

Summary
Platelets, the often-overlooked component of the immune system, have been shown to promote tumor growth. Nonalcoholic fatty liver disease (NAFLD) is a common disease in the Western world and rising risk for hepatocellular carcinoma (HCC). Unexpectedly, we observed that platelets can inhibit growth of established HCC in NAFLD mice. Through pharmacological inhibition and genetic depletion of P2Y12 as well as in vivo transfusion of WT or CD40L−/− platelets, we demonstrate that the anti-tumor function of platelets is mediated through P2Y12-dependent CD40L release, which leads to CD8+ T cell activation by the CD40 receptor. Unlike P2Y12 inhibition, blocking platelets with aspirin does not prevent platelet CD40L release nor accelerate HCC in NAFLD mice. Similar findings were observed in liver metastasis models. Together, our study reveals a complex role of platelets in tumor regulation. Anti-platelet treatment without inhibiting CD40L release could be considered for liver cancer patients with NAFLD.
Keywords: Platelets, HCC, NAFLD, CD40L, P2Y12, liver metastasis
eTOC Blurb:
Anti-platelet therapy is associated with reduced liver cancer. NAFLD, a rising risk for liver cancer, is known to activate platelets. Unexpectedly, Ma et al. found that platelets have a complex role in tumor regulation and can enhance CD8+ T cell-dependent anti-tumor immunity through P2Y12-dependent CD40L release from platelets.
Graphical Abstract
Introduction:
Hepatocellular carcinoma (HCC) is the most common type of primary liver malignancy and the third most common cause of cancer-related death worldwide (Llovet et al., 2021). Most HCC patients are diagnosed with unresectable disease and respond poorly to current treatment options. Despite the many advances in modern cancer therapy, HCC is among the few cancers with a rising mortality rate (Llovet et al., 2021).
Beyond the traditional view as a modulator of thrombosis and hemostasis, platelets have many other functions and are considered as an essential element of the immune system participating in both innate and adaptive immune responses (Franco et al., 2015; Sprague et al., 2008). Cancer patients often present with abnormal platelet counts or function, and emerging evidence suggests that platelets contribute to tumor metastasis and poor prognosis (Franco et al., 2015; Labelle et al., 2011; Schumacher et al., 2013). Recent clinical studies suggest a critical role of platelets in HCC where an association between the use of platelet inhibitor aspirin and a reduced HCC risk in patients with viral hepatitis has been found (Lee et al., 2017; Lee et al., 2019; Simon et al., 2020). Consistently, antiplatelet therapy reduces HCC in a mouse model of chronic hepatitis B through inhibition of immune-mediated liver injury by HBV-specific cytotoxic CD8+ T cells (Sitia et al., 2012). Interestingly, the mouse study remarks that the same antiplatelet therapy regimen had no effect on hepatotoxic chemical carbon tetrachloride (CCL4)-induced HCC(Sitia et al., 2012), suggesting the complex role of platelets in HCC regulation.
Non-alcoholic fatty liver disease (NAFLD), characterized by excessive lipid accumulation inside the liver, is a common liver disorder with close association with obesity and type 2 diabetes. NAFLD is a leading cause of liver-related morbidity in the US and globally, also an important risk factor for HCC (Michelotti et al., 2013; Wree et al., 2013). Patients with NAFLD often display an increased mean platelet volume (MPV), a marker for platelet turnover but also used clinically as a surrogate of platelet activation (Madan et al., 2016). Currently, there are no clinical studies investigating the effect of anti-platelet treatment in HCC patients with underlying NAFLD. Recently, platelets have been shown to become activated and to aggregate in the liver thereby promoting non-alcoholic steatohepatitis (NASH), a severe form of NAFLD, and accelerate subsequent NASH-driven HCC in mice (Malehmir et al., 2019). However, it should be noted that the study focused on the regulation by platelets of NASH progression, and HCC was studied using a purely NASH-driven HCC mouse model (Malehmir et al., 2019). Thus, attenuated NASH by platelet inhibition was expected to reduce HCC formation in these mice, and the study was not designed to look at the direct impact of platelet-regulated immunity on the progression of HCC(Diggs and Greten, 2019). It is still unclear whether platelets regulate the progression of established HCC in the context of NAFLD.
To address this question, we designed a mouse study to simplify the complex NAFLD-HCC process, and particularly focused on tumor growth regulation by platelets in the context of NAFLD. The use of orthotopic implantation of established HCC tumors or carcinogen- and oncogene-driven HCC models was done to ensure liver tumor formation, which helped exclude other factors of NAFLD-driven hepatocarcinogenesis. We made the unexpected observation that platelets inhibited growth of HCC and liver metastasis in NAFLD through P2Y12-dependent platelet CD40L release. Besides the reported HCC-promoting function by driving NASH pathology, platelets also have surprising anti-HCC activity against established HCC through enhancing anti-tumor immunity. Our study suggests the complex role of platelets in tumor regulation, and possible clinical interventions.
Results:
Platelet depletion or P2Y12 inhibition accelerates HCC in NAFLD.
To study whether platelets can regulate HCC growth in NAFLD, we injected orthotopically murine HCC Hep55.1c tumor cells into C57BL/6 congenic mice fed with a methionine choline deficient (MCD) diet. Mice were treated with antibodies targeting platelet glycoprotein Ibα (GPIbα) (Bergmeier and Boulaftali, 2014), a platelet membrane glycoprotein, to deplete platelets in tumor-bearing mice. Platelet depletion efficacy was confirmed (Figure S1A). Tumor growth was measured by microCT scan 7 days after tumor injection, and by weight at the end of the experiment (21 days). Unexpectedly, platelet depletion promoted liver tumor growth in NAFLD mice (Figure 1A, B). To support this finding, the study was repeated using clopidogrel, a widely used platelet function inhibitor antagonizing the P2Y12 receptor. The efficacy of clopidogrel treatment in mice was confirmed by measuring platelet surface expression of activation marker CD62P after ex vivo stimulation (Figure S1B–D). Similarly, clopidogrel treatment increased orthotopic Hep55.1c tumor growth in NAFLD mice (Figure 1C, Figure S1E).
Figure 1. Platelet depletion or P2Y12 inhibiton accleates HCC in NAFLD.
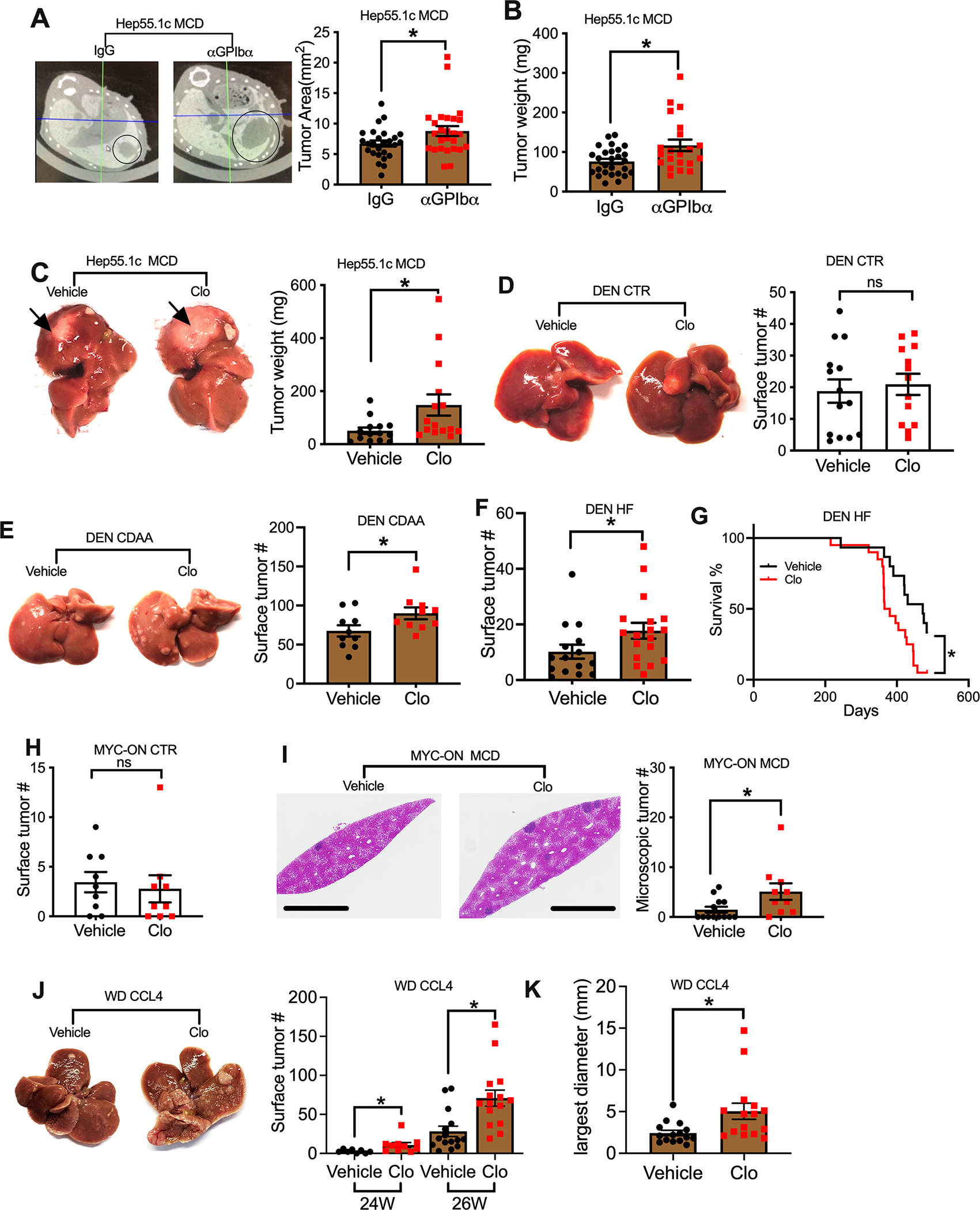
(A, B) Intrahepatic Hep55.1c tumor cells were implanted into MCD diet-fed mice. After tumor implantation, mice were given i.p. injection of 50 μg αGPIb antibody or IgG control every 3 days. Tumor growth was monitored by microCT scan at day 7 after tumor injection (A) or by weight at the experimental end point (B). Data are presented as mean ± s.e.m. from three independent experiments. n=26, *P <0.05, Student’s t-test.
(C) Hep55.1c tumor cells were implanted into livers of MCD diet-treated mice with or without clopidogrel (Clo) treatment for 3 weeks. Mice were euthanized, and liver tumor weight was measured. Representative images are shown. Data are presented as mean ± s.e.m. from two independent experiments. n=15, *P <0.05, Student’s t-test.
(D) DEN injected mice kept on control diet (CTR) were were treated with clopidogrel or vehicle for 6 months. Mice were euthanized and liver surface tumor nodules were counted. Representative liver images were shown. Cumulative data are presented as mean ± s.e.m, n=14.
(E) DEN mice fed with CDAA diet and clopidogrel or vehicle for 6 month. Liver surface tumor nodules were counted. Representative liver images are shown. Cumulative data are presented as mean ± s.e.m, n=10. *p<0.05, Student’s t-test.
(F) DEN mice fed with hight fat (HF) diet were treated with clopidogrel or vehicle for 5 months. Liver surface tumor nodules were counted. Cumulative data are presented as mean± s.e.m, n=15 for vehicle, 18 for Clopidogrel. *P <0.05, Student’s t-test.
(G) DEN mice were fed with hight fat (HF) diet for 5 months, then mice were given clopidogrel or vehicle. Survival of HCC bearing mice was monitored. n=20 for Clo and 15 for vehicle. *p<0.05, Log-rank Mantel-Cox test.
(H) MYC-ON mice fed on control diet were treated with clopidogrel or vehicle for 8 weeks. Liver surface tumor nodules were counted. Cumulative data are presented as mean ± s.e.m. n=9.
(I) MYC-ON mice fed with MCD diet were treated with clopidogrel or vehicle for 4 weeks. Representitave images of H&E staining of liver sections are shown. The scale bars represent 2 mm. Microscopic tumor leisions were counted, and cumulative data are presented as mean ± s.e.m. n=13 for vehicle, 10 for Clopidogrel. *P <0.05, Student’s t-test.
(J, K) Representative liver images and liver surface tumor count of western diet-CCL4 treated male C57BL/6 mice given clopidogrel or vehicle (J). Largest tumor diameter of each mouse was measured at 26-week time point (K). Data are presented as mean ± s.e.m. n=8 for vehicle 24W,10 for clopidogrel 24W, 15 for vehicle 16W or clopidogrel 26W. *P <0.05, Student’s t-test.
See also Figure S1.
Next, we extended these studies to carcinogen- or oncogene-driven HCC models with various NAFLD dietary regimen and used clopidogrel to inhibit platelet function. The use of carcinogen or oncogene HCC models ensures tumor development in the absence of NAFLD, which make it possible to compare the platelet tumor regulatory function in both conditions with or without NAFLD. First, a well-characterized diethylnitrosamine (DEN) carcinogen-induced murine model of HCC in C57BL/6 mice was used (Ma et al., 2016) and mice were treated with clopidogrel. Under the regular diet feeding condition, DEN-injected mice developed macroscopic liver surface tumor nodules but clopidogrel treatment had no effect on growth of liver tumors (Figure 1D). In contrast, when DEN injected mice were fed with a choline-deficient L-amino acid-defined (CDAA) diet to induce NAFLD (Ma et al., 2016), clopidogrel treatment promoted the growth of liver tumors (Figure 1E). To rule out a CDAA diet specific effect, DEN injected mice were placed on high fat (HF) diet. Again, clopidogrel treatment increased the HCC burden in DEN HF mice (Figure 1F). Furthermore, we studied the impact of clopidogrel treatment on survival of mice bearing established HCC, and clopidogrel treatment was started in DEN mice after feeding HF diet for 5 months when macroscopic liver tumors began to appear (Figure 1F). Indeed, clopidogrel treatment decreased survival in DEN HF mice (Figure 1G). To further confirm the findings, we also tested a third murine HCC model using FVB/N genetic background MYC-ON mice which express liver-specific MYC oncogene and develop HCC (Ma et al., 2016). Similarly, clopidogrel treatment had no effect on tumor growth in MYC-ON mice kept on a regular diet (Figure 1H), in contrast to those with diet-induced NAFLD, where clopidogrel treatment caused more tumor growth (Figure 1I).
Platelet tumor regulatory function was also assessed in a Western diet-CCL4 HCC mouse model which recapitulates the progression of NAFLD to HCC in humans (Tsuchida et al., 2018). Clopidogrel treatment was started after mice had been fed the Western diet plus weekly CCL4 injections for 21 weeks and when liver tumors began to appear. As seen in other models, more liver tumors were found in mice treated with clopidogrel (Figure 1J&K).
The liver is a common site for metastatic tumor formation, and the majority of liver cancer cases are secondary cancers derived from primary tumors in other organs (Disibio and French, 2008). Therefore, we tested whether the tumor growth regulatory function of platelets would also apply to metastatic liver tumors. Indeed, clopidogrel treatment enhanced the growth of B16-F10 tumors in livers of NAFLD mice following either intrahepatic tumor implantation (Figure S1F) or intrasplenic injection (Figure S1G). Similarly, more liver A20 tumors were found in clopidogrel treated NAFLD mice after i.v. injection of tumor cells (Figure S1H). Our results suggest that platelets can also suppress the growth of metastatic tumors inside NAFLD livers. Finally, we confirmed the baseline circulating platelet count in various mouse strains kept on different diets and in different housing conditions to rule out that the tumor regulatory function of platelets was due to unphysiological platelets numbers (Figure S1I).
In summary, by testing four different dietary NAFLD models with six different liver tumor models in three different mouse strains, our results demonstrate that platelets can inhibit tumor growth in mice with NAFLD independent of the specific tumor model.
Platelets release more CD40L in NAFLD.
Platelets are a major source of circulating CD40L(Nagasawa et al., 2005). Several studies have shown that platelet-derived CD40L is functional and can modulate adaptive immune responses (Elzey et al., 2003; Sprague et al., 2008). Therefore, we studied CD40L and platelets in NAFLD in more detail. First, we measured circulating CD40L in NAFLD mice. Plasma samples were used to exclude the influence of CD40L released by platelets during blood coagulation ex vivo. Indeed, higher levels of plasma CD40L were observed in C57BL/6 mice fed with either a CDAA or a Western diet (Figure 2A). Similarly, the increase of circulating CD40L was also found in C57BL/6 mice fed with the MCD diet as early as two weeks, and the increase became more prominent following NAFLD progression (Figure 2B). Similar results were observed in BALB/c mice on the MCD diet (Figure S2A) and in C57BL/6 mice bearing liver Hep55.1c tumors (Figure 2C). Next, platelet depletion was performed to test whether platelets were the source for elevated plasma CD40L in NAFLD mice. As expected, platelet depletion reduced plasma CD40L concentration in NALFD mice (Figure 2D), suggesting that platelets were the main source for elevated CD40L. NAFLD is closely associated with obesity and we asked whether platelet function would be altered in mice with NAFLD but without obesity. However, the increase of platelet-derived CD40L was still found in a non-obese NAFLD mouse model (Tu et al., 2017) by feeding a high fat, high cholesterol, cholate (HFHCC) diet (Figure S2B). It should also be noted that mice kept on the MCD diet also do not develop obesity (Rinella et al., 2008), but also demonstrated changes in platelet derived CD40L. Thus, our results suggest that NAFLD is sufficient to increase CD40L even in the absence of obesity.
Figure 2. Platelets release more CD40L in NAFLD.
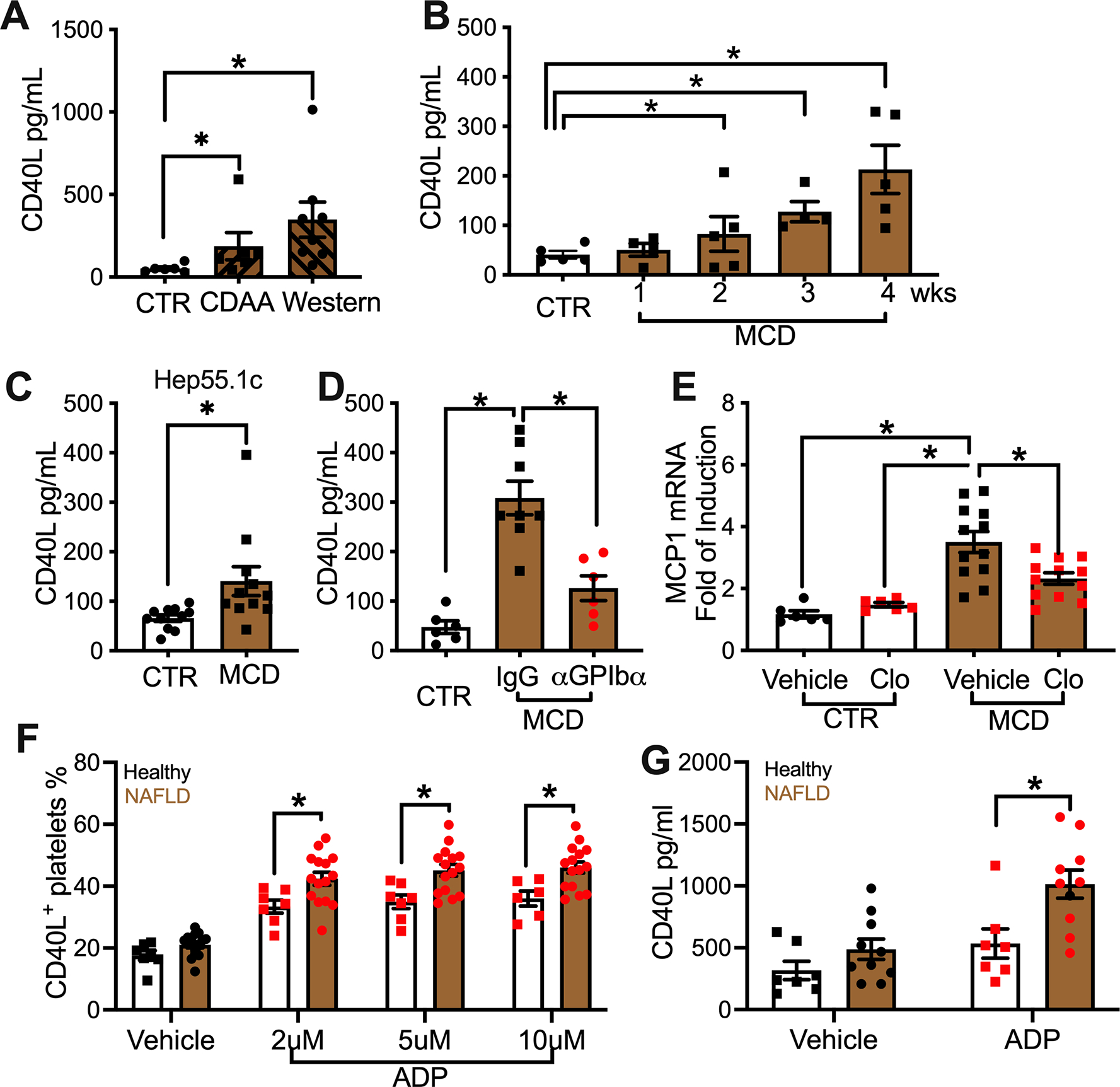
(A) C57BL/6 mice were fed with CDAA, western diet or control diet for 6 months. Plasma CD40L levels were measured. Data are presented as mean± s.e.m. from two independent experiments. n=7 for CTR and CDAA, 9 for Western. *P < 0.05, one-way ANOVA.
(B) Plasma CD40L levels of mice fed with MCD diet for different time periods. Cumulative data are presented as mean± s.e.m. n=5 for control, 4 for 1 week and 3 weeks, 5 for 2 and 4 weeks. *P < 0.05, one-way ANOVA.
(C) Plasma CD40L level of liver Hep55.1c tumor-bearing mice fed with MCD or control diet. Data are presented as mean± s.e.m. n=10, *P <0.05, Student’s t-test.
(D) Plasma CD40L level of MCD diet-fed mice given i.p. injection of α-GPIbα or IgG control. Data are presented as mean± s.e.m. from two independent experiments. n=6 for control, 8 for MCD IgG, and 6 for MCD α-GPIbα. *P < 0.05, one-way ANOVA.
(E) Control diet- or MCD diet-fed mice were treated with or without clopidogrel. Mouse plasma samples were collected and incubated with MS-1 cells. MCP1 mRNA expression of MS-1 cells was measured by real-time PCR. Data are mean± s.e.m of two independent experiments. n=6 for control diet with or without clopidogrel, n=12 for MCD diet with or without clopidogrel. *P < 0.05, one-way ANOVA.
(F) Freshly isolated platelets from NAFLD patients or healthy donors were incubated with different concentration of ADP for 15 minutes. Surface CD40L+ platelet percentage was measured by flow cytometry analysis. Cumulative data are presented as mean± s.e.m. n=15 for NAFLD, 7 for healthy. *P < 0.05, two-way ANOVA.
(G) Isolated platelets were incubated with or without 5 μM of ADP for 2 hours. After centrifugation, the released CD40L was measured. Cumulative data are presented as mean± s.e.m. n=10 for NAFLD, 7 for healthy. *P < 0.05, two-way ANOVA.
See also Figure S2.
To confirm that circulating CD40L was biologically functional, we utilized a reporter assay by measuring MCP1 expression level in MS1 cells as an indicator for CD40L activity(Sprague et al., 2008) (Figure S2C). Consistently, increased MCP1 expression was found when MS1 cells were incubated with plasma from NAFLD mice (Figure 2E). In this experiment CD40L−/− mice served as a control to confirm assay specificity (Figure S2D).
To see whether these observations applied to humans, plasma CD40L was measured in blood from NAFLD patients. Consistent with our observation in mice and a previous human study (Sookoian et al., 2010), elevated CD40L was found in NAFLD patient blood compared to the healthy donors (Figure S2E). Furthermore, activation and CD40L release of freshly isolated platelets were assessed after ex vivo stimulation. As expected, platelets gradually upregulated activation marker CD62P on their surface following the increased stimulation (Figure S2E). Compared to healthy donors, NAFLD patients’ platelets showed significantly higher CD62P at the highest stimulation given (Figure S2F), suggesting that NAFLD platelets are more susceptible to in vitro activation. This result is in line with previous reports that platelets are more active in NAFLD (Madan et al., 2016; Malehmir et al., 2019). During platelet activation, CD40L is translocated from α-granules to the platelet surface before it is released (Hermann et al., 2001). Indeed, activated platelets increased surface CD40L shortly after stimulation (Figure 2F, Figure.S2G). Interestingly, unlike CD62P, even at the lowest stimulation tested, significantly higher surfaced CD40L was already detected on platelets from NAFLD patients compared to of healthy donors (Figure 2F). In both patients and healthy donors, plasma CD40L correlated with the stimulation-induced surface CD40L translocation of platelets, (Figure S2H) supporting the conclusion that platelet-derived CD40L is the main source of circulating CD40L. As expected, after stimulation, higher levels of CD40L were released from platelets of NAFLD patients than from those of healthy donors (Figure 2G). These results suggest that platelets can release more CD40L in NAFLD patients.
Platelet-derived CD40L Inhibits HCC in NAFLD.
CD40L can be essential for generation of robust anti-tumor responses (Elgueta et al., 2009; Marigo et al., 2016), but its effect on anti-tumor immunity has never been studied in the context of NAFLD. The specific role of CD40L on HCC growth in NAFLD was investigated using an anti-CD40L neutralizing antibody. Compared to the IgG control, CD40L neutralizing antibody treatment increased HCC in MYC-ON NAFLD mice (Figure 3A). Consistent with the observation that blocking platelets increased HCC only in NAFLD (Figure 1A–H), no change of liver tumor burden was found in MYC-ON control mice given the CD40L neutralization treatment (Figure 3B).
Figure 3. Platelet-derived CD40L inhibits HCC in NAFLD.
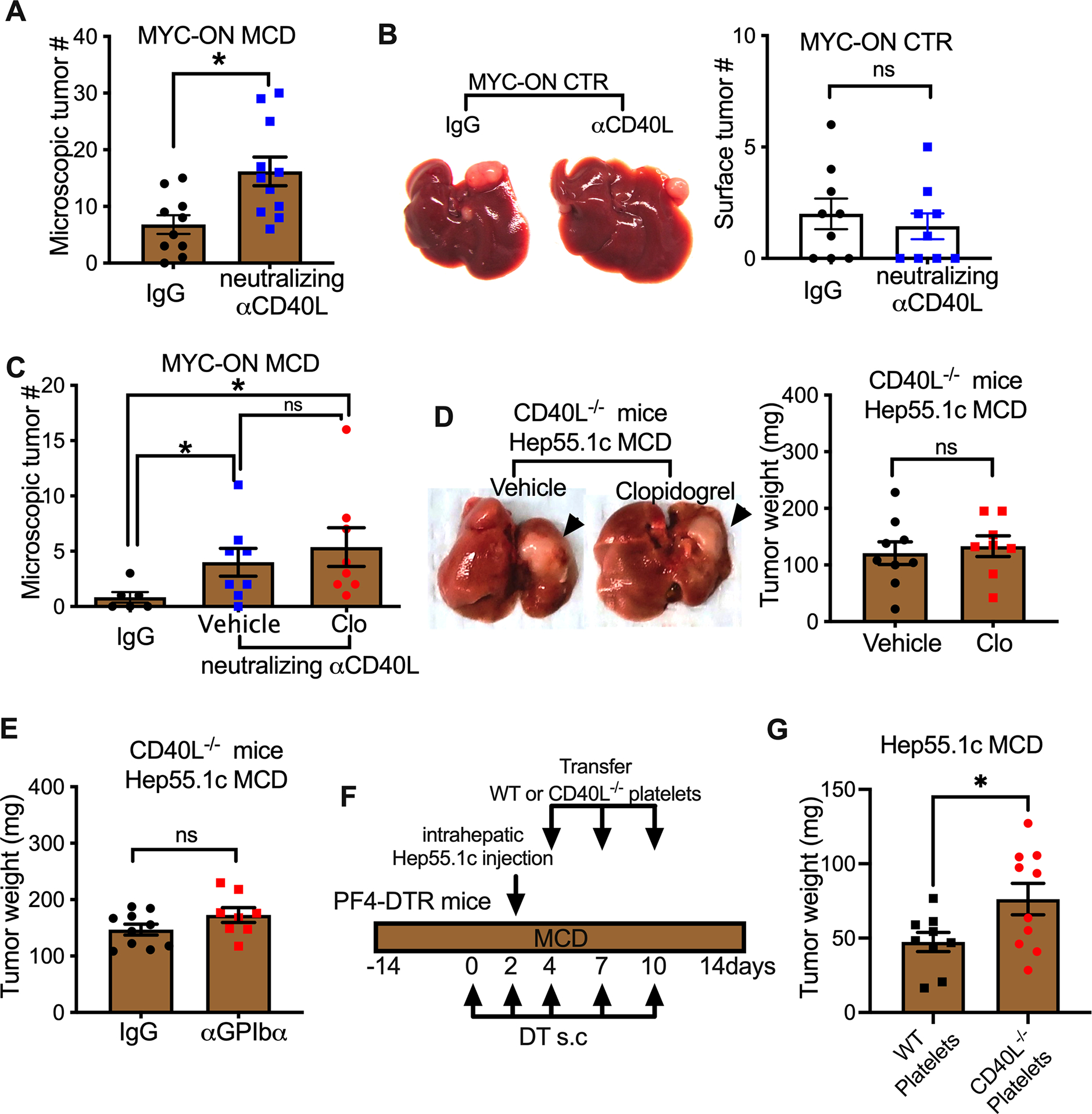
(A) MYC-ON MCD mice given i.p. injection of CD40L neutralizing antibody or IgG control. Fixed liver tissues were subjected to H&E staining. Microscopic liver tumor lesions were counted. Cumulative data are presented as mean± s.e.m. n=10 for IgG, 11 for αCD40L. *P <0.05, Student’s t-test.
(B) MYC-ON CTR mice were injected with CD40L-neutralizing antibody or IgG control. Surface liver tumor nodules were counted. Representative imagines are shown in d. Cumulative data are presented as mean± s.e.m. (e). n=10, *P <0.05, Student’s t-test.
(C) MYC-ON MCD mice were given i.p. injection of CD40L neutralizing antibody, control IgG or combination of CD40L neutralization with clopidogrel. Four weeks after treatment, mice were euthanized, and fixed liver tissues were subjected to H&E staining. Microscopic tumor lesions were counted. Cumulative data are presented as mean± s.e.m. n=6 for IgG, 8 for αCD40L, 8 for αCD40L+clopidogrel. *P <0.05, one-way ANOVA.
(D) Hep55.1c tumor cells were orthotopically injected into the livers of CD40L−/− mice fed with MCD diet together with or without clopidogrel. Three weeks later, mice were euthanized. Liver tumor burden was measured. Representative imagines are shown. Data are presented as mean± s.e.m. of two independent experiments. n=9 for vehicle and 8 for Clo.
(E) MCD diet-fed CD40L−/− mice were given intrahepatic injection of hep55.1c tumor cells. Tumor-bearing mice were injected i.p. with αGPIb antibody or IgG control. Liver tumor burden was measured at the end point. Data are presented as mean± s.e.m. of two independent experiments.
(F,G) MCD diet-fed PF4-DTR mice were given s.c. injection of diphtheria toxin (DT) to deplete endogenous platelets. Two days after DT treatment, mice were given intrahepatic injection of hep55.1c tumor cells followed by adoptive transfer of WT or CD40L−/− platelets isolated from MCD diet treated mice. Treatment scheme was depicted in F. At experimental end point, mice were euthanized, and liver tumors were weighed. Data are presented as mean± s.e.m. of two independent experiments. n=9 for wt platelets and 10 for CD40L−/− platelets. *P <0.05, Student’s t-test.
See also Figure S3.
Next, we examined whether the platelets’ anti-tumor function in NAFLD was mediated through CD40L. We first compared the effect of CD40L neutralization alone to that of combination of CD40L neutralization and clopidogrel. As shown before, CD40L neutralization treatment increased liver tumors in MYC-ON NAFLD mice (Figure 3C). However, the addition of clopidogrel did not provide any further increase of tumor growth (Figure 3C), suggesting that platelets inhibit NAFLD-promoted HCC through CD40L. Similar, CD40L neutralization-caused tumor promotion was seen when B16-F10 tumors were injected into the mouse livers with NAFLD (Figure S3A).
To support our findings, liver tumor progression was examined in CD40L-deficient mice. Unlike in wildtype C57BL/6 mice (Fig. 1C), anti-platelet treatment with clopidogrel failed to increase intrahepatic Hep55.1c tumor burden in CD40L-deficient mice with NAFLD (Fig. 3D, Extended Data Fig.S3A). Similarly, CD40L deficiency abolished the increase of Hep55.1c tumor growth in NAFLD livers after platelet depletion (Fig. 3E). In addition, in CD40L-deficient mice, clopidogrel treatment failed to increase B16-F10 tumor burden in NAFLD livers (Extended Data Fig.S3B, C), suggesting the same mechanism can also apply to other liver tumors including metastases.
To provide the direct evidence that platelet-derived CD40L controls HCC in NAFLD, we adopted a recently developed inducible platelet depletion model using PF4-DTR mice (Salzmann et al., 2020). The function of platelet-derived CD40L was determined by transferring wildtype or CD40L-deficient platelets into PF4-DTR mice lacking endogenous platelets, and then monitoring the impact on the growth of intrahepatic injected Hep55.1c tumors (Fig. 3F). Depletion of endogenous platelets by diphtheria toxin treatment and the delivery of donor platelets in PF4-DTR mice with NAFLD were confirmed (Extended Data Fig. S3D, E). Significantly larger liver Hep55.1c tumors were found in mice after transfer of CD40L−/− platelets than after transfer of wildtype platelets (Fig. 3G). These experiments provide direct evidence that platelet-derived CD40L controls liver tumor growth in NAFLD.
CD40 and CD8+ T cells mediate the platelet-dependent tumor inhibition.
CD40L is well known to bind its receptor CD40 for immune regulation(Elgueta et al., 2009). The role of CD40 in platelet-dependent regulation of tumor progression was investigated. First, the relevance of CD40 signaling in HCC was tested using an agonistic CD40 antibody to treat MYC-ON mice. As expected, activating CD40 signaling caused a strong reduction of liver tumors (Fig. 4A). Certain HCC tumors have been reported to express CD40(Sugimoto et al., 1999). However, no surface CD40 expression was detected on MYC tumor cells (Extended Data Fig.S4A), ruling out a direct effect. Similar tumor inhibition was seen when B16-F10 tumors were injected into the mouse livers (Extended Data Fig.S4B, C). In addition, we recently reported that activating CD40 also suppresses mouse cholangiocarcinoma (Diggs et al., 2020), suggesting that activating CD40 can induce a broad and potent anti-tumor response against liver cancer even in the absence of NAFLD. Reciprocally, the role of CD40 in NAFLD HCC progression was tested using CD40 knockout mice. Consistent with the finding using the CD40L-deficient mice, clopidogrel treatment failed to increase the growth of intrahepatic Hep55.1c tumor in CD40-deficient mice with NAFLD (Fig. 4B), indicating that the anti-tumor function of platelet derived CD40L acts through CD40.
Figure 4. CD40 and CD8+T cells mediate the platelet dependent tumor inhibition.
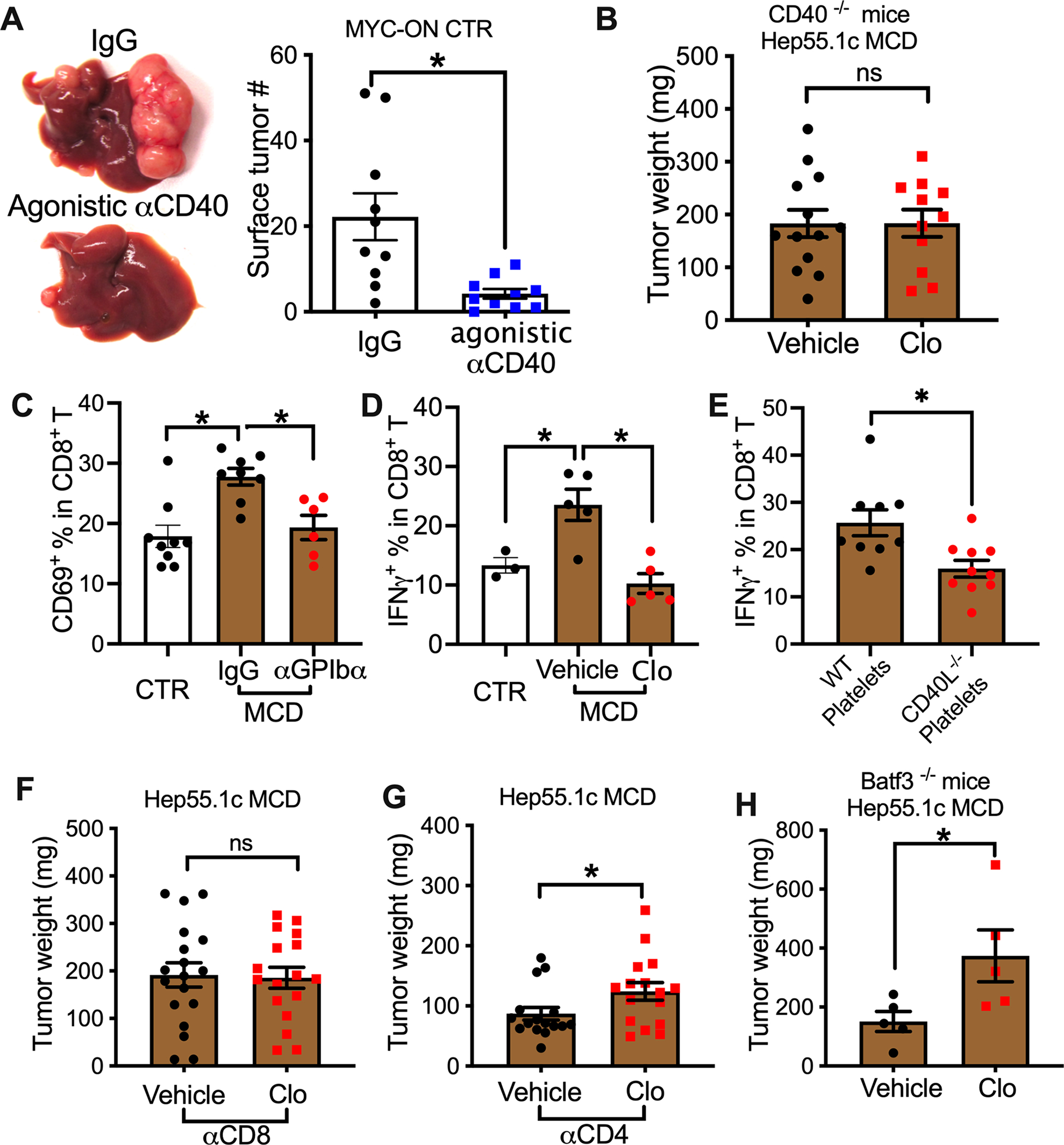
(A) Control diet fed-MYC-ON mice were treated with agonistic CD40 antibody or control IgG. Representative liver images are shown. Liver surface tumor numbers were measured. Cumulative data are shown as mean± s.e.m. n=10, *P <0.05, Student’s t-test.
(B) Hep55.1c tumor cells were orthotopically injected into the livers of CD40−/− mice fed with MCD diet together with or without clopidogrel. Three weeks later, mice were euthanized, and liver tumor weight was measured. Data are presented as mean± s.e.m. of two independent experiments. n=13 for vehicle and 11 for Clo.
(C) MCD diet-fed mice were injected i.p. with α-GPIbα or IgG control. CD69 expression on intrahepatic CD8+ T cells was measured by flow cytometry. Data are presented as mean± s.e.m. from two independent experiments. n=9 for control, 8 for MCD IgG, and 6 for MCD α-GPIbα. *P < 0.05, one-way ANOVA.
(D) IFNγ expression of intrahepatic CD8+ T cells from MCD diet-fed mice treated with or without clopidogrel. Data are presented as mean± s.e.m. n=3 for control, 5 for MCD vehicle, and 5 for MCD Clo. *P < 0.05, one-way ANOVA.
(E) IFNγ expression of intrahepatic CD8+ T cells was measured from the mice that received adoptively transferred platelets described in Fig. 3F. Data are presented as mean± s.e.m. of two independent experiments. n=9 for wt platelets and 10 for CD40L−/− platelets. *P <0.05, Student’s t-test.
(F,G) Hep55.1c tumor cells were implanted into livers of MCD diet-fed mice treated with or without clopidogrel, then tumor-bearing mice were injected i.p. with αCD8 (F) or αCD4 (G). Liver tumor burden was measured at end point. Data are presented as mean± s.e.m. from two independent experiments. n=20, *P <0.05, Student’s t-test.
(H) Hep55.1c tumor cells were orthotopically injected into the livers of Batf3−/− mice fed with the MCD diet together with or without clopidogrel. Liver tumor burden was measured at experimental end point. Data are presented as mean± s.e.m. n=5.
See also Figure S4.
CD40L/CD40 signaling leads to cytotoxic T cell activation(Elgueta et al., 2009). Both CD4+ and CD8+ T cells have been reported to have important anti-HCC function in NAFLD (Ma et al., 2016; Shalapour et al., 2017). Thus, the effect of platelets on T cells in NAFLD was investigated. Consistent with previous reports that NAFLD drives an activation of adaptive immune responses(Ghazarian et al., 2017; Ma et al., 2016), more intrahepatic CD69+CD4+ and CD69+CD8+ T cells were detected in NAFLD mice, but the increase was inhibited by platelet depletion (Fig. 4C, Extended Data Fig.S4D). Similarly, clopidogrel treatment also reversed the increase of CD69+CD4+ and CD69+CD8+ T cells in NAFLD mice (Extended Data Fig.S4E & F). In addition, clopidogrel attenuated the increased IFNγ production of intrahepatic T cells in NAFLD mice (Fig. 4D, Extended Data Fig.S4G). Consistently, CD40L deficiency reversed the inhibitory effect of platelet depletion on CD69 expression on T cells (Fig. S4H & I). Furthermore, in the platelet transfer study described above (Fig. 3F), higher IFNγ production was found in intrahepatic CD8+ T cells from mice after transfer of wildtype platelets than after transfer of CD40L−/− platelets (Fig. 4E), supporting that platelet-derived CD40L is critical for T cell-dependent anti-tumor immunity in NAFLD. To understand the role of different T cell subsets, CD4+ or CD8+ T cells were depleted to study their role in platelet-controlled HCC progression. Indeed, depletion of CD8+ T cells abrogated clopidogrel treatment-dependent growth promotion of intrahepatic Hep55.1c tumors in NAFLD mice (Fig. 4F). In contrast, CD4+ T cell depletion had no effect (Fig. 4G).
CD40L/CD40 licensing of conventional type 1 dendritic cells (cDC1s) is well characterized for priming CD8+ T cells(Elgueta et al., 2009). Importantly, a recent publication demonstrates that cDC1s are critical to prime CD8+ T cells and drive NAFLD pathology(Deczkowska et al., 2021). Therefore, Batf3-deficient mice, which lack cDC1s, were tested. However, clopidogrel treatment still caused bigger intrahepatic Hep55.1c tumors in Batf3-deficient NAFLD mice (Fig. 4H), suggesting that cDC1s are not critical for the platelet-dependent tumor inhibition. Together, our results show that the anti-tumor function of platelets is mediated by CD40/CD40L and CD8+ T cells.
P2Y12 controls platelet CD40L release and HCC growth.
To elucidate the mechanism, we asked how NAFLD mediates enhanced CD40L release from platelets. As expected, clopidogrel treatment reduced plasma CD40L levels to baseline in NAFLD mice (Fig. 5A). In contrast, treatment with aspirin, the most common antiplatelet medication that acts by inhibiting cyclooxygenases (COXs), had no effect on plasma CD40L (Fig. 5B). This result is in line with the report that aspirin did not affect platelet CD40L release, which does not involve COXs(Hermann et al., 2001). The differential effect between P2Y12 and COX inhibition on plasma CD40L indicates that P2Y12 is critical for the increased platelet CD40L release in NAFLD. Therefore, we tested P2Y12 knockout mice and saw that CD40L plasma concentrations were similar in mice with and without NALFD (Fig. 5C).
Figure 5. P2Y12 controls platelet CD40L release and HCC growth.
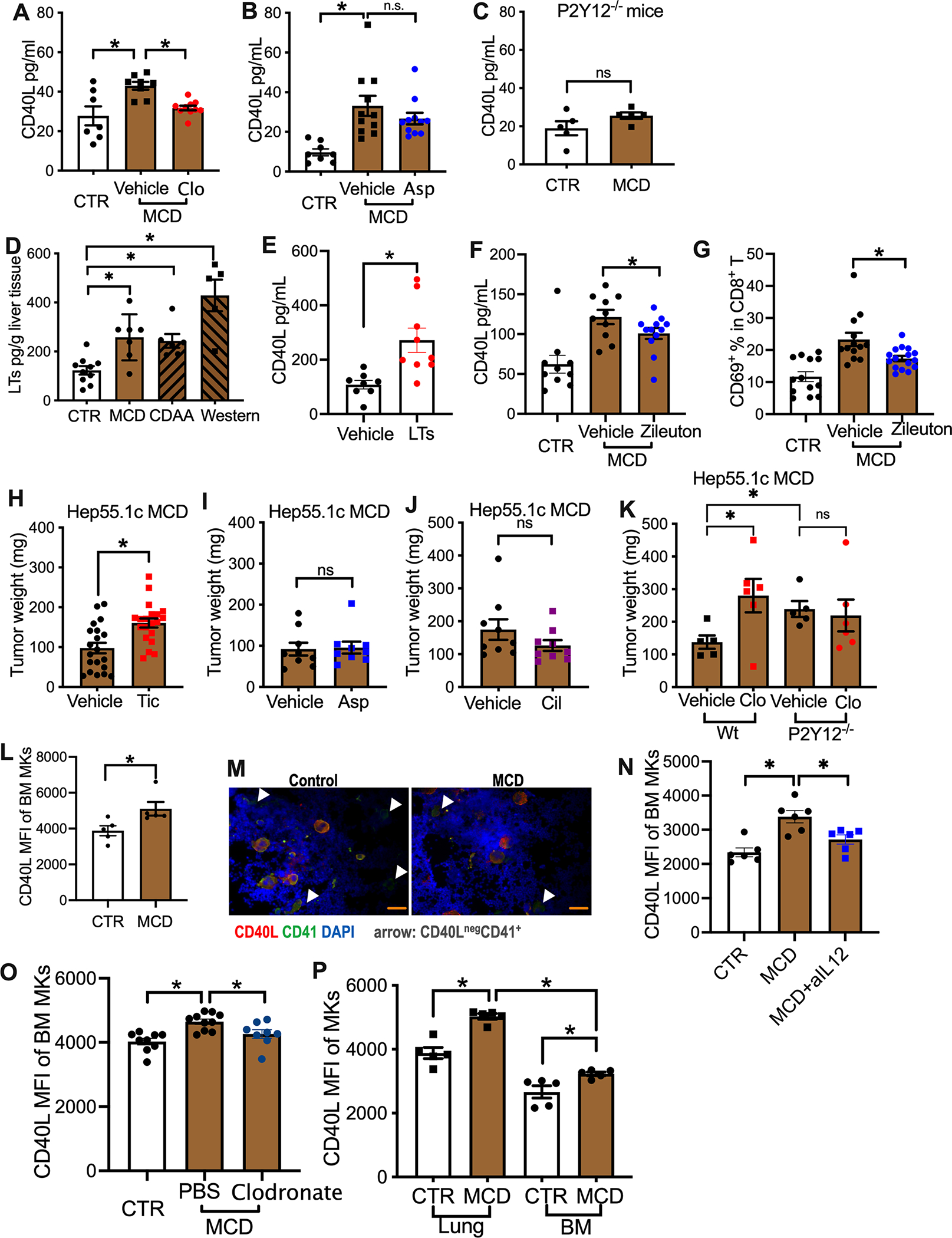
(A, B) Plasma CD40L levels of MCD-fed mice treated with or without clopidogrel (A) or aspirin (Asp) (B). Cumulative data are presented as mean± s.e.m. *P < 0.05, one-way ANOVA.
(C) P2Y12−/− mice were fed with MCD diet or control diet for 4 weeks. Plasma CD40L levels were measured. Cumulative data are presented as mean± s.e.m.
(D) Cysteinyl leukotrienes (LTs) levels in liver tissue lysates prepared from mice fed on MCD, CDAA or Western diet were measured by ELSIA. Data are presented as mean± s.e.m. n=10 for CTR, 5 for CDAA, 6 for Western, 7 for MCD. *P < 0.05, one-way ANOVA.
(E) Plasma CD40L levels of native mice injected i.v. with a mix of cysteinyl leukotrienes. Data are presented as mean± s.e.m. from two independent experiments. n=8 for vehicle, 9 for LTs. *P <0.05, Student’s t-test.
(F, G) Plasma CD40L levels (F) and intrahepatic CD8 T cell CD69 expression(G) of MCD diet-fed mice treated with or without zileuton. Data are presented as mean± s.e.m. *P < 0.05, one-way ANOVA.
(H-J) Hepatic hep55.1c tumor-bearing MCD diet-fed mice were treated with or without ticagrelor (Tic, H), aspirin (Asp, I) or cilostazol (Cil, J) for three weeks. Liver tumor burden was measured. Data are presented as mean± s.e.m. from two independent experiments. n=20 for ticagrelor study, n=9 for Aspirin or cilostazol study. *P <0.05, Student’s t-test.
(K) MCD diet-fed wildtype or P2Y12−/− mice with or without clopidogrel were given intrahepatic injection of hep55.1c tumor cells. Liver tumor burden was measured. Data are presented as mean± s.e.m. *P < 0.05, one-way ANOVA.
(L) CD40L expression levels of bone marrow megakaryocytes from mice fed with MCD or control diet were measured by flow cytometry. Data are presented as mean± s.e.m. from two independent experiments. *P <0.05, Student’s t-test.
(M) Bone marrow smears prepared from MCD or control diet-fed mice were subjected to immunohistochemistry staining of CD40L (red) and CD41 (green). Nuclei were stained by DAPI. Organ scale bars represent 200μM. Representative imagines are shown.
(N) MCD diet-fed mice were injected with IL-12-neutralizing antibody or IgG control. CD40L expression levels of bone marrow megakaryocytes were measured. Data are presented as mean± s.e.m. from two independent experiments. n=6, *P <0.05, one-way ANOVA.
(O) Clodronate liposome was used to deplete macrophage in MCD diet-fed mice. CD40L expression levels of bone marrow megakaryocytes were measured. Data are presented as mean± s.e.m. n=9 for CTR, 10 for MCD-PBS, 8 for MCD-Clodronate, *P <0.05, one-way ANOVA.
(P) Mice were fed with MCD diet or control diet. CD40L of lung and bone marrow megakaryocytes were measured. Data are presented as mean± s.e.m. from two independent experiments. n=5, *P <0.05, one-way ANOVA.
See also Figure S5.
Platelets express surface receptors for cysteinyl leukotrienes (leukotriene C4, D4 and E4) and can be directly activated by leukotrienes(Cummings et al., 2013). Interestingly, the P2Y12 receptor is necessary for the leukotriene-induced platelet activation via an autocrine ADP release-dependent mechanism(Cummings et al., 2013; Paruchuri et al., 2009). Importantly, elevated lipoxygenase activity and increased cysteinyl leukotrienes have been found in livers of NASH patients(Gorden et al., 2015; Puri et al., 2009). Indeed, higher levels of cysteinyl leukotrienes were found in livers of NAFLD mice kept on all three diets tested (WD, CDAA or MCD diet) (Fig. 5D). Therefore, we looked for a possible link between cysteinyl leukotrienes and CD40L release by platelets. Intravenous administration of a mix of cysteinyl leukotrienes increased plasma CD40L in mice kept on a normal diet (Fig. 5E), suggesting that cysteinyl leukotrienes can increase platelet CD40L release. Furthermore, blocking the production of cysteinyl leukotrienes using zileuton, a clinically used inhibitor of 5-lipoxygenase, partially inhibited the upregulation of plasma CD40L (Fig. 5F). As expected, zileuton treatment also decreased intrahepatic T cell activation in NAFLD mice (Fig. 5G. Fig.S5A–C). These results suggest that the NAFLD-increased platelet CD40L release is mediated by P2Y12, and in part through cysteinyl leukotrienes.
The findings that platelet CD40L is critical for the tumor growth inhibition by platelets, and clopidogrel but not aspirin blocked platelet CD40L release, suggest a unique platelet P2Y12-CD40L pathway for HCC regulation in NAFLD. Therefore, we evaluated the influence of several common platelet function inhibitors in murine HCC/NAFLD models. Similar to clopidogrel, treatment with ticagrelor, another drug targeting the P2Y12 receptor, increased intrahepatic Hep55.1c tumor burden in NAFLD mice (Fig. 5H). In contrast, COX inhibition by aspirin or blocking PDE3 by cilostazol did not affect the growth of intrahepatic Hep55.1c tumors (Fig. 5I,J). The impact of aspirin on spontaneous HCC development was also tested. In MYC-ON NAFLD mice, aspirin treatment caused actually a marginal decrease of HCC (Extended Data Fig.S5D). Combination treatment with aspirin and clopidogrel is commonly used in the clinic. Addition of aspirin reduced the tumor promoting effect of clopidogrel treatment in MYC-ON NAFLD mice (Extended Data Fig.S5E), but the combination treatment still accelerated HCC in DEN NAFLD mice (Extended Data Fig.S5F). To confirm the important role of platelet P2Y12 receptor in HCC regulation, P2Y12 knockout mice were used. Indeed, P2Y12 knockout mice grew bigger intrahepatic Hep55.1c tumors compared to wildtype mice under NAFLD conditions (Fig. 5K). In addition, unlike in wildtype mice, clopidogrel treatment did not cause further increase of liver tumors (Fig. 5K). Together, these results demonstrate that platelets exert anti-tumor function through a P2Y12 receptor-dependent CD40L release in the context of NAFLD.
These findings may also have clinical implications since according to the 2015–2016 National Health and Nutrition Examination Survey (NHANES) clopidogrel is more likely to be used in NAFLD patients (2.6%) than in non-NAFLD patients (1%, p=0.002, Table 1), consistent with the known association of NAFLD with cardiovascular disease.
Table 1.
Higher clopidogrel usage in NAFLD patients.
| Clopidogrel Use | No Clopidogrel Use | Total | |
|---|---|---|---|
| NAFLD | 22 (2.6%) | 815 (97.4%) | 837 |
| Non-NAFLD | 19 (1%) | 1922 (99%) | 1941 |
Footnote: data from the 2015–2016 National Health and Nutrition Examination Survey (NHANES) was analyzed for the presence of NAFLD (US fatty liver index >=30) and clopidogrel use. P=0.0017, Fisher’s exact test.
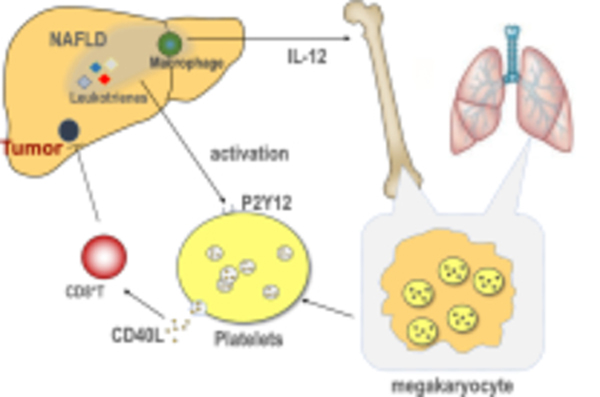
Finally, we studied megakaryocytes, the precursor cells to produce platelets, in mice with and without NAFLD. CD40L expression in bone marrow megakaryocytes was higher in NAFLD mice (Fig. 5L,M, Extended Data Fig.S5G). In addition, plasma from NAFLD mice increased CD40L expression in cultured megakaryocytes (Extended Data Fig.S5H). IL-12 is known to induce CD40L production(Peng et al., 1998). As reported(Sutti et al., 2014), increased IL-12 in liver was observed in NAFLD mice fed with Western, CDAA or MCD diets (Extended Data Fig.S5I) and injection of an IL-12-neutralizing antibody reduced CD40L expression in bone marrow megakaryocytes (Fig. 5N). Macrophages have been found to produce IL-12 in NAFLD mice (Kremer et al., 2010). Therefore, we tested the potential role of macrophages in regulating megakaryocyte CD40L expression. Consistent with a previous report (Kremer et al., 2010), depleting macrophages reduced liver IL-12 expression in NAFLD mice (Figure S5J). Importantly, removing macrophages reduced CD40L expression in bone marrow megakaryocytes (Figure 5O), suggesting that macrophage-released IL-12 promotes megakaryocyte CD40L expression. Recently, lung megakaryocytes have been discovered to be important for platelet generation (Lefrancais et al., 2017). Similar to the findings with bone marrow megakaryocytes, increased CD40L expression was also observed in lung megakaryocytes (Figure 5P).
Discussion
Pharmaceutical inhibition of platelet function using aspirin treatment and lower platelet numbers have previously been shown to correlate with better outcome in cancer patients suggesting that platelets may promote tumor growth and carcinogenesis (Lee et al., 2017; Lee et al., 2019; Simon et al., 2020). Interestingly, in patients with NAFLD, platelets show an increased volume indicating a state of activation (Madan et al., 2016) and giving rise to the question whether platelets control cancer growth in patients with HCC and NAFLD. Here, we decided to study the immunological function of platelets in carcinogen- or oncogene-induced HCC in mice with NAFLD. Using three different tumor models, four different dietary NAFLD models, pharmacological inhibition of the P2Y12 receptor and use of genetic knockout models, we describe that inhibition of the P2Y12 receptor on platelets promotes tumor growth via CD40L in mice with NAFLD. Although our observations described here at first sight seem contradictory to what has been shown before (Lee et al., 2017; Lee et al., 2019; Simon et al., 2020; Sitia et al., 2012) our results are in accordance with observations described earlier by others (Elzey et al., 2003; Iannacone et al., 2005). It is important to note that here we have studied the effect of platelets in models in which tumors and NASH were already established in contrast to others who have focused on NASH progression and HCC induction. As an example, Malehmir et al. studied antiplatelet treatment in a purely NASH-driven murine HCC model (Malehmir et al., 2019). In this model, mice are kept on a CDAA diet causing about 20% of all mice to develop HCC after a period of 12 months (Wolf et al., 2014). Inhibition of platelet function in this model impaired primarily NASH development and it was not possible to determine whether reduced HCC formation was an indirect result of altered NASH formation or platelet function (Malehmir et al., 2019). Therefore, we decided to use alternate tumor/NAFLD models, which allow answering this important question.
We used orthotopic implantation of established HCC tumors or DEN carcinogen- or MYC oncogene-driven mouse HCC models in combination with various commonly used dietary NAFLD models. Platelet functional studies were performed using several different approaches including pharmacological anti-platelet medications, antibody-dependent platelet depletion and transfusion of platelets with genetic modification. Our results demonstrate that platelets can indeed inhibit HCC growth in mice with NAFLD and that platelets play a much more complex role in controlling tumor growth than previously thought. In the context of NAFLD and HCC, platelets can show both pro-hepatocarcinogenesis function by driving NASH pathology, and anti-tumor activity against established tumors by driving CD8+ T cell responses.
It has previously been described that platelets can enhance adaptive immunity (Elzey et al., 2003; Iannacone et al., 2005). Activated platelets release various bioactive factors from α-granules, one of which is CD40L. This is particularly interesting because CD40L’s role in anti-tumor immunity is well known and platelets are the main source of circulating CD40L (Nagasawa et al., 2005; Vonderheide, 2007). However, the role of platelet-derived CD40L in tumor surveillance has not been studied. Here, we found greater release of platelet-derived CD40L in both NAFLD mouse models and patients with NASH. Further investigations using neutralizing antibody and CD40L genetic deficient mice suggest its critical anti-tumor function in NAFLD-HCC. More importantly, we provide the direct in vivo evidence that platelet-derived CD40L has anti-tumor function in NAFLD by conducting a technically challenging platelet transfusion study. In this setting we were able to directly compare the function of wildtype platelets versus CD40L−/− platelets. Emerging data suggest that platelet bioactive cargo is a critical modulator of anti-tumor immunity(Franco et al., 2015). Unlike the anti-tumor activity of platelet CD40L in this study, TGF-β from platelets has been reported to impair T cell anti-tumor function (Rachidi et al., 2017). The balance between anti- and pro-tumor functions of platelet bioactive factors and their interactions with the immune environment of various tumors with different inflammatory settings should be further studied.
The regulation of platelet-derived release of CD40L in NAFLD was further investigated. Interestingly, we found that blocking platelet function using COX inhibitors or the P2Y12 inhibitor clopidogrel had different effects on platelet CD40L release. The finding is in line with previous reports that COX inhibition fails to block platelet CD40L release which depends on internal Ca2+ and protein kinase C but does not involve tyrosine kinases, ERK or p38, and COXs are downstream of ERK and p38 during platelet activation (Barry et al., 1999; Hermann et al., 2001). The critical role of P2Y12 in NAFLD-induced platelet CD40L release was confirmed using P2Y12 genetic deficient mice. All these studies suggest that various platelet inhibition strategies can affect platelet CD40L release differently, causing opposite effects on HCC tumor growth in NAFLD. Indeed, P2Y12 inhibition-enhanced NAFLD-HCC could not be found using COX or PDE3 inhibitors. This may have potential clinical implication for treatment of HCC patients with NAFLD, since platelets contribute to NASH pathology (Malehmir et al., 2019) and NAFLD patients often present with cardiovascular diseases and often require anti-platelet treatment. Blocking platelet aggregation while sparing CD40L release using non-P2Y12 inhibitors such as aspirin may be more suitable for HCC patients with NAFLD. Further clinical studies are still needed to examine the effect of common anti-platelet medications on the outcome of NAFLD patients with liver tumors. It is also interesting that P2Y12 inhibitors have been suggested to associate with potential more cancer-related death in DAPT and TIMI 38 trials, although later meta-analysis of large patient cohorts failed to identify such correlation (Fierro et al., 2019; Mauri et al., 2014). Our results suggest that further analysis of impact of P2Y12 inhibitor use on cancer-related death should focus on conditions with increased platelet CD40L such as NAFLD.
Besides enhanced CD40L release, we also demonstrated that NAFLD increases CD40L production in megakaryocytes. In line with a previous report (Sutti et al., 2014), increased IL-12 expression was found in NAFLD mouse livers. An IL-12 dependent increase of CD40L production was found in bone marrow megakaryocytes. Further investigation demonstrated that macrophages caused an increase of liver IL-12 and CD40L production from bone marrow megakaryocytes. Thus, our results suggest a crosstalk between the liver and bone marrow in NAFLD. Indeed, the communication between liver and bone marrow is also supported by a recent study in NAFLD (Deczkowska et al., 2021), which demonstrates that expansion of hepatic dendritic cells in NASH was caused by increased production of dendritic cell progenitors inside the bone marrow and enhanced liver recruitment. Recently, the lung has been discovered to harbor megakaryocytes and recognized as an important organ for platelet biogenesis (Lefrancais et al., 2017). Similarly, we found that lung megakaryocytes produce higher CD40L in NAFLD mice. Interestingly, CD40L levels in lung megakaryocytes were even higher than in the bone marrow both at baseline and in mice kept on MCD diet. The reason for higher CD40L expression in lung megakaryocytes remains unknown, but the finding is consistent with a recent report which also observed higher CD40L in lung megakaryocytes compared to that in bone marrow (Pariser et al., 2021). Our results suggest that the lung may potentially also contribute to liver tumor regulation through modulating platelets in NAFLD. Due to the communication and potential reconstitution between lung and bone marrow megakaryocytes (Lefrancais et al., 2017), it is challenging to separate the contribution between lung and bone marrow megakaryocytes to circulating platelet CD40L level.
As anucleate cell fragments, platelets possess functional spliceosome and develop a unique functional regulation mechanism by generating mature messages from pre-mRNAs for protein translation through a signal-dependent splicing. We limited our studies to the analysis of CD40L protein and did not measure platelet CD154 mRNA. Therefore, we cannot exclude whether platelet protein translation also contributes to the increased platelet-derived CD40L in NAFLD. However, a recent study evaluating the platelet transcriptome from obese patients undergoing bariatric surgery-based weight reduction demonstrated differences in the expression of NAFLD related genes but not CD154 (Ezzaty Mirhashemi et al., 2021), indicating that NAFLD might preferentially induce CD40L protein production in megakaryocytes rather than NAFLD or tumors causing a transfer of CD40L pre-mRNA or mRNA into platelets.
Platelet heterogeneity is an interesting but unexplored area in this study. Platelet heterogeneity even occurs within one individual and has been recognized to link to platelet function and disease condition (Handtke and Thiele, 2020). Although controversial, increased platelet size has been considered as a character of younger and more reactive platelets. This information is interesting since NAFLD patients are known to have an increased mean platelet volume. In addition, the differential megakaryocyte CD40L expression levels indicate the potential existence of CD40L high and low platelet subsets. Further investigation of platelet heterogeneity is needed to better understand the role of platelets in tumor regulation.
CD40L is known for induction of strong CD8+ T cells responses through CD40 licensing of dendritic cells (Elgueta et al., 2009). Our results demonstrate that platelet-derived CD40L enhanced CD8+ T cell activation. Our finding is complementary to the observation that platelets enhance CD8+ T cell trafficking to liver in NAFLD (Malehmir et al., 2019). Importantly, depleting CD8+ T cells attenuated platelet inhibition-enhanced HCC in NAFLD, suggesting the anti-HCC platelet function was at least in part mediated by CD8+ T cells. The critical tumor inhibitory function of CD8+ T cells was also found in a high-fat-diet-fed MUP-uPA HCC mouse model, in which removing CD8+ T cells accelerated HCC (Shalapour et al., 2017).
In summary, our study demonstrates that platelets can mediate anti-tumor immunity. NAFLD increased P2Y12-dependent release of CD40L by platelets which led to enhanced anti-tumor immunity through CD40 and CD8+ T cells. P2Y12 inhibition accelerated HCC growth in several HCC-NAFLD mouse models. The finding is an important complement to the pro-tumor functions of platelets and suggests the complex role of platelets in tumor regulation, and also suggests possible therapeutic approaches for translation to the clinic.
STAR Methods
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Tim F. Greten (tim.greten@nih.gov)
Materials availability
The study did not generate new unique reagents, and all the reagents are commercially available.
Data and code availability
All data are available in the main text or the supplementary materials.
Experimental model and subject details
Human samples
Fasting blood was collected from adult subjects with NAFLD enrolled in a natural history study of liver diseases (Clinicaltrials.gov NCT00001971). NAFLD was diagnosed by imaging and/or liver biopsy. Subjects were excluded if they had decompensated cirrhosis, significant alcohol consumption, thrombocytopenia, or other liver disease and if they were using aspirin or clopidogrel. The study was approved by the NIDDK/NIAMS Institutional Review Board and all subjects provided written informed consent. Samples were processed within 60 minutes of collection.
Mice
C57BL/6, CD40L−/−, CD40−/−, Batf3−/−, PF4-cre and iDTR mice were purchased from The Jackson Laboratory. BALB/c mice were purchased from Charles River. LAP-tTA and TRE-MYC mice were used as described (Ma et al., 2016), and P2Y12−/− mice were provided by Dr. Long-Jun Wu (Mayo Clinic). All mice were maintained under special pathogen free condition without restation to food and water with 12-h light/12-h dark cycle at the NIH Bethesda CRC animal facility. Both female and male mice were used as indicated, and animal age was described for each type of mouse model. Whenever possible, animals were randomly assigned to various groups. All experiments were conducted according to local institutional guidelines and approved by the Animal Care and Use Committee of the National Institutes of Health, Bethesda, USA.
Tumor cell lines
Hep55.1c cells were purchased from Cell Lines Service (CLS, Germany). B16-F10 cells (CRL-6475), A20 cell (TIB-208), and MS-1 cells (CRL-2279) were purchased from ATCC. Cell lines were cultured in RPMI with 10% FBS and 1% penicillin-streptomycin (PS) at 37°C with 5% CO2. All cell lines were confirmed to be free of mycoplasma contamination.
Method details
DEN HCC model
Chemically induced HCC was established by intraperitoneal injection of diethylnitrosoamine (DEN) (Sigma) into 2-week-old male C57BL/6 pups at a dose of 20 μg/g body weight as described before (Ma et al., 2016). Starting from the age of 6 weeks, mice were fed with CDAA or HF diet to induce NAFLD. Anti-platelet therapy was performed by giving clopidogrel (20 μg/ml, dissolved in 0.003% HCl water final), aspirin (100 μg/ml in water final) or both in drinking water, as previously described. For survival assay, clopidogrel treatment was started in DEN mice which had already fed with HF diet for 5 months. At the time of sacrifice, liver tumors were measured.
MYC HCC model
MYC expression in the liver was activated by removing doxycycline treatment (100/μg ml) from the drinking water of 4-week-old double transgenic mice for both TRE-MYC and LAP-tTA as previously described (Ma et al., 2016). Starting from the age of 6 weeks, mice were fed on MCD diet. Antiplatelet treatment was performed by giving clopidogrel (20 μg/ml, dissolved in 0.003% HCl water final), aspirin (100 μg/ml in water final) or both in drinking water. CD40L neutralizing antibody (100 μg/mouse) or CD40 agonistic antibody (50 μg/mouse) were given by i.p. injection once per week for a total of 4 or 6 weeks, respectively. CD4+ or CD8+ T cells were depleted by i.p. injection of depleting antibody at the dose of 100 μg/mouse once per week for a total of 6 weeks. At the time of sacrifice, surface liver tumor nodules were counted. H&E staining was performed on fixed liver sections, and microscopic tumor lesions were counted (Ma et al., 2016).
Western-CCL4 HCC model
As described (Tsuchida et al., 2018), male C57BL/6 mice (8 weeks old) were fed with Western diet (TD.120528, Tekland) and high sugar solution containing 23.1 g/L d-fructose (Cat#F0127, Sigma) and 18.9g/L d-glucose (Cat#G8270, Sigma). Mice were given a weekly i.p. injection of 0.2 μl/g body weight of CCL4 (Cat#289116, Sigma) dissolved in olive oil (Cat#O1514, Sigma) in a 1:10 (v:v) ratio. After 21 weeks of Western diet feeding plus CCL4 injections, liver tumors began to appear. Then mice were given clopidogrel (20 μg/ml, dissolved in 0.003% HCl water final) or control water. At the experimental end point, mice were euthanized, and liver surface tumor nodules were counted.
Intrahepatic tumor injection model
To induce liver tumors, 2 × 105 Hep55.1c or 2.5 × 105 B16-F10 cells in 20 μL PBS and Matrigel 50:50 solution were injected under the liver capsule into the left liver lobe of anesthetized recipient mice after subcostal laparotomy as described before(Heinrich et al., 2021). Antiplatelet medication treatment was performed by giving clopidogrel (20 μg/ml, dissolved in 0.003% HCl water final), aspirin (100 μg/ml in water final), ticagrelor (40 μg/ml in water final) or cilostazol (300 μg/ml in water final). Platelet depletion was performed by i.p. injection of 50μg of GPIbα (Cat#R300) or isotype IgG control (Cat#C301) every 3 days start from 24 hours following surgery. Tumor growth was measured by microCT scan at day 7 after injection or by weighing at the time of sacrifice.
MicroCT scan
MicroCT scan was performed using the Quantum GX microCT Imaging System (PerkinElmer, Waltham, Ma). In order to enhance visualization of liver tumors, ExiTron nano 6000 (Miltenyi) contrast dye was used at a one-time dose of 100ul/mouse via tail vein injection on post-operative day 2. Scan images were analyzed and tumor sizes including area and largest diameter were determined using DragonFly 4.0 Software (Object Research Systems, Montreal, Canada).
Intrasplenic tumor injection model
To induce metastatic liver tumors in MCD diet-fed C57BL/6 mice, intrasplenic injections of B16-F10 tumor cells were performed as we previously described (Ma et al., 2018). Briefly, mouse spleen was exposed under anesthesia, and 5 × 105 B16-F10 cells in 50 μL PBS were injected into spleen. Injected mice were randomized and assigned to clopidogrel or vehicle group and kept on MCD diet. Three weeks later, mice were euthanized, and liver tumors were counted.
A20 liver metastasis model
BALB/c mice were fed with HFHCC diet (TD.88051, Teklad) 3 weeks to induce NAFLD. NAFLD mice were given i.v. injection of 1 × 106 A20 tumor cells in 100 μL PBS as we previously described (Ma et al., 2018). After tumor injection, mice were randomly divided to clopidogrel or vehicle group and kept on HFHCC diet. Three weeks later, mice were euthanized, and liver tumors were counted.
Depletion of endogenous platelets and platelet transfer in PF4-DTR mice
PF4-DTR mice were generated by crossing PF4-Cre and iDTR mice as described(Salzmann et al., 2020). MCD diet fed PF4-DTR mice were given s.c. injection of diphtheria toxin (3 times/week, 100ng/mouse first week and 200ng/mouse second week) for two weeks to eliminate endogenous platelets. Platelet depletion was confirmed by measuring peripheral blood platelet level using Flow Cytometry. Platelet transfer was performed as described(Salzmann et al., 2020). Briefly, fresh mouse blood was collected and ACD (Sigma) was added in a 1:10 ratio. Then blood was aliquoted in 500ul amount into 1.5 tubes, then added 300ul Tyrode Hepes (TH) buffer. After centrifuge 100×g 6 min in swing rotor at room temperature, the top platelet rich fraction was collected. Platelets were concentrated by centrifuge 1000×g 1.5min at room temperature at the presence of PGI2(0.1ug/ml), PGE1(1uM) and apyrase (0.5U/ml). Donor platelets were freshly prepared from MCD diet-fed wildtype or CD40L−/− mice. Each recipient mouse received ~4×108 donor platelets per transfer through tail vein injection. To test platelet transfer efficiency, donor platelets were labelled with CFSE (5uM) 10 min at room temperature. After transfer the donor and endogenous platelets were measured by Flow Cytometry.
ELISA
Plasma samples were prepared by mixing fresh blood sample with 3.8% sodium citrate (6/1, v/v) and centrifuged at 1500 g for 10 minutes at room temperature. The supernatant was transferred to a new tube and centrifuged at 2500 g for 10 minutes at room temperature and transferred to a new tube again. This step was repeated once more to get platelet free plasma. Plasma CD40L level was measured by using mouse or human CD40L ELISA kit (Thermo Fisher Scientific) following manufactory protocol, except mouse plasma samples were mixed with 5% BSA (final) and 1% Tween20 (final) before added to microwells.
MS1 cell stimulation and MCP1 real-time PCR
MS1 stimulation assay was performed according to a previous protocol with some modifications(Sprague et al., 2008). Briefly, 1X105 MS1 cells were plated into each well of a 24-well plate. The next day, 100 ul of mouse serum samples were added to each well of MS1 cells and incubated for 6 hours at 37° C. MS1 cells were collected and lysed for RNA isolation and cDNA synthesis. Total RNA was isolated using RNeasy Mini Kit (Cat. NO. 74104, Qiagen) according to manufacturer’s instructions. cDNA synthesis was performed using iScriptTM cDNA synthesis kit (Cat. NO. 170–8891, BIO-RAD). RT-PCR was performed using iQ SYBR Green Supermix (Cat. NO. 1708882, BIO-RAD). The following primers were used for RT-PCR: MCP1: Forward 5’-TGA TCC CAA TGA GTA GGC TGG AG, Reverse 5’- ATG TCT GGA CCC ATT CCT TCT TG-3’; GAPDH: Forward 5’-CCT GCA CCA CCA ACT GCT TA-3’, Reverse 5’-TCA TGA GCC CTT CCA CAA TG-3’. The relative expression levels of MCP1 were determined by 2−^^CT methods. GAPDH expression level was used as control.
Platelet isolation and ex vivo ADP activation
Platelet samples were prepared by mixing fresh blood sample with 3.8% sodium citrate (6/1, v/v) and centrifugation at 70×g for 8 minutes at room temperature with an acceleration of 3 and deceleration of 0. The supernatant was carefully transferred to a new tube and mixed very gently to get platelet-rich plasma. Approximately 1×107 platelets were activated in PBS with 2 mM CaCl2 and 5 μM ADP at 37° C for 10 minutes. The activated platelets were used for subsequent flow cytometry analysis, or the released CD40L was measured with a human CD40L ELISA kit.
In vivo leukotrienes and zileuton treatment
Mice were given i.v. injection of a mix of leukotrienes C4, D4 and E4 at the dose of 1μg/each leukotriene/mouse. Twenty-four hours later, mice were killed, and plasmas were prepared for ELISA analysis of CD40L. Zileuton stock solution was prepared in ethanol at the concentration of 120mg/ml and stored at −20°C, working solution was diluted in 0.1% carboxymethylcellulose before using. Mice were given oral gavage of zileuton twice/day at the dose of 30mg/kg body weight. Cysteinyl leukotriene levels in mouse livers were measured using cysteinyl leukotriene ELISA kit (Enzo life sciences) following manufactory protocol.
Flow cytometry
Liver infiltering mononuclear cells were prepared as previously reported. Briefly, livers were removed immediately after mice were sacrificed. After homogenization, debris was removed by passing through nylon mesh. Liver infiltrating cells were isolated by isotonic Percoll centrifugation (850×g, 25min). Red blood cells were lysed by using ACK lysing buffer. Cells were surface-labeled with indicated antibodies for 15 minutes at 4° C. Isolated platelets were surface-labeled with indicated antibodies for 15 minutes at room temperature. Intracellular staining was performed with a Foxp3/transcription factor staining buffer set (eBioscience) was used according to the manufacturer’s instructions. Flow cytometry was performed on CytoFLEX LX platforms and results were analyzed using FlowJo software version 10.4.2 (TreeStar Inc). Dead cells were excluded by using live/dead fixable near-IR dead cell staining kit (ThermoFisher scientific). The following antibodies were used for flow cytometry analysis: anti-mouse CD41(clone MWReg30), anti-mouse CD62P (clone RMP-1), anti-mouse CD45 (clone 30-F11), anti-mouse CD40 (clone 3/23), anti-mouse CD3(clone 17A2), anti-mouse CD4(clone GK1.5), anti-mouse CD4(clone RM4–4), anti-mouse CD8(clone 53–6.7), anti-mouse CD69 (clone H1.2F3), anti-mouse CD44 (clone BJ18), anti-mouse CD62L (clone MEL14), anti-mouse IFNγ (clone XMG1.2), anti-human CD42b (clone HIP1), anti-human CD40L (clone 24–31), anti-human CD62P-APC (clone AK4).
Use of clopidogrel in NAFLD
Demographic, anthropometric, laboratory, and medication use data were obtained from participants of the 2015–2016 National Health and Nutrition Examination Survey (NHANES), a health survey aiming to capture a multiethnic population representative of the noninstitutionalized civilian resident population of the United States. The US fatty liver index (USFLI) was calculated as described by Ruhl & Everhart(Ruhl and Everhart, 2015) in 2778 adult responders with sufficient data and the presence of NAFLD was defined by a USFLI >= 30. The significance of differences of frequency of use of clopidogrel in subjects with NAFLD compared to those without was tested using Fisher’s exact test.
Quantification and statistical analysis
Statistical analysis
Sample size for animal studies were guided by previous study in our laboratory in which the same NAFLD-HCC mouse models were used. For DEN- or MYC-tumor model, neither randomization nor blinding was performed. However, mice from same littermates were evenly distributed into control or treatment groups whenever possible. For intrahepatic tumor model randomization and blinding were used. Statistical analysis was performed with GraphPad Prism 8 (GraphPad Software). Significance of the difference between groups was calculated by Student’s unpaired t-test, one-way or two-way ANOVA (with Tukey’s and Bonferroni’s multiple comparison test). P<0.05 was considered as statistically significant.
Supplementary Material
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-mouse CD41-FITC (clone MWReg30) | Biolegend | Cat#133903 |
| anti-mouse CD62P-PE (clone RMP-1) | Biolegend | Cat#148305 |
| anti-mouse CD45-BV605 (clone 30-F11) | Biolegend | Cat#103139 |
| anti-mouse CD40-APC (clone 3/23) | Biolegend | Cat#124611 |
| anti-mouse CD3-BV510(clone 17A2) | Biolegend | Cat#100233 |
| anti-mouse CD4-AF700(clone GK1.5) | Biolegend | Cat#100429 |
| anti-mouse CD4-AF700(clone RM4-4) | Biolegend | Cat#116021 |
| anti-mouse CD8-BV421(clone 53-6.7) | Biolegend | Cat#100737 |
| anti-mouse CD69-FITC (clone H1.2F3) | Biolegend | Cat#104505 |
| anti-mouse CD44-PE/Cy7 (clone BJ18) | Biolegend | Cat#338815 |
| anti-mouse CD62L-PerCP/Cy5.5 (clone MEL14) | Biolegend | Cat#104431 |
| anti-mouse IFNg-PE (clone XMG1.2) | Biolegend | Cat#505807 |
| anti-human CD42b-FITC (clone HIP1) | Biolegend | Cat#303903 |
| anti-human CD40L-PE (clone 24–31) | Biolegend | Cat#310805 |
| anti-human CD62P-APC (clone AK4) | Biolegend | Cat#304910 |
| anti-mouse CD40L Ab | Abcam | Cat#ab2391 |
| anti-mouse CD41 Ab | Abcam | Cat#1b134131 |
| In vivo anti-mouse CD4 (clone GK1.5) | BioxCell | BE0003-1 |
| In vivo anti-mouse CD8a (clone 2.43) | BioxCell | BE0061 |
| In vivo CD40 agonistic Ab (clone FGK4.5) | BioxCell | BE0016-2 |
| In vivo CD40L neutralizing Ab (clone MR-1) | BioxCell | BE0017-1 |
| In vivo IL-12 neutralizing Ab (clone C17.8) | BioxCell | BE0051 |
| In vivo platelet depletion anti-mouse GPIba | Emfret analytics | Cat#R300 |
| Biological samples | ||
| Fasting blood from NAFLD patients | NIDDK | Registered as clinicaltrial.gov NCT00001971 |
| Chemicals, peptides, and recombinant proteins | ||
| Clopidogrel | Sigma | Cat#PHR1431 |
| Aspirin | Sigma | Cat#PHR1003 |
| Ticagrelor | Vest-Ward pharmaceuticals Corp | NDC 0186-0776-60 |
| Cilostazol | AstraZeneca | NDC 0002-4462-30 |
| Leukotriene C4 | Cayman Chemical | Cat#20210 |
| Leukotriene D4 | Cayman Chemical | Cat#20310 |
| Leukotriene E4 | Cayman Chemical | Cat#20410 |
| zileuton | Cayman Chemical | Cat#10006967 |
| ExiTron nano 6000 | Miltenyi Biotec | 130-095-146 |
| Clodronate liposomes and control liposomes | Liposoma research liposomes | Cat#CP-005-005 |
| Carbon tetrachloride (CCL4) | Sigma | Cat#289116 |
| Critical commercial assays | ||
| CD40L(soluble) mouse ELISA kit | ThermoFisher | BMS6010 |
| CD40L(soluble) Human ELISA kit | ThermoFisher | BMS239 |
| cysteinyl leukotriene ELISA kit | Enzo life sciences | ADI-900-070 |
| Experimental models: Cell lines | ||
| Hep55.1c | Cell Lines Service | Cat# 400201 |
| B16-F10 | ATCC | CRL-6475 |
| A20 | ATCC | TIB-208 |
| MS-1 | ATCC | CRL-2279 |
| Experimental models: Organisms/strains | ||
| Mouse: CD40L−/−, B6.129S2-Cd40lgtm1Imx/J | The Jackson Laboratory | Strain#002770 |
| Mouse: CD40−/−, B6.129P2-Cd40tm1Kik/J | The Jackson Laboratory | Strain#002928 |
| Mouse: Batf3−/−, B6.129S(C)-Batf3tm1Kmm/J | The Jackson Laboratory | Strain#013755 |
| Mouse: Pf4-cre, C57BL/6-Tg(Pf4-icre)Q3Rsko/J | The Jackson Laboratory | Strain#008535 |
| Mouse: iDTR, C57BL/6 Gt(ROSA)26Sortm1(HBEGF)Awai/J | The Jackson Laboratory | Strain#007900 |
| Mouse: LAP-tTA mice | From Dr. Dean Felsher | N/A |
| Mouse: TRE-MYC mice | From Dr. Dean Felsher | N/A |
| Mouse: P2Y12−/− mice | From Dr. Long-Jun Wu | N/A |
| Oligonucleotides | ||
| Primer MCP1 Forward TGATCCCAATGAGTAGGCTGGAG | This paper | N/A |
| Primer MCP1 Reverse ATGTCTGGACCCATTCCTTCTTG | This paper | N/A |
| Primer IL-12 Forward GGAAGCACGGCAGCAGAATA | This paper | N/A |
| Primer IL-12 Reverse AACTTGAGGGAGAAGTAGGAATGG | This paper | N/A |
| Primer GAPDH Forward CCTGCACCACCAACTGCTTA | This paper | N/A |
| Primer GAPDH Reverse TCATGAGCCCTTCCACAATG | This paper | N/A |
| Software and algorithms | ||
| FlowJo | TreeStar Inc | https://www.flowjo.com |
| GraphPad Prism | GraphPad Software | https://www.graphpad.com |
| DragonFly 4.0 | Object Research Systems | https://theobjects.com/dragonfly/new.html |
| Other | ||
| Methionine–choline-deficient (MCD) diet | Research Diets | Cat#A02082002BR |
| High fat diet (rodent diet with 60 kcal% fat) | Research Diets | Cat#D12492 |
| Choline-deficient and amino-acid-defined (CDAA) diet | Dyets Inc | Cat#518753 |
| High fat, high cholesterol, cholate (HFHCC) diet | Teklad | TD.88051 |
| Western diet | Teklad | TD.120528 |
Highlights:
Platelets enhance anti-tumor immune response in NAFLD through CD40L.
CD8+ T cells mediate the anti-tumor immune response.
P2Y12/leukotrienes control CD40L release from platelets in NAFLD.
Macrophages release IL12 to promote CD40L production by megakaryocytes.
Acknowledgments
We would like to thank Dr. Jay Berzofsky’s lab members for helpful discussions. We also want to thank Dr. Henrik Thorlacius, Joshua Welsh, Jennifer Jones, Bennett Elzey, Timothy Ratliff, Alice Assinger, Manuel Salzmann and Waltraud Schrottmaier for technical advice. T.F.G. was supported by the Intramural Research Program of the NIH, NCI. Y.R. was supported by the Intramural Research Program of NIH, NIDDK. S.W. was supported by the Deutsche Forschungsgemeinschaft (WA-4610/1–1). BR was supported by the International Liver Cancer Association (ILCA) fellowship award 2021.
Footnotes
Declaration of interests
Authors declare that they have no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barry OP, Kazanietz MG, Pratico D, and FitzGerald GA (1999). Arachidonic acid in platelet microparticles up-regulates cyclooxygenase-2-dependent prostaglandin formation via a protein kinase C/mitogen-activated protein kinase-dependent pathway. J Biol Chem 274, 7545–7556. 10.1074/jbc.274.11.7545. [DOI] [PubMed] [Google Scholar]
- Bergmeier W, and Boulaftali Y (2014). Adoptive transfer method to study platelet function in mouse models of disease. Thromb Res 133 Suppl 1, S3–5. 10.1016/j.thromres.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings HE, Liu T, Feng C, Laidlaw TM, Conley PB, Kanaoka Y, and Boyce JA (2013). Cutting edge: Leukotriene C4 activates mouse platelets in plasma exclusively through the type 2 cysteinyl leukotriene receptor. J Immunol 191, 5807–5810. 10.4049/jimmunol.1302187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deczkowska A, David E, Ramadori P, Pfister D, Safran M, At The B, Giladi A, Jaitin DA, Barboy O, Cohen M, et al. (2021). XCR1(+) type 1 conventional dendritic cells drive liver pathology in non-alcoholic steatohepatitis. Nat Med 27, 1043–1054. 10.1038/s41591-021-01344-3. [DOI] [PubMed] [Google Scholar]
- Diggs LP, and Greten TF (2019). The effects of platelet accumulation in fatty liver disease. Nat Rev Gastroenterol Hepatol 16, 393–394. 10.1038/s41575-019-0160-8. [DOI] [PubMed] [Google Scholar]
- Diggs LP, Ruf B, Ma C, Heinrich B, Cui L, Zhang Q, McVey JC, Wabitsch S, Heinrich S, Rosato U, et al. (2020). CD40-mediated immune cell activation enhances response to anti-PD-1 in murine intrahepatic cholangiocarcinoma. J Hepatol. 10.1016/j.jhep.2020.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disibio G, and French SW (2008). Metastatic patterns of cancers: results from a large autopsy study. Arch Pathol Lab Med 132, 931–939. 10.1043/1543-2165(2008)132[931:MPOCRF]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Elgueta R, Benson MJ, de Vries VC, Wasiuk A, Guo Y, and Noelle RJ (2009). Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev 229, 152–172. 10.1111/j.1600-065X.2009.00782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elzey BD, Tian J, Jensen RJ, Swanson AK, Lees JR, Lentz SR, Stein CS, Nieswandt B, Wang Y, Davidson BL, and Ratliff TL (2003). Platelet-mediated modulation of adaptive immunity. A communication link between innate and adaptive immune compartments. Immunity 19, 9–19. [DOI] [PubMed] [Google Scholar]
- Ezzaty Mirhashemi M, Shah RV, Kitchen RR, Rong J, Spahillari A, Pico AR, Vitseva O, Levy D, Demarco D, Shah S, et al. (2021). The Dynamic Platelet Transcriptome in Obesity and Weight Loss. Arterioscler Thromb Vasc Biol 41, 854–864. 10.1161/ATVBAHA.120.315186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierro JJ, Cave B, and Khouzam RN (2019). P2Y12 inhibitors: do they increase cancer risk? Ann Transl Med 7, 409. 10.21037/atm.2019.07.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco AT, Corken A, and Ware J (2015). Platelets at the interface of thrombosis, inflammation, and cancer. Blood 126, 582–588. 10.1182/blood-2014-08-531582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghazarian M, Revelo XS, Nohr MK, Luck H, Zeng K, Lei H, Tsai S, Schroer SA, Park YJ, Chng MHY, et al. (2017). Type I Interferon Responses Drive Intrahepatic T cells to Promote Metabolic Syndrome. Sci Immunol 2. 10.1126/sciimmunol.aai7616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorden DL, Myers DS, Ivanova PT, Fahy E, Maurya MR, Gupta S, Min J, Spann NJ, McDonald JG, Kelly SL, et al. (2015). Biomarkers of NAFLD progression: a lipidomics approach to an epidemic. J Lipid Res 56, 722–736. 10.1194/jlr.P056002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handtke S, and Thiele T (2020). Large and small platelets-(When) do they differ? J Thromb Haemost 18, 1256–1267. 10.1111/jth.14788. [DOI] [PubMed] [Google Scholar]
- Heinrich B, Brown ZJ, Diggs LP, Vormehr M, Ma C, Subramanyam V, Rosato U, Ruf B, Walz JS, McVey JC, et al. (2021). Steatohepatitis Impairs T-cell-Directed Immunotherapies Against Liver Tumors in Mice. Gastroenterology 160, 331–345 e336. 10.1053/j.gastro.2020.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann A, Rauch BH, Braun M, Schror K, and Weber AA (2001). Platelet CD40 ligand (CD40L)--subcellular localization, regulation of expression, and inhibition by clopidogrel. Platelets 12, 74–82. [DOI] [PubMed] [Google Scholar]
- Iannacone M, Sitia G, Isogawa M, Marchese P, Castro MG, Lowenstein PR, Chisari FV, Ruggeri ZM, and Guidotti LG (2005). Platelets mediate cytotoxic T lymphocyte-induced liver damage. Nat Med 11, 1167–1169. 10.1038/nm1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer M, Thomas E, Milton RJ, Perry AW, van Rooijen N, Wheeler MD, Zacks S, Fried M, Rippe RA, and Hines IN (2010). Kupffer cell and interleukin-12-dependent loss of natural killer T cells in hepatosteatosis. Hepatology 51, 130–141. 10.1002/hep.23292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labelle M, Begum S, and Hynes RO (2011). Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 20, 576–590. 10.1016/j.ccr.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, Chung GE, Lee JH, Oh S, Nam JY, Chang Y, Cho H, Ahn H, Cho YY, Yoo JJ, et al. (2017). Antiplatelet therapy and the risk of hepatocellular carcinoma in chronic hepatitis B patients on antiviral treatment. Hepatology 66, 1556–1569. 10.1002/hep.29318. [DOI] [PubMed] [Google Scholar]
- Lee TY, Hsu YC, Tseng HC, Yu SH, Lin JT, Wu MS, and Wu CY (2019). Association of Daily Aspirin Therapy With Risk of Hepatocellular Carcinoma in Patients With Chronic Hepatitis B. JAMA Intern Med 179, 633–640. 10.1001/jamainternmed.2018.8342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefrancais E, Ortiz-Munoz G, Caudrillier A, Mallavia B, Liu F, Sayah DM, Thornton EE, Headley MB, David T, Coughlin SR, et al. (2017). The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature 544, 105–109. 10.1038/nature21706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, Lencioni R, Koike K, Zucman-Rossi J, and Finn RS (2021). Hepatocellular carcinoma. Nat Rev Dis Primers 7, 6. 10.1038/s41572-020-00240-3. [DOI] [PubMed] [Google Scholar]
- Ma C, Han M, Heinrich B, Fu Q, Zhang Q, Sandhu M, Agdashian D, Terabe M, Berzofsky JA, Fako V, et al. (2018). Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science 360. 10.1126/science.aan5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Kesarwala AH, Eggert T, Medina-Echeverz J, Kleiner DE, Jin P, Stroncek DF, Terabe M, Kapoor V, ElGindi M, et al. (2016). NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature 531, 253–257. 10.1038/nature16969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan SA, John F, and Pitchumoni CS (2016). Nonalcoholic Fatty Liver Disease and Mean Platelet Volume: A Systemic Review and Meta-analysis. J Clin Gastroenterol 50, 69–74. 10.1097/MCG.0000000000000340. [DOI] [PubMed] [Google Scholar]
- Malehmir M, Pfister D, Gallage S, Szydlowska M, Inverso D, Kotsiliti E, Leone V, Peiseler M, Surewaard BGJ, Rath D, et al. (2019). Platelet GPIbalpha is a mediator and potential interventional target for NASH and subsequent liver cancer. Nat Med 25, 641–655. 10.1038/s41591-019-0379-5. [DOI] [PubMed] [Google Scholar]
- Marigo I, Zilio S, Desantis G, Mlecnik B, Agnellini AHR, Ugel S, Sasso MS, Qualls JE, Kratochvill F, Zanovello P, et al. (2016). T Cell Cancer Therapy Requires CD40-CD40L Activation of Tumor Necrosis Factor and Inducible Nitric-Oxide-Synthase-Producing Dendritic Cells. Cancer Cell 30, 377–390. 10.1016/j.ccell.2016.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauri L, Kereiakes DJ, Yeh RW, Driscoll-Shempp P, Cutlip DE, Steg PG, Normand SL, Braunwald E, Wiviott SD, Cohen DJ, et al. (2014). Twelve or 30 months of dual antiplatelet therapy after drug-eluting stents. N Engl J Med 371, 2155–2166. 10.1056/NEJMoa1409312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelotti GA, Machado MV, and Diehl AM (2013). NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol 10, 656–665. 10.1038/nrgastro.2013.183. [DOI] [PubMed] [Google Scholar]
- Nagasawa M, Zhu Y, Isoda T, Tomizawa D, Itoh S, Kajiwara M, Morio T, Nonoyama S, Shimizu N, and Mizutani S (2005). Analysis of serum soluble CD40 ligand (sCD40L) in the patients undergoing allogeneic stem cell transplantation: platelet is a major source of serum sCD40L. Eur J Haematol 74, 54–60. 10.1111/j.1600-0609.2004.00342.x. [DOI] [PubMed] [Google Scholar]
- Pariser DN, Hilt ZT, Ture SK, Blick-Nitko SK, Looney MR, Cleary SJ, Roman-Pagan E, Saunders J 2nd, Georas SN, Veazey J, et al. (2021). Lung megakaryocytes are immune modulatory cells. J Clin Invest 131. 10.1172/JCI137377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paruchuri S, Tashimo H, Feng C, Maekawa A, Xing W, Jiang Y, Kanaoka Y, Conley P, and Boyce JA (2009). Leukotriene E4-induced pulmonary inflammation is mediated by the P2Y12 receptor. J Exp Med 206, 2543–2555. 10.1084/jem.20091240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X, Remacle JE, Kasran A, Huylebroeck D, and Ceuppens JL (1998). IL-12 up-regulates CD40 ligand (CD154) expression on human T cells. J Immunol 160, 1166–1172. [PubMed] [Google Scholar]
- Puri P, Wiest MM, Cheung O, Mirshahi F, Sargeant C, Min HK, Contos MJ, Sterling RK, Fuchs M, Zhou H, et al. (2009). The plasma lipidomic signature of nonalcoholic steatohepatitis. Hepatology 50, 1827–1838. 10.1002/hep.23229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachidi S, Metelli A, Riesenberg B, Wu BX, Nelson MH, Wallace C, Paulos CM, Rubinstein MP, Garrett-Mayer E, Hennig M, et al. (2017). Platelets subvert T cell immunity against cancer via GARP-TGFbeta axis. Sci Immunol 2. 10.1126/sciimmunol.aai7911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinella ME, Elias MS, Smolak RR, Fu T, Borensztajn J, and Green RM (2008). Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline-deficient diet. J Lipid Res 49, 1068–1076. 10.1194/jlr.M800042-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhl CE, and Everhart JE (2015). Fatty liver indices in the multiethnic United States National Health and Nutrition Examination Survey. Aliment Pharmacol Ther 41, 65–76. 10.1111/apt.13012. [DOI] [PubMed] [Google Scholar]
- Salzmann M, Schrottmaier WC, Kral-Pointner JB, Mussbacher M, Volz J, Hoesel B, Moser B, Bleichert S, Morava S, Nieswandt B, et al. (2020). Genetic platelet depletion is superior in platelet transfusion compared to current models. Haematologica 105, 1738–1749. 10.3324/haematol.2019.222448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher D, Strilic B, Sivaraj KK, Wettschureck N, and Offermanns S (2013). Platelet-derived nucleotides promote tumor-cell transendothelial migration and metastasis via P2Y2 receptor. Cancer Cell 24, 130–137. 10.1016/j.ccr.2013.05.008. [DOI] [PubMed] [Google Scholar]
- Shalapour S, Lin XJ, Bastian IN, Brain J, Burt AD, Aksenov AA, Vrbanac AF, Li W, Perkins A, Matsutani T, et al. (2017). Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature 551, 340–345. 10.1038/nature24302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon TG, Duberg AS, Aleman S, Chung RT, Chan AT, and Ludvigsson JF (2020). Association of Aspirin with Hepatocellular Carcinoma and Liver-Related Mortality. N Engl J Med 382, 1018–1028. 10.1056/NEJMoa1912035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitia G, Aiolfi R, Di Lucia P, Mainetti M, Fiocchi A, Mingozzi F, Esposito A, Ruggeri ZM, Chisari FV, Iannacone M, and Guidotti LG (2012). Antiplatelet therapy prevents hepatocellular carcinoma and improves survival in a mouse model of chronic hepatitis B. Proc Natl Acad Sci U S A 109, E2165–2172. 10.1073/pnas.1209182109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sookoian S, Castano GO, Burgueno AL, Rosselli MS, Gianotti TF, Mallardi P, Martino JS, and Pirola CJ (2010). Circulating levels and hepatic expression of molecular mediators of atherosclerosis in nonalcoholic fatty liver disease. Atherosclerosis 209, 585–591. 10.1016/j.atherosclerosis.2009.10.011. [DOI] [PubMed] [Google Scholar]
- Sprague DL, Elzey BD, Crist SA, Waldschmidt TJ, Jensen RJ, and Ratliff TL (2008). Platelet-mediated modulation of adaptive immunity: unique delivery of CD154 signal by platelet-derived membrane vesicles. Blood 111, 5028–5036. 10.1182/blood-2007-06-097410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto K, Shiraki K, Ito T, Fujikawa K, Takase K, Tameda Y, Moriyama M, and Nakano T (1999). Expression of functional CD40 in human hepatocellular carcinoma. Hepatology 30, 920–926. 10.1002/hep.510300424. [DOI] [PubMed] [Google Scholar]
- Sutti S, Jindal A, Locatelli I, Vacchiano M, Gigliotti L, Bozzola C, and Albano E (2014). Adaptive immune responses triggered by oxidative stress contribute to hepatic inflammation in NASH. Hepatology 59, 886–897. 10.1002/hep.26749. [DOI] [PubMed] [Google Scholar]
- Tsuchida T, Lee YA, Fujiwara N, Ybanez M, Allen B, Martins S, Fiel MI, Goossens N, Chou HI, Hoshida Y, and Friedman SL (2018). A simple diet- and chemical-induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. J Hepatol 69, 385–395. 10.1016/j.jhep.2018.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu LN, Showalter MR, Cajka T, Fan S, Pillai VV, Fiehn O, and Selvaraj V (2017). Metabolomic characteristics of cholesterol-induced non-obese nonalcoholic fatty liver disease in mice. Sci Rep 7, 6120. 10.1038/s41598-017-05040-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonderheide RH (2007). Prospect of targeting the CD40 pathway for cancer therapy. Clin Cancer Res 13, 1083–1088. 10.1158/1078-0432.CCR-06-1893. [DOI] [PubMed] [Google Scholar]
- Wolf MJ, Adili A, Piotrowitz K, Abdullah Z, Boege Y, Stemmer K, Ringelhan M, Simonavicius N, Egger M, Wohlleber D, et al. (2014). Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell 26, 549–564. 10.1016/j.ccell.2014.09.003. [DOI] [PubMed] [Google Scholar]
- Wree A, Broderick L, Canbay A, Hoffman HM, and Feldstein AE (2013). From NAFLD to NASH to cirrhosis-new insights into disease mechanisms. Nat Rev Gastroenterol Hepatol 10, 627–636. 10.1038/nrgastro.2013.149. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the main text or the supplementary materials.