

Abstract
Therapy of molybdenum co-factor (Moco) deficiency has received FDA approval in 2021. While urothione, the urinary excreted catabolite of Moco, is used as diagnostic biomarker for Moco-deficiency, its catabolic pathway remains unknown. Here, we identified the urothione-synthesizing methyltransferase using mouse liver tissue by anion exchange/size exclusion chromatography and peptide mass fingerprinting. We show that the catabolic Moco S-methylating enzyme corresponds to thiopurine S-methyltransferase (TPMT), a highly polymorphic drug-metabolizing enzyme associated with drug-related haematotoxicity but unknown physiological role. Urothione synthesis was investigated in vitro using recombinantly expressed human TPMT protein, liver lysates from Tpmt wild-type and knock-out (Tpmt−/−) mice as well as human liver cytosol. Urothione levels were quantified by LC-MS/MS in kidney and urine of mice. TPMT-genotype/phenotype and excretion levels of urothione were investigated in human samples and validated in an independent population-based study. As Moco provides a physiological substrate (thiopterin) of TPMT, thiopterin-methylating activity was associated with TPMT activity determined with its drug substrate (6-thioguanin) in mice and humans. Urothione concentration was extremely low in kidney and urine of Tpmt−/− mice. Urinary urothione concentration in TPMT-deficient patients depends on common TPMT polymorphisms, with extremely low levels in homozygous variant carriers (TPMT*3A/*3A) but normal levels in compound heterozygous carriers (TPMT*3A/*3C) as validated in the population-based study. Our work newly identified an endogenous substrate for TPMT and shows an unprecedented link between Moco-catabolism and drug-metabolism. Moreover, the TPMT example indicates that phenotypic consequences of genetic polymorphisms may differ between drug- and endogenous substrates.
Keywords: drug metabolism, molybdenum co-factor, pharmacogenomics, thiopurine methyltransferase, urothione, thiopurines
Introduction
Molybdenum (Mo) is an essential trace element, forming the catalytic site of all molybdenum-dependent enzymes.1 More than 50 Mo-enzymes have been described in nature, catalyzing key reactions of the global carbon, nitrogen and sulfur cycles.2 With the exception of nitrogenase, in all other Mo-enzymes, molybdenum is coordinated by a tricyclic pterin forming the molybdenum cofactor (Moco), which is synthesized by an evolutionary ancient and conserved pathway.3 For humans with genetic defects leading to the loss of Moco early childhood death has been the usual outcome until recently.4 Following successful preclinical studies in a mouse model of Moco deficiency (MoCD),5,6 we reported a first successful therapy in men7 using the biosynthetic precursor of Moco, cyclic pyranopterin monophosphate (cPMP).8 A prospective study in 11 patients disclosed remarkable clinical improvement in patients that received treatment prior to disease manifestation9 leading to the approval of cPMP by the FDA in 2021. MoCD patients are diagnosed by the absence of urothione in their urine, which goes back to an early finding back in 1982.10 In contrast to the well-known pathway leading to the synthesis of Moco,4 the catabolism of this essential cofactor which ends with the excretion of urothione11 remained entirely unexplored. Once released from the enzyme, Moco is very unstable and rapidly and irreversibly oxidized under aerobic conditions12 forming a compound known as Form B showing remarkable similarities to urothione (Figure 1A). Notably, one sulfur atom of urothione is methylated and the phosphate group is absent when compared to Form B. Based upon the known structures of Moco, Form B and urothione, we propose a mechanism of Moco catabolism (Figure 1A), which involves at least two enzyme-catalyzed reaction steps: methylation and dephosphorylation.
Figure 1. Proposed scheme of molybdenum cofactor (Moco) catabolism and urothione-synthesizing activity in mouse crude protein extracts.

(A) The pathway involves at least two enzyme-catalyzed reactions methylation and dephosphorylation. The order of both reactions is unknown. Also, release of molybdenum, pyran ring opening and pterin oxidation, are also required for the formation of urothione and believed to take place in early steps of this pathway. Moco [1], a methylated intermediate [2], urothione [3], and Form B [4], the air oxidized product of Moco are shown. (B) HPLC analysis of in vitro generated urothione from mouse liver extract in the presence (black) and absence (gray) of SAM. (C) In vitro urothione synthesis using crude protein extracts (4 μg/μL) from liver, kidney and brain using in vitro synthesized thiopterin (2.72 μM) in the presence of 50 μM SAM. (D and E) Purification of urothione synthesizing activity from mice crude liver extracts using anion exchange and size exclusion chromatography. Fractions were taken and urothione synthesis was determined using in vitro synthesized molybdopterin (MPT) in the absence (D) and presence of alkaline phosphatase (E). Gray highlight indicates the fraction which was further purified (D) or analyzed for protein content (E). (F) Results of peptide mass finger printing derived from the sample with highest activity shown in panel E. (G) HPLC analysis of in vitro generated urothione from mouse liver extract in the presence of SAM (black) or betaine (gray).
Here, we report based on in vitro and in vivo evidence in mice and humans that the newly identified catabolic Moco S-methylating enzyme corresponds to thiopurine S-methyltransferase (TPMT). TPMT represents an important drug-metabolizing enzyme in humans and catalyzes the S-methylation of thiopurines, which play a crucial role in the therapy of inflammatory bowel diseases or in the maintenance therapy of children suffering from acute lymphoblastic leukemia. It is well known that polymorphisms in the TPMT gene result in an increased risk for thiopurine related hematotoxicity, e.g. myelosuppression can be expected in all patients with TPMT deficiency receiving standard thiopurine dosage.13,14 Thus, clinical implementation of TPMT genotyping has been proposed by the Clinical Pharmacogenetics Implementation Consortium,13 as well as the Dutch pharmacogenetics group15 in order to reduce thiopurine-related toxicity. The physiological role of TPMT however remained unknown until today.
Methods
Mouse samples und study approval
Wild type (WT) mice C57Bl/6J (Charles River) with a mean age of 54 weeks (44-60) with a weight range of 35 - 45 g were used. Organs were removed and immediately snap frozen in liquid nitrogen. For preparative purification of urothione-synthesizing activity a total of 10 g liver was used. Mice were held in IVC cages with 500 cm2 floor space in groups ≤5. Food and water were ad libitum. Cages were maintained in a climate-controlled room (20-24°C; with a relative humidity 45–65%) in a 12-h light/dark cycle. All procedures involving breeding, maintenance and humane killing of WT mice were approved by the local veterinary office (City of Cologne, Germany).
Tpmt knockout (Tpmt−/−) and wild-type (WT) mice on the 129X1/SvJ background are described in detail by Hartford C et al.16. These mice were backcrossed for 6 generations onto a BALB/cJ background (Jackson Laboratories (stock number 000651, Sacramento, CA) to generate Tpmt−/− and WT mice on a second background. Only male mice (mean age: 11.3 weeks [9.6-12.7]) were used during these experiments with a weight range of 23–30 g. Organs from seven Tpmt knockout (Tpmt−/−) and wild-type (WT) mice were removed and immediately snap frozen in liquid nitrogen (mean weight (g) liver: 1.18 (0.85-1.51), kidney: 0.45 (0.37-0.55)). For housing of mice transparent cages (<5 per cage) with nesting material were used. Cages were maintained in a climate-controlled room (temperature 19–24°C; relative humidity 45–65%) with a 12-h light/dark cycle (lights on at 7:00 am). Food and water were ad libitum. All efforts were made to minimize animal suffering. All experiments involving the production and use of the Tpmt−/− mice were approved by the Institutional Animal Care and Use Committee of St. Jude Children’s Research Hospital, USA.
Human samples and study approval
Histologically normal human liver samples as well as corresponding blood samples were collected as previously described17 from patients undergoing liver surgery at the Department of General, Visceral and Transplantation Surgery (University Medical Center Charité, Berlin, Germany). 20 liver samples were included in the present study. The study was approved by the ethics committees of the Charité, Humboldt University (Berlin, Germany) and the University of Tübingen (Tübingen, Germany) in accordance with the principles of the Declaration of Helsinki. The tissue samples were stored at −80°C and cytosolic fractions were prepared according to standard procedures. Urine as well as corresponding blood samples from 31 individuals participating to a prospectively designed clinical trial in cancer patients18 and healthy individuals19 were used. Both studies were approved by the ethics committee of the University Hospital Tuebingen, Germany, in accordance with the principles of the Declaration of Helsinki. An additional patient with thiopurine-related pancytopenia was derived from the clinical laboratory service for pharmacogenetic testing at the Dr Margarete Fischer-Bosch-Institute of Clinical Pharmacology, Stuttgart, Germany. Urine samples were stabilized using sodium azide and stored at −20°C. Genomic DNA from EDTA blood samples was purified as described previously.17,19 In addition, urine and corresponding DNA samples of 213 individuals (Table S1) from the population-based Study of Health in Pomerania (SHIP) cohort (SHIP-Trend) were included. Study details were described in detail previously.20-22 The study followed the recommendations of the Declaration of Helsinki. The study protocol was approved by the medical ethics committee of the University of Greifswald, and written informed consent was obtained from each study participant.
Preparation of organ extracts
Mouse brain, liver and kidney raw lysates were prepared by resuspension and homogenization in 0.1 M Tris/HCl, pH 7.5 containing protease inhibitor (Roche). Details see supplementary material.
Purification of urothione-synthesizing activity from mouse liver
For purification of urothione-synthesizing activity 10 g of mouse liver was homogenized in 0.1 M Tris/HCl, pH 7.5 using a Potter S homogenizer (1000 rpm passages, on ice, followed by centrifugation three times at 21,000 x g 20 min at 4°C). Ammonium sulfate was added to the lysate to 25%-saturation. Precipitated proteins were collected by centrifugation at 21,000 x g at 4°C. Urothione-synthesizing activity was detected in the pellet using HPLC. The pellet was resuspended in 50 mM Tris/HCl, pH 7.5 and subjected to dialysis followed by anion exchange chromatography (Source 15Q) by applying a gradient of 0–1 M NaCl in 50 mM Tris/HCl pH 7.5. Urothione-synthesizing activity was determined in all fractions collected. Fractions containing highest urothione-synthesizing activity were subjected to further purification by size exclusion chromatography (Superdex 200, GE Healthcare) equilibrated in 50mM Tris/HCl, 250 mM NaCl. Again, urothione-synthesizing activity was determined in all fractions. Note, to compensate for the loss of the predicted phosphates, we added alkaline phosphatase. The fraction with highest activity was subjected to peptide mass fingerprinting using an EASY nLC II nano-LC (Proxeon/Thermo Scientific) coupled to LTQ Orbitrap-mass spectrometer (LTQ Orbitrap Discovery, Thermo Scientific) using a 150 mm x 75 μm C18 reverse phase column (Dr. Maisch GmbH, Ammerbuch-Entringen). The peptides detected were identified using the MASCOT-algorithm 23 by comparison to the Uniprot database.
In vitro urothione synthesis assay
For in vitro synthesis of urothione two different sources of thiopterin were used: (i) in vitro generated thiopterin from cyclic pyranopterin monophosphate (cPMP) using bacterial molybdopteroin synthase (EC 2.8.1.12) 24 or thiopterin derived from recombinantly expressed Moco domain of human sulfite Oxidase (SOMo).25 The concentration of thiopterin in a given preparation was determined by HPLC analysis of the oxidation product Form A dephospho26.
A standard in vitro urothione synthesis consisted of two steps: First the reaction components thiopterin (1-3 μM in vitro synthesized molybdopterin or 15 μM sulfite oxidase Mo-domain), 250 μM DTT, SAM (50 μM–1 mM) and either TPMT (85-570 nM) or organ raw extract (1-5 μg/μL) were mixed and incubated for 2 h (TPMT) or 4 h (organ raw extracts) at 37°C in 50 mM Tris/HCl pH 7.5 in a total volume of 150 μL. Were indicated AP was added to dephosphorylate 2-MTPP. The reaction was stopped by heat denaturation using I2/KI (1/2%) in 1 M HCl. In a second step, the pH was adjusted to 8.3 by the addition of 1 M unbuffered Tris, 20 mM MgCl2 and 30 U alkaline phosphatase (Roche). The reactions were incubated for 4 h at 37°C and stopped by heat denaturation.
Detection of urothione and the reaction intermediate 2-MTPP
Urothione was quantified by HPLC using either a C4 Reprosil 100, 150 x 2.0 mm, 5 μm particle size column (Dr. Maisch) or C18 Hydrosphere, 250 x 4.0 mm, 5 μm particle size column (YMC) coupled to diode-array detection at 380 nm.10 For identification the resulting peak was compared to that of a urothione standard isolated from human urine as described by Goto.27 2-MTPP was found to be the product of TPMT-dependent thiopterin methylation. It was in vitro synthesized by the same method as that described for in vitro urothione synthesis, however without the addition of alkaline phosphatase (AP).
TPMT activity assay using recombinant TPMT protein
For the determination of kinetic parameters, recombinant TPMT (3 ng/μL) (details regarding preparation see supplementary material) was incubated with varying concentrations of thiopterin (0.5 −14 μM) or 6-MP (0.01 - 1 mM) in the presence of SAM (1 mM) and 250 μM DTT in 50 mM Tris/HCl, pH 7.5. The initial velocity of the respective reaction in a reaction time of 12 min was determined. The reaction was terminated by the addition of 2 mM TPMT inhibitor 3,4,5-triiodobenzoic acid (TIBA) followed by heat denaturation. While 6-MMP was analyzed directly, the reaction for urothione formation was treated with AP prior to HPLC analysis.
Measurement of TPMT activity
TPMT activity was measured in RBCs, as well as in liver cytosol through non-radioactive HPLC method using 6-TG as a substrate as previously described 19,28.
TPMT genotyping
Genotyping for genetic variants in the TPMT gene was performed using TaqMan technology and MALDI-TOF MS as described previously.29,30 Variants (rs1800462, rs1800460, rs1142345) defining the TPMT*2,*3A-C alleles were considered for definition of TPMT WT, HET or HOM carriers in human individuals and in human liver samples. Samples from the SHIP-Trend study were genotyped using Illumina Human Omni 2.5 [(Calling method: GenCall), (Sample and variant quality control: sample level QC: sample call rate ≥ 94%, genotyped sex different from recorded sex, duplicates by estimated IBD; SNP level QC: SNP call rate ≥ 95%, a deviation from HWE ≥ 1E-4, and MAF >0%. Excluded SNPs unable to liftover to Build 37, with mismatch in reference allele, or duplicate ID), (Imputation software: Eagle and minimac3 for pre-phasing and imputation, respectively (Michigan Imputation Server)), (Reference Panel: Haplotype Reference Consortium release 1.1)]. WT, HET or HOM carriers in the SHIP-Trend study were selected based on genotyping results of rs1142345. Moreover, genotyping results from rs1800462 were considered to exclude presence of TPMT*2 allele in WT and HET carriers. Homozygous variant allele carriers of rs1142345 were additionally genotyped for TPMT*3A-C.
Quantification of urothione by LC-MS-MS
Synthesis of the internal standard [13C2H3]urothione, sample preparation, the establishment of the novel LC-MS-MS method using an Agilent 6460 triple quadrupole mass spectrometer (Agilent, Waldbronn, Germany), and standardization and validation of the assay are described in detail in the supplementary material.
Non-targeted metabolomics
A non-targeted metabolomics approach was applied to analyze kidney samples derived from 4 wild-type and 4 Tpmt−/− mice using quadrupole time-of-flight mass spectrometry (6550 iFunnel Q-TOF LC/MS, Agilent Technologies, Germany) in negative and positive ionization mode. Details regarding standardization and validation of the assay and processing of mass spectral data and statistical analysis are described in detail in the supplementary material.
Statistics
Statistical analyses were performed using GraphPad Prism 9 software (GraphPad Software). P-values indicating *p<0.05, **p<0.01 and *** p<0.001, were determined by Mann-Whitney test. A P value of less than 0.05 was considered significant.
Results
In order to investigate urothione biosynthesis (Figure 1A), we first developed a method for the in vitro formation of urothione. As substrate – named thiopterin – we used either Moco or molybdopterin, which were either isolated from sulfite oxidase (Moco) or synthesized in vitro (molybdopterin), respectively. In vitro conversion of thiopterin to urothione was established with liver crude extracts given the high expression of Moco-containing enzymes in liver.31 We found that in vitro urothione synthesis was strictly dependent on the presence of S-adenosyl methionine (SAM) (Figure 1B). Highest urothione-synthesizing activity was found in liver, while significant activity was also detectable in brain (Figure 1C).
Enrichment and identification of the urothione-synthesizing activity from mouse liver was performed by ammonium sulfate precipitation, anion exchange (Figure 1D) and size exclusion chromatography (Figure 1E). Peptide mass fingerprinting identified five methyltransferases in the fraction with highest urothione-synthesizing activity (Figure 1F). Of these, only two enzymes, TPMT and betaine-homocysteine S-methyltransferase were known to methylate substrates at sulfur atoms.32 Given that TPMT is SAM-dependent and betaine was not able to promote urothione synthesis in vitro (Figure 1G), and that TPMT was identified with the highest score (Figure 1F) amongst five methyltransferases, we proposed TPMT as candidate enzyme for methylating urothione.
TPMT has been extensively studied in the past due to its important role in the detoxification of thiopurine medications 6-mercaptopurine (6-MP), 6-thioguanine (6-TG) and azathioprine, all of which are widely used for treatment of cancer and inflammatory diseases. However, the biological function and natural substrate of TPMT has remained unknown. In order to characterize the role of TPMT in urothione formation, His-tagged human TPMT was recombinantly expressed in E. coli and purified to homogeneity. In vitro urothione synthesis was performed with TPMT in the presence of alkaline phosphatase (AP), given that urothione formation requires a dephosphorylation step. Purified TPMT was able to catalyze the in vitro synthesis of urothione using increasing concentrations of thiopterin in a substrate-dependent manner (Figure S1).
Next, catalytic properties of TPMT in the generation of urothione were compared to the well-studied methylation of 6-MP to form 6-methylmercaptopurine (6-MMP).33 The enzymatic reaction followed a Michaelis-Menten type of kinetics (Figure 2A and 2B) and revealed a KMthiopterin of 1.86 ± 0.36 μM and KM6-MP of 73.95 ± 11.13 μM, indicating a much higher affinity of TPMT for thiopterin than for any of the so far known substrates. In addition, the kcat for thiopterin was 0.129 ± 0.01 s−1, again higher than the kcat for 6-MP of 0.048 ± 0.002 s−1. Both, KM and kcat indicate that the catalytic efficacy of TPMT in methylating thiopterin is approximately 200-fold higher than reported for 6-mercaptopurine, the best-characterized TPMT substrate.33
Figure 2. Characterization of TPMT activity.
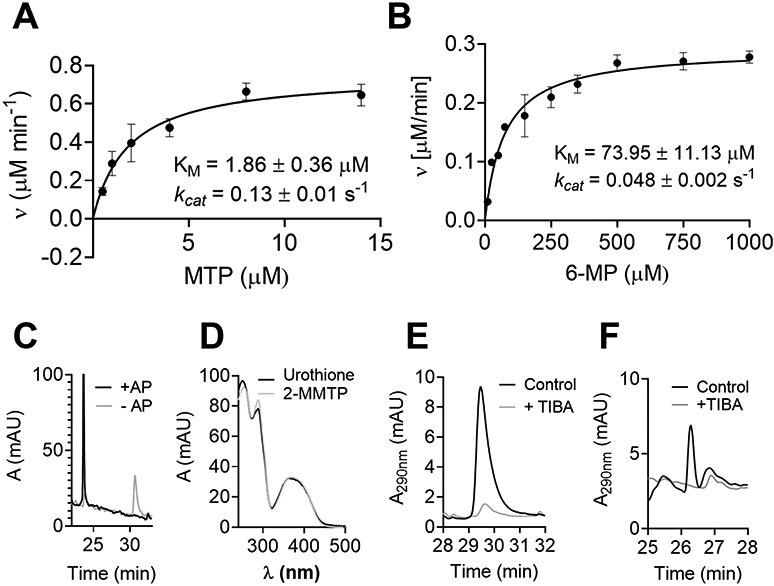
(A) HPLC-based steady-state kinetics of TPMT using 0-15 μM thiopterin. Hyperbolic regression analysis according to Michaelis-Menten resulted in a KMthiopterin of 1.86 ± 0.36 μM and kcat of 0.129 ± 0.01s−1. (B) The analysis for the well-known substrate 6-mercaptopurine resulted in a KM6-MP of 73.95 ± 11.13 μM and kcat of 0.048 ± 0.002 s−1. Values represent means of three different measurements; error bars indicate standard deviations from the mean. (C) HPLC profile of TPMT-dependent methylation of thiopterin in the presence or absence of AP. (D) UV-vis spectrum of urothione and the peak eluted at 31 min shown in panel C. (E) 2-MTPP produced by TPMT in the standard assay (black) or in the presence of 3,4,5-triiodobenzoic acid (TIBA) inhibitor (gray). (F) Urothione generated from mouse liver raw extract (black). In the presence of TIBA, no urothione was detected (gray).
Besides TPMT, AP, a phosphatase with broad substrate specificity, was required to produce urothione in our in vitro assay. In the absence of dephosphorylation we therefore aimed to detect a methylated, phosphorylated intermediate with structure [2] (Figure 1A). HPLC analysis of the TPMT reaction with thiopterin in the absence of alkaline phosphatase (AP) identified a substance eluting with a retention time of 30.2 min vs. 23.8 min for urothione (Figure 2C) exhibiting an UV-vis spectrum indistinguishable from urothione (Figure 2D). The molecular ion size in the MS-positive ion mode was 406 m/z, which matches with a threefold protonated form of structure [2] carrying a single positive charge (Figure S2). Similar spectroscopic properties between urothione and molecule [2] are consistent with a molecule that differs from urothione only in the presence of a phosphate group (Figure 1A). Molecule [2] was named 2-methyl thiophosphopterin (2-MTPP).
Next, we probed the effect of the non-competitive TPMT-inhibitor 3,4,5-triiodobenzoic acid (TIBA)34 on the in vitro generation of 2-MTPP by using recombinant TPMT (Figure 2E) and thiopterin. Addition of 2.5 μM TIBA resulted in a significant reduction of 2-MTPP synthesis. In addition, TPMT activity was inhibited by TIBA in crude mouse liver extract and the synthesis of urothione (in the presence of AP to convert 2-MTPP) was investigated (Figure 2F). Under these conditions, urothione synthesis was completely blocked by TIBA, further indicating that TPMT functions in urothione biosynthesis.
To obtain in vivo evidence that TPMT functions as a methyltransferase in urothione biosynthesis, liver lysates from seven Tpmt wild-type (wt) and knock-out (Tpmt−/−) mice (four mice with 129X1/SvJ background and three mice with Balb/cJ background) were investigated for their ability to synthesize urothione in vitro. These and all subsequent studies (if not stated otherwise) were conducted as blinded studies. As a result, TPMT phenotypes of the mice were confirmed through HPLC-based TPMT activity measurements in liver cytosol (Figure 3A) using 6-TG as substrate. Urothione was only detected in liver extracts from wildtype mice (Figure 3B). No HPLC peak with an urothione-specific UV-vis spectrum could be detected in Tpmt−/− samples. The remaining overlapping minor peak eluting at the expected position of urothione in Tpmt−/− samples was shown to be unrelated to urothione thus representing a co-eluting impurity (Figure S3). Next, we aimed to detect urothione levels in kidney tissue of mice. Given the expected low quantities of urothione, we developed a novel mass spectrometric method for urothione detection and found only very low concentrations of urothione below the limit of quantification in Tpmt−/− mice (n=4), whereas in Tpmt wildtype mice (n=4) urothione concentrations varied between 0.023 and 0.037 pmol/mg (Figure 3C).
Figure 3. TPMT activity in mice and men.
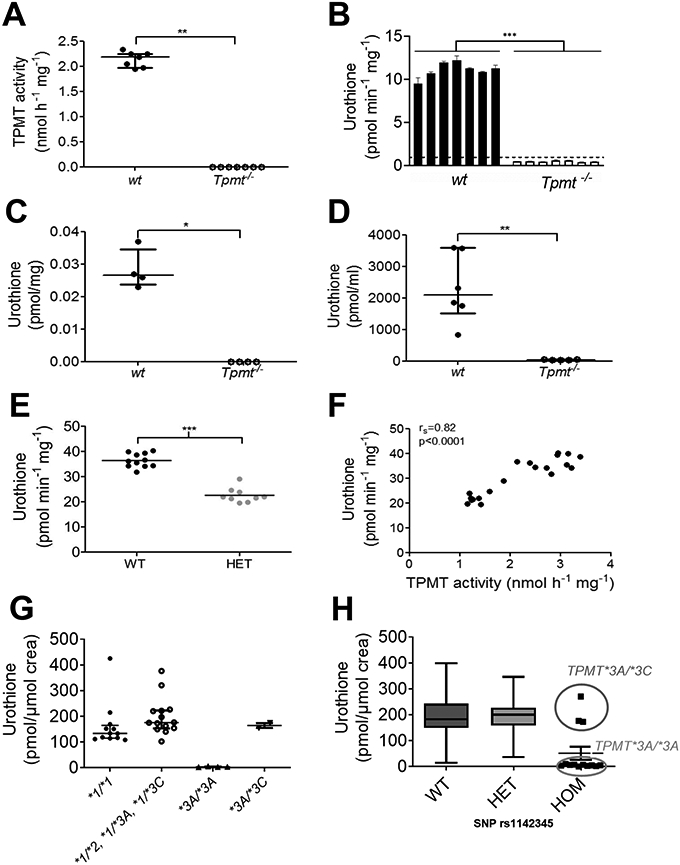
(A) TPMT activity in wt (n=7) and Tpmt−/− (n=7) mice liver protein extracts. (B) Urothione-synthesizing activity in liver crude protein extracts (1.65 μg/μL) of wt (n=7) and Tpmt−/− (n=7) mice in the presence of 1 mM SAM. Note, in contrast to Figure 1C, 30 units AP were added to maximize urothione synthesis. Values represent means of three different measurements; error bars indicate standard deviations from the mean. (C) Urothione concentration in kidney tissue of wt (n=4) and Tpmt−/− (n=4) mice. (D) Urinary urothione concentration in wt (n=6) and Tpmt−/− (n=5) mice. (E) Urothione-synthesizing activity in human liver extracts of wildtype (WT, n=11) and TPMT-heterozygous (HET, n=9 comprising eight TPMT*1/*3A and one TPMT*1/*2 carrier) individuals. (F) Correlation of hepatic urothione synthesizing activity and hepatic TPMT-activity in 20 humans (rs indicates Spearman correlation coefficient). (G) Urinary urothione excretion in TPMT wildtype (n=12) and heterozygous (n=14, comprising eleven TPMT*1/*3A, two TPMT*1/*3C and one TPMT*1/*2 carrier) individuals, as well as in 6 subjects with TPMT deficiency based on 6-TG methylation as activity measure and genotypes (4 cases with homozygous TPMT genotype (*3A/*3A) and 2 cases with compound heterozygous TPMT genotype (*3A/*3C). (H) Urinary urothione excretion in TPMT wildtype (n=100) and heterozygous (n=100) individuals, as well as in 13 subjects with TPMT deficiency based on genotypes (10 cases with the homozygous TPMT genotype *3A/*3A and 3 cases with the compound heterozygous TPMT genotype *3A/*3C) from the SHIP study. P-values indicate *p<0.05, **p<0.01 and *** p<0.001.
Finally, we also detected urinary urothione excretion in mice. Urinary concentrations of urothione were found to be very low (between 31 and 53 pmol/ml) in Tpmt−/− mice (n=5), whereas in Tpmt wildtype mice (n=6) urothione concentrations varied between 827 and 3602 pmol/mL (Figure 3D) in urine. Therefore, we conclude that TPMT is the only murine enzyme capable of catalyzing thiopterin methylation in vivo, an essential step in urothione synthesis.
Notably, as shown in Figure S4, metabolite levels do not differ between Tpmt−/− and Tpmt wildtype mice, indicating that the metabolome in general is not influenced by TPMT activity.
In a next step, we aimed to transfer our findings from mouse to the human situation. Since liver represents the predominant organ of drug metabolism, we investigated urothione synthesis in human liver cytosol samples derived from individuals with normal or reduced TPMT activity. All individuals were phenotyped through HPLC-based TPMT activity assessment with 6-TG as substrate and genotyped for relevant, non-functional TPMT alleles. As shown in Figure 3E, subjects with heterozygous TPMT genotype (HET, comprising eight TPMT*1/*3A and one TPMT*1/*2 carrier) produced significantly less urothione compared to individuals with normal TPMT activity (WT, n=11). In addition, urothione synthesis correlated significantly with hepatic TPMT activity (Figure 3F).
In order to determine a possible association between TPMT-genotype/phenotype and excretion levels of urothione, we investigated urine of individuals with either normal (TPMT*1/*1, n=12), reduced (TPMT*1/*2, n=1; TPMT*1/*3A, n=11; TPMT*1/*3C, n=2) or negligible TPMT activity (TPMT*3A/*3A, n=4; TPMT*3A/*3C, n=2) as determined using S-methylation of 6-TG (Figure 3G). Notably, TPMT*3A, which is the most common variant allele in Caucasians, comprises two nonsynonymous variants (G460A and A719G), whereas TPMT*3C, the most common variant allele in Africans and Asians, includes only the A719G variant. Urothione was detected in urine of individuals carrying at least one wildtype TPMT allele. Moreover, urothione was found to be near the limit of quantification (LOQ) in the urine of individuals with TPMT*3A/*3A genotype, including a TPMT-deficient patient suffering from acute life-threatening pancytopenia due to excessively high levels of active thiopurine metabolites (1905 pmol thioguanine nucleotides/8x108 red blood cells (RBC)) following treatment of Crohn’s disease with standard dosage of the 6-MP pro-drug azathioprine (150 mg/day). Urothione levels in urine from this patient were found to be very low near the LOQ (2.87 pmol/μmol creatinine). Thus, in this case TPMT deficiency assessed through TPMT activity measurement in RBC and genotyping (TPMT*3A/*3A) was confirmed through the lack of urothione in this patient. In contrast, normal urothione levels were found in compound heterozygous TPMT*3A/*3C individuals (Figure 3G) with negligible TPMT activity determined using S-methylation of 6-TG. Based on these results, differences in the phenotypic consequences of TPMT polymorphisms between the endogenous and drug substrates exist.
To further validate our human data, samples derived from an independent population-based study (SHIP) were analyzed 20,21. Here, 213 samples were selected based on the presence of the key variant (rs1142345, A719G) for TPMT*3A and *3C allele. Again, low urothione levels were only detected in ten TPMT*3A/*3A carriers and not in three TPMT*3A/*3C individuals (Figure 3H).
Discussion
Although therapy of molybdenum co-factor (Moco) deficiency has received approval by the FDA in 2021 and the excreted catabolite of Moco is used as diagnostic biomarker for Moco deficiency, the catabolic pathway of Moco leading to its urinary excreted catabolite urothione remained unknown. In the present study, we extensively studied the catabolic pathway and show that the newly identified catabolic Moco S-methylating enzyme corresponds to thiopurine S-methyltransferase (TPMT). So far, TPMT was only recognized as important drug metabolizing enzyme of thiopurine drugs like 6-mercaptopurine and 6-TG, but its physiological role was still not known. Our study clearly indicates that Moco provides a physiological substrate (thiopterin) of TPMT. Interestingly, the catalytic efficacy (KM and kcat) of TPMT in methylating thiopterin is approximately 200-fold higher than reported for the drug 6-mercaptopurine, the best-characterized TPMT substrate.33
Moreover, thiopterin-methylating activity was strongly associated with TPMT activity (determined with 6-TG as substrate) in mice and humans in our study. In line, urothione concentration was extremely low in renal tissue and urine of Tpmt−/− mice. However, metabolism in general did not differ between Tpmt−/− and Tpmt wildtype mice, indicating that the metabolome is not influenced by TPMT activity. This is consistent with no known physiological phenotype in TPMT-deficient mice and humans, unless treated with thiopurines.16,19 However, due to the role of TPMT as drug-metabolizing enzyme, individuals with TPMT deficiency (caused by homozygosity or compound heterozygosity for variant alleles) are at risk of serious life-threatening adverse events (e.g. myelosuppression) when treated with standard doses of thiopurines.13,14
In humans, currently more than 30 TPMT variant alleles have been identified and linked with altered TPMT activity, collectively resulting in a prevalence of TPMT deficiency of ~0.3 % in the Caucasian population.19,35 The most common variant allele in Caucasians comprising two nonsynonymous variants (G460A and A719G) is the TPMT*3A allele, whereas TPMT*3C is the most common variant allele in Africans and Asians including only the A719G variant. Interestingly, urinary urothione concentration in TPMT-deficient patients strongly depends on TPMT polymorphisms, with extremely low levels detected in TPMT*3A/*3A carriers and normal levels in compound TPMT*3A/*3C carriers, which was validated in an independent population-based study. Regarding the effect of other TPMT variant alleles like TPMT*2, further studies including homozygous or compound heterozygous variant allele carriers are warranted.
Generally, as known for many other genes encoding metabolic enzymes (e.g. CYP2D6 36), the presence of one functional allele appears to be sufficient for the excretion of urothione within a broad concentration range comparable to individuals carrying two intact copies of the gene. However, the identification of TPMT-deficient individuals with altered thiopurine metabolism based on extremely low concentration of urothione in their urine samples is dependent on TPMT variants. As most non-functional variant alleles of TPMT, including *3A and *3C, encode proteins that destabilize folding leading to aggregation and enhanced proteolysis but not complete loss of function,37-39 differences in remaining protein levels and subsequent substrate specificity of both alleles might be responsible for the observed differences in urothione levels. For the recombinant proteins, differences in enzyme kinetics with 6-MP or 6-TG have been reported. While TPMT*3A protein showed virtually complete loss of enzyme activity as a result of enhanced proteolysis, TPMT*3C retained a reduced activity with kinetic parameters comparable to the wild type protein.37,38,40,41 Whether discrepancies between TPMT*3A and TPMT*3C result from an increased stability of TPMT*3C protein, which is sufficient to metabolize the endogenous substrate despite its faster degradation compared to WT needs further investigation. In either case, it might be feasible to propose that due to the 200-fold higher catalytic efficacy of TPMT towards thiopterin, a reduction in active enzyme concentration in TPMT*3A/*3C carriers may be still tolerated, while in the thiopurine (6-MP or 6-TG) based loading test, the reduced total activity of enzyme is a more direct readout of available active TPMT. As a result, the remaining activity in TPMT*3A/*3C carriers may have been sufficient to produce significant amounts of urothione, which is still a minor metabolic excretion product. To which extend a loss of function of TPMT activity as observed in TPMT*3A/*3A carriers will impact Moco homeostasis remains to be elucidated. In vitro oxidation of Moco to Form B 10 has been reported and may serve as alternative exit route for the pterin following its dephosphorylation. Alternatively, increased activity of the Moco enzymes sulfite oxidase and xanthine oxidase may result in alteration of turnover of cysteine and hypoxanthine/xanthine, respectively the later could result in an increased prevalence to develop gout.1,3,42
In summary, we identified TPMT as an essential enzyme for urothione synthesis by providing multiple lines of in vitro and in vivo evidence for the methylation of Moco by TPMT. Most importantly, the extremely low urinary excretion of urothione in TPMT-deficient mice and human individuals with compromised TPMT activity reveals the first known endogenous substrate of TPMT and unambiguously demonstrates the physiological function of TPMT in Moco catabolism. Urothione excretion was similar between TPMT wildtype and heterozygotes with one non-functional TPMT allele, suggesting that TPMT is not rate-limiting in urothione excretion. Moreover, on the example of TPMT the prediction of phenotypic consequences of genetic polymorphisms in drug metabolizing enzymes for personalized therapy based on an endogenous substrate may differ and requires careful evaluation of each drug substrate and genetic variant.
Supplementary Material
Study Highlights.
What is the current knowledge on the topic?
Thiopurine S-methyltransferase (TPMT) is the key enzyme in metabolism of thiopurine drugs, contributing to individualized therapy. Although TPMT is one of the best-recognized examples in pharmacogenomics, its endogenous substrate remained unknown.
What question did this study address?
Urothione, the urinary excreted catabolite of molybdenum co-factor (Moco) is used as diagnostic biomarker for Moco-deficiency, but the catabolic pathway of Moco including the involved urothione-synthesizing methyltransferase remained entirely unexplored.
What does this study add to our knowledge?
TPMT was identified by an unbiased search for enzymes involved in molybdenum cofactor (Moco) catabolism. In vivo evidence in mice and men confirms the physiological function of TPMT in Moco catabolism.
How might this change clinical pharmacology or translational science?
Phenotypic consequences of TPMT genetic variation differ between drug- and endogenous substrates. Systematic analyses of phenotype-genotype correlation for endogenous substrates of other drug metabolizing enzymes are warranted.
Acknowledgments:
We appreciate the expert technical assistance of Simona Jansen, Joana Stegemann, Monika Laurin (Institute of Biochemistry, University of Cologne, Germany), Monika Elbl, Monika Seiler, Andrea Jarmuth (Dr. Margarete Fischer-Bosch-Institute of Clinical Pharmacology, Stuttgart, Germany), Anke Witt and Hanka Zobel (Institute of Clinical Chemistry and Laboratory Medicine, University Medicine Greifswald, Germany). We thank Tobias Lampkemeyer (CECAD Cologne) for peptide mass finger printing, Kathrin Klein and Ulrich Zanger (Dr. Margarete Fischer-Bosch-Institute of Clinical Pharmacology, Stuttgart) for the use of liver tissue samples and Mirjam Geditz (Dr. Margarete Fischer-Bosch-Institute of Clinical Pharmacology, Stuttgart) for supporting the establishment of LC-MS-MS assay. We thank Georg Heinkele (Dr. Margarete Fischer-Bosch-Institute of Clinical Pharmacology, Stuttgart) for his help to synthesize the labelled urothione.
Funding Information:
The work was supported, in part, by the Fonds der Chemischen Industrie (J.C., G.S.), by the Robert Bosch Stiftung, Stuttgart, Germany, the Federal Ministry for Education and Research (BMBF, Berlin, Germany) grants 0315755 and 031L0037, the Horizon 2020-PHC-2015 grant U-PGx 6683533, the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy - EXC 2180 – 390900677) and by US NIH National Cancer Institute grant R37 CA36401 (W.E.E., M.V.R.), R01 142665 (M.V.R.), NIH National Institute of General Medical Sciences Pharmacogenomics Research Network grant U01 GM92666 and P50 GM115279 (M.V.R., W.E.E.), Cancer Center Support Grant CA 21765 from the NCI and by the American Lebanese Syrian Associated Charities (ALSAC). SHIP is supported by the German Federal Ministry of Education and Research [Grants 01ZZ0403, 01ZZ0103, 01ZZ9603], the German Ministry for Education, Research and Cultural Affairs, as well as the Ministry of Social Affairs of the Federal State of Mecklenburg-West Pomerania.
Footnotes
Conflict of interest: The authors declared no competing interests for this work.
Data availability:
The data that support the findings of this study are available from the corresponding author upon reasonable request. Some data may not be made available because of privacy or ethical restrictions.
References
- 1.Schwarz G, Mendel RR & Ribbe MW Molybdenum cofactors, enzymes and pathways. Nature 460, 839–847 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Hille R The Mononuclear Molybdenum Enzymes. Chemical reviews 96, 2757–2816 (1996). [DOI] [PubMed] [Google Scholar]
- 3.Mayr SJ, Mendel R-R & Schwarz G Molybdenum cofactor biology, evolution and deficiency. Biochimica et biophysica acta. Molecular cell research 1868, 118883 (2021). [DOI] [PubMed] [Google Scholar]
- 4.Schwarz G Molybdenum cofactor biosynthesis and deficiency. Cellular and molecular life sciences : CMLS; 62, 2792–2810 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee H-J et al. Molybdenum cofactor-deficient mice resemble the phenotype of human patients. Human molecular genetics 11, 3309–3317 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Schwarz G et al. Rescue of lethal molybdenum cofactor deficiency by a biosynthetic precursor from Escherichia coli. Human molecular genetics 13, 1249–1255 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Veldman A et al. Successful treatment of molybdenum cofactor deficiency type A with cPMP. Pediatrics 125, e1249–54 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Santamaria-Araujo JA et al. The tetrahydropyranopterin structure of the sulfur-free and metal-free molybdenum cofactor precursor. The Journal of biological chemistry 279, 15994–15999 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Schwahn BC et al. Efficacy and safety of cyclic pyranopterin monophosphate substitution in severe molybdenum cofactor deficiency type A. A prospective cohort study. The Lancet 386, 1955–1963 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Johnson JL & Rajagopalan KV Structural and metabolic relationship between the molybdenum cofactor and urothione. Proceedings of the National Academy of Sciences of the United States of America 79, 6856–6860 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koschara W Uriothion, ein gelber, schwelfelreicher Farbstoff aus Menschenharn. Hoppe Seylers Z Physiol Chem 263, 78–79 (1940). [Google Scholar]
- 12.Kuper J, Palmer T, Mendel RR & Schwarz G Mutations in the molybdenum cofactor biosynthetic protein Cnx1G from Arabidopsis thaliana define functions for molybdopterin binding, molybdenum insertion, and molybdenum cofactor stabilization. Proceedings of the National Academy of Sciences 97, 6475 (2000) 6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Relling MV et al. Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing: 2013 update. Clinical Pharmacology and Therapeutics 93, 324–325 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Relling MV et al. Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. Journal of the National Cancer Institute 91, 2001–2008 (1999). [DOI] [PubMed] [Google Scholar]
- 15.Swen JJ et al. Pharmacogenetics. From bench to byte—an update of guidelines. Clinical Pharmacology and Therapeutics 89, 662–673 (2011). [DOI] [PubMed] [Google Scholar]
- 16.Hartford C et al. Differential effects of targeted disruption of thiopurine methyltransferase on mercaptopurine and thioguanine pharmacodynamics. Cancer Research 67, 4965–4972 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Nies AT et al. Expression of organic cation transporters OCT1 (SLC22A1) and OCT3 (SLC22A3) is affected by genetic factors and cholestasis in human liver. Hepatology 50, 1227–1240 (2009). [DOI] [PubMed] [Google Scholar]
- 18.Schwab M et al. Role of genetic and nongenetic factors for fluorouracil treatment-related severe toxicity: a prospective clinical trial by the German 5-FU Toxicity Study Group. Journal of Clinical Oncology 26, 2131–2138 (2008). [DOI] [PubMed] [Google Scholar]
- 19.Schaeffeler E et al. Comprehensive analysis of thiopurine S-methyltransferase phenotype-genotype correlation in a large population of German-Caucasians and identification of novel TPMT variants. Pharmacogenetics 14, 407–417 (2004). [DOI] [PubMed] [Google Scholar]
- 20.John U et al. Study of Health in Pomerania (SHIP): A health examination survey in an east German region: Objectives and design. Sozial- und Präventivmedizin 46, 186–194 (2001). [DOI] [PubMed] [Google Scholar]
- 21.Völzke H et al. Cohort profile. The study of health in Pomerania. International journal of epidemiology 40, 294–307 (2011). [DOI] [PubMed] [Google Scholar]
- 22.Völzke H et al. Cohort Profile Update: The Study of Health in Pomerania (SHIP). International journal of epidemiology (2022). doi: 10.1093/ije/dyac034 [DOI] [PubMed] [Google Scholar]
- 23.Perkins DN, Pappin DJC, Creasy DM & Cottrell JS Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 (1999). [DOI] [PubMed] [Google Scholar]
- 24.Gutzke G, Fischer B, Mendel RR & Schwarz G Thiocarboxylation of molybdopterin synthase provides evidence for the mechanism of dithiolene formation in metal-binding pterins. The Journal of biological chemistry 276, 36268–36274 (2001). [DOI] [PubMed] [Google Scholar]
- 25.Belaidi AA & Schwarz G Metal insertion into the molybdenum cofactor. Product-substrate channelling demonstrates the functional origin of domain fusion in gephyrin. The Biochemical journal 450, 149–157 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Johnson JL, Hainline BE, Rajagopalan KV & Arison BH The pterin component of the molybdenum cofactor. Structural characterization of two fluorescent derivatives. The Journal of biological chemistry 259, 5414–5422 (1984). [PubMed] [Google Scholar]
- 27.Goto M, Sakurai A, Ota K & Yamakami H Die Struktur des Urothions. Journal of biochemistry 65, 611–620 (1969). [DOI] [PubMed] [Google Scholar]
- 28.Schwab M et al. Azathioprine therapy and adverse drug reactions in patients with inflammatory bowel disease. Impact of thiopurine S-methyltransferase polymorphism. Pharmacogenetics 12, 429–436 (2002). [DOI] [PubMed] [Google Scholar]
- 29.Schaeffeler E, Zanger UM, Eichelbaum M, Asante-Poku S, Shin J-G & Schwab M Highly multiplexed genotyping of thiopurine S-methyltransferase variants using MALDI-TOF mass spectrometry: reliable genotyping in different ethnic groups. Clinical Chemistry 54, 1637–1647 (2008). [DOI] [PubMed] [Google Scholar]
- 30.Dilger K et al. Monitoring of thiopurine methyltransferase activity in postsurgical patients with Crohn’s disease during 1 year of treatment with azathioprine or mesalazine. Therapeutic Drug Monitoring 29, 1–5 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Woo WH, Yang H, Wong KP & Halliwell B Sulphite oxidase gene expression in human brain and in other human and rat tissues. Biochemical and Biophysical Research Communications 305, 619–623 (2003). [DOI] [PubMed] [Google Scholar]
- 32.Garrow TA Purification, kinetic properties, and cDNA cloning of mammalian betaine-homocysteine methyltransferase. The Journal of biological chemistry 271, 22831–22838 (1996). [DOI] [PubMed] [Google Scholar]
- 33.Woodson LC & Weinshilboum RM Human kidney thiopurine methyltransferase purification and biochemical properties. Biochemical Pharmacology 32, 819–826 (1983). [DOI] [PubMed] [Google Scholar]
- 34.Ames MM, Selassie CD, Woodson LC, van Loon JA, Hansch C & Weinshilboum RM Thiopurine methyltransferase. Structure-activity relationships for benzoic acid inhibitors and thiophenol substrates. Journal of medicinal chemistry 29, 354–358 (1986). [DOI] [PubMed] [Google Scholar]
- 35.Appell ML et al. Nomenclature for alleles of the thiopurine methyltransferase gene. Pharmacogenetics and genomics 23, 242–248 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zanger UM et al. Comprehensive analysis of the genetic factors determining expression and function of hepatic CYP2D6. Pharmacogenetics 11, 573–585 (2001). [DOI] [PubMed] [Google Scholar]
- 37.Tai H-L, Krynetski EY, Schuetz EG, Yanishevski Y & Evans WE Enhanced proteolysis of thiopurine S-methyltransferase (TPMT) encoded by mutant alleles in humans (TPMT*3A, TPMT*2): mechanisms for the genetic polymorphism of TPMT activity. Proceedings of the National Academy of Sciences 94, 6444–6449 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tai H-L et al. Enhanced proteasomal degradation of mutant human thiopurine S-methyltransferase (TPMT) in mammalian cells: mechanism for TPMT protein deficiency inherited by TPMT*2, TPMT*3A, TPMT*3B or TPMT*3C. Pharmacogenetics 9, 641–650 (1999). [DOI] [PubMed] [Google Scholar]
- 39.Wang L, Nguyen TV, McLaughlin RW, Sikkink LA, Ramirez-Alvarado M & Weinshilboum RM Human thiopurine S-methyltransferase pharmacogenetics. Variant allozyme misfolding and aggresome formation. Proceedings of the National Academy of Sciences of the United States of America 102, 9394–9399 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Salavaggione OE, Wang L, Wiepert M, Yee VC & Weinshilboum RM Thiopurine S-methyltransferase pharmacogenetics: variant allele functional and comparative genomics. Pharmacogenetics and genomics 15, 801–815 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Ujiie S, Sasaki T, Mizugaki M, Ishikawa M & Hiratsuka M Functional characterization of 23 allelic variants of thiopurine S-methyltransferase gene (TPMT*2 - *24). Pharmacogenetics and genomics 18, 887–893 (2008). [DOI] [PubMed] [Google Scholar]
- 42.Okamoto K, Kusano T & Nishino T Chemical nature and reaction mechanisms of the molybdenum cofactor of xanthine oxidoreductase. Current pharmaceutical design 19, 2606–2614 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request. Some data may not be made available because of privacy or ethical restrictions.