

Abstract
Pseudohypoparathyroidism type Ib (PHP1B) is characterized predominantly by resistance to parathyroid hormone (PTH) leading to hypocalcemia and hyperphosphatemia. These laboratory abnormalities are caused by maternal loss-of-methylation (LOM) at GNAS exon A/B, which reduces in cis expression of the stimulatory G protein α-subunit (Gsα). Paternal Gsα expression in proximal renal tubules is silenced through unknown mechanisms, hence LOM at exon A/B reduces further Gsα protein in this kidney portion, leading to PTH-resistance. In a previously reported PHP1B family, affected members showed variable LOM at exon A/B, yet no genetic defect was found by whole-genome sequencing despite linkage to GNAS. Using targeted long-read sequencing (T-LRS), we discovered an approximately 2,800-bp maternally inherited retrotransposon insertion nearly 1,200 bp downstream of exon XL not found in public databases or in 13,675 DNA samples analyzed by short-read whole-genome sequencing. T-LRS data furthermore confirmed normal methylation at exons XL, AS, and NESP and showed that LOM comprising exon A/B is broader than previously thought. The retrotransposon most likely causes the observed epigenetic defect by impairing function of a maternally derived NESP transcript, consistent with findings in mice lacking full-length NESP mRNA and in PHP1B patients with deletion of exon NESP and adjacent intronic sequences. In addition to demonstrating that T-LRS is an effective strategy for identifying a small disease-causing variant that abolishes or severely reduces exon A/B methylation, our data demonstrate that this sequencing technology has major advantages for simultaneously identifying structural defects and altered methylation.
Keywords: pseudohypoparathyroidism, Gs-alpha, GNAS, parent-specific GNAS methylation, targeted long-read sequencing, retrotransposon
INTRODUCTION
Pseudohypoparathyroidism type Ia (PHP1A; MIM #103580) is caused by maternally inherited variants in exons 1–13 of GNAS, which encode the α-subunit of the stimulatory G protein α-subunit (Gsα). This ubiquitously expressed signaling protein is required for mediating the actions of numerous G protein-coupled receptors, including those of the PTH/PTHrP receptor (PTH1R). Gsα is biallelically expressed in most tissues, with few exceptions, including the proximal renal tubules, where functional Gsα is derived predominantly from the maternal allele. Inactivating Gsα mutations involving this parental GNAS allele lead to parathyroid hormone (PTH) resistance at the PTH1R in this portion of the kidney and insufficient down-regulation of sodium-dependent phosphate co-transporters thereby impairing urinary phosphate excretion. In addition, reduced PTH-dependent 1,25(OH)2 vitamin D production impairs intestinal calcium absorption, contributing to the development of hypocalcemia and elevated PTH levels.(1–3)
In addition to Gsα, several alternative first exons and their promoters lead to the generation of additional mRNA variants that are transcribed from the GNAS locus (Figure 1A). These include the A/B and XL transcripts, which are derived only from the paternal allele because their maternal promoters are methylated at CpG islands that constitute differentially methylated regions (DMRs). Conversely, the NESP (neuroendocrine secretory peptide) transcript is derived from the maternal GNAS allele; hence, its DMR is methylated on the paternal allele. The A/B, XL, and NESP mRNAs differ because of unique first exons but are identical in their 3’-regions due to splicing onto GNAS exons 2–13. In addition to these three sense transcripts, an antisense (AS) RNA comprising at least five exons is derived from the nonmethylated AS exon 1 promoter on the paternal GNAS allele.(1–3)
Figure 1: Differential methylation of the STX16-GNAS region results in five different sense and antisense transcripts.
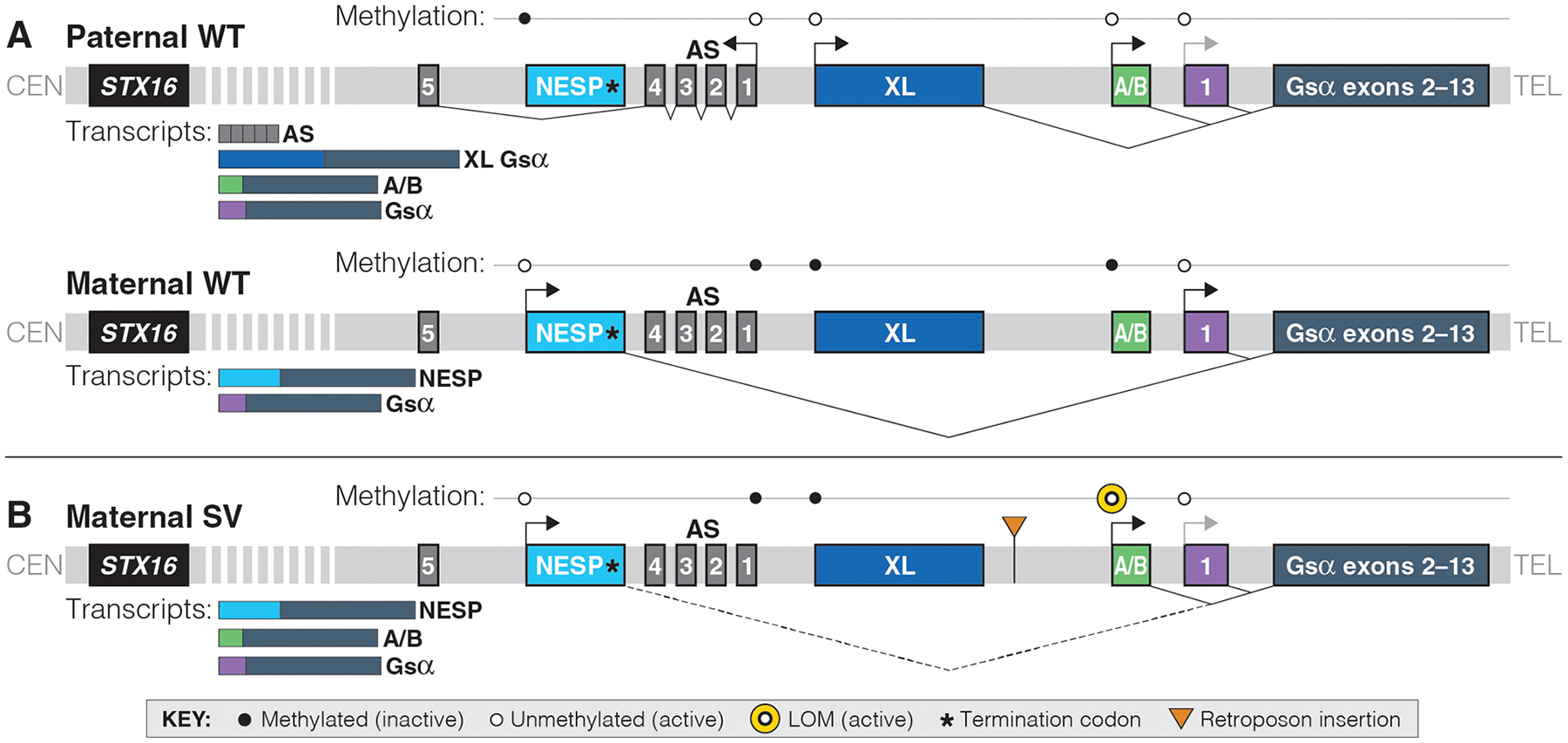
Not to genomic scale; bent arrows represent direction of transcription; light gray bent arrows represent reduced expression; thin lines represent splicing; dotted line represents generation of an aberrant transcript in the presence of the SVA_E insertion; CEN, centromere; TEL, telomere; WT, wild-type. A. Maternal methylation results in the XL, A/B, and AS transcripts being expressed only from the paternal allele. Paternal methylation of the NESP promoter results in NESP mRNA only being derived from the maternal allele. Gsα is expressed from both alleles. B. An SVA_E insertion in family 208 results in loss-of-methylation (LOM) at the GNAS_A/B locus and alters abundance of maternal transcripts. Previous work has shown that lack of the NESP exon in two PHP1B families leads to LOM at A/B alone. Thus, we hypothesize that a non-spliced RNA derived from NESP may be required for proper A/B methylation (dotted line).
In contrast to genetic defects that cause PHP1A through mutations involving the GNAS exons encoding Gsα, the different forms of pseudohypoparathyroidism type Ib (PHP1B; MIM #603233) are caused by methylation abnormalities at one or several GNAS DMRs. Most familial cases of autosomal dominant PHP1B (AD-PHP1B) are caused by a maternally inherited 3-kbp STX16 deletion, which leads to loss-of-methylation (LOM) at GNAS exon A/B alone.(1–3) The same epigenetic changes are also observed in patients with other maternal mutations, including several larger STX16 deletions(4–7), deletions of the GNAS exon NESP and adjacent intronic regions(6,8), a large inversion involving exon A/B and all of the exons encoding Gsα(9), and several different duplications/triplications involving the region centromeric of exon A/B.(10–12)
Additional AD-PHP1B variants are caused by inherited deletions comprising NESP and/or several AS exons that lead to a loss of the maternal methylation imprints and an apparent increase in methylation at the NESP DMR(13–15); similar epigenetic changes have also been described for a large inherited duplication of the entire GNAS locus.(12) However, the most frequent form of PHP1B is sporadic (sporPHP1B) and affected patients show LOM at the three maternal GNAS DMRs and gain-of-methylation (GOM) at exon NESP, which is unique to this disease variant.(5,7,15–18)
All PHP1B variants share LOM at the exon A/B DMR (Figure 1B). The resulting biallelic expression from the exon A/B promoter reduces Gsα expression(19) and causes Gsα deficiency in tissues such as the proximal renal tubules where this signaling protein is derived mainly from the maternal allele. Mice lacking the paternal exon 1A (the murine equivalent of human exon A/B) show normal methylation at the maternal exon 1A DMR but de-repressed paternal Gsα expression in brown fat and alleviated PTH-resistance in mice with an inactivating mutation in the maternal exon 6 of Gsα (Oed-Sml mice)(20). These findings are consistent with the conclusion that 1A and A/B transcripts have, independent of epigenetic GNAS changes, a silencing function that reduces the biological activity of Gsα. In addition, the A/B transcript generates, through the use of an initiator AUG located in GNAS exon 3, an amino-terminally truncated form of Gsα that is capable of reducing the responsiveness to various agonists.(21) Both mechanisms, namely reduced Gsα expression and generation of a Gsα antagonist through the A/B transcript, could thus contribute to hormonal resistance in PHP1B.
We recently reported a family with AD-PHP1B, in which the affected family members share the same GNAS allele telomeric of microsatellite marker D20S100 that is associated with LOM restricted to GNAS exon A/B (Figure 2). In addition, these patients had a significant GOM at a newly defined AS2–1 DMR located between AS exon 1 and the XL exon, while the adjacent AS2–2 DMR showed normal methylation.(22) Short-read whole-genome sequencing (WGS) failed to identify a disease-causing variant within the STX16-GNAS region associated with these methylation differences. To continue searching for a novel mutation, we further defined the linked region using additional family members and additional microsatellite markers (Figure 2) and then used targeted long-read sequencing (T-LRS) to identify a novel retrotransposon insertion associated with the methylation abnormalities seen in this family (Figure 3).
Figure 2: Pedigree and microsatellite markers of AD-PHP1B family 208.
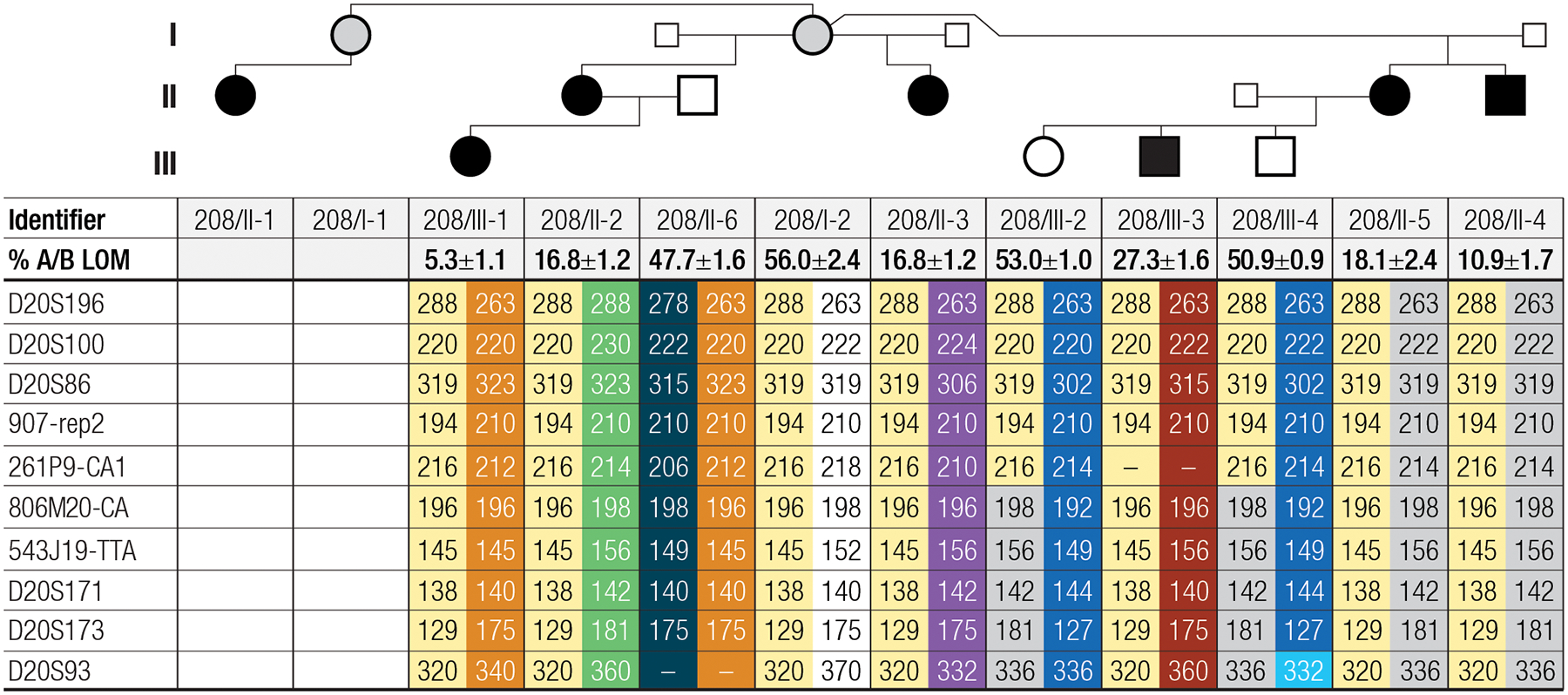
Analysis of microsatellites from this family reveals that the four affected children of unaffected female 208/I-2 share the same maternally inherited chromosome 20 microsatellites telomeric of STX16 (yellow). Two affected grandchildren (individuals 208/III-1 and 208/III-3) also carry the same allele while two unaffected grandchildren inherited a maternal allele not linked to the disease. Analysis of genomic DNA by MS-MLPA revealed LOM at GNAS exon A/B for all six affected individuals (shown as % A/B LOM), consistent with inheritance of the disease-associated allele from a female. Note that a recombination event was observed between 261P9-CA1 and 806M20-CA for the two unaffected children (208/III-2 and 208/III-4) of the affected female 208/II-5. Small squares indicate that genomic DNA was not available for these individuals; gray circles indicate an unaffected carrier of the mutation; dashes indicate that evaluation of the marker failed; genomic DNA was not available for individuals 208/I-1 and 208/II-1.
Figure 3: T-LRS identified an SVA_E insertion that alters methylation at the exon A/B DMR.
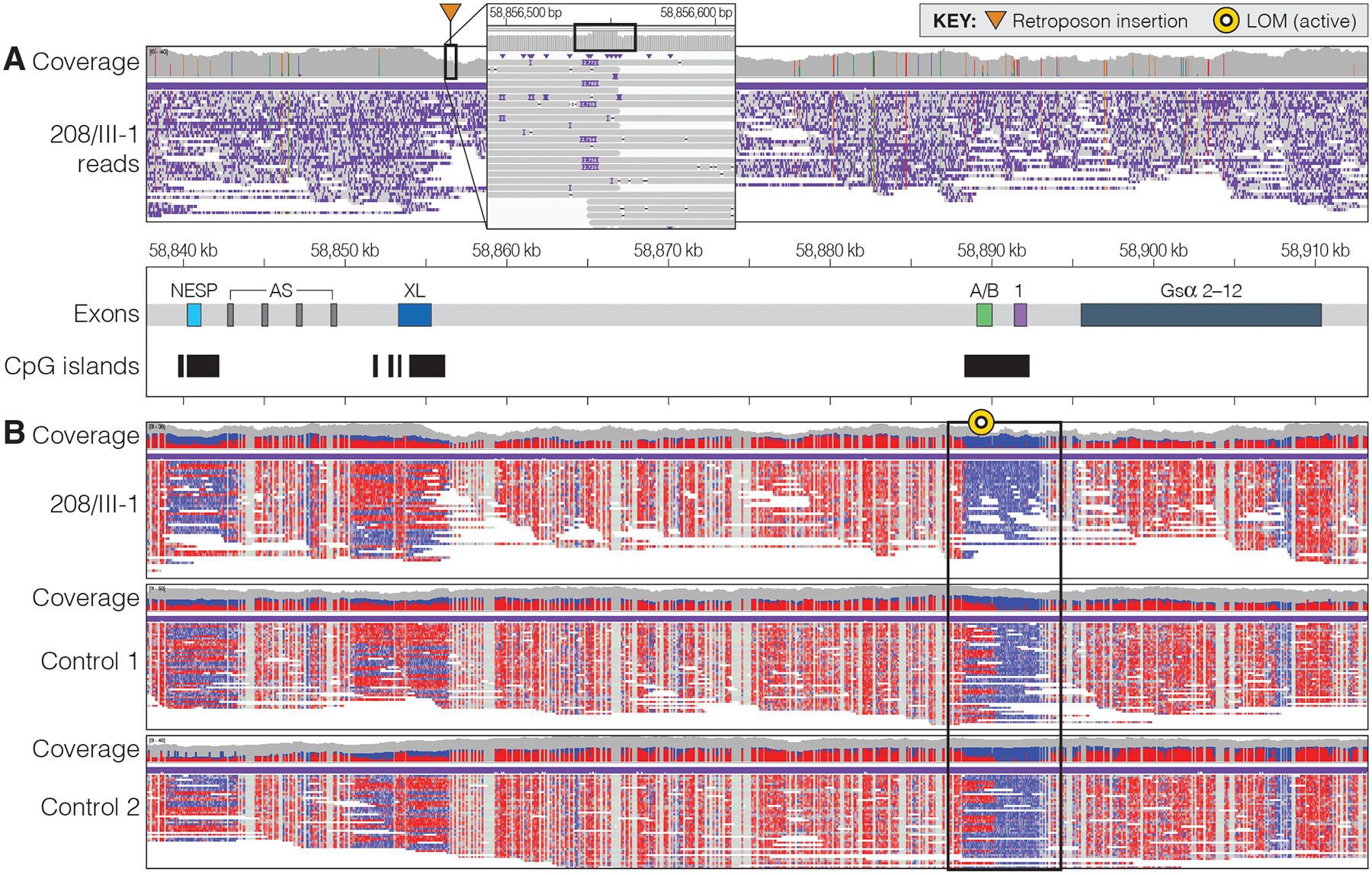
A. IGV view of the GNAS region in individual 208/III-1. Analysis of the region revealed an approximately 2,800-bp insertion on the telomeric side of exon XL that was determined to be an SVA_E insertion. The callout box shows per-read details at the insertion site. Increased coverage in the insert is associated with a target site duplication (box). B. Analysis of methylation at the DMRs within GNAS revealed no difference in 208/III-1 from controls at the NESP and XL exons but did reveal that the individual has no methylation at the A/B exon whereas unrelated controls 1 and 2 have approximately 50% methylation at this region. Note that all three individuals have no methylation at GNAS exon 1, the first exon encoding Gsα, next to exon A/B. Methylated CpGs are denoted in red, unmethylated CpGs are blue.
MATERIALS and METHODS
Microsatellite marker analyses, Multiplex Ligation-dependent Probe Amplification (MLPA), and Methylation-Sensitive MLPA (MS-MLPA)
The following microsatellite markers in the chromosome 20q13.3 region were investigated: D20S178, D20S196, D20S100, D20S86, 907-rep2, 261P9-CA, 806M20-CA, 543J19-TTA, D20S171, D20S173, and D20S93.(15,23) All markers were analyzed at the Center for Human Genetic Research of the Massachusetts General Hospital. MLPA and MS-MLPA analyses were performed using ME034, as previously described.(23)
Targeted long-read sequencing of the GNAS region
We performed T-LRS using Adaptive Sampling on the Oxford Nanopore Technologies (ONT) platform. Briefly, 1.5 μg of genomic DNA from individual 208/III-1 (Figure 2)(22) was sheared to approximately 10,000 bp using a Covaris g-TUBE by centrifuging at 6,000 rpm and inverting twice. Sequencing libraries were prepared using the ONT SQK-LSK110 kit (ONT) following the manufacturer’s instructions with the following modifications: the sample was held at 60°C for 30 minutes instead of 5 minutes to allow the DNA repair step to proceed longer, and the ligation step was allowed to proceed at room temperature for 1 hour instead of 10 min.
Target regions for sequencing were defined as chr20:56,000,000–64,444,000 (GRCh38 coordinates) for the GNAS region and three control regions (chr17:50,150,000–50,250,000, chrX:50,150,000–50,250,000, and chrX:147,850,000–148,000,000). Adaptive sampling on the ONT platform works by selectively sequencing DNA molecules that reside within predefined regions provided by the user. Molecules are base called in real time by the sequencer and after approximately 500 bp of sequence are aligned to a reference genome. Sequencing continues if the molecule being sequenced lies within the region defined by the user sequencing; if it does not, the current on the pore is reversed, the molecule is ejected, and a new molecule is selected for sequencing. This occurs simultaneously and independently for all 512 channels on the ONT MinION or GridION flow cell. An 896 ng aliquot of the prepared library was loaded onto a R9.4.1 flow cell and run for 24 hr on a GridION running MinKNOW software version 21.10.8. After 24 hr, the flow cell was washed, and a second library aliquot was loaded and run for an additional 48 hrs. FASTQ files were generated from the raw sequencing data with Guppy 5.0.12 (ONT) using the superior model. Methylation was called using Megalodon (version 2.3.5, ONT), and modified BAM files were generated using Megalodon.
Nucleotide sequence analysis of the GNAS region
FASTQ files were aligned to GRCh38 using minimap2.(24) Single-nucleotide (SNV) and insertion/deletion (indel) variants for the GNAS target region were called and phased with Clair3 (version v0.1-r10).(25) Because Clair3 was unable to phase the GNAS region into separate haplotypes, variants were also called and phased with medaka (version 1.4.2, ONT) which was also unable to phase the region into separate haplotypes. SNVs and indels called with Clair3 were annotated with VEP (version 103.1).(26) Variants within the target region with allele frequency <0.01 or that had never been observed before were isolated for further analysis. Structural variants (SVs) were called using Sniffles, SVIM, and CuteSV.(27–29) SVs within GNAS were then manually evaluated using IGV.(30) CpG methylation of the GNAS locus was visually analyzed with IGV.
Assembly of the GNAS target region
To better evaluate the SINE-VNTR-Alu (SVA) insertion identified within GNAS, we assembled all FASTQ reads using Flye 2.9,(31) which produced one 8.5-Mbp contig and several smaller contigs (Supplemental File 1). Because the GNAS target region was 8.4 Mbp in size, we suspected that our assembly resulted in a single contig of the target region and confirmed this by comparison to the reference genome (Supplemental Figure 1). The SVA insertion within the assembly was isolated by searching for the 50 bp flanking either side of the insertion within the assembly and then isolating reads between the flanking reads. This resulted in identification of a 2,795-bp insertion that aligned with greater than 98% identity to other SVA_E retrotransposon sequences.
Estimating the population frequency of the identified SVA insertion
To determine if the observed SVA insertion was present in WGS data from other individuals, we analyzed 13,675 genomes from families in which at least one individual is affected with autism.(32) Briefly, split and discordant reads from a 2-kbp region surrounding the SVA insertion that had been identified in family 208 (chr20:58,856,000–58,857,000; GRCh38) were isolated. No individual in this dataset had more than five split or discordant reads within the 2-kbp region suggesting that none of those individuals carried an insertion within this window. To confirm that our method would detect an insertion within this region, we performed our analysis on short-read WGS data from family 208 and identified the insertion in all cases.
Confirmation of the insertion telomeric of GNAS exon XL
To determine the telomeric boundary of the insertion, two primer pairs were designed (PrimerBlast) for multiplex PCR amplification of genomic DNA. A 783-bp amplicon comprising the SVA insertion was obtained when using forward primer 5’-GAAGAGCCCGAGGTAAGCCA-3’ and reverse primer 5 ‘-TATCCCAGACGATGGGTGG-3’; a 271-bp amplicon derived from the wild-type allele when using forward primer 5’-GAAGAGCCCGAGGTAAGCCA-3’ and reverse primer 5’-CCTTTGACAAGCCTCTTTGCC-3’. PCR was performed with QIAGEN Taq DNA polymerase (QIAGEN, Valencia, CA) following the manufacturer’s protocol; cycler program: denaturation at 94°C for 5 min followed by 30 cycles at 95°C for 20s, 58°C for 15s, and 72°C for 25s, followed by an additional elongation step at 72°C for 10 min. All PCR products were purified using ExoSap-IT (Affymetrix, Santa Clara, CA), which were sequenced at the DNA core facility of the Massachusetts General Hospital in Boston, MA.
Consent
Written consent was obtained from each individual or guardian from this family, as previously described.(22)
RESULTS
Despite LOM restricted to GNAS exon A/B, we had previously excluded STX16 deletions as the cause of the PTH-resistant hypocalcemia and hyperphosphatemia for the affected members of this family (AD-PHP1B family 208), who had been available at the time.(22) Here, we have expanded this kindred by four members, including an unaffected female (208/I-2) who has four affected children with three different partners (Figure 2). One affected daughter (208/II-2) has one affected child (208/III-1) with LOM of the exon A/B DMR only. Another affected daughter (208/II-5) has one affected son (208/III-3) with LOM restricted to the exon A/B DMR as well as an unaffected daughter (208/III-2) and an unaffected son (208/III-4) with normal GNAS methylation. Furthermore, individual 208/I-1 (the full sister of individual 208/I-2) has a single affected daughter (208/II-1) for whom DNA was not available for testing. That all affected individuals in family 208 were born to affected or unaffected females was consistent with the mode of inheritance previously established for several different AD-PHP1B variants and suggested that the disease-causing genetic defect could reside within the STX16-GNAS region.(2,3)
AD-PHP1B associated with LOM at GNAS exon A/B alone is frequently caused by a maternally inherited 3-kbp deletion in STX16.(2,3) However, analysis of microsatellite marker 261P9-CA1, which is located within intron 7 (GRCh38) of STX16, revealed that the 216 allele is shared not only by six affected members in family 208, but also by the two unaffected siblings of individual 208/III-3 (Figure 2, Supplemental Figure 2). This suggested that the genetic defect in this family is located outside of STX16. We therefore analyzed several additional markers for all available family members. Analysis of the unaffected individuals 208/III-2 and 208/III-4 revealed that the disease-causing variant in family 208 resides telomeric of marker 261P9-CA1. While marker 806M20-CA was not fully informative, four additional microsatellites (543J19-TTA, D20S171, D20S173, and D20S93) that are located telomeric of 261P9-CA1 are consistent with linkage to the region downstream of STX16. In fact, D20S171, D20S173, and D20S93 were fully informative, including the data for the unaffected individuals 208/III-2 and 208/III-4, for whom a recombination event had occurred between markers 261P9-CA1 and 806M20-CA. While these data suggest that the genetic defect leading to AD-PHP1B in kindred 208 resides telomeric of STX16, analysis of previous short-read WGS for individuals 208/III-1 and 208/II-5 did not identify a disease-causing variant in this region.(22)
T-LRS identifies a retrotransposon insertion in GNAS
To search for a genetic defect telomeric of STX16 that may have been missed or difficult to detect using short-read WGS, we performed T-LRS using Adaptive Sampling on the ONT platform with genomic DNA from affected individual 208/III-1 (Figure 2).(33) Targeting of an 8.4-Mbp region resulted in average depth of coverage of 29x from a single ONT R9.4.1 flow cell. Visual analysis of GNAS revealed a small spike in coverage approximately 1,200 bp telomeric of GNAS exon XL. Analysis of this region suggested the presence of an approximately 2,800-bp insertion in half of the reads (Figure 3A). No other SNVs or SVs identified in this region were predicted to be pathogenic. The insertion site was not flanked by repetitive or low-complexity sequence and, thus, might have been detectable in short-read WGS data. We confirmed that this insertion was present in short-read WGS data previously generated for this individual and was not present in gnomAD or from 13,675 whole-genome samples, suggesting that it is very rare. To determine the sequence of the 2,800-bp insertion, we performed de novo assembly of all reads with quality scores greater than 7 using Flye.(31) This resulted in a single contig with a length of 8.5 Mbp, strikingly similar to our target region size of 8.4 Mbp. Comparison of this contig to the reference genome revealed that the two are nearly identical without major structural differences (Supplemental Figure 1). The inserted sequence was isolated and BLAT(34) was used to identify this sequence as an SVA_E retrotransposon.
Multiplex PCR allowed amplification of the SVA_E insertion with a reverse primer that is located in the centromeric portion of the retrotransposon combined with a forward primer located in GNAS just upstream of the insertion (783 bp), as well as amplification of the wild-type allele (271 bp) due to a second reverse primer derived from the wild-type genomic sequence just downstream of the retrotransposon insertion. Amplification of genomic DNA from all six affected family members revealed two bands: the 783-bp amplicon derived from the mutant allele across the centromeric insertion site and the 271-bp amplicon derived from the wild-type allele (Figure 4). In contrast, the retrotransposon insertion was not found in genomic DNA from three unaffected family members: 208/III-2 and 208/III-4, who are the children of affected female 208/II-5, and individual 208/II-6, the unrelated husband of 208/II-2 (Figure 2). Individual 208/I-2, the mother of the four affected individuals in the second generation, is also a carrier of the insertion; however, she is unaffected and analysis of her genomic DNA by MS-MLPA revealed normal methylation pattern at GNAS exon A/B. Because her full sister has an affected daughter, this suggests that both sisters inherited the SVA_E insertion from their father although no DNA is available from him for confirmation (Figure 2).
Figure 4: Affected individuals carry the SVA_E insertion identified by T-LRS.
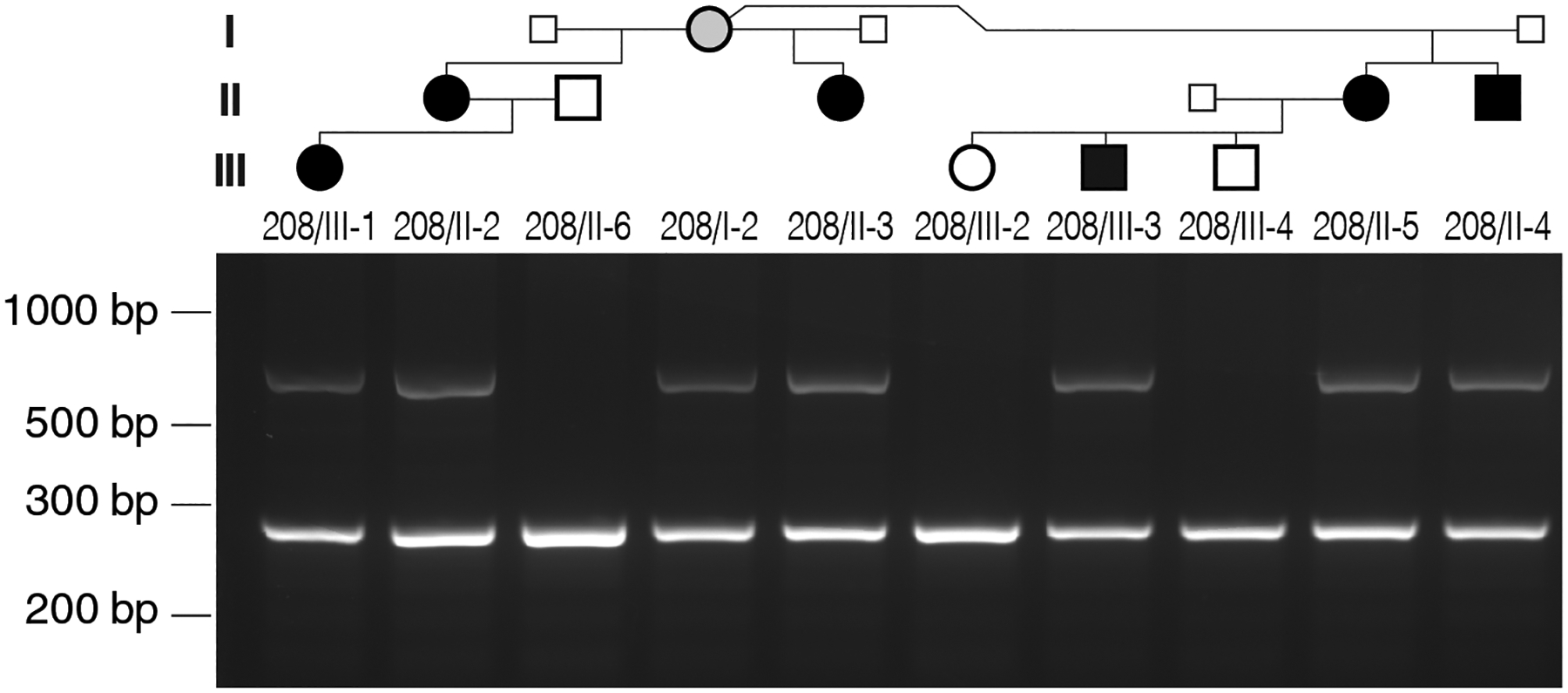
Several primers were designed using the assembled sequence of the SVA_E insertion for multiplex PCR (see Materials and Methods). The 783-bp amplicon is derived in the six affected family members from the maternal allele carrying the retrotransposon insertion, while the 271-bp amplicon is derived from the wild-type GNAS allele and is observed in both affected and unaffected individuals. Small squares indicate that genomic DNA was not available for these males; gray circle indicates unaffected carrier of the mutation.
Methylation analysis of the GNAS locus
Because SVs such as deletions and inversions at the GNAS locus have been shown to alter local methylation patterns, we sought to determine if the SVA_E insertion affected methylation at any of the CpG islands in this region. Megalodon was used to call 5mC methylation from the T-LRS data from which we generated a modified BAM file for visualization. Analysis of the GNAS region in individual 208/III-1 revealed LOM at the CpG island comprising the GNAS A/B exon and the adjacent sequences (Figure 3B); unrelated control cases had approximately 50% methylation at this region. No evidence for epigenetic changes was observed for individual 208/III-1 by T-LRS at the other three GNAS DMRs. These data are consistent with our MS-MLPA findings that demonstrated LOM restricted to the GNAS A/B locus not only for 208/III-1, but also for the other affected members in family 208(22), all of whom inherited the retrotransposon from a female carrier (Figure 2). Two independent approaches thus confirmed that the SVA_E insertion on the maternal GNAS allele results in loss of the maternal methylation imprint only at the exon A/B DMR.(22)
DISCUSSION
Deletions, duplications, and one inversion involving the STX16-GNAS region have previously been identified as causes of AD-PHP1B associated either with LOM at the exon A/B DMR alone or the loss of all three maternal methylation imprints.(3,6) Here, we report a novel cause of AD-PHP1B, namely the insertion of a retrotransposon approximately 1200 bp telomeric of GNAS exon XL, which is associated with variable LOM at the exon A/B DMR, but not at the other maternal DMRs. SVA retrotransposons are known to be active in the human genome, are frequently associated with disease(35,36), and have been shown to alter local methylation patterns.(37) A similar insertion at this position was not found in public genome databases, or after analysis of nearly 14,000 whole short-read genomes of unrelated individuals.
The unaffected female (208/I-2) in our AD-PHP1B family has three affected daughters and one affected son with three different partners, as well as two affected grandchildren through two of her daughters. All six affected individuals share the same haplotype with individual 208/I-2 that extends from microsatellite marker 806M20-CA to the telomere of chromosome 20q while the two unaffected grandchildren do not carry the disease-associated allele for this marker (Figure 2). Despite linkage to the region telomeric of STX16, we were previously unable to identify the underlying genetic cause by short-read WGS of two of the affected family members(22). T-LRS of individual 208/III-1 allowed us to identify and fully assemble an approximately 2800-bp SVA_E insertion. Using the single long-read dataset we were also able to determine the methylation status of each of the DMRs in the GNAS region and confirm the previous finding that LOM is restricted to the GNAS A/B DMR (Figure 3). We were unable to phase this region into maternal and paternal haplotypes because of a paucity of polymorphisms between the SVA_E insertion and the GNAS A/B DMR, as well as shearing required to increase coverage when using T-LRS. However, the retrotransposon was identified only in the affected family members, who had all inherited the insertion from a female and showed LOM at the GNAS exon A/B DMR. The unaffected female 208/I-2 is also a carrier of the retrotransposon but was found to have normal methylation at GNAS exon A/B and has a full unaffected sister who has an affected daughter (Figure 2). Together, this indicates that both individuals most likely inherited the disease-causing variant from their father. Based on our genetic, epigenetic, and laboratory findings, we conclude that the SVA_E insertion is most likely responsible for AD-PHP1B in this family.
It remains unknown how the SVA_E insertion downstream of exon XL alters, reduces, or abolishes methylation at the exon A/B DMR alone (Figure 3B). Transcription is thought to play an important role in the establishment of germline DMRs within the GNAS locus. Indeed, Chotalia and collages demonstrated that disruption of the transcript encoding the neuroendocrine secretory peptide Nesp in mice prevents the establishment of normal methylation at the DMRs within this region.(38) These authors demonstrated that pups inheriting from their mother a Gnas allele in which a polyadenylation signal had been placed downstream of Nesp (Nesptrun mice) made no spliced transcripts extending from exon Nesp to Gnas exon 2. This suggested that complete LOM at exon 1A (the murine equivalent of human exon A/B) and the variable epigenetic changes at exons XL and AS, which the authors had confirmed through methylation analysis(38), is due to the lack of an intact Nesp mRNA. Thus, transcripts derived from the Nesp promoter, which are readily detectable at the different stages of oocyte development appear to be necessary for re-establishing methylation on the maternal exon 1A DMR during oocyte development.(39) In contrast, methylation of the paternal Nesp DMR occurs post-fertilization in mice and humans.(40,41)
However, maternal inheritance of a Nesp allele with ablation of 26 nucleotides in Nesp exon 2 that includes the AUG codon for translation initiation eliminate the Nesp protein but these animals provided no evidence for methylation changes at the exon 1A DMR.(42) This raises the possibility that the Nesp protein itself is not required for re-establishing the methylation imprint at exon 1A after complete demethylation has occurred at the Gnas locus in oogonia.(39) Instead, a long Nesp preRNA or an as-yet-unknown alternatively spliced Nesp mRNA may be required during oogenesis for re-establishing methylation at the exon 1A DMR. Several AATAAA motifs are located in the affected members of family 208 at the telomeric end of the retrotransposon immediately before the poly-A stretch. It is therefore conceivable that NESP-derived mRNAs can be truncated similar to previous studies in mice with the engineered polyadenylation signal downstream of Nesp exon 2(38) and that this altered mRNA is unable to facilitate re-methylation at the A/B DMR during oogenesis.
Consistent with the findings in the Nesptrun mice, AD-PHP1B patients with deletions involving the entire exon NESP, but not any of the AS exons, show LOM at exon A/B and subsequently develop PTH-resistant hypocalcemia and hyperphosphatemia.(6,8) However, humans affected by PHP1B due to deletions involving exon NESP and adjacent intronic sequences show, besides PTH-resistance, no apparent abnormalities in growth and development; this is different from Nesptrun mice that die shortly after birth.(38) Similarly, no evidence for increased lethality has been noted for patients affected by AD-PHP1B due to deletions involving the maternal exons NESP and AS 2–4.(13,15) In contrast, mice with a maternal deletion extending from exon AS2 to AS4, which includes the two Nesp exons, die by 5 days of life.(43) Mice and humans differ significantly with regards to the importance of the NESP/AS 2–4 region for regulating epigenetic modifications.
Like the two overlapping deletions that are limited to GNAS exon NESP and adjacent intronic sequences(6,8), three different duplications involving regions that extend as far telomeric as exon A/B also lead to isolated LOM at the exon A/B DMR.(10–12) The mechanisms through which these large duplications prevent normal methylation at exon A/B are unknown. It is possible that, for each of these variants, changes in chromatin structure and/or NESP preRNA folding may prevent methyltransferases or other unknown adaptor proteins from properly attaching to the CpG island at exon A/B and establishing the necessary maternal methylation pattern. Likewise, NESP transcripts are likely unable to reach the exon A/B CpG island due to an inversion comprising approximately 1.8 Mbp of genomic DNA with the centromeric breakpoint residing between exons A/B and XL.(9) This large inversion thus places the DMR at exon A/B, in opposite orientation, far telomeric of its normal position, thereby preventing a putative NESP transcript from allowing the establishment of A/B methylation during oogenesis. LOM at exon A/B alone is also associated with deletions involving STX16—by far the most frequent causes of AD-PHP1B.(2,3,6) However, the mechanisms leading to these restricted epigenetic changes are even less well understood as these deletions are 200 kbp or more centromeric of exon A/B.
In summary, we show that T-LRS can be used as a single data source for comprehensively evaluating the complex GNAS region. This approach has significant cost and materials savings because a single sequencing method can be used to identify SNVs, indels, SVs, and differences of methylation within a specific region of interest. Our results suggest that this technique is a reasonable next best genetic approach after traditional genetic evaluation of this region failed to resolve PHP1B cases.
Supplementary Material
ACKNOWLDGEMENTS
We thank all members of family 208 for participating in this study. We thank A. Miller for help with figure preparation and editorial assistance and T. Brown for editorial assistance. This work was supported, in part, by the US National Institutes of Health (NIH) grant R01MH101221 (E.E.E.), a Trainee Award from the Brotman Baty Institute for Precision Medicine (D.E.M.), NIH grants DK046718 (H.J.) and PO1-DK11794 (subproject III) (H.J.) as well as support provided by the K20 Association (https://www.associationk20.com). Analysis of the sequence data was supported by NHGRI grants U01 HG011744 and UM1 HG006493 (University of Washington Center for Rare Disease Research). E.E.E. is an investigator of the Howard Hughes Medical Institute.
DISCLOSURES
D.E.M. has received travel support from Oxford Nanopore Technologies (ONT) to speak on their behalf. D.E.M. is a paid consultant for and holds stock options in MyOme. D.E.M. and E.E.E. are engaged in a research agreement with ONT. E.E.E. is a scientific advisory board (SAB) member of Variant Bio, Inc. The authors declare no other conflict of interests.
Footnotes
ETHICS STATEMENT
Written consent was obtained from all individuals or their guardians under IRB number 2001P000648.
DATA AVAILABILITY
Data that support the findings of this study are available upon request from the corresponding authors.
REFERENCES
- 1.Zhu Y, He Q, Aydin C, Rubera I, Tauc M, Chen M, Weinstein LS, Marshansky V, Jüppner H, Bastepe M. Ablation of the Stimulatory G Protein α-Subunit in Renal Proximal Tubules Leads to Parathyroid Hormone-Resistance With Increased Renal Cyp24a1 mRNA Abundance and Reduced Serum 1,25-Dihydroxyvitamin D. Endocrinology. 2016;157(2):497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mantovani G, Bastepe M, Monk D, Sanctis L de, Thiele S, Usardi A, Ahmed SF, Bufo R, Choplin T, Filippo GD, Devernois G, Eggermann T, Elli FM, Freson K, Ramirez AG, Germain-Lee EL, Groussin L, Hamdy N, Hanna P, Hiort O, Jüppner H, Kamenický P, Knight N, Kottler M-L, Norcy EL, Lecumberri B, Levine MA, Mäkitie O, Martin R, Martos-Moreno GÁ, Minagawa M, Murray P, Pereda A, Pignolo R, Rejnmark L, Rodado R, Rothenbuhler A, Saraff V, Shoemaker AH, Shore EM, Silve C, Turan S, Woods P, Zillikens MC, Nanclares GP de, Linglart A. Diagnosis and management of pseudohypoparathyroidism and related disorders: first international Consensus Statement. Nat Rev Endocrinol. 2018;14(8):476–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jüppner H Molecular Definition of Pseudohypoparathyroidism Variants. J Clin Endocrinol Metabolism. 2021;106(6):1541–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Linglart A, Gensure RC, Olney RC, Jüppner H, Bastepe M. A Novel STX16 Deletion in Autosomal Dominant Pseudohypoparathyroidism Type Ib Redefines the Boundaries of a cis-Acting Imprinting Control Element of GNAS. Am J Hum Genetics. 2005;76(5):804–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elli FM, Sanctis L de, Bollati V, Tarantini L, Filopanti M, Barbieri AM, Peverelli E, Beck-Peccoz P, Spada A, Mantovani G. Quantitative Analysis of Methylation Defects and Correlation With Clinical Characteristics in Patients With Pseudohypoparathyroidism Type I and GNAS Epigenetic Alterations. J Clin Endocrinol Metabolism. 2014;99(3):E508–17. [DOI] [PubMed] [Google Scholar]
- 6.Danzig J, Li D, Beur SJ de, Levine MA. High-throughput Molecular Analysis of Pseudohypoparathyroidism 1b Patients Reveals Novel Genetic and Epigenetic Defects. J Clin Endocrinol Metabolism. 2021;106(11):e4603–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao P, Liu J, Cheng J, Li Q, Fu S, Wang Y, Yang X, Ma X. Analysis of defects in GNAS and STX16 genes in a Chinese family with pseudohypoparathyroidism. Minerva Endocrinol. 2020; [DOI] [PubMed] [Google Scholar]
- 8.Richard N, Abeguilé G, Coudray N, Mittre H, Gruchy N, Andrieux J, Cathebras P, Kottler M-L. A New Deletion Ablating NESP55 Causes Loss of Maternal Imprint of A/B GNAS and Autosomal Dominant Pseudohypoparathyroidism Type Ib. J Clin Endocrinol Metabolism. 2012;97(5):E863–7. [DOI] [PubMed] [Google Scholar]
- 9.Grigelioniene G, Nevalainen PI, Reyes M, Thiele S, Tafaj O, Molinaro A, Takatani R, Ala‐Houhala M, Nilsson D, Eisfeldt J, Lindstrand A, Kottler M, Mäkitie O, Jüppner H. A Large Inversion Involving GNAS Exon A/B and All Exons Encoding Gsα Is Associated With Autosomal Dominant Pseudohypoparathyroidism Type Ib (PHP1B). J Bone Miner Res. 2017;32(4):776–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakamura A, Hamaguchi E, Horikawa R, Nishimura Y, Matsubara K, Sano S, Nagasaki K, Matsubara Y, Umezawa A, Tajima T, Ogata T, Kagami M, Okamura K, Fukami M. Complex Genomic Rearrangement Within the GNAS Region Associated With Familial Pseudohypoparathyroidism Type 1b. J Clin Endocrinol Metabolism. 2016;101(7):2623–7. [DOI] [PubMed] [Google Scholar]
- 11.Reyes M, Kagami M, Kawashima S, Pallotta J, Schnabel D, Fukami M, Jüppner H. A Novel GNAS Duplication Associated With Loss‐of‐Methylation Restricted to Exon A/B Causes Pseudohypoparathyroidism Type Ib (PHP1B). J Bone Miner Res. 2021;36(3):546–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perez-Nanclares G, Velayos T, Vela A, Muñoz-Torres M, Castaño L. Pseudohypoparathyroidism Type Ib Associated with Novel Duplications in the GNAS Locus. Plos One. 2015;10(2):e0117691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bastepe M, Jüppner H. GNAS Locus and Pseudohypoparathyroidism. Horm Res Paediat. 2005;63(2):65–74. [DOI] [PubMed] [Google Scholar]
- 14.Chillambhi S, Turan S, Hwang D-Y, Chen H-C, Juüppner H, Bastepe M. Deletion of the Noncoding GNAS Antisense Transcript Causes Pseudohypoparathyroidism Type Ib and Biparental Defects of GNAS Methylation in cis. J Clin Endocrinol Metabolism. 2010;95(8):3993–4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takatani R, Molinaro A, Grigelioniene G, Tafaj O, Watanabe T, Reyes M, Sharma A, Singhal V, Raymond FL, Linglart A, Jüppner H. Analysis of Multiple Families With Single Individuals Affected by Pseudohypoparathyroidism Type Ib (PHP1B) Reveals Only One Novel Maternally Inherited GNAS Deletion. J Bone Miner Res. 2016;31(4):796–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rochtus A, Martin-Trujillo A, Izzi B, Elli F, Garin I, Linglart A, Mantovani G, Nanclares GP de, Thiele S, Decallonne B, Geet CV, Monk D, Freson K. Genome-wide DNA methylation analysis of pseudohypoparathyroidism patients with GNAS imprinting defects. Clin Epigenetics. 2016;8(1):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bastepe M, Fröhlich LF, Hendy GN, Indridason OS, Josse RG, Koshiyama H, Körkkö J, Nakamoto JM, Rosenbloom AL, Slyper AH, Sugimoto T, Tsatsoulis A, Crawford JD, Jüppner H. Autosomal dominant pseudohypoparathyroidism type Ib is associated with a heterozygous microdeletion that likely disrupts a putative imprinting control element of GNAS. J Clin Invest. 2003;112(8):1255–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maupetit‐Méhouas S, Azzi S, Steunou V, Sakakini N, Silve C, Reynes C, Nanclares GP de, Keren B, Chantot S, Barlier A, Linglart A, Netchine I. Simultaneous Hyper‐ and Hypomethylation at Imprinted Loci in a Subset of Patients with GNAS Epimutations Underlies a Complex and Different Mechanism of Multilocus Methylation Defect in Pseudohypoparathyroidism Type 1b. Hum Mutat. 2013;34(8):1172–80. [DOI] [PubMed] [Google Scholar]
- 19.Bastepe M, Fröhlich LF, Linglart A, Abu-Zahra HS, Tojo K, Ward LM, Jüppner H. Deletion of the NESP55 differentially methylated region causes loss of maternal GNAS imprints and pseudohypoparathyroidism type Ib. Nat Genet. 2005;37(1):25–7. [DOI] [PubMed] [Google Scholar]
- 20.Williamson CM, Ball ST, Nottingham WT, Skinner JA, Plagge A, Turner MD, Powles N, Hough T, Papworth D, Fraser WD, Maconochie M, Peters J. A cis-acting control region is required exclusively for the tissue-specific imprinting of Gnas. Nat Genet. 2004;36(8):894–9. [DOI] [PubMed] [Google Scholar]
- 21.Puzhko S, Goodyer CG, Kerachian MA, Canaff L, Misra M, Jüppner H, Bastepe M, Hendy GN. Parathyroid hormone signaling via Gαs is selectively inhibited by an NH2‐terminally truncated Gαs: Implications for pseudohypoparathyroidism. J Bone Miner Res. 2011;26(10):2473–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hanna P, Francou B, Delemer B, Jüppner H, Linglart A. A novel familial PHP1B variant with incomplete loss-of-methylation at GNAS-A/B and enhanced methylation at GNAS-AS2. J Clin Endocrinol Metabolism. 2021;106(9):dgab136-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Milioto A, Reyes M, Hanna P, Kiuchi Z, Turan S, Zeve D, Agarwal C, Grigelioniene G, Chen A, Mericq V, Frangos M, Ten S, Mantovani G, Salusky IB, Tebben P, Jüppner H. Lack of GNAS Remethylation During Oogenesis May Be a Cause of Sporadic Pseudohypoparathyroidism Type Ib. J Clin Endocrinol Metabolism. 2021;107(4):e1610–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018. May 10;3(18):321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng Z, Li S, Su J, Leung AW-S, Lam T-W, Luo R. Symphonizing pileup and full-alignment for deep learning-based long-read variant calling. Biorxiv. 2021;2021.12.29.474431. [DOI] [PubMed] [Google Scholar]
- 26.McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, Flicek P, Cunningham F. The Ensembl Variant Effect Predictor. Genome Biol. 2016;17(1):122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heller D, Vingron M. SVIM: Structural Variant Identification using Mapped Long Reads. Bioinformatics. 2019;35(17):btz041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sedlazeck FJ, Rescheneder P, Smolka M, Fang H, Nattestad M, Haeseler A von, Schatz MC. Accurate detection of complex structural variations using single-molecule sequencing. Nat Methods. 2018;15(6):461–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang T, Liu Y, Jiang Y, Li J, Gao Y, Cui Z, Liu Y, Liu B, Wang Y. Long-read-based human genomic structural variation detection with cuteSV [Internet]. 2020. [cited 2020 Aug 4]. Available from: https://genomebiology.biomedcentral.com/track/pdf/10.1186/s13059-020-02107-y [DOI] [PMC free article] [PubMed]
- 30.Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013. Mar 20;14(2):178–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kolmogorov M, Yuan J, Lin Y, Pevzner PA. Assembly of long, error-prone reads using repeat graphs. Nat Biotechnol. 2019;37(5):540–6. [DOI] [PubMed] [Google Scholar]
- 32.Wilfert AB, Turner TN, Murali SC, Hsieh P, Sulovari A, Wang T, Coe BP, Guo H, Hoekzema K, Bakken TE, Winterkorn LH, Evani US, Byrska-Bishop M, Earl RK, Bernier RA, Consortium TS, Zhou X, Feliciano P, Hall J, Astrovskaya I, Xu S, Shu C, Obiajulu J, Brueggeman L, Wright J, Marchenko O, Fleisch C, Chang TS, Snyder LG, Barns SD, Han B, Harvey W, Nishida A, Doan R, Soucy A, O’Roak BJ, Yu TW, Geschwind D, Michaelson J, Volfovsky N, Shen Y, Chung WK, Zody MC, Eichler EE. Recent ultra-rare inherited variants implicate new autism candidate risk genes. Nat Genet. 2021;53(8):1125–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miller DE, Sulovari A, Wang T, Loucks H, Hoekzema K, Munson KM, Lewis AP, Fuerte EPA, Paschal CR, Walsh T, Thies J, Bennett JT, Glass I, Dipple KM, Patterson K, Bonkowski ES, Nelson Z, Squire A, Sikes M, Beckman E, Bennett RL, Earl D, Lee W, Allikmets R, Perlman SJ, Chow P, Hing AV, Wenger TL, Adam MP, Sun A, Lam C, Chang I, Zou X, Austin SL, Huggins E, Safi A, Iyengar AK, Reddy TE, Majoros WH, Allen AS, Crawford GE, Kishnani PS, Genomics U of WC for M, King M-C, Cherry T, Chong JX, Bamshad MJ, Nickerson DA, Mefford HC, Doherty D, Eichler EE. Targeted long-read sequencing identifies missing disease-causing variation. Am J Hum Genetics. 2021; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kent WJ. BLAT—The BLAST-Like Alignment Tool. Genome Res. 2002;12(4):656–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ostertag EM, Goodier JL, Zhang Y, Kazazian HH. SVA Elements Are Nonautonomous Retrotransposons that Cause Disease in Humans. Am J Hum Genetics. 2003;73(6):1444–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang H, Xing J, Grover D, Hedges DJ, Han K, Walker JA, Batzer MA. SVA Elements: A Hominid-specific Retroposon Family. J Mol Biol. 2005;354(4):994–1007. [DOI] [PubMed] [Google Scholar]
- 37.Hall A, Moore AK, Hernandez DG, Billingsley KJ, Bubb VJ, Quinn JP, Consortium) N (North ABE. A SINE-VNTR-Alu in the LRIG2 Promoter Is Associated with Gene Expression at the Locus. Int J Mol Sci. 2020;21(22):8486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chotalia M, Smallwood SA, Ruf N, Dawson C, Lucifero D, Frontera M, James K, Dean W, Kelsey G. Transcription is required for establishment of germline methylation marks at imprinted genes. Gene Dev. 2009;23(1):105–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gahurova L, Tomizawa S, Smallwood SA, Stewart-Morgan KR, Saadeh H, Kim J, Andrews SR, Chen T, Kelsey G. Transcription and chromatin determinants of de novo DNA methylation timing in oocytes. Epigenet Chromatin. 2017;10(1):25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Okae H, Chiba H, Hiura H, Hamada H, Sato A, Utsunomiya T, Kikuchi H, Yoshida H, Tanaka A, Suyama M, Arima T. Genome-Wide Analysis of DNA Methylation Dynamics during Early Human Development. Plos Genet. 2014;10(12):e1004868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu J, Yu S, Litman D, Chen W, Weinstein LS. Identification of a Methylation Imprint Mark within the Mouse Gnas Locus. Mol Cell Biol. 2000;20(16):5808–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Plagge A, Isles AR, Gordon E, Humby T, Dean W, Gritsch S, Fischer-Colbrie R, Wilkinson LS, Kelsey G. Imprinted Nesp55 Influences Behavioral Reactivity to Novel Environments. Mol Cell Biol. 2005;25(8):3019–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fröhlich LF, Mrakovcic M, Steinborn R, Chung U-I, Bastepe M, Jüppner H. Targeted deletion of the Nesp55 DMR defines another Gnas imprinting control region and provides a mouse model of autosomal dominant PHP-Ib. Proc National Acad Sci. 2010;107(20):9275–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data that support the findings of this study are available upon request from the corresponding authors.