

Abstract
Structural isomers of sialylated N-glycans contribute to the diversity of the N-glycome and to a range of biological functions. Sialyl linkage isomers can be readily distinguished by mass spectrometry with mass differences between α2,3- and α2,6-linkages generated by a two-step sialic acid linkage-specific alkylamidation. To improve identification of N-glycans from complex mixtures, we added a delactonization step after the first alkylamidation step, which regenerates negatively charged carboxylic acids on α2,3-sialic acids. N-glycan isomers with α2,3-sialic acids are then fractionated by ion-exchange chromatography prior to the second alkylamidation step. With this modified alkylamidation method, sialylated N-glycans were enriched and stabilized for structural characterization by capillary electrophoresis-mass spectrometry and tandem mass spectrometry. We identified 52 sialylated N-glycan structures, including 107 linkage isomers, in human serum and confirmed the presence of positional isomers of specific sialyl linkage isomers. Due to the reduced sample complexity after the ion-exchange fractionation and CE separation, substructural features of N-glycans were rapidly evaluated in human serum and included diagnostic ions corresponding to core- and antenna-fucosylation and poly-lactosamine.
Keywords: capillary electrophoresis, N-glycans, ion exchange, mass spectrometry, serum
1. Introduction
Protein glycosylation plays major roles in protein folding and function, signaling, and cell-to-cell recognition [1–2] and their consequences for human health and disease. Within various complexities of the human glycoproteome, different asparagine-linked oligosaccharides (N-glycans) are often recognized as biological determinants of differences between health and disease. With rapidly maturing analytical methodologies and approaches to understand the human glycoproteome [3–6], there are now numerous studies documented in the literature to correlate complex N-glycomic profiles with diseases and their progression, particularly in the search for cancer biomarkers [7–14] through mass spectrometry (MS) and related techniques. While a number of these studies implicate sialylated structures as pathologically significant, information on the extent of sialylation isomerism in these sugar residues is still scarce.
From the routinely profiled and reported range of biantennary to tetra-antennary N-glycans, various structures can be differentially sialylated, giving significant rise to N-glycomic structural and potentially functional diversity. Sialic acid residues are commonly linked to galactose residues via α2,3- or α2,6-linkages and rarely as disialic (α2,8) chain extensions. This structural heterogeneity of N-glycans increases the difficulty of accurate biomarker assessment, making further development of new analytical methods to differentiate various isomeric combinations and microscale isolation of specific types of isomers both popular and necessary.
Due to their labile nature, sialic acid residues can be derivatized and stabilized prior to analysis. A few derivatization strategies that can convert sialyl linkage isomers into different derivatives have been developed [9,15–19]. These linkage-specific derivatizations typically involve lactonization of α2,3-sialic acids and either esterification or amidation of α2,6-sialic acids [9,16–19]. Consequently, a mass difference between α2,3- and α2,6-sialic acids can be generated and used for the differentiation of the two sialyl linkages by MS. As an example, Borelli et al. [16] employed lactonization/ethyl esterification in a study of the plasma N-glycome of patients suffering from Down Syndrome, and they confirmed a decrease in ten α2,3-sialylated structures and increase in two α2,6-sialylated structures. Holst and co-workers [17] used a similar lactonization/ethyl esterification procedure with subsequent matrix-assisted laser desorption/ionization (MALDI) MS imaging to characterize N-glycan expression in heterogeneous tissues. Li et al. [18] developed a two-step derivatization to methylamidate α2,3-sialic acids and ethyl-esterificate α2,6-sialic acids. While successfully differentiating N-glycomic profiles of pancreatic cancer patients from those of healthy controls, Nishikaze et al. [19] systematically evaluated a variety of linkage-specific derivatization procedures and demonstrated high-conversion specificity and efficiency with sialic acid linkage-specific alkylamidation (SALSA), in which α2,3- and α2,6-sialic acids were converted into different alkylamides that were more stable than lactones and ethyl esters.
Most of the current efforts employed MALDI-MS, which provides simultaneous analyses of N-glycans, but often lacks complete discrimination of their positional isomers [20–24]. In contrast, conventional and microfluidic liquid chromatography (LC) and capillary electrophoresis (CE) can readily resolve N-glycan structural isomers [25–28]. Although microfluidic CE provides rapid analysis of N-glycans with high separation efficiency, the lack of compositional information of the analytes and co-migrating components with overlapping electrophoretic mobilities result in ambiguous assignments. Conventional CE is readily interfaced with mass spectrometry through electrospray ionization (ESI) [29–32], bringing together isomeric separation and unambiguous compositional MS assignment of the N-glycans [33–34]. Tandem MS fragmentation for detailed structural elucidation is also feasible through coupling CE with MS [35–36].
Although linkage-specific derivatization can convert sialyl linkage isomers into products with different masses, the sample complexity increases after derivatization. Derivatives from different precursor structures can exhibit comparable masses, complicating the data analysis, especially for complex biological samples. Moreover, signal intensities for highly sialylated structures are lower after derivative formation, because individual structures with distinct sialyl linkage combinations from a single precursor structure are converted into multiple forms. One method to improve the confidence in structural assignment is to fractionate a preconcentrated sample into groups. Sialylated N-glycan structures can be fractionated through ion-exchange chromatography [37] based on the number of anionic groups (e.g., number of sialic acids) to reduce sample complexity of the individual fractions and determine a relative abundance for each component.
Herein, we describe a sample derivatization strategy used to fractionate and preconcentrate sialylated N-glycans in a linkage-specific manner. Our sample preparation procedure has been modified from the reported SALSA derivatization [19]. As outlined in Scheme 1, the sialic acids with both α2,3- and α2,6-linkages are converted to lactones and isopropylamides, respectively, during the first alkylamidation step. Subsequently, carboxyl groups of α2,3-sialic acids are recovered from their labile lactones under basic conditions and used for the fractionation of the N-glycans through ion-exchange chromatography based on the number of regenerated carboxyl groups. After fractionation, the carboxyl groups of α2,3-sialic acids are then neutralized and stabilized in the second alkylamidation step to form stable methylamides. Our CE-MS analysis detected 52 sialylated N-glycan structures, including 107 linkage isomers, with numerous positional isomers separated from each other. The combination of microscale derivatization, fractionation, and enrichment of glycans with specific sialyl linkages reduced sample complexity and aided in identification and further structural characterization through CE-MS/MS analysis. Overall, this approach enabled evaluation of substructural features of N-glycans (e.g., fucosylation and poly-lactosamine (LacNAc)) in a biological sample as complex as human serum.
Scheme 1.

Schematic diagram of derivatization and fractionation of sialylated N-glycans with a modified SALSA reaction and ion-exchange fractionation. The first alkylamidation reaction converts α2,3- and α2,6-sialic acids into lactones and isopropylamides, respectively. A mild basic solution (pH 10.2) composed of 50 mM Na2CO3/NaHCO3 is added to the lactonized α2,3-sialic acids and isopropylamidated α2,6-sialic acids to hydrolyze the lactones and convert α2,3-sialic acids to their native form, while leaving isopropylamidated α2,6-sialic acids unchanged. Subsequently, the mixture is separated by ion-exchange and fractionated by the number of recovered α2,3-sialic acid residues. After fractionation and a subsequent cleanup, each fraction is treated with the second alkylamidation, i.e., methylamidation reaction, to neutralize and stabilize the remaining α2,3-sialic acids.
2. Materials and Methods
2.1. Materials.
The following materials and chemicals were used: acrylamide and sodium dodecyl sulfate (SDS) from Bio-Rad Laboratories, Inc. (Hercules, CA); ammonium hydroxide from J.T. Baker (Phillipsburg, NJ); high-purity 8-aminopyrene-1,3,6-trisulfonic acid (APTS) from Beckman Coulter, Inc. (Indianapolis, IN); HPLC grade water, sodium acetate anhydrous, sodium carbonate monohydrate, and trifluoroacetic acid (TFA) from EMD Chemicals, Inc. (Gibbstown, NJ); sodium hydroxide from Fisher Scientific Co. (Indianapolis, IN); peptide-N-glycosidase F (PNGase F) of Chryseobacterium menigosepticum (EC 3.2.2.18) from Prozyme, Inc. (Santa Clara, CA); Nonidet P-40 from Roche Diagnostics (Indianapolis, IN); active charcoal and amino (NH2) micro-spin columns and 1000 Da cutoff cellulose dialysis tubes from Harvard Apparatus (Holliston, MA); ammonium acetate, hydrogen peroxide, hydrochloric acid, and isopropyl alcohol (IPA) from Macron Fine Chemicals, Inc. (Radnor, PA); isopropylamine hydrochloride (iPA-HCl) from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan); 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC-HCl) from Thermo Fisher Scientific, Inc. (Waltham, MA); HPLC grade acetonitrile from Mallinckrodt Baker, Inc. (Phillipsburg, NJ); Microposit MF-319 developer from MicroChem Corp. (Westborough, MA); chromium etchants 8002-A and 1020 and buffered oxide etchant from Transene Co., Inc. (Danvers, MA); 353NDT epoxy from Epoxy Technology, Inc. (Billerica, MA); B270 mask blanks and cover plates from Telic Co. (Valencia, CA), and pooled female human serum samples from BioreclamationIVT (Westbury, NY). All other chemicals were purchased from Sigma-Aldrich Co. (St. Louis, MO).
2.2. Preparation of N-Glycan Samples.
N-Glycans were enzymatically cleaved from human serum (5–10 μL) with PNGase F and purified with solid-phase extraction [12]. Briefly, aliquots of serum were suspended in 10 μL phosphate buffer (pH 7.5) with 0.2% β-mercaptoethanol and 0.1% SDS and thermally denatured at 60 °C for 60 min. The solution was cooled to room temperature prior to the addition of a 1-μL aliquot of 10% Nonidet P-40 and allowed to equilibrate for 10 min at room temperature. A 1-μL aliquot of PNGase diluted 1:10 in phosphate buffer was subsequently added to the solution, and the reaction mixture was incubated at 37 °C overnight.
2.3. Sialic Acid Linkage-Specific Alkylamidation and Purification.
Native sialyllactose samples and N-glycans derived from human serum were neutralized in a linkage-specific manner through a two-step derivatization [19] with a few modifications. In the first alkylamidation, N-glycans were dissolved in 20-μL aliquots of DMSO containing 2 M isopropylamine hydrochloride (iPA-HCl), 0.5 M 1-hydroxybenzotriazole hydrate (HOBt) and 0.5 M 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC-HCl) and incubated at room temperature for 1 h. After the first alkylamidation reaction, the lactonized/isopropylamidated N-glycans were recovered with amino micro-spin columns. After loading the N-glycan samples that were diluted in 240 μL of 15:85 water/acetonitrile, the columns were washed twice with 200 μL of 15:85 water/acetonitrile. N-Glycans were then eluted in 200 μL of 80:20 water/acetonitrile. Prior to the second methylamidation step, some of the samples were delactonized and fractionated through an ion-exchange step (see next section). In the second alkylamidation step, aliquots of a 10-μL DMSO solution containing 1 M methylamine hydrochloride, 0.5 M 4-methylmorpholine, and 0.05 M 7-azabenzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyAOP) were added to the samples and incubated at room temperature for 1 h. After the second alkylamidation, the methylamidated/isopropylamidated N-glycans were again recovered with amino micro-spin columns.
2.4. Fractionation of Sialyl Linkage Isomers.
After the first alkylamidation reaction, some of N-glycan samples were fractionated through ion-exchange chromatography. Dried, lactonized/isopropylamidated N-glycans were incubated with 20 μL of 50 mM Na2CO3/NaHCO3 (pH 10.2) at room temperature (23 °C) for 2 h to hydrolyze the lactone and regenerate the carboxylic acid. After the incubation, the native/isopropylamidated N-glycans were fractionated with an Oasis Max (3 mL) column and eluted by gravity [37]. The glycans were diluted with 3 mL of 95% acetonitrile and passed through the column three times, followed by elution of uncharged N-glycans with 6–8 mL of 50% acetonitrile (fraction A). The charged glycans were eluted into four fractions with increasing concentrations of sodium acetate: 3 mL 10 mM sodium acetate (fraction B), 3 mL 20 mM sodium acetate (fraction C), 3 mL 50 mM sodium acetate (fraction D), and 3 mL 100 mM sodium acetate (fraction E). The fractions eluted with sodium acetate were desalted with active charcoal micro-spin columns [37], eluted in 200 μL of 50:50:0.1 water/acetonitrile/trifluoroacetic acid, and dried in a speedvac concentrator. All fractions were then derivatized with the second alkylamidation step, i.e., methylamidation, and recovered through amino micro-spin columns.
2.5. Glycan Labeling.
Derivatized N-glycans were labeled with 8-aminopyrene-1,3,6-trisulfonic acid (APTS) to impart a negative charge for electrophoresis and negative-ion MS and to serve as a fluorescent label for laser-induced fluorescence (LIF) detection with microfluidic CE [12,38–39]. N-Glycans prepared for positive-mode CE-MS were labeled with Girard’s reagent T (GT) to impart a positive charge for electrophoresis and positive-ion MS detection [40].
2.6. CE-MS Analysis.
A bare-fused silica capillary cartridge (30 μm i.d. × 90 cm length; Opti-MS, B07367, SCIEX Separations, Framingham, MA) and a neutral capillary cartridge (30 μm i.d. × 90 cm length; Opti-MS, B07368, SCIEX Separations) were conditioned according to manufacturer protocols before use in the CESI 8000 instrument (SCIEX Separations). Dried samples were reconstituted in 20 μL 1:19 10% acetic acid (BGE):water prior to analysis. Hydrostatic injections at 1 psi for 5–25 s introduced the sample into the capillaries before a separation voltage of 25 kV was applied. Constant pressures of 0.1 and 2 psi were applied to bare-fused silica and neutral separation capillaries, respectively, to generate stable fluid flow for electrospray at 20 °C. For CE-MS analysis in positive-ion mode with a delayed flow, supplemental pressure was not applied to the neutral capillary until 60 to 80 min after electrophoresis started. The capillaries were interfaced with the nanospray source of an LTQ Orbitrap mass spectrometer (Thermo Fisher Scientific, Inc., Waltham, MA) through an adapter kit. Electrospray was initiated by applying 1.05–1.1 kV for 10 s, then stepping the potential down to 0.97–1.02 kV (e.g., 80 V lower) for the remainder of the separation through the Xcalibur software (Version 2.2, Thermo Fisher Scientific, Inc.). Data were collected over a range of 100–2000 m/z at a maximum injection time of 125 ms with 15K resolution on the FTMS analyzer. Source-induced dissociation (SID) fragmentation spectra were collected with the FTMS analyzer with an in-source energy of 35–45 V. Spectra were manually evaluated to determine low abundance species with the mass tolerance of 50 ppm and analyzed with the Qual Brower of the Xcalibur software (Version 2.2, Thermo Fisher Scientific, Inc.). Extracted ion electropherograms (EIEs) for individual m/z values (± 0.05 Da) were boxcar-averaged (7 points), exported, and plotted with OriginPro 2018 (OriginLab Corp., Northhampton, MA).
2.7. Fabrication and Operation of Microfluidic Devices.
Microfluidic devices with a 22-cm serpentine channel and asymmetrically tapered, 180° turns were fabricated in glass substrates [12,41]. To minimize electroosmotic flow and sample adsorption, the microchannels were coated with linear poly(acrylamide). Modified pinched injections [38] were used to introduce samples into the analysis channel. Laser-induced fluorescence with excitation from the 454-nm line of an argon ion laser (Melles Griot, Inc., Carlsbad, CA) was used for detection of APTS-labeled N-glycans.
3. Results and Discussion
3.1. Microfluidic CE and CE-MS Analysis of Alkylamidated Serum N-Glycans.
Sialyl linkage isomers contribute greatly to the structural diversity of N-glycans in complex biological samples, such as human serum. Here, we employed a two-step sialic acid linkage-specific alkylamidation (SALSA) [19] to convert α2,3-sialic acids and α2,6-sialic acids to methylamides and isopropylamides, respectively, for the differentiation of sialyl linkage isomers of the N-glycans derived from serum. We evaluated the specificity of SALSA with glycan standards, 3’-sialyllactose (3’-SL) and 6’-sialyllactose (6’-SL) with microfluidic CE (Figure S1a in Supporting Information). Because the charge on both α2,3- and α2,6-linked sialic acids was neutralized, the migration order of glycans depended solely on their hydrodynamic volumes during electrophoresis. In addition, the 28.03 Da mass difference generated between α2,3- and α2,6-sialic acids identified the type of sialyl linkage isomers by MS. An extremely low mis-conversion rate was observed when analyzing the sialic acid derivatives through CE-MS (Figure S1b–c in Supporting Information), demonstrating the high specificity of this derivatization.
After evaluating the specificity of SALSA, the derivatization strategy was applied to the N-glycans derived from human serum. Shifts in migration time for the structures containing α2,6-sialic acids in the electropherograms were observed when the profile of the structures derivatized with SALSA was compared to the structures derivatized through methylamidation (Figure S2 in Supporting Information). Consequently, the migration shifts aided in assigning several α2,6-sialylated structures (Figure 1a). Although microfluidic CE provided fast analysis of a complex set of N-glycans with isomeric resolution, the lack of direct compositional/mass information of N-glycans, as well as the presence of potentially co-migrating species, limited the ability to make definitive structural assignments.
Figure 1.

(a) An electropherogram of SALSA-derivatized, APTS-labeled N-glycans that were derived from human serum and analyzed by microfluidic CE. N-Glycan structures were identified with electrophoretic mobility standards and linkage-specific derivatization with molecular weight determined by mass spectrometry. (b)-(c) Extracted ion electropherograms (EIEs) of SALSA-derivatized N-glycans derived from human serum and analyzed by CE-MS. In panel (b), the N-glycans were labeled with 8-aminopyrene-1,3,6-trisulfonic acid (APTS) and detected in negative-ion mode, and in panel (c) the N-glycans were labeled with Girard’s reagent T (GT) and detected in positive-ion mode.
CE-MS is capable of separating N-glycan structural isomers and collecting mass information of the N-glycans [33–34]. Negative-ion CE-MS is compatible with the APTS-labeled N-glycans, which were previously analyzed by microfluidic CE, allowing direct comparison of electropherograms generated by microfluidic CE and CE-MS [33]. With a bare-fused silica capillary and acidic background electrolyte (BGE, 10% acetic acid, pH 2.3), the electroosmotic flow (EOF) generated at a separation potential of 25 kV in normal polarity was used as the main source of the fluid flow, which was essential for a stable electrospray. Negatively charged analytes migrated against the EOF, effectively increasing the separation length and, thus, facilitating isomeric separation of the N-glycan structures (Figure 1b). By combining CE-MS and SALSA, low-abundance structures that had previously been masked by high-abundance structures with similar electrophoretic mobilities were detected, while sialyl linkage isomers were differentiated from one another based on their unique mass fingerprints.
To detect the derivatives of large N-glycan structures present in low abundance, we used the commercially available SCIEX neutral capillary cartridge, which is sequentially coated with a hydrophobic coating and hydrophilic poly(acrylamide) coating, to minimize sample adsorption and electroosmotic flow. However, the separation efficiencies of negatively charged APTS-labeled N-glycans on the neutral capillary cartridge were low, and severely distorted peaks resulted (data not shown). The reason for the poor separation performance is not clear, but may be due to sample interactions with the binary coating on the capillary surface or lack of stability of the electrospray with the cartridge. Typically, electrophoretic separations of APTS-labeled N-glycans on poly(acrylamide)-coated microchannels and capillaries exhibit extremely high separations efficiencies (see Figure 1a).
Consequently, we labeled the N-glycans with Girard’s reagent T (GT) to impart a +1 charge for electrophoresis and positive-ion MS detection. To enhance separative performance, supplemental pressure, which is needed for stable electrospray, was not applied to the separation capillary during the first 80 min of the separation. As a result, many positively charged, derivatized N-glycan isomers were separated (Figure S3 in Supporting Information). Moreover, electrospray generated with positive-ion MS showed better stability than with negative-ion MS and, thus, led to higher signal intensities, allowing the detection of N-glycans as large as a tetra-antennary, tetra-sialylated structure with multiple LacNAc repeat units (Figure 1c). In total, 63 N-glycan structures, including 48 sialylated structures with 92 linkage isomers, were detected by CE-MS in positive-ion mode (Figure S4–1 in Supporting Information).
3.2. Fractionation of Sialyl Linkage Isomers of Sialylated N-Glycans.
Many highly sialylated structures co-migrated during microfluidic CE, which made characterization of these larger N-glycan structures challenging. Moreover, some structures with different compositions exhibited similar electrophoretic mobilities as well as similar m/z values in the CE-MS analysis. For example, a tri-antennary structure, A3G3S3 (A: N-acetylglucosamine, G: galactose, S: sialic acid), which contained three α2,6-sialic acids, and a tetra-antennary structure, A4G4S2, which contained one α2,3-sialic acid and one α2,6-sialic acid, had overlapping isotopic clusters and electrophoretic migration after derivatization by SALSA (Figure S5a in Supporting Information).
To deconvolute the data and avoid false assignment, we modified the SALSA derivatization [18–19] by adding ion-exchange fractionation as an intermediate step to collect various sialyl linkage isomers into different fractions (see Scheme 1). First, native, underivatized N-glycans had their α2,3- and α2,6-sialic acids converted into lactones and isopropylamides, respectively. After the first alkylamidation step and prior to the delactonization step, the phosphorylated N-glycans were removed by passing the sample through an amino spin column. Then, prior to the second alkylamidation step, a mild basic solution (pH 10.2) composed of 50 mM Na2CO3/NaHCO3 was added to the samples to hydrolyze the lactones and convert α2,3-sialic acids into their native form, while leaving isopropylamidated α2,6-sialic acids unchanged. Subsequently, the samples containing native α2,3-sialic acids and isopropylamidated α2,6-sialic acids were fractionated through ion-exchange chromatography [37], which led to pre-separation of sialyl linkage isomers based on the number of recovered α2,3-sialic acid residues. After fractionation and subsequent cleanup, each fraction was treated with the methylamidation reagent to neutralize and stabilize the remaining α2,3-sialic acids. The fractions were then labeled with APTS or GT for microfluidic CE and CE-MS analyses.
Figure 2 compares microfluidic electropherograms of unfractionated N-glycans to fractions derivatized with the modified SALSA reaction. The majority of the N-glycans derived from serum were eluted in fraction A, indicating that most of the N-glycan structures were neutral or only contained α2,6-sialic acids, which agreed with our previous assignments. Two intense peaks at 142–145 s, which were absent in fraction A, were now prominent in fraction B. These two peaks correspond to tri-antennary, tri-sialylated structures containing one α2,3-sialic acid and two α2,6-sialic acids and its fucosylated counterpart. Similarly, the peak at 127 s, which was absent in fractions A and B, was in relatively high abundance in fraction C and corresponded to a bi-antennary, di-sialylated, fucosylated structure containing two α2,3-sialic acids. Sialyl linkage isomers of tetra-antennary, tetra-sialylated structures were observed in fractions D and E.
Figure 2.
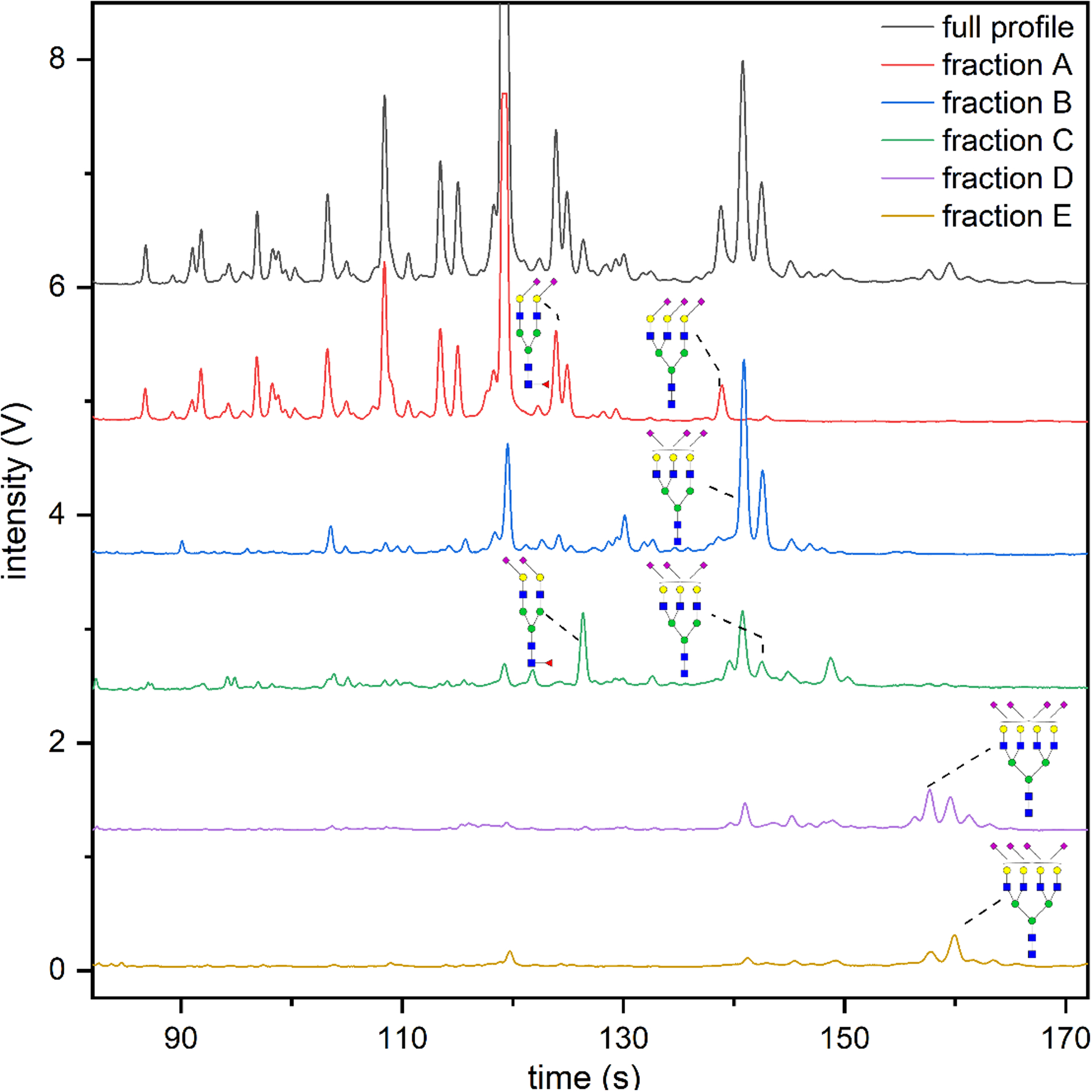
Electropherograms of N-glycans derived from human serum, derivatized by SALSA, labeled with APTS, and analyzed by microfluidic CE. N-Glycans were fractionated by ion exchange and separated by the number of remaining α2,3-sialic acids after hydrolysis of the lactone. Ion-exchange fractions A, B, C, D, and E contained N-glycan structures in which the majority had 0, 1, 2, 2, and 3 α2,3-sialic acids, respectively. Selected structures with the same compositions, but different sialyl linkages are assigned to the corresponding peaks.
3.3. Efficiency of Lactone Hydrolysis for α2,3-Sialic Acids.
To avoid false assignments, we evaluated the effectiveness of the delactonization step with CE-MS. If hydrolysis of the lactone was incomplete, fraction A would contain structures associated with one or more α2,3-sialic acids, which would subsequently be converted from the lactones to methylamides later in the sample preparation process and detected by CE-MS. The structural conformation of A3G3S3 that contained one α2,3-sialic acid and two α2,6-sialic acids at m/z 1030.10 was selected to evaluate efficiency of the hydrolysis step, because this structural conformation was the most abundant among all the α2,3-sialylated N-glycans. For N-glycans derived from 5 μL serum, ions at m/z 1030.10 were not detected in fraction A, implying near-complete hydrolysis of lactonized α2,3-sialic acids. For N-glycans derived from 10 μL serum, a low ion intensity at m/z 1030.10 was detected in fraction A, constituting 2.8% of the total amount of this isomer eluted in fractions A and B combined. Therefore, we used N-glycans derived from 5 μL of serum for the rest of the experiments to ensure the accurate structural assignments.
3.4. Enrichment of Sialyl Linkage Isomers of Sialylated N-Glycans in Individual Fractions.
Each ion-exchange fraction was analyzed by CE-MS to determine N-glycan compositions and evaluate fractionation efficiency. Generally, fraction A contained neutral and sialylated structures with just isopropylamidated α2,6-sialic acids. Fraction B contained sialylated structures with one α2,3-sialic acid. Fraction C had primarily structures with two α2,3-sialic acids and a small fraction of structures with one α2,3-sialic acid. Fractions D and E had a mixture of structures with two or three α2,3-sialic acids, whereas fraction E contained more structures with three α2,3-sialic acids than fraction D.
Because complexity of each sample decreased after fractionation, an additional fifteen α2,3-sialylated N-glycan structures, corresponding to four compositions, were identified. These structures were in low abundance and had not been previously identified in the unfractionated sample (Figure S4–2 in Supporting Information), resulting in a more comprehensive characterization of the serum N-glycome. Furthermore, co-migrating components that had overlapping isotopic clusters were separated into different fractions (Figure S5b in Supporting Information), making some previous assignments less ambiguous.
In addition to the detection of previously unidentified structures and differentiation of structures with similar electrophoretic mobilities and m/z values, specific sialyl linkage isomers were enriched in individual fractions. A tetra-antennary, tetra-sialylated structure, A4G4S4, was selected to evaluate enrichment, because all five sialyl linkage isomers of this structure were detected in the unfractionated sample. Figure 3 shows the relative abundances of sialyl linkage isomers of A4G4S4 in the unfractionated sample and fractions A to E. The sialyl linkage isomers were enriched as follows: the isomer with four α2,6-sialic acids in fraction A (77%), the isomer with one α2,3-sialic acid and three α2,6-sialic acids in fraction B (92%), the isomer with two α2,3-sialic acids and two α2,6-sialic acids in fraction C (88%), and the isomer with two α2,3-sialic acids and two α2,6-sialic acids and the isomer with three α2,3-sialic acids and one α2,6-sialic acid in both fractions D and E (≥ 60%). Notably, the isomer with four α2,3-sialic acids was enriched 12-fold in fraction E (3.1%).
Figure 3.

Relative abundances of sialyl linkage isomers of the structure A4G4S4 with four sialic acids in unfractionated samples and fractions A to E. The subscripts after α2,3 and α2,6 denote the number of those linkages in each structure. The predominant structures have one to three α2,3-sialic acids whereas a negligible fraction of the structure contains four α2,3-sialic acids.
3.5. Source-Induced Fragmentation.
MS/MS analysis provided information on glycan sequence, linkage isomers, and positional isomers [42–46]. Combining the ion-exchange fractionation and CE-MS/MS with source-induced fragmentation generated significant information about the glycan structure and sialyl linkages simultaneously. Selectivity was not applied to the mass filter to isolate the precursor ions, but instead, fragmentation selectivity was ensured by the reduced sample complexity due to the ion-exchange fractionation and high resolution of the CE separation. Fragmentation spectra of N-glycan structures with distinguishable migration times could then be extracted without significant overlap with other structures.
Figure 4 shows fragmentation spectra of several structures that were eluted in fraction B and had different structural features obtained through fragmentation with an in-source energy of 45 V during CE analysis. For this analysis, an 80-min delay time was used to achieve the desired separation resolution. Labeling the N-glycans with GT at the reducing end further enabled us to differentiate fragment ions generated from the non-reducing end from those from the reducing end, because fragments from the reducing end would have an additional mass of 113.15 Da. Thus, the presence of m/z 481.25, 670.27, and 1170.49, and the absence of m/z 512.19 and 816.33 in Figure 4a confirmed core-fucosylation of a bi-antennary, fucosylated, α2,3-sialylated structure, FA2G2S1, at migration time 96.45 min. Similarly, the presence of m/z 512.19, 538.27 and 816.33, and the absence of m/z 481.25 in Figure 4b confirmed antenna-fucosylation of a tri-antennary, mono-fucosylated structure with one α2,3-sialic acid and one α2,6-sialic acid, FA3G3S2, at migration time 100.73 min. Fragmentation spectra of large, tetra-antennary structures were also collected through in-source fragmentation. Figure 4c shows fragment ions corresponding to a poly-LacNAc motif detected at m/z 731.27 and 1063.43 at migration time 106.06 min. Interestingly, α2,3-sialylated poly-LacNAc fragment ions were not detected, indicating that the majority of the poly-LacNAc repeat units were probably associated with just α2,6-sialic acids.
Figure 4.

Fragmentation spectra of SALSA-derivatized and GT-labeled N-glycans derived from human serum, collected in ion-exchange fraction B, and analyzed by CE-MS/MS. Selectivity of the precursor ion relied on the high separation efficiency of CE and reduced sample complexity after ion-exchange fractionation. Fragmentation spectra of (a) structure FA2G2S1 with one α2,3-sialic acid at migration time 96.45 min, (b) structure FA3G3S2 with one α2,3-sialic acid and one α2,6-sialic acid at migration time 100.73 min, and (c) structure A5G5S4 with one α2,3-sialic acid and three α2,6-sialic acids at migration time 106.06 min. Fragment ions were singly charged unless otherwise specified. Precursor ions were identified by the presence of [M + nH]n+ ions.
3.6. Evaluation of Fucose Rearrangement.
In positive-ion MS/MS analysis, fucose rearrangement, which potentially leads to a false assignment, has been reported for protonated N-glycan ions and proposed to be associated with mobile protons [47–49]. We evaluated the degree of fucose rearrangement with fragment ions at m/z 816.33 and 846.36, which corresponded to the [Neu5Ac + Gal + GlcNAc + Fuc]+ ion with α2,3- and α2,6-sialylation, respectively. The presence of m/z 846.36 indicated the occurrence of fucose rearrangement, because antennary fucosylation is expected to be associated with the α2,3-sialylated antenna based on the literature and established biosynthetic pathways [50]. The abundance of m/z 846.36 was approximately 7% relative to that of m/z 816.33 in the fragmentation spectra of unfractionated samples (data not shown). Consequently, a set of fragment ions, including non-fucosylated fragments generated from the reducing end and antenna-fucosylated fragments, were monitored to verify fucosylation. With multiple criteria applied, the minor abundance of fragment ions containing rearranged fucose did not raise a major concern in characterizing the N-glycan structures.
3.7. Rapid Evaluation of Structural Features.
Combining fractionation and in-source fragmentation also allowed rapid evaluation of some substructural features of the N-glycans, e.g., fucosylation and sialylated poly-LacNAc, from the corresponding EIEs. In Figure S6 in Supporting Information and Figure 5, delay times of 60 and 80 min were used to analyze fractions A and B, respectively, to separate the N-glycans while retaining information of the neutral structures in fraction A. In fraction A, an abundant m/z 481.25, which corresponded to the [GT + GlcNAc + Fuc]+ ion, was detected (Figure S6b), whereas only a small amount of m/z 512.19, which indicated antenna-fucosylation, was observed (Figure S6c). These m/z values demonstrated that most of the neutral and completely α2,6-sialylated structures were core-fucosylated. The fragment corresponding to antenna fucosylation at m/z 512.19 (Figure S6c) was likely cleaved from the structure FA3G3S2 that contained two α2,6-sialic acids at migration time 82.7 min, because the fucose on this structure was possibly located on the non-sialylated antenna. Rapid evaluation of sialylated structures and high-mannose structures can be seen in Figure S6d and S6e, respectively.
Figure 5.
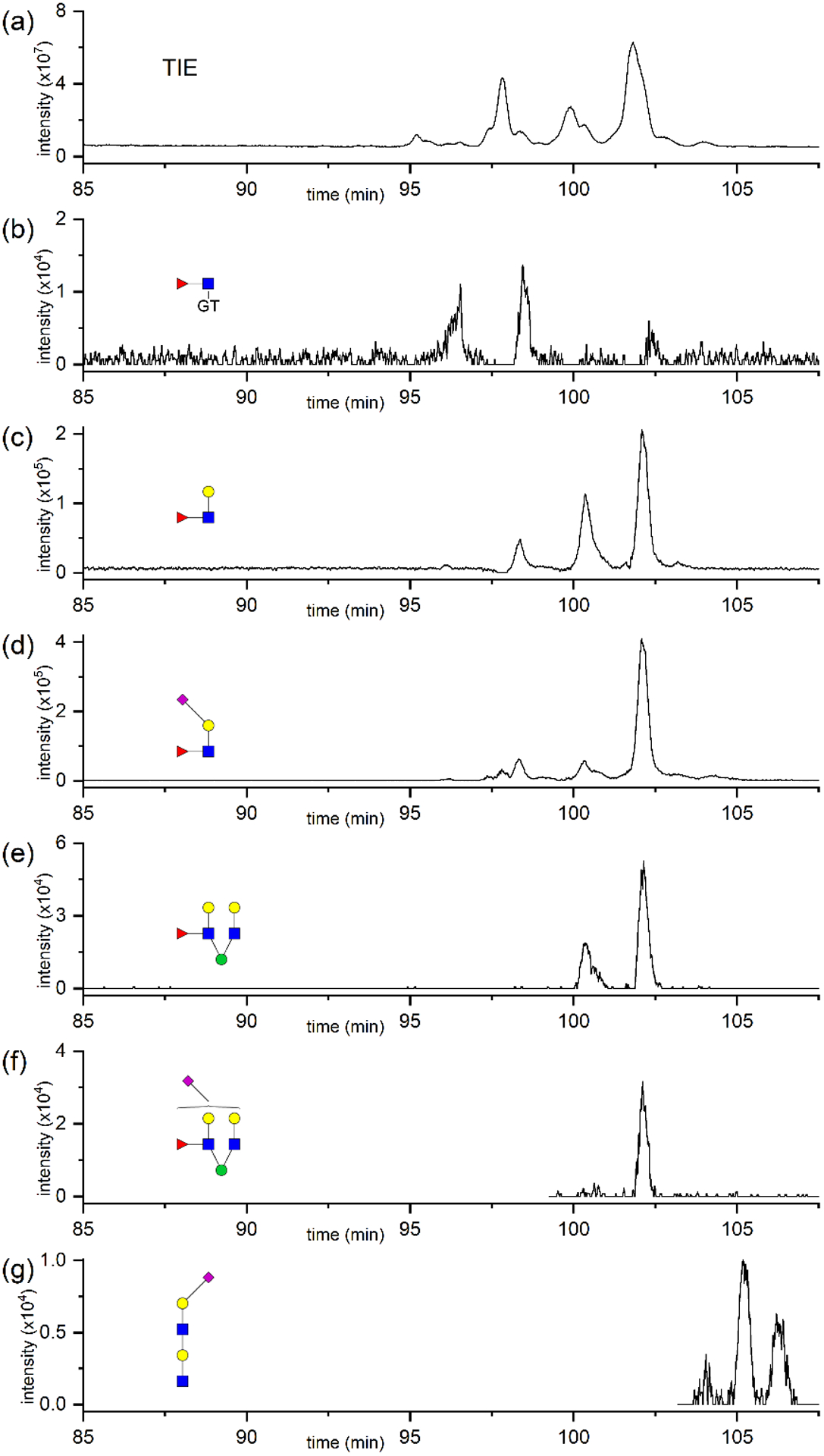
(a) Total ion electropherogram (TIE) of SALSA-derivatized and GT-labeled N-glycans derived from human serum and collected in ion-exchange fraction B. (b-g) EIEs of fragment ions corresponding to N-glycan structural features in fraction B. All ion species were detected as the protonated form.
On the other hand, m/z 481.25 (Figure 5b) had minor ion intensity in fraction B, whereas m/z 512.19 (Figure 5c) and m/z 816.33 (Figure 5d) had abundant intensities in the same fraction, which indicated antennary fucosylation. Notably, higher relative abundance of m/z 512.19 compared to m/z 816.33 was observed at migration time 100.7 min, possibly indicating antennary fucosylation of the non-sialylated antenna of the structure FA3G3S2 that contained one α2,3-sialic acid and one α2,6-sialic acid (Figure 4b). Similarly, the difference in abundance between the fragments shown in Figure 5e and 5f at migration time 100.7 min also confirmed such interpretation. These observations also revealed that the fucosylated antenna was likely one of the two antennae attached to the same mannose of the chitobiose core for this structure FA3G3S2. Fragment ions corresponding to sialylated poly-LacNAc were detected at migration times >103 min (Figure 5g), confirming the presence of structures featuring LacNAc repeating units.
4. Concluding Remarks
In this study, we developed a sample derivatization strategy that combined a modified SALSA derivatization with ion-exchange fractionation to reduce sample complexity and enrich certain sialyl linkage isomers. By combining CE-MS with the fractionation of sialyl linkage isomers, we identified 52 sialylated structures, including 107 linkage isomers. N-Glycans with various structural features in a sample as complex as serum were rapidly evaluated through the combination of fractionation and in-source fragmentation, aiding in the assignment of certain structural types. Enrichment of specific sialyl linkage isomers, achieved through the fractionation, could be beneficial for future discovery of potential N-glycan biomarkers. The analytical methodology proposed here can easily be extended to N-glycans derived from other complex sample sets, e.g., human urinary exosomes, for comprehensive structural characterization of the N-glycome.
Supplementary Material
Acknowledgment.
This work was supported in part by NIH R01 GM106084 and NIH R35 GM141922.
Non-standard abbreviations:
- APTS
8-aminopyrene-1,3,6-trisulfonic acid
- EIE
extracted ion electropherogram
- GT
Girard’s reagent T
- LacNAc
N-acetyllactosamine
- SALSA
sialic acid linkage-specific alkylamidation
- SID
source-induced dissociation
- TIE
total ion electropherogram
Footnotes
Conflict of Interest Statement. The authors declare no conflict of interest.
Availability of Data.
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
References
- 1.Varki A, Lowe JB, in: Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME (Eds.), Essentials of Glycobiology. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY: 2009, 2nd Ed., Ch. 6. [PubMed] [Google Scholar]
- 2.Ohtsubo K, Marth JD. Glycosylation in cellular mechanisms of health and disease. Cell. 2006;126:855–67. [DOI] [PubMed] [Google Scholar]
- 3.Kolarich D, Lepenies B, Seeberger PH. Glycomics, glycoproteomics and the immune system. Curr Opin Chem Biol. 2012;16:214–20. [DOI] [PubMed] [Google Scholar]
- 4.Thaysen-Andersen M, Packer NH, Schulz BL. Maturing glycoproteomics technologies provide unique structural insights into the N-glycoproteome and its regulation in health and disease. Mol Cell Proteomics. 2016;15:1773–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gaunitz S, Nagy G, Pohl NLB, Novotny MV. Recent advances in the analysis of complex glycoproteins. Anal Chem. 2017;89:389–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ruhaak LR, Xu GG, Li QY, Goonatilleke E, Lebrilla CB. Mass spectrometry approaches to glycomic and glycoproteomic analyses. Chem Rev. 2018;118:7886–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goetz JA, Mechref Y, Kang P, Jeng MH, Novotny MV. Glycomic profiling of invasive and non-invasive breast cancer cells. Glycoconjugate J. 2009;26:117–31. [DOI] [PubMed] [Google Scholar]
- 8.Alley WR Jr., Madera M, Mechref Y, Novotny MV. Chip-based reversed-phase liquid chromatography-mass spectrometry of permethylated N-linked glycans: A potential methodology for cancer-biomarker discovery. Anal Chem. 2010;82:5095–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alley WR Jr., Novotny MV. Glycomic analysis of sialic acid linkages in glycans derived from blood serum glycoproteins. J Proteome Res. 2010;9:3062–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alley WR, Vasseur JA, Goetz JA, Svoboda M, Mann BF, Matei DE, Menning N, Hussein A, Mechref Y, Novotny MV. N-linked glycan structures and their expressions change in the blood sera of ovarian cancer patients. J Proteome Res. 2012;11:2282–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hecht ES, Scholl EH, Walker SH, Taylor AD, Cliby WA, Motsinger-Reif AA, Muddiman DC. Relative quantification and higher-order modeling of the plasma glycan cancer burden ratio in ovarian cancer case-control samples. J Proteome Res. 2015;14:4394–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mitra I, Alley WR, Goetz JA, Vasseur JA, Novotny MV, Jacobson SC. Comparative profiling of N-glycans isolated from serum samples of ovarian cancer patients and analyzed by microchip electrophoresis. J Proteome Res. 2013;12:4490–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Biskup K, Braicu EI, Sehouli J, Fotopoulou C, Tauber R, Berger M, Blanchard V. Serum glycome profiling: A biomarker for diagnosis of ovarian cancer. J Proteome Res. 2013;12:4056–63. [DOI] [PubMed] [Google Scholar]
- 14.Zahradnikova M, Ihnatova I, Lattova E, Uhrik L, Stuchlikova E, Nenutil R, Valik D, Nalezinska M, Chovanec J, Zdrahal Z, Vojtesek B, Hernychova L, Novotny MV. N-glycome changes reflecting resistance to platinum-based chemotherapy in ovarian cancer. J Proteomics. 2021;230:103964. [DOI] [PubMed] [Google Scholar]
- 15.Vasseur JA, Goetz JA, Alley WR, Novotny MV. Smoking and lung cancer-induced changes in N-glycosylation of blood serum proteins. Glycobiology. 2012;22:1684–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Borelli V, Vanhooren V, Lonardi E, Reiding KR, Capri M, Libert C, Garagnani P, Salvioli S, Franceschi C, Wuhrer M. Plasma N-glycome signature of down syndrome. J Proteome Res. 2015;14:4232–45. [DOI] [PubMed] [Google Scholar]
- 17.Holst S, Heijs B, de Haan N, van Zeijl RJM, Briaire-de Bruijn IH, van Pelt GW, Mehta AS, Angel PM, Mesker WE, Tollenaar RA, Drake RR, Bovee J, McDonnell LA, Wuhrer M. Linkage-specific in situ sialic acid derivatization for N-glycan mass spectrometry imaging of formalin-fixed paraffin-embedded tissues. Anal Chem. 2016;88:5904–13. [DOI] [PubMed] [Google Scholar]
- 18.Li HH, Gao WJ, Feng XJ, Liu BF, Liu X. MALDI-MS analysis of sialylated N-glycan linkage isomers using solid-phase two step derivatization method. Anal Chim Acta. 2016;924:77–85. [DOI] [PubMed] [Google Scholar]
- 19.Nishikaze T, Tsumoto H, Sekiya S, Iwamoto S, Miura Y, Tanaka K. Differentiation of sialyl linkage isomers by one-pot sialic acid derivatization for mass spectrometry-based glycan profiling. Anal Chem. 2017;89:2353–60. [DOI] [PubMed] [Google Scholar]
- 20.Ferdosi S, Rehder DS, Maranian P, Castle EP, Ho TH, Pass HI, Cramer DW, Anderson KS, Fu L, Cole DEC, Le T, Wu XF, Borges CR. Stage dependence, cell-origin independence, and prognostic capacity of serum glycan fucosylation, beta 1–4 branching, beta 1–6 branching, and alpha 2–6 sialylation in cancer. J Proteome Res. 2018;17:543–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kammeijer GSM, Nouta J, de la Rosette J, de Reijke TM, Wuhrer M. An in-depth glycosylation assay for urinary prostate-specific antigen. Anal Chem. 2018;90:4414–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Drabik A, Bodzon-Kulakowska A, Suder P, Silberring J, Kulig J, Sierzega M. Glycosylation changes in serum proteins identify patients with pancreatic cancer. J Proteome Res. 2017;16:1436–44. [DOI] [PubMed] [Google Scholar]
- 23.Snyder CM, Alley WR, Campos MI, Svoboda M, Goetz JA, Vasseur JA, Jacobson SC, Novotny MV. Complementary glycomic analyses of sera derived from colorectal cancer patients by MALDI-TOF-MS and microchip electrophoresis. Anal Chem. 2016;88:9597–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reiding KR, Blank D, Kuijper DM, Deelder AM, Wuhrer M. High-throughput profiling of protein N-glycosylation by MALDI-TOF-MS employing linkage-specific sialic acid esterification. Anal Chem. 2014;86:5784–93. [DOI] [PubMed] [Google Scholar]
- 25.Ni WQ, Bones J, Karger BL. In-depth characterization of N-linked oligosaccharides using fluoride-mediated negative ion microfluidic chip LC-MS. Anal Chem. 2013;85:3127–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bunz S-C, Rapp E, Neusuess C. Capillary electrophoresis/mass spectrometry of APTS-labeled glycans for the identification of unknown glycan species in capillary electrophoresis/laser-induced fluorescence systems. Anal Chem. 2013;85:10218–24. [DOI] [PubMed] [Google Scholar]
- 27.Mitra I, Snyder CM, Zhou XM, Campos MI, Alley WR, Novotny MV, Jacobson SC. Structural characterization of serum N-glycans by methylamidation, fluorescent labeling, and analysis by microchip electrophoresis. Anal Chem. 2016;88:8965–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khatri K, Klein JA, Haserick JR, Leon DR, Costello CE, McComb ME, Zaia J. Microfluidic capillary electrophoresis mass spectrometry for analysis of monosaccharides, oligosaccharides, and glycopeptides. Anal Chem. 2017;89:6645–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jeong JS, Kim SK, Park SR. Capillary electrophoresis mass spectrometry with sheathless electrospray ionization for high sensitivity analysis of underivatized amino acids. Electrophoresis. 2012;33:2112–21. [DOI] [PubMed] [Google Scholar]
- 30.Wojcik R, Dada OO, Sadilek M, Dovichi NJ. Simplified capillary electrophoresis nanospray sheath-flow interface for high efficiency and sensitive peptide analysis. Rapid Commun Mass Spectrom. 2010;24:2554–60. [DOI] [PubMed] [Google Scholar]
- 31.Maxwell EJ, Zhong XF, Zhang H, van Zeijl N, Chen DDY. Decoupling CE and ESI for a more robust interface with MS. Electrophoresis. 2010;31:1130–7. [DOI] [PubMed] [Google Scholar]
- 32.Shi LH, Jin YX, Moon DC, Kim SK, Park SR. A sheathless CE/ESI-MS interface with an ionophore membrane-packed electro-conduction channel. Electrophoresis. 2009;30:1661–9. [DOI] [PubMed] [Google Scholar]
- 33.Song W, Zhou X, Benktander JD, Gaunitz S, Zou GZ, Wang Z, Novotny MV, Jacobson SC. In-depth compositional and structural characterization of N-glycans derived from human urinary exosomes. Anal Chem. 2019;91:13528–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Snyder CM, Zhou XM, Karty JA, Fonslow BR, Novotny MV, Jacobson SC. Capillary electrophoresis-mass spectrometry for direct structural identification of serum N-glycans. J Chromatogr A. 2017;1523:127–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kammeijer GSM, Jansen BC, Kohler I, Heemskerk AAM, Mayboroda OA, Hensbergen PJ, Schappler J, Wuhrer M. Sialic acid linkage differentiation of glycopeptides using capillary electrophoresis-electrospray ionization-mass spectrometry. Sci Rep. 2017;7:3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhong XF, Chen ZW, Snovida S, Liu Y, Rogers JC, Li LJ. Capillary electrophoresis-electrospray ionization-mass spectrometry for quantitative analysis of glycans labeled with multiplex carbonyl-reactive tandem mass tags. Anal Chem. 2015;87:6527–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zou G, Benktander JD, Gizaw ST, Gaunitz S, Novotny MV. Comprehensive analytical approach toward glycomic characterization and profiling in urinary exosomes. Anal Chem. 2017;89:5364–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhuang Z, Starkey JA, Mechref Y, Novotny MV, Jacobson SC. Electrophoretic analysis of N-glycans on microfluidic devices. Anal Chem. 2007;79:7170–5. [DOI] [PubMed] [Google Scholar]
- 39.Ruhaak LR, Hennig R, Huhn C, Borowiak M, Dolhain R, Deelder AM, Rapp E, Wuhrer M. Optimized workflow for preparation of APTS-labeled N-glycans allowing high-throughput analysis of human plasma glycomes using 48-channel multiplexed cge-lif. J Proteome Res. 2010;9:6655–64. [DOI] [PubMed] [Google Scholar]
- 40.Gil GC, Kim YG, Kim BG. A relative and absolute quantification of neutral N-linked oligosaccharides using modification with carboxymethyl trimethylammonium hydrazide and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Anal Biochem. 2008;379:45–59. [DOI] [PubMed] [Google Scholar]
- 41.Zhuang Z, Mitra I, Hussein A, Novotny MV, Mechref Y, Jacobson SC. Microchip electrophoresis of N-glycans on serpentine separation channels with asymmetrically tapered turns. Electrophoresis. 2011;32:246–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harvey DJ. Fragmentation of negative ions from carbohydrates: Part 1. Use of nitrate and other anionic adducts for the production of negative ion electrospray spectra from N-linked carbohydrates. J Am Soc Mass Spectrom. 2005;16:622–30. [DOI] [PubMed] [Google Scholar]
- 43.Harvey DJ. Fragmentation of negative ions from carbohydrates: Part 2. Fragmentation of high-mannose N-linked glycans. J Am Soc Mass Spectrom. 2005;16:631–46. [DOI] [PubMed] [Google Scholar]
- 44.Harvey DJ. Fragmentation of negative ions from carbohydrates: Part 3. Fragmentation of hybrid and complex N-linked glycans. J Am Soc Mass Spectrom. 2005;16:647–59. [DOI] [PubMed] [Google Scholar]
- 45.Harvey DJ. Collision-induced fragmentation of negative ions from N-linked glycans derivatized with 2-aminobenzoic acid. J Mass Spectrom. 2005;40:642–53. [DOI] [PubMed] [Google Scholar]
- 46.Zhao J, Li SY, Li C, Wu SL, Xu W, Chen YT, Shameem M, Richardson D, Li HJ. Identification of low abundant isomeric N-glycan structures in biological therapeutics by LC/MS. Anal Chem. 2016;88:7049–59. [DOI] [PubMed] [Google Scholar]
- 47.Nwosu C, Yau HK, Becht S. Assignment of core versus antenna fucosylation types in protein N-glycosylation via procainamide labeling and tandem mass spectrometry. Anal Chem. 2015;87:5905–13. [DOI] [PubMed] [Google Scholar]
- 48.Wuhrer M, Koeleman CAM, Hokke CH, Deelder AM. Mass spectrometry of proton adducts of fucosylated N-glycans: Fucose transfer between antennae gives rise to misleading fragments. Rapid Commun Mass Spectrom. 2006;20:1747–54. [DOI] [PubMed] [Google Scholar]
- 49.Harvey DJ, Mattu TS, Wormald MR, Royle L, Dwek RA, Rudd PM. “Internal residue loss”: Rearrangements occurring during the fragmentation of carbohydrates derivatized at the reducing terminus. Anal Chem. 2002;74:734–40. [DOI] [PubMed] [Google Scholar]
- 50.Stanley P, Cummings RD, in: Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, Darvill AG, Kinoshita T, Packer NH, Prestegard JH, Schnaar RL, Seeberger PH (Eds.), Essentials of Glycobiology. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY: 2015, 3rd Ed., Ch. 14. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors upon reasonable request.