

Abstract
The preparation of heterobenzylic amines by a Ni-catalyzed reductive cross-coupling between heteroaryl imines and C(sp3) electrophiles is reported. This umpolung-type alkylation proceeds under mild conditions, avoids the pre-generation of organometallic reagents, and exhibits good functional group tolerance. Mechanistic studies are consistent with the imine substrate acting as a redox-active ligand upon coordination to a low-valent Ni center. The resulting bis(2-imino)heterocycle·Ni complexes can engage in alkylation reactions with a variety of C(sp3) electrophiles, giving heterobenzylic amine products in good yields.
Keywords: cross-coupling, electrochemistry, imines, nickel-catalysis, alkylation
Graphical Abstract
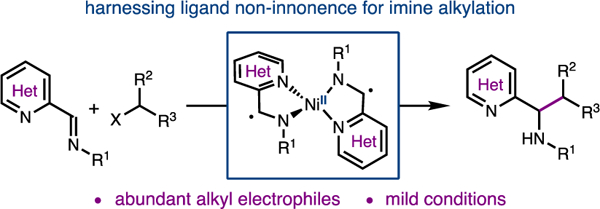
A Ni-catalyzed reductive cross-coupling of heteroaryl imines with C(sp3) electrophiles for the preparation of heterobenzylic amines is reported. Mechanistic studies are consistent with the imine substrate acting as a redox-active ligand upon coordination to a low-valent Ni center.
Introduction
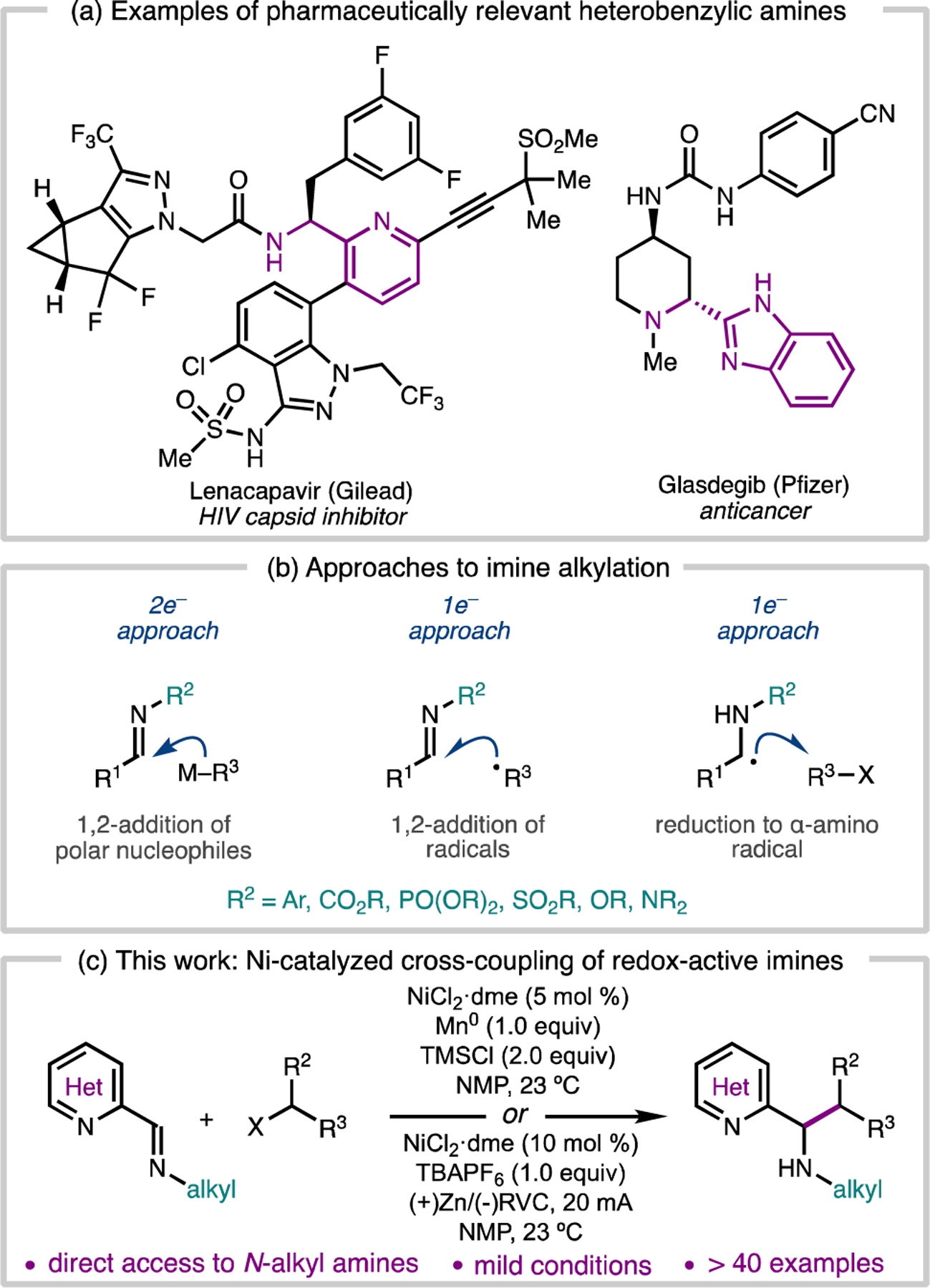
Benzylic amines are common substructures in a variety of natural products, agrochemicals, and pharmaceuticals.1 In particular, heterobenzylic amines serve as important nitrogen-containing scaffolds in medicinal chemistry. Two representative examples are Gilead’s Phase II/III HIV capsid inhibitor Lenacapavir2 and Pfizer’s commercial anticancer agent Glasdegib3 (Figure 1a). Due to broad interest in this structural motif, a variety of synthetic approaches to prepare benzylic amines have been developed. Of these methods, the 1,2-addition of organometallic reagents to imines is one of the most well-established;4 however, pre-generation of sensitive and reactive organometallic reagents and use of activated imine derivatives is typically required (Figure 1b). When simple N-alkylimines are employed, stoichiometric Lewis acid additives can be necessary to enhance the reactivity. Moreover, α-deprotonation of the imine substrate by the basic nucleophiles can be problematic.
Figure 1.
Context for development of Ni-catalyzed reductive imine alkylation.
In order to improve access to benzylic amines, chemists have explored complimentary single electron reactions of imines, including the 1,2-addition of organic radicals to imines5,6,7 and the reductive alkylation of imines via α-amino radicals.8 These reactions often exhibit improved functional group tolerance by avoiding the use of organometallic reagents; however, they typically require activated imine derivatives (e.g. sulfinyl imines, N-arylimines, oximes, hydrazones, or phosphoryl imines) to stabilize the resulting N-centered radicals or facilitate imine reduction. As part of our efforts to broaden the scope of electrophiles for cross-electrophile coupling, we became interested in a mechanistically distinct transition metal-catalyzed reductive alkylation of heterocyclic imines9,10 that leverages the redox non-innocence of 2-iminoheterocycles as ligands on first-row transition metals. This strategy allows for the mild activation of imines for single electron alkylation and provides direct access to N-alkyl heterobenzylic amines by the equivalent of a C(sp3)–C(sp3) coupling reaction. In this report, we describe the development of this method, which provides access to a variety of heterobenzylic N-alkylamines in good yields.
Conjugated nitrogen ligands such as diiminopyridines, α-diimines, and bi- and terpyridines can be electronically non-innocent: their π-systems are able to accept one or two electrons when bound to first-row transition metals.11 For example, spectroscopic, electrochemical, and computational investigations conducted by Wieghardt and coworkers demonstrated that low-valent Cr, Mn, Fe, Co, Ni, and Zn bis(2-imino)pyridine complexes possess ligand-centered radicals (Figure 2a).12 Although the alkylation of ligand backbones has been observed previously,13 this reactivity has not been leveraged for a catalytic cross-coupling.
Figure 2.
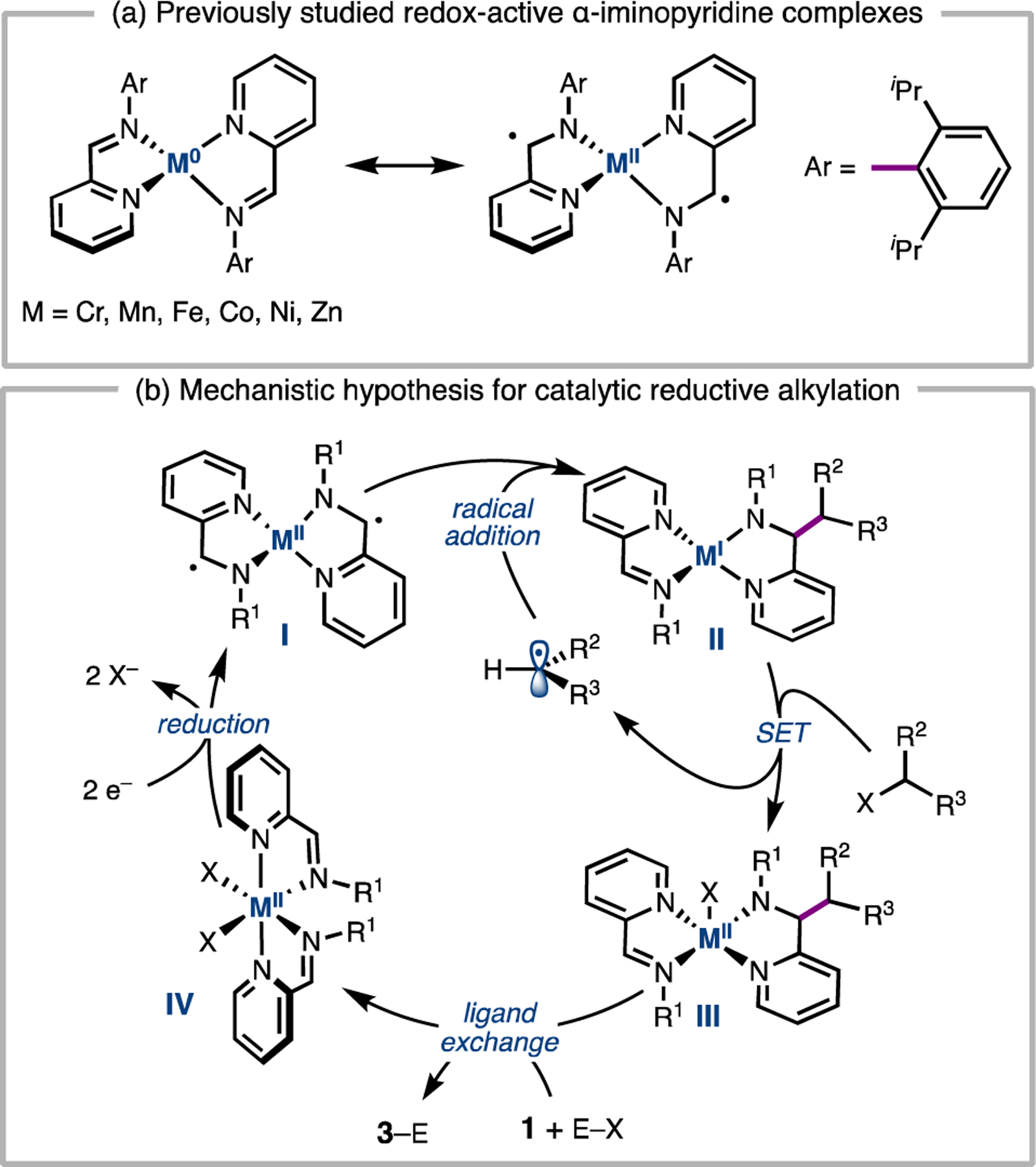
(a) Redox activity of bis(2-imino)pyridine transition metal complexes as candidates for catalysts. (b) Mechanistic framework for catalytic reaction design.
We hypothesized that these redox-active complexes could be considered persistent α-amino radicals, which might react with alkyl radicals to give metal-coordinated imine alkylation products (Figure 2b, I to II). This process could be rendered catalytic if 1) the alkylated product-metal complex II could activate a C(sp3) electrophile to generate an alkyl radical, 2) the product could be liberated from complex III by exchange with imine 1, and 3) the bis(2-iminoheterocycle)MIIX2 complex IV could be reduced by a terminal reductant to regenerate the low-valent complex I. We envisioned that turnover might be facilitated by a Brönsted acid (H–X) or electrophilic reagent (E–X) able to sequester the anionic nitrogen of III.
Results and Discussion
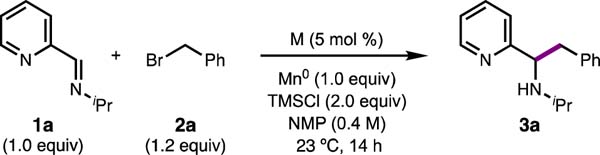
Our investigations commenced with the coupling between (E)-N-isopropyl-1-(pyridin-2-yl)methanimine (1a) and benzyl bromide (2a) in the presence of Mn0 as a stoichiometric reductant, N-methylpyrrolidone (NMP) as the solvent, and trimethylsilyl chloride (TMSCl) as an additive. Product 3a was formed in varying yields for a series of metal dihalide salts (Table 1, entries 1–6). Of the metals evaluated, NiCl2·dme was found to be optimal, providing 3a in 87% yield (Table 1, entry 1). Interestingly, when TMSCl is used, the reaction proceeds in the absence of exogenous catalyst (Table 1, entry 7). It is likely that the combination of Mn0 and TMSCl generates MnCl2, which was previously shown by Wieghardt12 to form a redox-active complex with a similar heteroaryl imine. Use of MnCl2 gives no improvement over just Mn0 and provides 3a in lower yield than NiCl2·dme (entry 6).13,14,15 When TMSCl was omitted from the reaction, 3a was formed in only 39% yield (entry 9). Protic additives such as hexafluoroisopropanol (HFIP) (entry 10) and AcOH (entry 11) were also beneficial, but inferior to TMSCl.16
Table 1.
Optimization of reaction conditionsa
| |||
|---|---|---|---|
| entry | M catalyst | deviation from standard conditions | yield (%)b |
| 1 | NiCl2·dme | none | 87 |
| 2 | CrCl2 | none | 25 |
| 3 | FeBr2 | none | 50 |
| 4 | ZnCl2 | none | 62 |
| 5 | CoCl2 | none | 80 |
| 6 | MnCl2 | none | 68 |
| 7 | none | none | 66 |
| 8 | none | no TMSCl | 19 |
| 9 | NiCl2·dme | no TMSCl | 39 |
| 10 | NiCl2·dme | NMP/HFIP (4:1), no TMSCl | 67 |
| 11 | NiCl2·dme | AcOH (1 equiv), no TMSCl | 69 |
| 12 | NiCl2·dme | Zn0 (2 equiv), no Mn0 | 45 |
| 13 | NiCl2·dme | TDAE (1.5 equiv), no Mn0 | 12 |
| 14 | NiCl2·dme | 1 mol % catalyst | 83 |
| 15 | NiCl2·dme | 0.1 mol % catalyst | 62 |
| 16c,d | NiCl2·dme | Zn anode, RVC cathode, TBAPF6 (1 equiv), 20 mA, no Mn0 or TMSCl | 76 |
| 17c,d | MnCl2 | Zn anode, RVC cathode, TBAPF6 (1 equiv), 20 mA, no Mn0 or TMSCl | 23 |
Reactions conducted under inert atmosphere on 0.3 mmol scale.
Determined by 1H NMR versus an internal standard.
1.2 mmol scale.
1.5 equiv of 2a.
Alternative reductants such as Zn0 and tetrakis(N,N- dimethylamino)ethylene (TDAE) did not perform as well as Mn0 (entries 12–13). The catalyst loading could be dropped to 1 mol % with only a small decrease in yield (entry 14); however, lowering the catalyst loading to 0.1 mol % significantly reduced the yield and showed no improvement over the background Mn-mediated reaction (entry 15 vs. entry 7). To investigate the reaction in the absence of Mn0, a constant current electrolysis protocol was explored for both Ni and Mn salts. The Ni-catalyzed electrolysis provided 3a in good yield (entry 16) while the Mn-catalyzed reaction provided drastically lower yield of 3a (entry 17). Although the reaction could be performed with just Mn0, the addition of NiCl2·dme resulted in higher yields of the imine alkylation product. As a result, the conditions from entry 1 were used to evaluate the scope of the reaction using Mn0 as the terminal reductant.
The scope of the heteroaryl imine coupling partner was investigated using benzyl bromide as the electrophile (Scheme 1). Sterically diverse N-substitution on the imine was well tolerated, affording the products containing nBu, iPr, and tBu groups in high yields (3a–3c). Imines bearing cyclopropyl and cyclobutyl groups, two increasingly popular fragments in drug development,17 provided the coupled products in 67% yield (3d) and 70% yield (3e), respectively. Use of the chiral imine derived from (R)-1-phenylethylamine gave product 3f in good yield, albeit with poor diastereoselectivity. The use of a ketimine substrate did result in product formation (3g); however, the yield was low, likely due to the increased steric hindrance at the site of C–C bond formation.
Scheme 1.
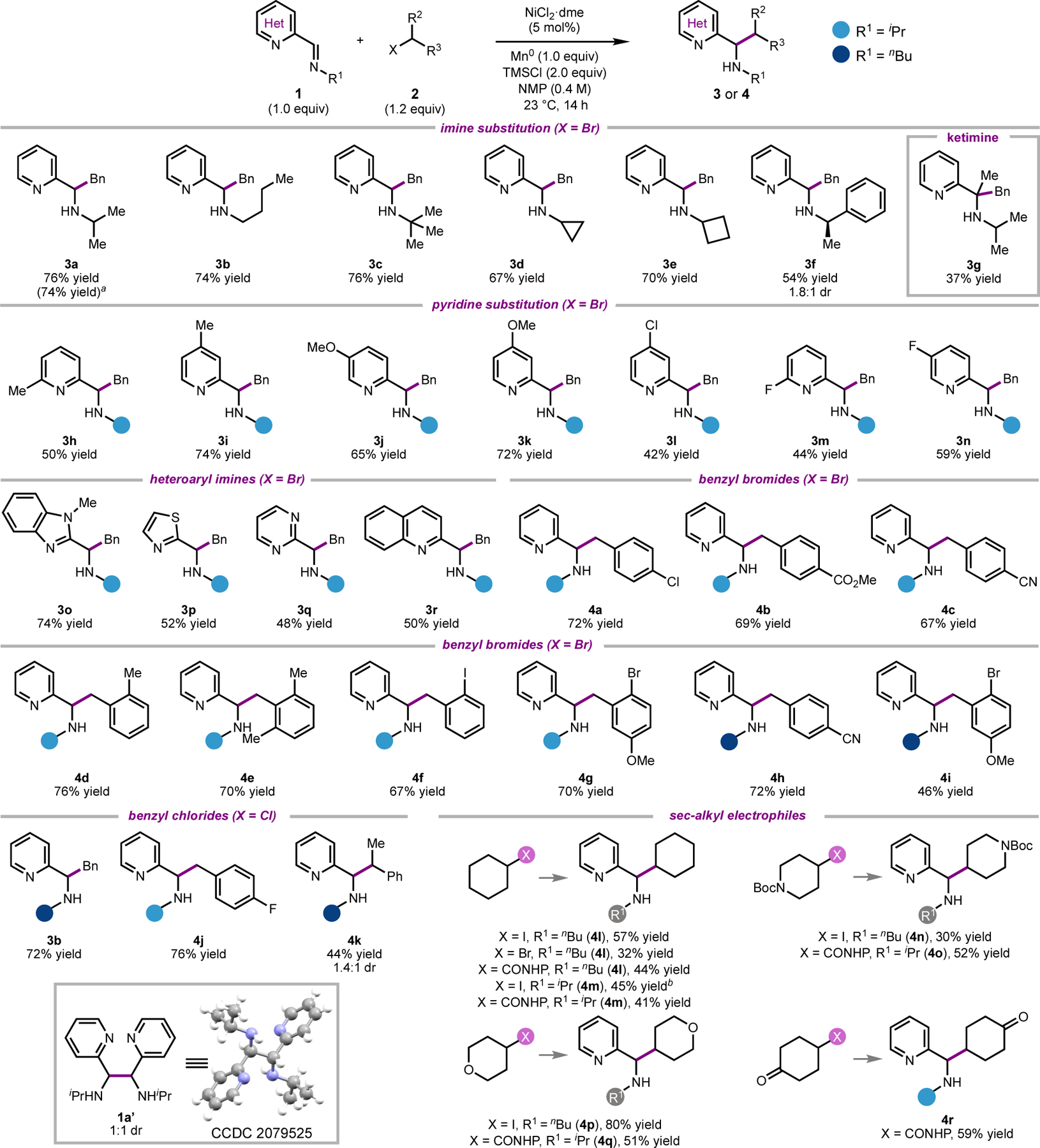
Substrate scope. Reactions were conducted under inert atmosphere on 0.3 mmol scale with isolated yields reported as average of 2 runs. a1.0 mmol scale. b50% yield of homocoupling product 1a’.
Electron donating substituents at the 4- and 5-position of the pyridine were tolerated, furnishing the desired products in generally good yields (3i–3k). Substitution at the 6-position afforded the products in lower yields (3h and 3m), possibly because the substituent hinders coordination of the imine to the Ni-catalyst. In general, substrates bearing electron withdrawing groups at the 5-position gave lower yields of the product. In addition to 2-iminopyridines, several other heterocyclic imines can be employed, including the corresponding benzimidazole (3o), thiazole (3p), pyrimidine (3q), and quinoline (3r).
A range of substituted benzylic bromides could be coupled with imine 1a. Ortho-substituted benzylic bromides coupled smoothly, affording products 4d–4g in good yield. In addition, the reaction exhibits chemoselectivity for the benzylic halide in the presence of aryl iodides and bromides (4f and 4g); these functionalities are frequently incompatible with standard organometallic reagents. Benzylic chlorides perform comparably under standard reaction conditions (3b, X = Cl and 4j). A secondary benzylic chloride also underwent the alkylation, although in reduced yield and with poor diastereoselectivity (4k).
Non-benzylic alkyl halides were also investigated (Scheme 1), which revealed that the reaction yield is influenced by the identity of both the imine and the alkyl electrophile. N-nBu imine 1b could be coupled with cyclohexyl iodide and cyclohexyl bromide to furnish 4l in 57% yield and 32% yield, respectively. Coupling of the N-iPr imine (1a) with cyclohexyl iodide gave 4m in 45% yield; however, it was accompanied by 50% yield of the imine homocoupling product 1a’.18 19 In contrast, use of the corresponding N-hydroxyphthalimide (NHP) ester20 gave 4m in 41% yield but with minimal formation of 1a’. Reaction of 1a or 1b with pyranyl and piperdinyl electrophiles furnished products 4n–4q in modest to good yields. Taken together, these scope studies demonstrate a generally high tolerance for nitrile, ketone, ester, and halide functional groups, which are often incompatible with organomagnesium and organolithium reagents.
Given that deleterious imine homodimerization was observed in some reactions when Mn0 was used as a reductant (Table S1), we sought to drive the reaction electrochemically to eliminate the need for Mn0. Moreover, an electroreductive system removes the mechanistic ambiguity about the identity of the active catalyst (Ni vs. Mn). Under constant current electrolysis using reticulated vitreous carbon (RVC) foam as the cathode and Zn0 metal as a sacrificial anode, alkylation of 1a with 2a proceeded smoothly (Scheme 2). We were pleased to find that several substrates that gave low yields under the Mn0 conditions performed better under the electroreductive conditions. For example, when 1a was coupled with iodocyclohexane under standard conditions, product 4m was formed in 45% yield and was accompanied by 50% yield (Figure S2) of imine dimer 1a’ (see Scheme 1). Under the electroreductive conditions, 4m was produced in 59% yield on a 1.2 mmol scale; no 1a’ was observed. Alkylation products from primary (4s, 4v, and 4w) and tertiary (4u) iodides, could also be formed in good yield under the electroreductive conditions (Scheme 2). Both reactions proceeded in <20% yield when Mn0 was used as a reductant.
Scheme 2.
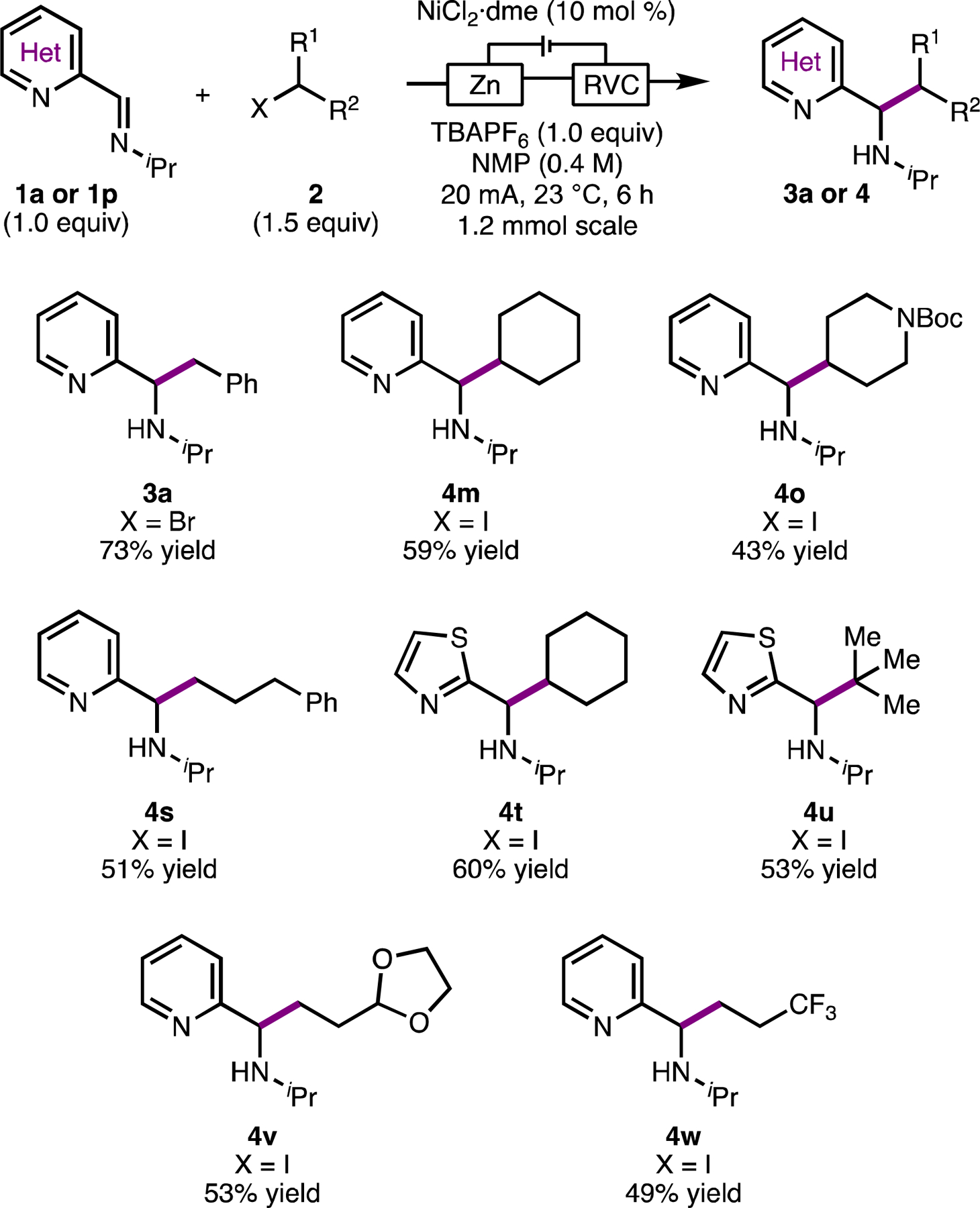
Representative scope of electroreductive imine alkylation. Reactions conducted under inert atmosphere on a 1.2 mmol scale.
Since the electroreductive coupling demonstrates that Ni salts can catalyze the alkylation of 2-iminopyridines, we carried out a series of mechanistic experiments studying the Ni system. Initial mechanistic investigations focused on the substrate-catalyst complexes proposed to be key catalytic intermediates (Scheme 3). Non-chelating substrates like benzaldehyde-derived imine 5 and isomeric pyridyl imine 6 failed to couple under standard conditions, demonstrating the importance of forming a bidentate substrate-metal complex (Scheme 3a). Bis(2-iminopyridine)·Ni complex 9 was prepared by the addition of imine 1a (2.0 equiv) to Ni(cod)2 (1.0 equiv);12 subsequent addition of benzyl bromide provided 3a in 53% yield, providing support for reduced Ni complex 9 as a competent species in the catalytic cycle (Scheme 3b).
Scheme 3.
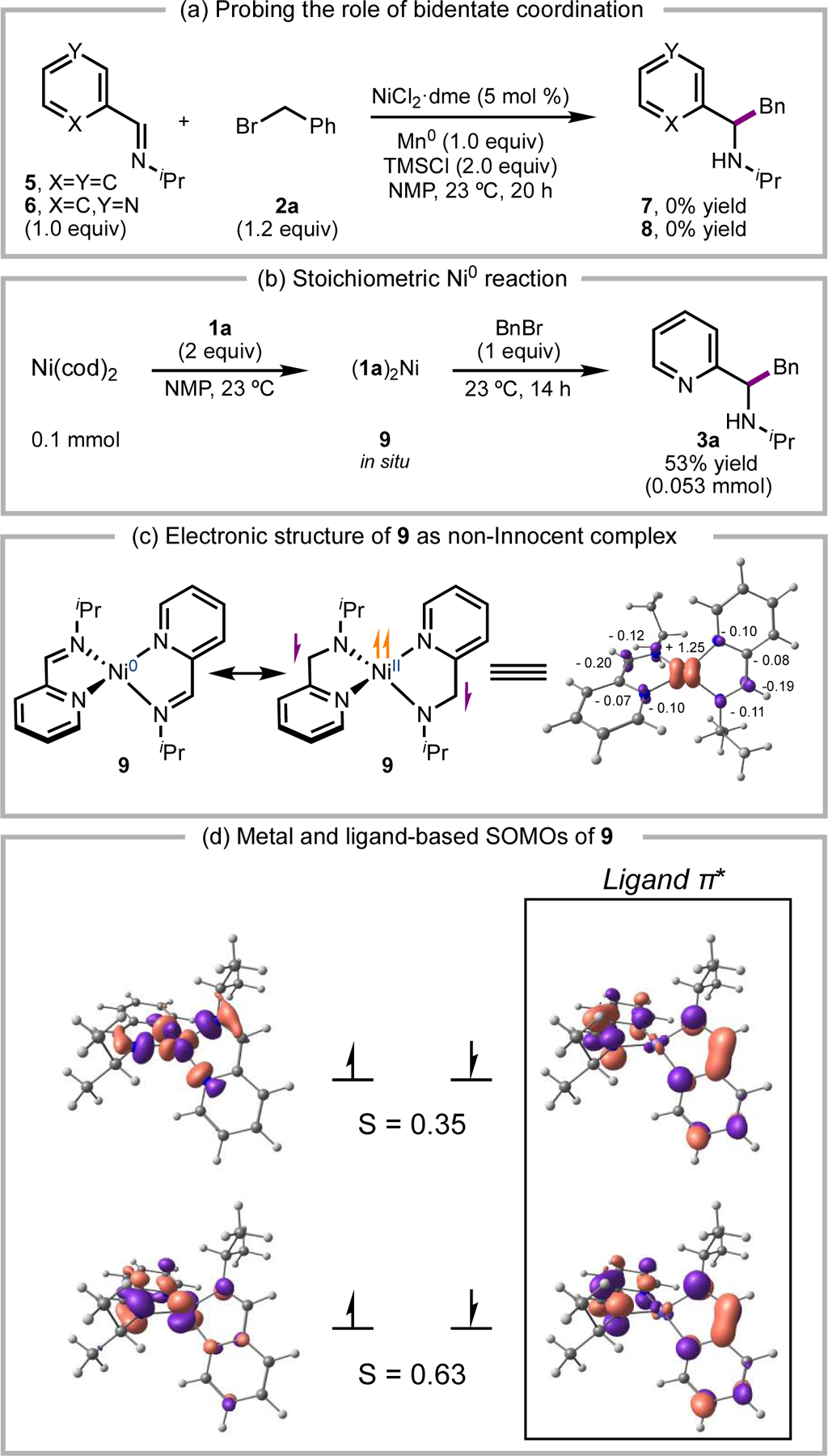
(a) Attempted alkylation with substrates that cannot undergo bidentate coordination to Ni catalyst. (b) Stoichiometric alkylation reaction with 9 prepared from Ni(COD)2. (c) Calculated electronic structure of 9 with spin density plot labelled with Loewdin spin population values for atoms with significant radical character. (d) Qualitative molecular orbital diagram of BS(2,2) 9 and corresponding magnetic orbitals with corresponding spatial overlap (S) for orbitals with S < 0.999.
In agreement with Wieghardt and coworkers,12 computational studies suggest that the electronic structure of the formally Ni0 complex 9 is best described as a NiII center with antiferromagnetically coupled ligand-based radicals. DFT calculations of 9 at the B3LYP/def2-TZVP level of theory show the broken symmetry solution BS(2,2) being lower in energy than the closed-shell or high spin solutions (Scheme 3c).21,22 A qualitative molecular orbital diagram of the magnetic orbitals reveals seven orbitals with significant d contribution (Figure S30). Upon closer examination of the electronic structure, there are two ligand-based singly-occupied molecular orbitals (SOMOs) as the imine π* orbitals (Scheme 3d). Using the Yamaguchi equation, the spin-spin coupling constant (J) between the metal-based SOMOs and the ligand-based SOMOs was calculated to be J = −777 cm-1.23 These data support our hypothesis that the ligand non-innocence of reduced catalyst-substrate complexes such as 9 allows for facile access to persistent α-amino radical intermediates (Figure 2b).
We sought to investigate the redox properties of (1a)2NiCl2 (10) to confirm that reduction to the low-valent complex 9 is possible under the reaction conditions. Using cyclic voltammetry (CV), the reduction potential of free 1a was compared to the reduction potentials of corresponding in situ generated complexes (1a)2NiCl2 (10) and (1a)2MnCl2 (11) (Figure 3a). Complex 11 (E1/2 = −1.82 V vs. Fc/Fc+ in NMP) is more challenging to reduce than Ni complex 10 (E1/2 = −1.43 V vs. Fc/Fc+ in NMP). The free imine 1a has a reduction potential (Ep/2) of −2.65 V vs. Fc/Fc+ in NMP, which is significantly more negative than that of either complex 10 or 11. Complexation of 1a with a non-redox-active Lewis acid such as MgBr2 does not significantly change the potential of imine reduction (Ep/2 = −2.55 V vs. Fc/Fc+ in NMP) (Figure 3a). The significant anodic shift of the reduction potentials and the increased reversibility of the redox events demonstrate that imine coordination to Ni and Mn facilitates reduction and stabilizes the ligand-centered radicals. We note that reduction of 10 is 420 mV more anodic than 11 indicating the formation of proposed intermediate I (Figure 2b) is more thermodynamically favorable, which may correlate with the improved product yields when catalytic Ni is included.
Figure 3.
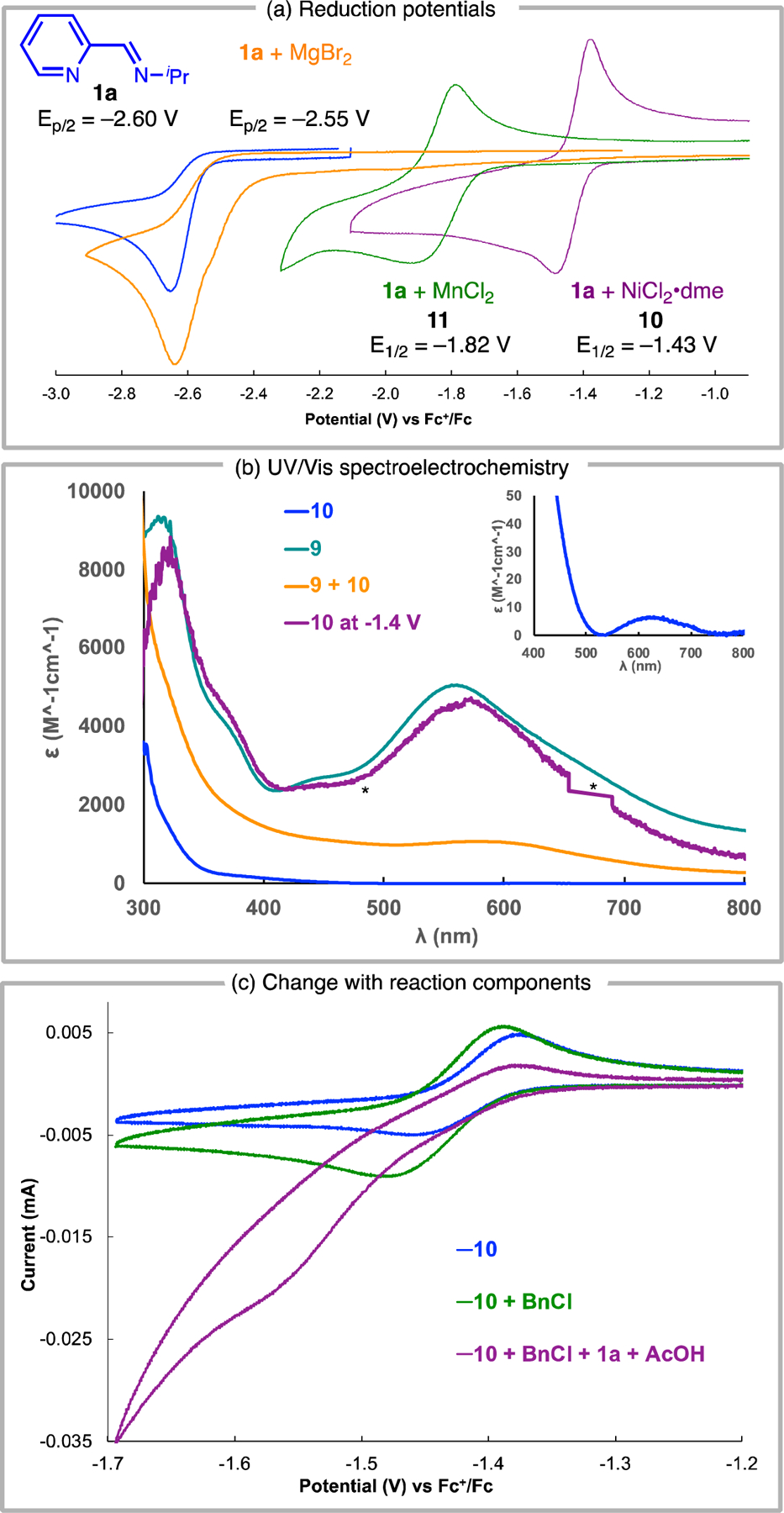
Electroanalytical investigations. (a) CV of 1a (blue) and complexes with Ni (1 mM NiCl2·dme and 20 mM 1a, purple), Mn (1 mM MnCl2 and 20 mM 1a, green) and Mg (2 mM MgCl2 and 100mM 1a, orange). CVs measured with 0.1 M TBAPF6 in NMP at 25°C with 100 mV/s scan rate. (b) UV/Vis spectra of 10 (blue); 10 after holding at −1.4 V (vs. Fc/Fc+; 0.1 M TBAPF6 in NMP, purple) for 2.5 minutes; independently synthesized 9 (teal); NiI complex obtained from comproportionation of 9 and 10 (orange, 9ox). Spectra were baseline corrected to be zero at 860 nm, and * indicates signal saturation inherent to the light source and detector used for the experiment. Inset: enlargement of 400–800 nm region for 10. See Supporting Information for individual spectra and calculations of ε. (c) 10 (1.0 mM, blue); 10 with 100 mM benzyl chloride (green); 10 with 40 mM 1a and 150 mM AcOH (purple). CVs acquired with 0.1 M TBAPF6 in NMP at 25°C with 50 mV/s scan rate.
It was unclear from the CV alone whether the observed reduction of (1a)2NiCl2 (10) corresponded to a one-electron or a two-electron process.24 To investigate the identity of the species generated upon reduction, UV/Vis spectroelectrochemical analysis of 10 was performed at varying potentials (Figure S24). At −1.4 V vs. Fc/Fc+, a species develops with a UV/Vis spectrum that is consistent with that of an independently prepared sample of (1a)2Ni (9) (Figure 3b). Alternatively, mixing 1 equiv each of 9 and 10 results in comproportionation to the NiI complex; this species has a different spectroscopic profile, and consistent with Wieghardt’s prior studies,12 computational and EPR studies suggest that this complex does have not significant radical character on the ligand backbone (Figure S3). These experiments suggest that at potentials accessible under the catalytic reaction conditions, complex 10 undergoes two electron reduction to generate 9.25,26
To probe whether reductively generated 9 can react with alkyl electrophiles, CVs of complex 10 in the presence of benzyl chloride were acquired. Scanning in the negative direction, the CV of a mixture of 10 (1 equiv) and benzyl chloride (100 equiv) shows a cathodic shift and increase in peak current relative to complex 10 alone (Figure 3c). The cathodic shift indicates that, upon reduction, complex 10 does not react with benzyl chloride through a simple EC mechanism, but instead through a mechanism that likely involves intermediate chemicals steps such as loss of chloride ligands. Kinetic analysis of the reaction with benzyl chloride reveals a second order rate constant k = 1.8×10−1 M−1s−1 (Supporting Information section 6.3).27 Addition of AcOH (150 equiv) and additional 1a (50 equiv) results in a catalytic wave (Figure 3c) that is not observed in the absence of BnCl or excess 1a (Figure S13). AcOH was used for these studies because it was found to give reasonable alkylation yields (Table 1, entry 11) and had greater stability than TMSCl in the electrochemical cell.
Conclusion
In conclusion, the Ni-catalyzed reductive cross-coupling of (2-imino)heterocycles with C(sp3) alkyl electrophiles has been reported. The reaction occurs under mild conditions and is tolerant of a variety of functional groups, including N- and S-heterocyclic imine coupling partners. Mechanistic studies support the formation of low-valent bis(2-imino)pyridine·Ni complexes as persistent ligand-centered radical species that can react with alkyl electrophiles and be leveraged for catalytic C–C bond formation.
Supplementary Material
Acknowledgements
Dr. Scott Virgil and the Caltech Center for Catalysis and Chemical Synthesis are gratefully acknowledged for access to analytical equipment. Fellowship support was provided by the Swiss National Science Foundation (M. B.). S.E.R. acknowledges financial support from the NIH (R35GM118191). The authors would also like to thank Dr. Nathan Dalleska and the Resnick Sustainability Institute’s Water and Environmental Lab for elemental analysis of commercial manganese; Dr. Mona Shahgholi for assistance with mass spectrometry measurements; Dr. Paul Oyala for assistance with X-band EPR measurements; Dr. David E. Hill for invaluable assistance with electroanalytical and spectroelectrochemical experiments; as well as Z. Jaron Tong for helpful discussions on DFT calculations and non-innocent ligand complexes.
Footnotes
This paper is dedicated in memory of our colleague and friend Prof. Robert H. Grubbs.
Institute and/or researcher Twitter usernames: @sarah_reisman
References
- 1.a) Lawrence SA, Amines : Synthesis, Properties and Applications, Cambridge University Press, 2004; [Google Scholar]; b) Lewis JR, Nat. Prod. Rep 2001, 18, 95–128; [DOI] [PubMed] [Google Scholar]; c) Carey JS, Laffan D, Thomson C, Williams MT, Org. Biomol. Chem 2006, 4, 2337–2347; [DOI] [PubMed] [Google Scholar]; d) Hili R, Yudin AK, Nat Chem Biol 2006, 2, 284–287. [DOI] [PubMed] [Google Scholar]
- 2.Link JO, Rhee MS, Tse WC, Zheng J, Somoza JR, Rowe W, Begley R, Chiu A, Mulato A, Hansen D, Singer E, Tsai LK, Bam RA, Chou C-H, Canales E, Brizgys G, Zhang JR, Li J, Graupe M, Morganelli P, Liu Q, Wu Q, Halcomb RL, Saito RD, Schroeder SD, Lazerwith SE, Bondy S, Jin D, Hung M, Novikov N, Liu X, Villaseñor AG, Cannizzaro CE, Hu EY, Anderson RL, Appleby TC, Lu B, Mwangi J, Liclican A, Niedziela-Majka A, Papalia GA, Wong MH, Leavitt SA, Xu Y, Koditek D, Stepan GJ, Yu H, Pagratis N, Clancy S, Ahmadyar S, Cai TZ, Sellers S, Wolckenhauer SA, Ling J, Callebaut C, Margot N, Ram RR, Liu Y-P, Hyland R, Sinclair GI, Ruane PJ, Crofoot GE, McDonald CK, Brainard DM, Lad L, Swaminathan S, Sundquist WI, Sakowicz R, Chester AE, Lee WE, Daar ES, Yant SR, Cihlar T, Nature 2020, 584, 614–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Munchhof MJ, Li Q, Shavnya A, Borzillo GV, Boyden TL, Jones CS, LaGreca SD, Martinez-Alsina L, Patel N, Pelletier K, Reiter LA, Robbins MD, Tkalcevic GT, ACS Med. Chem. Lett 2012, 3, 106–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Bloch R, Chem. Rev 1998, 98, 1407–1438; [DOI] [PubMed] [Google Scholar]; b) Marcantoni E, Petrini M, in Comprehensive Organic Synthesis (Second Edition) (Ed.: Knochel P), Elsevier, Amsterdam, 2014, pp. 344–364. [Google Scholar]
- 5.a) For reviews on radical additions to imines under thermal conditions, see: Friestad GK, Tetrahedron 2001, 57, 5461–5496; [Google Scholar]; b) Miyabe H, Yoshioka E, Kohtani S, Current Organic Chemistry 2010, 14, 1254–1264. [Google Scholar]; (c) Tauber J, Imbri D, Opatz T, Molecules 2014, 19, 16190–16222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) For reviews on addition to imines under photoredox catalysis, see: Cullen STJ, Friestad GK, Synthesis 2021, 53, 2319–2341; [Google Scholar]; b) Zhao J-J, Zhang H-H, Yu S, Synthesis 2021, 53, 1706–1718. [Google Scholar]
- 7.For a recent method involving free radical addition to N-alkyliminium ions, see: Kumar R, Flodén NJ, Whitehurst WG, Gaunt MJ, Nature 2020, 581, 415–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For a review on the use of photoredox catalysis to generate α-amino radicals from imines, see: Leitch JA, Rossolini T, Rogova T, Maitland JAP, Dixon DJ, ACS Catal 2020, 10, 2009–2025. [Google Scholar]
- 9.For a complementary Ni-catalyzed imine alkylation, see Heinz C, Lutz JP, Simmons EM, Miller MM, Ewing WR, Doyle AG, J. Am. Chem. Soc 2018, 140, 2292–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.For a Ni-catalyzed addition of free radicals to glyoxylate-derived sulfinnimines, see: Ni S, Garrido-Castro AF, Merchant RR, de Gruyter JN, Schmitt DC, Mousseau JJ, Gallego GM, Yang S, Collins MR, Qiao JX, Yeung K-S, Langley DR, Poss MA, Scola PM, Qin T, Baran PS, Angew. Chem. Int. Ed 2018, 57, 14560–14565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Jacquet J, Desage-El Murr M, Fensterbank L, ChemCatChem 2016, 8, 3310–3316; [Google Scholar]; b) Luca OR, Crabtree RH, Chem. Soc. Rev 2013, 42, 1440–1459; [DOI] [PubMed] [Google Scholar]; c) Praneeth VKK, Ringenberg MR, Ward TR, Angew. Chem. Int. Ed 2012, 51, 10228–10234; [DOI] [PubMed] [Google Scholar]; d) Lyaskovskyy V, de Bruin B, ACS Catal 2012, 2, 270–279; [Google Scholar]; e) Chirik PJ, Wieghardt K, Science 2010, 327, 794–795. [DOI] [PubMed] [Google Scholar]
- 12.Lu CC, Bill E, Weyhermüller T, Bothe E, Wieghardt K, J. Am. Chem. Soc 2008, 130, 3181–3197. [DOI] [PubMed] [Google Scholar]
- 13.a) Bailey PJ, Dick CM, Fabre S, Parsons S, Yellowlees LJ, Dalton Trans 2006, 1602–1610; [DOI] [PubMed] [Google Scholar]; b) Riollet V, Copéret C, Basset J-M, Rousset L, Bouchu D, Grosvalet L, Perrin M, Angew. Chem. Int. Ed 2002, 41, 3025–3027; [DOI] [PubMed] [Google Scholar]; c) Kaupp M, Stoll H, Preuss H, Kaim W, Stahl T, Van Koten G, Wissing E, Smeets WJJ, Spek AL, J. Am. Chem. Soc 1991, 113, 5606–5618. [Google Scholar]
- 14.(a) Solomon MB, Chan B, Kubiak CP, Jolliffe KA, D’Alessandro DM, Dalton Trans 2019, 48, 3704–3713; [DOI] [PubMed] [Google Scholar]; b) Morrison MM, Sawyer DT, Inorg. Chem 1978, 17, 333–337; [Google Scholar]; c) Rao JM, Hughes MC, Macero DJ, Inorg. Chim. Acta 1976, 18, 127–131. [Google Scholar]
- 15. Trace metal analysis by ICP-MS found that the Mn0 source contains 54 ppm total Ni species. However, this would represent a very low concentration of Ni catalyst (<0.005 mol %). See Supporting Information section 11 for ICP-MS sample preparation and calculations.
- 16. Alternative additives such as phenol, acetic anhydride, trifluoroacetic anhydride, and benzoic acid led to no further improvement.
- 17.Talele TT, J. Med. Chem 2016, 59, 8712–8756. [DOI] [PubMed] [Google Scholar]
- 18.a) Less reactive alkyl halides with hindered imines produce varying quantities of 1a’ (see Figure S2 for 1a’ production across several substrates). Using a radical precursor that is more facile to reduce, like NHP esters, enhances the rate of alkyl radical generation and favors cross-coupling over sp2–sp2 homocoupling. For examples, see: Faugeroux V, Genisson Y, Current Organic Chemistry 2008, 12, 751–773; [Google Scholar]; b) Hulley EB, Wolczanski PT, Lobkovsky EB, J. Am. Chem. Soc 2011, 133, 18058–18061; [DOI] [PubMed] [Google Scholar]; c) Inoue S, Yan Y-N, Yamanishi K, Kataoka Y, Kawamoto T, Chem. Commun 2020, 56, 2829–2832; [DOI] [PubMed] [Google Scholar]; d) Pokhriyal D, Heins SP, Sifri RJ, Gentekos DT, Coleman RE, Wolczanski PT, Cundari TR, Fors BP, Lancaster KM, MacMillan SN, Inorg. Chem 2021, 60, 18662–18673. [DOI] [PubMed] [Google Scholar]
- 19. All data for X-ray crystal structures have been deposited in the CCDC, under the following deposition numbers: 1a’ = 2079525, 10 = 2117478, 11 = 2117477.
- 20.a) Huihui KMM, Caputo JA, Melchor Z, Olivares AM, Spiewak AM, Johnson KA, DiBenedetto TA, Kim S, Ackerman LKG, Weix DJ, J. Am. Chem. Soc 2016, 138, 5016–5019; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Suzuki N, Hofstra JL, Poremba KE, Reisman SE, Org. Lett 2017, 19, 2150–2153;. [DOI] [PMC free article] [PubMed] [Google Scholar]; For a recent review of Ni-catalyzed cross-couplings with NHP esters, see: Konev MO, Jarvo ER, Angew. Chem. Int. Ed 2016, 55, 11340–11342. [DOI] [PubMed] [Google Scholar]
- 21.a) For additional investigations into the electronic structure of redox-active first-row transition metal bis-iminopyridine complexes, see reference 12 as well as: Lu CC, Bill E, Weyhermüller T, Bothe E, Wieghardt K, Inorg. Chem 2007, 46, 7880–7889; [DOI] [PubMed] [Google Scholar]; b) Mondal A, Weyhermüller T, Wieghardt K, Chem. Commun 2009, 6098–6100; [DOI] [PubMed] [Google Scholar]; c) Tsvetkov NP, Chen C-H, Andino JG, Lord RL, Pink M, Buell RW, Caulton KG, Inorg. Chem 2013, 52, 9511–9521; [DOI] [PubMed] [Google Scholar]; d) Sengupta D, Ghosh P, Chatterjee T, Datta H, Paul ND, Goswami S, Inorg. Chem 2014, 53, 12002–12013. [DOI] [PubMed] [Google Scholar]
- 22. DFT calculations were performed using the ORCA software package at the B3LYP/def2-TZVP level of theory. See Supporting Information for optimization, frequency, broken symmetry, and property calculations.
- 23.Soda T, Kitagawa Y, Onishi T, Takano Y, Shigeta Y, Nagao H, Yoshioka Y, Yamaguchi K, Chem. Phys. Lett 2000, 319, 223–230. [Google Scholar]
- 24. The peak-to-peak separation was determined to be ~106 mV for 10. Given this deviation from theoretical, we cannot draw a conclusion from the CV alone about whether the reduction is a one or two electron process.
- 25.Mn0(S) -> MnII(NMP) is estimated to be ~ −1.8 V vs. Fc/Fc+ by converting the known value of −1.185 V vs. SHE for Mn to MnII. The reduction of 10 was found to occur at −1.4 V vs. Fc/Fc+, and therefore should be in the reducing window of Mn0. (a) For standard reduction potential values, see: Haynes WM, CRC Handbook of Chemistry and Physics. [Electronic Resource] : A Ready-Reference Book of Chemical and Physical Data, CRC Press, 2018. [Google Scholar]; b) For converting SCE potentials to 0.1 M TBAPF6 in DMF vs. Fc/Fc+, see: Lin Q, Dawson G, Diao T, Synlett 2021, 32, 1606–1620. [Google Scholar]
- 26.The facile two electron reduction of 10 is in contrast to recent studies of (Phen)NiBr2 complexes, which undergo one electron reduction at similar potentials: Lin Q, Diao T, J. Am. Chem. Soc 2019, 141, 17937–17948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sandford C, Fries LR, Ball TE, Minteer SD, Sigman MS, J. Am. Chem. Soc 2019, 141, 18877–18889. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.