

Abstract
The genetic etiology of autism spectrum disorder (ASD) is multifactorial, but how combinations of genetic factors determine risk is unclear. In a large family sample, we show that genetic loads of rare and polygenic risk are inversely correlated in cases and greater in females than in males, consistent with a liability threshold that differs by sex. De novo mutations (DNMs), rare-inherited variants and polygenic scores were associated with various dimensions of symptom severity in children and parents. Parental age effects on risk for ASD in offspring were attributable to a combination of genetic mechanisms, including DNMs that accumulate in the paternal germline and inherited risk that influences behavior in parents. Genes implicated by rare variants were enriched in excitatory and inhibitory neurons compared to genes implicated by common variants. Our results suggest that a phenotypic spectrum of ASD is attributable to a spectrum of genetic factors that impact different neurodevelopmental processes.
The major risk factors for autism spectrum disorder (ASD) are genetic and include a variety of rare and common alleles, including rare de novo copy number variants (CNVs)1 or protein-truncating SNPs and indels of large effect2 and common polygenic risk that is measured as the sum of thousands of common alleles with small effects3. Despite the success in identifying and characterizing multiple types of genetic risk, there is no one variant, gene or polygenic score (PS) that has a high predictive value for an ASD diagnosis. Even CNVs with large effect sizes (OR > 30) for ASD present with variable psychiatric traits4, and risk is attributable to a combination of rare and common variation5,6.
Sex is also a major genetic factor that influences ASD risk. Males are diagnosed with ASD more frequently than are females at a ratio of 4:1. A small proportion of cases are associated with X-linked variants7, but the male preponderance of ASD is not largely explained by genetic variation on sex chromosomes. We and others have hypothesized that it may instead be explained by sex differences in the effects of autosomal variants8–10. This hypothesis is supported by previous studies showing that females with ASD have a greater burden of rare CNVs1,11,12 and gene mutations13,14. However, gene-by-sex interactions in ASD have not been examined systematically.
Previous genetic studies have been focused on defining new categories of rare variant risk from DNA sequencing or by improving the statistical power of genome-wide association studies (GWAS). How combinations of multiple genetic factors contribute to risk and clinical presentation is not known. Here we investigate, in a large dataset of whole genomes and exomes, the combined contributions of de novo, rare inherited and polygenic risk to ASD. We show that the genetic architecture of ASD varies as a spectrum of rare and common variation, each having distinct phenotypic correlates and differential effects in males and females.
Results
Defining multiple components of genetic risk.
We investigated the combined effects of multiple genetic factors, detectable by genome sequencing or a combination of exome sequencing and SNP genotyping, on risk for ASD. We focused on several factors that have established associations with case status, such as de novo protein-truncating (dnLoF) and missense (dnMIS) mutations1,2 and rare inherited variants15,16 that disrupt genes (inhLoF) and polygenic scoring models that have been associated with ASD case status, including PS for ASD (PSASD), schizophrenia (PSSZ) and educational attainment (PSEA)17,18 (see Methods for details on the selection of genetic factors).
We confirmed genetic associations by whole genome analysis of 37,375 individuals from 11,313 ASD families (12,270 cases, 5,190 typically-developing siblings, and 19,917 parents). The sample was composed of three datasets, including whole genome sequencing (WGS) of cohorts from UCSD (https://sebatlab.org/reach-project) and the Simons Simplex Collection (SSC) and exomes and SNP genotyping from the SPARK study19 (see Methods and Supplementary Tables 1 and 2). SNP, indel, structural variant (SV), and DNM calling, and calculation of ancestry principal components were performed using functionally equivalent pipelines for each dataset as described in the Methods, and PSs were calculated using the polygenic scoring method SbayesR20. Rare variants were annotated for gene functional constraint. Analysis of protein-coding loss of function (LoF) and cis-regulatory (CRE) variants was restricted to variant-intolerant genes (LOEUF < 0.37) and analysis of missense variants was restricted to those with missense badness (MPC) scores > 2.
Association tests were performed for case-control differences in DNM burden. Association of inherited risk was tested by transmission disequilibrium test (TDT)15. Common variant associations were tested by a polygenic TDT (pTDT) that measures overtransmission of risk alleles as the deviation of the offspring PS from the average PS of the parents17. We confirmed that de novo synonymous variants were not associated with ASD in the combined sample (Extended Data Fig. 1a), and rates of DNMs were not influenced by batch effects or other confounders (Extended Data Fig. 1b,c). Results confirm significant contributions from genetic factors, including de novo loss of function (dnLoF) and missense (dnMIS) mutations (Fig. 1a and Supplementary Tables 3–6). TDT confirmed the associations of rare inherited protein-truncating SNVs (inhLoF) and SVs (LoFSV) (Fig. 1b and Supplementary Tables 7–9). SVs that disrupt cis-regulatory variants (CRE-SVs) of constrained genes showed differential transmission in cases and controls, but the TDT test did not reach statistical significance in cases. Polygenic scores PSASD, PSSZ and PSEA were all significantly associated with ASD (Fig. 1c and Supplementary Table 10), and the polygenic contribution to ASD was consistent across all three cohorts (Supplementary Table 11).
Figure 1 |. Risk for ASD is attributable to multiple genetic factors including DNMs, rare inherited variants and polygenic risk.
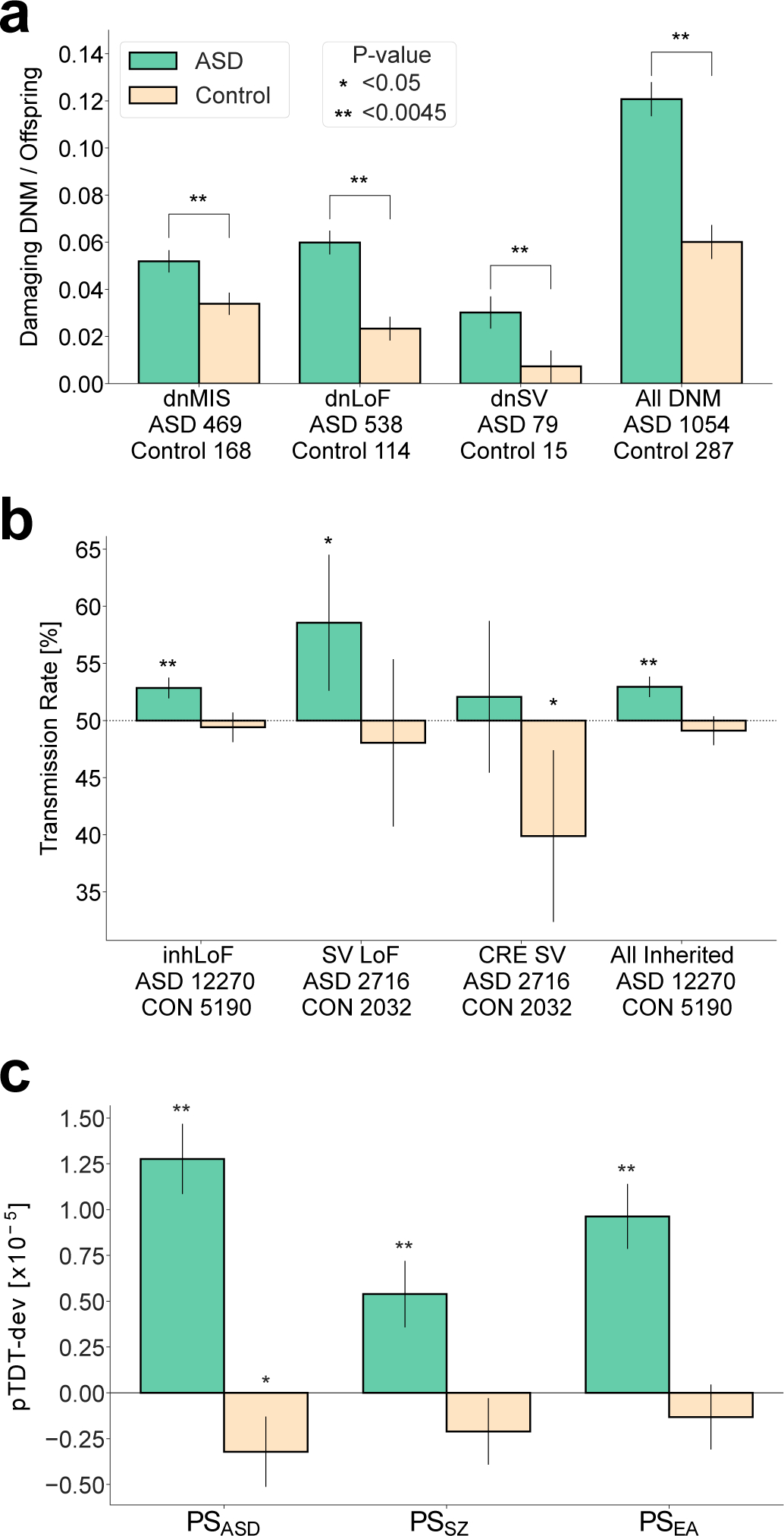
Multiple genetic factors that have been previously associated with ASD were confirmed in our combined sample. “**” denotes associations that were significant after correction for 11 tests (P < 0.0045). Error bars represent the 95% confidence intervals. a, Damaging DNMs in genes that are functionally constrained (LOEUF < 0.37 and MPC ≥ 2), including missense variants (dnMIS), and protein-truncating SNVs and indels (dnLoF) and SVs (dnSV), occur at higher frequencies in cases than in sibling controls. P-values were based on two-sided t-tests. b, Protein-truncating SNVs and indels (inhLOF) and SVs (SVLoF) and non-coding SVs that disrupt cis-regulatory elements (CRE-SVs) were associated with ASD based on a TDT test. c, Polygenic TDT (pTDT) was significant for all three polygenic scores for autism (PSASD), schizophrenia (PSSZ), and educational attainment (PSEA). Rare variant associations (a,b) were tested in the full sample (n = 37,375). Polygenic pTDT association was tested in samples of European ancestry (n = 25,391). Results for a-c and full lists of rare de novo and inherited variants in constrained genes are provided in Supplementary Tables 3–10.
We examined the combined effects of rare and common variation. To ensure that genetic factors were ascertained consistently across the three cohorts, analysis was restricted to six categories that are detectable in exome and WGS with comparable sensitivity: dnLoF, dnMIS, inhLoF and polygenic scores (PSASD, PSSZ, PSEA). SVs and CNVs, variant types that cannot be ascertained comparably in exome and WGS datasets were not included. To minimize ancestry as a confounder in PSs, analysis was restricted to a subset of 7,181 families (n = 25,391 individuals) with parents and offspring that have confirmed European ancestry.
The contribution of each factor individually and the additive contributions of multiple factors was estimated by multivariable regression (Fig. 2a). The variance explained by individual genetic factors in this study was consistent with previous studies. Polygenic risk explained 2% of the variance in case status in the combined sample (Supplementary Table 12), consistent with the ~2% of variance explained by polygenic risk in the most recent GWAS meta-analysis3. The combined contribution of rare variants was similar, also explaining 2% of the variance in case status (Fig. 2a). Our results indicate that rare variants and polygenic risk form two major components of the genetic architecture of ASD, and the additive effects of all factors combined could be quantified in a single model (r2 = 4%, Fig. 2a and Supplementary Table 12). We applied the estimates of the multivariable regression to create composite genetic risk scores of multiple factors, including a rare variant risk score (RVRS) for the combination of dnMIS, dnLoF and inhLoF, a common variant risk score (CVRS) for the combination of PSASD, PSSZ and PSEA, and a genomic risk score (GRS) for the combination of all six genetic factors. For each, we calculated the case-control odds ratios at multiple score thresholds (Fig. 2b and Supplementary Table 13), and we found that, across the full distribution of risk scores, the GRS detects an effect size that is 40% stronger than effect sizes for RVRS or CVRS (Supplementary Table 14).
Figure 2 |. Multivariable regression of six genetic factors to create a composite genomic risk score.
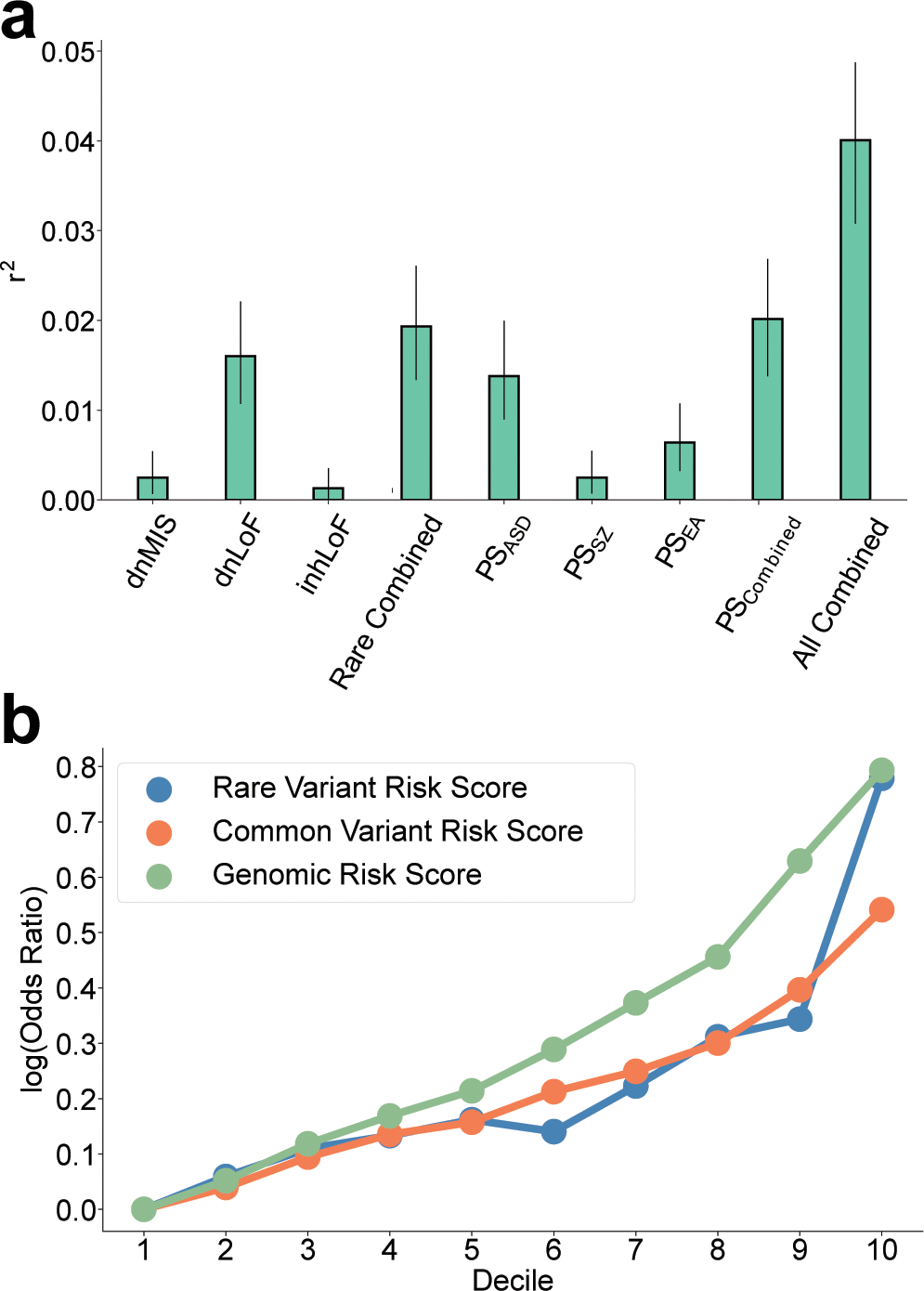
a, Variance in case status explained (r2 and 95% CI) by each genetic factor individually and in combination. Combined effects of rare variants (Rare combined) polygenic scores (PS combined) and all genetic factors (All combined) were estimated in the European-ancestry sample (n = 25,391) by multivariable logistic regression controlling for sex, cohort and principal components. b, log10 odds-ratios of case/control proportions for the composite genetic risk scores RVRS, CVRS, and GRS at multiple thresholds (deciles). Across all thresholds, effect sizes for the GRS was 41–42% greater than for RVRS or CVRS alone. See results in Supplementary Tables 13 and 14.
Sex differences in genetic load.
Sex differences in genetic load were evident for both polygenic and rare variant risk (Fig. 3a,b). Female cases had significantly increased CVRS (P = 5.96 × 10−4; Fig. 3b) and RVRS (P = 4.32 × 10−7; Fig. 3a) compared to male cases. A similar trend was seen for polygenic risk in controls, with female controls having a greater CVRS than males (P = 0.026; Fig. 3b). These results are consistent with a “female protective effect” in which females in the general population tolerate a greater genetic load of ASD risk, and likewise a greater genetic load is required for females to meet diagnostic criteria for ASD case status21. The full distribution of GRS is skewed upward in females compared to males (Fig. 3c), which is further highlighted by a fill plot comparing the densities of distributions of GRS between groups (Fig. 3d). As expected, the distribution of GRS is bimodal, with a subset of DNM carriers having the highest scores and the greatest enrichment of female cases.
Figure 3 |. Increased genetic load in females with ASD compared to males.
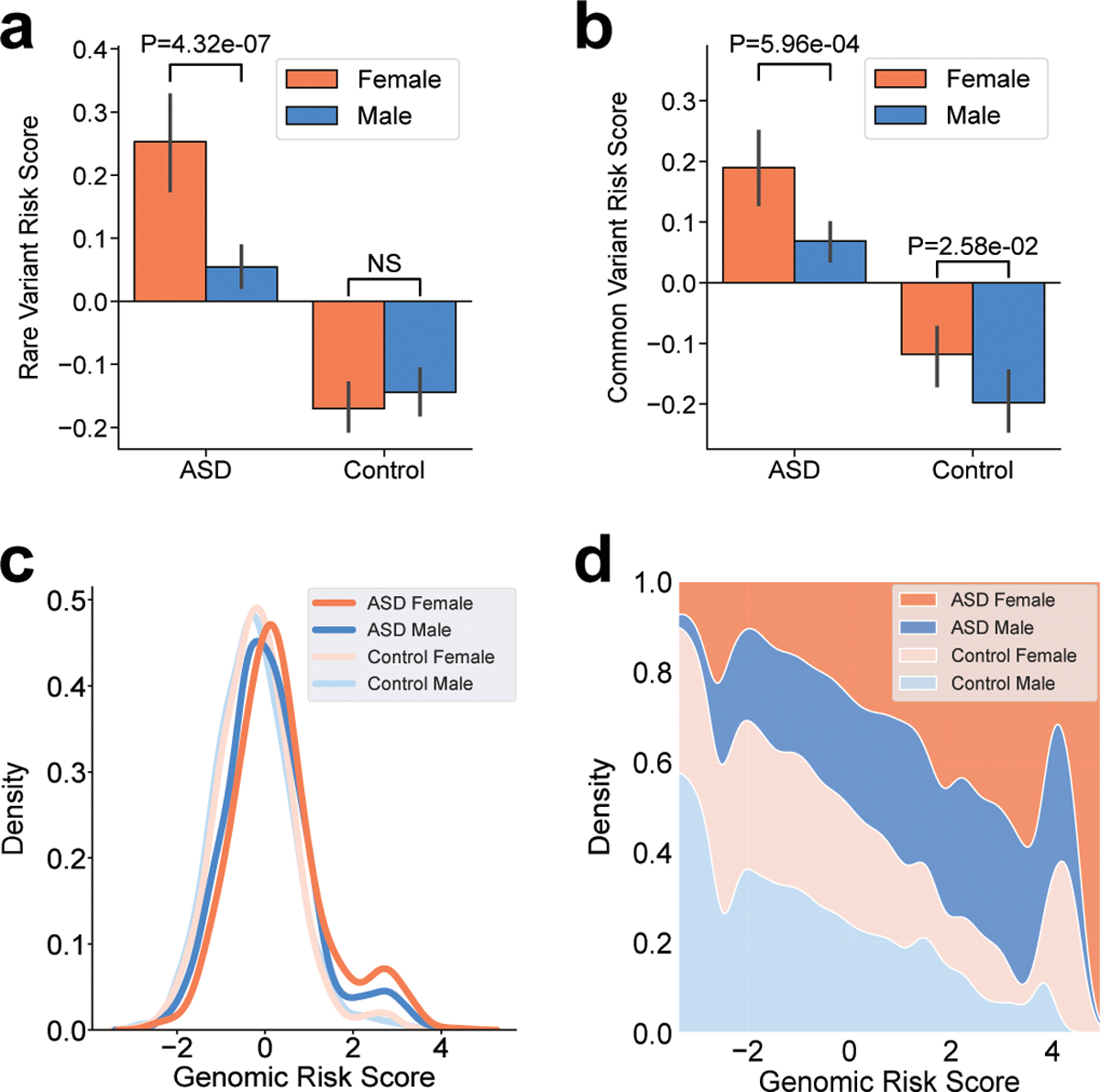
a,b, Increased burden of genetic risk in female cases compared to male cases is evident for combined rare de novo and inherited variants (RVRS) (a) and combined polygenic scores (CVRS) (b). P-values from a two-sample t-test are shown. Participants consisted of 5,247 cases (4,256 males and 991 females) and 3,054 controls (1,504 males and 1,550 females) of European ancestry. c, Sex differences in the combined genetic load (GRS) is evident across the full distribution. d, A fill plot comparing the densities of distributions illustrates that the GRS of females (cases and controls) are skewed upward relative to males.
According to a liability-threshold model for ASD22,23, a total genetic load sufficient to meet diagnostic criteria can be reached through differing combinations of rare and common variation. Subjects having a greater rare variant load may require less polygenic load5 and vice versa. In this study, cases who carry de novo damaging mutations (dnLoF or dnMIS) had a combined polygenic load that was reduced compared to cases that do not carry damaging DNMs (Fig. 4). A similar trend was seen in both sexes, but the effect was not statistically significant in females. Thus, in the presence of a damaging DNM, less polygenic risk is required to meet diagnostic criteria for ASD. The negative correlation of the composite risk scores RVRS and CVRS (P = 0.0037, Pearson correlation = −0.015) was stronger than for the pairwise correlations of individual factors (Fig. 4b and Supplementary Table 15), consistent with liability being attributable to the additive effects of multiple rare and common genetic factors. Also consistent with a liability threshold model, rare inherited variants (inhLoF) were negatively correlated with DNMs (P = 0.03; Extended Data Fig. 2a).
Figure 4 |. Negative correlation of rare variants and polygenic risk is consistent with a liability threshold model.
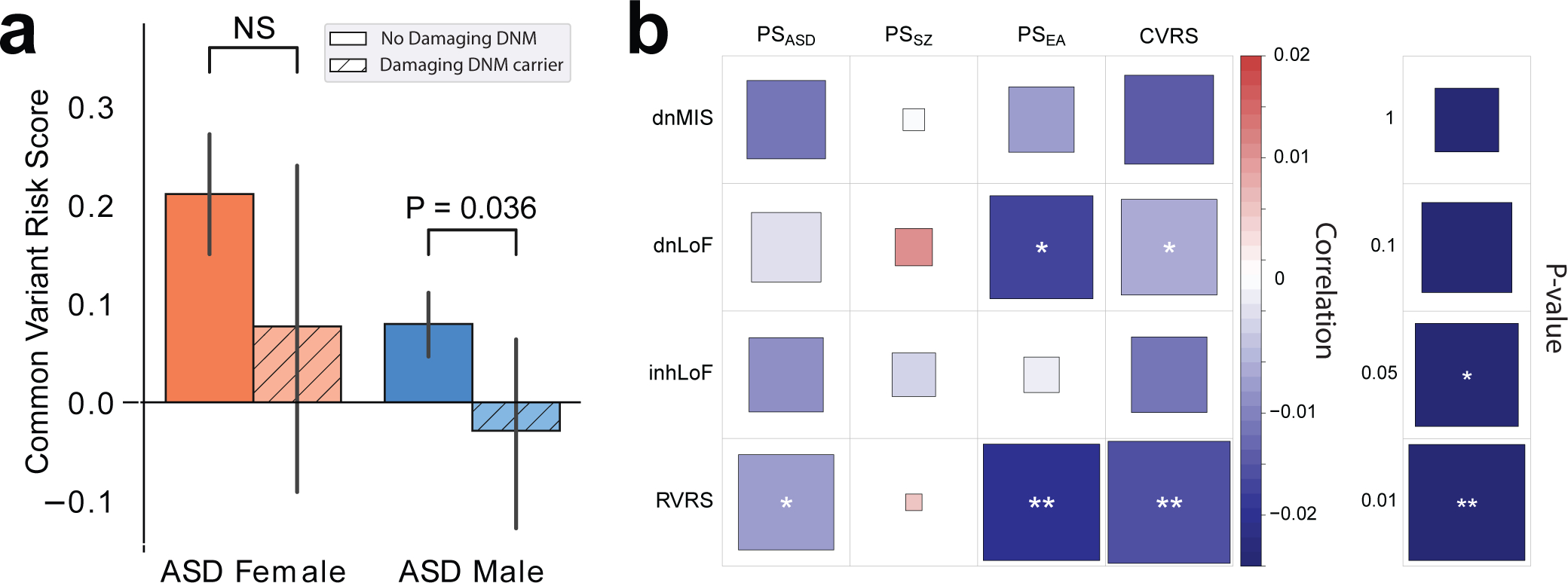
a, Transmission of polygenic risk (pTDT) is reduced to cases that carry damaging DNMs (dnLoF and dnMIS combined), but the result was not significant in females. P-values were based on two-sided t-tests. n = 4,256 male cases (423 DNM and 3,833 no DNM) and 991 females (1,504 DNM and 1,550 no DNM) of European ancestry. b, A heatmap displaying the strength of the correlations between polygenic scores and rare variants. P-values were derived from linear regression. Results are provided in Supplementary Table 15.
The strength of the threshold effect in Figure 4b did not differ significantly by sex. This is in contrast to our previous analysis of this dataset using the polygenic scoring method PRSice, which found evidence that the anti-correlation of CVRS and RVRS was stronger in males24 than in females (Extended Data Fig. 3). Evidence for sex differences in the strength of this negative correlation is therefore not robust across multiple polygenic scoring methods. Evidence for sex-biased transmission of rare inhLoF variants within families was similarly weak. For instance, we did not observe a biased transmission of risk from the more “protected” parent (mothers) to the more susceptible offspring (male cases) (Extended Data Fig. 2b), as we have previously hypothesized8. Thus, we do not find evidence that gene-by-sex effects result in dramatic biases in the transmission of risk from parent to child (see Supplementary Note for additional discussion).
Differential effects of genetic factors on behavioral traits.
We hypothesize that the differences in genetic architecture that we observe in this study could underlie broad variation in clinical phenotype across the autism spectrum. DNMs have been associated with a more severe clinical presentation of ASD characterized by greater intellectual impairment2,25 and delays in meeting developmental milestones 26,27. PSs for cognitive traits have been associated with a clinical subtype of high-functioning “Asperger” cases18. We investigated behavioral correlates of genetic factors in quantitative phenotype data on cases, sibling controls and parents that were available in the SSC and SPARK cohorts. Phenotypic measures in offspring included repetitive behavior (RBS), social responsiveness (SRS), social communication (SCQ), adaptive behavior (VABS) and motor coordination (DCDQ). Behavioral traits in parents, included ASD symptoms (SRS, BAPQ), educational attainment (EA), and parental age at birth of the proband (Supplementary Table 16). Genetic effects were tested by linear regression controlling for cohort, age, sex and principal components, and effects were also tested for gene-by-sex interactions (Supplementary Table 17).
Multiple genetic risk factors contributed to dimensions of ASD symptom severity in cases and in their typically developing sibling and parents. Six gene-trait correlations were significant after Bonferroni correction for 72 tests (Fig. 5a), and 18 showed nominal associations (P ≤ 0.05). Social deficits (SCQ, SRS) in offspring were associated with polygenic risk (PSASD) and de novo mutations (dnLoF), and the same factors influenced social behavior (SRS, BAPQ) in parents (Fig. 5b), with PSASD associated with social deficits and dnLoF correlated with reduced symptom severity in parents consistent with a de novo etiology. Deficits in adaptive behavior (VABS) in offspring were weakly correlated with dnLoF and polygenic risk (PSASD, PSSZ). Deficits in motor coordination (DCDQ) were associated with rare variants (dnMIS, dnLoF, inhLoF) but not with polygenic scores. PSEA was protective for core ASD symptoms of repetitive behavior (RBS) and social communication deficits (SCQ) in offspring and was also associated with reduced symptom severity in parents (BAPQ, EA). Intriguingly, multiple inherited genetic factors in parents (inhLoF, PSEA, PSSZ) were associated with parental age.
Figure 5 |. Differential effects of rare and common variation on behavioral traits in cases, sibling controls and parents.
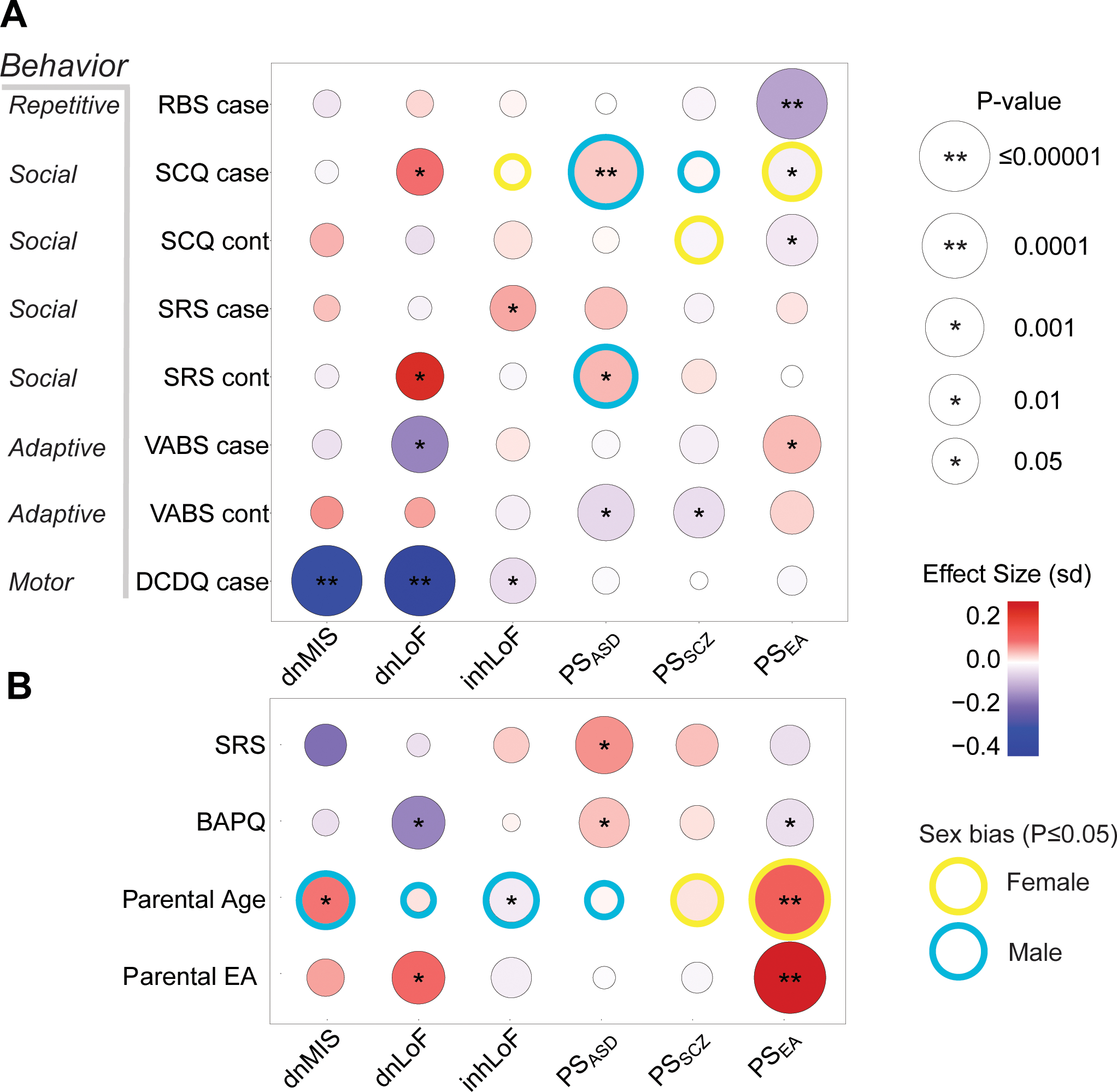
a, The effects of genetic factors were tested on five phenotype measures in children: repetitive behavior (RBS), social responsiveness (SRS), social communication (SCQ), vineland adaptive behavior (VABS) and developmental motor coordination (DCDQ). Note that RBS, SRS, SCQ and BAPQ are measures of “deficit”; thus, in the heatmap, red corresponds to increased severity. VABS and DCDQ are measures of “skill”; thus, blue corresponds to increased severity on these two instruments. Gene-phenotype correlations were tested by linear regression controlling for sex, age, cohort and PCs. Effect size is given as standard deviation (sd) of phenotype per unit of genetic factor. b, Genetic effects on parental behavior were tested for autism-related symptoms (BAPQ, SRS), educational attainment and parental age. In total, six gene-trait correlations were significant after Bonferroni correction for 72 tests (**P ≤ 0.0007), 18 were nominally significant (*P ≤ 0.05), and 11 showed evidence of sex-biased effects (gene-by-sex interaction P ≤ 0.05). Male or female sex bias indicates which sex had the greatest absolute value of effect size. Sample sizes for each phenotype ranged from 3,429 to 11,485. Sample numbers and results are summarized in Supplementary Tables 16 and 17. Analysis was restricted to individuals of European ancestry.
The correlations of genetic factors with behavioral traits were weakly sex biased. Eleven gene-trait relationships showed nominal evidence for an interaction by sex, but none were statistically significant after correction for multiple testing. These results suggest that the effects of most genetic factors on behavioral traits were similar in females and males. Among the weak interactions that were observed, a majority of gene-by-sex effects (8/11) were observed in controls or in parents. This may be attributable to a reduced power to detect sex differences in case samples that are predominantly male, or it could be due to the homogenizing effects of clinical ascertainment of ASD cases. Sex-differences in genetic effects were not exclusively male-biased (5/11 had stronger effects in females). For example, genetic effects on social communication (SCQ) in cases included two factors that were male-biased (PSASD and PSSZ) and two that were female-biased (inhLoF and PSEA) (Fig. 5a). Perhaps the most striking example of gene-by-sex interaction was that all six factors showed evidence for differential effects on maternal and paternal age (Fig. 5b).
Multiple genetic factors contribute to parental-age effects.
We and others have demonstrated that advanced paternal age correlates with increased rates of germline mutation in offspring28–30, consistent with parental age effects being attributable in part to de novo mutations that accumulate in the paternal germline. An alternative model by Gratten et al. has postulated that advanced paternal age could itself be a trait that is directly influenced by genetic liability for ASD that is carried by the father31. A recent study has found evidence that PSASD is positively correlated with paternal age32, providing support for the Gratten et al. model.
Our results demonstrate that the genetic basis of the parental age effect in ASD is highly multifactorial with contributions from de novo mutation, rare-inherited variants and polygenic risk. For example, common (PSEA) and rare (inhLoF) variation in fathers were associated with older and younger paternal age respectively (Fig. 6a), and the correlation of PSEA with advanced parental age was even stronger for mothers (Fig. 6a and Supplementary Table 18). As expected, the rate of de novo SNVs increased with paternal age in the combined dataset (r2paternal = 0.42, r2maternal = 0.27; Extended Data Fig. 4), and dnLoF and dnMIS variants mirror this effect (Fig. 6a).
Figure 6 |. The genetic basis of parental-age effects on ASD risk in offspring is multifactorial.
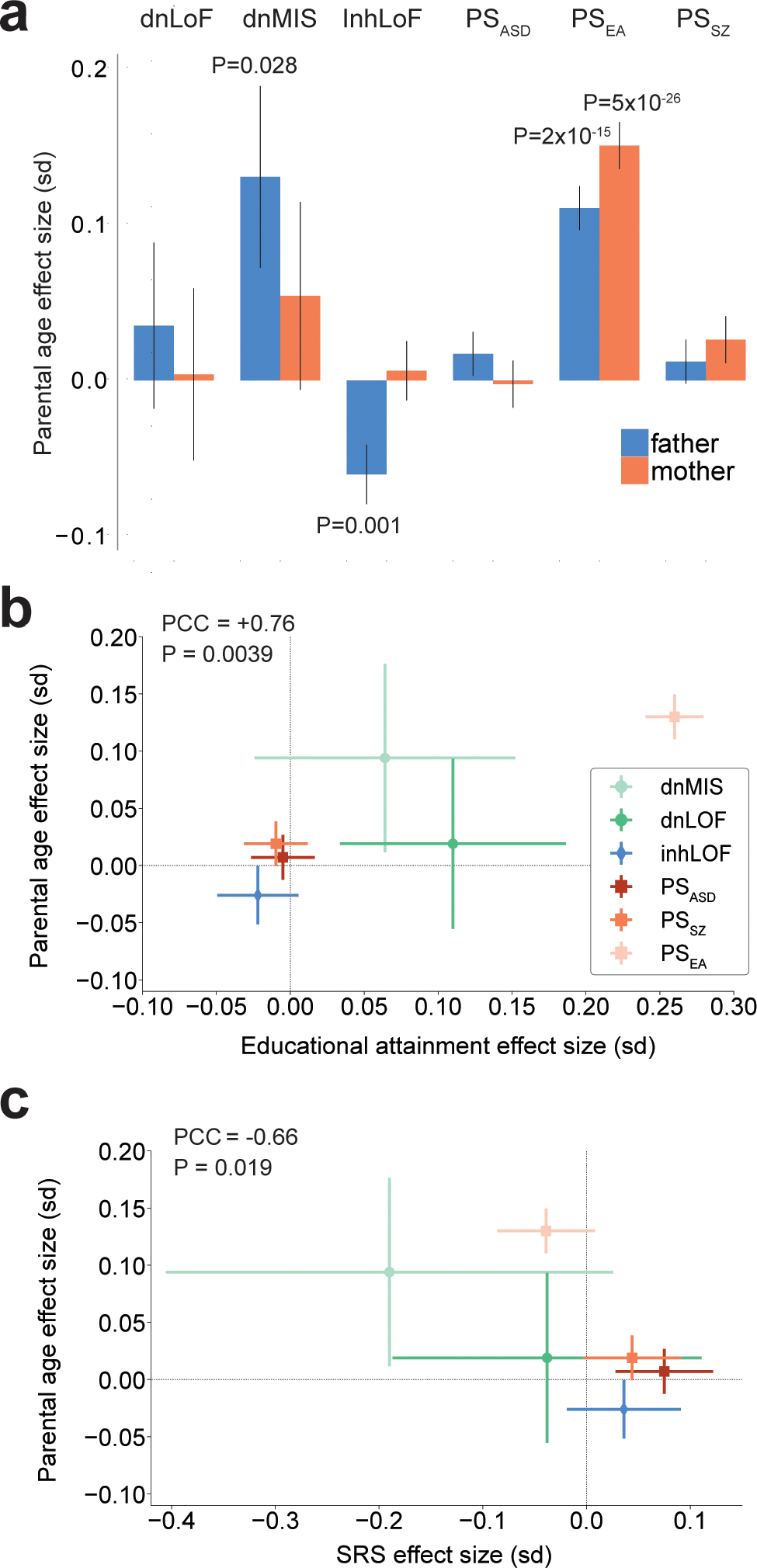
a, Multiple genetic risk factors for ASD are correlated with parental age with effects that differ by sex. Correlations of genetic factors with parental age (standard deviation of age per unit of genetic load) were estimated for 11,485 individuals (5,749 mothers and 5,736 fathers). P-values based on linear regression are given for individual effects with P < 0.05. Sex-stratified results for genetic effects on parental age are in Supplementary Table 18. b, The effects of six genetic factors on parental age were positively correlated with their effects on educational attainment, and the strongest correlate of parental age was PSEA. c, The effects of six genetic factors on parental age were negatively correlated with their effects on the SRS in parents. P-values were derived from linear regression. Whiskers represent 95% confidence intervals.
The single strongest inherited factor that influenced parental age was PSEA (r2 = 0.017; Supplementary Table 17), while PSASD and PSSZ showed much weaker correlations (r2 ≤ 0.0006). Consistent with these results, parents’ levels of education were significantly correlated with parental age in our sample and maternally biased (r2maternal = 0.066, r2paternal = 0.023), but social deficits in parents were not correlated with parental age (Supplementary Table 19). To further examine what behavioral traits in parents may explain inherited mechanisms of parental age effects, we compared the relative effects of genetic factors on the age, education and social behavior of parents (Fig. 6b,c). The effects of six genetic factors on parental age were positively correlated with their effect sizes for educational attainment of parents (PCC = +0.76, P = 0.0039; Fig. 6b) and negatively correlated with their effect sizes for social deficits (PCC = −0.66, P = 0.016; Fig. 6c). These results suggest that inherited mechanisms of parental age effects on ASD risk in offspring may be driven by genetic effects on learning and education in parents rather than by effects on parental social behavior.
Rare variant risk is enriched in neurons of the fetal cortex.
ASD susceptibility genes are preferentially expressed in the developing brain18,27. We hypothesize that differences in effect sizes and associated phenotypes between common variants and rare variants may be attributable, in part, to differences in the brain expression of their respective genes. Here we confirmed that ASD susceptibility genes are enriched in fetal cortex and cortical cell types, and compared the degree of enrichment between protein-coding genes implicated by rare variants or by GWAS.
We applied a rare-variant transmission and de novo association (TADA) test33 to the combined data in this study to define a set of 125 ASD susceptibility (TADA genes) with FDR < 0.05, and we obtained a set of 114 high-confidence protein-coding genes identified in a previous GWAS by Grove et al.18 (GWAS genes). To define a null distribution of expression values across developmental periods and cell types, 1,000 protein-coding genes were randomly sampled from the expression datasets. The three gene lists are provided in Supplementary Table 20. The expression of TADA genes and GWAS genes were then compared to the null distribution in cortex bulk tissue data from the BrainSpan transcriptome atlas34 and cell-type expression data obtained from the Cortical development expression (CoDEx) resource35.
In bulk human cortex, GWAS genes were more highly expressed (expression across all cortex samples and periods) compared to the null distribution, and TADA genes were further enriched (Fig. 7a). After normalizing cortex expression of each gene across periods, GWAS genes show increased relative expression during fetal development (Fig. 7b) compared to the null, and again TADA genes showed a further enrichment in fetal cortex. At the level of cortical cell types, the expression of TADA genes was significantly (~2 fold) greater than the null in excitatory and inhibitory neurons (Fig. 7c and Supplementary Table 21), and GWAS genes did not show a significant enrichment of expression by cell type. These results are consistent with rare variants of large effect impacting genes that have key roles in early fetal brain development.
Figure 7 |. ASD susceptibility genes implicated by rare variants are enriched in neuronal cell types of the developing brain.
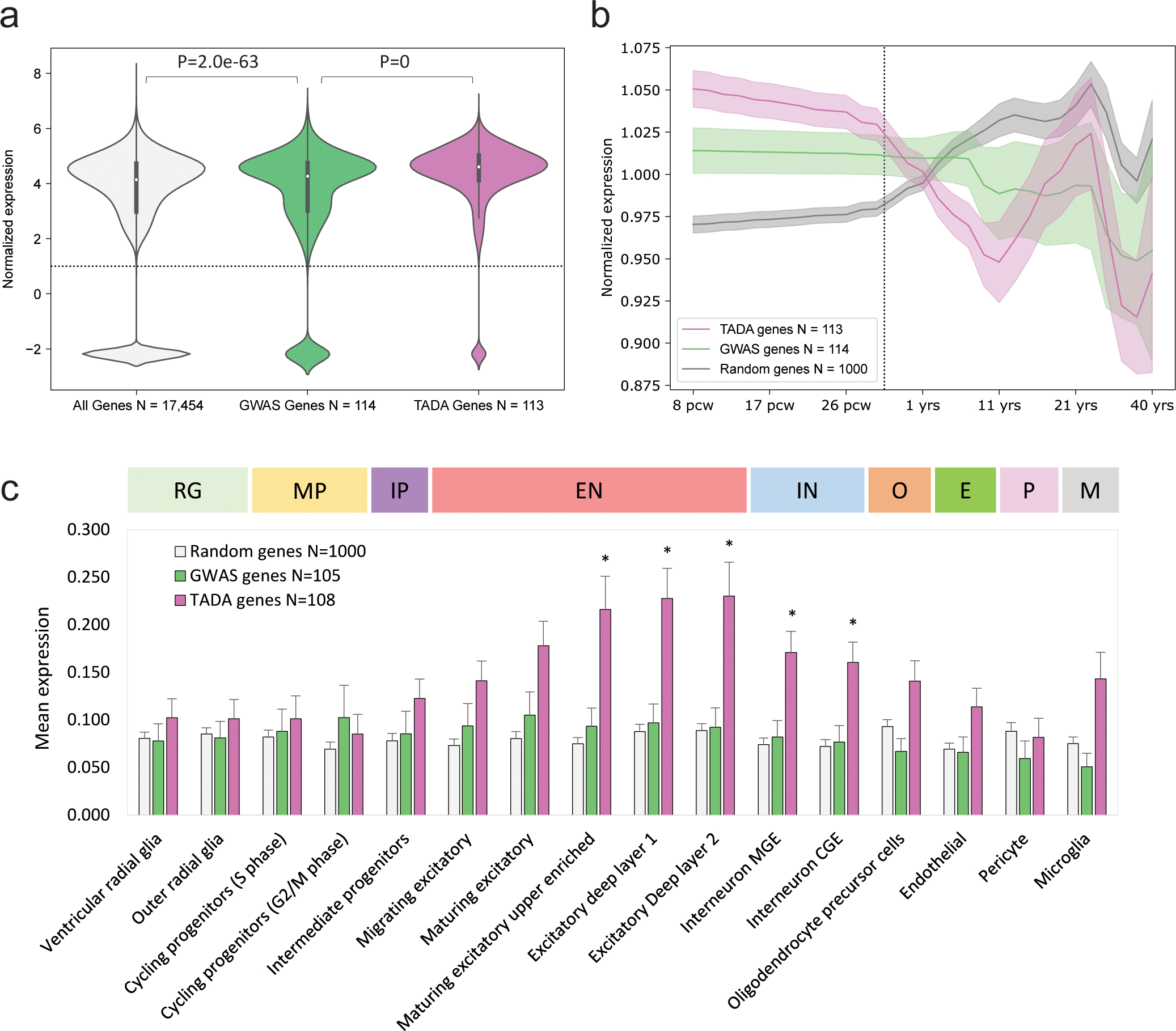
Expression levels of protein-coding genes in bulk tissue (BrainSpan) and in 16 cortical cell types (CoDEx) were compared between 115 genes identified with a rare variant association test in this study (TADA) and 114 genes implicated by common variants in Grove et al.18 (GWAS). a, Expression of GWAS genes across all periods and brain regions was enriched relative to the full distribution, and the expression of TADA genes was further enriched relative to GWAS. Boxes and whiskers represent the interquartile range (IQR) and 1.5*IQR, respectively. b, The expression of ASD genes in the developing cortex (after normalizing genes in BrainSpan to mean expression of 1 across periods) was enriched during prenatal development relative to null distribution consisting of 1,000 randomly protein-coding genes, with TADA genes being enriched to a greater extent. Shaded regions represent mean of the 95% CI from lowess smoothing. c, Mean expression of the GWAS and TADA genes were estimated within 16 cell types in the CoDEx datset and compared to the null distribution of randomly sampled genes (811/1,000 genes that were included in CoDEx) by a two-sample t-test. After Bonferroni correction for 32 tests (*P ≤ 0.0016), expression of TADA genes was significantly increased relative to the null in five neuronal cell types. Error bars represent standard error of the mean (s.e.m.), and P-values were derived from two-sided t-test. Gene sets and cell-type expression results are provided in Supplementary Tables 20 and 21. RG, radial glia; MP, mitotic progenitor; IP, intermediate progenitor; EN, excitatory neuron; IN, interneuron; O, oligodendrocyte precursor; E, endothelial cell; P, pericyte; M, microglia.
Discussion
Whole genome analysis of a large ASD family cohort demonstrates how the genetic basis of ASD consists of multiple genetic components, including DNMs, rare inherited variants and polygenic scores for psychiatric and behavioral traits. In this study, the predictive accuracies of polygenic scores and rare variants were similar, each explaining 2% of variance in case status. As new sequencing technologies continue to chip away at the missing heritability of ASD, additional genetic factors could be incorporated into the composite GRS to further improve upon this simple model. Furthermore, when WGS sample sizes become larger, more accurate estimates of the heritability explained by rare and common variants36 could be feasible.
The genetic architectures of ASD vary across cases, which is evident by an inverse correlation of rare variants and polygenic scores, consistent with a liability threshold model. This suggests that the genetic architectures of cases represent a spectrum of genetic loadings that span between extremes of polygenicity and monogenic disease. Furthermore, female cases have a significantly greater overall genetic load of polygenic and rare variation than male cases, confirming that a “female protective effect”, in which females display a greater tolerance for ASD risk alleles, applies generally to all components of the genetic architecture.
The spectrum of genetic architectures that we observe contributes to phenotypic variation across the cohort. Multiple genetic factors influence ASD symptom severity in cases and in their typically developing siblings and parents, with each factor having a different pattern of trait-association. Considering core symptom domains such as social deficits and repetitive behavior, PSASD and dnLoF were associated with severity in social deficits, and PSEA was protective for these traits. Several factors were weakly correlated with adaptive behavior, and deficits in developmental motor coordination were attributable solely to rare variants. For most gene-trait relationships, genetic effects on symptom severity paralleled their effects on case status. The one exception was PSEA, which was negatively correlated with symptom severity in offspring and parents but was positively correlated with case status. Thus, the association of PSEA with ASD could not be explained by any of the behavioral traits that were measured in this study. Potentially, SNPs that are captured by PSEA may influence dimensions of social cognition that were not tested in this study, or they may contribute to a clinically distinct subtype of high-functioning ASD. Consistent with the latter hypothesis, Grove et al. reported that the effect size for PSEA was strongest in the “Asperger syndrome” clinical subtype18.
Based on the evidence for a “female-protective effect” on the genetic load in cases, one might predict that genetic effects on social behavior would be stronger in males than in females. However, gene-trait relationships did not consistently follow this pattern. Most gene-trait correlations did not differ by sex. Genetic effects on social communication in cases consisted of two factors with evidence of a male bias (PSASD and PSSZ) and two with evidence of a female bias (inhLoF and PSEA). Genetic correlations with parental age consisted of four factors that were paternally biased (dnMIS, dnLoF, inhLoF and PSASD) and two that were maternally biased (PSEA and PSSZ). The observation that gene-by-sex effects go both ways is consistent with studies that have found preliminary evidence that some ASD genes are prevalent in female cases and others are prevalent in males37. Caution is warranted when interpreting gene-by-sex interactions. Given that all ASD GWASs have included case samples that were predominantly male, PSASD may be over-represented in male-biased SNP effects. In addition, genetic effects that differ by sex could reflect the influences of social factors or clinical ascertainment38. For example, a female bias in the effects of inhLoF variants might be expected if the clinical ascertainment of females is biased toward subjects with greater symptom severity and greater rare variant load39.
Multiple genetic factors were associated with parental age with effects that differed by sex. These results provide new insights into the genetic mechanisms of parental-age effects on ASD risk in offspring40. Parental age effects are attributable to multiple mechanisms, including: (1) a de novo mutation mechanism (dnMIS, dnLoF) in which new mutations accumulate with age in the germline as fathers age28,41; (2) inherited rare-variants that directly contribute to parental age behavior in fathers, and; (3) a polygenic mechanism that influences parental age in mothers and fathers31 with PSEA having by far the strongest effect. Our genetic findings support a model in which the combined effects of inhLoF, DNMs and polygenic scores contribute to a U-shaped effect of parental age and genetic risk for ASD. This model is consistent with several previous studies that have found evidence for a U-shaped relationship of parental age and risk for ASD or other developmental disorders in offspring42–46.
The effects of genetic factors on parental age were positively correlated with their effects on educational attainment. Rare inhLoF variants were associated with early paternal age, and fathers that carried inhLoFs had reduced educational attainment, but this association was not statistically significant (P < 0.058; Supplementary Table 18). The single strongest predictor of advanced parental age, particularly for mothers, was PSEA. We confirmed in our dataset a significant correlation of parental education and parental age47 that was stronger for mothers (r2 = 0.06, P = 3.5 × 10−52; Supplementary Table 19) than for fathers (r2 = 0.03, P = 1.3 × 10−23). By contrast, measures of social impairment in parents (SRS, BAPQ) were not associated with advanced parental age. Our results support a hypothesis that inherited mechanisms of parental-age effects are mediated by genetic effects on learning and education in parents.
Differences in cognitive traits associated with rare variants and polygenic risk may be in part attributable to expression patterns of the respective genes during fetal development. By comparing the expression of GWAS and TADA genes in transcriptome data from bulk tissue and single cells of the developing cortex, genes implicated by rare variants were more strongly enriched during fetal development, specifically within neurons. These results are consistent with polygenic models in which rare variants impact genes that play key roles in neurodevelopment, while the effects of common risk alleles are distributed more broadly across genetic regulatory networks48,49. Given that much of the polygenic risk influences non-coding regulatory elements of genes50, it is possible that the brain and cell-type enrichment of common variant effects may be greater for the underlying regulatory elements than for the transcripts as a whole. However, these results do highlight one aspect of the genetic architecture: polygenic risk for ASD is not restricted to a narrowly defined brain region, cell type or pathway.
The results described here highlight how an integrated analysis of multiple genetic factors can improve our understanding of the genetic basis of ASD. While most of the heritability of ASD remains unexplained, the expanding arsenal of sequencing platforms and methods of variant detection promise to expand the range of genetic factors that can be captured from a genome. The growing cohorts of ASD19 as well as individual rare diseases51 promise to improve knowledge of the effects of risk alleles on psychiatric traits and how their combined effects determine clinical outcome.
Methods
Our research complies with all relevant ethical regulations as approved by the institutional review board (IRB) of the UC San Diego School of Medicine.
Datasets.
The sample was comprised of three datasets, including whole genome sequencing of cohorts from the REACH project at UCSD (https://sebatlab.org/reach-project) and the Simons Simplex Collection (SSC) and a dataset of exomes and SNP genotyping from the SPARK study19. The combined sample of 11,313 ASD families consisted of a total 37,375 individuals, including 12,270 cases, 5,190 typically developing siblings, and 19,917 parents (Supplementary Tables 1 and 2). All categories of genetic risk to be evaluated in this study were confirmed previously within smaller cohorts of this study (REACH or SSC). Thus, the combined sample provides improved power to determine effect sizes for the same genetic factors. See Data and Code Availability for details on data access.
Processing of DNA sequence data.
Each of the three datasets consisted of Illumina paired-end sequence data, which were processed by BWA alignment and variant calling using GATK best practices. Specific differences between datasets include library prep (PCR vs. PCR free, WGS vs. exome) and differences in software version. Details are provided in the sections below. Analysis was carried out with SNP, indel and SV variant calls mapped to GRCh38. All calls were generated from sequence aligned to GRCh38. Jointly called VCFs from the REACH cohort were lifted over from GRCh37 to GRCh38 prior to annotation and analysis.
REACH cohort.
Whole genome sequencing was performed on blood-derived genomic DNA as described in our previous publication52. Standard quality control steps were carried out to ensure proper relatedness and genetic sex concordance with the sample manifest. Sequencing reads were aligned to the GRCh37 reference genome using bwa-mem (v0.7.12). Subsequent processing of the alignments followed GATK Best Practices guidelines including sorting, marking duplicate reads, indel realignment, and base quality score recalibration.
To ensure functional equivalency with other cohorts in our dataset, we applied the same SNV/indel variant calling pipeline used on the SSC cohort (see SSC section below for details). We utilized GATK HaplotypeCaller (v4.1) to first call SNVs and indels in individual samples. GRCh38 GVCFs were then combined using CombineGVCF and jointly genotyped. Variants quality score recalibration (VQSR) was then performed on the joint VCF. The VQSR model was trained with the parameter “maxGaussians=8” for SNVs and “maxGaussians=4” for indels. Variant scores were recalibrated with the truth sensitivity level of 99.8% for SNVs and 99.0% for indels. Sample-level filtering converted genotypes to noncalls (“./.”) if the GQ < 20 or the DP < 10. Before proceeding with variant annotation, variants were lifted over from GRCh37 to GRCh38 with the GATK LiftoverVcf command.
Simons Simplex cohort.
Whole genome sequencing was performed at the New York Genome Center (NYGC) on an Illumina HiSeq X10 sequencer using 150-bp paired-end reads to an average depth of 40x. Reads were aligned to the GRCh38 reference genome using bwa-mem with subsequent processing of alignments in line with GATK Best Practices for functional equivalence.
Jointly-called vcfs containing SNV and indel calls were provided by the NYGC (dated 2019–03-21). Briefly, variant calling was performed using GATK (v3.5). Variant discovery implemented HaplotypeCaller in GVCF mode. Variant quality scores were recalibrated by VQSR with the truth sensitivity level of 99.8% for SNVs and 99.0% for indels. Low quality genotype calls were defined as GQ < 20 or DP < 10 and were converted to missing genotypes (“./.”). Only variants that had “PASS” entries in the FILTER column were considered for analysis of inherited variants. Further details on the generation of the SSC SNV and indel joint calls can be found in the PDF accompanying the data release from the Simons Foundation. WGS SNP genotypes and GWAS imputed genotypes were subsequently merged in PLINK 1.953 for generation of PCs and polygenic scores.
SPARK cohort.
The publicly available SPARK dataset consisted of SNP genotyping (Illumina global screening array GSA-24v1–0) and exomes (IDT xGen capture sequenced on the Illumina NovaSeq 6000 using 2/S4 flow cells). Imputation of SNP genotypes was performed using the RICOPILI pipeline (https://sites.google.com/a/broadinstitute.org/ricopili/imputation)54.
Downstream processing of exome data was performed as follows. Per-sample GVCFs were obtained from the SPARK September 2019 data release in which GVCFs had been generated with GATK v4.1.2.0 HaplotypeCaller from CRAM files aligned to GRCh38 with bwa-mem. Joint genotyping and QC of SNP and indel variant calls was performed at UCSD in batches of 100 families the same GATK pipeline that was used for the REACH and SSC WGS. Variants with “PASS” in the FILTER column were retained for analysis. Likewise, indel calls with QD < 7.5 were omitted.
Principal components (PCs) calculation.
Genotype data was LD pruned to a set of 100,370 unambiguous markers with minor allele frequency > 5% in PLINK 1.9, using the --indep-pairwise command with a 200 variant window, shifting the window 100 variants at a time, and pruning variants with r2 > 0.2. KING version 2.2.4 (https://doi.org/10.1093/bioinformatics/btq559) was used to identify a set of unrelated individuals (1st and 2nd degree relatives removed). PCs were calculated in the unrelated individuals based on LD pruned data using FlashPCA2 (https://doi.org/10.1093/bioinformatics/btx299) and related individuals were then projected onto the PCs.
Polygenic score (PS) calculation.
PSSZ was calculated based on current schizophrenia summary statistics from the psychiatric genomics consortium (https://www.med.unc.edu/pgc/download-results/). PSASD was calculated from summary statistics in Grove et al.18 after excluding the SSC dataset used in this study. PSEA was calculated from summary statistics of the recent GWAS meta-analysis of educational attainment by Lee et al.55.
Two polygenic scoring methods were evaluated, and the method with the greatest prediction accuracy for ASD case status was selected for all analyses described here. Prior to manuscript submission, polygenic scores were calculated from summary statistics using the method PRSice (version 2.3.0)56, and the results of this analysis are posted to MedRxiv24. As recommended during peer review, PSs were recalculated using a newer method SbayesR20. The recalculated polygenic scores, particularly PSASD, had greater predictive value for case status (Extended Data Fig. 5), and overall results were highly consistent between both PS methods. A comparison of the two is discussed in further detail in the Supplementary Note. SBayesR polygenic scores were used for all analyses presented here.
SBayesR.
Polygenic scores were calculated using SBayesR20, a polygenic scoring method that provides an advantage over “clumping and thresholding” methods such as the method PRSice56. SBayesR utilizes all SNPs, and SNP effect sizes are re-scaled using a Bayesian (multiple regression) posterior inference model. SBayesR was implemented according to default settings as described in the software tutorial https://cnsgenomics.com/software/gctb/#Tutorial using the Banded LD matrix provided https://cnsgenomics.com/software/gctb/#LDmatrices. We used the --exclude-mhc argument, which excludes variants in the Major Histocompatibility Complex. Polygenic risk scores were calculated from the SBayesR summary statistics using PLINK.
PRSice version 2.3.0 (https://doi.org/10.1093/gigascience/giz082).
Only unambiguous variants with MAF > 1% in the reference dataset were included. Variants were LD clumped over a 250-kb window with an r2 value of 0.1. PSs were calculated at multiple P-value thresholds (0.01–0.9) to determine the optimal threshold. The best fitting PS for each trait was selected based on significance level of a TDT test carried out in autism cases (P-value threshold = 0.1 for ASD and SCZ, P-value threshold = 0.05 for EA). The best fitting PS was carried forward for all subsequent statistical analyses.
SV calling.
SV calls were only produced for the WGS datasets REACH and SSC. Our SV calling and filtering workflow has been described in detail in our previous publication52. Briefly, we ran ForestSV, LUMPY, and Manta on each sample calling deletions and duplications. ForestSV mainly relies on coverage as a feature to call SVs, resulting in segmented calls for large events that span repetitive elements such as segmental duplications. Because of this, we applied a stitching algorithm to ForestSV calls, combining calls of the same SV class if they were ≤10 kb apart. As a preliminary filter, we omitted any variant that overlapped more than two-thirds of the SV length to centromeres, telomeres, segmental duplications, regions with low mappability with 100-bp reads, antibody parts, T-cell receptors, and other assembly gaps.
The resulting calls were genotyped using SV2 and SVTyper within each sample. SVs and genotypes were then collapsed for overlapping calls. The collapsing algorithm first prioritized the breakpoint confidence intervals if both the start and end confidence intervals provided by LUMPY and/or Manta overlapped. For ForestSV calls, the confidence interval was defined as +/− 100 bp from the start and end positions. The consensus position determined for a collapsable cluster was determined by the SV position with the highest number of overlaps. In the case of a tie, the median position was recorded. This method allows for collapsing of common SVs that “tile” across a region, which rarely occurs outside of variable regions such as the HLA locus. The resulting calls were then subject to a further round of collapsing, this time reducing calls to a consensus position if they overlapped 80% reciprocally with each other. This method was applied recursively until no more calls could be collapsed. As for confidence interval collapsing, the consensus position reported was the SV with the highest number of overlaps. Variant level genotype likelihood scores were generated with SV2 by pooling all features from REACH and SSC samples. If the SV2 variant score was not “PASS” then the SVTyper or Manta genotypes were recorded, as previously described52. Samples without a genotype call were considered as missing (“./.”).
DNM calling.
DNMs were called using the synthDNM software57. SynthDNM is a random forest (RF)-based classifier which uses only a pedigree file (PED/FAM) and VCF files as input and can be readily optimized for different technologies or variant calling pipelines. For WGS datasets (REACH and SSC), we used the default SynthDNM classifier (SSC1 GATK), which was trained on GATK variant calls from >30X Illumina WGS data. This default classifier had high accuracy (AUC = 0.997) for detecting a truth-set of orthogonally validated de novo SNVs and indels from SSC57. For the exome dataset (SPARK), we trained an additional four classifiers, one for each set of variant calls: DeepVariant, WeCall, SPARK GATK, and SSC GATK. To maximize sensitivity while controlling for false positives, we retained DNMs if they were called by three out of the five classifiers. To further confirm the accuracy of SPARK DNM calls, we compared the de novo SNV and indel calls on the SPARK dataset to a set of validated DNMs that were confirmed by Sanger sequencing from a previous pilot study58. For SNVs, the recall rate for SNVs ranged from 92.6% to 98.2% (n = 117), while for indels the recall range from 98.6 to 100% (n = 107). For further details of the methodology and performance of SynthDNM, refer to our companion paper57.
De novo SVs were defined as events with heterozygous genotypes in offspring and homozygous reference calls in parents. We only considered variants that passed the stringent “DENOVO_FILTER” filter produced by SV259. We applied our standard filtering guidelines detailed below to omit variants present in regions known to produce spurious calls. We also supplemented our de novo calls with the de novo CNV calls generated from microarrays in SSC samples from Sanders et al.11 since many of these calls are likely to be missed by paired-end SV callers. We then manually inspected the list of de novo SVs and stitched calls together if they were separated by segmental duplications greater than 10 kb (the maximum stitching requirement for ForestSV calls detailed in the section below).
Variant annotation.
Variant Effect Predictor (VEP) v97 along with transcript annotations from Gencode v31 were used in annotation of SNVs and indels. Variants were flagged as “LoF” if the functional consequence one of the following: “transcript_ablation”, “splice_acceptor_variant”, “splice_donor_variant”, “stop_gained”, “frameshift_variant”, “stop_loss”, “start_loss”. LoF variants exclusive to nonsense mediated decay transcripts were omitted from subsequent analysis. SVs were annotated by overlap to exons and proximal cis-regulatory elements including 5’UTRs, transcription start sites, and fetal brain promoters. Since the list of annotated proximal cis-regulatory elements were in GRCh37, we lifted over the GRCh38 SV calls to GRCh37 for all subsequent analysis.
We assigned gnomAD LOEUF scores (v2.1.1) to each LoF variant and CRE-SV. If a variant overlapped more than one gene, as in the case for large SVs, we recorded the minimum (most constrained) LOEUF score to that variant. Constraint was quantified for missense variants using the “Missense Badness, PolyPhen-2, and Constraint” (MPC) scores60. These scores are available for the GRCh37 build of the human genome and were transposed to GRCh38 for analysis. Missense variants without MPC scores due to updates to the reference genome were not used in subsequent analysis. The recommended cutoffs to enrich for the top tier of constraint (LOEUF < 0.37; MPC > 2) was applied to de novo and rare inherited LoF variants.
Association tests.
Selection of variant types to be tested.
For this study, we sought to define several major categories of rare variant and common variant risk and to investigate their combined effects on ASD risk and behavioral traits. We settled on six categories (three rare and three common) that all have a strong prior evidence for their contribution to ASD.
Rare variants.
The major categories of rare variants that have been reproducibly associated with associated with ASD include: (1) de novo protein truncating/loss-of-function mutations (dnLoF), a category where genetic association is concentrated within genes that are loss-of-function intolerant61; (2) de novo missense variants (dnMIS), a category where genetic association is concentrated within genes that show missense constraint60 and; (3) inherited loss-of-function (inhLoF) variants in LOF-intolerant genes16,52. In our analysis of genetic association, we confirmed the association of SNPs, indels and SVs within the above categories (Fig. 1). Analysis of the interactions between factors and correlations of genetic factors with behavioral traits across multiple cohorts was restricted to SNP and indel variants that can be detected across cohorts with comparable sensitivity.
Polygenic scores.
Across a series of studies, schizophrenia62,63 and educational attainment63,64 have stood out as traits that are correlated with polygenic risk for ASD. PSSZ and PSEA may not be the psychiatric trait scores that are most highly correlated with PSASD, but they are among the most well powered GWASs. For this reason, these polygenic scores were selected for the first family-based study that applied a polygenic TDT test (also used in this study) to demonstrate an overtransmission of PSASD, PSSZ and PSEA to cases17. This established for us the proof of concept for their inclusion in this study. Given the high intercorrelation of genetic risk for a variety of other psychiatric disorders and traits62, a rationale could be made for examining several more polygenic scores, but given the wide variety of equally valid and highly correlated traits and polygenic scores to choose from, we sought to err on the side of simplicity and included these three as the main polygenic factors of interest.
Definitions of variant types
. All rare variant categories described below consisted of private variants in which the alt allele was present in only one family in this study and had an allele frequency <1% in gnomAD (v2.1.1). Target categories included only variants in functionally constrained genes as defined below.
dnMIS variants were defined as all private de novo missense SNVs with MPC scores > 2. dnLoF variants were defined as variant calls that were predicted to result in loss of protein function (truncation of a protein) and included stop-gain, frameshift, splice site and exonic deletion in a loss-of-function intolerant gene defined as LOEUF < 0.37, per recommendations from gnomAD. SVs that intersected more than one gene were assigned minimum LOEUF score (corresponding to the most constrained gene). De novo synonymous variants (dnSyn), and this category included all private de novo synonymous SNVs.
InhLoF variants were defined as private SNV or indel variants with a “PASS” entry in the “FILTER” column and we removed variants with ≥5% missing calls across the cohorts. For inhLoF variants in the SPARK dataset, we applied one additional filter removing indel variants with QD scores less than 7.5.
For dnSVs and inherited LOF SVs, we included only private exonic SVs > 50 bp in length and CRE-SVs ≥ 2.5 kb that passed the “DENOVO_FILTER” from the SV2 software which is a stringent filter recommended for ultra-rare variants.
De novo association.
The burden of damaging DNMs (dnLoF, dnMIS) was compared between cases and controls by a two-sample independent t-test reporting the two-sided P-values. Results are provided for the set of de novo mutations in the combined sample. In addition, to evaluate the consistency of DNM ascertainment between the REACH, SSC and SPARK cohorts, dnLoF, dnMIS and dnSyn variants in all cohorts were compared by restricting DNMs to a common set of exome targets that was used in a previous publication by Iossifov et al.2. dnSyn variants did not differ significantly between cases and controls in the combined sample (Extended Data Fig. 1a). In the SPARK cohort we observed a 1.1-fold excess of synonymous variants in cases (OR = 1.1, P = 0.02). This trend could be attributable to other factors, including chance or true causal noncoding variants enriched in cases. No quality metrics that were tested were correlated with case status in the SPARK dataset including coverage, transition:transversion (Ti/Tv) ratio, ratio of heterozygous to homozygous genotypes (Extended Data Fig. 1b) and paternal age (Extended Data Fig. 1c). Thus, variables could not be identified that explain a subtle baseline difference in dnSyn burden, which could be included as covariates in a regression model. However, this very subtle effect does not contribute to a bias in the combined sample and cannot explain the strong associations reported for other categories of de novo mutation (Fig. 1).
Inherited rare variant association.
The number of transmissions and nontransmissions from parent to offspring was obtained using plink’s “--tdt poo” command (v1.9). Pooling of transmission and nontransmission counts for the transmission disequilibrium test (TDT) was done using the pytdt python package (https://github.com/sebatlab/pytdt). This package takes as input a data table containing a unique variant ID and counts for transmissions and nontransmissions in fathers and mothers for both cases and controls. Pytdt performs the pooling or group-wise analysis of private LoF variants and CRE-SVs by summing the counts of transmissions and non-transmissions for all variants encompassing a group. The package also reports odds ratios, confidence intervals, and other statistics commonly used for TDT analysis. We also conditioned the TDT according to damaging DNM burden in the offspring using a binomial test for statistical significance of transmission distortion of private variants to cases or controls separately. For a summary of the TDT results and a list of all the private variants tested in the analysis, see Supplementary Tables 7–9.
Polygenic TDT.
Per methods from Weiner et al.17, trio-based association of polygenic scores (PSASD, PSSZ, PSEA) with ASD was tested with the polygenic TDT (pTDT), which tests the significance of the deviation of the child PS from the average PS of the parents.
P-value was then calculated with a one-sample t-test of pTDT-dev (Fig. 1c) with a population mean of 0. Results of the pTDT are reported in Supplementary Table 10.
Calculating composite risk scores RVRS, CVRS and GRS.
We used multivariable regression to capture the combined effects of multiple genetic factors on case status. For rare variant factors, the predictor variables in the model consisted of rare variant burden counts for dnMIS, dnLoF and inhLoF. For polygenic scores PSASD, PSSZ and PSEA, the predictor variables consisted of the pTDT-dev values of the trios. To calculate a composite genetic risk score, each predictor variable was first residualized for PCs and sex. Then estimates were calculated from a generalized linear model as follows
where y is case status and x1, x2 and x3 are residualized predictor variable for three genetic factors. PCs for all regression models consisted of the first 10 principal components from the PCA. Then, the composite risk score (RS) is calculated using r as
Each RS was than standardized by Z-transformation. Predictor variables (x1, x2, x3, etc.) for each risk score consisted of
To compare the effect sizes on case status for the genetic factors and the composite risk scores (Fig. 2b), Nagelkerke’s r2 values were calculated for each of the residualized predictor variables and for each composite risk score.
Pairwise correlations of rare variants and polygenic risk.
To test the correlations between rare variants and polygenic risk, we constructed pairwise linear models
where the variable y is a polygenic score (PSASD, PSSZ, PSEA or CVRS) and x is a measure of rare variant load (dnLoF, dnMIS, inhLoF or RVRS). Gene-by-sex interaction was then tested in the following model.
Supplementary Table 15 contains the full results for all pairwise correlation of rare and polygenic risk conditioned on sex.
Effects of genetic factors on behavioral traits.
The effects of genetic factors on behavioral traits were investigated in the SSC and SPARK cohorts using clinical phenotype data available from SFARI (see Data and Code Availability). To eliminate confounders due to ancestry, only individuals of European ancestry confirmed by PCA were included. Clinical measures of ASD symptoms and related behaviors were selected that were available for cases, typically developing sibling, or parents. Phenotype measures consisted of the summary scores from the developmental coordination disorder questionnaire (DCDQ) of motor function and the Repetitive Behavior Scale (RBS) that were available on cases; the Vineland Adaptive Behavior Scale (VABS), Social Communication Questionnaire (SCQ) and Social Responsiveness Scales (SRS) that were available on both cases and TD siblings. Behavioral phenotypes available on parents included the Broad Autism Phenotype Questionnaire (BAPQ), parental educational attainment (from the background history questionnaire) and parental age at birth (for the children with ASD diagnosis). Phenotype measures that were available for both the SSC and SPARK cohorts were normalized within cohort by Z-transformation, then combined, and cohort was included as a covariate in the downstream analyses. A summary of the sample sizes available for each phenotype measure is provided (Supplementary Table 16).
Association of genetic factors with developmental traits was tested by linear regression controlling for sex, cohort and principal components. In addition, a gene-by-sex interaction was tested to determine if genetic effects on cognitive traits differed for males and females. Phenotypes in offspring (cases and siblings) were tested using the model
where y is the phenotype variable and x is the genetic factor (DNMs, inhLoFs and PSs). In addition, a gene-by-sex interaction was then tested in this model.
Brain and cell-type expression of ASD susceptibility genes.
The lists of TADA, GWAS and randomly selected protein-coding genes are provided in Supplementary Table 20. The expression of TADA genes and GWAS genes were compared in the developing human brain using the publicly available gene expression matrix from BrainSpan34. The two gene sets were also compared across 16 cell types in the human cortex using cell type expression data available from the CoDEx dataset35.
TADA genes.
We defined a set of genes implicated by rare variants with the transmission and de novo association test (TADA)33 in our combined sample, using the recommended parameters for ASD relative risk and using mutational rates for LoF and missense variants calculated by Samocha et al.65. TADA genes were defined as a set of 113 ASD genes that were associated with ASD at an FDR < 0.05.
GWAS genes.
We obtained the list of high confidence genes that were implicated by GWAS associations and described by Grove et al.18 (GWAS genes). Briefly, genes that are likely contributors to GWAS associations were defined with H-MAGMA, a method that assigns noncoding SNPs to their genes based on long-range interactions detected by Hi-C in fetal and adult brain66. A list of 121 GWAS genes was provided by the authors (Hyejung Won, personal communication). To facilitate a valid comparison of genes implicated by rare variants and common variants, the GWAS gene set was restricted to a subset of 114 genes that were protein coding according grch38 Ensembl gene annotations.
Random genes.
Patterns of expression across developmental periods (Fig. 7b) and cell types (Fig. 7c) for GWAS genes and TADA genes were compared to null distributions obtained by randomly sampling 1,000 protein-coding genes from the BrainSpan and CoDEx datasets.
Analysis of gene expression in bulk tissue (BrainSpan).
The Developmental transcriptome dataset was downloaded from BrainSpan (https://www.brainspan.org/static/download.html), which consisted of normalized gene expression data from 26 brain structures (including 21 within the cortex) across 31 developmental time periods. Overall expression of GWAS and TADA genes in the developing cortex were compared by combining expression values across cortex samples, and gene sets were compared to the null distribution by Student’s t-test. Likewise, patterns of expression in cortex across developmental time periods was compared between gene sets by first normalizing the cortex expression of each gene to its mean across cortex samples, and then fitting the expression values of each gene set by lowess smoothing using the “lowess” function described here https://james-brennan.github.io/posts/lowess_conf/.
Analysis of gene expression in 16 cell types from fetal cortex (CoDEx).
Analysis was performed on cell type gene expression values provided in the CoDEx dataset35, which consisted of single cell RNA-seq (scRNA-seq) obtained by DropSeq analysis of sections of germinal zone and ventricular zone tissue from mid-gestation fetal cortex67. Briefly, in Polioudakis et al., raw counts were normalized and cells were clustered using Seurat (v2.3.4)68, and mean gene expression values per cell were calculated for genes in 16 cortical cell types. Cell-type expression values were obtained from the “Genes” table on the CoDEx web interface (http://solo.bmap.ucla.edu/shiny/webapp/) for TADA genes, GWAS genes, and these were compared to a random sampling of 1,000 protein-coding genes.
Extended Data
Extended Data Fig. 1. Rates of de novo mutations stratified by cohort and evaluation of potential confounders.
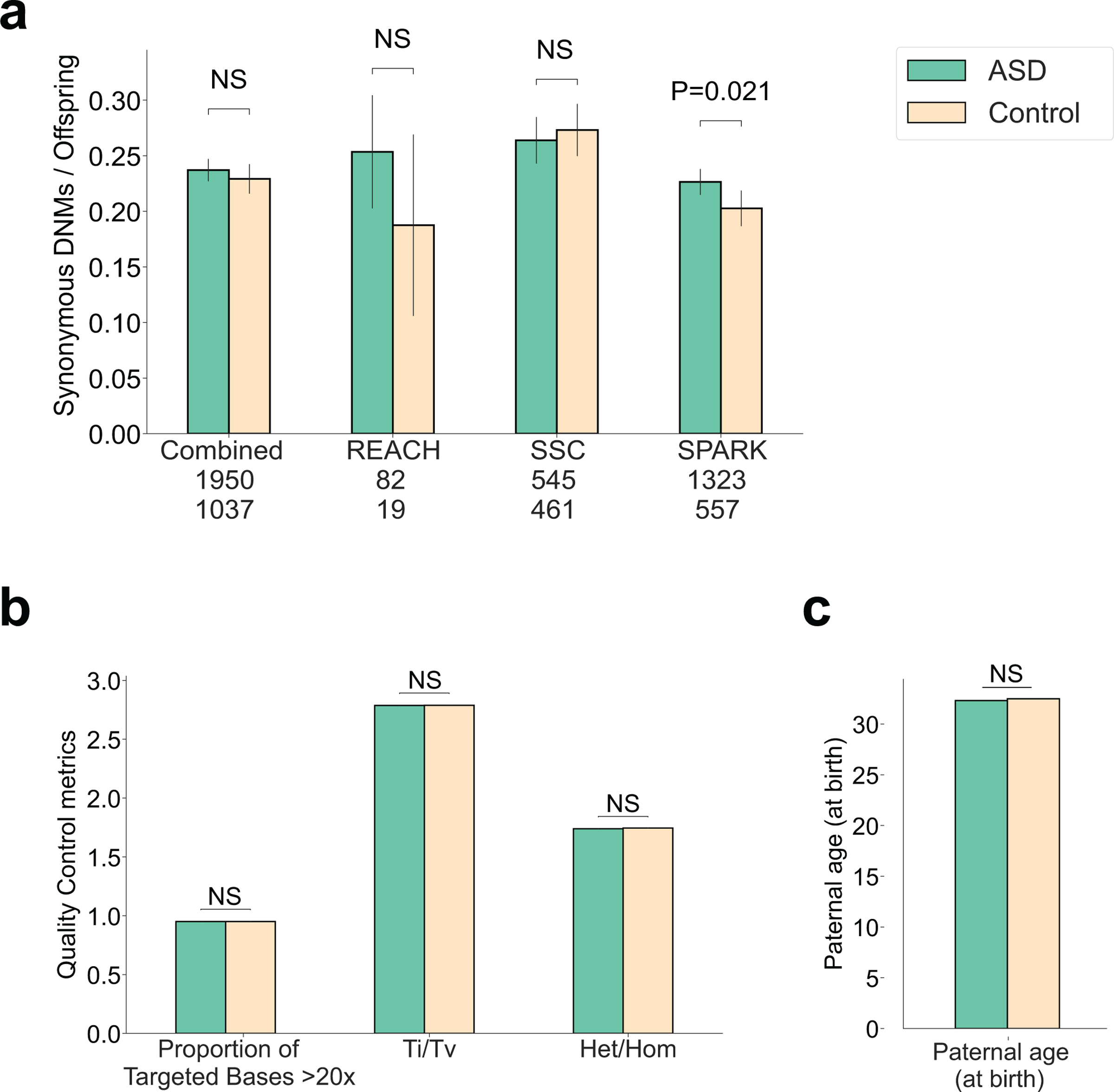
a, Rates of de novo synonymous (dnSyn) variants were not associated with ASD in the combined sample, but were enriched 1.1-fold in the SPARK cohort (P = 0.021). b, We evaluated whether quality metrics or other confounders could explain the slight excess of dnSyn variants in SPARK cases. Quality metrics did not differ in cases and controls including coverage, transition:transversion ratio (Ti/Tv) or ratio of heterozygous calls (Het/Hom). c, Paternal age did not differ significantly between cases and controls.
Extended Data Fig. 2. The combined effects of dnLoF, inhLoF and sex on the transmission of rare variants in families.
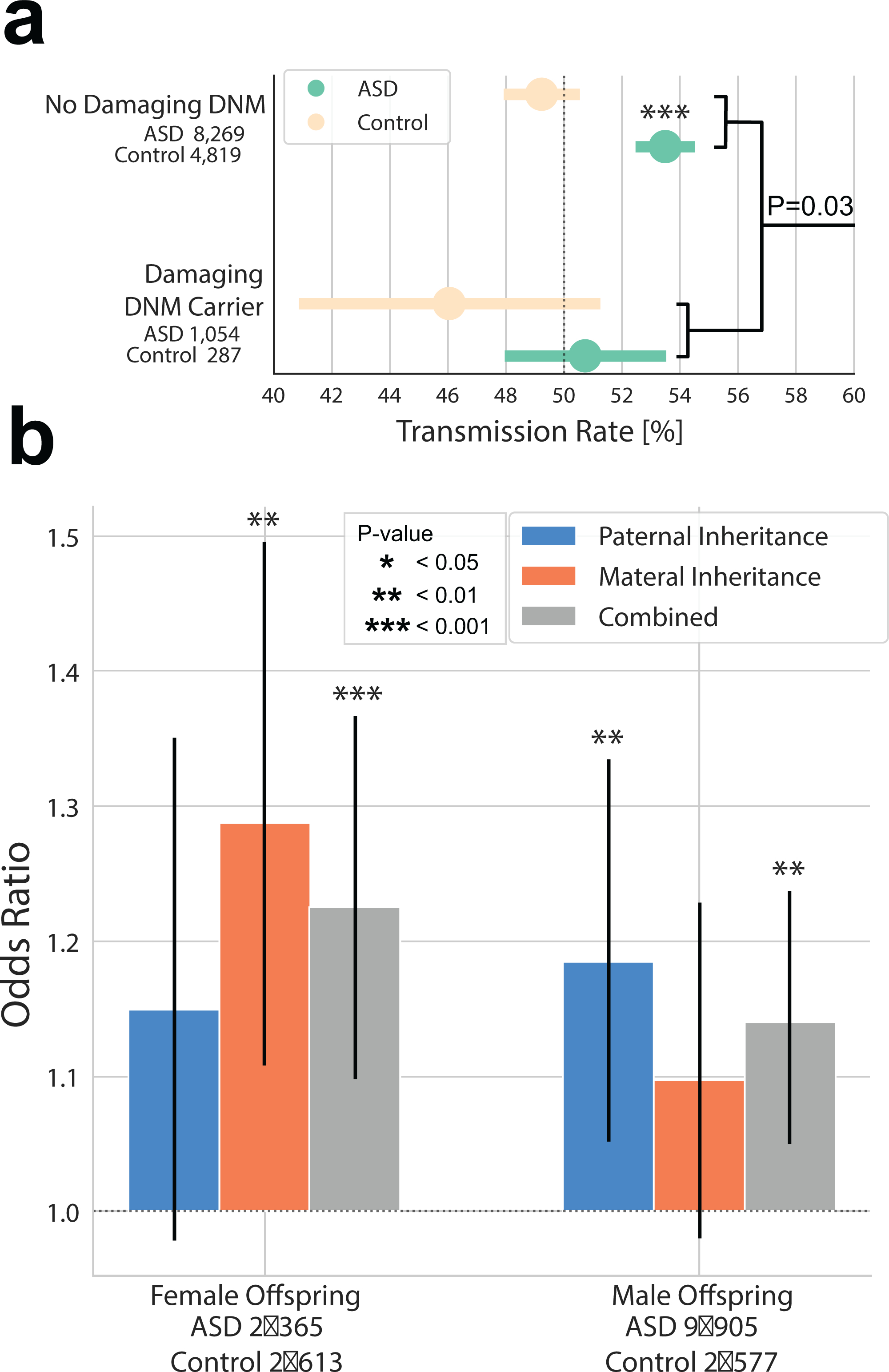
a, A significant liability threshold for rare variants was evident based on a negative correlation of dnLoF and inhLoF (linear regression P = 0.03), and this effect did not differ significantly by sex. b, Case-control odds ratios were compared for the transmission rates in families by sex (father-daughter, mother-daughter, father-son, mother-son). Both maternal and paternal rare variants contribute to ASD with a significant over-transmission from mother to daughter and from father to son. We did not observe a significant sex bias in the transmission of rare variants in families. In particular, we did not observe an enriched transmission from mother to male cases as we have previously hypothesized8.
Extended Data Fig. 3. Sex differences in the correlation of rare variant and common variant risk was not robust across multiple polygenic scoring methods.
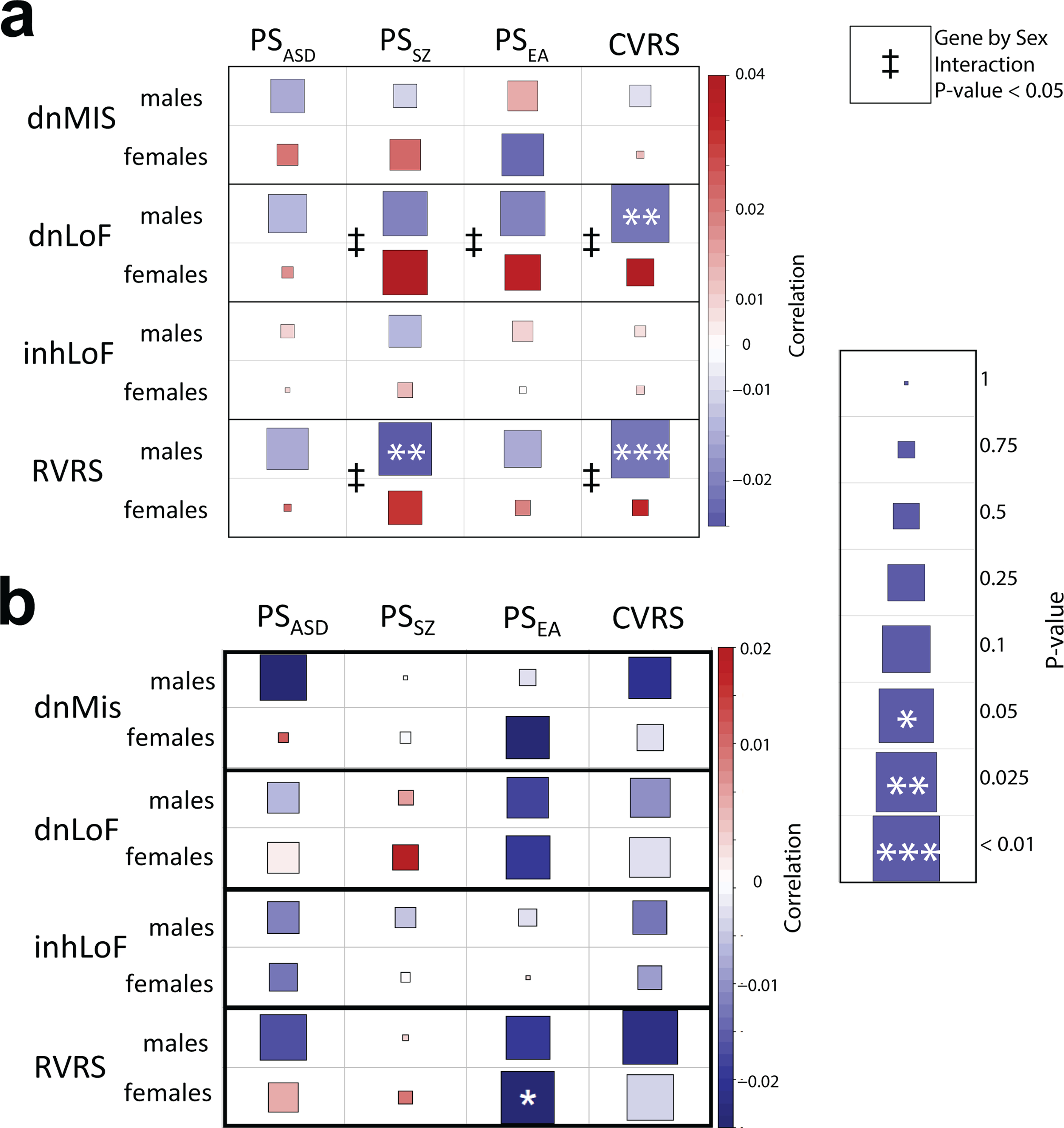
a, An early analysis of this dataset using polygenic score estimates from PRSice observed that the negative correlation of RVRS and CVRS was stronger in males than in females, consistent with males having less tolerance of genetic risk. The heatmap displays the correlations between polygenic scores and rare variants in males and females separately. Correlations were tested by linear regression controlling for cohort, case status and ancestry PCs, and a gene-by-sex interaction was tested in the combined sample (ǂgene-by-sex P < 0.05). b, With polygenic scores calculated using SBayesR, there was a similar trend with the correlation of CVRS and RVRS being stronger in males; however, the gene-by-sex interaction was not statistically significant.
Extended Data Fig. 4. Correlation of de novo mutation rate with parental age.
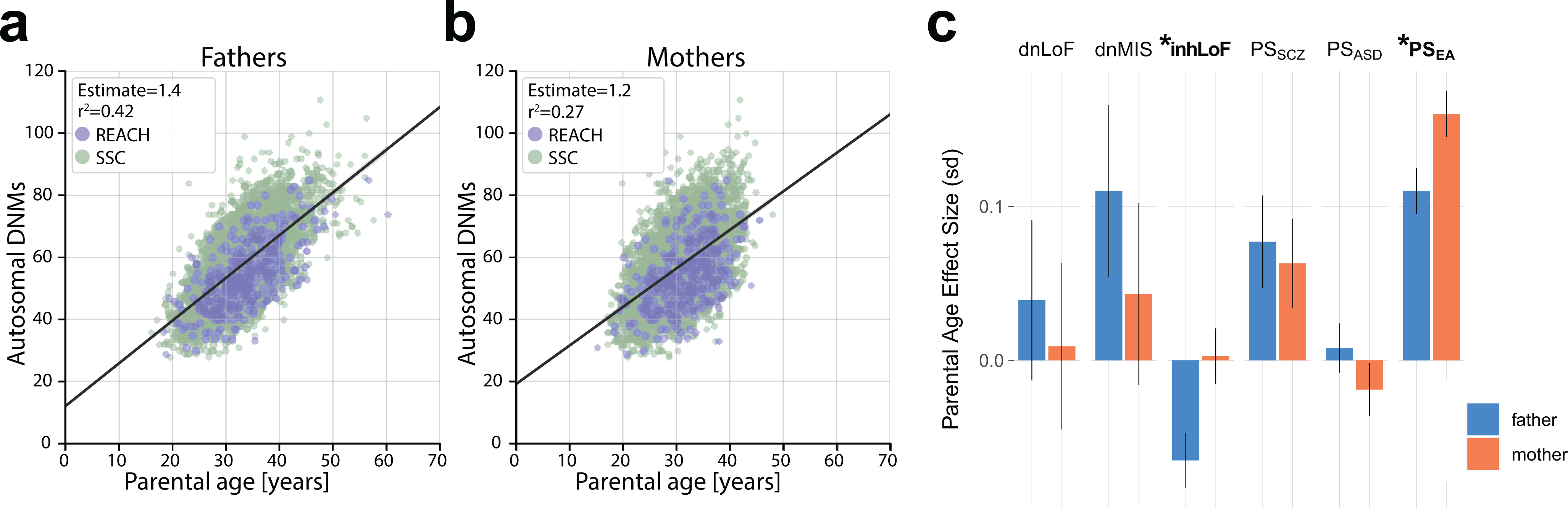
a,b, Correlation of total autosomal de novo SNVs with age of fathers (a) and mothers (b). See also Figure 6a. n = 4,518 trios for which age-at-birth was available for the mother and father.
Extended Data Fig. 5. Comparison of the predictive values of polygenic scoring methods PRSice and SBayesR.
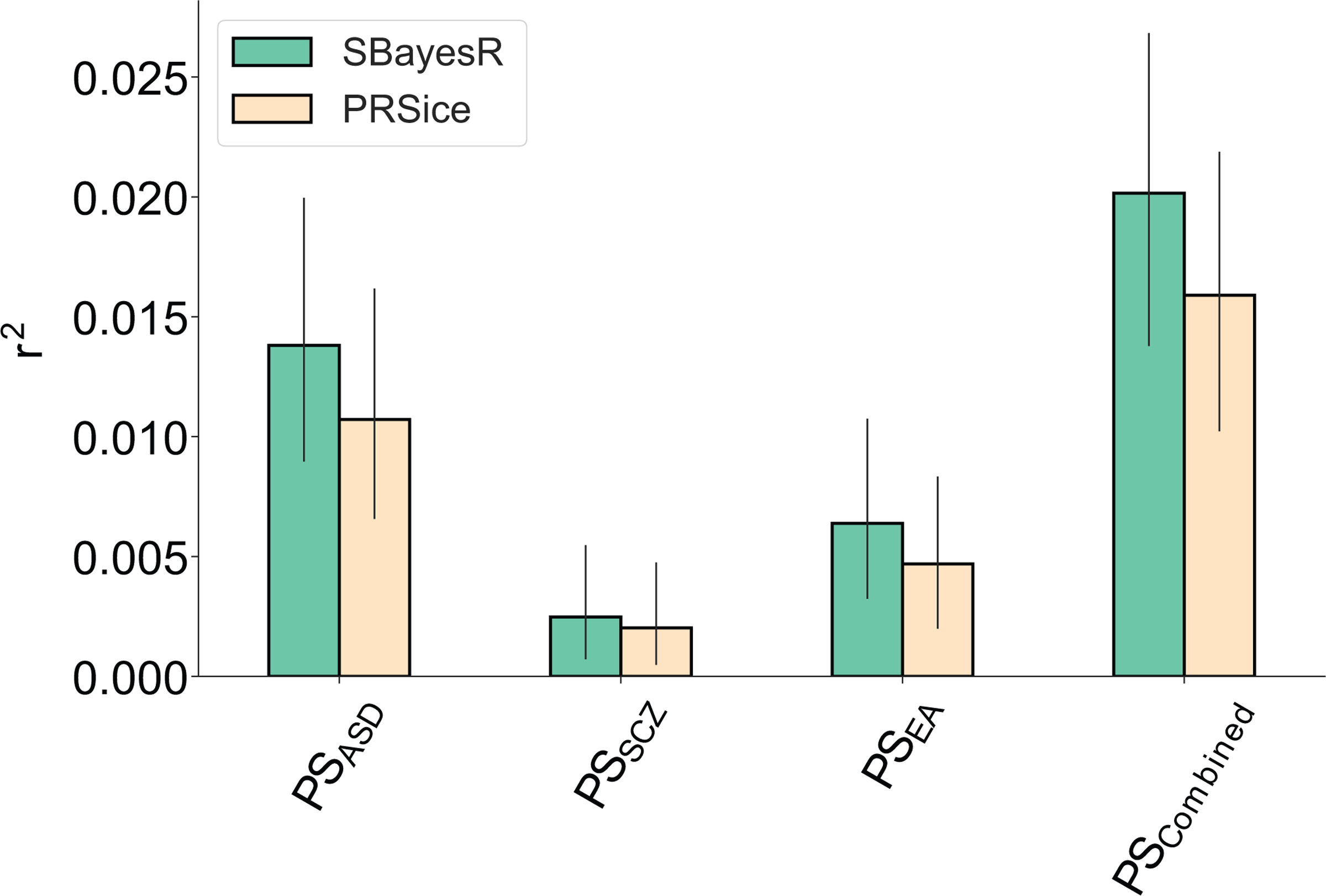
Polygenic scores calculated using SBayesR had greater predictive value for polygenic scores for ASD (PSASD), schizophrenia (PSSZ) and educational attainment (PSEA).
Supplementary Material
Acknowledgements
We thank the families who participated in genetic studies of the REACH, SSC and SPARK cohorts. The large samples used in this study were made possible through the strong commitment to sharing of individual level data by the Simons Foundation Autism Research Initiative (SFARI), NIH, UCSD, and the sharing of summary-level data by the Psychiatric Genomics Consortium. We thank W. Pfeiffer, the San Diego Supercomputer Center, and Amazon Web Services for hosting the computing infrastructure necessary for completing this project. We thank Wendy Chung and SFARI for providing data and materials for validation of DNM calls. We thank Nikolas Baya for assistance with data processing. We thank Hyejung Wong for providing the list of high-confidence GWAS genes. This work was supported by grants to J.S. from the Simons Foundation Autism Research initiative (SFARI 606768), National Institutes of Health (MH113715, MH119746, 1MH109501) and the Escher fund for Autism (20171603) and grant to C.M.N. from the National Institutes of Health (MH106595). D.A. was supported by a T32 training grant from the NIH (GM008666). M.K. was supported by a Rubicon grant from the Dutch Research Council (NWO 45219212). L.M.I. was supported by the NIH (MH109885, MH108528, MH105524, MH104766) and by the Simons Foundation for Autism Research (SFARI 345469).
Footnotes
Competing Interests
A.R.M. is a co-founder and has an equity interest in TISMOO, a company focusing on applications of genetics and human brain organoids to personalized medicine. The terms of this arrangement have been reviewed and approved by the University of California, San Diego, in accordance with its conflict of interest policies. The remaining authors declare no competing interests.
Data Availability
WGS data from the SSC and Exome and SNP genotyping data from SPARK are available from the Simons Foundation Autism Research Initiative (SFARI) (https://www.sfari.org/resource/autism-cohorts). Summary genetic data from WGS and exomes including individual counts for dnLoF, dnMIS, inhLoF and polygenic scores for all subjects in this study and input data files for all analysis code are also available from SFARI. WGS data from the REACH project are available from the NIMH Data Archive (NDA), including the structural variant callset, and raw sequence (FASTQ), alignment (BAM) and variant call (VCF) files from the REACH cohort (https://nda.nih.gov/edit_collection.html?id=2019). GWAS summary statistics are available from the Psychiatric Genomics Consortium (ASD and SZ) (https://www.med.unc.edu/pgc/download-results/) and the Social Science Genetic Association Consortium (EA) (http://www.thessgac.org/data). Bulk tissue expression data on ASD susceptibility genes was obtained from the Brainspan developmental transcriptome dataset (v10; https://www.brainspan.org/api/v2/well_known_file_download/267666525). Cell type expression levels of ASD susceptibility genes in fetal cortex were obtained through the web interface of the Cortical development expression viewer (CoDEx) (http://solo.bmap.ucla.edu/shiny/webapp/).
Code Availability
Analysis code for all major statistical genetic analyses in the paper and for generating Figures 1- 7 is available as a Google Colab notebook on Github (https://github.com/sebatlab/Antaki2021).
References
- 1.Sebat J et al. Strong association of de novo copy number mutations with autism. Science 316, 445–449 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iossifov I et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grove J et al. Common risk variants identified in autism spectrum disorder. bioRxiv (2017). [Google Scholar]
- 4.Sebat J, Levy DL & McCarthy SE Rare structural variants in schizophrenia: one disorder, multiple mutations; one mutation, multiple disorders. Trends Genet. 25, 528–535 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bergen SE et al. Joint contributions of rare copy number variants and common SNPs to risk for schizophrenia. Am. J. Psychiatry 176, 29–35 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davies RW et al. Using common genetic variation to examine phenotypic expression and risk prediction in 22q11.2 deletion syndrome. Nat. Med. 26, 1912–1918 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lim ET et al. Rare complete knockouts in humans: population distribution and significant role in autism spectrum disorders. Neuron 77, 235–242 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao X et al. A unified genetic theory for sporadic and inherited autism. Proc. Natl. Acad. Sci. USA 104, 12831–12836 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Werling DM & Geschwind DH Recurrence rates provide evidence for sex-differential, familial genetic liability for autism spectrum disorders in multiplex families and twins. Mol. Autism 6, 27 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robinson EB, Lichtenstein P, Anckarsater H, Happe F & Ronald A Examining and interpreting the female protective effect against autistic behavior. Proc. Natl. Acad. Sci. USA 110, 5258–5262 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanders SJ et al. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron 87, 1215–1233 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Desachy G et al. Increased female autosomal burden of rare copy number variants in human populations and in autism families. Mol. Psychiatry 20, 170–175 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Jacquemont S et al. A higher mutational burden in females supports a “female protective model” in neurodevelopmental disorders. Am. J. Hum. Genet. 94, 415–425 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Rubeis S et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brandler WM et al. Paternally inherited noncoding structural variants contribute to autism. bioRxiv (2017). [Google Scholar]
- 16.Krumm N et al. Excess of rare, inherited truncating mutations in autism. Nat. Genet. 47, 582–588 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weiner DJ et al. Polygenic transmission disequilibrium confirms that common and rare variation act additively to create risk for autism spectrum disorders. Nat. Genet. 49, 978–985 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grove J et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 51, 431–444 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spark Consortium. SPARK: a US cohort of 50,000 families to accelerate autism research. Neuron 97, 488–493 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lloyd-Jones LR et al. Improved polygenic prediction by Bayesian multiple regression on summary statistics. Nat. Commun. 10, 5086 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Werling DM The role of sex-differential biology in risk for autism spectrum disorder. Biol. Sex. Differ. 7, 58 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Falconer DS Inheritance of liability to certain diseases estimated from incidence among relatives. Ann. Hum. Genet. 29, 51-& (1965). [Google Scholar]
- 23.Reich T, Morris CA & James JW Use of multiple thresholds in determining mode of transmission of semi-continuous traits. Ann. Hum. Genet. 36, 163-& (1972). [DOI] [PubMed] [Google Scholar]
- 24.Antaki D et al. A phenotypic spectrum of autism is attributable to the combined effects of rare variants, polygenic risk and sex. medRxiv, 2021.03.30.21254657 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robinson EB et al. Genetic risk for autism spectrum disorders and neuropsychiatric variation in the general population. Nat. Genet. 48, 552–555 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buja A et al. Damaging de novo mutations diminish motor skills in children on the autism spectrum. Proc. Natl. Acad. Sci. USA 115, E1859–E1866 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Satterstrom FK et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 180, 568–584.e23 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Michaelson JJ et al. Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell 151, 1431–1442 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kong A et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature 488, 471–475 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goriely A & Wilkie AO Missing heritability: paternal age effect mutations and selfish spermatogonia. Nat. Rev. Genet. 11, 589 (2010). [DOI] [PubMed] [Google Scholar]
- 31.Gratten J et al. Risk of psychiatric illness from advanced paternal age is not predominantly from de novo mutations. Nat. Genet. 48, 718–724 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Mullins N et al. Reproductive fitness and genetic risk of psychiatric disorders in the general population. Nat. Commun. 8, 15833 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.He X et al. Integrated model of de novo and inherited genetic variants yields greater power to identify risk genes. PLoS Genet. 9, e1003671 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li M et al. Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science 362, eaat7615 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Polioudakis D et al. A single-cell transcriptomic atlas of human neocortical development during mid-gestation. Neuron 103, 785–801.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wainschtein P et al. Assessing the contribution of rare variants to complex trait heritability from whole-genome sequence data. Nat. Genet. 54, 263–273 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Turner TN et al. Sex-based analysis of de novo variants in neurodevelopmental disorders. Am. J. Hum. Genet. 105, 1274–1285 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Russell G, Steer C & Golding J Social and demographic factors that influence the diagnosis of autistic spectrum disorders. Soc. Psychiatry Psychiatr. Epidemiol. 46, 1283–1293 (2011). [DOI] [PubMed] [Google Scholar]
- 39.Werling DM & Geschwind DH Sex differences in autism spectrum disorders. Curr. Opin. Neurol. 26, 146–153 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.D’Angelo D et al. Defining the effect of the 16p11.2 duplication on cognition, behavior, and medical comorbidities. JAMA Psychiatry 73, 20–30 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kong A et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature 488, 471–475 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Malaspina D et al. Paternal age and intelligence: implications for age-related genomic changes in male germ cells. Psychiatr. Genet. 15, 117–125 (2005). [DOI] [PubMed] [Google Scholar]
- 43.Lyall K et al. The association between parental age and autism-related outcomes in children at high familial risk for autism. Autism Res. 13, 998–1010 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Frans EM et al. Autism risk across generations: a population-based study of advancing grandpaternal and paternal age. JAMA Psychiatry 70, 516–521 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lampi KM et al. Parental age and risk of autism spectrum disorders in a Finnish national birth cohort. J. Autism Dev. Disord. 43, 2526–2535 (2013). [DOI] [PubMed] [Google Scholar]
- 46.Lundstrom S et al. Trajectories leading to autism spectrum disorders are affected by paternal age: findings from two nationally representative twin studies. J. Child Psychol. Psychiatry 51, 850–856 (2010). [DOI] [PubMed] [Google Scholar]
- 47.Fulco CJ, Henry KL, Rickard KM & Yuma PJ Time-varying outcomes associated with maternal age at first birth. Journal of Child and Family Studies 29, 1537–1547 (2020). [Google Scholar]
- 48.Boyle EA, Li YI & Pritchard JK An expanded view of complex traits: from polygenic to omnigenic. Cell 169, 1177–1186 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu X, Li YI & Pritchard JK Trans effects on gene expression can drive omnigenic inheritance. Cell 177, 1022–1034.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maurano MT et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jacquemont S et al. Genes to mental health (G2MH): a framework to map the mombined effects of rare and common variants on dimensions of cognition and psychopathology. Am J Psychiatry. 179, 189–203 (2022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brandler WM et al. Paternally inherited cis-regulatory structural variants are associated with autism. Science 360, 327–331 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Purcell S et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lam M et al. RICOPILI: Rapid Imputation for COnsortias PIpeLIne. Bioinformatics 36, 930–933 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee JJ et al. Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat. Genet. 50, 1112–1121 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Choi SW & O’Reilly PF PRSice-2: polygenic risk score software for biobank-scale data. Gigascience 8, giz082 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lian A, Guevara J, Xia K & Sebat J Customized de novo mutation detection for any variant calling pipeline: SynthDNM. Bioinformatics 37, 3640–3641 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Feliciano P et al. Exome sequencing of 457 autism families recruited online provides evidence for autism risk genes. npj Genom. Med. 4, 19 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Antaki D, Brandler WM & Sebat J SV2: accurate structural variation genotyping and de novo mutation detection from whole genomes. Bioinformatics 34, 1774–1777 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Samocha KE et al. Regional missense constraint improves variant deleteriousness prediction. bioRxiv, 148353 (2017). [Google Scholar]
- 61.Kosmicki JA et al. Refining the role of de novo protein-truncating variants in neurodevelopmental disorders by using population reference samples. Nat. Genet. 49, 504–510 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Consortium Brainstorm et al. Analysis of shared heritability in common disorders of the brain. Science 360, eaap8757 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hagenaars SP et al. Shared genetic aetiology between cognitive functions and physical and mental health in UK Biobank (N =112 151) and 24 GWAS consortia. Mol. Psychiatry 21, 1624–1632 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Clarke TK et al. Common polygenic risk for autism spectrum disorder (ASD) is associated with cognitive ability in the general population. Mol. Psychiatry 21, 419–425 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Samocha KE et al. A framework for the interpretation of de novo mutation in human disease. Nat. Genet. 46, 944–950 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sey NYA et al. A computational tool (H-MAGMA) for improved prediction of brain-disorder risk genes by incorporating brain chromatin interaction profiles. Nat. Neurosci. 23, 583–593 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Macosko EZ et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Butler A, Hoffman P, Smibert P, Papalexi E & Satija R Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
WGS data from the SSC and Exome and SNP genotyping data from SPARK are available from the Simons Foundation Autism Research Initiative (SFARI) (https://www.sfari.org/resource/autism-cohorts). Summary genetic data from WGS and exomes including individual counts for dnLoF, dnMIS, inhLoF and polygenic scores for all subjects in this study and input data files for all analysis code are also available from SFARI. WGS data from the REACH project are available from the NIMH Data Archive (NDA), including the structural variant callset, and raw sequence (FASTQ), alignment (BAM) and variant call (VCF) files from the REACH cohort (https://nda.nih.gov/edit_collection.html?id=2019). GWAS summary statistics are available from the Psychiatric Genomics Consortium (ASD and SZ) (https://www.med.unc.edu/pgc/download-results/) and the Social Science Genetic Association Consortium (EA) (http://www.thessgac.org/data). Bulk tissue expression data on ASD susceptibility genes was obtained from the Brainspan developmental transcriptome dataset (v10; https://www.brainspan.org/api/v2/well_known_file_download/267666525). Cell type expression levels of ASD susceptibility genes in fetal cortex were obtained through the web interface of the Cortical development expression viewer (CoDEx) (http://solo.bmap.ucla.edu/shiny/webapp/).