

Abstract
Congenital diaphragmatic hernia (CDH) can occur in isolation or in conjunction with other birth defects (CDH+). A molecular etiology can only be identified in a subset of CDH cases. This is due, in part, to an incomplete understanding of the genes that contribute to diaphragm development. Here, we used clinical and molecular data from 36 individuals with CDH+ who are catalogued in the DECIPHER database to identify genes that may play a role in diaphragm development and to discover new phenotypic expansions. Among this group, we identified individuals who carried putatively deleterious sequence or copy number variants affecting CREBBP, SMARCA4, UBA2, and USP9X. The role of these genes in diaphragm development was supported by their expression in the developing mouse diaphragm, their similarity to known CDH genes using data from a previously published and validated machine learning algorithm, and/or the presence of CDH in other individuals with their associated genetic disorders. Our results demonstrate how data from DECIPHER, and other public databases, can be used to identify new phenotypic expansions and suggest that CREBBP, SMARCA4, UBA2, and USP9X play a role in diaphragm development.
Keywords: Congenital diaphragmatic hernia, diaphragmatic eventration, CREBBP, SMARCA4, UBA2, USP9X, DECIPHER database
INTRODUCTION
Congenital diaphragmatic hernia (CDH) is a life-threatening structural birth defect with an incidence of approximately 1 in 4,000 (Langham et al., 1996). CDH is defined as an abnormal opening in the diaphragm that allows the abdominal viscera to protrude into the thorax. Here we will use an expanded definition that also includes diaphragmatic eventrations in which the diaphragm continuity is not disrupted, yet the diaphragm is abnormally elevated, often due to a defect in diaphragmatic muscularization (Clugston & Greer, 2007). In isolated cases, CDH is the only defect identified. In contrast, individuals with CDH+ have both a diaphragmatic defect and one or more, non-hernia-related anomalies. Advances in copy number variant detection and next generation sequencing technologies have made it possible to identify a molecular etiology in many individuals with isolated CDH and an even higher percentage of individuals with CDH+ (Scott et al., 2021; Wat et al., 2011).
In some CDH cases, failure to identify a molecular etiology is due to an incomplete understanding of the genes that can contribute to diaphragm development. Genes that are associated with high penetrance of CDH can be readily identified through standard gene discovery methods in which recurrent clinical phenotypes identified in cohorts of patients carrying deleterious changes affect a specific candidate gene (Qi et al., 2018). Low penetrance CDH genes are much harder to identify using these methods, particularly if the genetic disorders caused by these gene are relatively rare. In such case, additional evidence that a gene contributes to the development of CDH may come from mouse models, individual case reports, expression data, and/or bioinformatic studies (Scott et al., 2021).
Here we use publicly available clinical and molecular genetic data from individuals cataloged in the DECIPHER database (https://www.deciphergenomics.org/) to identify phenotypic expansions involving CDH (Firth et al., 2009). Our results provide evidence that deleterious variants in CREBBP, SMARCA4, UBA2, and USP9X are associated with CDH+.
MATERIALS AND METHODS
Ethical Approval
Publication of anonymized data from Subjects 1–16 and 19–38 was approved by the Institutional Review Board of Baylor College of Medicine (protocol H-47546). Subjects 17 and 18 were accrued into a research protocol approved by Institutional Review Board of Columbia University (protocol AAAB2063) after obtaining written informed consent.
In all cases, research was performed in accordance with international standards and was conducted in accordance with the ethical standards of this institution’s committee on human research and international standards.
Database analysis and clinical review
We performed a phenotype-based search of the DECIPHER database to identify individuals with “congenital diaphragmatic hernia” and/or “diaphragmatic eventration” who carried single nucleotide variants and copy number variants (Firth et al., 2009). Contact was made with each of the submitting centers who then approved the publication of their patient’s clinical and molecular data.
Sequencing studies
Whole exome sequencing was performed for Subjects 17 and 18 as previously described (Qi et al., 2018).
Literature and database searches
We searched for reports in which CDH candidate genes and/or their associated genetic disorders were mentioned in conjunction with key words such as “diaphragm”, “diaphragmatic hernia”, “CDH”, or “diaphragmatic eventration”.
Machine learning
We hypothesize that genes that cause CDH, 1) will have similar patterns of annotation, 2) will be associated with the development of similar defects in mouse models, 3) will interact with each other physically or within the same molecular pathways, 4) will be regulated by the same microRNAs, 5) will be expressed in similar tissues, 6) will be regulated by similar transcription factors, and 7) will have similar epigenetic modification patterns. We have previously used a machine learning algorithm to rank all RefSeq genes based on their similarity in each of these domains to a set of 31, manually curated human genes known to cause CDH, or whose mouse homolog causes CDH in mice, (CHAT, DNASE2, EFEMP2, EFNB1, FBN1, FGFRL1, FREM1, FZD2, GATA4, GLI2, GLI3, HLX, HOXB4, LOX, LRP2, MET, MSC, NIPBL, NR2F2, PAX3, PBX1, PDGFRA, RARA, RARB, ROBO1, SLIT3, SOX7, STRA6, TCF21, WT1, and ZFPM2) which acted as our CDH training gene set (Callaway et al., 2018).
Briefly, our machine learning algorithm integrated data from corresponding large-scale genomic knowledge including 1) Gene Ontology (GO), 2) Mouse Genome Informatics (MGI) phenotype annotations, 3) the Protein Interaction Network Analysis (PINA), 4) the Kyoto Encyclopedia of Genes and Genomics (KEGG) molecular interaction network data and 5) microRNA (miRNA) targeting data, 5) the GeneAtlas expression distribution, and 6) transcription factor binding and 7) epigenetic histone modification data from the NIH Roadmap Epigenomics Mapping Consortium, to generate CDH-specific pathogenicity scores ranging from (0–100%) for each RefSeq gene (Callaway et al., 2018; Campbell et al., 2013). These scores represent how closely each gene’s annotation, mouse defect, protein/pathway interaction, microRNA regulation, transcription factor regulation, and epigenetic modification patterns compared to those of the 31 genes in the CDH training set.
To validate our procedure, we performed a leave-one-out cross-validation in which each gene from the training set was individually removed and the remainder of the training set genes were used to fit the machine learning procedure. After each fitting, all of the genes in the genome were ranked based on their similarity to the remaining training set genes. The scores for each of the training set genes were then plotted to characterize the performance of the procedure by tabulating the fraction of test genes with score percentiles exceeding each cutoff. This generated positive receiver operation (ROC)-style curves where the effectiveness of the procedure corresponds to the area under the curve and above a diagonal line which represents the result that would be generated by chance alone. These studies generated positive ROC curves based on data from each knowledge source, and an omnibus score representing the average of the scores across all knowledge sources (Callaway et al., 2018). This demonstrated the ability of our scoring procedure to identify CDH training set genes more efficiently than random chance.
We then performed as second validation in which we identified 35 CDH-related/CDH candidate gene from reviews authored by Donahoe et al and Kardon et al. that were not part of the training set and determined their centile ranks in comparison with all RefSeq genes (Donahoe et al., 2016; Kardon et al., 2017). The median CDH-specific pathogenicity score of these CDH-related genes was 95.5% and all but one gene had ranks >50%. This analysis provided additional evidence that our procedure was able to detect CDH-related genes more efficiently than random chance (Callaway et al., 2018).
Having scored all RefSeq genes with regards to their similarity to the 31 CDH genes in the training set, we then used these CDH-specific pathogenicity scores to prioritize the CDH candidate genes within the CDH critical region on chromosome 4p16 (Callaway et al., 2018). We subsequently used the same scores to evaluate the CDH candidate genes identified using data from a clinical database (Scott et al., 2021). Here, we apply these same CDH-specific pathogenicity scores to a new dataset from the DECIPHER database.
Statistical analysis
Box plots were generated using the Alcula.com Statistical Calculator: Box Plot program (http://www.alcula.com/calculators/statistics/box-plot/). For comparisons of CDH penetrance between reported cohorts and the general population, we used a two-tailed Fisher’s exact tests performed using a 2 × 2 contingency table calculator available through GraphPad QuickCalcs (https://www.graphpad.com/quickcalcs/contingency1/).
To correct for multiple comparisons, we determined an adjusted p value of 0.00179 using the Statology.org Bonferroni Correction Calculator (https://www.statology.org/bonferroni-correction-calculator/) with n = 28, representing all of the genes listed in Tables 1, 2, and 3.
Table 1.
Genes carrying sequence variants for which there is sufficient evidence to support a phenotypic expansion involving CDH/DE
| Gene (pLI) | Disorder† (MIM number) | Subject ID; variant; ACMG interpretation | Expressed in the developing diaphragm?‡ | CDH Pathogenicity Score§ | CDH previously reported for this gene/disorder in humans? | References |
|---|---|---|---|---|---|---|
| CREBBP (pLI = 1) | Rubinstein-Taybi syndrome 1 (MIM: 180849) | Subject 4; c.3779+1G>A [NM_004380.2], p.(?), SpliceAI: ΔS donor loss; Pathogenic | Yes | 92% | No/Yes | (Benjamin et al., 1988; Scott et al., 2021) |
| SMARCA4 (pLI = 1) | Coffin-Siris syndrome 4 (MIM: 614609) | Subject 10; c.2453G>A [NM_001128849.2] p.(Trp818*); Likely pathogenic | Yes | 94% | Yes/Yes | (Delvaux et al., 1998; Fleck et al., 2001; Scott et al., 2021; Sweeney et al., 2018) |
| UBA2 (pLI = 1) | UBA2-Related Disorder | Subject 17; c.364C>T [NM_005499.3], p.(Arg122*); Pathogenic Subject 23, Partial deletion | Yes | 42% | No/No | (Schnur et al., 2021) |
| USP9X (pLI = 1) | Mental retardation, X-linked 99, syndromic, female-restricted (MIM: 300968) | Subject 14; c.1812_1815delTCAA [NM_001039590.3], p.(Gln605Phefs*7); Pathogenic Subject 16; c.5186A>G [NM_001039590.3], p.(His1729Arg); Likely Pathogenic | Yes | 66% | Yes/Yes | (Reijnders et al., 2016) |
Disorder associated with the gene of interest that most closely fits the phenotype descriptions of the individuals in which CDH+ was identified.
Is the mouse orthologue expressed in the developing diaphragm at embryonic day (E)11.5, E12.5 and E16.5 based on whole-transcriptome expression profiles published by Russell et al. (Russell et al., 2012)? This pattern is not seen in ~ 20% of the genes evaluated by Russell et al.
A machine learning-generated centile score that ranks the similarity of this gene to a set of 31 known CDH gene in comparison to all RefSeq genes (Callaway et al., 2018).
Table 2.
Genes carrying sequence variants for which there is currently insufficient evidence to support a phenotypic expansion involving CDH/DE
| Gene | Disorder (MIM number) | Subject ID; variant; ACMG interpretation | Homolog expressed in the developing mouse diaphragm?† | CDH Pathogenicity Score‡ | Other cases of CDH/DE reported for this gene/disorder in humans |
|---|---|---|---|---|---|
| CDK13 § | Congenital heart defects, dysmorphic facial features, and intellectual developmental disorder (MIM: 617360) | Subject 1; c.2524A>G [NM_003718.5], p.(Asn842Asp); Pathogenic | Yes | 75% | No/No |
| CHAMP1 | Mental retardation, autosomal dominant 40 (MIM: 616579) | Subject 2; c.1741delG [NM_001164144.3], p.(Glu581Asnfs*4); Pathogenic | Yes | 81% | No/No |
| CHD8 | Autism, susceptibility to, 18 (MIM: 615032); CHD8-related intellectual disability and overgrowth | Subject 4; c.6394A>C [NM_001170629.2], p.(Asn2132His); VUS | Yes | 52% | No/No |
| PIGL | CHIME syndrome (MIM: 280000) | Subject 6 ; c.[500T>C];[660+1G>T] [NM_004278.4], p.[(Leu167Pro)];[(?)], SpliceAI: ΔS donor loss; Pathogenic/Likely Pathogenic | Yes | 82% | No/No |
| SACS | Spastic ataxia, Charlevoix-Saguernay type (MIM: 270550) | Subject 8; c.[10298C>G];[c.3655C>T] [NM_014363.6], p.[(Pro1219Ser)];[(Thr3433Arg)]; VUS/Likely Benign | Yes | 86% | No/No |
| SETD5 ¶ | Mental retardation, autosomal dominant 23 (MIM: 615761) | Subject 9; c.2302C>T [NM_001080517.3], p.(Arg768*); Pathogenic | Yes | 38% | No/No |
| GNAI1 β | N/A (MIM: 139310) | Subject 9; c.134G>T [NM_002069.6], p.(Gly45Val); VUS | Yes | 93% | No/No |
| TBX3 | Ulnar-mammary syndrome (MIM: 181450) | Subject 12; c.337T>G [NM_005996], p.(Trp113Gly); Likely Pathogenic | Yes | 95% | No/No |
| TCF4 | Pitt-Hopkins syndrome (MIM: 610954) | Subject 13; c.1570C>T [NM_001083962.2], p.(Gln524*); Pathogenic | Yes | 93% | No/No |
Is the mouse orthologue expressed in the developing diaphragm at embryonic day (E)11.5, E12.5 and E16.5 based on whole-transcriptome expression profiles published by Russell et al. (Russell et al., 2012)? This pattern is not seen in ~ 20% of the genes evaluated by Russell et al.
A machine learning-generated centile score that ranks the similarity of this gene to a set of 31 known CDH gene in comparison to all RefSeq genes (Callaway et al., 2018).
The individual with the CDK13 variant was previously reported as Subject 12 by Hamilton et al. (Hamilton et al., 2018).
The individual with the SETD5 and GNAI1 variants was previously reported by Rawlins et al. (Rawlins et al., 2017). N/A = Not applicable
Table 3.
Genes with high pLI scores (> 0.8) that are deleted or potentially disrupted by a duplication for which there is currently insufficient evidence to support a phenotypic expansion involving CDH/DE
| Gene (pLI score) | Disorder (MIM number) | Subject, CNV type | Homolog expressed in the developing mouse diaphragm?† | CDH Pathogenicity Score‡ | Other cases of CDH reported for this gene/disorder in humans |
|---|---|---|---|---|---|
| ACACA (pLI = 1) | Acetyl-CoA carboxylase deficiency (MIM: 613933) | Subject 18, Deletion | Yes | 81% | No/No |
| ATP7A (pLI = 1) | Occipital horn syndrome (MIM: 304150); Menkes disease (MIM: 309400) | Subject 19, Deletion | Yes | 82% | Yes/No§ |
| HMGB1 (pLI = 0.82) | N/A | Subject 20¶, Deletion | Yes | 81% | No/N/A |
| HNF1B (pLI = 1) | Renal cysts and diabetes syndrome (MIM: 137920) | Subject 18, Deletion | Yes | 97% | No/No |
| HSPH1 (pLI = 1) | N/A | Subject 20¶, Deletion | Yes | 72% | No/N/A |
| INTS2 (pLI = 1) | N/A | Subject 21, Deletion | Yes | 81% | No/N/A |
| MED13 (pLI =1) | Intellectual developmental disorder 61 (MIM: 618009) | Subject 21, Deletion | Yes | 72% | No/No |
| SYNRG (pLI = 0.93) | N/A | Subject 18, Deletion | Yes | 9% | No/N/A |
| PACC1 (pLI = 0.84) | N/A | Subject 22, Deletion | Yes | 23% | No/N/A |
| ARHGAP6 (pLI = 1) | N/A | Subject 27, Breakpoint of Duplication | Yes | 92% | No/N/A |
| DLC1 (pLI = 1) | N/A | Subject 28, Breakpoint of Duplication | N/D | 84% | No/N/A |
| FRMPD4 (pLI = 1) | Intellectual developmental disorder, X-linked 104 (MIM: 300983) | Subject 27, Breakpoint of Duplication | Yes | 99% | No/No |
| ILRUN (pLI = 0.85) | N/A | Subject 31, Breakpoint of Duplication | N/D | N/D | No/N/A |
| KANSL1 (pLI = 1) | Koolen-De Vries syndrome (MIM: 610443) | Subject 19, Breakpoint of Duplication | Yes | 57% | No/No |
| TENM1 (pLI = 1) | N/A | Subject 30, Breakpoint of Duplication | N/D | 57% | No/N/A |
Is the mouse orthologue expressed in the developing diaphragm at embryonic day (E)11.5, E12.5 and E16.5 based on whole-transcriptome expression profiles published by Russell et al. (Russell et al., 2012)? This pattern is not seen in ~ 20% of the genes evaluated by Russell et al.
A machine learning-generated centile score that ranks the similarity of this gene to a set of 31 known CDH gene in comparison to all RefSeq genes (Callaway et al., 2018).
Subject 19 is a female with additional phenotypes that are not typical for X-linked Menkes disease.
Previously published as Pt2 by Bartholdi et al. (Bartholdi et al., 2014).
RESULTS
To identify genes that may play a role in diaphragm development, and to discover new phenotypic expansions, we searched the DECIPHER database to identify individuals with CDH who carried single nucleotide variants and rare (not commonly seen in the Database of Genomic Variants), small (< 1.5 Mb and containing 1–20 protein-coding genes) copy number variants (CNVs) (Firth et al., 2009; MacDonald et al., 2014). We obtained permission to publish the clinical and molecular data from 36 individuals with CDH who met these criteria; 15 individuals with sequence variants (Subjects 1–15; Supplemental Information Table S1), and 21 individuals with CNVs (Subjects 18–38; Supplemental Information Table S2). Two additional individuals with CDH, Subjects 16 and 17 were accrued through a research study. Subjects 1, 9, and 20 were previously published (Bartholdi et al., 2014; Hamilton et al., 2018; Rawlins et al., 2017).
For DECIPHER patients with deletions, we focused on genes that are predicted to have high loss-of-function intolerance (pLI >0.8 in gnomAD) since they are the most likely to be able to cause CDH when deleted (Karczewski et al., 2020). For DECIPHER patients with duplications, we focused on genes that are predicted to have high loss-of-function intolerance (pLI >0.8 in gnomAD) whose function may have been disrupted by the duplications based on the presence of one or more breakpoints within the gene. For example, Subjects 29 and 30, a male child and his mother, respectively, carried identical intergenic duplications that involved exons 3–6 of the GPC3 gene [NM_001164617]. Loss of GPC3 function causes Simpson-Golabi-Behmel syndrome, type 1 (MIM: 312870), an X-linked overgrowth syndrome that has been previously shown to be associated with the development of CDH that can affect both males and females (Chong et al., 2018; Vaisfeld et al., 2017).
Five individuals in the cohort carried sequence changes in genes that have already been associated with CDH including CHRNG (n = 1), GATA4 (n = 1), RARB (n = 1), STAG2 (n = 1) and WT1 (n =1) (Aoi et al., 2020; Devriendt et al., 1995; Jordan et al., 2018; Morgan et al., 2006; Srour et al., 2013; Yu et al., 2015; Yu et al., 2013). The remaining 31 individuals carried sequence or CNVs affecting genes whose association with CDH has not been clearly defined. To determine the likelihood that these genes contribute to the development of CDH, we used an approach previously employed by Scott et al. (Scott et al., 2021). Briefly, we determined whether each gene was: 1) expressed in the developing mouse diaphragm at embryonic day (E)11.5, E12.5, and E16.5 based on whole-transcriptome expression profiles published by Russell et al. (Russell et al., 2012), 2) had a high similarity to previously published CDH genes based on a high CDH-specific pathogenicity scores (≥85% rank compared to all RefSeq genes) previously generated using a validated machine learning procedure (Callaway et al., 2018; Campbell et al., 2013), and/or 3) had been previously reported in association with CDH in humans, or is known to cause a genetic syndrome previously associated with CDH. The results of these evaluations are summarized in Tables 1–3.
Genes for which there is sufficient evidence to support a phenotypic expansion including CDH are shown in Table 1 and include CREBBP (Subject 4), SMARCA4 (Subject 10), UBA2 (Subjects 17 and 23) and USP9X (Subjects 14 and 16). Genes affected by sequence variants or CNVs for which there is currently insufficient evidence to suggest that they are associated with the development of CDH are shown in Tables 2 and 3, respectively.
Including the individuals reported in this manuscript, there have now been four individuals reported with CDH associated with causative variants in SMARCA4, three individuals reported with CDH associated with causative variants in UBA2, and three females reported with CDH associated with causative variants in USP9X (Reijnders et al., 2016; Scott et al., 2021; Wild et al., 2022). The human disorders associated with these genes were only described in 2012, 2021, and 2016, respectively (Reijnders et al., 2016; Schnur et al., 2021; Tsurusaki et al., 2012). Hence, it is reasonable to assume that less than 650 individuals with Coffin-Siris syndrome 4 caused by variants in SMARCA4, less than 330 individuals with UBA2-related disorder, and less than 330 individuals with intellectual developmental disorder, X-linked 99, syndromic, female-restricted caused by variants in USP9X have been reported to date. If this assumption is true, then the CDH rate in individuals reported with these disorders is significantly higher (p ≤ 0.0017; two-tailed Fisher’s exact tests) than what would be expected based on the rate of CDH in the general population (1 in 4,000), even when taking into account multiple comparisons (Bonferroni adjusted p value = 0.00179).
As a means of assessing the performance of the CDH-specific pathogenicity scores used in this manuscript, we compared the median scores of the 31 CDH genes used to train our machine learning algorithm and the genes in Tables 1–3 (Figure 1). As expected, the training set had the highest median (98.8%). The medians of the genes listed in Table 1 (78.7%), Table 2 (81.5%), and Table 3 (76.4%) were lower, but all exceeded the median for all RefSeq genes (50%), indicating that each of these groups are enriched for genes that are similar to the known CDH genes in the training set.
Figure 1. Genes identified in this study are more similar to a set of 31 genes known to cause CDH than would be expected by chance.
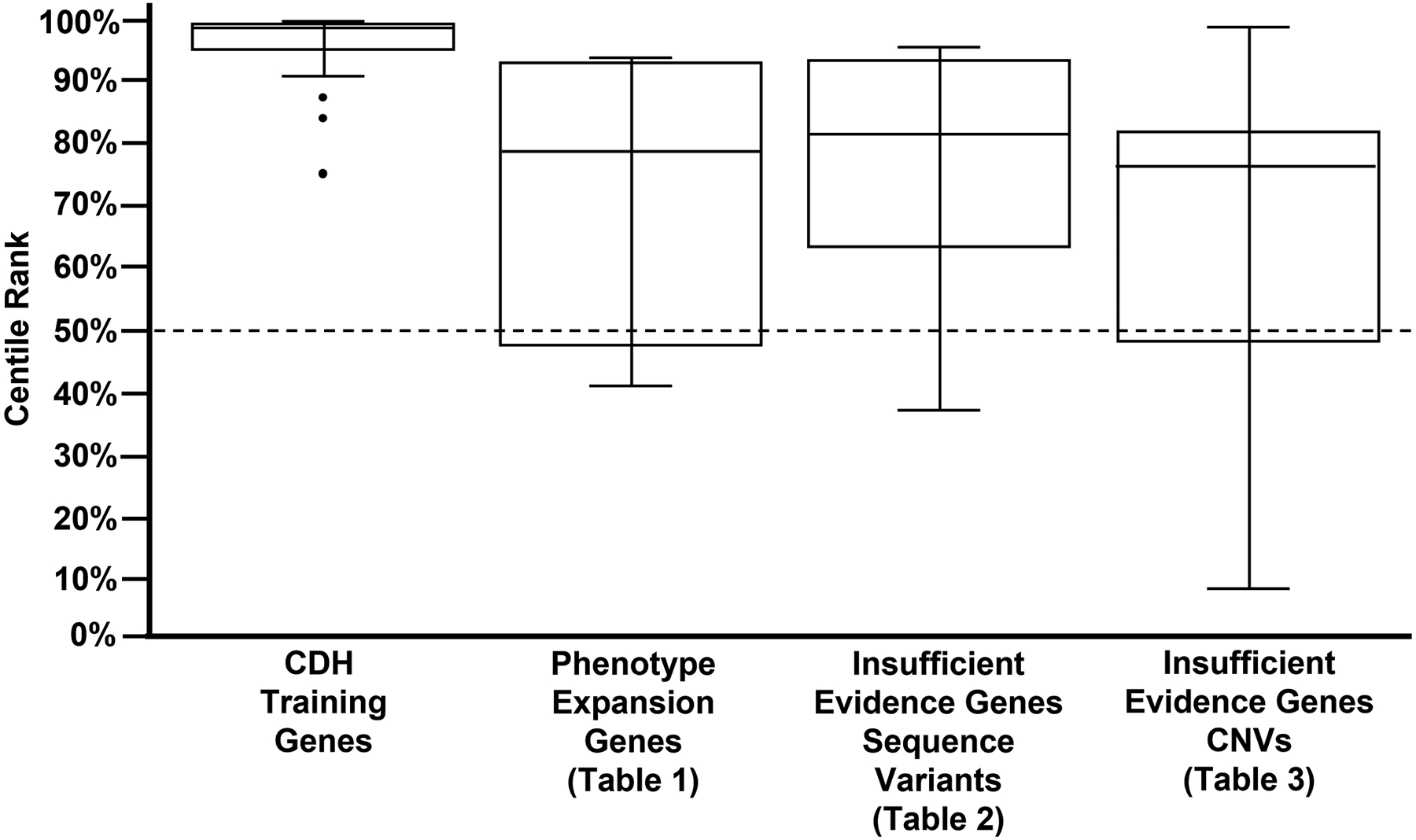
Box plots generated based on the CDH-specific pathogenicity scores of, 1) the 31 known CDH genes used to train our machine learning algorithm (Callaway et al., 2018), 2) genes for which there is sufficient evidence to support a phenotype expansion involving CDH (Table 1) and, 3) genes for which there is currently insufficient evidence to support a phenotype expansion involving CDH (Tables 2 and 3). As expected, the medians of the genes listed in Table 1 (78.7%), Table 2 (81.5%) and Table 3 (76.4%) were lower that of the training genes (99.8%) but exceeded the median for all RefSeq genes (50%; chance) represented by the dashed line. This indicates that each of these groups are enriched for genes that are similar to the 31 known CDH genes in the training set.
DISCUSSION
CDH is a relatively common birth defect which may be associated with multifactorial inheritance risk (Edwards, 1960; Norio et al., 1984; Wolff, 1980). It is possible, therefore, that some individuals with pathogenic variants in known disease genes develop CDH through an unrelated mechanism. In other cases, the pathogenic variants may be causative. Distinguishing between a non-causal and causal relationship can be difficult, especially since CDH is often found to be an incompletely penetrant phenotype of genetic disorders.
In this manuscript, we searched the DECIPHER database to identify individuals with CDH. We then evaluated whether the genes affected by sequence changes or CNVs they carried were likely to be causative of their CDH based on: 1) the gene’s expression in the developing mouse diaphragm, 2) the gene’s similarity to known CDH genes based on CDH-specific pathogenicity scores generated by a previously published and validated machine learning procedure, and/or 3) whether the gene, or its associated genetic syndrome, has been previously associated with CDH in the literature. Based on these criteria, there is sufficient evidence to suggest that pathogenic variants in CREBBP, SMARCA4, UBA2, and USP9X can predispose an individual to develop CDH.
CREBBP encodes a nuclear transcriptional coactivator protein that regulates gene activity throughout the body (Chrivia et al., 1993). Pathogenic variants of the CREBBP cause Rubinstein-Taybi syndrome 1 (MIM: 180849) (Petrij et al., 1995). This syndrome is characterized by neurodevelopmental phenotypes, distinct facial features, broad thumbs and first toes, short stature, and a high rate of congenital heart defects and renal anomalies (Milani et al., 2015). Although individuals with clinically diagnosed Rubinstein-Taybi syndrome or Rubinstein-Taybi syndrome 2 caused by pathogenic variants in EP300 have been reported to have CDH, CREBBP has not been previously implicated in the development of CDH (Benjamin et al., 1988; Scott et al., 2021). Subject 4 is a male with CDH who carries a de novo c.3779+1G>A [NM_004380.2] pathogenic variant in CREBBP. CREBBP’s putative role in diaphragm development is also supported by the expression of its mouse homolog in the developing diaphragm and CREBBP’s high CDH-specific pathogenicity score of 92% (Callaway et al., 2018; Russell et al., 2012). Subject 4 also carried a de novo c.6394A>C [NM_001170629.2], p.(Asn2132His) VUS in CHD8, a gene associated with autism, susceptibility to, 18 (MIM: 615032), and more recently implicated as a cause of intellectual disability and overgrowth (Ostrowski et al., 2019). We cannot exclude the possibility that this variant also contributed to the development of CDH in this individual.
SMARCA4 encodes a catalytic subunit of SWI/SNF complexes which serves to regulate gene expression by altering nucleosome conformation through chromatin remodeling (Barutcu et al., 2016). Pathogenic variants in this gene cause Coffin-Siris syndrome 4 (MIM: 614609) (Tsurusaki et al., 2012). The majority of SMARCA4 variants associated with Coffin-Siris syndrome 4 are missense or small in-frame deletions and have been hypothesized to exert dominant-negative or gain-of-function effects (Kosho et al., 2014). However, recent reports have indicated that inactivating variants may cause a milder version of this syndrome (Errichiello et al., 2017; Li et al., 2020; Mitrakos et al., 2020). Subject 10 is a male with a right-sided CDH who died shortly after birth. He carried a likely pathogenic c.2453G>A [NM_001128849.2], p.(Trp818*) stop gain variant in SMARCA4 that was inherited from an asymptomatic father. SMRCA4’s homolog is expressed in the developing mouse diaphragm, and it has a CDH-specific pathogenicity score of 94%. Recently, three other individuals with CDH were reported to carry de novo, pathogenic (c.2936G>A [NM_001128849.2], p.(R979Q)), or likely pathogenic (c.3595G>A NM_001128849.3], p.(Val1199Met); c.3728G>A [NM_001128844.3], p.(Arg1243Gln)), variants in SMARCA4 (Scott et al., 2021; Wild et al., 2022). The association of SMARCA4 with CDH is also supported by the fact that CDH has been seen previously in individuals with Coffin-Siris syndrome (Delvaux et al., 1998; Fleck et al., 2001; Russell et al., 2012; Sweeney et al., 2018).
UBA2 binds to SAE1 to form a heterodimer that plays a key role in the SUMOylation of proteins by activating SUMO1 (Desterro et al., 1999; Okuma et al., 1999). SUMOylation, in turn, plays an essential role in a variety of biological processes including cell growth, migration, and cellular responses to stress (Yang et al., 2017). Heterozygous pathogenic variants in UBA2 are associated with a recently described genetic disorder characterized by highly variable neurologic, cardiac, renal, skeletal, dermatologic and extremity phenotypes (Schnur et al., 2021). UBA2 is partially deleted in Subject 23 by an ~175 kb interstitial deletion of chromosome 19q13.11. She has a variety of structural birth defects including a large 5 cm × 5 cm, left-sided CDH with only a thin rim of tissue posteriorly. Her identical twin, who presumably carries the same deletion, had a similar large, left-sided CDH. Subject 17, who has a cleft lip and a Morgagni type CDH diagnosed at 1 year of age, carries a de novo pathogenic c.364C>T [NM_005499.3], p.(Arg122*) variant in UBA2. The identification of three individuals with CDH associated with variants affecting UBA2 provides strong evidence for its association with CDH. As expected, the mouse homolog of UBA2 is expressed in the developing mouse diaphragm (Russell et al., 2012). Since UBA2’s function in SUMOylation is distinct from that of any other CDH-related gene, it is not surprising that its CDH-specific pathogenicity score is relatively low at 42% (Callaway et al., 2018).
USP9X encodes a large substrate-specific deubiquitylating enzyme similar to ubiquitin-specific proteases (Wood et al., 1997). Pathogenic hemizygous and heterozygous variants in USP9X cause intellectual developmental disorder, X-linked 99 (MIM: 300919) and intellectual developmental disorder, X-linked 99, syndromic, female-restricted (MIM: 300968), respectively (Homan et al., 2014; Reijnders et al., 2016). Reijnders et al. reported a female with intellectual developmental disorder, X-linked 99, syndromic, female-restricted and CDH who carried a de novo c.2554C>T [NM_001039590.3], p.(Arg852*) stop gain variant in USP9X (Reijnders et al., 2016). Subject 14 is a female with CDH who carries a de novo, pathogenic c.1812_1815delTCAA [NM_001039590.3], p.(Gln605Phefs*7) variant in USP9X, and Subject 16 is a female with a left-sided CDH who carries a de novo, likely pathogenic c.5186A>G [NM_001039590.3], p.(His1729Arg) variant in USP9X. This variant is considered “Disease Causing” by MutationTaster and has a CADD score of 25 (Rentzsch et al., 2021; Schwarz et al., 2014). Subject 14 has abnormal cerebral white matter and global developmental delay, and Subject 16 has partial agenesis of the corpus callosum, speech delay, and hypotonia, consistent with a diagnosis of intellectual developmental disorder, X-linked 99, syndromic, female-restricted. The identification of three CDH cases with putatively damaging variants in USP9X provides strong evidence for its association with CDH. USP9X’s mouse homolog is expressed in the developing mouse diaphragm (Russell et al., 2012). USP9X has a relatively low CDH-specific pathogenicity score of 66%, possibly due to its unique role in ubiquitination, which is uncommon among CDH-related genes.
In conclusion, our results suggest that CREBBP, SMARCA4, UBA2, and USP9X play a role in diaphragm development and that individuals affected by their associated genetic disorders—Rubinstein-Taybi syndrome 1, Coffin-Siris syndrome 4, UBA2-related disorder, and mental retardation, X-linked 99, syndromic, female-restricted—can present with CDH. Currently, there is insufficient evidence to support an association between CDH and the other genes affected by sequence variants or CNVs reported here. Many of these genes are expressed in the developing mouse diaphragm, and several have high similarity to genes implicated in the development of CDH as evidenced by their high CDH-specific pathogenicity scores. Hence, it is possible that they will be implicated in the development of CDH in the future.
Supplementary Material
ACKNOWLEDGEMENTS
This study makes use of data generated by the DECIPHER community. A full list of centers who contributed to the generation of the data is available from https://deciphergenomics.org/about/stats and via email from contact@deciphergenomics.org. We note that those who carried out the original analysis and collection of the DECIPHER data bear no responsibility for the further analysis or interpretation of the data.
FUNDING:
This research was funded in part by NIH/NICHD grants HD098458 to D.A.S and P01HD068250 to W.K.C. This work was also supported by CPRIT RP160283 - Baylor College of Medicine Comprehensive Cancer Training Program. Funding for the DECIPHER project was provided by Wellcome.
CONFLICT OF INTEREST:
Baylor College of Medicine (BCM) and Miraca Holdings Inc. have formed a joint venture with shared ownership and governance of Baylor Genetics (BG), which performs genetic testing and derives revenue. P. L. is an employee of BCM and derives support through a professional services agreement with BG. The Department of Molecular & Human Genetics at Baylor College of Medicine receives revenue from clinical genetic testing completed at BG.
DATA AVAILABILITY STATEMENT
The data generated during this study can be found within the published article and its supplementary files. Variants described in this manuscript have been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/).
REFERENCES
- Aoi H, Lei M, Mizuguchi T, Nishioka N, Goto T, Miyama S, Suzuki T, Iwama K, Uchiyama Y, Mitsuhashi S, Itakura A, Takeda S, & Matsumoto N (2020). Nonsense variants of STAG2 result in distinct congenital anomalies. Hum Genome Var, 7, 26. 10.1038/s41439-020-00114-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartholdi D, Stray-Pedersen A, Azzarello-Burri S, Kibaek M, Kirchhoff M, Oneda B, Rodningen O, Schmitt-Mechelke T, Rauch A, & Kjaergaard S (2014). A newly recognized 13q12.3 microdeletion syndrome characterized by intellectual disability, microcephaly, and eczema/atopic dermatitis encompassing the HMGB1 and KATNAL1 genes. Am J Med Genet A, 164A, 1277–1283. 10.1002/ajmg.a.36439 [DOI] [PubMed] [Google Scholar]
- Barutcu AR, Lajoie BR, Fritz AJ, McCord RP, Nickerson JA, van Wijnen AJ, Lian JB, Stein JL, Dekker J, Stein GS, & Imbalzano AN (2016). SMARCA4 regulates gene expression and higher-order chromatin structure in proliferating mammary epithelial cells. Genome Res, 26, 1188–1201. 10.1101/gr.201624.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin DR, Juul S, & Siebert JR (1988). Congenital posterolateral diaphragmatic hernia: associated malformations. J Pediatr Surg, 23, 899–903. 10.1016/s0022-3468(88)80380-4 [DOI] [PubMed] [Google Scholar]
- Callaway DA, Campbell IM, Stover SR, Hernandez-Garcia A, Jhangiani SN, Punetha J, Paine IS, Posey JE, Muzny D, Lally KP, Lupski JR, Shaw CA, Fernandes CJ, & Scott DA (2018). Prioritization of Candidate Genes for Congenital Diaphragmatic Hernia in a Critical Region on Chromosome 4p16 using a Machine-Learning Algorithm. J Pediatr Genet, 7, 164–173. 10.1055/s-0038-1655755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell IM, Rao M, Arredondo SD, Lalani SR, Xia Z, Kang SH, Bi W, Breman AM, Smith JL, Bacino CA, Beaudet AL, Patel A, Cheung SW, Lupski JR, Stankiewicz P, Ramocki MB, & Shaw CA (2013). Fusion of large-scale genomic knowledge and frequency data computationally prioritizes variants in epilepsy. PLoS Genet, 9, e1003797. 10.1371/journal.pgen.1003797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong K, Saleh M, Injeyan M, Miron I, Fong K, & Shannon P (2018). Nonisolated diaphragmatic hernia in Simpson-Golabi-Behmel syndrome. Prenat Diagn, 38, 117–122. 10.1002/pd.5198 [DOI] [PubMed] [Google Scholar]
- Chrivia JC, Kwok RP, Lamb N, Hagiwara M, Montminy MR, & Goodman RH (1993). Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature, 365, 855–859. 10.1038/365855a0 [DOI] [PubMed] [Google Scholar]
- Clugston RD, & Greer JJ (2007). Diaphragm development and congenital diaphragmatic hernia. Semin Pediatr Surg, 16, 94–100. 10.1053/j.sempedsurg.2007.01.004 [DOI] [PubMed] [Google Scholar]
- Delvaux V, Moerman P, & Fryns JP (1998). Diaphragmatic hernia in the Coffin-Siris syndrome. Genet Couns, 9, 45–50. https://www.ncbi.nlm.nih.gov/pubmed/9555587 [PubMed] [Google Scholar]
- Desterro JM, Rodriguez MS, Kemp GD, & Hay RT (1999). Identification of the enzyme required for activation of the small ubiquitin-like protein SUMO-1. J Biol Chem, 274, 10618–10624. 10.1074/jbc.274.15.10618 [DOI] [PubMed] [Google Scholar]
- Devriendt K, Deloof E, Moerman P, Legius E, Vanhole C, de Zegher F, Proesmans W, & Devlieger H (1995). Diaphragmatic hernia in Denys-Drash syndrome. Am J Med Genet, 57, 97–101. 10.1002/ajmg.1320570120 [DOI] [PubMed] [Google Scholar]
- Donahoe PK, Longoni M, & High FA (2016). Polygenic Causes of Congenital Diaphragmatic Hernia Produce Common Lung Pathologies. Am J Pathol, 186, 2532–2543. 10.1016/j.ajpath.2016.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards JH (1960). The simulation of mendelism. Acta Genet Stat Med, 10, 63–70. http://www.ncbi.nlm.nih.gov/pubmed/13725809 [DOI] [PubMed] [Google Scholar]
- Errichiello E, Mustafa N, Vetro A, Notarangelo LD, de Jonge H, Rinaldi B, Vergani D, Giglio SR, Morbini P, & Zuffardi O (2017). SMARCA4 inactivating mutations cause concomitant Coffin-Siris syndrome, microphthalmia and small-cell carcinoma of the ovary hypercalcaemic type. J Pathol, 243, 9–15. 10.1002/path.4926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, Van Vooren S, Moreau Y, Pettett RM, & Carter NP (2009). DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am J Hum Genet, 84, 524–533. 10.1016/j.ajhg.2009.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleck BJ, Pandya A, Vanner L, Kerkering K, & Bodurtha J (2001). Coffin-Siris syndrome: review and presentation of new cases from a questionnaire study. Am J Med Genet, 99, 1–7. [DOI] [PubMed] [Google Scholar]
- Hamilton MJ, Caswell RC, Canham N, Cole T, Firth HV, Foulds N, Heimdal K, Hobson E, Houge G, Joss S, Kumar D, Lampe AK, Maystadt I, McKay V, Metcalfe K, Newbury-Ecob R, Park SM, Robert L, Rustad CF, … Suri M (2018). Heterozygous mutations affecting the protein kinase domain of CDK13 cause a syndromic form of developmental delay and intellectual disability. J Med Genet, 55, 28–38. 10.1136/jmedgenet-2017-104620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homan CC, Kumar R, Nguyen LS, Haan E, Raymond FL, Abidi F, Raynaud M, Schwartz CE, Wood SA, Gecz J, & Jolly LA (2014). Mutations in USP9X are associated with X-linked intellectual disability and disrupt neuronal cell migration and growth. Am J Hum Genet, 94, 470–478. 10.1016/j.ajhg.2014.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan VK, Beck TF, Hernandez-Garcia A, Kundert PN, Kim BJ, Jhangiani SN, Gambin T, Starkovich M, Punetha J, Paine IS, Posey JE, Li AH, Muzny D, Hsu CW, Lashua AJ, Sun X, Fernandes CJ, Dickinson ME, Lally KP, … Scott DA (2018). The role of FREM2 and FRAS1 in the development of congenital diaphragmatic hernia. Hum Mol Genet, 27, 2064–2075. 10.1093/hmg/ddy110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, Brand H, Solomonson M, Watts NA, Rhodes D, Singer-Berk M, England EM, Seaby EG, Kosmicki JA, … MacArthur DG (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581, 434–443. 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kardon G, Ackerman KG, McCulley DJ, Shen Y, Wynn J, Shang L, Bogenschutz E, Sun X, & Chung WK (2017). Congenital diaphragmatic hernias: from genes to mechanisms to therapies. Dis Model Mech, 10, 955–970. 10.1242/dmm.028365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosho T, Okamoto N, & Coffin-Siris Syndrome International C (2014). Genotype-phenotype correlation of Coffin-Siris syndrome caused by mutations in SMARCB1, SMARCA4, SMARCE1, and ARID1A. Am J Med Genet C Semin Med Genet, 166C, 262–275. 10.1002/ajmg.c.31407 [DOI] [PubMed] [Google Scholar]
- Langham MR Jr., Kays DW, Ledbetter DJ, Frentzen B, Sanford LL, & Richards DS (1996). Congenital diaphragmatic hernia. Epidemiology and outcome [Review]. Clin Perinatol, 23, 671–688. http://www.ncbi.nlm.nih.gov/pubmed/8982563 [PubMed] [Google Scholar]
- Li D, Ahrens-Nicklas RC, Baker J, Bhambhani V, Calhoun A, Cohen JS, Deardorff MA, Fernandez-Jaen A, Kamien B, Jain M, McKenzie F, Mintz M, Motter C, Niles K, Ritter A, Rogers C, Roifman M, Townshend S, Ward-Melver C, & Schrier Vergano SA (2020). The variability of SMARCA4-related Coffin-Siris syndrome: Do nonsense candidate variants add to milder phenotypes? Am J Med Genet A, 182, 2058–2067. 10.1002/ajmg.a.61732 [DOI] [PubMed] [Google Scholar]
- MacDonald JR, Ziman R, Yuen RK, Feuk L, & Scherer SW (2014). The Database of Genomic Variants: a curated collection of structural variation in the human genome. Nucleic Acids Res, 42, D986–992. 10.1093/nar/gkt958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milani D, Manzoni FM, Pezzani L, Ajmone P, Gervasini C, Menni F, & Esposito S (2015). Rubinstein-Taybi syndrome: clinical features, genetic basis, diagnosis, and management. Ital J Pediatr, 41, 4. 10.1186/s13052-015-0110-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitrakos A, Lazaros L, Pantou A, Mavrou A, Kanavakis E, & Tzetis M (2020). Coffin-Siris Syndrome 4-Related Spectrum in a Young Woman Caused by a Heterozygous SMARCA4 Deletion Detected by High-Resolution aCGH. Mol Syndromol, 11, 141–145. 10.1159/000508563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan NV, Brueton LA, Cox P, Greally MT, Tolmie J, Pasha S, Aligianis IA, van Bokhoven H, Marton T, Al-Gazali L, Morton JE, Oley C, Johnson CA, Trembath RC, Brunner HG, & Maher ER (2006). Mutations in the embryonal subunit of the acetylcholine receptor (CHRNG) cause lethal and Escobar variants of multiple pterygium syndrome. Am J Hum Genet, 79, 390–395. 10.1086/506256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norio R, Kaariainen H, Rapola J, Herva R, & Kekomaki M (1984). Familial congenital diaphragmatic defects: aspects of etiology, prenatal diagnosis, and treatment. Am J Med Genet, 17, 471–483. 10.1002/ajmg.1320170210 [DOI] [PubMed] [Google Scholar]
- Okuma T, Honda R, Ichikawa G, Tsumagari N, & Yasuda H (1999). In vitro SUMO-1 modification requires two enzymatic steps, E1 and E2. Biochem Biophys Res Commun, 254, 693–698. 10.1006/bbrc.1998.9995 [DOI] [PubMed] [Google Scholar]
- Ostrowski PJ, Zachariou A, Loveday C, Beleza-Meireles A, Bertoli M, Dean J, Douglas AGL, Ellis I, Foster A, Graham JM, Hague J, Hilhorst-Hofstee Y, Hoffer M, Johnson D, Josifova D, Kant SG, Kini U, Lachlan K, Lam W, … Tatton-Brown K (2019). The CHD8 overgrowth syndrome: A detailed evaluation of an emerging overgrowth phenotype in 27 patients. Am J Med Genet C Semin Med Genet, 181, 557–564. 10.1002/ajmg.c.31749 [DOI] [PubMed] [Google Scholar]
- Petrij F, Giles RH, Dauwerse HG, Saris JJ, Hennekam RC, Masuno M, Tommerup N, van Ommen GJ, Goodman RH, Peters DJ, & et al. (1995). Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature, 376, 348–351. 10.1038/376348a0 [DOI] [PubMed] [Google Scholar]
- Qi H, Yu L, Zhou X, Wynn J, Zhao H, Guo Y, Zhu N, Kitaygorodsky A, Hernan R, Aspelund G, Lim FY, Crombleholme T, Cusick R, Azarow K, Danko ME, Chung D, Warner BW, Mychaliska GB, Potoka D, … Shen Y (2018). De novo variants in congenital diaphragmatic hernia identify MYRF as a new syndrome and reveal genetic overlaps with other developmental disorders. PLoS Genet, 14, e1007822. 10.1371/journal.pgen.1007822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlins LE, Stals KL, Eason JD, & Turnpenny PD (2017). De novo SETD5 nonsense mutation associated with diaphragmatic hernia and severe cerebral cortical dysplasia. Clin Dysmorphol, 26, 95–97. 10.1097/MCD.0000000000000144 [DOI] [PubMed] [Google Scholar]
- Reijnders MR, Zachariadis V, Latour B, Jolly L, Mancini GM, Pfundt R, Wu KM, van Ravenswaaij-Arts CM, Veenstra-Knol HE, Anderlid BM, Wood SA, Cheung SW, Barnicoat A, Probst F, Magoulas P, Brooks AS, Malmgren H, Harila-Saari A, Marcelis CM, … Kleefstra T (2016). De Novo Loss-of-Function Mutations in USP9X Cause a Female-Specific Recognizable Syndrome with Developmental Delay and Congenital Malformations. Am J Hum Genet, 98, 373–381. 10.1016/j.ajhg.2015.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentzsch P, Schubach M, Shendure J, & Kircher M (2021). CADD-Splice-improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med, 13, 31. 10.1186/s13073-021-00835-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell MK, Longoni M, Wells J, Maalouf FI, Tracy AA, Loscertales M, Ackerman KG, Pober BR, Lage K, Bult CJ, & Donahoe PK (2012). Congenital diaphragmatic hernia candidate genes derived from embryonic transcriptomes [Research Support, Non-U.S. Gov’t]. Proc Natl Acad Sci U S A, 109, 2978–2983. 10.1073/pnas.1121621109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnur RE, Yousaf S, Liu J, Chung WK, Rhodes L, Marble M, Zambrano RM, Sobreira N, Jayakar P, Pierpont ME, Schultz MJ, Pichurin PN, Olson RJ, Graham GE, Osmond M, Contreras-Garcia GA, Campo-Neira KA, Penaloza-Mantilla CA, Flage M, … Hufnagel RB (2021). UBA2 variants underlie a recognizable syndrome with variable aplasia cutis congenita and ectrodactyly. Genet Med, 23, 1624–1635. 10.1038/s41436-021-01182-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Cooper DN, Schuelke M, & Seelow D (2014). MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods, 11, 361–362. 10.1038/nmeth.2890 [DOI] [PubMed] [Google Scholar]
- Scott TM, Campbell IM, Hernandez-Garcia A, Lalani SR, Liu P, Shaw CA, Rosenfeld JA, & Scott DA (2021). Clinical exome sequencing data reveal high diagnostic yields for congenital diaphragmatic hernia plus (CDH+) and new phenotypic expansions involving CDH. J Med Genet, 59, 270–278. 10.1136/jmedgenet-2020-107317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srour M, Chitayat D, Caron V, Chassaing N, Bitoun P, Patry L, Cordier MP, Capo-Chichi JM, Francannet C, Calvas P, Ragge N, Dobrzeniecka S, Hamdan FF, Rouleau GA, Tremblay A, & Michaud JL (2013). Recessive and dominant mutations in retinoic acid receptor beta in cases with microphthalmia and diaphragmatic hernia. Am J Hum Genet, 93, 765–772. 10.1016/j.ajhg.2013.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney NM, Nahas SA, Chowdhury S, Campo MD, Jones MC, Dimmock DP, Kingsmore SF, & Investigators R (2018). The case for early use of rapid whole-genome sequencing in management of critically ill infants: late diagnosis of Coffin-Siris syndrome in an infant with left congenital diaphragmatic hernia, congenital heart disease, and recurrent infections. Cold Spring Harb Mol Case Stud, 4. 10.1101/mcs.a002469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsurusaki Y, Okamoto N, Ohashi H, Kosho T, Imai Y, Hibi-Ko Y, Kaname T, Naritomi K, Kawame H, Wakui K, Fukushima Y, Homma T, Kato M, Hiraki Y, Yamagata T, Yano S, Mizuno S, Sakazume S, Ishii T, … Matsumoto N (2012). Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat Genet, 44, 376–378. 10.1038/ng.2219 [DOI] [PubMed] [Google Scholar]
- Vaisfeld A, Pomponi MG, Pietrobono R, Tabolacci E, & Neri G (2017). Simpson-Golabi-Behmel syndrome in a female: A case report and an unsolved issue. Am J Med Genet A, 173, 285–288. 10.1002/ajmg.a.38003 [DOI] [PubMed] [Google Scholar]
- Wat MJ, Veenma D, Hogue J, Holder AM, Yu Z, Wat JJ, Hanchard N, Shchelochkov OA, Fernandes CJ, Johnson A, Lally KP, Slavotinek A, Danhaive O, Schaible T, Cheung SW, Rauen KA, Tonk VS, Tibboel D, de Klein A, & Scott DA (2011). Genomic alterations that contribute to the development of isolated and non-isolated congenital diaphragmatic. J Med Genet, 48, 299–307. 10.1136/jmg.2011.089680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild KT, Schindewolf E, Hedrick HL, Rintoul NE, Hartman T, Gebb J, Moldenhauer JS, Zackai EH, & Krantz ID (2022). The Genomics of Congenital Diaphragmatic Hernia: A 10 Year Retrospective Review. J Pediatr. Apr 14;S0022-3476(22)00326-2. doi: 10.1016/j.jpeds.2022.04.012. [DOI] [PubMed] [Google Scholar]
- Wolff G (1980). Familial congenital diaphragmatic defect: review and conclusions [Review]. Hum Genet, 54, 1–5. http://www.ncbi.nlm.nih.gov/pubmed/6993337 [DOI] [PubMed] [Google Scholar]
- Wood SA, Pascoe WS, Ru K, Yamada T, Hirchenhain J, Kemler R, & Mattick JS (1997). Cloning and expression analysis of a novel mouse gene with sequence similarity to the Drosophila fat facets gene. Mech Dev, 63, 29–38. 10.1016/s0925-4773(97)00672-2 [DOI] [PubMed] [Google Scholar]
- Yang Y, He Y, Wang X, Liang Z, He G, Zhang P, Zhu H, Xu N, & Liang S (2017). Protein SUMOylation modification and its associations with disease. Open Biol, 7. 10.1098/rsob.170167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Sawle AD, Wynn J, Aspelund G, Stolar CJ, Arkovitz MS, Potoka D, Azarow KS, Mychaliska GB, Shen Y, & Chung WK (2015). Increased burden of de novo predicted deleterious variants in complex congenital diaphragmatic hernia. Hum Mol Genet, 24, 4764–4773. 10.1093/hmg/ddv196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Wynn J, Cheung YH, Shen Y, Mychaliska GB, Crombleholme TM, Azarow KS, Lim FY, Chung DH, Potoka D, Warner BW, Bucher B, Stolar C, Aspelund G, Arkovitz MS, & Chung WK (2013). Variants in GATA4 are a rare cause of familial and sporadic congenital diaphragmatic. Hum Genet, 132(3), 285–292. 10.1007/s00439-012-1249-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated during this study can be found within the published article and its supplementary files. Variants described in this manuscript have been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/).