

Abstract
Biologically active small molecules can impart modulatory effects, in some cases providing extended long-term memory. In a screen of biologically active small molecules for regulators of tumor necrosis factor (TNF) induction, we identify several compounds with the ability to induce training effects on human macrophages. Rutaecarpine shows acute and long-term modulation, enhancing lipopolysaccharide (LPS)-induced pro-inflammatory cytokine secretion and relieving LPS tolerance in human macrophages. Rutaecarpine inhibits β-glucan-induced H3K4Me3 marks at the promoters of several pro-inflammatory cytokines, highlighting the potential of this molecule to modulate chromosomal topology. Syk kinase inhibitor (SYKi IV), another screen hit, promotes an enhanced response to LPS similar to that previously reported for β-glucan-induced training. Macrophages trained with SYKi IV show a high degree of resistance to influenza A, multiple variants of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), and OC43 coronavirus infection, highlighting a potential application of this molecule and other SYKis as prophylactic treatments for viral susceptibility.
Keywords: innate immune training, SARS-CoV-2, Syk inhibition, rutaecarpine, LPS tolerance, NFAT-2
Graphical abstract
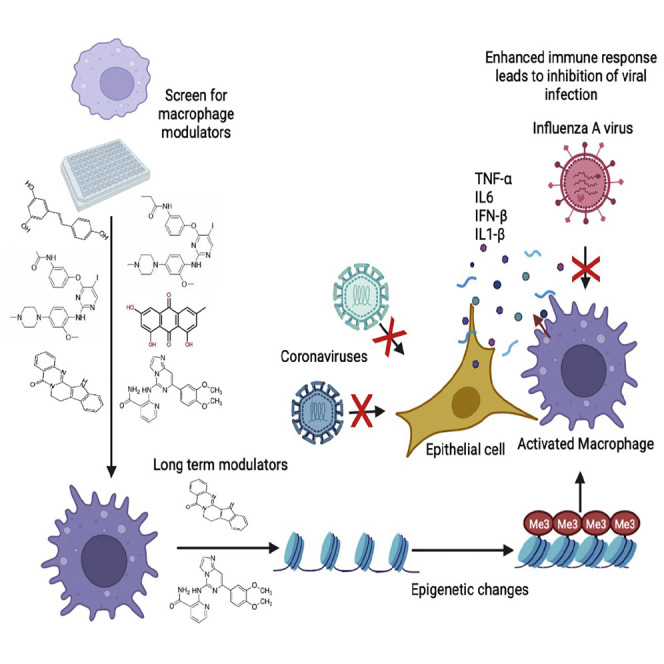
Innate immune training of macrophages can impart long-term modulation. John et al. show that Syk inhibition and rutaecarpine treatment can impart similarly long-term modulatory effects in macrophages. Using a treatment/resting regime, Syk inhibitor reduces susceptibility to viruses, including SARS-CoV-2, while rutaecarpine reverses LPS tolerance and β-glucan induced training.
Introduction
The innate immune system establishes a barrier to pathogen invasion through multiple mechanisms (Gasteiger et al., 2017). Though innate immune defenses in mammals encompass most tissues and cells, specialized myeloid cells initiate and exert a program of immune activation in response to tissue damage, infection, or genotoxic stress (Mosser and Edwards, 2008). Macrophages, a key myeloid cell lineage, detect danger and infectious signals through a range of pattern recognition receptors, leading to the secretion of pro-inflammatory cytokines such as tumor necrosis factor (TNF), interleukin-6 (IL-6), and IL-1β (Arango Duque and Descoteaux, 2014). A tight regulation of the immune response is essential to avoid sustained or prolonged activation of the immune system, which, if not controlled appropriately, disrupts immunological homeostasis and can lead to acute or chronic inflammatory disease (Yoshimura et al., 2018). Biologics that modulate TNF, a primary effector of macrophage-driven inflammation, have shown the potential to treat many inflammatory conditions (Croft et al., 2013; Kalliolias and Ivashkiv, 2016). Small-molecule modulators of the innate immune response therefore have considerable potential both in treating immune diseases (Song and Buchwald, 2015; Savvides and Elewaut, 2020) and enhancing immunogenicity as vaccine adjuvants (Lacey, 2019).
Some microbial molecules that activate innate immune responses have been shown to rearrange the host cell chromatin topology and metabolic state in ways that can escalate the immune response to a second stimulus, a phenomenon termed “trained immunity” (Mantovani and Netea, 2020). Stimulation with such “innate immune training” molecules imparts an epigenetic memory on the promoters and enhancers of pro-inflammatory cytokines (Quintin et al., 2012; Netea et al., 2016; Mulder et al., 2019). Histone modifications such as H3K4 trimethylation (H3K4Me3), H3K27Ac (acetylation), and H3K4M1 (mono-methylation) have been shown to correlate with Bacille-Guerin (BCG)-induced and β-glucan-induced trained immunity (Mulder et al., 2019; Saeed et al., 2014; Novakovic et al., 2016). Though monocytes only have a life span of a few days (Patel et al., 2017), trained immunity has been observed in these cells for many months to years in patients (Kleinnijenhuis et al., 2014). Further studies have also demonstrated that consumption of a “Western” diet drives a low-grade inflammatory response that can establish a detrimental innate immune trained state in myeloid cells (Christ et al., 2018). Such long-term training is mediated through epigenetic and metabolic changes in hematopoietic stem and progenitor cells (HSPCs), potentially through the action of the pro-inflammatory cytokine IL-1 (Mulder et al., 2019; Moorlag et al., 2020). Studies have also shown that trained HSPCs enter into the circulation, live longer, and re-engraft into bone marrow niches, thus promoting long-term innate immune memory (Wright et al., 2001). BCG-trained HSPCs generate epigenetically modified macrophages that provide significantly better protection against virulent M. tuberculosis infection than naive macrophages (Kaufmann et al., 2018). Identifying biologics that modulate macrophage-driven inflammation and innate immune training would therefore be of significant therapeutic potential. Targeting host immunity rather than a specific pathogen for therapeutic development has the added advantage of protecting the host against multiple infectious agents, and newly evolving variants, through a single intervention.
In addition to microbial ligands, endogenous biomolecules such as uric acid, lipoprotein A, and oxidized low-density lipoprotein (oxLDL) have been shown to induce trained immunity (Bekkering et al., 2014; Crisan et al., 2016). A common feature among the molecules that induce innate immune training is that they activate signaling pathways leading to transcription of immune effector genes (Bekkering et al., 2016; van der Heijden et al., 2018). Molecules that induce innate immune training have several putative applications (Mourits et al., 2018; Mulder et al., 2019). For example, immunotolerant states in sepsis and cancer could possibly be reversed by inducing trained immunity (Mourits et al., 2018). On the other hand, inhibiting trained immunity could dampen hyperinflammatory conditions such as atherosclerosis (Mourits et al., 2018). Despite considerable progress in the characterization of trained innate immunity, few studies have attempted to identify and characterize small-molecule modulators of this process. To address this gap, we optimized a previously described human macrophage TNF reporter system (Li et al., 2015) for screening in 1,536-well format to identify innate immune modulators that can also impart trained immunity. We identify both Syk kinase inhibitors and the compound rutaecarpine (RUT) as promising molecules for therapeutic regulation of macrophage training and inflammatory output.
Results
A screen of pharmacologically active molecule library identifies both inhibitors and activators of TNF in human macrophage cells
TNF is a cytokine that is induced multifold in response to infection through the activation of pattern recognition receptor signaling pathways (Johnston and Conly, 2006). We have previously reported the creation and utilization of a human macrophage TNF reporter cell system in THP1 cells for use in genetic perturbation screens (John et al., 2018; Li et al., 2015). To adopt this cell line for small-molecule screening (Figure 1A), we first titrated the optimal cell number to attain a robust lipopolysaccharide (LPS)-induced TNF response in 1,536-well plate format (Figure S1A). Using a chosen plating of 6,000 cells per well, we then tested three known inhibitors of the TLR4 pathway, SB 203580 (a MAPK [p38] inhibitor), Bay 11-7082 (an inhibitor of I-κB kinase [IKK]), and IRAK1/4 inhibitor I (Pubchem: 11983295). The half-maximal inhibitory concentration (IC50) values of 2, 354, and 272 nM, respectively, were within the expected range (Figure S1B), with low toxicity over a broad concentration range (Figure S1C).
Figure 1.

Identification of bioactive modulators of the innate immune response
(A) Schematic of the reporter human macrophage cell line and assay protocol for the bioactive molecule screen. Matched plates were prepared for both the TNF reporter assay and cytotoxicity measurement.
(B) Schematic of hit compound classification from the primary screen based on phenotypic activity curve classes (see STAR Methods).
(C) Percentage of compounds identified from the primary screen classified as inhibitors and activators.
(D) Number of compounds in different curve sub classes.
Data in (C) and (D) show representative data from a screen performed over a broad range of compound doses.
We then screened the Sigma LOPAC1280 pharmacologically active molecule library in a serial dilution format with concentrations ranging from 10 mM to 100 pM (Figure 1A). Compounds were added 30 min prior to stimulation with LPS for 4 h, and those with modulatory effects were classified based on previously described “curve classes” (Inglese et al., 2006) (Figure 1B). We identified 17 inhibitors with curve class response (CCR) scores of −1.1 or −1.2 and 39 activators with scores of +1.1 or +1.2 in the primary screen (Figures 1B, S1D, and S1E). Of the 1,280 compounds we screened, 192 and 195 compounds showed inhibitory or activatory effects on TNF, respectively (Figures 1C and 1D).
Modulators of the TNF transcriptional reporter regulate TNF and IL-6 secretion from primary macrophage cells
The most potent compounds (11 inhibitors and 20 activators) were chosen for follow up screening in primary human macrophages. We first evaluated primary macrophage viability in response to these molecules and those that were cytotoxic at higher concentrations were omitted from further analysis (Figure S2). Most inhibitors identified in the THP1 cell line TNF reporter screen also led to inhibition of secretion of both TNF and IL-6 from primary human macrophages (Figures 2A and 2B), an important primary cell validation of the initial THP1 cell line screen.
Figure 2.

Inhibitors and activators differentially modulate secretion of TNF and IL-6 in LPS-stimulated primary macrophages
Non-cytotoxic compounds scoring in curve classes +1.1 and −1.1 were chosen to assay for effects on cytokine secretion in primary human macrophages.
(A and B) Cells were pre-treated with 10 μM compounds for 0.5, 4, and 24 h before challenge with 100 ng/mL LPS. TNF (A) and IL-6 (B) secretion were measured by ELISA at 4 and 24 h post-LPS.
(C and D) Screen to identify training molecules. Cells were pre-treated with 10 μM compounds for 24 h, then washed and rested for 3 days before challenge with 100 ng/mL LPS. TNF (C) and IL-6 (D) secretion were measured by ELISA at 4 and 24 h post-LPS. Average of 3 replicates was normalized to the respective DMSO control. p values for each compound were calculated in comparison to the DMSO control and are shown on the right side of each treatment.
(E) Schematic summarizing the primary cell validation screens performed for acute modulators of cytokine secretion (in A and B) and for long-term modulators (training molecules) of cytokine secretion (in C and D).
In contrast to the inhibitory compounds from the primary screen, we found that few of the compounds that activated the TNF transcriptional reporter in THP1 cells had a comparable activating effect on TNF secretion from primary macrophages (Figure 2A; Table S1). Some of the compounds showed a slight activation of IL-6 secretion, but these effects were modest and isolated to a few compounds (Figure 2B; Table S1). The compound that most significantly upregulated both TNF and IL-6 was RUT (15892), and its effects were strongest after extended treatment of macrophages up to 24 h (Figures 2A and 2B).
Wash and rest protocol uncovers long-term modulatory effects of select compounds
While the initial screen identified acute modulators of TNF expression, we noted the stronger effects of several compounds after longer incubations with primary cells. We therefore tested their ability to sustain their effects after a washout and 3 day rest period. Most inhibitors from the primary screen lost their inhibitory effect when the cells were treated for a day, washed, and rested for 3 days (Figures 2C and 2D). However, several compounds, including 94419 and 186039, which are inhibitors of cysteine proteases and Syk kinase, respectively, showed increased secretion of both TNF and IL-6 (Figures 2C and 2D). The most substantial enhancement was observed with the inhibitor of Syk kinase (186039, SYKi IV).
We then evaluated the effect of SYKi IV treatment for a day and resting for 3 days on the transcriptional response to LPS in human primary macrophages (Figures 3A and 3B). SYKi IV pre-treatment followed by resting led to an enhanced LPS response for many genes (Figures 3A and 3B), with an enrichment for inflammatory cytokines, antiviral genes, and host responses to infection (Figures S3A and S3B). Both qPCR and ELISA analysis from SYKi IV treated and rested cells confirmed significant upregulation of both IL-6 and TNF (Figures 3C and 3D). Consistent with the upregulated antiviral genes in the SYKi IV-treated cells, the LPS-induced expression of IFNB1 was also elevated by this compound (Figure 3C). We tested another inhibitor of Syk kinase, R406, which is the active component of fostamatinib, an FDA-approved drug for use in adult patients with chronic immune thrombocytopenia (Mullard, 2018). Similar to SYKi IV treatment, R406 also produced similar upregulation of inflammatory cytokines upon treatment of macrophages for a day and resting for 3 days (Figure S3C). These data suggest that Syk kinase inhibition, followed by a resting phase, leads to an enhanced macrophage inflammatory cytokine response to LPS.
Figure 3.

SYKi IV treatment followed by resting trains human macrophages for enhanced responses to LPS
Primary human macrophages were pre-treated ±10 μM SYK IV for 24 h and rested for 3 days prior to 100 ng/mL LPS challenge.
(A and B) RNA-seq analysis after 4 and 24 h LPS.
(A) Heatmap of 1,985 genes induced >1.0 (log2) by 4 h LPS are shown for all conditions.
(B) Volcano plot comparing differentially expressed genes in SYKi IV + LPS versus LPS alone at 4 h.
(C) qPCR analysis of TNF, IL6, and IFNB1 mRNA.
(D) ELISA assay of secreted TNF and IL-6.
(E) Western blot analysis of NFAT2 in the cytosolic (GAPDH-enriched) and nuclear (hnRNPL-enriched) fractions of SYKi IV- (10 μM) or DMSO-treated THP1 cells. Quantitation of the band intensity is shown below as the normalized value to the loading controls, GAPDH for cytosol and hnRNPL for nuclear samples.
(F) Quantification of NFAT2 nuclear intensity in THP1 cells following addition of SYKi IV (10 μM) from imaging data in Figure S3E.
(G) U937 cells were treated with DMSO or SYKi IV for 24 h and rested for 3 days, and H3K4Me3 enrichment was measured by ChIP-qPCR at the promoters of TNF, IL-6, and IL-1β.
(H) Schematic of experimental design for data in (A)–(G).
Data in (A)–(G) are representative of three independent experiments. Experiments in (F) are mean ± SD of 10 fields imaged in each of 3 independent experiments. (C and D) Area expressed as mean ± SD; ∗∗p < 0.01, ∗∗∗p < 0.001 (two-way ANOVA followed by Sidak’s multiple comparison test).
The long-term modulatory effect of Syk inhibition resembles the recently described phenomenon of innate immune training, where macrophages are primed to respond more robustly to a secondary infectious challenge (Netea et al., 2016). Molecules that alter innate immune training have potential applications in disease states such as hyperinflammation and immune-paralysis (Mourits et al., 2018). Syk kinase is known to regulate proteins in the Toll-like receptor (TLR signaling pathway, including the TLR receptors and the MAPKs ERK and JNK (Yi et al., 2014). We therefore tested TLR pathway activation in SYKi IV pre-treated cells compared with the pro-typical innate training stimulus β-glucan (van der Meer et al., 2015). SYKi IV or β-glucan pre-treatment led to no significant change in phosphorylation of p65/nuclear factor κB (NF-κB) or p38 MAPK; however, ERK phosphorylation was significantly enhanced in the SYKi IV pre-treated cells at 20–60 min post-LPS stimulation, while β-glucan pre-treated cells showed a pattern of sustained ERK activation (Figure S3D). These data suggest that enhanced ERK activation may contribute to the enhanced cytokine response observed in SYKi IV- and β-glucan-treated macrophages.
SYKi IV treatment leads to nuclear localization of NFAT2 in human macrophage cells and enhanced H3K4Me3 marks at the promoters of immune genes
Nuclear factor of activated T cells (NFAT) has been proposed to mediate β-glucan-driven innate immune training by upregulating immune gene-priming long non-coding RNAs (IP-lncRNAs), which increases levels of H3K4me3 at the promoters of trained genes (Fanucchi et al., 2019; Quintin et al., 2012). Upon Dectin-1 binding by β-glucan, NFAT is dephosphorylated, exposing the nuclear localization signal, which leads to nuclear translocation (Hogan et al., 2003; Okamura et al., 2000). Phosphorylated NFAT proteins are maintained in the cytoplasm by the coordinated action of three kinases: casein kinase 1 (CK1), glycogen synthase kinase 3 (GSK3), and dual specificity tyrosine phosphorylation regulated kinase (DYRK) (Arron et al., 2006; Gwack et al., 2006; Okamura et al., 2000, 2004). Syk inhibition has been reported to reduce GSK3 activity (Paris et al., 2014). We therefore tested the effect of long term (>1 h) Syk inhibition on nuclear localization of the predominant macrophage NFAT isoform NFAT2 in THP1-differentiated macrophages. We observed enhanced nuclear presence of NFAT2 at 1 and 2 h post-SYKi IV treatment (Figure 3E). To further validate this observation, we performed confocal imaging and found that the nuclear intensity of NFAT2 was significantly enhanced upon treatment with SYKi IV, peaking at 1 h and remaining elevated thereafter (Figures 3F, left, and S3E). A similar pattern of nuclear enhancement of NFAT2 was also observed in SYKi IV-treated primary macrophages (Figure 3F, right).
We then tested if SYKi IV treatment/resting leads to epigenetic chromosomal alterations. One of the hallmarks of innate immune training is the deposition of H3K4Me3 marks at the promoters of trained genes prior to any PRR activation (Saeed et al., 2014). We performed chromatin immunoprecipitation (ChIP)-PCR analysis for H3K4Me3 enrichment at the TNF, IL-6, and IL-1β promoters in PMA-differentiated U937 macrophages, a commonly used human macrophage cell line employed for innate immune training studies. Consistent with previous innate immune training reports (Quintin et al., 2012), H3K4Me3 was significantly elevated at all three gene loci after training with SYKi IV alone in the absence of LPS challenge (Figure 3G). These data suggested that SYKi IV treatment and resting leads to epigenetic rewiring and innate immune training in macrophage cells.
RUT reduces the β-glucan-mediated training in macrophage cells and H3K4Me3 marks at the promoters of TNF and IL-6
To further investigate whether the molecules identified in our initial screen could modulate macrophage behavior, we sought to test their effect on the enhanced response that is observed in β-glucan-trained cells. First, we optimized the β-glucan training protocol for screening in TNF reporter THP1 cells, demonstrating a signal to background ratio of 4.2 and a Z value of 0.29 (Figures 4A and S4A). Six inhibitors and 20 activators from the initial screen were tested for their ability to modulate β-glucan-induced training (Figure 4A). We identified several compounds enhancing or inhibiting the β-glucan-mediated training effect (Figure 4B). The compounds 25125 and 15892 had the most substantial inhibitory effects, with 15892 (RUT) showing the most potent reversal of β-glucan-induced training (Figures 4B and S4B). This inhibitory effect was also observed when RUT was added together with β-glucan (Figure S4C).
Figure 4.

RUT inhibits β-glucan-induced training in differentiated THP1 cells
(A) Assay design to test modulation of β-glucan-induced innate immune training. TNF reporter THP1 cells were treated with 5 ng/mL β-glucan during 10 ng/mL PMA differentiation, then compounds at 10 μM before challenge with 100 ng/mL LPS.
(B) TNF reporter luciferase readings were normalized to the β-glucan + LPS control.
(C) ChIP-qPCR analysis from THP1 cells treated as in (A) with 10 μM RUT at the compound step. Nuclear lysates were immunoprecipitated with H3K4Me3 antibody before qPCR with TNF and IL-6 promoter-specific primers.
(D) THP1 cells were trained with 10 μM RUT for 24 h, washed, and rested for 3 days before 5 h challenge with 100 ng/mL LPS. Nuclear lysates were immunoprecipitated with H3K4Me3 antibody before qPCR with TNF and IL-6 promoter-specific primers.
Data in (B) are averaged from three replicates. Data in (C and D) are representative of three independent experiments expressed as mean ± SD; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 (two-way ANOVA followed by Tukey’s, C, and Sidak’s, D, multiple comparison test).
β-Glucan-treated and rested THP1 cells showed elevated H3K4Me3 marks at the promoters of both TNF and IL-6, with further increases upon LPS stimulation (Figure 4C). When RUT was added 1 h prior to LPS stimulation, the H3K4Me3 marks were diminished at the promoters of TNF and IL-6 both before and after LPS stimulation (Figure 4C). This is consistent with the inhibitory effect of RUT on β-glucan training in the screen and shows its ability to modulate H3K4Me3 marking in innate immune cells. We also evaluated the effects of RUT on LPS-induced H3K4Me3 deposition in the absence of β-glucan and observed a RUT-mediated increase in H3K4Me3 marks at the promoters of both TNF and IL-6 (Figure 4D). This suggests that while RUT can interfere with β-glucan-induced epigenetic modification associated with training, it may itself have some innate immune training capacity.
RUT upregulates transcriptional activation of inflammatory cytokines and shows characteristics of other innate immune training molecules
Since RUT was identified as an acute activator of LPS-induced TNF transcription in our initial compound screen, we assessed its effect on LPS signaling responses. We found that when primary macrophages were pre-treated with RUT prior to LPS stimulation, there was no significant change in the activation of NF-κB p65 or p38 MAPK phosphorylation (Figure 5A). This suggested that the augmented transcriptional response observed with RUT pre-treatment is not due to enhanced signaling activation upstream of NF-κB and MAPK. We also tested TNF mRNA transcription in the presence of RUT by real-time qPCR. Consistent with the screen reporter assay, we found that RUT pre-treatment leads to enhanced LPS-induced transcription of TNF mRNA in primary macrophages (Figure 5B). To assess globally which genes are differentially activated in RUT-treated cells, we performed an RNA sequencing (RNA-seq) analysis in primary macrophages treated with RUT or LPS alone for 4 and 24 h. While LPS induced a much more robust transcriptional response, a number of LPS-induced genes were also induced by RUT, including TNF and IL1B (Figures 5C and S5A). Pathway analysis of RUT-induced genes showed enrichment for chemotaxis at 4 h and cytokine-cytokine receptor interaction at 24 h (Figure S5B).
Figure 5.

RUT selectively enhances transcription and secretion of pro-inflammatory cytokines in primary macrophages
(A and B) Primary human macrophages were pre-treated with 10 μM RUT prior to 100 ng/mL LPS challenge.
(A) Western blot analysis of phosphorylated p65/NF-κB and p38/MAPK. Quantitation of the band intensity is shown below as the normalized value to the loading control, GAPDH.
(B) qPCR analysis of TNF mRNA.
(C) RNA-seq analysis of primary human macrophages treated with 100 ng/mL LPS or 10 μM RUT for the indicated times.
(D) RNA-seq analysis of primary human macrophages treated with 100 ng/mL LPS ±10 μM RUT. RUT+ cells were treated and rested for 3 days prior to LPS challenge.
For (C) and (D), 1,572 genes induced >1.0 (log2) by 4 h LPS are shown.
(E) Secreted cytokine measurements from primary human macrophages treated with 100 ng/mL LPS ±10 μM RUT. RUT+ cells were treated and rested for 3 days prior to LPS challenge.
(F) RUT releases the tolerance induced by LPS. qPCR (left) and ELISA (right) analysis of LPS-tolerant human primary macrophages upon treatment with RUT. Primary human macrophages were treated ±100 ng/mL LPS for 24 h (I), rested for 24 h, then treated ±10 μM RUT for 24 h before the second ±100 ng/mL LPS challenge (II) for 4 h.
Data in (B), (F), and (G) are representative of three independent experiments, expressed as mean ± SD; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 (two-way ANOVA followed by Sidak’s multiple comparison test). Data in (C) and (E) are average of 3 replicates normalized to an untreated control.
We next tested whether RUT has any effect on TLR4 signaling in treated and rested cells. Similar to the acute treatment data (Figure 5A), RUT pre-treatment/resting did not substantially alter the LPS-induced activation of NF-κB or p38 MAPK (Figure S5D). RNA-seq analysis showed that while RUT pre-treatment/resting had a modest overall effect on the LPS-induced gene program (Figure 5D), several inflammatory genes, including TNF, were enhanced (Figure S5E). This pattern was reflected at the secretion level for the cytokines TNF, IL-6, and IL-12 p40 (Figure 5E).
Pathway analysis of LPS-induced genes enhanced by RUT showed enrichment for cell adhesion and cellular response to IL-1 at 4 h LPS and genes associated with leukocyte migration at 24 h LPS (Figure S5C). Conversely, a diverse set of LPS-induced genes were downregulated in RUT-treated and rested cells associated with pathways for cytokine-cytokine interaction and responses to interferon gamma (Figure S5F). This observation may relate to previously reported anti-inflammatory functions of RUT (Choi et al., 2006).
Since RUT treatment and resting led to the enhanced transcription of established inflammatory genes (Figure S5E), and an increase in the innate training-associated histone marker deposition at the TNF and IL-6 promoters (Figure 4D), we analyzed the RNA-seq data for enhanced expression of lncRNAs, another characteristic feature of innate immune training (Fanucchi et al., 2019). We identified several lncRNAs that were preferentially expressed upon RUT treatment (Table S2), providing putative candidates for future study. We further compared the RUT-induced gene expression profile with BCG (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE162729), another established immune training agent (Netea et al., 2016). We used both IPA and MAGIC analyses to predict enriched transcription factor binding motifs and found that 12 and 15 transcription factors (TFs) were common between the two stimuli from IPA and MAGIC analyses, respectively (Table S3). Among these TFs, FOSL2, SMARCA4, and STAT3 were enriched by both analysis methods. This suggests that RUT treatment promotes activation of a transcriptional response with features common to previously characterized immune training activators.
Since innate immune training stimuli have been shown to reverse LPS tolerance through epigenetic rewiring of macrophage cells (Novakovic et al., 2016), we sought to test the effect of RUT on this phenomenon. We tolerized primary human macrophage cells with 100 ng/mL LPS for 24 h, then treated ±RUT for 24 h prior to LPS re-stimulation for 4 h. RUT substantially reduced LPS tolerance in primary human macrophages as measured by TNF mRNA induction and protein secretion (Figure 5F), consistent with previous studies using β-glucan (Novakovic et al., 2016).
RUT and SYKi IV treatment and resting leads to inhibition of SARS-CoV-2, OC43 coronavirus, and influenza A viral replication in human macrophages
Trained immunity has the potential for reducing host susceptibility to viral infection and disease severity (Netea et al., 2020b). The human coronavirus strain OC43 (HCoV-OC43) along with 3 other coronaviruses, HcoV-229E, HcoV-NL63, and HcoV-HKU1, cause approximately 15% of common cold infections acquired globally each year (Sariol and Perlman, 2020). HcoV-OC43 and HcoV-HKU1 belong to the β coronavirus subgroup, which also includes more pathogenic coronaviruses such as SARS-CoV-2, the agent responsible for the ongoing COVID-19 pandemic (Devarakonda et al., 2020). Considering the augmented innate immune response in macrophages pre-treated with RUT or SYKi IV, we evaluated their effect on viral infection both in acute and rested conditions in THP1-differentiated macrophage cells. In the acute regime of 1 h pre-treatment, SYKi IV had no effect on OC43 coronavirus infection, while RUT reduced infection at concentrations >3 mM (Figures 6A and 6B). RUT was also more effective than SYKi IV in limiting influenza infection in an acute pre-treatment regime (Figures S6A and S6B). In contrast, when THP1 macrophages were treated and rested for 3 days, we found that both OC43 and influenza virus infection were strongly attenuated by SYKi IV pre-treatment (Figures 6C, 6D, S6C, and S6D). RUT pre-treatment and resting also protected against both viruses, although to a lesser extent than SYKi IV (Figures 6C, 6D, S6C, and S6D). These results may reflect augmented cytokine responses in macrophages trained by SYKi IV and RUT, and the more potent effect of SYKi IV is consistent with more robust immune effector enhancement in these cells (Figures 3A–3D).
Figure 6.

SYKi IV-induced macrophage training can restrict coronavirus infection
(A) Percentage infection of OC43 in differentiated THP1 cells treated ±SYKi IV or RUT in a dose range of 25 μM to 3 nM 1 h prior to infection with OC43 coronavirus for 24 h.
(B) Representative images of the OC43 infection at 6.25 μM SYKi IV and RUT from (A).
(C) Percentage infection of OC43 in differentiated THP1 cells treated ±SYKi IV or RUT in a dose range of 25 μM to 3 nM for 24 h and rested for 3 days prior to infection with OC43 coronavirus for 24 h.
(D) Representative image of the OC43 infection at 6.25 μM SYKi IV and RUT from (C).
Images in (B) and (D) show representative data from 48 imaged fields. Quantified data in (A) and (C) are representative of three independent experiments, expressed as mean + SD. p values are shown as ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 (ordinary two-way ANOVA).
In contrast to OC43 coronavirus and influenza virus, SARS-CoV-2 does not productively infect macrophage cells, with ACE2-expressing epithelial cells considered to be the primary target (Garcia-Nicolas et al., 2021). To assess the impact of macrophage training on SARS-CoV-2 epithelial infection, we developed a co-culture system of primary human macrophages with hACE2-expressing A549 epithelial cells (Figure 7A). The D614G and UK variants of SARS-CoV-2 productively infected A549-hACE2 cells with distinct syncytia formation (Figures 7B–7D, left panels). Macrophage training with RUT showed a slight reduction of SARS-CoV-2 infection of A549-hACE2 (Figures 7B–7D). In contrast, A549-hACE2 cells co-cultured with macrophages that were trained with either SYKi IV or R406 showed substantially reduced infection with SARS-CoV-2 with no syncytia formation (Figures 7B–7D, right panels). To further assess the specificity of Syk inhibition leading to the training and inhibition of SARS-CoV-2 infection, we tested 7 additional Syk inhibitors (Table S4) in the co-culture assay. Aside from Piceatannol, all Syk inhibitors tested significantly reduced SARS-CoV-2 infection in the co-culture system for both the D614G and UK variants (Figures S7A–S7C).
Figure 7.

SYKi IV-induced macrophage training restricts SARS-CoV-2 infection in co-culture system of A549-hACE2 and macrophages
(A) Schematic of the co-culture system for SARS-CoV-2 infection. Primary human macrophages were treated ±6.25 μM compounds for 24 h and rested for 3 days before plating with A549-hACE2 cells for 16 h prior to infection with SARS-CoV-2 variants at a multiplicity of infection (MOI) 1.0 for 24 h.
(B) Percentage of cells infected with SARS-CoV-2 in co-culture of A549 cells together with macrophages trained with SYKi IV, RUT, and R406 at 6.25 μM.
(C and D) Representative images of SARS-CoV-2 variants D614G (C) and UK (D) infection from experiment in (B).
Images in (C) and (D) show representative data from 48 imaged fields. Quantified data in (B) are representative of three independent experiments, expressed as mean + SD. p values for significant changes are shown as ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 (ordinary two-way ANOVA).
Discussion
Innate immune cells like macrophages play a key role in controlling microbial infections and are also implicated in eliciting autoimmune and other inflammatory diseases (Atri et al., 2018; Ma et al., 2019). Recent discoveries that innate immune cells can exhibit a sustained trained state after exposure to certain stimuli have opened up new potential therapeutic opportunities for both infectious and immune-related diseases. Here, we have used a small-molecule screening strategy to identify modulators of the macrophage inflammatory state. Among the compounds identified, Syk kinase inhibitors and RUT exhibited an ability to establish long-term modulatory effects similar to previously reported innate immune training stimuli.
Syk kinase has been targeted for the development of therapeutics in several diseases including inflammatory diseases and cancer (Geahlen, 2014). R406 (fostamatinib), a highly potent inhibitor of Syk kinase, was recently approved by the FDA to treat chronic immune thrombocytopenia (Mullard, 2018). Our studies suggest another potential use for the Syk inhibitor SYKi IV in combating viral infection. In a treatment/resting protocol, SYKi IV promoted substantial inhibition of both coronavirus and influenza A virus infection of macrophages. Syk is highly expressed in several hematopoietic cells and can regulate pattern recognition receptor (PRR) pathways activated by innate training stimuli such as TLR ligands, β-glucan derived from yeast or bacterial cell walls, and the anti-mycobacterial vaccine BCG (Lee et al., 2009; Rogers et al., 2005; Villasenor et al., 2019; Yanagi et al., 1995, 2001). We show that SYKi IV treatment followed by resting leads to the elevated activation of ERK, which may contribute to enhanced TLR4 signaling and subsequent cytokine induction in the trained macrophages. We also observe upregulation of IFNB1 in SYKi IV-trained cells, which, together with the enhanced cytokine expression, may contribute to the observed inhibition of viral replication.
Syk inhibition with R406 was also recently shown to activate genes involved in amino acid metabolism, leading to metabolic reprograming of pluripotent stem cells (Wang et al., 2021). Innate immune training is associated with metabolic reprogramming and elevated amino acid synthesis (Ferreira et al., 2022), suggesting another possible basis for the observed training effect of Syk inhibition on the macrophage response. Accordingly, we show that NFAT2, which has been implicated as a key TF in the gene program promoting the aforementioned metabolic reprogramming (Wang et al., 2021), undergoes nuclear translocation in macrophages treated with SYKi IV. Nuclear enrichment of NFAT2, and the enhanced training-specific epigenetic marks observed at the SYKi IV-treated immune gene promoters, suggests that Syk inhibition can lead to activation of NFAT2 and generation of an innate immune training state similar to that induced by classic stimuli such as β-glucan and BCG (Saeed et al., 2014). While additional studies will be required to fully elucidate the mechanism by which Syk kinase inhibition mediates its macrophage training effect, targeting of this kinase is emerging as a promising therapeutic approach in a number of diseases.
RUT has previously been characterized as an anti-inflammatory molecule where intraperitoneal injection reduced sepsis-induced peritoneal macrophage recruitment and inflammation (Li et al., 2019). Consistent with this previous observation, our studies find that several genes in inflammatory pathways such as “regulation of chemokine production” were substantially downregulated when primary macrophages were treated with RUT prior to LPS challenge. In contrast, our global analysis of gene expression found that RUT could also induce certain pro-inflammatory genes directly while also boosting the LPS activation of major pro-inflammatory cytokines such as TNF, IL-6, and IL12 p40. While these findings demonstrate that RUT treatment has a complex modulatory effect on the expression of immune response genes, the net effect of RUT appears similar to a typical innate immune training stimulus, as RUT was able to reduce viral infection of macrophages in a dose-dependent manner. Further characteristic features of innate immune training induced by RUT include H3K4Me3 deposition at cytokine promoters, increased lncRNA expression, and upregulation of IL1B (a key player in innate immune training) (Moorlag et al., 2020). In addition, analysis of TF enrichment among differentially expressed genes induced by RUT and the established training stimulus BCG (GEO: GSE162729) found substantial overlap, including the SMARCA4 component of the SWI/SNF nucleosome remodeler complex (Wilson and Roberts, 2011). The role of SWI/SNF in inducing innate immune training is actively under investigation (Kar and Joosten, 2020). A somewhat counterintuitive observation was that RUT inhibited β-glucan-induced H3K4Me3 deposition at the promoters of IL-6 and TNF. However, this is not without precedent, as training induced by BCG, measles, smallpox, and oral polio vaccines can be reversed if followed by administration of a non-live vaccine (Netea et al., 2020a; Aaby et al., 2003; Blok et al., 2020).
Molecules that enhance immune responses have the potential to be developed as vaccine adjuvants and therapeutics for infectious diseases. We have shown that both RUT and SYKi IV can impart long-term modulatory effects on human macrophages. These effects emerged following a treatment and resting regime and showed features characteristic of established innate immune training molecules. ChIP-PCR analysis showed that RUT has potential as an epigenetic modifier to train certain gene loci and to modulate epigenetic marks, with potential applications in relieving endotoxin tolerance, immune paralysis, or other immunotolerant states. SYKi IV treatment and resting led to a more robust enhancement of both transcription and secretion of inflammatory cytokines and effectively controlled the replication of both influenza and coronaviruses. Syk kinase inhibition may therefore have the potential to be developed as a pan prophylactic therapeutic against infectious agents, including different variants of the SARS-CoV-2 coronavirus, the pathogen responsible for the ongoing COVID-19 pandemic.
Trained immunity, and the associated enhanced immune responses, can also be beneficial in immunotolerant states occurring in sepsis or cancer (Mourits et al., 2018). In sepsis-induced tolerance, macrophages have been shown to have altered epigenetic and metabolic states (Cheng et al., 2014; Saeed et al., 2014). Studies have shown that training molecules like β-glucan can reverse the tolerized state in ex vivo analysis of monocytes from volunteers with experimental endotoxemia, leading to the reversal of histone modifications and enhanced expression of previously tolerized immune effector genes (Novakovic et al., 2016). Moreover, since tumor-associated macrophages have well-known immunosuppressive effects such as inhibition of CD8+ cytotoxic cells (Chanmee et al., 2014), screening strategies on a larger scale than those employed here could be used to identify training molecules that could polarize tumor-associated macrophages into a more inflammatory and immune activatory state.
Limitations of the study
Here, we first used a screen to identify acute modulators of macrophage cytokine activation, then a follow-up screen was conducted to identify training molecules that have longer-term effects. Though this approach identified two potent training molecules, future studies that directly screen for training molecules in the primary screen have the potential to identify many more therapeutic candidates. We believe our study highlights strategies to identify training molecules with potentially broad applications in the medical sciences.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-H3K4Me3 | Abcam | ab8580; RRID:AB_306649 |
| Anti-Influenza A | Abcam | ab128193; RRID:AB_11143769 |
| Anti-Coronavirus | Millipore | MAB9013; RRID:AB_95425 |
| Anti-mouse-Alexa 488 | Invitrogen | A21202; PRID: AB_141607 |
| Anti-NFAT-2 | Santa Cruz | sc7294; RRID:AB_2152503 |
| Anti-rabbit -Alexa 488 | Cell Signaling | 4412; RRID:AB_1904025 |
| Anti-SARS CoV-2 Spike | GeneTex | GTX632604; RRID:AB_2864418 |
| Anti-phospho-p38 | Cell Signaling | 4511S; RRID:AB_2139682 |
| Anti-phospho-p65 | Cell Signaling | 3033S; RRID:AB_331284 |
| Anti-GAPDH | Cell Signaling | 97166; RRID:AB_2756824 |
| Anti-phospho-ERK | Cell Signaling | 4370; RRID:AB_2315112 |
| Anti-hnRNPL | Santa Cruz | sc-32317; RRID:AB_627736 |
| Anti-mouse-HRP | Invitrogen | A16011; RRID:AB_2534685 |
| Anti-rabbit-HRP | Invitrogen | A16023; RRID:AB_2534697 |
| Bacterial and virus strains | ||
| OC43 Coronavirus | ATCC | VR-1558 |
| Influenza A (Texas/36/91) | Gift from Dr. Ronald Germain, NIAID | N/A |
| SARS CoV2 (D614G) | NIAID SARS Corona Virus Core Lab | N/A |
| SARS CoV2 (U.K) | NIAID SARS Corona Virus Core Lab | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| LOPAC library | Sigma | 200-664-3 |
| Phorbol 12-myristate 13-acetate | Sigma | P8139 |
| GM-CSF | R&D Systems | 215-GM-050/CF |
| Hygromycin B | Thermofisher Scientific | 10687010 |
| LPS from Salmonella | Enzo Life Sciences | ALX-581-008-L002 |
| DMSO | Sigma | D2650-100ML |
| Rutaecarpine | Sigma | R3277-5MG |
| Syk inhibitor IV | Sigma | 574714-2MG |
| β-glucan | David Williams | East Tennessee State University |
| TaqMan™ Gene Expression Master Mix | Thermofisher Scientific | 4369016 |
| Paraformaldehyde (Formaldehyde) Aqueous Solutio | Electron Microscopy Sciences | 15714-S |
| Protease Inhibitor Cocktail powder | Sigma | P2714-1BTL |
| Pierce™ Protein A/G Agarose | Thermofisher Scientific | 20421 |
| Proteinase K Solution | Thermofisher Scientific | 25530049 |
| UltraPure™ Phenol:Chloroform:Isoamyl Alcohol | Thermofisher Scientific | 15593031 |
| Critical commercial assays | ||
| West Dura Extended Duration Substrate reagents | Thermofisher Scientific | 34076 |
| Luciferase Assay System | Promega | E1500 |
| Dual-Luciferase® Reporter Assay System | Promega | E1960 |
| CellTiter-Glo® Luminescent Cell Viability Assay | Promega | G7570 |
| RNeasy Plus Mini Kit | Qiagen | 74134 |
| iScript™ Reverse Transcription Supermix for RT-qPCR | Bio-rad | 1708841 |
| Human TNF ELISA Set | BD Biosciences | 555212 |
| Human IL-6 ELISA Set | BD Biosciences | 555220 |
| Human IL-12/IL-23p40 Flex | BD Biosciences | 560154 |
| NE-PER™ Nuclear and Cytoplasmic Extraction Reagents | Thermofisher Scientific | 78833 |
| Deposited data | ||
| RNA Sequencing Data | GEO | GSE210476 |
| Experimental models: Cell lines | ||
| THP1 monocytes | ATCC | TIB-202 |
| U937 monocytes | ATCC | CRL-1593.2 |
| THP1 B5 reporter cells | Home | (Li et al., 2015) |
| Primary monocytes | NIH blood bank | NIH |
| A549-hACE2 | Dr. Jonathan Yewdell, | NIAID, NIH |
| Oligonucleotides | ||
| TNF Promoter forward primer-ChIP qPCR | IDTDNA | ACACACAAATCAGTCAGTGG |
| TNF Promoter reverse primer-ChIP qPCR | IDTDNA | CTTCTGTCTCGGTTTCTTCTC |
| IL6 Promoter forward primer- ChIP qPCR | IDTDNA | GGAGCAGTGGCTTCGTTTCA |
| IL6 Promoter reverse primer- ChIP qPCR | IDTDNA | CGGTGGGATTCTTTGGTGTG |
| IL1B Promoter forward primer-ChIP qPCR | IDTDNA | AAAGAAGTGAATGAAGAAAAGT |
| IL1B Promoter reverse primer-ChIP qPCR | IDTDNA | AGTTGATGTCCACATTAAAATA |
| IL6 reverse-qPCR | IDTDNA | GCAACTGGACCGAAGGC |
| IL6 forward-qPCR | IDTDNA | AAGCTCTATCTCCCCTCCAGGA |
| IL6 PROBE | IDTDNA | 56-FAMCTTCTCCAC/ZEN/ AAGGGCCT |
| HPRT forward-qPCR | IDTDNA | CTGGAAAGAATGTCTTGA TTGTGG |
| HPRT reverse-qPCR | IDTDNA | CTTGCGACCTTGACCATCTT |
| HPRT probe-qPCR | IDTDNA | 56-FAM/AGACTTTGCTT/ZEN/ TCCTTGGTCAGGCA |
| TNF forward-qPCR | IDTDNA | CCAGGGACCTCTCTCTAATCA |
| TNF reverse-qPCR | IDTDNA | TCAGCTTGAGGGTTTGCTAC |
| TNF PROBE | IDTDNA | 56-FAM/AGTGACAAG/ZEN/ CCTGTAGCCCA |
| IFNB1 forward- qPCR | IDTDNA | TGAGCAGTCTGCACCTGAA |
| IFNB1 reverse-qPCR | IDTDNA | ACAGTGACTGTACTCCTTGG |
| Human IFNB1 PROBE | IDTDNA | 56-FAM/ATTCTGCAT/ZEN/ TACCTGAAG |
| Software and algorithms | ||
| Graphpad Prism v9 | GraphPad | https://www.graphpad.com |
| Bioconductor tool | Bioconductor.org | https://bioconductor.org/packages/release/bioc/html/EnhancedVolcano.html |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Sinu P. John (sinu.john@nih.gov).
Materials availability
This study did not generate new unique reagents.
Experimental methods and details
Cell culture and stimulation
THP1 monocytes (ATCC), U937 monocytes (ATCC), THP1 B5 reporter cells (Li et al., 2015) and human blood derived monocytes were propagated in RPMI media with 10% FBS, 10 mM HEPES, and β-mercaptoethanol in 5% CO2 at 37°C. Human peripheral blood monocyte samples from screened, healthy donors were obtained under the NIH Clinical Center IRB-approved protocol 99-CC-0168 from the NIH Department of Transfusion Medicine. THP1 and U937 cells were differentiated into a macrophage-like state with 10 ng/mL PMA (Sigma) for three days. Human primary monocytes were differentiated into macrophages with 10 ng/ml GM-CSF (R&D) for 7 days. A549-hACE2 cells were a generous gift from Dr. Jonathan Yewdell, NIAID, NIH and were maintained in DMEM 10% FBS+ 250 μg/mL hygromycin (ThermoFisher, Cat# 10687010). Hygromycin was omitted during viral infection.
Compound library
LOPAC library (Sigma Aldrich) was used, which is a collection of 1280 pharmacologically active compounds consisting of biologically annotated collection of inhibitors, receptor ligands, pharma-developed tools, and approved drugs impacting most signaling pathways and covering all major drug target classes.
Primary screen
Primary screen was performed at the high throughput screening facility at NCATS, NIH. THP1 cells expressing TNF promoter-driven firefly luciferase were batch differentiated in T150 flasks for 3 days. Cells were lifted using cell strip buffer and scraping, counted and plated into 1536 well plates (catalog # 789173-F) using a small cassette and a Multidrop Combi dispenser (Thermo Fisher Scientific Inc.,Waltham, MA) in 4 μL volume/well at a density of 6,000 cells per well. Following overnight incubation, compounds were added using a Kalypsys pintool (Columns 5–48) in serial dilution over a 7-point dose range. After 30 min, cells were stimulated with LPS (100 ng/mL) for 4 h at 37°C, 5% CO2, 95% humidity covered with low evaporation stainless steel lids from Kalypsys. Cells were lysed in 5 μL of lysis buffer (Promega) following LPS stimulation and firefly luciferase reading was measured by the luciferase assay system (Promega) with a ViewLux (PerkinElmer) containing a luminescent filter following the manufacturers’s methods.
Relative luminescence units (RLU) for each well were normalized to the median RLUs from the control wells with LPS stimulation (Col 2) as 100% signal and median RLUs from wells without LPS (Col 1) as 0% signal. Data was analyzed using a curve class classification algorithm which classifies dose responses in four categories (Class 1, 2, 3 and 4) based on the quality of curve fit to the data (r2), the magnitude of the response (efficacy), and the number of asymptotes to the calculated curve classes’ (Inglese et al., 2006). Compounds with a curve class of either −1.1, −1.2, +1.1, +1.2 in the primary assay were chosen for follow up studies. Compounds in class 1a curves were well fit (r 2 ≥ 0.9), showing a full dose response (efficacy >80%), exhibiting both upper and lower asymptotes. Class 1b curves were the same as Class 1a except with a lower efficacy value (30–80%) and a shallow curve. Class 2 curves were incomplete and contained only one asymptote that were divided into two subclasses. Class 2a had a good fit (r 2 ≥ 0.9) and a sufficient response (efficacy >80%) to calculate an inflection point, whereas Class 2b characterized a weaker response (efficacy <80% and r 2 < 0.9).
Dual luciferase secondary screen
The human THP1 B5 cell clone (Li et al., 2015) was differentiated into a macrophage-like state. Compounds were added 30 min before stimulating with LPS (100 ng/mL) as described above. Firefly and renilla luciferase activity in the cell lysates was determined using the Dual-Luciferase Reporter Assay System (Promega, E1960), following the manufacturers protocol, and the ratio of firefly luminescence to renilla luminescence was used to reflect the cell response to LPS stimulation.
Cell titer glow for viability assay
Differentiated primary macrophage cells were plated in 384 well plates at a density of 5000 cells per well. Compounds were added in a 2 fold serial dilution starting from 10 μM concentration in DMSO in a total of 25 μL volume. After 24h LPS stimulation, 25 μL cell titer glow reagent (Promega) was added, the plates were incubated for 15 min at room temperature, and the luminescence was measured following the manufacture’s protocol using a ViewLux (PerkinElmer) containing a luminescent filter. Relative luminescence units (RLU) for each well were normalized to the median RLUs from the DMSO control wells.
TLR ligands
LPS derived from Salmonella was from ENZO Life Sciences, Minnesota. 100 ng/mL LPS was used in all the experiments. Cells were plated and differentiated in 12-well plates for ELISA, qPCR and RNA seq experiments. Ligands were diluted in culture media and added onto differentiated cells at the respective time points.
Quantitative PCR
Total RNA was extracted with the RNeasy Plus Mini Kit (QIAGEN). cDNA was reverse transcribed from 1 μg RNA using the iScript Reverse Transcription kit (Bio-rad). For qPCR, 200 ng RNA equivalent of cDNA was used per reaction with gene specific primers and FAM conjugated probes (IDT DNA) and qPCR Solaris mix (Thermofisher Scientific). PCR reactions were performed in an Quantstudio6 (Thermofisher Sci) with the following thermal cycles: 95°C for 15 min, (95°C for 15 s, 60°C for 60 s) × 40 cycles. The Ct values were analyzed with Quant studio 6 software.
Measurement of cytokine secretion
For secondary screening cytokine assays, differentiated primary macrophages were plated in 384-well plate at a density of 5000 cells/well. Compounds were added at 10 μM final concentration in DMSO 0.5 h, 4 h or 24 h prior to LPS stimulation for either 4 h or 24h. Supernatants were collected for assaying the secreted cytokines IL-6 and TNF. For follow up assays on the long term effect of RUT and SYKi IV, differentiated primary macrophage cells were treated with either RUT, SYKi IV or DMSO in 6-well plates for the corresponding longer incubations (24 h, or 24 h followed by wash and resting for 3 days) and lifted by scrapping and replated at a density of 20,000 cells per well into 96-well plate or 3 × 105 cells/well into 12-well plates the day prior to assay. After LPS treatment, the supernatants were collected and the concentration of TNF-α or IL-6 was quantitated by ELISA and IL-12 p40 by CBA assay. ELISA kits (OptEIA) and CBA assay were purchased from BD bioscience and performed according to the manufacture protocol.
Macrophage training
Macrophage cells were trained with β-glucan by treating cells with β-glucan 5 μg/mL for a day, followed by media change and rest for 3 days. THP1 cells were first differentiated with PMA prior to training with β-glucan. Training of THP1 cells with SYKI IV or RUT were done together with PMA (20 ng/mL) for a day followed by changing media and resting for 3 days. Human primary blood derived monocytes were first GM-CSF differentiated prior to training with either, β-glucan, RUT or SYKi IV. Cells were treated with SYKi IV or RUT at 10 μM for a day, washed and rested for 3 days prior to stimulation or infection with LPS or viruses. Supernatant were collected for ELISA and CBA measurement of secreted cytokines and cells were lyzed in RLT buffer for total RNA isolation for RNA sequencing and qPCR.
RNA sequencing analysis
Differentiated primary macrophage cells (4.5×105 per well) were plated in 12-well plates. Cells were treated with SYKi IV, RUT or DMSO for 24 h followed by resting for 3 days or treated with RUT alone for 0h, 4 h or 24 h. Rested cells were then stimulated with 100 ng/mL LPS for 0 h, 4 h, and 24 h. RNA was isolated with an RNeasy Mini Kit (QIAGEN) following the manufacturer’s protocol and eluted in RNase free TE buffer.
RNAseq libraries were created in triplicate for all cell types using Illumina TruSeq Stranded mRNA Library Prep, pooled, and sequenced on a NextSeq producing paired-end 75 base-pair reads. RNA-seq data was processed using the Pipeliner workflow (https://github.com/CCBR/Pipeliner). Reads were trimmed using cutadapt v1.18 (Martin, 2011). and aligned to the human hg38 reference genome and Gencode release28 using STAR v2.5.2b (Dobin et al., 2013). RSEM v1.3.0 (Li and Dewey, 2011) was used for gene-level expression quantification, and limma v 3.40 (Ritchie et al., 2015) was used for voom quantile normalization and paired differential expression analysis. Genes induced >1.0 (log2) normalized to the untreated by 4 h LPS are shown for all the heatmaps. Volcano plots were generated using the Bioconductor tool, https://bioconductor.org/packages/release/bioc/html/EnhancedVolcano.html.
Chromatin immunoprecipitation-PCR (ChIP-PCR)
THP1 or U937 cells were treated with PMA together with RUT, SYKi IV or β-glucan for a day and rested for 3 days. After LPS stimulation, cells were fixed in 0.8% PFA (final) for 10 min at 37°C and quenched with 125 mM (final) glycine for 10 min at 37°C. Cells were lysed in 1 mL Lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl pH 8.1 and protease inhibitors (Sigma)). Lysate was then sonicated ten times for 30 s (with 30 s breaks) at 4°C and centrifuged at 20,000×g for 10 min at 4°C to remove cell debris. A fraction of the lysate was used to quantitate chromatin DNA. Lysates were diluted in Dilution Buffer to 250 ug chromatin/mL (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl pH 8.1, 167 mM NaCl 1 and protease inhibitors). Lysates were precleared with Protein A/G Dynabeads (Life Technologies) for 1 h at 4°C. 4 μL of anti-H3K4Me3 (Abcam, ab8580) was used to bind Dyna Beads (80 μL per sample) in Dilution Buffer for 1 h at RT. Beads were then washed three times with 1 mL Dilution Buffer and incubated with the lysate overnight at 4°C. After incubation, beads were washed 2 times in 500 μL each of, Low Salt Buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl pH 8, 150 mM NaCl), High Salt Buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl pH 8.1, 500 mM NaCl), LiCl Buffer (0.25 M LiCl, 1% NP-40,1% deoxycholate, 1 mM EDTA, 10 mM Tris-HCl pH 8.1), and 1X TE Buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8). The beads were then suspended in 400 μL elution buffer (1% SDS, 10 mM Tris-HCL pH 8., EDTA 5 mM, 300 mM NaCl) and incubated with 20 U Proteinase K at 65°C overnight. For normalization, 50 μL input sample was mixed with 350 μL elution buffer. DNA was extracted from both input and ChIP elution samples with Phenol:Chloroform:Amyl alcohol (Life Technology) followed by ethanol precipitation. PCR primers were designed based on known promoter sequences (https://epd.vital-it.ch/human/human_database.php and http://www.ag-rehli.de/NGSdata.htm). qPCR reactions on the precipitated DNA were performed with Sybr green dye and the following primers:
TNF forward primer (ACACACAAATCAGTCAGTGG).
TNF reverse primer (CTTCTGTCTCGGTTTCTTCTC).
IL6 forward primer (GGAGCAGTGGCTTCGTTTCA).
IL6 reverse primer (CGGTGGGATTCTTTGGTGTG).
IL1B forward primer (AAAGAAGTGAATGAAGAAAAGT).
IL1B reverse primer (AGTTGATGTCCACATTAAAATA).
Viral assay
For rested cells, THP1 cells were co-differentiated with PMA (20 ng/mL) and RUT or SYKi IV (10 μM) dissolved in DMSO for a day followed by washing and resting for three days. For acute treatment, cells were first differentiated for 3 days in PMA (10 ng/mL) and incubated with RUT or SYKi IV one hour prior to infection. Cells were infected with Influenza A virus (Texas/36/91 strain, obtained from Dr. Ronald Germain, (Brandes et al., 2013) and Coronavirus OC43 (ATCC) diluted in RPMI media in a serial dilution starting with an MOI of 1.0 for 24 h. 16 h (Influenza A) and 24h (OC43) post infection, cells were washed with PBS and fixed in 50 μL 4% PFA for 10 min and washed, permeabilized in 0.25% Triton X-100, blocked in BSA and stained with anti influenza (Abcam: ab128193) or coronavirus (Millipore:MAB9013) antibody at 1:1000 dilution in 1% BSA followed by anti-mouse antibody coupled with Alexa 488 flourophore (Invitrogen, A21202).
Confocal imaging
Primary macrophages and PMA-differentiated THP1 cells were plated in 96-well plates at a density of 3×104 cells/well. Where indicated, SYKi IV was added to cells at a concentration of 10 μM for the indicated duration. Cells were fixed with 4% formaldehyde, permeabilized in 0.5% Triton X-100 for 15 min at RT, and blocked with 4% BSA for 1 h at RT. Cells were then incubated overnight at 4°C with an anti-NFAT-2 antibody (Santa Cruz, sc7294) at 1:250 dilution. Cells were washed in PBST three times and probed with secondary (anti-rabbit) antibody coupled with Alexa 488 flourophor (Cell Signaling, 4412) for 1 h at 1:1000 dilution, followed by washing with PBST and nuclear staining with Hoechst 33,342 (Sigma, 14,533). Images were acquired on a Leica SP8 confocal microscope equipped with a 63X/1.4NA oil objective, an Argon 488 nm laser and a 405 nm diode laser, as well as HyD detectors. Four fields of view were collected for each sample with a voxel size of approximately 360 nm. The fields collected encompassed more than 30 cells. Nuclear co-localization of NFAT-2 was calculated based on the mean fluorescent intensity in the nucleus using the software Imaris (Bitplane).
Nuclear and cytoplasmic fractionation
Nuclear and cytosolic fractions were isolated from cells using NE-PER nuclear and cytosolic fractionation kit (Thermo Fisher, 78,833). THP1 cells (7×106 cells per sample) were PMA differentiated for three days and treated with DMSO control and SYKi IV for the indicated duration. Fractionation was conducted following the manufacturer’s protocol.
Co-culture system and assay for SARS CoV-2
Human primary macrophage cells (5000 cells/well) in 384-well plates were treated with DMSO, SYKi IV or R406 at 10 μM for 24h followed by changing media and resting for 3 days. A549-hACE2 cells (5000 cells/well) were plated onto the primary macrophages in RPMI (10% FBS + HEPES) 16 h prior to infection with SARS CoV-2 variants at an MOI of 0.5 for 24h. Infections with SARS CoV-2 were performed at the SARS CoV-2 core laboratory of NIAID following the approved BSL-3 standard operating procedures. Cells were 4% PFA fixed for 20 min at RT, permeabilized with 0.1% Triton X-100 for 10 min and blocked in 5% BSA for 1 h. SARS CoV-2 infection was measured by staining with anti-Spike antibody (GeneTex, GTX632604) at 1:1000 over-night at 4°C followed by incubation of anti-mouse antibody coupled with Alexaflour 488 (Invitrogen, A21202) at 1:1000 dilutions for 1 h and Hoechst (Life technology, H21492) at 1:10,000 dilutions for 10 min at RT.
Image quantitation of viral infections
Images were captured on a CellInsight CX7 high content imager (Thermo Fisher Scientific) at 20X magnification. In channel 1, nuclear stained objects were identified to establish a mask identifying single cells. In channel 2, the viral protein antibody stained intensity was measured. The channel 2 intensity minimum threshold was set at the median of uninfected cells. Infected cells were scored on the basis of significant channel 2 intensity in a 2 pixel wide ring region drawn around each nuclei from channel 1. Percentage of infected cells were calculated based on the ratio of the number of objects that were scored above the intensity threshold in channel 2 to total number of objects (cell nuclei) in channel 1. Images were captured from 16 different fields of 4 replicate wells done in 2–3 independent experiments.
Western blotting
Differentiated primary human macrophages or THP1 cells at a density of 3 × 105 cells/well were plated in 12-well plates and treated with small molecules and/or LPS as described in figure legends. Cells after treatment were lyzed in 1X SDS-PAGE loading buffer with protease and phosphatase inhibitor cocktails (Roche). Samples were resolved on a 4%–20% gradient SDS-PAGE gel. Following transfer of the proteins, the nitrocellulose membrane was blocked in 5% milk for 1 h and probed with the following antibodies overnight at 4°C: Rabbit anti-phospho-p38 (Cell Signaling, 4511S), Rabbit anti-phospho-p65 (Cell Signaling, 3033S), Mouse anti-GAPDH (Cell Signaling, 97,166), Rabbit anti-phospho-ERK (Cell Signaling, 4370), Mouse anti-NFAT2 (Santa Cruz, sc7294), Mouse anti-hnRNPL (Santa Cruz, sc-32317). Western blots were incubated with respective HRP-conjugated secondary antibodies (anti mouse-HRP, Invitrogen, A16011 and anti-rabbit-HRP, Invitrogen, A16023) and visualized using SuperSignal West Dura Extended Duration Substrate reagents (Thermo Fisher Scientific) in Chemidoc MP gelimager (Bio-rad).
Identification of transcription factors
Transcriptional factors potentially responsible for the observed gene expression changes were identified by two complimentary methods, IPA (The Ingenuity Pathway Analysis software, Qiagen, Redwood City, CA, USA) (Kramer et al., 2014) and MAGIC (Mining Algorithm for Genetic Controllers) (Roopra, 2020). IPA identifies TFs by querying the differentially expressed genes (DEGs) against experimentally validated TF-gene interactions in the Ingenuity Knowledge Base. MAGIC uses data derived from ChIP-seq tracks archived at ENCODE to decipher which TFs are most likely to preferentially bind lists of DEGs. The reference genome used for MAGIC analysis included 19,328 protein-coding genes downloaded from HGNC (2020-11-09, https://www.genenames.org/). The results were compared, and the common TFs identified by both methods were given higher priorities for further verification analysis.
Quantification and statistical analysis
Statistical analyses were performed using Prism software version 9.0 for Mac OS X (GraphPad Software). For PCR, infection and ELISA experiments, relative values were compared either by a paired t test or by two-way ANOVA followed by Sidak’s multiple comparison test or Tukey’s multiple comparison test. All the statistical details of experiments, including the statistical tests used, are listed within each figure legend. Data presented are representative of three or more independent experiments, unless otherwise stated in the figure legend, and are expressed as mean ± SD ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Acknowledgments
We thank Mark Henderson, Matthew Hall, and Min Shen from NCATS for support in setting up the primary small-molecule screen and providing compounds for follow-up studies and Justin Lack at the NIAID Collaborative Bioinformatics Resource for oversight of the RNA-seq experiments as well as the Center for Cancer Research Sequencing Facility and staff at the National Cancer Institute at Fredrick for performing sequencing and initial data processing. We thank Reed Johnson, Bernard Lafont, and Nicole Lackameyer of the NIAID SARS Virology Core (SVC) for providing BSL3 training and SARS-CoV-2 virus stocks. We thank the NIH Department of Transfusion Medicine for providing human blood-derived monocytes and colleagues at the Laboratory of Immune System Biology for helpful discussions. β-glucan used in this study was a generous gift from David Williams of East Tennesse State University. This work was generously supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases and National Center for Advancing Translational Sciences. C.L., Z.T., S.B.-L., and M.J.P. were supported through the NIH Community College Program and Special Projects of the Office of Intramural Training and Education at the NIH.
Author contributions
S.P.J. and I.D.C.F. designed the study. S.P.J., A.S., J.S., M.J.P., L.A., C.L., Z.T., S.B.-L., C.J.B., and M.S. performed the experiments. S.P.J., A.S., J.S., T.E.M., M.S., and I.D.C.F. analyzed the data. S.P.J. and I.D.C.F. wrote the paper. All authors reviewed results and approved the final version of the manuscript.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper received support from a program designed to increase minority representation in their field of research.
Published: October 4, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111441.
Supplemental information
Data and code availability
All data generated or analyzed during this study are included in this article, supplemental information, and source data files.
This paper does not report original code.
The raw NGS data have been deposited to the Gene Expression Omnibus (GEO) and are publicly available (GSE210476).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Aaby P., Jensen H., Samb B., Cisse B., Sodemann M., Jakobsen M., Poulsen A., Rodrigues A., Lisse I.M., Simondon F., Whittle H. Differences in female-male mortality after high-titre measles vaccine and association with subsequent vaccination with diphtheria-tetanus-pertussis and inactivated poliovirus: reanalysis of West African studies. Lancet. 2003;361:2183–2188. doi: 10.1016/S0140-6736(03)13771-3. [DOI] [PubMed] [Google Scholar]
- Arango Duque G., Descoteaux A. Macrophage cytokines: involvement in immunity and infectious diseases. Front. Immunol. 2014;5:491. doi: 10.3389/fimmu.2014.00491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arron J.R., Winslow M.M., Polleri A., Chang C.P., Wu H., Gao X., Neilson J.R., Chen L., Heit J.J., Kim S.K., et al. NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature. 2006;441:595–600. doi: 10.1038/nature04678. [DOI] [PubMed] [Google Scholar]
- Atri C., Guerfali F.Z., Laouini D. Role of human macrophage polarization in inflammation during infectious diseases. Int. J. Mol. Sci. 2018;19:1801. doi: 10.3390/ijms19061801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekkering S., Blok B.A., Joosten L.A., Riksen N.P., Van Crevel R., Netea M.G. In vitro experimental model of trained innate immunity in human primary monocytes. Clin. Vaccine Immunol. 2016;23:926–933. doi: 10.1128/CVI.00349-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekkering S., Quintin J., Joosten L.A., Van Der Meer J.W., Netea M.G., Riksen N.P. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler. Thromb. Vasc. Biol. 2014;34:1731–1738. doi: 10.1161/ATVBAHA.114.303887. [DOI] [PubMed] [Google Scholar]
- Blok B.A., De Bree L.C.J., Diavatopoulos D.A., Langereis J.D., Joosten L.A.B., Aaby P., Van Crevel R., Benn C.S., Netea M.G. Interacting, nonspecific, immunological effects of Bacille calmette-guerin and tetanus-diphtheria-pertussis inactivated polio vaccinations: an explorative, randomized trial. Clin. Infect. Dis. 2020;70:455–463. doi: 10.1093/cid/ciz246. [DOI] [PubMed] [Google Scholar]
- Brandes M., Klauschen F., Kuchen S., Germain R.N. A systems analysis identifies a feedforward inflammatory circuit leading to lethal influenza infection. Cell. 2013;154:197–212. doi: 10.1016/j.cell.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanmee T., Ontong P., Konno K., Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers. 2014;6:1670–1690. doi: 10.3390/cancers6031670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S.C., Quintin J., Cramer R.A., Shepardson K.M., Saeed S., Kumar V., Giamarellos-Bourboulis E.J., Martens J.H., Rao N.A., Aghajanirefah A., et al. mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. 2014;345:1250684. doi: 10.1126/science.1250684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y.H., Shin E.M., Kim Y.S., Cai X.F., Lee J.J., Kim H.P. Anti-inflammatory principles from the fruits of Evodia rutaecarpa and their cellular action mechanisms. Arch Pharm. Res. (Seoul) 2006;29:293–297. doi: 10.1007/BF02968573. [DOI] [PubMed] [Google Scholar]
- Christ A., Gunther P., Lauterbach M.A.R., Duewell P., Biswas D., Pelka K., Scholz C.J., Oosting M., Haendler K., Bassler K., et al. Western diet triggers NLRP3-dependent innate immune reprogramming. Cell. 2018;172:162–175.e14. doi: 10.1016/j.cell.2017.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisan T.O., Cleophas M.C., Oosting M., Lemmers H., Toenhake-Dijkstra H., Netea M.G., Jansen T.L., Joosten L.A. Soluble uric acid primes TLR-induced proinflammatory cytokine production by human primary cells via inhibition of IL-1Ra. Ann. Rheum. Dis. 2016;75:755–762. doi: 10.1136/annrheumdis-2014-206564. [DOI] [PubMed] [Google Scholar]
- Croft M., Benedict C.A., Ware C.F. Clinical targeting of the TNF and TNFR superfamilies. Nat. Rev. Drug Discov. 2013;12:147–168. doi: 10.1038/nrd3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devarakonda C.K.V., Meredith E., Ghosh M., Shapiro L.H. Coronavirus receptors as immune modulators. J. Immunol. 2020;206:923–929. doi: 10.4049/jimmunol.2001062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanucchi S., Fok E.T., Dalla E., Shibayama Y., Borner K., Chang E.Y., Stoychev S., Imakaev M., Grimm D., Wang K.C., et al. Immune genes are primed for robust transcription by proximal long noncoding RNAs located in nuclear compartments. Nat. Genet. 2019;51:138–150. doi: 10.1038/s41588-018-0298-2. [DOI] [PubMed] [Google Scholar]
- Ferreira A.V., Domiguez-Andres J., Netea M.G. The role of cell metabolism in innate immune memory. J. Innate Immun. 2022;14:42–50. doi: 10.1159/000512280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Nicolas O., V'kovski P., Zettl F., Zimmer G., Thiel V., Summerfield A. No evidence for human monocyte-derived macrophage infection and antibody-mediated enhancement of SARS-CoV-2 infection. Front. Cell. Infect. Microbiol. 2021;11:644574. doi: 10.3389/fcimb.2021.644574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger G., D'osualdo A., Schubert D.A., Weber A., Bruscia E.M., Hartl D. Cellular innate immunity: an old game with new players. J. Innate Immun. 2017;9:111–125. doi: 10.1159/000453397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geahlen R.L. Getting Syk: spleen tyrosine kinase as a therapeutic target. Trends Pharmacol. Sci. 2014;35:414–422. doi: 10.1016/j.tips.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwack Y., Sharma S., Nardone J., Tanasa B., Iuga A., Srikanth S., Okamura H., Bolton D., Feske S., Hogan P.G., Rao A. A genome-wide Drosophila RNAi screen identifies DYRK-family kinases as regulators of NFAT. Nature. 2006;441:646–650. doi: 10.1038/nature04631. [DOI] [PubMed] [Google Scholar]
- Hogan P.G., Chen L., Nardone J., Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003;17:2205–2232. doi: 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- Inglese J., Auld D.S., Jadhav A., Johnson R.L., Simeonov A., Yasgar A., Zheng W., Austin C.P. Quantitative high-throughput screening: a titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc. Natl. Acad. Sci. USA. 2006;103:11473–11478. doi: 10.1073/pnas.0604348103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John S.P., Sun J., Carlson R.J., Cao B., Bradfield C.J., Song J., Smelkinson M., Fraser I.D.C. IFIT1 exerts opposing regulatory effects on the inflammatory and interferon gene programs in LPS-activated human macrophages. Cell Rep. 2018;25:95–106.e6. doi: 10.1016/j.celrep.2018.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston B., Conly J. Tumour necrosis factor inhibitors and infection: what is there to know for infectious diseases physicians? Can. J. Infect Dis. Med. Microbiol. 2006;17:209–212. doi: 10.1155/2006/385789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalliolias G.D., Ivashkiv L.B. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat. Rev. Rheumatol. 2016;12:49–62. doi: 10.1038/nrrheum.2015.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar U.K., Joosten L.A.B. Training the trainable cells of the immune system and beyond. Nat. Immunol. 2020;21:115–119. doi: 10.1038/s41590-019-0583-y. [DOI] [PubMed] [Google Scholar]
- Kaufmann E., Sanz J., Dunn J.L., Khan N., Mendonca L.E., Pacis A., Tzelepis F., Pernet E., Dumaine A., Grenier J.C., et al. BCG educates hematopoietic stem cells to generate protective innate immunity against tuberculosis. Cell. 2018;172:176–190.e19. doi: 10.1016/j.cell.2017.12.031. [DOI] [PubMed] [Google Scholar]
- Kleinnijenhuis J., Quintin J., Preijers F., Benn C.S., Joosten L.A., Jacobs C., Van Loenhout J., Xavier R.J., Aaby P., Van Der Meer J.W., et al. Long-lasting effects of BCG vaccination on both heterologous Th1/Th17 responses and innate trained immunity. J. Innate Immun. 2014;6:152–158. doi: 10.1159/000355628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer A., Green J., Pollard J., Jr., Tugendreich S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics. 2014;30:523–530. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey C.J. HPV vaccination in HIV infection. Papillomavirus Res. 2019;8:100174. doi: 10.1016/j.pvr.2019.100174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y.G., Chain B.M., Cho J.Y. Distinct role of spleen tyrosine kinase in the early phosphorylation of inhibitor of kappaB alpha via activation of the phosphoinositide-3-kinase and Akt pathways. Int. J. Biochem. Cell Biol. 2009;41:811–821. doi: 10.1016/j.biocel.2008.08.011. [DOI] [PubMed] [Google Scholar]
- Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N., Sun J., Benet Z.L., Wang Z., Al-Khodor S., John S.P., Lin B., Sung M.H., Fraser I.D. Development of a cell system for siRNA screening of pathogen responses in human and mouse macrophages. Sci. Rep. 2015;5:9559. doi: 10.1038/srep09559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Yang M., Peng Y., Gao M., Yang B. Rutaecarpine ameliorated sepsis-induced peritoneal resident macrophages apoptosis and inflammation responses. Life Sci. 2019;228:11–20. doi: 10.1016/j.lfs.2019.01.038. [DOI] [PubMed] [Google Scholar]
- Ma W.T., Gao F., Gu K., Chen D.K. The role of monocytes and macrophages in autoimmune diseases: a comprehensive review. Front. Immunol. 2019;10:1140. doi: 10.3389/fimmu.2019.01140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A., Netea M.G. Trained innate immunity, epigenetics, and covid-19. N. Engl. J. Med. 2020;383:1078–1080. doi: 10.1056/NEJMcibr2011679. [DOI] [PubMed] [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.J. 2011;17:10–12. [Google Scholar]
- Moorlag S., Khan N., Novakovic B., Kaufmann E., Jansen T., Van Crevel R., Divangahi M., Netea M.G. Beta-glucan induces protective trained immunity against Mycobacterium tuberculosis infection: a key role for IL-1. Cell Rep. 2020;31:107634. doi: 10.1016/j.celrep.2020.107634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosser D.M., Edwards J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourits V.P., Wijkmans J.C., Joosten L.A., Netea M.G. Trained immunity as a novel therapeutic strategy. Curr. Opin. Pharmacol. 2018;41:52–58. doi: 10.1016/j.coph.2018.04.007. [DOI] [PubMed] [Google Scholar]
- Mulder W.J.M., Ochando J., Joosten L.A.B., Fayad Z.A., Netea M.G. Therapeutic targeting of trained immunity. Nat. Rev. Drug Discov. 2019;18:553–566. doi: 10.1038/s41573-019-0025-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullard A. FDA approves first-in-class SYK inhibitor. Nat. Rev. Drug Discov. 2018;17:385. doi: 10.1038/nrd.2018.96. [DOI] [PubMed] [Google Scholar]
- Netea M.G., Dominguez-Andres J., Barreiro L.B., Chavakis T., Divangahi M., Fuchs E., Joosten L.A.B., Van Der Meer J.W.M., Mhlanga M.M., Mulder W.J.M., et al. Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 2020;20:375–388. doi: 10.1038/s41577-020-0285-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netea M.G., Giamarellos-Bourboulis E.J., Dominguez-Andres J., Curtis N., Van Crevel R., Van De Veerdonk F.L., Bonten M. Trained immunity: a tool for reducing susceptibility to and the severity of SARS-CoV-2 infection. Cell. 2020;181:969–977. doi: 10.1016/j.cell.2020.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netea M.G., Joosten L.A., Latz E., Mills K.H., Natoli G., Stunnenberg H.G., O'neill L.A., Xavier R.J. Trained immunity: a program of innate immune memory in health and disease. Science. 2016;352:aaf1098. doi: 10.1126/science.aaf1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novakovic B., Habibi E., Wang S.Y., Arts R.J.W., Davar R., Megchelenbrink W., Kim B., Kuznetsova T., Kox M., Zwaag J., et al. Beta-glucan reverses the epigenetic state of LPS-induced immunological tolerance. Cell. 2016;167:1354–1368.e14. doi: 10.1016/j.cell.2016.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura H., Aramburu J., Garcia-Rodriguez C., Viola J.P., Raghavan A., Tahiliani M., Zhang X., Qin J., Hogan P.G., Rao A. Concerted dephosphorylation of the transcription factor NFAT1 induces a conformational switch that regulates transcriptional activity. Mol. Cell. 2000;6:539–550. doi: 10.1016/s1097-2765(00)00053-8. [DOI] [PubMed] [Google Scholar]
- Okamura H., Garcia-Rodriguez C., Martinson H., Qin J., Virshup D.M., Rao A. A conserved docking motif for CK1 binding controls the nuclear localization of NFAT1. Mol. Cell Biol. 2004;24:4184–4195. doi: 10.1128/MCB.24.10.4184-4195.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paris D., Ait-Ghezala G., Bachmeier C., Laco G., Beaulieu-Abdelahad D., Lin Y., Jin C., Crawford F., Mullan M. The spleen tyrosine kinase (Syk) regulates Alzheimer amyloid-beta production and Tau hyperphosphorylation. J. Biol. Chem. 2014;289:33927–33944. doi: 10.1074/jbc.M114.608091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A.A., Zhang Y., Fullerton J.N., Boelen L., Rongvaux A., Maini A.A., Bigley V., Flavell R.A., Gilroy D.W., Asquith B., et al. The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J. Exp. Med. 2017;214:1913–1923. doi: 10.1084/jem.20170355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintin J., Saeed S., Martens J.H.A., Giamarellos-Bourboulis E.J., Ifrim D.C., Logie C., Jacobs L., Jansen T., Kullberg B.J., Wijmenga C., et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe. 2012;12:223–232. doi: 10.1016/j.chom.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers N.C., Slack E.C., Edwards A.D., Nolte M.A., Schulz O., Schweighoffer E., Williams D.L., Gordon S., Tybulewicz V.L., Brown G.D., Reis E Sousa C. Syk-dependent cytokine induction by Dectin-1 reveals a novel pattern recognition pathway for C type lectins. Immunity. 2005;22:507–517. doi: 10.1016/j.immuni.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Roopra A. MAGIC: a tool for predicting transcription factors and cofactors driving gene sets using ENCODE data. PLoS Comput. Biol. 2020;16:e1007800. doi: 10.1371/journal.pcbi.1007800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed S., Quintin J., Kerstens H.H., Rao N.A., Aghajanirefah A., Matarese F., Cheng S.C., Ratter J., Berentsen K., Van Der Ent M.A., et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. 2014;345:1251086. doi: 10.1126/science.1251086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sariol A., Perlman S. Lessons for COVID-19 immunity from other coronavirus infections. Immunity. 2020;53:248–263. doi: 10.1016/j.immuni.2020.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savvides S.N., Elewaut D. Small-molecule inhibitors get pro-inflammatory TNF into shape. Nat. Rev. Rheumatol. 2020;16:189–190. doi: 10.1038/s41584-020-0388-2. [DOI] [PubMed] [Google Scholar]
- Song Y., Buchwald P. TNF superfamily protein-protein interactions: feasibility of small- molecule modulation. Curr. Drug Targets. 2015;16:393–408. doi: 10.2174/1389450116666150223115628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Heijden C., Noz M.P., Joosten L.A.B., Netea M.G., Riksen N.P., Keating S.T. Epigenetics and trained immunity. Antioxid. Redox Signal. 2018;29:1023–1040. doi: 10.1089/ars.2017.7310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meer J.W., Joosten L.A., Riksen N., Netea M.G. Trained immunity: a smart way to enhance innate immune defence. Mol. Immunol. 2015;68:40–44. doi: 10.1016/j.molimm.2015.06.019. [DOI] [PubMed] [Google Scholar]
- Villasenor T., Madrid-Paulino E., Maldonado-Bravo R., Perez-Martinez L., Pedraza-Alva G. Mycobacterium bovis BCG promotes IL-10 expression by establishing a SYK/PKCalpha/beta positive autoregulatory loop that sustains STAT3 activation. Pathog Dis. 2019;77:ftz032. doi: 10.1093/femspd/ftz032. [DOI] [PubMed] [Google Scholar]
- Wang W., Ren S., Lu Y., Chen X., Qu J., Ma X., Deng Q., Hu Z., Jin Y., Zhou Z., et al. Inhibition of Syk promotes chemical reprogramming of fibroblasts via metabolic rewiring and H2 S production. EMBO J. 2021;40:e106771. doi: 10.15252/embj.2020106771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson B.G., Roberts C.W. SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer. 2011;11:481–492. doi: 10.1038/nrc3068. [DOI] [PubMed] [Google Scholar]
- Wright D.E., Wagers A.J., Gulati A.P., Johnson F.L., Weissman I.L. Physiological migration of hematopoietic stem and progenitor cells. Science. 2001;294:1933–1936. doi: 10.1126/science.1064081. [DOI] [PubMed] [Google Scholar]
- Yanagi S., Inatome R., Takano T., Yamamura H. Syk expression and novel function in a wide variety of tissues. Biochem. Biophys. Res. Commun. 2001;288:495–498. doi: 10.1006/bbrc.2001.5788. [DOI] [PubMed] [Google Scholar]
- Yanagi S., Kurosaki T., Yamamura H. The structure and function of nonreceptor tyrosine kinase p72syk expressed in hematopoietic cells. Cell. Signal. 1995;7:185–193. doi: 10.1016/0898-6568(94)00088-s. [DOI] [PubMed] [Google Scholar]
- Yi Y.S., Son Y.J., Ryou C., Sung G.H., Kim J.H., Cho J.Y. Functional roles of Syk in macrophage-mediated inflammatory responses. Mediators Inflamm. 2014;2014:270302. doi: 10.1155/2014/270302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura A., Ito M., Chikuma S., Akanuma T., Nakatsukasa H. Negative regulation of cytokine signaling in immunity. Cold Spring Harb. Perspect. Biol. 2018;10:a028571. doi: 10.1101/cshperspect.a028571. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this article, supplemental information, and source data files.
This paper does not report original code.
The raw NGS data have been deposited to the Gene Expression Omnibus (GEO) and are publicly available (GSE210476).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.