Competent E. coli (High Efficiency) | New England BioLabs Inc | Cat#C3019H |
| BL21(DE3) Competent E. coli | New England BioLabs Inc | Cat#C2527H |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Dulbecco’s Modified Eagle Medium (DMEM) | Gibco | Cat#11965–092 |
| Penicillin/streptomycin | Gibco | Cat#15140–122 |
| Fetal Bovine Serum (FBS) | Gibco | Cat#16140–071 |
| Z56965384 | Enamine | Cat#Z56965384 |
| 3-methyl-1-phenyl-1H-spiro[benzo[4,5]thiazolo[3,2-a]pyrazolo[3,4-d]pyrimidine-4,3’-indolin]-2’-one (AV-9606–27) | This paper | PubChem CID: 2998850 |
| 6’-amino-3’-isopropyl-2-oxo-2’H-spiro[indoline-3,4’-pyrano[2,3-c]pyrazole]-5’-carbonitrile (AV-9606-41) | This paper | PubChem CID: 3624054 |
| (2’R,3R,3’S)-2’-(4-fluorobenzoyl)-3’-(thiophene-2-carbonyl)-2’,3’,8’,8a’-tetrahydro-7’H-spiro[indoline-3,1’-indolizin]-2-one (AD-10942–8) | This paper | N/A |
| 5-bromo-3’-phenyl-1’H-spiro[indoline-3,2’-quinazoline]-2,4’(3’H)-dione (AV-9606–99) | This paper | PubChem CID: 3487576 |
| 2-(3,4-dichlorophenyl)-N-(5-(N,N-dimethylsulfamoyl)-2-methylphenyl)acetamide (AV-9606–129) | This paper | PubChem CID: 3985183 |
| 5-(5-(3-chlorobenzyl)-1,2,4-oxadiazol-3-yl)pyridin-2(1H)-one (AV-11324–75) | This paper | PubChem CID: 136087907 |
| 5-(5-(3,4-dichlorobenzyl)-1,2,4-oxadiazol-3-yl)pyridin-2(1H)-one (AV-9606–180) | This paper | N/A |
| N-(3,4-dimethoxyphenethyl)-4-propyl-2,3-dihydro-1H-cyclopenta[4’,5’]pyrido[3’,2’:4,5]thieno[3,2-d]pyrimidin-7-amine (bin-01–07-07) | This paper | PubChem CID: 2019582 |
| (S)-N-(1-((5-(N-isopropylsulfamoyl)naphthalen-1-yl)amino)-1-oxo-3-phenylpropan-2-yl)benzamide (AV-11324–5) | {Kluge, 2018 #22} This paper | PubChem CID: 4383255 |
| Ubiquitin-Rhodamine 110 (Ub-Rho) | R&D Systems | Cat#U-555–050 |
| Ubiquitin-propargylamine (Ub-PA) | UbiQ | Cat#UbiQ-076 |
| Ubiquitin-vinyl methyl ester (Ub-VME) | UbiQ | Cat#UbiQ-054 |
| Critical Commercial Assays | ||
| DUBprofiler™ | Ubiquigent | N/A |
| Deposited Data | ||
| Raw mass spectrometry data files for TMT 6-Plex activity-based protein profiling | Mass Spectrometry Interactive Virtual Environment (MassIVE) https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp | Dataset Identifier: MSV000087089 |
| Primary and dose-response confirmation screening data | PubChem | https://pubchem.ncbi.nlm.nih.gov/bioassay/1645869 |
| Experimental Models: Cell Lines | ||
| HEK293T cells | ATCC | Cat# CRL-3216; RRID:CVCL_0063 |
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| Recombinant DNA | ||
| pOPINK-OTUD3 (OTU+UBA, aa 52–275) | {Mevissen, 2013 #122} | Addgene plasmid# 61411 |
| pET28aLIC-USP7 (His-tagged, aa 207–532) | This paper | N/A |
| pET28aLIC-USP8 (His-tagged, aa 742–1110) | This paper | N/A |
| pET28PP-USP10 (His-tagged, aa 376–798) | This paper | N/A |
| pET28b-USP17 (His-tagged, aa 1–530) | This paper | N/A |
| pET28a USP25 (His-tagged, aa 207–532) | This paper | N/A |
| pET28aLIC-USP28 (His-tagged, aa 207–532) | This paper | N/A |
| pET28PP-USP30 (His-tagged, aa 207–532) | This paper | N/A |
| pGEX6P1-UCHL1 (GST-tagged, aa 1–223) | This paper | N/A |
| Software and Algorithms | ||
| Prism 8 | GraphPad | https://www.graphpad.com/scientific-software/prism/;RRID:SCR_002798 |
| Image Studio Lite | LiCOR | https://www.licor.com/bio/image-studio-lite/;RRID:SCR_013715 |
| Other | ||
| UpSetR code | {Conway, 2017 #26} | https://github.com/hms-dbmi/UpSetR |
| Multiplierz v2.0 | {Alexander, 2017 #74} | RRID:SCR_012058 |
| Mascot 2.6.1 | Matrix Science | RRID:SCR_014322 |
Significance.
Deubiquitinating enzymes (DUBs) are an integral part of the ubiquitin-proteasome system and have been reported to regulate many key biological pathways. DUBs represent an emerging target class that have been implicated in many different diseases. The lack of well validated and selective probe compounds for specific DUBs has presented an obstacle for full elucidation of their role in biological processes and diseases. In this study, we optimized and deployed a high-throughput screen of a 50,000 compound library against a panel of eight DUBs spanning three of the DUB families, with an emphasis on identifying selective hits for each DUB early on in the process. We demonstrate that this approach has led to the identification of selective inhibitors for a subset of the DUBs included in the screen and has generated a robust dataset that will be useful in DUB inhibitor development. Identification of these selective compounds can serve as the basis of medicinal chemistry campaigns geared towards the optimization of potency for the development of high-quality DUB inhibitors.
Highlights.
High throughput screen of ~50k compound library against 8 deubiquitinases
Identification and validation of selective inhibitors for individual DUBs
Study demonstrates importance of validation experiments as part of hit triage
Generation of robust dataset of compound activities against selected DUBs
Acknowledgements
Funding acknowledgments: S.J.B. and J.A.M. acknowledges generous support from the Mark Foundation ASPIRE program, NIH CA233800, and NIH CA247671.
Declaration of Interests
J.A.M. serves on the SAB of 908 Devices and receives sponsored research funding from Vertex and AstraZeneca. S.J.B. serves on the SAB of Adenoid Cystic Carcinoma Foundation. The authors have submitted patent applications related to compounds in this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- ABDUL REHMAN SA, KRISTARIYANTO YA, CHOI SY, NKOSI PJ, WEIDLICH S, LABIB K, HOFMANN K.& KULATHU Y.2016. MINDY-1 Is a Member of an Evolutionarily Conserved and Structurally Distinct New Family of Deubiquitinating Enzymes. Mol Cell, 63, 146–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALEXANDER WM, FICARRO SB, ADELMANT G.& MARTO JA 2017. multiplierz v2.0: A Python-based ecosystem for shared access and analysis of native mass spectrometry data. Proteomics, 17. [DOI] [PubMed] [Google Scholar]
- AMERIK AY & HOCHSTRASSER M.2004. Mechanism and function of deubiquitinating enzymes. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, 1695, 189–207. [DOI] [PubMed] [Google Scholar]
- ASKENAZI M, MARTO JA & LINIAL M.2010. The complete peptide dictionary--a meta-proteomics resource. Proteomics, 10, 4306–10. [DOI] [PubMed] [Google Scholar]
- BAELL JB & HOLLOWAY GA 2010. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. Journal of Medicinal Chemistry, 53, 2719–2740. [DOI] [PubMed] [Google Scholar]
- BINGOL B, TEA JS, PHU L, REICHELT M, BAKALARSKI CE, SONG Q, FOREMAN O, KIRKPATRICK DS & SHENG M.2014. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature, 510, 370–5. [DOI] [PubMed] [Google Scholar]
- BORODOVSKY A, OVAA H, KOLLI N, GAN-ERDENE T, WILKINSON KD, PLOEGH HL & KESSLER BM 2002. Chemistry-Based Functional Proteomics Reveals Novel Members of the Deubiquitinating Enzyme Family. Chemistry & Biology, 9, 1149–1159. [DOI] [PubMed] [Google Scholar]
- BOSELLI M, LEE B-H, ROBERT J, PRADO MA, MIN S-W, CHENG C, SILVA MC, SEONG C, ELSASSER S, HATLE KM, GAHMAN TC, GYGI SP, HAGGARTY SJ, GAN L, KING RW & FINLEY D.2017. An inhibitor of the proteasomal deubiquitinating enzyme USP14 induces tau elimination in cultured neurons. The Journal of Biological Chemistry, 292, 19209–19225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN J., JIAN X., DENG S., MA Z., SHOU X., SHEN Y., ZHANG Q., SONG Z., LI Z., PENG H., PENG C., CHEN M., LUO C., ZHAO D., YE Z., SHEN M., ZHANG Y., ZHOU J., FAHIRA A., WANG Y., LI S., ZHANG Z., YE H., LI Y., SHEN J., CHEN H., TANG F., YAO Z., SHI Z., CHEN C., XIE L., WANG Y., FU C., MAO Y., ZHOU L., GAO D., YAN H., ZHAO Y., HUANG C. & SHI Y. 2018. Identification of recurrent USP48 and BRAF mutations in Cushing’s disease. Nat Commun, 9, 3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CIECHANOVER A.2005. Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat Rev Mol Cell Biol, 6, 79–87. [DOI] [PubMed] [Google Scholar]
- CONWAY JR, LEX A.& GEHLENBORG N.2017. UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics, 33, 2938–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COTTO-RIOS XM, BÉKÉS M, CHAPMAN J, UEBERHEIDE B.& HUANG TT 2012. Deubiquitinases as a signaling target of oxidative stress. Cell reports, 2, 1475–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’ARCY P, WANG X.& LINDER S.2015. Deubiquitinase inhibition as a cancer therapeutic strategy. Pharmacology & Therapeutics, 147, 32–54. [DOI] [PubMed] [Google Scholar]
- DAINA A, MICHIELIN O.& ZOETE V.2017. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Scientific Reports, 7, 42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAS S, RAMAKRISHNA S.& KIM K-S 2020. Critical Roles of Deubiquitinating Enzymes in the Nervous System and Neurodegenerative Disorders. Molecules and Cells, 43, 203–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DING W-X, NI H-M, GAO W, CHEN X, KANG JH, STOLZ DB, LIU J.& YIN X-M 2009. Oncogenic transformation confers a selective susceptibility to the combined suppression of the proteasome and autophagy. Molecular cancer therapeutics, 8, 2036–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FARSHI P, DESHMUKH RR, NWANKWO JO, ARKWRIGHT RT, CVEK B, LIU J.& DOU QP 2015. Deubiquitinases (DUBs) and DUB inhibitors: a patent review. Expert Opinion on Therapeutic Patents, 25, 1191–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FICARRO SB, ZHANG Y, CARRASCO-ALFONSO MJ, GARG B, ADELMANT G, WEBBER JT, LUCKEY CJ & MARTO JA 2011. Online nanoflow multidimensional fractionation for high efficiency phosphopeptide analysis. Mol Cell Proteomics, 10, O111 011064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FLUGEL D, GORLACH A.& KIETZMANN T.2012. GSK-3beta regulates cell growth, migration, and angiogenesis via Fbw7 and USP28-dependent degradation of HIF-1alpha. Blood, 119, 1292–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FRAILE JM, QUESADA V, RODRÍGUEZ D, FREIJE JMP & LÓPEZ-OTÍN C.2012. Deubiquitinases in cancer: new functions and therapeutic options. Oncogene, 31, 2373–2388. [DOI] [PubMed] [Google Scholar]
- FU A, ALVAREZ-PEREZ JC, AVIZONIS D, KIN T, FICARRO SB, CHOI DW, KARAKOSE E, BADUR MG, EVANS L, ROSSELOT C, BRIDON G, BIRD GH, SEO HS, DHE-PAGANON S, KAMPHORST JJ, STEWART AF, JAMES SHAPIRO AM, MARTO JA, WALENSKY LD, JONES RG, GARCIA-OCANA A.& DANIAL NN 2020. Glucose-dependent partitioning of arginine to the urea cycle protects beta-cells from inflammation. Nat Metab, 2, 432–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAVORY G, O’DOWD CR, HELM MD, FLASZ J, ARKOUDIS E, DOSSANG A, HUGHES C, CASSIDY E, MCCLELLAND K, ODRZYWOL E, PAGE N, BARKER O, MIEL H.& HARRISON T.2018. Discovery and characterization of highly potent and selective allosteric USP7 inhibitors. Nat Chem Biol, 14, 118–125. [DOI] [PubMed] [Google Scholar]
- GERSCH M, WAGSTAFF JL, TOMS AV, GRAVES B, FREUND SMV & KOMANDER D.2019. Distinct USP25 and USP28 Oligomerization States Regulate Deubiquitinating Activity. Molecular Cell, 74, 436–451.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOPINATH P, OHAYON S, NAWATHA M.& BRIK A.2016. Chemical and semisynthetic approaches to study and target deubiquitinases. Chem Soc Rev, 45, 4171–98. [DOI] [PubMed] [Google Scholar]
- GUBLER H., CLARE N., GALAFASSI L., GEISSLER U., GIROD M. & HERR G. 2018. Helios: History and Anatomy of a Successful In-House Enterprise High-Throughput Screening and Profiling Data Analysis System. SLAS Discov, 23, 474–488. [DOI] [PubMed] [Google Scholar]
- HAAHR P, BORGERMANN N, GUO X, TYPAS D, ACHUTHANKUTTY D, HOFFMANN S, SHEARER R, SIXMA TK & MAILAND N.2018. ZUFSP Deubiquitylates K63-Linked Polyubiquitin Chains to Promote Genome Stability. Mol Cell, 70, 165–174 e6. [DOI] [PubMed] [Google Scholar]
- HARRIGAN JA, JACQ X, MARTIN NM & JACKSON SP 2018. Deubiquitylating enzymes and drug discovery: emerging opportunities. Nat Rev Drug Discov, 17, 57–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HEMELAAR J, GALARDY PJ, BORODOVSKY A, KESSLER BM, PLOEGH HL & OVAA H.2004. Chemistry-based functional proteomics: mechanism-based activity-profiling tools for ubiquitin and ubiquitin-like specific proteases. J Proteome Res, 3, 268–76. [DOI] [PubMed] [Google Scholar]
- HERMANNS T, PICHLO C, WOIWODE I, KLOPFFLEISCH K, WITTING KF, OVAA H, BAUMANN U.& HOFMANN K.2018. A family of unconventional deubiquitinases with modular chain specificity determinants. Nat Commun, 9, 799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HEWINGS DS, HEIDEKER J, MA TP, AHYOUNG AP, EL OUALID F, AMORE A, COSTAKES GT, KIRCHHOFER D, BRASHER B, PILLOW T, POPOVYCH N, MAURER T, SCHWERDTFEGER C, FORREST WF, YU K, FLYGARE J, BOGYO M.& WERTZ IE 2018. Reactive-site-centric chemoproteomics identifies a distinct class of deubiquitinase enzymes. Nat Commun, 9, 1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HJORTLAND NM & MESECAR AD 2016. Steady-state kinetic studies reveal that the anti-cancer target Ubiquitin-Specific Protease 17 (USP17) is a highly efficient deubiquitinating enzyme. Arch Biochem Biophys, 612, 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HU H.& SUN S-C 2016. Ubiquitin signaling in immune responses. Cell Research, 26, 457–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUGHES CS, FOEHR S, GARFIELD DA, FURLONG EE, STEINMETZ LM & KRIJGSVELD J.2014. Ultrasensitive proteome analysis using paramagnetic bead technology. Molecular Systems Biology, 10, 757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAPURIA V, PETERSON LF, FANG D, BORNMANN WG, TALPAZ M.& DONATO NJ 2010. Deubiquitinase inhibition by small-molecule WP1130 triggers aggresome formation and tumor cell apoptosis. Cancer Research, 70, 9265–9276. [DOI] [PubMed] [Google Scholar]
- KATEGAYA L, DI LELLO P, ROUGE L, PASTOR R, CLARK KR, DRUMMOND J, KLEINHEINZ T, LIN E, UPTON JP, PRAKASH S, HEIDEKER J, MCCLELAND M, RITORTO MS, ALESSI DR, TROST M, BAINBRIDGE TW, KWOK MCM, MA TP, STIFFLER Z, BRASHER B, TANG Y, JAISHANKAR P, HEARN BR, RENSLO AR, ARKIN MR, COHEN F, YU K, PEALE F, GNAD F, CHANG MT, KLIJN C, BLACKWOOD E, MARTIN SE, FORREST WF, ERNST JA, NDUBAKU C, WANG X, BERESINI MH, TSUI V, SCHWERDTFEGER C, BLAKE RA, MURRAY J, MAURER T.& WERTZ IE 2017. USP7 small-molecule inhibitors interfere with ubiquitin binding. Nature, 550, 534–538. [DOI] [PubMed] [Google Scholar]
- KERSCHER O, FELBERBAUM R.& HOCHSTRASSER M.2006. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu Rev Cell Dev Biol, 22, 159–80. [DOI] [PubMed] [Google Scholar]
- KLEIGER G.& MAYOR T.2014. Perilous journey: a tour of the ubiquitin-proteasome system. Trends Cell Biol, 24, 352–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KLUGE AF, LAGU BR, MAITI P, JALEEL M, WEBB M, MALHOTRA J, MALLAT A, SRINIVAS PA & THOMPSON JE 2018. Novel highly selective inhibitors of ubiquitin specific protease 30 (USP30) accelerate mitophagy. Bioorg Med Chem Lett, 28, 2655–2659. [DOI] [PubMed] [Google Scholar]
- KULATHU Y, GARCIA FJ, MEVISSEN TE, BUSCH M, ARNAUDO N, CARROLL KS, BARFORD D.& KOMANDER D.2013. Regulation of A20 and other OTU deubiquitinases by reversible oxidation. Nat Commun, 4, 1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KWASNA D, ABDUL REHMAN SA, NATARAJAN J, MATTHEWS S, MADDEN R, DE CESARE V, WEIDLICH S, VIRDEE S, AHEL I, GIBBS-SEYMOUR I.& KULATHU Y.2018. Discovery and Characterization of ZUFSP/ZUP1, a Distinct Deubiquitinase Class Important for Genome Stability. Mol Cell, 70, 150–164 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAMBERTO I., LIU X., SEO HS., SCHAUER NJ., IACOB RE., HU W., DAS D., MIKHAILOVA T., WEISBERG EL., ENGEN JR., ANDERSON KC., CHAUHAN D., DHE-PAGANON S. & BUHRLAGE SJ. 2017. Structure-Guided Development of a Potent and Selective Non-covalent Active-Site Inhibitor of USP7. Cell Chem Biol, 24, 1490–1500 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAWSON AP, BAK DW, SHANNON DA, LONG MJC, VIJAYKUMAR T, YU R, OUALID FE, WEERAPANA E.& HEDSTROM L.2017. Identification of deubiquitinase targets of isothiocyanates using SILAC-assisted quantitative mass spectrometry. Oncotarget, 8, 51296–51316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEE BH, LEE MJ, PARK S, OH DC, ELSASSER S, CHEN PC, GARTNER C, DIMOVA N, HANNA J, GYGI SP, WILSON SM, KING RW & FINLEY D.2010. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature, 467, 179–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEE JG, BAEK K, SOETANDYO N.& YE Y.2013. Reversible inactivation of deubiquitinases by reactive oxygen species in vitro and in cells. Nat Commun, 4, 1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEESON PD & SPRINGTHORPE B.2007. The influence of drug-like concepts on decision-making in medicinal chemistry. Nature Reviews Drug Discovery, 6, 881–890. [DOI] [PubMed] [Google Scholar]
- LEGER PR, HU DX, BIANNIC B, BUI M, HAN X, KARBARZ E, MAUNG J, OKANO A, OSIPOV M, SHIBUYA GM, YOUNG K, HIGGS C, ABRAHAM B, BRADFORD D, CHO C, COLAS C, JACOBSON S, OHOL YM, POOKOT D, RANA P, SANCHEZ J, SHAH N, SUN M, WONG S, BROCKSTEDT DG, KASSNER PD, SCHWARZ JB & WUSTROW DJ 2020. Discovery of Potent, Selective, and Orally Bioavailable Inhibitors of USP7 with In Vivo Antitumor Activity. J Med Chem, 63, 5398–5420. [DOI] [PubMed] [Google Scholar]
- LEROY E, BOYER R, AUBURGER G, LEUBE B, ULM G, MEZEY E, HARTA G, BROWNSTEIN MJ, JONNALAGADA S, CHERNOVA T, DEHEJIA A, LAVEDAN C, GASSER T, STEINBACH PJ, WILKINSON KD & POLYMEROPOULOS MH 1998. The ubiquitin pathway in Parkinson’s disease. Nature, 395, 451–452. [DOI] [PubMed] [Google Scholar]
- LIM K-H, JOO J-Y & BAEK K-H 2020. The potential roles of deubiquitinating enzymes in brain diseases. Ageing Research Reviews, 61, 101088. [DOI] [PubMed] [Google Scholar]
- LIPINSKI CA, LOMBARDO F, DOMINY BW & FEENEY PJ 2001. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev, 46, 3–26. [DOI] [PubMed] [Google Scholar]
- LIU B, SUREDA-GOMEZ M, ZHEN Y, AMADOR V.& REVERTER D.2018. A quaternary tetramer assembly inhibits the deubiquitinating activity of USP25. Nat Commun, 9, 4973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOR LA, SCHNECK J, MCNULTY DE, DIAZ E, BRANDT M, THRALL SH & SCHWARTZ B.2007. A Simple Assay for Detection of Small-Molecule Redox Activity. Journal of Biomolecular Screening, 12, 881–890. [DOI] [PubMed] [Google Scholar]
- MATYSKIELA ME, LU G, ITO T, PAGARIGAN B, LU C-C, MILLER K, FANG W, WANG N-Y, NGUYEN D, HOUSTON J, CARMEL G, TRAN T, RILEY M, NOSAKA LA, LANDER GC, GAIDAROVA S, XU S, RUCHELMAN AL, HANDA H, CARMICHAEL J, DANIEL TO, CATHERS BE, LOPEZ-GIRONA A.& CHAMBERLAIN PP 2016. A novel cereblon modulator recruits GSPT1 to the CRL4 CRBN ubiquitin ligase. Nature, 535, 252–257. [DOI] [PubMed] [Google Scholar]
- MEIER F, BECK S, GRASSL N, LUBECK M, PARK MA, RAETHER O.& MANN M.2015. Parallel Accumulation-Serial Fragmentation (PASEF): Multiplying Sequencing Speed and Sensitivity by Synchronized Scans in a Trapped Ion Mobility Device. J Proteome Res, 14, 5378–87. [DOI] [PubMed] [Google Scholar]
- MEIER F, BRUNNER AD, KOCH S, KOCH H, LUBECK M, KRAUSE M, GOEDECKE N, DECKER J, KOSINSKI T, PARK MA, BACHE N, HOERNING O, COX J, RATHER O.& MANN M.2018. Online Parallel Accumulation-Serial Fragmentation (PASEF) with a Novel Trapped Ion Mobility Mass Spectrometer. Mol Cell Proteomics, 17, 2534–2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MEVISSEN TE., HOSPENTHAL MK., GEURINK PP., ELLIOTT PR., AKUTSU M., ARNAUDO N., EKKEBUS R., KULATHU Y., WAUER T., EL OUALID F., FREUND SM., OVAA H. & KOMANDER D. 2013. OTU deubiquitinases reveal mechanisms of linkage specificity and enable ubiquitin chain restriction analysis. Cell, 154, 169–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MEVISSEN TET & KOMANDER D.2017. Mechanisms of Deubiquitinase Specificity and Regulation. Annu Rev Biochem, 86, 159–192. [DOI] [PubMed] [Google Scholar]
- NAVIA MA & CHATURVEDI PR 1996. Design principles for orally bioavailable drugs. Drug Discovery Today, 1, 179–189. [Google Scholar]
- NDUBAKU C.& TSUI V.2015. Inhibiting the deubiquitinating enzymes (DUBs). J Med Chem, 58, 158–195. [DOI] [PubMed] [Google Scholar]
- OGATA K.& ISHIHAMA Y.2020. Extending the Separation Space with Trapped Ion Mobility Spectrometry Improves the Accuracy of Isobaric Tag-Based Quantitation in Proteomic LC/MS/MS. Anal Chem, 92, 8037–8040. [DOI] [PubMed] [Google Scholar]
- OLIVEIRA AM, HSI BL, WEREMOWICZ S, ROSENBERG AE, DAL CIN P, JOSEPH N, BRIDGE JA, PEREZ-ATAYDE AR & FLETCHER JA 2004. USP6 (Tre2) fusion oncogenes in aneurysmal bone cyst. Cancer Res, 64, 1920–3. [DOI] [PubMed] [Google Scholar]
- PETZOLD G, FISCHER ES & THOMÄ NH 2016. Structural basis of lenalidomide-induced CK1α degradation by the CRL4(CRBN) ubiquitin ligase. Nature, 532, 127–130. [DOI] [PubMed] [Google Scholar]
- PINTO-FERNANDEZ A, DAVIS S, SCHOFIELD AB, SCOTT HC, ZHANG P, SALAH E, MATHEA S, CHARLES PD, DAMIANOU A, BOND G, FISCHER R.& KESSLER BM 2019. Comprehensive Landscape of Active Deubiquitinating Enzymes Profiled by Advanced Chemoproteomics. Front Chem, 7, 592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PINTO-FERNANDEZ A.& KESSLER BM 2016. DUBbing Cancer: Deubiquitylating Enzymes Involved in Epigenetics, DNA Damage and the Cell Cycle As Therapeutic Targets. Frontiers in Genetics, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POPOV N, WANZEL M, MADIREDJO M, ZHANG D, BEIJERSBERGEN R, BERNARDS R, MOLL R, ELLEDGE SJ & EILERS M.2007. The ubiquitin-specific protease USP28 is required for MYC stability. Nat Cell Biol, 9, 765–74. [DOI] [PubMed] [Google Scholar]
- PRIETO-GARCIA C, HARTMANN O, REISSLAND M, BRAUN F, FISCHER T, WALZ S, SCHULEIN-VOLK C, EILERS U, ADE CP, CALZADO MA, ORIAN A, MARIC HM, MUNCH C, ROSENFELDT M, EILERS M.& DIEFENBACHER ME 2020. Maintaining protein stability of Np63 via USP28 is required by squamous cancer cells. EMBO Mol Med, 12, e11101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAMAKRISHNA S, SURESH B.& BAEK K-H 2011. The role of deubiquitinating enzymes in apoptosis. Cellular and molecular life sciences: CMLS, 68, 15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REINCKE M, SBIERA S, HAYAKAWA A, THEODOROPOULOU M, OSSWALD A, BEUSCHLEIN F, MEITINGER T, MIZUNO-YAMASAKI E, KAWAGUCHI K, SAEKI Y, TANAKA K, WIELAND T, GRAF E, SAEGER W, RONCHI CL, ALLOLIO B, BUCHFELDER M, STROM TM, FASSNACHT M.& KOMADA M.2015. Mutations in the deubiquitinase gene USP8 cause Cushing’s disease. Nat Genet, 47, 31–8. [DOI] [PubMed] [Google Scholar]
- RICHARDSON PG, HIDESHIMA T.& ANDERSON KC 2003. Bortezomib (PS-341): a novel, first-in-class proteasome inhibitor for the treatment of multiple myeloma and other cancers. Cancer Control, 10, 361–9. [DOI] [PubMed] [Google Scholar]
- RITORTO MS, EWAN R, PEREZ-OLIVA AB, KNEBEL A, BUHRLAGE SJ, WIGHTMAN M, KELLY SM, WOOD NT, VIRDEE S, GRAY NS, MORRICE NA, ALESSI DR & TROST M.2014. Screening of DUB activity and specificity by MALDI-TOF mass spectrometry. Nat Commun, 5, 4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAKAMOTO KM, KIM KB, KUMAGAI A, MERCURIO F, CREWS CM & DESHAIES RJ 2001. Protacs: Chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation. Proceedings of the National Academy of Sciences of the United States of America, 98, 8554–8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAUER F, KLEMM T, KOLLAMPALLY RB, TESSMER I, NAIR RK, POPOV N.& KISKER C.2019. Differential Oligomerization of the Deubiquitinases USP25 and USP28 Regulates Their Activities. Molecular Cell, 74, 421–435.e10. [DOI] [PubMed] [Google Scholar]
- SCHAUER NJ., LIU X., MAGIN RS., DOHERTY LM., CHAN WC., FICARRO SB., HU W., ROBERTS RM., IACOB RE., STOLTE B., GIACOMELLI AO., PERERA S., MCKAY K., BOSWELL SA., WEISBERG EL., RAY A., CHAUHAN D., DHE-PAGANON S., ANDERSON KC., GRIFFIN JD., LI J., HAHN WC., SORGER PK., ENGEN JR., STEGMAIER K., MARTO JA. & BUHRLAGE SJ. 2020a. Selective USP7 inhibition elicits cancer cell killing through a p53-dependent mechanism. Sci Rep, 10, 5324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHAUER NJ, MAGIN RS, LIU X, DOHERTY LM & BUHRLAGE SJ 2020b. Advances in Discovering Deubiquitinating Enzyme (DUB) Inhibitors. J Med Chem, 63, 2731–2750. [DOI] [PubMed] [Google Scholar]
- SCHLIERF A, ALTMANN E, QUANCARD J, JEFFERSON AB, ASSENBERG R, RENATUS M, JONES M, HASSIEPEN U, SCHAEFER M, KIFFE M, WEISS A, WIESMANN C, SEDRANI R, EDER J.& MARTOGLIO B.2016. Targeted inhibition of the COP9 signalosome for treatment of cancer. Nat Commun, 7, 13166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SENFT D, QI J.& RONAI ZEA 2018. Ubiquitin ligases in oncogenic transformation and cancer therapy. Nature reviews. Cancer, 18, 69–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STOLTE B, INIGUEZ AB, DHARIA NV, ROBICHAUD AL, CONWAY AS, MORGAN AM, ALEXE G, SCHAUER NJ, LIU X, BIRD GH, TSHERNIAK A, VAZQUEZ F, BUHRLAGE SJ, WALENSKY LD & STEGMAIER K.2018. Genome-scale CRISPR-Cas9 screen identifies druggable dependencies in TP53 wild-type Ewing sarcoma. J Exp Med, 215, 2137–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TURCU FER, VENTII KH & WILKINSON KD 2009. Regulation and Cellular Roles of Ubiquitin-specific Deubiquitinating Enzymes. Annual review of biochemistry, 78, 363–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TURNBULL AP, IOANNIDIS S, KRAJEWSKI WW, PINTO-FERNANDEZ A, HERIDE C, MARTIN ACL, TONKIN LM, TOWNSEND EC, BUKER SM, LANCIA DR, CARAVELLA JA, TOMS AV, CHARLTON TM, LAHDENRANTA J, WILKER E, FOLLOWS BC, EVANS NJ, STEAD L, ALLI C, ZARAYSKIY VV, TALBOT AC, BUCKMELTER AJ, WANG M, MCKINNON CL, SAAB F, MCGOURAN JF, CENTURY H, GERSCH M, PITTMAN MS, MARSHALL CG, RAYNHAM TM, SIMCOX M, STEWART LMD, MCLOUGHLIN SB, ESCOBEDO JA, BAIR KW, DINSMORE CJ, HAMMONDS TR, KIM S, URBE S, CLAGUE MJ, KESSLER BM & KOMANDER D.2017. Molecular basis of USP7 inhibition by selective small-molecule inhibitors. Nature, 550, 481–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VALERO R, BAYÉS M, FRANCISCA SÁNCHEZ-FONT M, GONZÁLEZ-ANGULO O, GONZÀLEZ-DUARTE R.& MARFANY G.2001. Characterization of alternatively spliced products and tissue-specific isoforms of USP28 and USP25. Genome Biology, 2, research0043.1-research0043.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG H, MENG Q, DING Y, XIONG M, ZHU M, YANG Y, SU H, GU L, XU Y, SHI L, ZHOU H.& ZHANG N.2021. USP28 and USP25 are downregulated by Vismodegib in vitro and in colorectal cancer cell lines. FEBS J, 288, 1325–1342. [DOI] [PubMed] [Google Scholar]
- WARING MJ 2010. Lipophilicity in drug discovery. Expert Opinion on Drug Discovery, 5, 235–248. [DOI] [PubMed] [Google Scholar]
- WERTZ IE, O’ROURKE KM, ZHOU H, EBY M, ARAVIND L, SESHAGIRI S, WU P, WIESMANN C, BAKER R, BOONE DL, MA A, KOONIN EV & DIXIT VM 2004. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature, 430, 694–699. [DOI] [PubMed] [Google Scholar]
- WINTER GE, BUCKLEY DL, PAULK J, ROBERTS JM, SOUZA A, DHE-PAGANON S.& BRADNER JE 2015. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science, 348, 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WRIGLEY JD., GAVORY G., SIMPSON I., PRESTON M., PLANT H., BRADLEY J., GOEPPERT AU., ROZYCKA E., DAVIES G., WALSH J., VALENTINE A., MCCLELLAND K., ODRZYWOL KE., RENSHAW J., BOROS J., TART J., LEACH L., NOWAK T., WARD RA., HARRISON T. & ANDREWS DM. 2017. Identification and Characterization of Dual Inhibitors of the USP25/28 Deubiquitinating Enzyme Subfamily. ACS Chem Biol, 12, 3113–3125. [DOI] [PubMed] [Google Scholar]
- ZHOU F, LU Y, FICARRO SB, ADELMANT G, JIANG W, LUCKEY CJ & MARTO JA 2013. Genome-scale proteome quantification by DEEP SEQ mass spectrometry. Nat Commun, 4, 2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHOU F, SIKORSKI TW, FICARRO SB, WEBBER JT & MARTO JA 2011. Online nanoflow reversed phase-strong anion exchange-reversed phase liquid chromatography-tandem mass spectrometry platform for efficient and in-depth proteome sequence analysis of complex organisms. Anal Chem, 83, 6996–7005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZINNGREBE J, MONTINARO A, PELTZER N.& WALCZAK H.2014. Ubiquitin in the immune system. EMBO Reports, 15, 28–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The primary and dose-response screening datasets generated during this study are available at PubChem (PubChem AID: 1645869). https://pubchem.ncbi.nlm.nih.gov/bioassay/1645869
Raw mass spectrometry data files for TMT 6-Plex activity-based protein profiling have been deposited in MassIVE (Mass spectrometry Interactive Virtual Environment, https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp). Dataset Identifier: MSV000087089