

Graphical abstract
Keywords: Ion binding proteins, Feature extraction, Random forest, Predictor
Abstract
Ion binding proteins (IBPs) can selectively and non-covalently interact with ions. IBPs in phages also play an important role in biological processes. Therefore, accurate identification of IBPs is necessary for understanding their biological functions and molecular mechanisms that involve binding to ions. Since molecular biology experimental methods are still labor-intensive and cost-ineffective in identifying IBPs, it is helpful to develop computational methods to identify IBPs quickly and efficiently. In this work, a random forest (RF)-based model was constructed to quickly identify IBPs. Based on the protein sequence information and residues’ physicochemical properties, the dipeptide composition combined with the physicochemical correlation between two residues were proposed for the extraction of features. A feature selection technique called analysis of variance (ANOVA) was used to exclude redundant information. By comparing with other classified methods, we demonstrated that our method could identify IBPs accurately. Based on the model, a Python package named IBPred was built with the source code which can be accessed at https://github.com/ShishiYuan/IBPred.
1. Introduction
Ion binding proteins (IBPs) are proteins that selectively and non-covalently interact with ions, charged atoms, or groups of atoms. Usually, most ion binding proteins bind to metal ions, and a small portion of the proteins can bind non-metal ions. Of course, there are also existing proteins that can bind to both. These ions can directly or allosterically regulate the catalysis and maintain the structural stability of proteins, thereby enriching and diversifying proteins’ structures and functions [1]. For example, the zinc finger proteins, which always bind DNA in several processes, bind to Zn2+ to form more stable space structures [2]. In phages, these IBPs can control and regulate a wide variety of biological processes, such as viral entry into host cell [3], viral tail assembly [3], cytolysis [4], DNA synthesis [5], RNA synthesis [6], and even neurotransmitter secretion [7]. IBPs have been studied for a long time and can be used to develop treatments for diseases caused by drug-resistant bacteria due to their important role in biological processes [8], [9]. Therefore, the identification of IBPs in phages can be helpful for drug development.
Although biochemical experiments are an effective approach for accurately identifying IBPs, they are slightly inferior in terms of time, labor, and material consumption. Owing to the convenience and high efficiency, computational methods are a good choice for identifying IBPs. Many machine learning algorithms, such as support vector machine (SVM) [10], [11], [12], deep learning (DL) [13], [14], [15], [16], [17], [18], [19], extreme boosting algorithm (XGBoost) [20], [21], [22], [23], [24], and stacking ensemble models [25], [26], [27], [28], [29], [30], etc., have been developed for protein function, structure, subcellular localization, and even other biological processes. Different feature descriptors such as amino acid composition (AAC) [31], [32], [33], reduced amino acid composition [34], [35], [36], g-gap dipeptide composition [37], [38], and secondary structure features [39], etc., were adopted to represent protein sequences. While there is still no computational method to identify IBPs in phages, this study aims to design a novel model for IBP prediction.
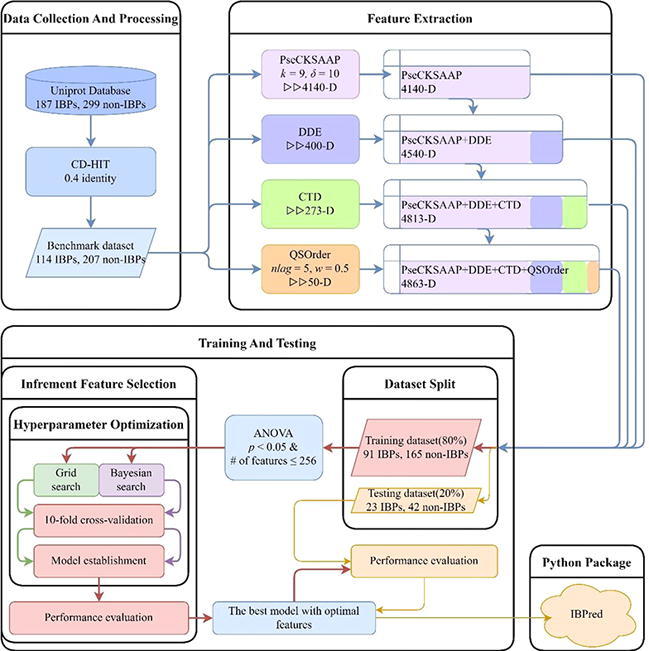
The following five steps were completed in this work to establish a Python package for the identification of IBPs. Firstly, the sequences of IBPs were collected to construct an objective benchmark dataset to train and test the model. Secondly, several feature extraction techniques were used for obtaining sample features. Thirdly, the ANOVA-based technique was applied in feature selection [40]. Fourthly, the RF [41] was selected to construct prediction models. Finally, based on the proposed model, a free and easy-to-use Python package called IBPred was established for the identification of IBPs. The workflow chart is shown in Fig. 1. Notably, the IBPred tool is only suitable for identifying IBPs from phages. Although IBPred can give probabilities of IBPs for protein sequences from other species, it lacks such accurate discrimination capacity as in phage.
Fig. 1.

The flow of the model building.
2. Material and methods
2.1. Benchmark dataset and independent dataset
We collected the proteins that meet our search results in the Universal Protein Resource (UniProt) [42], to establish prediction models. Manually annotated and reviewed sequences were chosen with query keywords “phage” in the Organism and “binding [0005488]” in the Gene Ontology (GO) [43]. Protein sequences that contain ambiguous residues, such as “B”, “J”, “O”, “U”, “X”, and “Z”, should be excluded. The raw data were divided into 187 IBPs and 299 non-IBPs according to the ion binding proteins tagged with the annotation term “ion binding [0043167]” in GO. Subsequently, the CD-HIT program [44] with a sequence identity threshold of 40 % was utilized to remove redundant sequences. 114 IBPs and 207 non-IBPs were retained as the benchmark dataset. To make the prediction more reliable, stratified sampling was used to split the benchmark dataset into the training set and testing set (8:2). Eventually, 91 IBPs and 165 non-IBPs were used for training, and 23 IBPs and 42 non-IBPs were used for independent testing.
2.2. Feature extraction
To extract characteristics of target proteins, transforming target protein sequences into numeric vectors is the key procedure in machine learning modeling. A protein P sequence with L amino acid residues can be expressed as follows:
where Ri (i = 1, 2, …, L) represents the i-th amino acid residue of the protein sequence. By using feature extraction methods, P can be converted to numeric vectors. Here, four types of feature extraction methods were utilized to describe protein sequences.
2.2.1. Pseudo composition of k-spaced amino acid pairs (PseCKSAAP)
The pseudo amino acid composition (PseAAC) [45], [46] is a feature extraction method that computes AAC and their physicochemical properties correlation. The composition of k-spaced amino acid pairs (CKSAAP) [47] is another feature descriptor calculating frequencies of dipeptides separated by various amino acid residues. By replacing AAC features with CKSAAP features, we developed a novel method called PseCKSAAP for sequence features extraction. Details of PseCKSAAP are described as follows.
The PseCKSAAP describes a feature vector with 400(k + 1) + nδ dimension which can be formulated as:
where T is the transposition of the vector; k denotes the number of spaced residues between the paired residues; δ represents the rank of correlation, and is numerically shown as the position difference value between paired residues (theoretically δ < L); n is the rank value of physicochemical properties; fi can be represented as:
where φi denotes the frequency of the j-th dipeptide (j = 1, 2, …, 400) separated by k residues and formulated as:
The symbol εi in Eq. (3) is the δ-tier sequence correlation factor calculated by the following formulas:
where is the correlation function of physicochemical properties between two residues Rx and Ry, it can be calculated by the following formula:
where ρn(Rx) and ρn(Ry) denote the n-th kind physicochemical property value of Rx and Ry. To obtain a high-quality feature set, all physicochemical properties were subjected to a standard conversion as below:
where is the n-th kind physicochemical property original value of residue Rx. The values of the 14 types of physicochemical properties used in this work are listed in Table S1 [48], [49].
Due to the time-consuming of searching parameters and the consideration of the length of protein sequences, we set k = 9 and δ = 10. According to the parameters, the dimension of the extracted feature vector is 400 × (9 + 1) + 14 × 10 = 4140.
2.2.2. Dipeptide deviation from expected mean (DDE)
DDE is a feature descriptor about the fixed composition of dipeptides, which considers the coding diversity of codons [50]. Since amino acids can be determined by combinations of 3 bases, the occurrence frequencies of dipeptides in sequences are innated varied. For a given sequence, we can get the DDE values by standardization (or called z-score normalization) of directly calculated dipeptide composition.
DCi (i = 1, 2, …, 400) describes the dipeptide composition and is given by:
where ni stands for the number of the i-th dipeptide in protein P. TMi denotes the theoretical mean, and can be calculated by:
where is the number of codons that code for the first amino acid residue and is the number of codons that code for the second amino acid residue in the given dipeptide “rs”; =61, is the total number of possible codons, excluding the three stop codons. TVi, the theoretical variance of the dipeptide “rs”, is given by:
Finally, the DDE feature vector can be calculated by the following formula:
2.2.3. Composition transition distribution (CTD)
The CTD is a feature extraction method that was first proposed for protein folding class prediction [51]. In the CTD, 13 types of physicochemical properties were further breakdown into 3 subgroups: polar, neutral, and hydrophobic to generate features (Table S2) [52]. Thus, amino acids were divided into a total of 13 × 3 = 39 groups. The “Composition” (called CTDC) of the CTD method represents the composition percentage of each group in sequence and can produce three features per physicochemical property, which is given by:
where ni,j is the number of residues in the i-th group of the j-th physicochemical property. The “Transition” (called CTDT) of the CTD method denotes the transition probability between two neighboring amino acid residues belonging to two different groups, and can be calculated by:
where nrs and nsr are the numbers of dipeptides “rs” and “sr” respectively, while “r” and “s” are amino acids in the i-th group and not. The “Distribution” (called CTDD) of the CTD method means the relative location in one sequence-represented distribution of residues of given groups. We can use denoting the number of residues of p% (p = 0, 25, 50, 75, 100) of the total number of residues in the i-th group of the j-th physicochemical property, and it can be calculated by:
and if < 1, it will be equaled to 1. Then, the feature vector of CTDD can be represented as:
where denote the location at the sequence that the occurrence number of residues of a given group reaches . Finally, we can get a 39 × (2 + 5) = 273 dimension of feature vector by concatenating , and in CTD.
2.2.4. Quasi-sequence-order (QSOrder)
The quasi-sequence-order descriptor [53] can be defined as:
where fr is the normalized occurrence of amino acid type r; w is a weighting factor; nlag denotes the maximum value of the lag, which is a parameter decided by the user; τq represents the q-th rank sequence-order-coupling number, and can be calculated as follows:
where dp,p+q denotes the item in a given distance matrix that describes the distance between two amino acids at position p and p + q of the protein. Both the Schneider-Wrede physicochemical distance matrix used by Chou [53] and the chemical distance matrix used by Grantham [54] are used to calculate the features. Here, a moderate setting of nlag = 5 and w = 0.5 was adopted. Therefore, (20 + 5) × 2 = 50 dimension of feature vector can be accessed from the QSOrder descriptor.
2.3. Feature selection
Generally, features contribute unequally to the prediction model. Some features make key contributions, some make minor contributions, and some might even reduce the performance of the model [55], [56], [57], [58], [59]. Therefore, feature selection is a vital step to improve classification performance.
To evaluate the classification contribution of each feature, ANOVA [40], [60] was used to score features in this work. The F-score for each feature is defined as follows:
where F(i) is the F-score of the i-th feature; and denote the sample variance between groups (means square between, MSB) and the sample variable within groups (means square within, MSW), respectively. They can be expressed as:
where K = 2, represents the number of groups; fs,j(i) denotes the feature value of the i-th feature of the s-th sample in the j-th group; mj is the number of samples in the j-th group.
It is obvious that the larger the F(i) value, the greater contribution of the i-th feature has. To eliminate the redundant features, all features were ranked according to their F-scores from high to low. Subsequently, incremental feature selection (IFS) was used to determine the optimal number of features. At the beginning, the performance of the first feature subset—which contains only the feature with the largest F-score, was examined. Then, the second feature subset that contains the top two features was evaluated. The process was repeated until all candidate features were added. The RF was used to evaluate the performance of each feature subset. The feature subset with the maximum AUC (the Area Under the receiver operating characteristic (ROC) Curve) was considered to be the optimal feature subset that does not contain redundant features. It should be noted that overfitting may occur when the dimension of the features is greater than the samples size. To avoid this problem, the top 256 feature subsets were used in this work.
2.4. Random forest and cross-validation
The RF is a classification algorithm for supervised machine learning [41], [61], [62], [63], [64]. As an ensemble method, the RF has good interpretability and a prominent advantage on small datasets. The forest consists of many decision trees, and each tree is built by the bootstrap sampling from the training dataset [65]. Additionally, the features are also randomly chosen during the tree construction. By averaging the predicted probabilities of the decision trees, the RF can achieve lower variance and more stable predictions. In this work, the scikit-learn (v1.0.1) package in Python (v3.9.7) was used to implement in RF [66].
In cross-validation methods, n-fold cross-validation, jackknife cross-validation, and independent data test are often used to measure the performance of prediction models [67], [68], [69], [70], [71]. Although jackknife cross-validation can produce a unique outcome, the time-consuming problem can be more serious. For the reliability of results, 10-fold cross-validation and independent test were adopted to evaluate the model performance.
2.5. Grid search and Bayesian search
Grid search (from scikit-learn) and Bayesian search (from scikit-optimize v0.9.0) methods were applied for hyperparameter optimization [72]. The search space of parameters and the number of parameter combinations in the two search methods are listed in Table 1.
Table 1.
The search spaces of search methods and the number of attempts.
| Parameters | Grid Search | Bayesian Search |
|---|---|---|
| “criterion” | Gini, Entropy | Gini, Entropy |
| “max_depth” | 5, 40, 75, 110, 145 | 5, 6, …, 150 |
| “min_samples_split” | 2, 7, 12, 17, 22, 27 | 2, 3, …, 30 |
| “n_estimators” | 10, 25, 63, 158, 398, 1000 | 10x, x∈[1], [3] |
| “min_samples_leaf” | 5 | 1, 2, …, 10 |
| “max_leaf_nodes” | 100 | 50, 51, …, 150 |
| “ccp_alpha” | 0.001 | 10x, x ∈ [-10, 0] |
| # of attempts | 360 | 64 (Our setting) |
The search spaces of the two search strategies are quite different. The grid search tries all combinations of parameters, while its counterpart tries a given number of parameter settings. Here, “min_samples_leaf”, “max_leaf_nodes”, and “ccp_alpha” are not very important parameters and can be set as constant values. Since there are 2–6 options for each other parameters, the grid search method would try 360 times to find the best model. By using the Gaussian process model to approximate the result function, Bayesian search reduces the uncertainty of a given type (category, real number, integer, or in log-scale) and ranges of parameters. A total of 64 trials were conducted to reduce time consumption.
2.6. Performance evaluation
Six assessment criteria were used to evaluate the performance of the prediction models[73], [74], [75]: 1) sensitivity (Sn) and 2) specificity (Sp), were used to evaluate a model’s ability to correctly predict positive and negative samples, respectively; 3) Mathew’s correlation coefficient (MCC), was used to evaluate the reliability of the algorithm; 4) average accuracy (AA), was a combination of the prediction accuracy of positive and negative samples; 5) overall accuracy (OA), reflected the probability of the correct predicted samples in the entire dataset; and 6) area under the receiver operating characteristic (ROC) curve (AUC), was the embodiment of comprehensive performance of the model. The first five metrics are defined as:
where TP and TN are the numbers of IBPs and non-IBPs that were correctly predicted, respectively; FP denotes the number of non-IBPs that were predicted as IBPs, while FN denotes the number of IBPs that were predicted as non-IBPs. The ROC curve is a type of comprehensive index that is drawn from the continuous variable of (1 – Sp) and Sn, which are the abscissa and the ordinate, respectively. The AUC could quantitatively evaluate the performance of the model. The greater the AUC, the better the performance of the prediction models is.
3. Results and discussion
3.1. Performance evaluation based on 10-fold cross-validation test and the independent data test
To find an appropriate way to represent the protein sequences, we investigated the performances of four feature extraction strategies: PseCKSAAP, PseCKSAAP + DDE, PseCKSAAP + DDE + CTD, and PseCKSAAP + DDE + CTD + QSOrder. The ANOVA combined with the IFS technique was used to evaluate and select the optimal features. Since there are four feature extraction strategies and two search methods (grid search and Bayesian search) for hyperparameter optimization, the IFS process was run eight times and eight models were constructed. The AUC of each feature subset was investigated using RF with 10-fold cross-validation on the training dataset. The feature subset that could produce the maximum AUC on the training dataset was regarded as the best features and was used to construct the model. After that, the performance of the models was examined on the testing dataset.
In the IFS curves using grid search (Fig. 2A), with the number of features increasing, the average AUCs rapidly rise to above 0.8 and then stabilize between 0.8 and 0.9. The curve of PseCKSAAP rises to the platform fastest in the first 25 features, and PseCKSAAP + DDE is slightly worse. The other two curves are very similar to each other. Before about 40 features, their AUCs fluctuate about 0.8 and rise rapidly to about 0.85. However, they are lower than the curve of PseCKSAAP + DDE. In the IFS curves using Bayesian search (Fig. 2B), the average AUCs also rapidly rise to more than 0.8. For PseCKSAAP, its AUCs slowly stabilize at about 0.9, and other AUCs of other strategies stabilize between 0.85 and 0.9. These IFS curves are in a similar tendency to the same feature extraction strategy in Fig. 2A.
Fig. 2.

The IFS curves of different search methods and feature extraction strategies on the 10-fold cross-validation test on the training dataset. The data in brackets are the best results of IFS curves that reached the highest average AUCs. (A) Grid search. (B) Bayesian search.
The results in Fig. 2 also showed that the IFS curves of PseCKSAAP are higher than other curves obtained from other features on the training dataset. By comparing the Bayesian search with the grid search, we found that PseCKSAAP achieves the maximum AUC of 0.922 when the feature dimension is 113 by using Bayesian search (Fig. 2B), whereas the maximum AUC of 0.904 when the feature dimension is 122 by using grid search (Fig. 2A). It indicates the Bayesian method can search for more feasible parameter settings and gain better performance. The result is not surprising because the Bayesian search method can explore wider spaces in a shorter time and gain better returns with some probability.
To further examine the robustness of the 8 optimal models obtained by different combinations and optimized by the two search methods, independent data was used. Results were recorded in Table 2. The ROC curves can demonstrate the predictive capability of the proposed method across the entire range of decision values. Thus, the ROC curves of eight models were plotted in Fig. S1.
Table 2.
The performance comparison of models on the training dataset and testing dataset using different search methods and feature extraction strategies.
| Search Method | Features | AUCtraining | AUCtest | OA(%) | MCC | Sn(%) | Sp(%) | AA(%) |
|---|---|---|---|---|---|---|---|---|
| Grid | PseCKSAAP (122D) | 0.904 ± 0.042 | 0.757 | 70.77 | 0.430 | 78.26 | 66.67 | 72.46 |
| PseCKSAAP + DDE (146D) | 0.891 ± 0.048 | 0.808 | 75.38 | 0.517 | 82.61 | 71.43 | 77.02 | |
| PseCKSAAP + DDE + CTD (173D) | 0.871 ± 0.027 | 0.836 | 81.54 | 0.596 | 73.91 | 85.71 | 79.81 | |
| PseCKSAAP + DDE + CTD + QSOrder (148D) | 0.871 ± 0.044 | 0.804 | 78.46 | 0.515 | 60.87 | 88.10 | 74.48 | |
| Bayesian | PseCKSAAP (112D) | 0.922 ± 0.028 | 0.751 | 73.85 | 0.558 | 95.65 | 61.90 | 78.78 |
| PseCKSAAP + DDE (193D) | 0.911 ± 0.029 | 0.865 | 76.92 | 0.578 | 91.30 | 69.05 | 80.18 | |
| PseCKSAAP + DDE + CTD (242D) | 0.895 ± 0.042 | 0.774 | 67.69 | 0.480 | 95.65 | 52.38 | 74.02 | |
| PseCKSAAP + DDE + CTD + QSOrder (239D) | 0.899 ± 0.038 | 0.805 | 75.38 | 0.486 | 73.91 | 76.19 | 75.05 | |
Note: Values are expressed as mean ± standard deviation in AUCtraining metric that indicates the results on the training dataset. The values highlighted in bold denote the best performance value for each metric across search methods and feature extraction strategies.
From Table 2, we noticed that 193 optimal features obtained from PseCKSAAP + DDE by Bayesian search displays the best performance (AUC = 0.865) on the test dataset suggesting that the model has the best generalization ability, though optimal features from PseCKSAAP has the better result on the training dataset. By comparing the AUCs on independent data with it on the training dataset, we could notice a wide disparity, implying an overfitting problem. Since RF is a kind of ensemble method, it weakens the effects of some abnormal trees and strengthens the stability of the forest. Otherwise, the problem would be worse. In addition, due to the small sample size, it is not ideal to split the benchmark dataset into three datasets for training, validation, and independent test. It is possible to adjust the model and avoid overfitting by reducing the error rate on the validation dataset. However, the independent data test could provide enough information to examine models’ performance. Thus, we considered the model based on the selected 193 features as the best model for predicting ion binding protein.
We thought that if features of the model included CTD features and QSOrder features, the performance of the model would be improved. But surprisingly, the addition of these features reduces the prediction performance of the model. This may be because the dataset has some abnormal samples that are difficult to distinguish, or the training dataset is not enough to provide sufficient samples, resulting in insufficient learning and the ability of the model to distinguish between IBPs and non-IBPs worse. This demonstrates that it is not true that the more features, the better the performance of the model.
3.2. Performance comparison of different algorithms
The comparison with other algorithms could provide more information and confidence for developing better models. Because of the best results based on PseCKSAAP + DDE as shown in Table 2, these features were inputted into various algorithms. However, due to the high time cost of the grid search, we only used Bayesian search. Here, we only investigated the prediction performance of random forest (RF), support vector machine (SVM), decision tree (DT), Naïve Bayes (NB), and AdaBoost (AB), which were listed in Table 3.
Table 3.
The performance comparison of different algorithms on the training dataset and testing dataset using Bayesian search and PseCKSAAP + DDE for feature extraction.
| Algorithm | AUCtraining | AUCtest | OA(%) | MCC | Sn(%) | Sp(%) | AA(%) |
|---|---|---|---|---|---|---|---|
| SVM (229D) | 0.962 ± 0.028 | 0.769 | 69.23 | 0.500 | 95.65 | 54.76 | 75.21 |
| DT (11D) | 0.775 ± 0.059 | 0.698 | 60.00 | 0.386 | 95.65 | 40.84 | 68.06 |
| NB (243D) | 0.950 ± 0.033 | 0.693 | 63.08 | 0.458 | 100.00 | 42.86 | 71.43 |
| RF (193D) | 0.911 ± 0.029 | 0.865 | 76.92 | 0.578 | 91.30 | 69.05 | 80.18 |
| AB (254D) | 0.926 ± 0.041 | 0.671 | 60.00 | 0.386 | 95.65 | 40.48 | 68.06 |
Note: Values are expressed as mean ± standard deviation in AUCtraining metric that indicates the results on the training dataset. The values highlighted in bold denote the best performance value for each metric across search methods and feature extraction strategies.
According to Table 3, the RF model is the best since it has the highest AUC on test data. Although the SVM, NB, and AB models can achieve relatively high AUCs on the training dataset, their AUCtest are lower than that of the RF model, suggesting that there are overfitting problems in these models. The ROCs of these models on test data were plotted in Fig. S2, and the results also demonstrate that the RF model outperforms other models. In the IBPred package, the optimum RF model with 193 features was set as the default predictor. Users can also manually select other models, including those recorded in Table 2.
3.3. Feature analysis
In the IFS process, the ANOVA was utilized to assess the significance of features. The F-scores can explain why the model has such a performance. Fig. 3 depicts basic statistical information about the 193 features with F-scores that are involved in building the optimal RF model.
Fig. 3.

The basic statistical information about the features of the optimal model. The PseCKSAAP(Pse) means the features extracted from the physicochemical properties in PseCKSAAP and the PseCKSAAP(CKSAAP) represents the features extracted from the CKSAAP part. (A) The sum of F-scores and the total number of features. (B) The F-scores of features are colored to indicate that they were applied in the optimal model. With a line at the median, the box stretches from the first quartile (Q1) to the third quartile (Q3) of F-scores. And the whiskers extend from the box by 1.5 times the inter-quartile range (IQR), which equals Q3 - Q1. The grey dots represent the features that were not involved in the optimal model construction (that is, were not contained in the optimal feature subset). (C) The counts of selected features and their sum of F-scores based on different gaps. “G0” to “G9” denote the 0–9 gap dipeptides, respectively. (D) The ranked features and their F-scores, as well as the cumulative sum of F-scores from 0 to all features used in the optimal model sequentially.
Among all the features involved in the construction of the optimal model, the physicochemical features are the least, the CKSAAP features are the most, and the DDE features are in the middle (Fig. 3A). Physicochemical features are not only rare, but also have low F-scores (Fig. 3B, D). Therefore, it should try more methods and strategies to extract useful physicochemical features that enhance the physicochemical property representation of proteins. The DDE method is the most efficient since 12.50 % of the original features (50 / 400) were applied to build the optimal model. In comparison, around 1.43 % and 3.53 % of features were selected from properties and CKSAAP, respectively. Most of the top-15 features (Fig. 3D) are DDE features (Fig. 3B), which further demonstrates that the DDE method is the most efficient.
The sum of F-scores of each “gap feature” suggests that their average contributions to the model’s discriminability are close (Fig. 3C). “G3” has the maximum sum of F-scores, whereas “G9” has the least. “G0”, “G3”, and “G6” features provide hints that their graphic points of sums of F-scores exceed the points of counts further than their counterparts. It is easy to infer that the composition of 0, 3, or 6-gap dipeptides in IBPs is significantly different from those in non-IBPs. One possibility is that amino acids attract or repel each other, and the local sequence forms a fragile structure. The different compositions of 0, 3, and 6-gap dipeptides between IBPs and non-IBPs lead to the disparity formation of specific structures for the acceptance of ions. However, amino acids can also attract or repel ions by their structure and physicochemical properties, thus affecting the structure formation. Therefore, it is necessary to further confirm the effect of these gap dipeptides.
Since both CKSAAP and DDE are dipeptide-based methods, the intersection of dipeptide features is most noteworthy. The heatmaps of F-scores of CKSAAP and DDE features extracted from the training dataset have been plotted in Fig. S3. All significant dipeptide features (p ≤ 0.001) of MAX_CKSAAP (a series of maximum scores of dipeptide features across the range of 0–9 gaps, Fig. S3) and DDE in the training dataset are recorded in Table 4.
Table 4.
Significant dipeptide features (p ≤ 0.001) in MAX_CKSAAP and DDE of training dataset.
| MAX_CKSAAP | Intersection | DDE |
|---|---|---|
| NI, ND, DI, VF, QA, SD, CD, RK, IN, CN, AH, GM, NL, VW, CT, IS, GI, DW, CV |
EC, II, YD | RL, VR, SR, ID, YD, RS, PP, PR, KF, FM, RV, KN |
The dipeptides “EC”, “II”, and “YD” are in the intersection (Table 4), and their corresponding dipeptide features’ F-scores rank among the top few of all features. These dipeptide features deserve further research. Most significant CKSAAP features contain cysteine (C), aspartic acid (D), isoleucine (I), and asparagine (N), while DDE features contain more arginine (R) (Table 4). Additionally, The MAX_CKSAAP (Fig. S3) gathers strong signals at the C cluster, D cluster, I cluster, and N cluster (the clusters mean the dipeptides starting with C, D, I, or N), while the DDE has an obvious R cluster. Because the composition of dipeptides is related to the theoretical composition level that is fixed by codon numbers, the R becomes more significant in DDE features.
Glutamate (E), D and R are hydrophilic negatively charged amino acids, while R has a relatively long group. I and tyrosine (Y) are hydrophobic neutral amino acids, while Y has an aromatic ring. C and N are hydrophilic neutral amino acids, while C has sulfur to form disulfide bonds and fold protein sequences. “EC”, “II”, and “YD” (Table 4) corresponding significant features are “EC.0”, “II.0”, “II.6”, and “YD.0”. We believe that C and D in “EC.0” and “YD.0” can attract positively charged ions. However, we do not know the significance of I in “II.0”, “II.6”. One possible answer is that “II.0” and “II.6” have higher composition in non-IBPs. For E, D, R, I, Y, C, and N, their structure and physicochemical properties contribute to the binding of ions. By manually checking the known ion-binding sites of IBPs in the benchmark dataset, C, D, and histidine (H) are the most ion-binding residues. There is a possible relationship between two amino acid distributions. However, the relatively high frequency of H that bind ions was not captured by CKSAAP and DDE.
3.4. Performance on the extra dataset and limitation of IBPred
The benchmark dataset used for the construction of IBPred are all collected from phages. Thus, the model is specific to identifying IBPs in phages. To investigate whether IBPred can be used to identify IBPs in other species, such as human, mouse, etc, we collected IBPs and non-IBPs from other species with similar conditions to the benchmark dataset. There are two points different from the condition of benchmark dataset collection: 1) “NOT” query keywords “phage” in Organism; 2) The Protein Existence is “Evidence at protein level” to reduce the number of proteins. The CD-HIT [44], with an identity threshold of 40 %, was also utilized to remove redundant sequences. Finally, 14,101 IBPs and 12,924 non-IBPs were retained as the extra independent dataset. The dataset is much larger than benchmark dataset. Then, we tested several models from the IBPred package on the extra dataset (Table 5).
Table 5.
The performance of different models on extra dataset.
| Search Method | Model | AUC | OA(%) | MCC | Sn(%) | Sp(%) | AA(%) |
|---|---|---|---|---|---|---|---|
| Grid | RF-P (122D) | 0.548 | 52.46 | 0.033 | 99.35 | 1.29 | 50.32 |
| RF-PD (146D) | 0.617 | 59.15 | 0.179 | 74.56 | 42.33 | 58.45 | |
| RF-PDC (173D) | 0.543 | 52.32 | 0.016 | 97.22 | 3.33 | 50.28 | |
| RF-PDCQ (148D) | 0.448 | 46.88 | −0.072 | 56.00 | 36.94 | 46.47 | |
| Bayesian | RF-P (112D) | 0.576 | 52.33 | 0.028 | 99.82 | 0.51 | 50.16 |
| RF-PD (193D) | 0.604 | 57.74 | 0.149 | 69.58 | 44.82 | 57.20 | |
| RF-PDC (242D) | 0.499 | 52.18 | 0.000 | 100.00 | 0.00 | 50.00 | |
| RF-PDCQ (239D) | 0.488 | 51.67 | −0.017 | 95.77 | 3.56 | 49.66 | |
| Bayesian | SVM-PD (229D) | 0.633 | 60.82 | 0.214 | 75.21 | 45.11 | 60.16 |
| DT-PD (11D) | 0.542 | 53.71 | 0.068 | 93.21 | 10.62 | 51.91 | |
| NB-PD (243D) | 0.597 | 58.89 | 0.180 | 82.63 | 32.99 | 57.81 | |
| ABC-PD (254D) | 0.624 | 58.13 | 0.179 | 89.48 | 23.93 | 56.70 |
Note: “P” is PseCKSAAP, “PD” is PseCKSAAP + DDE, “PDC” is PseCKSAAP + DDE + CTD, “PDCQ” is PseCKSAAP + DDE + CTD + QSOrder. The values highlighted in bold denote the best performance value for each metric across models.
The results in Table 5 show that SVM-PD (229D) has the best performance (AUC = 0.633) on extra dataset, while RF-PD (193D), the optimal model on test dataset from phage, is not satisfactory. It is surprising but reasonable that these models do not have good performances on the extra dataset as the test dataset. This indicates that the protein sequences in other species are quite different from those in phages. Since our features are extracted from phage proteins, these features could not well represent IBPs of other species. Additionally, the extra dataset is much larger than benchmark dataset. If we used the extra dataset as a new benchmark dataset to train models, the performance would be better. However, it is not this work’s theme. For a larger dataset, we need to consider neural networks, and the training time would be much longer.
Therefore, our proposed model cannot predict IBP in other species well. We recommend that users only use IBPred to identify IBPs in phages. If users want to identify IBPs in human, mouse, or other species, please try more models, such as SVM-PD (229D), ABC-PD (254D), etc., and consider the results comprehensively. In the future, we may try larger benchmark datasets, including more species and more methods in feature processing.
4. Conclusion
A random forest-based model was constructed for the accurate prediction of IBPs in phages. In this model, PseCKSAAP, DDE, CTD, and QSOrder were adopted to extract features. During the feature selection, the ANOVA was used to rank the importance of features, and then IFS was employed to determine the optimal feature subset. The RF model with the best performance was set as the default predictor. High AUCs indicated that the proposed method was an effective tool for predicting ion binding proteins. Based on the proposed method, a free and easy-to-use Python package has been built and is accessible at GitHub: https://github.com/ShishiYuan/IBPred, where the source code was also submitted.
Funding
This work has been supported by the grant from National Natural Science Foundation of China (62102067) and the Natural Science Foundation of the Inner Mongolia Autonomous Region (2018BS03021).
CRediT authorship contribution statement
Shi-Shi Yuan: Methodology, Software, Validation, Formal analysis, Data curation, Writing – original draft, Project administration. Dong Gao: Formal analysis, Investigation, Visualization. Xue-Qin Xie: Formal analysis, Investigation, Visualization. Cai-Yi Ma: Formal analysis, Investigation, Visualization. Wei Su: Writing – review & editing. Zhao-Yue Zhang: Writing – review & editing, Funding acquisition. Yan Zheng: Resources, Writing – review & editing, Funding acquisition. Hui Ding: Conceptualization, Methodology, Resources, Writing – review & editing, Supervision, Project administration, Funding acquisition.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2022.08.053.
Contributor Information
Zhao-Yue Zhang, Email: zyzhang@uestc.edu.cn.
Yan Zheng, Email: 348830527@qq.com.
Hui Ding, Email: hding@uestc.edu.cn.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Sippel K.H., Quiocho F.A. Ion-dipole interactions and their functions in proteins. Protein Sci. 2015;24(7):1040–1046. doi: 10.1002/pro.2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Isernia C., et al. 12. Zinc Fingers, in Transition Metals and Sulfur – A Strong Relationship for Life. De Gruyter; 2020. pp. 415–436. [Google Scholar]
- 3.Harada K., et al. Crystal structure of the C-terminal domain of Mu phage central spike and functions of bound calcium ion. Biochim Biophys Acta. 2013;1834(1):284–291. doi: 10.1016/j.bbapap.2012.08.015. [DOI] [PubMed] [Google Scholar]
- 4.Zhang X., Studier F.W. Multiple roles of T7 RNA polymerase and T7 lysozyme during bacteriophage T7 infection. J Mol Biol. 2004;340(4):707–730. doi: 10.1016/j.jmb.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 5.Kulczyk A.W., et al. An interaction between DNA polymerase and helicase is essential for the high processivity of the bacteriophage T7 replisome. J Biol Chem. 2012;287(46):39050–39060. doi: 10.1074/jbc.M112.410647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takeshita D., Tomita K. Molecular basis for RNA polymerization by Qbeta replicase. Nat Struct Mol Biol. 2012;19(2):229–237. doi: 10.1038/nsmb.2204. [DOI] [PubMed] [Google Scholar]
- 7.Fujinaga Y., et al. Molecular construction of Clostridium botulinum type C progenitor toxin and its gene organization. Biochem Biophys Res Commun. 1994;205(2):1291–1298. doi: 10.1006/bbrc.1994.2805. [DOI] [PubMed] [Google Scholar]
- 8.Azam A.H., Tanji Y. Bacteriophage-host arm race: an update on the mechanism of phage resistance in bacteria and revenge of the phage with the perspective for phage therapy. Appl Microbiol Biotechnol. 2019;103(5):2121–2131. doi: 10.1007/s00253-019-09629-x. [DOI] [PubMed] [Google Scholar]
- 9.Ao C., Yu L., Zou Q. Prediction of bio-sequence modifications and the associations with diseases. Brief Funct Genomics. 2021;20(1):1–18. doi: 10.1093/bfgp/elaa023. [DOI] [PubMed] [Google Scholar]
- 10.Kang J., et al. The identification of children with autism spectrum disorder by SVM approach on EEG and eye-tracking data. Comput Biol Med. 2020;120 doi: 10.1016/j.compbiomed.2020.103722. [DOI] [PubMed] [Google Scholar]
- 11.Joshi P., Masilamani V., Ramesh R. An ensembled SVM based approach for predicting adverse drug reactions. Curr Bioinform. 2021;16(3):422–432. [Google Scholar]
- 12.Jiang Q., et al. Predicting human microRNA-disease associations based on support vector machine. Int J Data Min Bioinform. 2013;8(3):282–293. doi: 10.1504/ijdmb.2013.056078. [DOI] [PubMed] [Google Scholar]
- 13.Lv, H., F.Y. Dao, and H. Lin, DeepKla: An attention mechanism‐based deep neural network for protein lysine lactylation site prediction. iMeta, 2022. 1(1). [DOI] [PMC free article] [PubMed]
- 14.Wang X., et al. DeepFusion-RBP: using deep learning to fuse multiple features to identify RNA-binding protein sequences. Curr Bioinform. 2021;16(8):1089–1100. [Google Scholar]
- 15.Muhammad Usman S., Khalid S., Bashir S. A deep learning based ensemble learning method for epileptic seizure prediction. Comput Biol Med. 2021;136 doi: 10.1016/j.compbiomed.2021.104710. [DOI] [PubMed] [Google Scholar]
- 16.Wu X., Yu L. EPSOL: sequence-based protein solubility prediction using multidimensional embedding. Bioinformatics. 2021;37(23):4314–4320. doi: 10.1093/bioinformatics/btab463. [DOI] [PubMed] [Google Scholar]
- 17.Zhao X., et al. Identifying plant pentatricopeptide repeat proteins using a variable selection method. Front Plant Sci. 2021;12 doi: 10.3389/fpls.2021.506681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu Z., et al. DLpTCR: an ensemble deep learning framework for predicting immunogenic peptide recognized by T cell receptor. Brief Bioinform. 2021;22(6) doi: 10.1093/bib/bbab335. [DOI] [PubMed] [Google Scholar]
- 19.Hasan M.M., et al. Deepm5C: a deep-learning-based hybrid framework for identifying human RNA N5-methylcytosine sites using a stacking strategy. Mol Ther. 2022 doi: 10.1016/j.ymthe.2022.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zulfiqar H., et al. Identification of cyclin protein using gradient boost decision tree algorithm. Comput Struct Biotechnol J. 2021;19:4123–4131. doi: 10.1016/j.csbj.2021.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Q., Wan J., Wang G. A survey on computational methods in discovering protein inhibitors of SARS-CoV-2. Brief Bioinform. 2022;23(1) doi: 10.1093/bib/bbab416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Teng Z., et al. ReRF-Pred: predicting amyloidogenic regions of proteins based on their pseudo amino acid composition and tripeptide composition. BMC Bioinf. 2021;22(1):545. doi: 10.1186/s12859-021-04446-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H., et al. dPromoter-XGBoost: detecting promoters and strength by combining multiple descriptors and feature selection using XGBoost. Methods. 2022 doi: 10.1016/j.ymeth.2022.01.001. [DOI] [PubMed] [Google Scholar]
- 24.Zhang J., et al. Rapid antibiotic resistance serial prediction in staphylococcus aureus based on large-scale MALDI-TOF data by applying XGBoost in multi-label learning. Front Microbiol. 2022;13 doi: 10.3389/fmicb.2022.853775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malik A., et al. SortPred: the first machine learning based predictor to identify bacterial sortases and their classes using sequence-derived information. Comput Struct Biotechnol J. 2022;20:165–174. doi: 10.1016/j.csbj.2021.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wei L., et al. Computational prediction and interpretation of cell-specific replication origin sites from multiple eukaryotes by exploiting stacking framework. Brief Bioinform. 2021;22(4) doi: 10.1093/bib/bbaa275. [DOI] [PubMed] [Google Scholar]
- 27.Li F., et al. Computational prediction and interpretation of both general and specific types of promoters in Escherichia coli by exploiting a stacked ensemble-learning framework. Brief Bioinform. 2021;22(2):2126–2140. doi: 10.1093/bib/bbaa049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Charoenkwan P., et al. StackIL6: a stacking ensemble model for improving the prediction of IL-6 inducing peptides. Brief Bioinform. 2021;22(6) doi: 10.1093/bib/bbab172. [DOI] [PubMed] [Google Scholar]
- 29.Basith S., et al. Integrative machine learning framework for the identification of cell-specific enhancers from the human genome. Brief Bioinform. 2021;22(6) doi: 10.1093/bib/bbab252. [DOI] [PubMed] [Google Scholar]
- 30.Wu H., et al. StackTADB: a stacking-based ensemble learning model for predicting the boundaries of topologically associating domains (TADs) accurately in fruit flies. Brief Bioinform. 2022;23(2) doi: 10.1093/bib/bbac023. [DOI] [PubMed] [Google Scholar]
- 31.Zhang D., et al. iCarPS: a computational tool for identifying protein carbonylation sites by novel encoded features. Bioinformatics. 2021;37(2):171–177. doi: 10.1093/bioinformatics/btaa702. [DOI] [PubMed] [Google Scholar]
- 32.Awais M., et al. iTSP-PseAAC: identifying tumor suppressor proteins by using fully connected neural network and PseAAC. Curr Bioinform. 2021;16(5):700–709. [Google Scholar]
- 33.Muller-Xing R., et al. Polycomb proteins control floral determinacy by H3K27me3-mediated repression of pluripotency genes in Arabidopsis thaliana. J Exp Bot. 2022;73(8):2385–2402. doi: 10.1093/jxb/erac013. [DOI] [PubMed] [Google Scholar]
- 34.Zuo Y., et al. PseKRAAC: a flexible web server for generating pseudo K-tuple reduced amino acids composition. Bioinformatics. 2017;33(1):122–124. doi: 10.1093/bioinformatics/btw564. [DOI] [PubMed] [Google Scholar]
- 35.Zheng L., et al. RAACBook: a web server of reduced amino acid alphabet for sequence-dependent inference by using Chou's five-step rule. Database (Oxford) 2019 doi: 10.1093/database/baz131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zheng L., et al. RaacLogo: a new sequence logo generator by using reduced amino acid clusters. Brief Bioinform. 2021;22(3) doi: 10.1093/bib/bbaa096. [DOI] [PubMed] [Google Scholar]
- 37.Tang H., et al. HBPred: a tool to identify growth hormone-binding proteins. Int J Biol Sci. 2018;14(8):957–964. doi: 10.7150/ijbs.24174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang L.W., et al. Identification of cancerlectins by using cascade linear discriminant analysis and optimal g-gap tripeptide composition. Curr Bioinform. 2020;15(6):528–537. [Google Scholar]
- 39.Leyi W., et al. An improved protein structural classes prediction method by incorporating both sequence and structure information. IEEE Trans Nanobiosci. 2015;14(4):339–349. doi: 10.1109/TNB.2014.2352454. [DOI] [PubMed] [Google Scholar]
- 40.Lin H., Ding H. Predicting ion channels and their types by the dipeptide mode of pseudo amino acid composition. J Theor Biol. 2011;269(1):64–69. doi: 10.1016/j.jtbi.2010.10.019. [DOI] [PubMed] [Google Scholar]
- 41.Breiman L. Random forests. Machine Learn. 2001;45(1):5–32. [Google Scholar]
- 42.UniProt, C., The Universal Protein Resource (UniProt). Nucleic Acids Res, 2007. 35(Database issue): p. D193-7. [DOI] [PMC free article] [PubMed]
- 43.The Gene Ontology, C., The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res, 2019. 47(D1): p. D330-D338. [DOI] [PMC free article] [PubMed]
- 44.Fu L., et al. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28(23):3150–3152. doi: 10.1093/bioinformatics/bts565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chou K.C. Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins. 2001;43(3):246–255. doi: 10.1002/prot.1035. [DOI] [PubMed] [Google Scholar]
- 46.Qian Y.Q., et al. Identification of DNA-binding proteins via hypergraph based laplacian support vector machine. Curr Bioinform. 2022;17(1):108–117. [Google Scholar]
- 47.Chen K., Kurgan L., Rahbari M. Prediction of protein crystallization using collocation of amino acid pairs. Biochem Biophys Res Commun. 2007;355(3):764–769. doi: 10.1016/j.bbrc.2007.02.040. [DOI] [PubMed] [Google Scholar]
- 48.Tang H., Chen W., Lin H. Identification of immunoglobulins using Chou's pseudo amino acid composition with feature selection technique. Mol Biosyst. 2016;12(4):1269–1275. doi: 10.1039/c5mb00883b. [DOI] [PubMed] [Google Scholar]
- 49.Kawashima S., Kanehisa M. AAindex: amino acid index database. Nucleic Acids Res. 2000;28(1):374. doi: 10.1093/nar/28.1.374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saravanan V., Gautham N. Harnessing computational biology for exact linear B-cell epitope prediction: a novel amino acid composition-based feature descriptor. OMICS. 2015;19(10):648–658. doi: 10.1089/omi.2015.0095. [DOI] [PubMed] [Google Scholar]
- 51.Dubchak I., et al. Prediction of protein folding class using global description of amino acid sequence. Proc Natl Acad Sci U S A. 1995;92(19):8700–8704. doi: 10.1073/pnas.92.19.8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen Z., et al. iFeature: a Python package and web server for features extraction and selection from protein and peptide sequences. Bioinformatics. 2018;34(14):2499–2502. doi: 10.1093/bioinformatics/bty140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chou K.C. Prediction of protein subcellular locations by incorporating quasi-sequence-order effect. Biochem Biophys Res Commun. 2000;278(2):477–483. doi: 10.1006/bbrc.2000.3815. [DOI] [PubMed] [Google Scholar]
- 54.Grantham R. Amino acid difference formula to help explain protein evolution. Science. 1974;185(4154):862–864. doi: 10.1126/science.185.4154.862. [DOI] [PubMed] [Google Scholar]
- 55.Zhao-Yue ZHANG, Z.-J.S., Yu-He YANG, Hao LIN, Towards a better prediction of subcellular location of long non-coding RNA. Front. Comput. Sci., 2022. 16(5): p. 165903-${article.jieShuYe}.
- 56.Han Y.M., et al. Risk prediction of diabetes and pre-diabetes based on physical examination data. Mathemat Biosci Eng. 2022;19(4):3597–3608. doi: 10.3934/mbe.2022166. [DOI] [PubMed] [Google Scholar]
- 57.Dao F.Y., et al. BDselect: a package for k-mer selection based on the binomial distribution. Curr Bioinform. 2022;17(3):238–244. [Google Scholar]
- 58.Yang H., et al. Risk prediction of diabetes: big data mining with fusion of multifarious physical examination indicators. Inform Fusion. 2021;75:140–149. [Google Scholar]
- 59.Long J., et al. Integrated biomarker profiling of the metabolome associated with impaired fasting glucose and type 2 diabetes mellitus in large-scale Chinese patients. Clini Trans Med. 2021;11(6):e432. doi: 10.1002/ctm2.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang X.F., et al. Predicting LncRNA subcellular localization using unbalanced pseudo-k nucleotide compositions. Curr Bioinform. 2020;15(6):554–562. [Google Scholar]
- 61.Ao C., Zou Q., Yu L. NmRF: identification of multispecies RNA 2'-O-methylation modification sites from RNA sequences. Brief Bioinform. 2022;23(1) doi: 10.1093/bib/bbab480. [DOI] [PubMed] [Google Scholar]
- 62.Nakayama J.Y., et al. Predictors of progression through the cascade of care to a cure for hepatitis C patients using decision trees and random forests. Comput Biol Med. 2021;134 doi: 10.1016/j.compbiomed.2021.104461. [DOI] [PubMed] [Google Scholar]
- 63.Ozgode Yigin B., Algin O., Saygili G. Comparison of morphometric parameters in prediction of hydrocephalus using random forests. Comput Biol Med. 2020;116 doi: 10.1016/j.compbiomed.2019.103547. [DOI] [PubMed] [Google Scholar]
- 64.Huang Y., et al. Prediction of transcription factors binding events based on epigenetic modifications in different human cells. Epigenomics. 2020;12(16):1443–1456. doi: 10.2217/epi-2019-0321. [DOI] [PubMed] [Google Scholar]
- 65.Efron, B. and R.J. Tibshirani, An introduction to the bootstrap. 1994: CRC press.
- 66.Pedregosa F., et al. Scikit-learn: machine learning in python. J Mach Learn Res. 2011;12:2825–2830. [Google Scholar]
- 67.Ao C., Zou Q., Yu L. RFhy-m2G: identification of RNA N2-methylguanosine modification sites based on random forest and hybrid features. Methods. 2021 doi: 10.1016/j.ymeth.2021.05.016. [DOI] [PubMed] [Google Scholar]
- 68.Ahmed F.F., et al. Prediction of protein-protein interactions in arabidopsis thaliana using partial training samples in a machine learning framework. Curr Bioinform. 2021;16(6):865–879. [Google Scholar]
- 69.Mullick B., et al. Understanding mutation hotspots for the SARS-CoV-2 spike protein using shannon entropy and k-means clustering. Comput Biol Med. 2021;138 doi: 10.1016/j.compbiomed.2021.104915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yu L., et al. Predicting therapeutic drugs for hepatocellular carcinoma based on tissue-specific pathways. PLoS Comput Biol. 2021;17(2):e1008696. doi: 10.1371/journal.pcbi.1008696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang L., Xiao X., Xu Z.C. iPromoter-5mC: a novel fusion decision predictor for the identification of 5-methylcytosine sites in genome-wide DNA promoters. Front Cell Dev Biol. 2020;8:614. doi: 10.3389/fcell.2020.00614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Snoek, J., H. Larochelle, and R.P. Adams, Practical Bayesian optimization of machine learning algorithms, in Proceedings of the 25th International Conference on Neural Information Processing Systems - Volume 2. 2012, Curran Associates Inc.: Lake Tahoe, Nevada. p. 2951–2959.
- 73.Yu L., Xia M., An Q. A network embedding framework based on integrating multiplex network for drug combination prediction. Brief Bioinform. 2022;23(1) doi: 10.1093/bib/bbab364. [DOI] [PubMed] [Google Scholar]
- 74.An Q., Yu L. A heterogeneous network embedding framework for predicting similarity-based drug-target interactions. Brief Bioinform. 2021;22(6) doi: 10.1093/bib/bbab275. [DOI] [PubMed] [Google Scholar]
- 75.Zhang Q., et al. Exosomal non-coding RNAs: new insights into the biology of hepatocellular carcinoma. Curr Oncol. 2022;29(8):5383–5406. doi: 10.3390/curroncol29080427. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.