

Abstract
Schwann cells (SCs) are the most abundant cell type in the nerves in the peripheral nervous system and compose a family of subtypes that are endowed with a variety of different functions. SCs facilitate the transmission of neural impulses, provide nutrients and protection for neurons, guide axons in nerve repair, and regulate immune functions. In the context of cancer, recent studies have revealed an active role of SCs in promoting cancer cell invasion, modulating immune responses, and transmitting pain sensation.
Keywords: cancer, nerve repair, reprogramming, schwann cells
Graphical Abstract
1. Introduction
Schwann cells (SCs) are glial cells that ensheath axons of neuron in the peripheral nervous system and are necessary for their maintenance and function. They originate from neural crest cells that then form several subtypes of SCs[1–3] (Figure 1). At an adult stage, SCs can be either myelinating or nonmyelinating. Myelinating SCs form the insulating myelin sheaths of peripheral axons. They produce myelin, a lipid-rich membrane with specific proteins, and facilitate the transmission of neuronal depolarization at high speed. Nonmyelinating SCs serve different functions. What is conventionally named non-myelinating SCs are Remak SCs, SCs that ensheath multiple small-caliber axons in “Remak bundles.” There are other non-myelinating SCs. They include terminal SCs and nerve repair SCs. Terminal SCs are present at the neuromuscular junction and provide chemical and physical support to the neuro-muscular synapse. Nerve repair SCs are generated at sites of nerve injury through transcriptional reprogramming of myelinating and Remak SCs in response to trauma. The ability of SCs to reprogram at an adult stage reflects the highly plastic nature of these cells. In this review, we report on the expanding recognition of SCs as active contributors to cancer progression.
Figure 1.
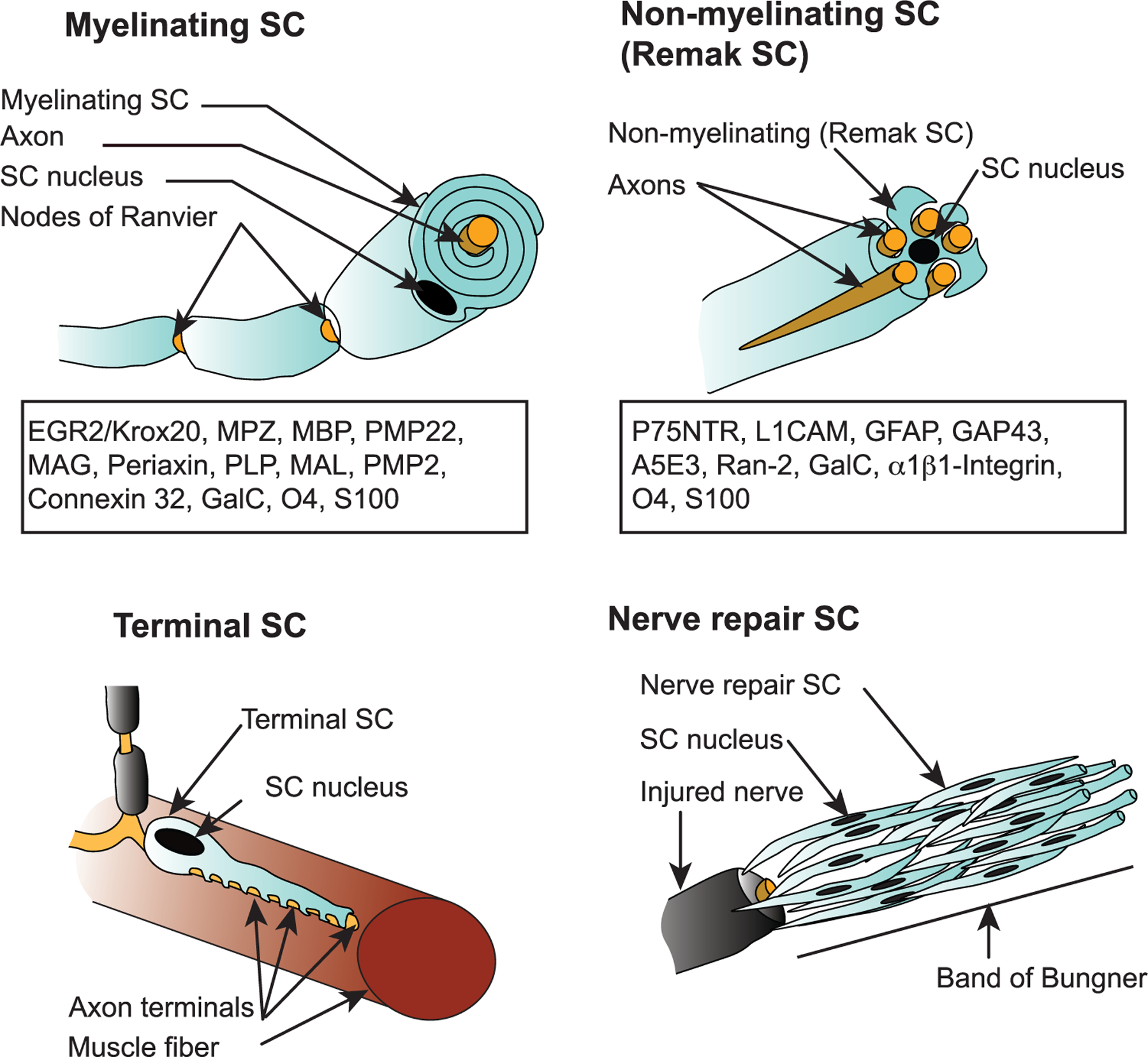
SC subtypes and their markers. Mature SCs comprise two major subtypes: myelinating and nonmyelinating SCs. Myelinating SCs produce myelin sheaths around large caliber axons and express characteristic markers, such as myelin protein zero (MPZ), myelin associated glycoprotein (MAG), and myelin basic protein (MBP). Nonmyelinating SCs or Remak SCs ensheath several smaller caliber axons and present markers corresponding to more immature state of SCs such as low affinity neurotrophin receptor p75 (p75NTR/NGFR) and L1 cell adhesion molecule (L1CAM). Terminal SCs are nonmyelinating SCs that surround axon terminals, for example at the neuromuscular junctions. Nerve repair SCs are nonmyelinating SCs that derive from myelinating and Remak SCs at nerve injury. They organize into bands of Bungner where they direct axonal regeneration. SCs in cancer tissue share strong similarities with nerve repair SCs. Early growth response 2: EGR2 (previously named Krox20): peripheral myelin protein 22: PMP22, proteolipid protein:PLP, Myelin and lymphocye protein: MAL, peripheral myelin protein 2: PMP2, Connexin 32, galactocerebroside: GalC, O4, glial fibrillary acidic protein: GFAP, Growth Associated Protein 43: GAP43, S100 calcium binding protein: S100, Oligodendrocyte marker O4:O4, GTP-binding nuclear protein Ran-2: Ran-2.[1]
1.1. Reprogramming of SCs in Cancer
It is now well accepted that “cancer is a wound that will not heal,” as first reported by Rudolph Virchow in the 19th century.[4] Nerve injury described in cancer is part of tumor-induced injury. Among the wound repair mechanisms, the mechanisms of nerve repair occur during cancer and include SC reprogramming. The reprogramming of SCs was first described during nerve repair after nerve injury, then during mycobacterium leprae infection, and more recently in the context of cancer. The ability to reprogram adult stage SCs to a more undifferentiated SC type reflects the very plastic and dynamic nature of SCs.
SC reprogramming during nerve repair involves a change in the expression of ≈4000 genes, and is controlled by c-Jun, Notch, Sox2, and Mitogen-activated protein kinase (MAPK) signaling in addition to other factors.[5,6] Nerve repair SCs are very motile, release neurotrophic factors and chemokines, recruit immune cells, clear myelin by autophagy and phagocytosis, and reorganize into cellular tracks called Büngner bands, forming a pathway to guide the regeneration of damaged axons.[7]
Reprogrammed SCs have been found in close association with a variety of cancer types including pancreatic cancer,[8,9] thyroid cancer,[8] colon cancer,[9] lung cancer,[10] neuroblastic cancer,[11] and skin cancer.[12] SCs in association with cancers reprogram to express high level of glial fibrillary acidic protein (GFAP), similar to nerve repair SCs that also undergo reprogramming.[8,9] GFAP+ SCs are found at premalignant stage of pancreatic and colon cancer and migrate toward the cancer cells.[9] GFAP+ SCs closely associate with cancer cells, dispersing them and stimulating their migration and invasion.[8] These SCs also attract and activate immune cells.[11,12]
In patients with melanoma, GFAP+ SCs increase in number in the tissue adjacent to the melanoma tumor.[12] Transcriptome analysis of such micro-dissected tissue showed increased expression of genes participating in neuro-regeneration and known to be upregulated after nerve injury. These genes are enriched in axon guidance and immune response pathways. Cultured SCs in melanoma-conditioned medium exhibit a gene expression profile resembling that of repair SCs, with an increased expression of genes such as Id2, Egr1, c-Jun, and Sox2, and a downregulation of genes promoting myelination such as Sox10, Egr2, and Oct6. Erk and Akt signaling pathways are activated in these cells as in nerve repair SCs. In addition, similar to nerve repair SCs, SCs cultured with melanoma-conditioned medium increase the expression of genes involved in nerve repair, extracellular matrix (ECM) reorganization, immune modulation, chemotaxis, and myelin phagocytosis, such as MMP1, FGF2, VEGF, IL6, MIF, AREG, BDNF, GDNF, Galectin-3, and TGF-β1.[12]
Transcriptome and proteomic analysis of human stromal SCs in ganglioneuromas shows the expression of nerve repair-associated genes.[11] As with nerve repair SCs, these SCs show altered expression of genes involved in axon guidance, lipid/myelin degradation/metabolism, basement membrane formation/ECM (re-)organization, phagocyte attraction, and MHC-II mediated immune regulation. These SCs also produce EGFL8, a matricellular protein that promotes neuritogenesis. SC EGFL8 induces neurite outgrowth and neuronal differentiation of neuroblastoma cells in culture, acting as a neuritogen and rewires cellular signaling by activating kinases involved in neurogenesis.
The factors that specifically induce SC reprogramming following nerve trauma and in cancer invasion remain undefined. Shurin et al. proposed that SC reprogramming is prompted directly by melanoma cells, which induce nerve injury in the tissue adjacent to the tumor.[12] In pancreatic cancer, Demir et al. proposed that hypoxia, cancer cells, and neuroinflammation activate the SCs, which depend on IL-6.[13]
However, IL-6 that is also produced during nerve repair and is involved in SC ability to recruit macrophages[14] is not critical in the overall process of nerve repair since IL-6 KO mice do not lose their ability to undergo nerve repair.[15]
In the cancer context, SCs conserve their nerve repair functions that include the ability to promote axon regrowth and to attract immune cells. In addition, SCs promote cancer invasion. In most cancer types, SCs and reprogrammed SCs have a pro-tumorigenic effect, through processes we describe below (cancer growth, cancer migration, and immune function). In contrast, SCs in neuroblastoma are proposed to provide an anti-tumorigenic effect through their neuroprotective and neuritogenic factors and because of their presence in the less aggressive forms of neuroblastoma.[11] These effects can be exploited for therapy of the more severe form of neuroblastoma that lacks stromal SCs.[11]
In contrast to reprogramming into nerve repair SCs, SCs are also sensitive to factors that induce myelination in cancer. Bressy et al. describe an IL6-related stem cell-promoting factor LIF as a factor released by fibroblasts in pancreatic cancer and whose effect on SCs induces cell migration and differentiation with an increase in the expression of POU3F2 and S100, as assessed in in vitro assays by immunofluorescence, immunoblotting, and PCR using the SC line sNF96.2.[16]
1.2. SCs Induce Cancer Cell Proliferation, Migration, and Guidance
SCs affect cancer progression at primary tumor sites and contribute to metastatic spread. Perineural invasion (PNI) is the invasion of cancer cells in or around the nerves. PNI is considered a form of metastasis since cancers propagate away from the primary site following neural pathways. Several studies implicate the role for SCs in the process of PNI that involves both cancer growth and cancer migration. In addition, SCs affect the conventional circulatory metastatic route. A recent study implicated SCs in enabling the progression of highly metastatic palmitate-treated oral cancer cells.[17] SCs in cancer invasion exert pro-tumorigenic effects by affecting cancer growth and cancer migration through a variety of processes.
2. Cancer Growth
SCs directly affect cancer growth by secreting a variety of molecules including growth factors, cytokines, and chemokines (Figure 2). Nerve repairing SCs after nerve injury produce the neurotrophic factors NGF, BDNF, and NT3[18] that are also secreted by SCs from pancreatic cancer.[19] The NGF receptors p75NTR and TrkA are present in PDAC and have higher expression in pancreatic cancers as compared to the normal pancreatic tissue.[19,20] Furthermore, P75NTR is associated with PNI in pancreatic cancer[21] and is also associated with PNI in cutaneous squamous cell carcinoma.[22,23] P75NTR is expressed in cancer stem cells of head and neck squamous cell carcinoma. Preventing NGF binding to p75NTR using blocking antibodies inhibits cell proliferation and tumor formation.[24]
Figure 2.
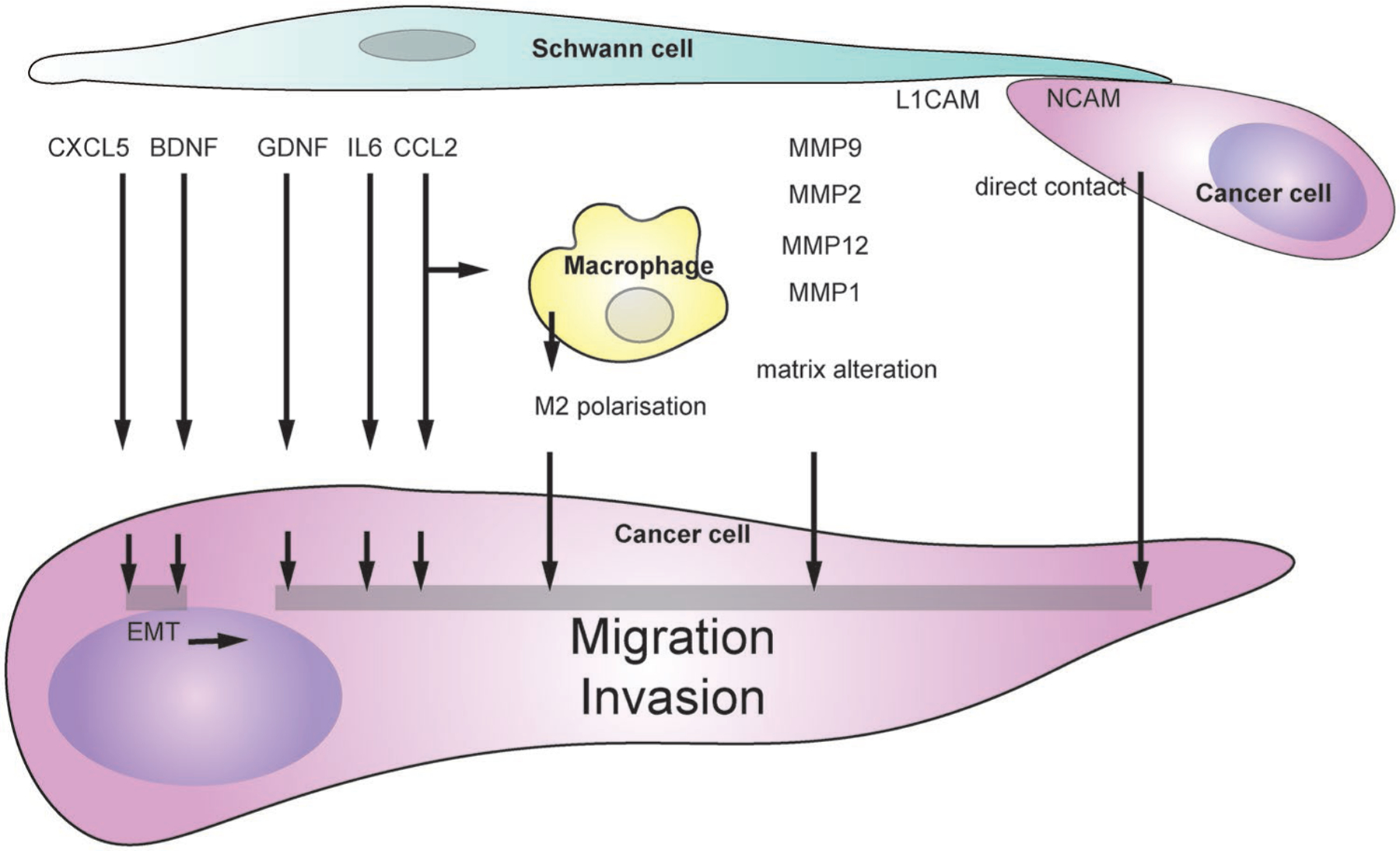
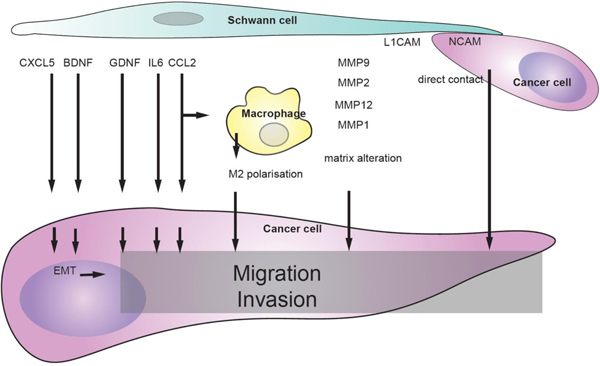
Cancer migration and invasion induced by SCs. SC-derived factors inducing cancer migration and invasion include CXCL5 and BDNF that stimulate epithelial to mesenchymal transition, GDNF, IL6, and CCL2 that stimulate cancer migration and invasion directly or indirectly through macrophages, MMPs that modify extracellular matrix. SCs the direct contact between SCs and cancer cells induced cancer invasion. SC-derived factors inducing cancer migration and invasion. SCs release several factors that stimulate cancer migration and invasion directly or indirectly through macrophages.
Similarly, p75NTR knock-down in melanoma cancer cells prevents tumor formation in a xenograft mouse model. The loss of p75NTR in these cells is associated with a loss of stemness marker.[25] P75NTR is also present in a population of breast cancer stem cells, in which it mediates a self-renewal effect by NGF.[26] It might be possible that SC NGF binds P75NTR on these breast cancer stem cells and mediates a similar effect as breast cancer stem cell NGF.
BDNF binds to its receptor TrkB and promotes the self-renewal of cancer stem cells in breast cancer. TrkB is present in the cancer stem cells of recurrent triple-negative breast cancer and mediates the effect of BDNF in promoting their expansion.[26] TrkB is also a receptor for NT3. It is possible that SC BDNF or SC NT3 may also stimulate cancer growth. The inflammatory cytokine TNF-α is another factor released by SCs that promotes cancer growth. TNFα stimulates the proliferation of human oral squamous cell carcinoma cells.[27]
SC-derived chemokine CCL2 also affects cancer growth by directly enhancing cancer proliferation of the cervical cancer cell lines HeLa and ME-180 in co-culture experiments[28] and by indirectly modulating the immune environment. This indirect effect of SC-released cytokines occurs by attracting macrophages and polarizing them into M2 phenotype, which are macrophages with immunosuppressive function, therefore allowing tumor growth.[12] In pancreatic cancer, SCs released CCL2 that recruit monocytes at sites of PNI that enhance invasion through cathepsin B secretion.[29] SCs allow M2 polarization of macrophages that stimulate proliferation of lung cancer cells A549 and H1299.[30] SCs activated by melanoma cells are more effective in chemoattracting CD11+ dendritic cells in an in vitro assay as compared to control SCs.[31]
SCs exert an indirect effect on melanoma growth. B16 melanoma cells failed to grow in presence of SCs in vitro, but grew tumors in vivo with Schwann cells. The authors proposed that melanoma-activated SCs modulate the immune system and the extracellular matrix to contribute to the melanoma growth.[12]
3. Cancer Migration and Invasion
During PNI, cells migrate along the nerves. Factors stimulating cancer migration in PNI include the glial cell line-derived neurotrophic factor GDNF,[32] an SC derived neurotrophic factor that has increased expression in SCs during nerve repair.[5] GDNF attracts pancreatic cancer MiaPaCa2 cells expressing the GDNF receptors RET and GFRα1 both in vitro and in an in vivo model of PNI. The GDNF receptors RET and GFRα1 are expressed in pancreatic cancer cells and have a higher expression than in nonmalignant cells in adjacent normal tissues.[32]
Other factors released by SCs that stimulate cancer migration include the chemokine CCL2 and the cytokine IL6. As with nerve repair,[18] these molecules are secreted by SCs during cancer invasion. In addition to attracting and polarizing macrophages into an M2 phenotype, CCL2 also attracts cancer cells.[28,33] CCL2 promotes PNI in prostate cancer through the CCR2 receptor[33] and in cervical cancer as well.[28]
The cytokine IL6, released by SCs, leads to pancreatic cancer cell migration through STAT3 activation and signaling.[34] This effect of IL6 has been demonstrated in vitro with wound healing, migration, and invasion assays using MiaPaCa-2 and AsPC-1 cells exposed to SC conditioned medium with or without blocking antibody to IL6. In this study, IL6 is secreted by SCs among other cytokines after NF-κB activation following IL1 β secretion by cancer cells.[34]
Another study demonstrated a role of TGF-β signaling in SC-induced cancer invasion.[35] Invasion by the pancreatic cancer cell line Capan-2 was induced by SC conditioned medium through TGF-β signaling, and is reduced by a TGF-β type1 receptor inhibitor. TGF-β is secreted by SCs as well as other members of the TGF-β superfamily, including GDNF and activin.
Cells may increase their migratory and invasive abilities during epithelial-to-mesenchymal transition (EMT), one of the critical steps in the acquisition of invasive and metastatic potential. SC-derived BDNF promotes EMT in salivary adenoid cystic carcinoma.[36] The adenoid cystic carcinoma cell line SACC-83 co-cultured with SCs change their morphology from an epithelial to a mesenchymal shape, increase their N-cadherin and vimentin expression and decrease their expression of E-cadherin. Pharmacologic inhibition of the BDNF receptor TrkB reduces SC-induced migration of SACC-83 cells. Another study showed that SCs increase the expression of the EMT markers Snail and Twist in two murine lung cancer cell lines through the SC-derived chemokine CXCL5 and its receptor CXCR2.[37] These studies demonstrated that SCs may increase EMT, migration, and invasion of cancer cells.
3.1. Matrix Alteration
Cell migration and invasion depend in part on the surrounding matrix. Cancer cells themselves and other surrounding cells in the tumor microenvironment including SCs can contribute to ECM modification. SCs seeded in Matrigel are able to digest the matrix and create pathways enabling cancer cell invasion in a 3D invasion assay.[8] Cancer cell activated SCs, as they do in nerve repair, produce metalloproteinases (MMPs), secreted proteolytic enzymes that can degrade matrix. Such SC MMPs include MMP2, MMP9, and MMP12, in cervical cancer,[28] MMP1 in melanoma.[12] SCs can also regulate cancer cell expression of MMP2 and MMP9 by releasing L1CAM and thus enhance perineural invasion. L1CAM is expressed in both SCs and cancer cells, and treatment of PDAC in KPC mice with an anti-L1CAM antibody leads to diminished nerve density and neural invasion.[38]
The components of the extracellular matrix made by SCs, such as laminin and collagen, are important during cancer migration. The transcriptome analysis of reprogrammed SCs in cancer shows a change in ECM components and factors involved in the matrix reorganization in melanoma[12] and neuroblastoma.[11] In addition to collagen and laminin, the matricellular protein EGFL8 has been identified as a neuritogenic factor released by stromal SCs in neuroblastoma.[11] Furthermore, analysis of proteins secreted by SCs stimulated by pancreatic cancer cells also identified ECM components and enzymes participating in matrix reorganization including cathepsin-D and MMP2.[39]
SC extracellular-matrix components also play an important role in transmitting oral cancer metastasis.[17] Cells pretreated with palm oil acquire a metastatic behavior and carry epigenetic memories for this feature. RNA-seq analysis showed high levels of ECM components formed by tumor-associated SCs. Furthermore, degrading SC-extracellular components by expressing a bacterial chondroitinase ABC in the cancer cells reduces their metastatic potential.[17]
3.2. Protrusion and Guidance
In a 3D in vitro assay, SCs seeded in Matrigel enhance cancer cell invasion into the matrix as compared with fibroblasts or with no SCs present. The presence of SC conditioned media, SC at the bottom of the gel matrix, or tunnels alone within the Matrigel after SCs are killed with puromycin is each insufficient to induce MiaPaCa-2 cancer invasion.[8] The physical presence of live SCs is necessary for enhanced cancer invasion. Time-lapse microscopy showed that SCs induce cancer migration by dispersing cancer cell clusters by direct cell contact. Cultured SCs intercalate between the cancer cells through their long processes. Although the role of GFAP has not been investigated in SCs, it is interesting to note that GFAP is abundant in astrocytes, glial cells from brain, in which a role in cell shape, motility, and long processes formation has been reported,[40,41] suggesting that GFAP might also have a role during the physical interaction between SCs and cancer cells by modulating SC processes functions.
The remodeling of cancer cell clusters by SCs also occurs in neuroblastoma since the presence of SCs is associated with cancer cell alignment.[11] In addition, at the site of contact, a cancer cell develops a protrusion in the direction of cell migration. The formation of a protrusion is a critical step for cells initiating cell migration. The cell adhesion molecule NCAM1 in SCs is involved in the process of protrusion generation in cancer cells and cancer displacement. In vitro and in vivo models of PNI show diminished PNI in absence of NCAM1 in SCs.[8]
Thus, the mechanisms of cancer guidance by SCs share strong similarities with those of SCs facilitating axonal regeneration during nerve repair. SCs allow axonal regeneration through the release of axonal guidance molecules, direct contact of axons through adhesion molecules, and by modifying the extracellular matrix to favor neurite extension.
4. Immune Functions of SCs
In the nerve repair setting, SCs display immune functions that include the recognition of antigens, the presentation of antigens and the transmission of an immune response. In the cancer setting, the role of SCs is less clear, but interest in the neuroimmune axis is growing. Whereas recent studies investigate the role of neurons in immune modulation, SCs might also play a role in this axis.
SCs can recognize antigens. They express Toll-like receptors (TLRs), molecules commonly expressed on T cells, and on antigen-presenting cells (APCs). TLRs play a key role in the activation of innate immunity by recognizing highly conserved molecules expressed by pathogens. TLRs also recognize endogenous ligands that are normally not present in the extracellular matrix called alarmins, which are released by cells upon tissue injury or cell death. High levels of alarmin production are also seen in many types of cancer such as breast, colon, pancreatic cancer, melanoma, and glioblastoma.[42,43]
One example of an alarmin is high mobility group box 1 (HMGB1). HMGB1 released by chemotherapy from cancer cells binds to TLR4 and induce antitumor T-cell immunity.[44] HMGB1 is released at site of injury after sciatic nerve axotomy and increases the expression of antigen receptors including TLRs in SCs.[45]
SCs have been described as antigen presenting cells (APCs). Conventional APCs include macrophages, dendritic cells, and B lymphocyte. APCs phagocytose exogenous antigens and degrade them into peptides, which are then presented extracellularly by MHC class II molecules. SCs demonstrate phagocytosis activity with macrophages during Wallerian degeneration after nerve injury by internalizing myelin debris.[46] SCs express MHC class II molecules in immune-mediated disorders of the peripheral nervous system[47,48] and with Mycobacterium Leprae infection.[49] SCs can present foreign and exogenous autoantigens to antigen-specific T cells. Foreign antigens include tuberculine purified protein derivative[50] and Leprae antigen.[49,51] Exogenous autoantigens include myelin basic protein[46,50] and myelin component P2.[52]
SCs induce a T cell response to antigen presentation. Presentation of the endogenous myelin protein P2 by SCs through MHC class II molecules can restimulate resting antigen-specific CD4+ T cell lines.[52] CD4+ T cells can also be activated by SCs presenting M. Leprae antigen and are involved in the elimination of these infected SCs.[49] Furthermore, SCs can act as an APC in an animal model.[53] Conditional deletion of MHC-II β-chain from myelinating SCs in mice leads to MHC-II expression by SCs and post-traumatic CD4+ T cell infiltration and axonal degeneration in vivo.[53]
SCs act as “non-professional” APCs: they are cells that behave as APCs but are less efficient. SCs were shown to need high concentration of myelin basic protein (MBP) antigen to activate autoimmune T cell lines in an in vitro experiment. These SCs needed ≈100-fold higher concentrations of MBP than professional APCs.[50] In addition, MHC class II proteins are constitutively expressed in conventional APCs, but not in SCs. Human and rat SCs in vitro constitutively express low levels of MHC class I but not MHC class II.[54–59] However, upon stimulation with the proinflammatory cytokine IFN-γ, MHC class II molecules are detected in SCs and can be increased by the addition of TNF-α .[57–62]
Other markers of APCs present on SCs include the antigen-presenting molecules CD74,[63] CD1a,[63] CD1b,[63] and CD1d,[64] the co-stimulatory molecules B7–1 (CD80)[63] and BB-1,[65] and the cell adhesion molecule CD58[63] Elements indicate that SCs might act as APCs in the cancer context. Transcriptome analysis of SCs in neuroblastoma revealed MHC-II expression from stromal SCs.[11]
SCs also regulate the immune response by releasing pro- and anti-inflammatory mediators. SCs produce a large variety of cytokines, which can act as immunomodulators.[66] These include interleukin (IL)-1, a cytokine involved in the initiation of an immune response by T cell activation,[67,68] the proinflammatory cytokines IL-6,[69,70] TNF-α ,[70,71] TGF-β ,[72–74] leukemia inhibitory factor (LIF), and CCL2 (MCP1). Other proinflammatory and immunoregulatory mediators, produced by SCs include prostaglandin E2, thromboxane A2, and leuktriene C4.[75,76]
SCs also express nuclear transcription factor-kB (NF-kB), which controls the expression of a variety of immunological mediators including cytokines, their receptors, and components of their signal transduction. Two NF-kB complexes, p65/p50 and p50/p50, can be activated and regulated in human SCs.[77] A natural inhibitor of NF-kB, IkB has also been detected in large amounts in SCs.[78]
In the context of cancer, SCs modulate the immune response by releasing some of these mediators. SCs release CCL2 that attracts monocytes to sites of cancer invasion that differentiate into protumorigenic macrophages.[12,29] SCs also release TNF-α. SCs treated with melanoma cells attract bone marrow-derived myeloid-derived suppressor cells (MDSCs) and CD11+ dendritic cells significantly stronger than control SCs.[31,79] The authors mention that SCs treated with a variety of tumor cell lines in vitro or from tumor-bearing mice have increased expression of various immunomodulator factors such as IL-1Ra, TNF-α, CCL3 (MIP-1 α), CCL4 (MIP-1β), CXCL2 (MIP-2), CXCL12 (SDF-1), and CXCL13 (BCA-1).[79]
5. Pain
Pain can be subdivided into two types: nociceptive pain and neuropathic pain. Nociceptive pain results from the detection of intense or noxious stimuli by specialized sensory neurons, the nociceptors, and this information is then transmitted to the spinal cord and brain. Neuropathic pain, such as pain after nerve injury, occurs in the absence of any stimulus or with reduced nociceptive thresholds, such that normally innocuous stimuli produce pain. Pain is highly prevalent in cancer and contributes to the morbidity of the disease. Cancer can induce both nociceptive and neuropathic pain. Neuropathic pain is found in cancers that induce nerve damage, such as with perineural invasion, or that increase nerve density and nerve hypertrophy in the tumor microenvironment.
Pancreatic ductal adenocarcinoma is associated with several of neuropathic features, including PNI, pancreatic neuroinflammation (neuritis), nerve hypertrophy, and nerve density. These features are associated with severity of abdominal pain syndrome in patients.[80] The concomitant association of these features with the presence of nerve repair SCs[8,9,13] and the reported contribution of SCs in the development and persistence of pain[81] suggest a role for these SCs in cancer.
SCs can act as pain modulator by acting on cancer cells and immune cells that transmit pain. As SCs stated in the previous section SCs modulate the immune response and recruit macrophages at sites of cancer.[29,79] Inflammation causes pain and several actors of the inflammation process including macrophages contribute to pain transmission.[82] In rat models of nerve injury, tactile hypersensitivity correlates with the number of macrophages at the site of injury.[83]
Cancer releases algogenic factors, such as NGF, TNF-α, and BDNF (Figure 3). These mediators excite and sensitize primary afferent neurons.[84] By increasing cancer growth, SCs contribute to pain increase. SCs can also release pain mediators.[81,85] A recent study showed that in oral cancer, SCs activated by cancer cells or TNF-α release the pain mediators NGF and TNF-α .[27] NGF and GDNF have also been reported to contribute to the initiation of pain in response to nerve injury.[81,86] Other molecules generated by SCs such as CCL3, CCL4, and CXCL2 can also contribute to neuropathic pain after nerve injury.[31,81] Interestingly, SCs also produce factors that can counteract some of these cytokines such as IL-10, erythropoietin, and erythropoietin-receptor[85] (Figure 3).
Figure 3.
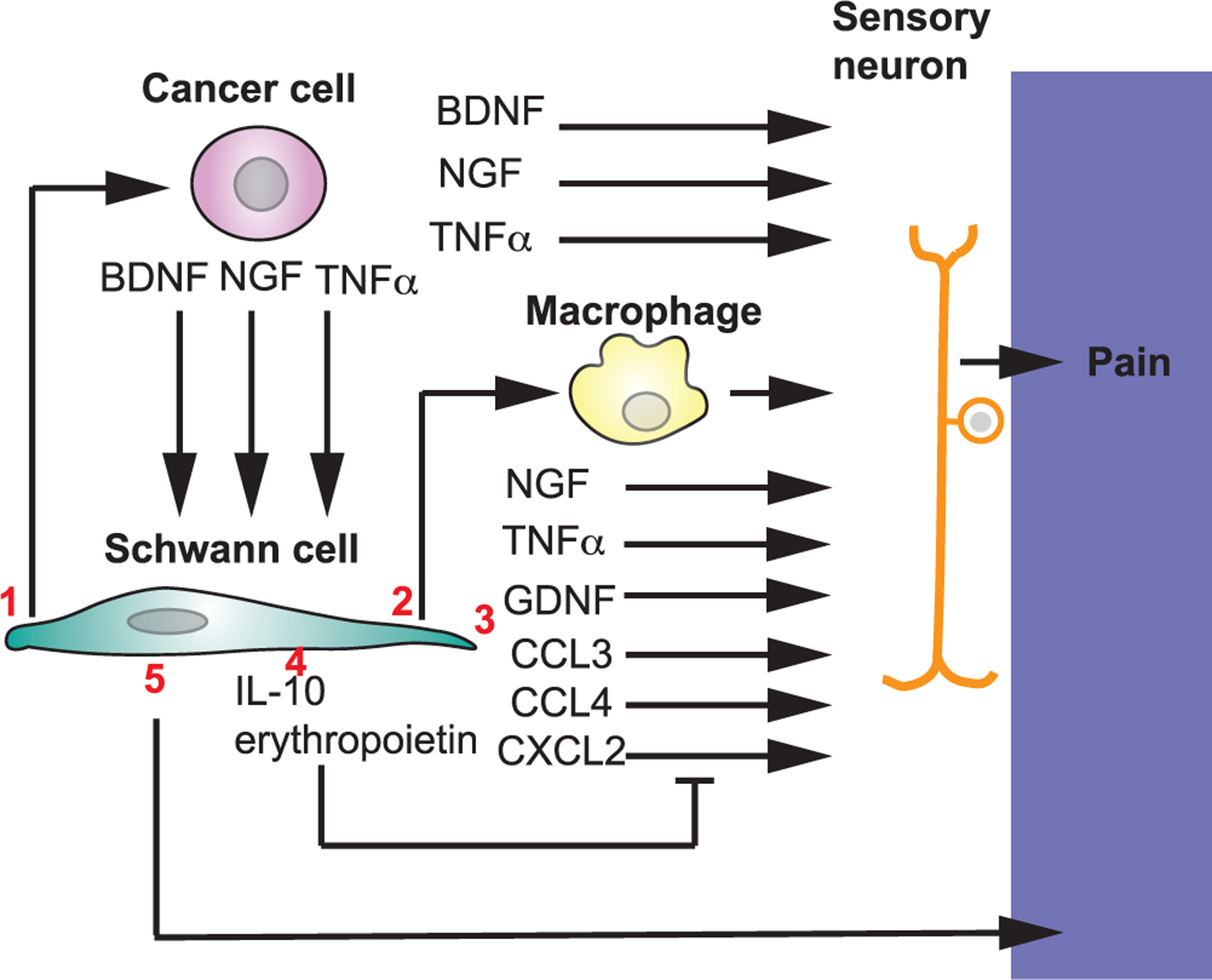
SCs and pain in cancer. 1) SCs increase cancer growth. Cancer cells induce pain by releasing factors that stimulate sensory neurons. 2) SCs recruit macrophages at cancer site. Macrophages in inflammation contribute to pain. 3) SCs can release factors inducing pain and 4) factors that counteract these factors. 5) SCs can act as nociceptors and transmit pain.
Experiments using transgenic conditional mice demonstrate that SCs are involved in pain transmission.[87–90] Mice with deficits in SCs are highly sensitive to touch. The deletion of the gene encoding LDL receptor-related protein-1 in SCs induces abnormalities in axon myelination and in axon ensheathment, inducing an increase in the pain after partial sciatic nerve ligation.[87] Deletion of the GABA-B receptor in myelinating SCs induces morphological and molecular changes in peripheral myelin and causes hyperalgesia and tactile allodynia.[88] Disruption of Neuregulin1-ErbB receptor signaling in nonmyelinating SCs induces SC proliferation and death, decreases of unmyelinated axons, and a loss of thermal sensitivity.[89] Similarly, mice with deletion of gene encoding the NMDA receptor GluN1 sub-unit in Remak SC present hypersensitivity in pain.[90]
In addition to generating stimuli for nociceptor neurons, SCs can act as nociceptor cells themselves to transmit pain. A recent study shows that SCs are mechanosensitive cells. SCs present at the dermo-epidermal junction are associated with unmyelinated nociceptive nerves and transmit nociceptive information.[91] In the cancer context, SCs could also act directly as nociceptor cells.
Interestingly, a study reveals a role for SCs in reducing pain in pancreatic cancer. In a mouse model of pancreatic cancer, the activation of SCs by IL-6 has been associated with reduced pain and with a decline of spinal astroglia and/or microglia.[13] IL-6 is released under hypoxia, and hypoxia decreases the level of human SC mRNA transcripts for the nociceptive receptors P2RX7 and P2RX3 as well as the nociception-related adenosine A1 receptor (ADORA1).[13] Thus, SCs could be a key element in the analgesic response to hypoxia by reducing its nociceptive molecules expression. This finding might explain why some patients with pancreatic cancer remain asymptomatic and their cancers remain undetected at early stages of the disease.
Therefore, SC actively modulates pain through a variety of molecular and cellular mechanisms. SCs potentiate cancer cells effects on pain by stimulating cancer growth. SCs modulate the immune microenvironment population that can contribute to pain. SCs release factors inducing pain by stimulating sensory neurons and release other molecules that counteract these factors. SCs can also transmit pain by acting as nociceptors whose expression can be downregulated to reduce pain transmission.
6. Conclusion
SCs undergo transcriptional reprogramming in the context of cancer invasion, similar to the dedifferentiation that SCs undergo following nerve injury, and acquire a wide variety of functions (Figure 4) that may impact cancer growth, migration, invasion, immune regulation, and pain. Important advances have been made recently in uncovering how SCs function in cancer and their parallels with other distinctive behaviors found following nerve trauma or infection. It is clear that our understanding of SC activity in cancer remains underdeveloped, and much will be discovered in the near future.
Figure 4.
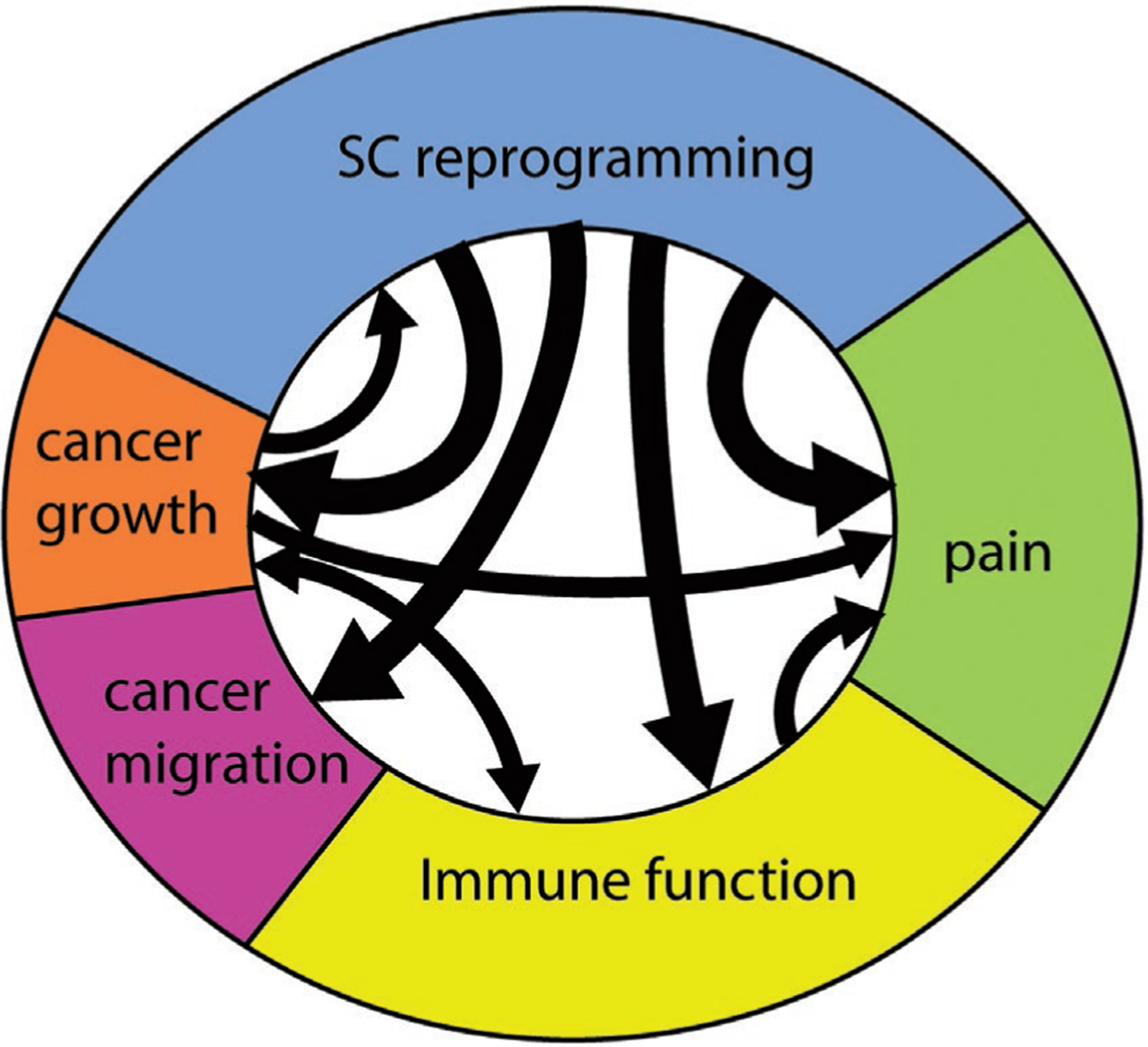
SC features in cancer. SC reprogramming occurs in cancer and is associated with several effects including cancer growth, cancer migration and invasion, immune regulation, and pain. These functions are interrelated. Cancer invasion induces SC reprogramming. Immune regulation and pain transmission can be directly controlled by cancer cells or indirectly via reprogrammed SCs in the microenvironment.
Acknowledgements
This work was supported by a grant from the National Institutes of Health (R01CA219534).
Biographies

Sylvie Deborde is an Assistant Attending Biologist at Memorial Hospital and an Assistant Laboratory Member at Memorial Sloan Kettering Cancer Center, New York. She is a cell biologist with a background in microscopy. She obtained her PhD in Molecular Pathology at University College London in UK. She completed a post-doctoral fellowship in Cell Biology at Weill Medical College Cornell University in New York City. Her current research activity is focused on the cellular and molecular mechanisms of cancer invasion induced by nerves and more specifically by Schwann cells.

Richard Wong is the Chief of Head and Neck Surgery at Memorial Sloan Kettering Cancer Center and Professor of Otolaryngology at Cornell Weill Medicine. His laboratory studies interactions between cancer cells and nerves. He is a member of the Tumor Progression and Metastasis Study Section at the National Institutes of Health. He completed his B.S. and B.A. at Stanford University, M.D. at Harvard Medical School, residency at the Harvard Combined Training Program in Otolaryngology, and fellowship in Head and Neck Surgical Oncology at Memorial Sloan Kettering Cancer Center.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Contributor Information
Sylvie Deborde, Department of Surgery, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA; David M. Rubenstein Center for Pancreatic Cancer Research, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA.
Richard J. Wong, Department of Surgery, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA; David M. Rubenstein Center for Pancreatic Cancer Research, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA.
References
- [1].Jessen KR, Mirsky R, J. Anat 2002, 200, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Jessen KR, Mirsky R, Lloyd AC, Cold Spring Harbor Perspect. Biol 2015, 7, a020487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Stierli S, Imperatore V, Lloyd AC, Glia 2019, 67, 2203. [DOI] [PubMed] [Google Scholar]
- [4].Byun JS, Gardner K, Am. J. Pathol 2013, 182, 1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Arthur-Farraj PJ, Latouche M, Wilton DK, Quintes S, Chabrol E, Banerjee A, Woodhoo A, Jenkins B, Rahman M, Turmaine M, Wicher GK, Mitter R, Greensmith L, Behrens A, Raivich G, Mirsky R, Jessen KR, Neuron 2012, 75, 633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Jessen KR, Arthur-Farraj P, Glia 2019, 67, 421. [DOI] [PubMed] [Google Scholar]
- [7].Jessen KR, Mirsky R, Front. Cell. Neurosci 2019, 13, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Deborde S, Omelchenko T, Lyubchik A, Zhou Y, He S, McNamara WF, Chernichenko N, Lee S-Y, Barajas F, Chen C-H, Bakst RL, Vakiani E, He S, Hall A, Wong RJ, J. Clin. Invest 2016, 126, 1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Demir IE, Boldis A, Pfitzinger PL, Teller S, Brunner E, Klose N, Kehl T, Maak M, Lesina M, Laschinger M, Janssen K-P, Algül H, Friess H, Ceyhan GO, Natl J. Cancer Inst 2014, 106, dju184. [DOI] [PubMed] [Google Scholar]
- [10].Silva VM, Gomes JA, Tenório LPG, de Omena Neta GC, da Costa Paixão K, Duarte AKF, da Silva GCB, Ferreira RJS, Koike BDV, de Sales Marques C, da Silva Miguel RD, de Queiroz AC, Pereira LX, de Carvalho Fraga CA, Oncotarget 2019, 10, 7288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Weiss T, Taschner-Mandl S, Janker L, Bileck A, Rifatbegovic F, Kromp F, Sorger H, Kauer MO, Frech C, Windhager R, Gerner C, Ambros PF, Ambros IM, Nat. Commun 2021, 12, 1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Shurin GV, Kruglov O, Ding F, Lin Y, Hao X, Keskinov AA, You Z, Lokshin AE, LaFramboise WA, Falo LD, Shurin MR, Bunimovich YL, Cancer Res 2019, 79, 2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Demir IE, Tieftrunk E, Schorn S, Saricaoglu ÖC, Pfitzinger PL, Teller S, Wang K, Waldbaur C, Kurkowski MU, Wörmann SM, Shaw VE, Kehl T, Laschinger M, Costello E, Algül H, Friess H, Ceyhan GO, Gut 2016, 65, 1001. [DOI] [PubMed] [Google Scholar]
- [14].Tofaris GK, Patterson PH, Jessen KR, Mirsky R, J. Neurosci 2002, 22, 6696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Inserra MM, Yao M, Murray R, Terris DJ, Archiv. Otolaryngol. Head Neck Surg 2000, 126, 1112. [DOI] [PubMed] [Google Scholar]
- [16].Bressy C, Lac S, Nigri J, Leca J, Roques J, Lavaut M-N, Secq V, Guillaumond F, Bui T-T, Pietrasz D, Granjeaud S, Bachet J-B, Ouaissi M, Iovanna J, Vasseur S, Tomasini R, Cancer Res 2018, 78, 909. [DOI] [PubMed] [Google Scholar]
- [17].Pascual G, Domínguez D, Elosúa-Bayes M, Beckedorff F, Laudanna C, Bigas C, Douillet D, Greco C, Symeonidi A, Hernández I, Gil SR, Prats N, Bescós C, Shiekhattar R, Amit M, Heyn H, Shilatifard A, Benitah SA, Nature 2021, 599, 485. [DOI] [PubMed] [Google Scholar]
- [18].Jessen KR, Mirsky R, J Physiol 2016, 594, 3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sakamoto Y, Kitajima Y, Edakuni G, Sasatomi E, Mori M, Kitahara K, Miyazaki K, Oncol. Rep 2001, 8, 477. [DOI] [PubMed] [Google Scholar]
- [20].Dang C, Zhang Y, Ma Q, Shimahara Y, J. Gastroenterol. Hepatol 2006, 21, 850. [DOI] [PubMed] [Google Scholar]
- [21].Ketterer K, Rao S, Friess H, Weiss J, Büchler MW, Korc M, Clin. Cancer Res 2003, 9, 5127. [PubMed] [Google Scholar]
- [22].Chen-Tsai CP, Colome-Grimmer M, Wagner RF, Dermatol. Surg 2004, 30, 1009. [DOI] [PubMed] [Google Scholar]
- [23].Kelso RL, Colome-Grimmer MI, Uchida T, Wang HQ, Wagner RF, Dermatol. Surg 2006, 32, 177. [DOI] [PubMed] [Google Scholar]
- [24].Murillo-Sauca O, Chung MK, Shin JH, Karamboulas C, Kwok S, Jung YH, Oakley R, Tysome JR, Farnebo LO, Kaplan MJ, Sirjani D, Divi V, Holsinger FC, Tomeh C, Nichols A, Le QT, Colevas AD, Kong CS, Uppaluri R, Lewis JS, Ailles LE, Sunwoo JB, Oncotarget 2014, 5, 6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Redmer T, Welte Y, Behrens D, Fichtner I, Przybilla D, Wruck W, Yaspo M-L, Lehrach H, Schäfer R, Regenbrecht CRA, PLoS One 2014, 9, e92596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tomellini E, Touil Y, Lagadec C, Julien S, Ostyn P, Ziental-Gelus N, Meignan S, Lengrand J, Adriaenssens E, Polakowska R, Bourhis XL, Stem Cells 2015, 33, 342. [DOI] [PubMed] [Google Scholar]
- [27].Salvo E, Tu NH, Scheff NN, Dubeykovskaya ZA, Chavan SA, Aouizerat BE, Ye Y, 11, 1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Huang T, Fan Q, Wang Y, Cui Y, Wang Z, Yang L, Sun X, Wang Y, Frontiers Oncol 2020, 10, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bakst RL, Xiong H, Chen C-H, Deborde S, Lyubchik A, Zhou Y, He S, McNamara W, Lee S-Y, Olson OC, Leiner IM, Marcadis AR, Keith JW, Al-Ahmadie HA, Katabi N, Gil Z, Vakiani E, Joyce JA, Pamer E, Wong RJ, Cancer Res 2017, 77, 6400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zhou Y, Li J, Han B, Zhong R, Zhong H, Cell. Immunol 2020, 357, 104211. [DOI] [PubMed] [Google Scholar]
- [31].Zhang SH, Shurin GV, Khosravi H, Kazi R, Kruglov O, Shurin MR, Bunimovich YL, Cancer Immunol. Immunother 2019, 594, 3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Gil Z, Cavel O, Kelly K, Brader P, Rein A, Gao SP, Carlson DL, Shah JP, Fong Y, Wong RJ, Natl J. Cancer Inst 2010, 102, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].He S, He S, Chen C-H, Deborde S, Bakst RL, Chernichenko N, McNamara WF, Lee S-Y, Barajas F, Yu Z, Al-Ahmadie HA, Wong RJ, Mol. Cancer Res 2015, 13, 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Su D, Guo X, Huang L, Ye H, Li Z, Lin L, Chen R, Zhou Q, Theranostics 2020, 10, 5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Roger E, Martel S, Bertrand-Chapel A, Depollier A, Chuvin N, Pommier RM, Yacoub K, Caligaris C, Cardot-Ruffino V, Chauvet V, Aires S, Mohkan K, Mabrut J-Y, Adham M, Fenouil T, Hervieu V, Broutier L, Castets M, Neuzillet C, Cassier PA, Tomasini R, Sentis S, Bartholin L, Cell Death Dis 2019, 10, 886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Shan C, Wei J, Hou R, Wu B, Yang Z, Wang L, Lei D, Yang X, Oncol. Rep 2015, 35, 427. [DOI] [PubMed] [Google Scholar]
- [37].Zhou Y, Shurin GV, Zhong H, Bunimovich YL, Han B, Shurin MR, Cancer Res 2018, 78, canres.1702.2018. [DOI] [PubMed] [Google Scholar]
- [38].Na’ara S, Amit M, Gil Z, Oncogene 2018, 107, 219. [Google Scholar]
- [39].Ferdoushi A, Li X, Griffin N, Faulkner S, Jamaluddin MFB, Gao F, Jiang CC, van Helden DF, Tanwar PS, Jobling P, Hondermarck H, Front. Oncol 2020, 10, 1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Duffy PE, Huang Y-Y, Rapport MM, Exp. Cell Res 1982, 139, 145. [DOI] [PubMed] [Google Scholar]
- [41].Elobeid A, Bongcam-Rudloff E, Westermark B, Nistér M, J. Neurosci. Res 2000, 60, 245. [DOI] [PubMed] [Google Scholar]
- [42].Urban-Wojciuk Z, Khan MM, Oyler BL, Fåhraeus R, Marek-Trzonkowska N, Nita-Lazar A, Hupp TR, Goodlett DR, Front Immunol 2019, 10, 2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Yang D, Han Z, Oppenheim JJ, Immunol Rev 2017, 280, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, Yang H, Amigorena S, Ryffel B, Barrat FJ, Saftig P, Levi F, Lidereau R, Nogues C, Mira J-P, Chompret A, Joulin V, Clavel-Chapelon F, Bourhis J, André F, Delaloge S, Tursz T, Kroemer G, Zitvogel L, Nat. Med 2007, 13, 1050.17704786 [Google Scholar]
- [45].Man L, Liu F, Wang Y, Song H, Xu H, Zhu Z, Zhang Q, Wang Y, Neural Regen Res 2015, 10, 1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Bigbee JW, Yoshino JE, DeVries GH, J. Neurocytol 1987, 16, 487. [DOI] [PubMed] [Google Scholar]
- [47].Pollard JD, Baverstock J, McLeod JG, Ann. Neurol 1987, 21, 337. [DOI] [PubMed] [Google Scholar]
- [48].Pollard JD, McCombe PA, Baverstock J, Gatenby PA, McLeod JG, J. Neuroimmunol 1986, 13, 123. [DOI] [PubMed] [Google Scholar]
- [49].Spierings E, de Boer T, Wieles B, Adams LB, Marani E, Ottenhoff TH, J. Immunol 2001, 166, 5883. [DOI] [PubMed] [Google Scholar]
- [50].Wekerle H, Schwab M, Linington C, Meyermann R, Eur. J. Immunol 1986, 16, 1551. [DOI] [PubMed] [Google Scholar]
- [51].Harboe M, Aseffa A, Leekassa R, Leprosy Rev 2005, 76, 5. [PubMed] [Google Scholar]
- [52].Lilje O, Cell. Molecul. Life Sci 2002, 59, 2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Hartlehnert M, Derksen A, Hagenacker T, Kindermann D, Schäfers M, Pawlak M, Kieseier BC, Horste GMZ, Sci. Rep 2017, 7, 12518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Bergsteinsdóttir K, Kingston A, Jessen KR, J Neurocytol 1992, 21, 382. [DOI] [PubMed] [Google Scholar]
- [55].Samuel NM, Jessen KR, Grange JM, Mirsky R, J Neurocytol 1987, 16, 281. [DOI] [PubMed] [Google Scholar]
- [56].Samuel NM, Mirsky R, Grange JM, Jessen KR, Clin. Exp. Immunol 1987, 68, 500. [PMC free article] [PubMed] [Google Scholar]
- [57].Armati PJ, Pollard JD, Gatenby P, Muscle Nerve 1990, 13, 106. [DOI] [PubMed] [Google Scholar]
- [58].Gold R, Toyka KV, Hartung HP, Cell. Immunol 1995, 165, 65. [DOI] [PubMed] [Google Scholar]
- [59].Lilje O, Armati PJ, J. Neuroimmunol 1997, 77, 75. [DOI] [PubMed] [Google Scholar]
- [60].Kingston AE, Bergsteinsdottir K, Jessen KR, Meide PHDV, Colston MJ, Mirsky R, Eur. J. Immunol 1989, 19, 177. [DOI] [PubMed] [Google Scholar]
- [61].Tsai CP, Pollard JD, Armati PJ, J. Neuroimmunol 1991, 31, 133. [DOI] [PubMed] [Google Scholar]
- [62].Horste GMZ, Heidenreich H, Lehmann HC, Ferrone S, Hartung H-P, Wiendl H, Kieseier BC, Glia 2010, 58, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Rhijn IV, den Berg LHV, Bosboom WMJ, Otten HG, Logtenberg T, Brain 2000, 123, 2020. [DOI] [PubMed] [Google Scholar]
- [64].Im JS, Tapinos N, Chae G-T, Illarionov PA, Besra GS, DeVries GH, Modlin RL, Sieling PA, Rambukkana A, Porcelli SA, J. Immunol 2006, 177, 5226. [DOI] [PubMed] [Google Scholar]
- [65].Murata K, Dalakas MC, Brain 2000, 123, 1660. [DOI] [PubMed] [Google Scholar]
- [66].Lisak RP, Skundric D, Bealmear B, Ragheb S, J Infect Dis 1997, 176, S173. [DOI] [PubMed] [Google Scholar]
- [67].Skundric DS, Lisak RP, Rouhi M, Kieseier BC, Jung S, Hartung H-P, J. Neuroimmunol 2001, 116, 74. [DOI] [PubMed] [Google Scholar]
- [68].Bergsteinsdottir K, Kingston A, Mirsky R, Jessen KR, J. Neuroimmunol 1991, 34, 15. [DOI] [PubMed] [Google Scholar]
- [69].Bolin LM, Verity AN, Silver JE, Shooter EM, Abrams JS, J. Neurochem 1995, 64, 850. [DOI] [PubMed] [Google Scholar]
- [70].Murwani R, Hodgkinson S, Armati P, J. Neuroimmunol 1996, 71, 65. [DOI] [PubMed] [Google Scholar]
- [71].Wagner R, Myers RR, Neuroscience 1996, 73, 625. [DOI] [PubMed] [Google Scholar]
- [72].Scherer SS, Kamholz J, Jakowlew SB, Glia 1993, 8, 265. [DOI] [PubMed] [Google Scholar]
- [73].Stewart HJ, Rougon G, Dong Z, Dean C, Jessen KR, Mirsky R, Glia 1995, 15, 419. [DOI] [PubMed] [Google Scholar]
- [74].Kiefer R, Funa K, Schweitzer T, Jung S, Bourde O, Toyka KV, Hartung HP, Am J Pathol 1996, 148, 211. [PMC free article] [PubMed] [Google Scholar]
- [75].Constable AL, Armati PJ, Hartung H-P, J Neurol Sci 1999, 162, 120. [DOI] [PubMed] [Google Scholar]
- [76].Constable AL, Armati PJ, Toyka KV, Hartung H-P, Brain Res 1994, 635, 75. [DOI] [PubMed] [Google Scholar]
- [77].Pereira RMS, Calegari-Silva TC, Hernandez MO, Saliba AM, Redner P, Pessolani MCV, Sarno EN, Sampaio EP, Lopes UG, Biochem. Bioph. Res. Co 2005, 335, 20. [DOI] [PubMed] [Google Scholar]
- [78].Andorfer B, Kieseier BC, Mathey E, Armati P, Pollard J, Oka N, Hartung HP, J. Neuroimmunol 2001, 116, 226. [DOI] [PubMed] [Google Scholar]
- [79].Martyn GV, Shurin GV, Keskinov AA, Bunimovich YL, Shurin MR, Cancer Immunol., Immunother 2019, 68, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Ceyhan GO, Bergmann F, Kadihasanoglu M, Altintas B, Demir IE, Hinz U, Müller MW, Giese T, Büchler MW, Giese NA, Friess H, Gastroenterology 2009, 136, 177. [DOI] [PubMed] [Google Scholar]
- [81].Scholz J, Woolf CJ, Nat. Neurosci 2007, 10, 1361. [DOI] [PubMed] [Google Scholar]
- [82].Ren K, Dubner R, Nat. Med 2010, 16, 1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Cui J, Holmin S, Mathiesen T, Meyerson BA, Linderoth B, Pain 2000, 88, 239. [DOI] [PubMed] [Google Scholar]
- [84].Schmidt BL, Neurosci 2014, 20, 546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Campana WM, Brain Behav. Immun 2007, 21, 522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Malin SA, Molliver DC, Koerber HR, Cornuet P, Frye R, Albers KM, Davis BM, J. Neurosci. Official J. Soc. Neurosci 2006, 26, 8588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Orita S, Henry K, Mantuano E, Yamauchi K, Corato AD, Ishikawa T, Feltri ML, Wrabetz L, Gaultier A, Pollack M, Ellisman M, Takahashi K, Gonias SL, Campana WM, J. Neurosci 2013, 33, 5590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Faroni A, Castelnovo LF, Procacci P, Caffino L, Fumagalli F, Melfi S, Gambarotta G, Bettler B, Wrabetz L, Magnaghi V, Glia 2014, 62, 548. [DOI] [PubMed] [Google Scholar]
- [89].Chen S, Rio C, Ji R-R, Dikkes P, Coggeshall RE, Woolf CJ, Corfas G, Nat. Neurosci 2003, 6, 1186. [DOI] [PubMed] [Google Scholar]
- [90].Brifault C, Romero H, Van-Enoo A, Pizzo D, Azmoon P, Kwon H, Nasamran C, Gonias SL, Campana WM, J. Neurosci 2020, 40, 9121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Abdo H, Calvo-Enrique L, Lopez JM, Song J, Zhang M-D, Usoskin D, Manira AE, Adameyko I, Hjerling-Leffler J, Ernfors P, Science 2019, 365, 695. [DOI] [PubMed] [Google Scholar]