

Abstract
Background:
Loss-of-function mutations in GRN are a cause of familial frontotemporal dementia, and common variants within the gene have been associated with an increased risk of developing Alzheimer’s disease and Parkinson’s disease. While TDP-43-positive inclusions are characteristic of GRN-related neurodegeneration, Lewy body co-pathology has also been observed in many GRN mutation carriers.
Methods:
We analyzed whole-genome sequence data generated for 2,591 European-ancestry Lewy body dementia (LBD) cases and 4,032 neurologically healthy controls to identify disease-causing mutations in GRN.
Results:
We identified six heterozygous exonic GRN mutations in seven study participants (cases: n=6; controls: n=1). Each variant was predicted to be pathogenic or likely pathogenic. We found significant enrichment of GRN loss-of-function mutations in LBD patients compared to controls (SKAT-O p-value = 0.0162). Immunohistochemistry in three definite LBD cases demonstrated Lewy body pathology and TDP-43-positive neuronal inclusions.
Conclusions:
Our findings suggest that deleterious GRN mutations are a rare cause of familial LBD.
Keywords: Lewy body dementia (LBD), frontotemporal lobar degeneration (FTLD), GRN mutations, Progranulin, Neurodegeneration
Graphical Abstract
Progranulin is a broadly expressed and secreted growth factor that plays a role in several biological processes, including neural circuit development, regulation of cell growth, wound healing, lysosomal homeostasis, and neuroinflammation.1–3 Progranulin is encoded by the GRN gene located on chromosome 17q21.31. Heterozygous mutations in this gene are the second most common cause of frontotemporal lobar degeneration (FTLD), accounting for 10% of all FTLD cases.4, 5 These loss-of-function GRN mutations result in haploinsufficiency of progranulin with subsequent lysosomal dysfunction and neurodegeneration.
Recently, genome-wide association studies identified common variants within the GRN locus as being associated with other age-related neurodegenerative diseases, such as Alzheimer’s disease and Parkinson’s disease.6–8 Moreover, neuropathological studies in patients carrying pathogenic GRN variants found frequent Lewy body co-pathology in addition to the expected TDP-43-positive inclusions, suggesting a potential molecular link between Lewy body diseases and frontotemporal dementia.9, 10 Interestingly, rare incidental GRN mutations have already been reported in Lewy body dementia (LBD) cases, but the relevance of these observations remains unclear.11, 12 Based on this prior evidence, we postulated that GRN mutations might give rise to LBD. The objective of our study was to assess an LBD case-control cohort for pathogenic variants in GRN and test whether there is an enrichment of damaging mutations among LBD patients.
METHODS
Subjects
We examined 2,591 LBD cases and 4,032 neurologically healthy controls in which we recently performed whole-genome sequencing.13 All study participants were of European-ancestry. Patients were diagnosed with pathologically definite (n = 1,789, 69.0%) or clinically probable LBD (n = 802, 31.0%), according to consensus criteria.14, 15 One hundred and twenty-five (4.8%) of LBD cases had the clinical subtype of Parkinson’s disease dementia, and the remainder had the clinical subtype of dementia with Lewy bodies. As expected for this form of dementia, nearly two thirds of LBD cases (63.4%) were males. Control subjects had no history of cognitive decline or neurological deficits on neurological examination. No evidence of significant neurodegenerative disease was detected on histopathological examination, performed on 15% (n = 605) of controls. Further details about the study demographics have been described elsewhere.13
Genome sequencing
All cases and controls underwent whole-genome sequencing using PCR-free library preparations followed by 150 base pair, paired-end sequencing on an Illumina HiSeq X Ten platform. The average coverage per genome was 35x. Sequencing, alignment (reference genome build GRCh38), variant calling, and quality control procedures (sample-level and variant-level) followed a standardized workflow described elsewhere.13
GRN variant annotation, filtering, and statistical analysis
To identify pathogenic variants in GRN (NM_002087), we extracted the GRN sequence from the genome data and annotated variants within this gene using Annovar (version 2018-04-16).16 We next filtered variants by allele frequency, applying a minor allele frequency threshold of <0.01. We examined frameshift mutations, stop-gain mutations, and previously described pathogenic missense variants in the resulting data and reviewed the clinicopathological information of mutation carriers. To test whether loss-of-function mutations in GRN are enriched in our LBD cases compared to controls, we performed the Optimized Sequence Kernel Association Test (SKAT-O) with a gene-wide significance threshold of 0.05.17 This analysis was performed in RVTESTS (vs.2.1.0) using sex, age, and five principal components as covariates.18
Data availability
Individual-level genome sequence data for the LBD and resource control genomes are available at dbGaP (https://www.ncbi.nlm.nih.nih.gov/gap/; accession no. phs001963.v1.p1 NIA DementiaSeq) and AMP-PD (https://amp-PD.org).
RESULTS
We examined coding variants in the GRN gene in 2,591 LBD cases and 4,032 neurologically healthy controls in which we recently performed whole-genome sequencing.13 This identified six unique, heterozygous, exonic GRN mutations in a total of seven study participants (six LBD cases and one neurologically healthy control, Figure 1A). These mutations included three frameshift mutations (p.Q130Sfs*124, p.T382Sfs*29, p.E498Dfs*11), two stop-gain mutations (p.R493X, p.R535X [identified in a control]), and one missense mutation (p.A9V) located in a highly conserved region of GRN (Figure 1B, Table S1). All five frameshift and stop-gain mutations were predicted to be pathogenic and have been previously reported to be disease-causing mutations in patients with frontotemporal dementia or Alzheimer’s disease.4, 5, 9, 19 The p.A9V missense variant (observed in an LBD patient) was classified as ‘likely pathogenic’ according to the ACMG consensus classification. None of identified GRN mutation carriers had a pathogenic variant in the genes GBA, SNCA, APP, PSEN1, PSEN2 that may otherwise explain the disease in these subjects. Additionally, none of the GRN mutation carriers had an APOE ε4 risk allele (Table S1). Gene burden analysis confirmed a significant enrichment of loss-of-function mutations in GRN in LBD cases compared to healthy controls (SKAT-O; p-value = 0.0162). As all pathogenic variant carriers were diagnosed with dementia with Lewy bodies, we performed a subanalysis in dementia with Lewy body cases (excluding 125 patients diagnosed with Parkinson’s disease dementia) compared to all controls, which again was found a significant association (SKAT-O; p-value = 0.0147).
Figure 1. Schematic illustration of GRN mutations.
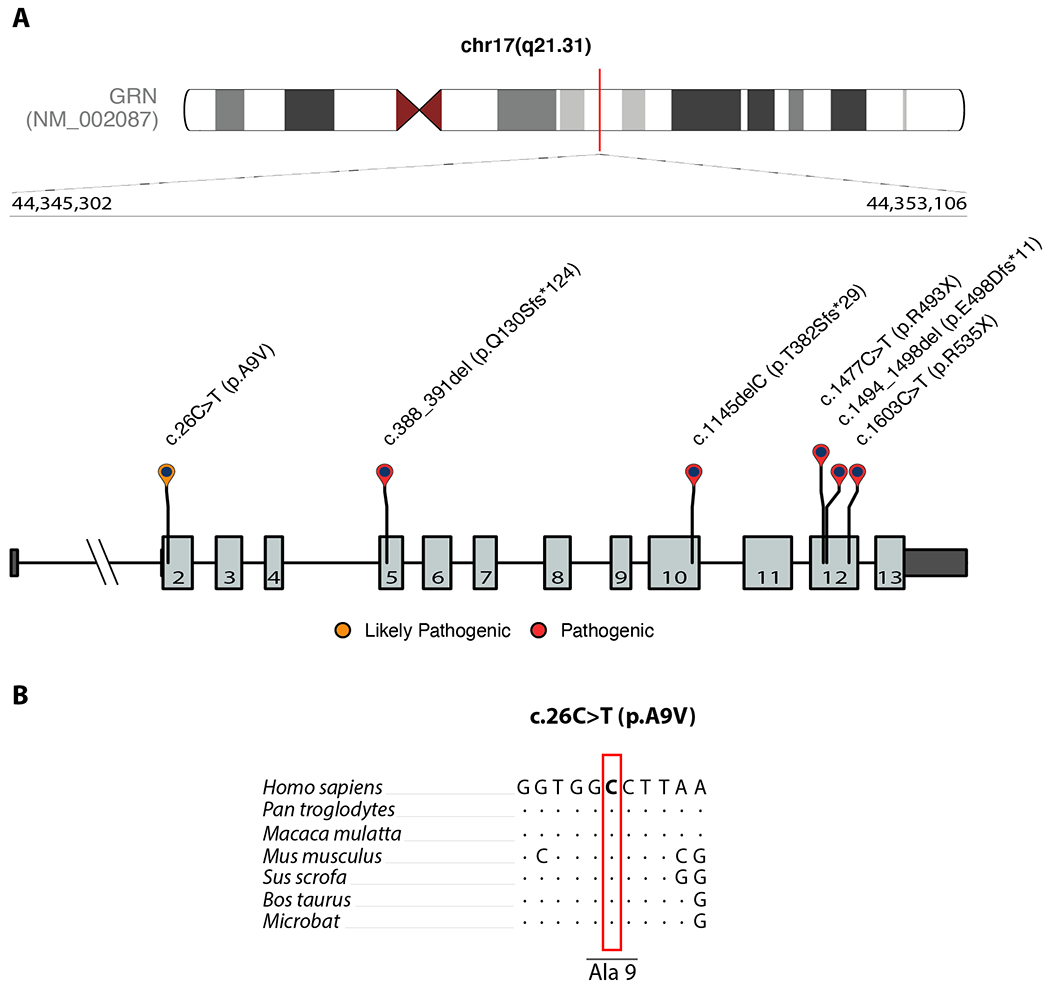
Schematic representation of GRN mutations identified in this study. Untranslated regions are illustrated by dark gray boxes, and exons are shown as light gray boxes. The bottom panel shows the nucleotide conservation for the novel coding mutation (c.26C>T = p.A9V) in seven mammals (B). A red box highlights the location of the mutated nucleotide. Dots indicate nucleotides that are identical to the human reference genome sequence.
The clinicopathological characteristics of all seven GRN mutation carriers are summarized in Table S1. The average age at death of LBD cases was 76.8 years (range: 63–83 years), while a neurologically healthy control, carrying the pathogenic p.R535X stop-gain variant, was still alive and cognitively well at age 74. Neuropathological confirmation was available for all LBD cases, all of whom fulfilled pathological consensus criteria for limbic (n = 3) or neocortical (n = 3) dementia with Lewy bodies.15 Family history information was available for five out of seven mutation carriers, of which two reported a strong family history of dementia. An in-depth review of the histopathology associated with three LBD cases carrying pathogenic frameshift variants revealed widespread α-synuclein-positive Lewy bodies and Lewy neuritis as well as TDP-43-positive neuronal cytoplasmic inclusions and short dystrophic neurites, consistent with FTLD with TDP-43 type A.20 Figure 2 showcases the immunohistochemical findings of a representative GRN-related LBD patient. Notably, this particular case (subject 2 in Table S1, carrying the pathogenic p.Q130Sfs*124 variant) reported a positive clinical history of dementia in several family members (a detailed description of the patient’s clinical history and disease course is documented in the Supplementary Note).
Figure 2. Representative images of α-synuclein and phosphorylated-TDP-43 immunohistochemistry.
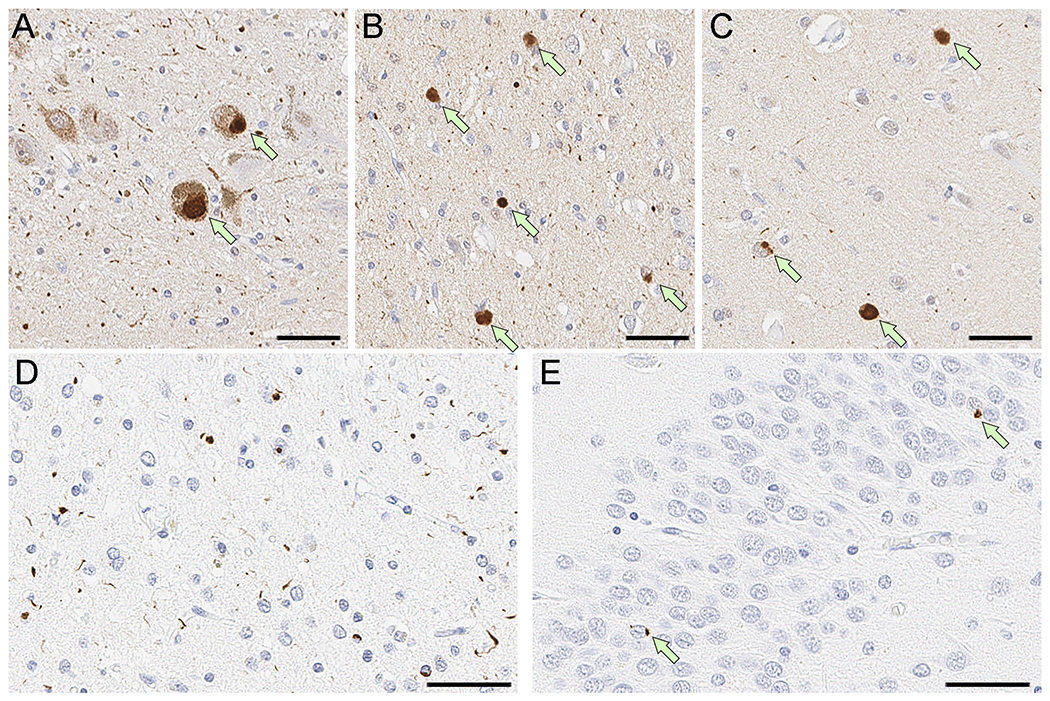
Shown are the immunohistochemical staining results in a GRN mutation carrier (p.Q130Sfs*124) illustrating α-synuclein-positive and TDP-43-positive co-pathologies. Immunohistochemistry for α-synuclein shows Lewy bodies (arrows) in the substantia nigra (A), cingulate gyrus (B), and superior temporal gyrus (C), consistent with diffuse Lewy body disease. Immunohistochemistry for phosphorylated-TDP-43 shows numerous neuronal cytoplasmic inclusions and short dystrophic neurites predominantly in layer II of the middle frontal gyrus (D), consistent with type A pathology. Dentate gyrus has a few compact neuronal cytoplasmic inclusions (arrows in E). Scale bars = 50μm.
DISCUSSION
Mutations in GRN are the second most common cause of autosomal dominant FTLD with TDP-43-positive inclusions. Recent evidence has implicated genetic variation at the GRN locus in the pathogenesis of a broader spectrum of neurodegenerative diseases, including Alzheimer’s disease and Parkinson’s disease, suggesting pleiotropic effects for this locus.6–8 Here, we provide genetic and histopathological evidence linking GRN mutations and LBD based on a large whole-genome sequence case-control cohort.
First, we identified six LBD cases carrying heterozygous exonic GRN mutations, five of which led to a truncated protein and have been already reported as disease-causing.4, 5, 9, 19 Second, we identified significant gene-wide enrichment of GRN loss-of-function mutations in LBD patients, supporting the notion that haploinsufficiency due to damaging mutations can also be a rare cause of familial Lewy body dementia. Third, immunohistochemistry of loss-of-function mutation carriers confirmed the presence of widespread Lewy body disease in addition to TDP-43 pathology in three out of six LBD cases, suggesting a molecular link between synuclein and TDP-43 proteinopathic changes and emphasizing the complexity of neurodegenerative disease related to GRN mutations. Taken together, our observations support the hypothesis that GRN mutations could be a rare cause of LBD.
Importantly, patient 2, who was found to carry the p.Q130Sfs*124 mutation, had a notable family history of dementia, which was reported as clinically diagnosed Alzheimer’s disease affecting his brother, father, and paternal grandfather (Supplementary Note). Although we could not test the other family members, we can assume that the frameshift variant is the disease-causing mutation in this particular family, underlining the role of deleterious GRN mutations in familiar LBD.
Our evaluation further identified a missense variant (c.26C>T = p.A9V), located at a highly conserved residue within the signal sequence of the protein (Figure 1) and predicted to be ‘likely pathogenic’. Another missense variant at the same amino acid residue (p.A9D = rs63751243) has been previously described as pathogenic and limited evidence suggests that the p.A9V mutation results in cytoplasmic missorting. It is difficult to interpret the clinical relevance of this mutation, as this patient is also homozygous for the protective TMEM106B allele.21–24 Further evidence is needed to draw conclusions about the pathogenicity of this coding variant in LBD.
It is also noteworthy that a stop-gain mutation (p.R535X) was identified in a 74-year neurologically healthy subject who is followed longitudinally (Table S1). Reduced penetrance of pathogenic variants is a well-described phenomenon in late-onset neurodegenerative diseases.25 Although predicted as pathogenic and previously reported as disease-causing,9 the variant leads to a truncated protein where 90% of the wild-type sequence is intact, and non-sensitivity to nonsense-mediated decay may explain why the damaging mutation did not cause disease in this particular case. We cannot exclude that the control subject is actually a pre-symptomatic patient who will phenoconvert in the future.
Our study expands our understanding of the genetic architecture underlying LBD. It also adds to the increasing literature of pleiotropic genes as important drivers for multi-proteinopathic changes in adult neurodegenerative diseases with heterogeneous clinical presentations.13, 26–29 Although GRN mutations are overall rare (0.23% of LBD patients), genetic testing is increasingly available in specialty clinics and allows clinicians to rapidly screen for damaging mutations, particularly in individuals with a family history of dementia. As targeted therapies for GRN-related neurodegeneration are already studied in clinical trials (Clinicaltrials.gov: NCT04408625, NCT04747431, NCT04374136, NCT03987295; accessed on 01/21/22), identifying these individuals may open new avenues for research and treatment in LBD, an otherwise fatal form of dementia.
CONCLUSION
In summary, our findings implicate loss-of-function mutations in GRN in the pathogenesis of LBD. Molecular screening for damaging GRN mutations should be considered in LBD patients to establish a molecular diagnosis, especially in patients with a family history of dementia. Although rare, this information may be clinically relevant, as targeted GRN treatment trials are already ongoing.
Supplementary Material
ACKNOWLEDGMENTS
Writing of the first draft: PR. Bioinformatic & statistical analysis: PR, ZS, RC. Data collection: SWS, CLD, The American Genome Center. Clinicopathological characterization: SK, RR, BFB, TGB, DWD, OAR. Conceptualization and design: SWS. Supervision: SWS. Manuscript review: all authors.
Funding sources:
This research was supported in part by the Intramural Research Program of the National Institutes of Health (National Institute on Aging, National Institute of Neurological Disorders and Stroke; project numbers: 1ZIAAG000935, 1ZIANS003154).
Financial Disclosure/Conflicts of Interest:
We are grateful to the Banner Sun Health Research Institute Brain and Body Donation Program of Sun City, Arizona, for the provision of human brain tissue and data. The Brain and Body Donation Program is supported by the National Institute of Neurological Disorders and Stroke (U24 NS072026 National Brain and Tissue Resource for Parkinson’s Disease and Related Disorders), the National Institute on Aging (P30 AG19610 Arizona Alzheimer’s Disease Core Center), the Arizona Department of Health Services (contract 211002, Arizona Alzheimer’s Research Center), the Arizona Biomedical Research Commission (contracts 4001, 0011, 05-901 and 1001 to the Arizona Parkinson’s Disease Consortium) and the Michael J. Fox Foundation for Parkinson’s Research. The Mayo Clinic Brain Bank is supported by U54 NS110435 (PI Dennis W. Dickson, MD) and The Mangurian Foundation Lewy Body Dementia Program. Sample collection and characterization is also supported by the Mayo Clinic Functional Genomics of LBD Program, The Little Family Foundation, and the Mayo Clinic Lewy Body Dementia Association (LBDA) Research Center of Excellence. Data used in the preparation of this article were obtained from the Accelerating Medicine Partnership (AMP) Parkinson’s Disease (AMP PD) Knowledge Platform. For up-to-date information on the study, visit https://www.amp-pd.org. A complete list of acknowledgments for individual studies/contributors to the AMP PD platform is given in the supplementary materials. S.W.S. serves on the Scientific Advisory Council of the Lewy Body Dementia Association. We thank the Mayo Clinic Brain Bank for contributing DNA samples and data from patients with Lewy body dementia, which is supported by U54 NS110435 (PI Dennis W. Dickson, MD), U01 NS100620 (PI Kejal Kantarci, MD), P30 AG62677 (PI Ronald Petersen, PhD, MD) and The Mangurian Foundation Lewy Body Dementia Program. Sample collection and characterization is also supported by the Mayo Clinic Functional Genomics of LBD Program, NINDS R01 NS78086 (PI Owen Ross, PhD), Mayo Clinic Center for Individualized Medicine, and The Little Family Foundation and the Mayo Clinic Lewy Body Dementia Association (LBDA) Research Center of Excellence.
REFERENCES
- 1.He Z, Ong CH, Halper J, Bateman A. Progranulin is a mediator of the wound response. Nat Med 2003;9(2):225–229. [DOI] [PubMed] [Google Scholar]
- 2.Kao AW, McKay A, Singh PP, Brunet A, Huang EJ. Progranulin, lysosomal regulation and neurodegenerative disease. Nat Rev Neurosci 2017;18(6):325–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lui H, Zhang J, Makinson SR, et al. Progranulin Deficiency Promotes Circuit-Specific Synaptic Pruning by Microglia via Complement Activation. Cell 2016;165(4):921–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baker M, Mackenzie IR, Pickering-Brown SM, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 2006;442(7105):916–919. [DOI] [PubMed] [Google Scholar]
- 5.Gass J, Cannon A, Mackenzie IR, et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet 2006;15(20):2988–3001. [DOI] [PubMed] [Google Scholar]
- 6.Nalls MA, Blauwendraat C, Sargent L, et al. Evidence for GRN connecting multiple neurodegenerative diseases. Brain Commun 2021;3(2):fcab095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nalls MA, Blauwendraat C, Vallerga CL, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol 2019;18(12):1091–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wightman DP, Jansen IE, Savage JE, et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat Genet 2021;53(9):1276–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brouwers N, Nuytemans K, van der Zee J, et al. Alzheimer and Parkinson diagnoses in progranulin null mutation carriers in an extended founder family. Arch Neurol 2007;64(10):1436–1446. [DOI] [PubMed] [Google Scholar]
- 10.Kelley BJ, Haidar W, Boeve BF, et al. Prominent phenotypic variability associated with mutations in Progranulin. Neurobiol Aging 2009;30(5):739–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keogh MJ, Wei W, Wilson I, et al. Genetic compendium of 1511 human brains available through the UK Medical Research Council Brain Banks Network Resource. Genome Res 2017;27(1):165–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Orme T, Hernandez D, Ross OA, et al. Analysis of neurodegenerative disease-causing genes in dementia with Lewy bodies. Acta Neuropathol Commun 2020;8(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chia R, Sabir MS, Bandres-Ciga S, et al. Genome sequencing analysis identifies new loci associated with Lewy body dementia and provides insights into its genetic architecture. Nat Genet 2021;53(3):294–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Emre M, Aarsland D, Brown R, et al. Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Mov Disord 2007;22(12):1689–1707; quiz 1837. [DOI] [PubMed] [Google Scholar]
- 15.McKeith IG, Boeve BF, Dickson DW, et al. Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology 2017;89(1):88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010;38(16):e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee S, Emond MJ, Bamshad MJ, et al. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am J Hum Genet 2012;91(2):224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhan X, Hu Y, Li B, Abecasis GR, Liu DJ. RVTESTS: an efficient and comprehensive tool for rare variant association analysis using sequence data. Bioinformatics 2016;32(9):1423–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Le Ber I, Camuzat A, Hannequin D, et al. Phenotype variability in progranulin mutation carriers: a clinical, neuropsychological, imaging and genetic study. Brain 2008;131(Pt 3):732–746. [DOI] [PubMed] [Google Scholar]
- 20.Mackenzie IR, Neumann M, Baborie A, et al. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol 2011;122(1):111–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cannon A, Fujioka S, Rutherford NJ, et al. Clinicopathologic variability of the GRN A9D mutation, including amyotrophic lateral sclerosis. Neurology 2013;80(19):1771–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mukherjee O, Pastor P, Cairns NJ, et al. HDDD2 is a familial frontotemporal lobar degeneration with ubiquitin-positive, tau-negative inclusions caused by a missense mutation in the signal peptide of progranulin. Ann Neurol 2006;60(3):314–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shankaran SS, Capell A, Hruscha AT, et al. Missense mutations in the progranulin gene linked to frontotemporal lobar degeneration with ubiquitin-immunoreactive inclusions reduce progranulin production and secretion. J Biol Chem 2008;283(3):1744–1753. [DOI] [PubMed] [Google Scholar]
- 24.Wider C, Uitti RJ, Wszolek ZK, et al. Progranulin gene mutation with an unusual clinical and neuropathologic presentation. Mov Disord 2008;23(8):1168–1173. [DOI] [PubMed] [Google Scholar]
- 25.Murphy NA, Arthur KC, Tienari PJ, Houlden H, Chio A, Traynor BJ. Age-related penetrance of the C9orf72 repeat expansion. Sci Rep 2017;7(1):2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dewan R, Chia R, Ding J, et al. Pathogenic Huntingtin Repeat Expansions in Patients with Frontotemporal Dementia and Amyotrophic Lateral Sclerosis. Neuron 2021;109(3):448–460 e444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ishikawa A, Piao YS, Miyashita A, et al. A mutant PSEN1 causes dementia with Lewy bodies and variant Alzheimer’s disease. Ann Neurol 2005;57(3):429–434. [DOI] [PubMed] [Google Scholar]
- 28.Johnson JO, Mandrioli J, Benatar M, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 2010;68(5):857–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zimprich A, Biskup S, Leitner P, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004;44(4):601–607. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Individual-level genome sequence data for the LBD and resource control genomes are available at dbGaP (https://www.ncbi.nlm.nih.nih.gov/gap/; accession no. phs001963.v1.p1 NIA DementiaSeq) and AMP-PD (https://amp-PD.org).