

Abstract
Circadian rhythm disruption is implicated in the initiation and progression of many diseases, including cancer. External stimuli, such as sunlight, serve to synchronize physiological processes and cellular functions to a 24-h cycle. The immune system is controlled by circadian rhythms, and perturbation of these rhythms can potentially alter the immune response to infections and tumors. The effect of circadian rhythm disruption on the immune response to tumors remains unclear. Specifically, the effects of circadian disruption (CD) on immunosuppressive cell types within the tumor, such as myeloid-derived suppressor cells (MDSCs), are unknown. In this study, a shifting lighting schedule is used to disrupt the circadian rhythm of mice. After acclimation to lighting schedules, mice are inoculated with 4T1 or B16-F10 tumors. Tumor growth is increased in mice housed under circadian disrupting lighting conditions compared to standard lighting conditions. Analysis of immune populations within the spleen and tumor shows an increased accumulation of MDSCs within these tissues, suggesting that MDSC mediated immunosuppression plays a role in the enhanced tumor growth caused by circadian disruption. This paves the way for future studies of the effects of CD on immunosuppression in cancer.
Keywords: cancer, circadian rhythm, immunology, immunosuppression, myeloid-derived suppressor cells, tumor immunology
1. Introduction
Circadian regulation allows organisms to anticipate daily environmental changes, temporally regulating many key processes essential for the function of an organism. Circadian rhythms are able to maintain periodicity within the 24-h period even when environmental conditions are kept constant, but the phase of a circadian cycle is determined by external environmental stimuli, such as sunlight. These external cues, known as zeitgebers, serve to synchronize internal rhythms with periodic external events. Exposure to nonperiodic or unnatural zeitgebers can disrupt circadian rhythms, alter neuroendocrine stress pathways, and impact a wide range of biological processes. Disruption of circadian rhythm has been implicated in the development of metabolic disease, cardiovascular disease, mental disorders, and cancer.[1]
The role of clock genes and circadian disruption (CD) in cancer progression, metastasis, and angiogenesis has been studied in vitro and in vivo.[2] Clinical trials have utilized melatonin in the treatment of cancer patients to correct circadian rhythm disruptions and have also investigated the effects of delivering pharmacological therapies at specific points of the circadian cycle in order to improve their efficacy and tolerance. Although these initial studies are provocative, they do not solidify our mechanistic understanding of how CD influences cancer development and progression.
Research in recent years has shown that immune functions are regulated by circadian rhythms. The concentration of immune mediating hormones and cytokines oscillate throughout the day, and circulating hematopoietic cells fluctuate rhythmically in both number and cell type.[3] This regulation of immunologic processes has the potential to be of particular importance to cancer research because it is now accepted that the immune system plays a pivotal role in eliminating and suppressing malignancies. Any perturbation of the balance between anti-tumor immune cells and pro-tumor immunosuppressive cells has the potential to allow cancer cells to escape immune control, resulting in disease progression.[4]
Two populations of immune cells that play a critical role in this balance are T cells and myeloid-derived suppressor cells (MDSCs). Although there are many distinct types of T cells involved in the immune response to tumors, CD8+ T cells generally act as the primary anti-tumor immune cell responsible for selectively killing tumor cells.[5] MDSCs on the other hand, function to quench the immune response, limit inflammation, and reduce the capacity of T cells to kill cancer cells.[6]
We have previously shown that several types of stress, including cool housing temperature and elevated levels of β-adrenergic receptor signaling, increase tumor growth rates and alter the balance of immune cell populations in tumors by increasing the accumulation and immunosuppressive function of MDSCs.[7] Here, we sought to determine whether the stress that results from circadian rhythm disruption is another avenue through which the anti-tumor immune response is hampered, allowing for accelerated tumor progression. Using two distinct in vivo models of murine cancer, we found that circadian rhythm disruption increases tumor growth rates while also increasing the accumulation of MDSCs.
2. Results
In order to perturb the circadian rhythm of mice, we housed them in an environment capable of precisely maintaining alternative light schedules which were programmed into the lighting system. CD mice were exposed to a light schedule that alternated between two days of T1 (light from 8 am to 8 pm) and three days of T2 (light from 2 am to 2 pm). This means that the dark period would be shortened by 6 h when transitioning from T1 to T2, and the dark period would be extended by 6 h when transitioning from T2 to T1, as shown in Figure 1. Since mice are nocturnal, the dark period would be considered the “active” phase of the day.
Figure 1.
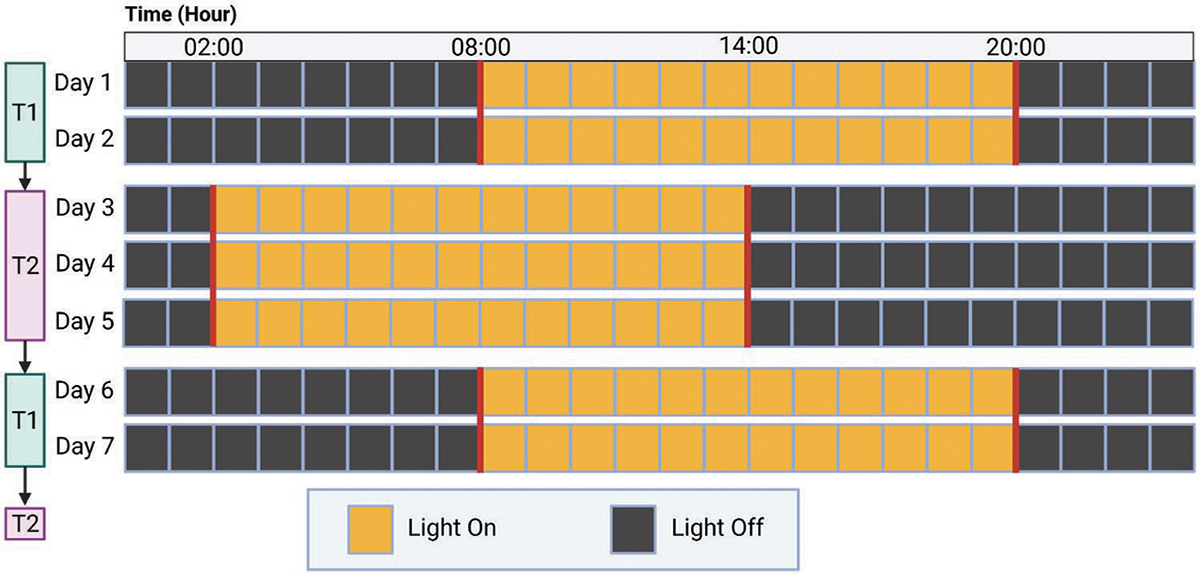
Circadian Disruption Lighting Schedule. CD schedule is shown. Lighting schedule would alternate between T1 and T2 schedules for the entirety of the experiment. SL schedule followed the same light timing as T1 and the light on and off times did not change at any point.
The model of CD used in this study is based upon models often described as “chronic jet lag” in the literature. These models of CD have been previously characterized to show that an altered light schedule is sufficient to cause disruption of circadian genes and chronicity of physiologic processes and behavior. Chronic jet lag models have been confirmed to induce CD by causing alterations in the chronicity and amplitude of clock genes within the suprachiasmatic nucleus (SCN)[8] and peripheral tissues.[8–9] This model has been shown to cause physiologic changes such as desynchronization of body temperature,[8b,10] chronic inflammation in the liver,[8a] altered metabolism,[11] reduced glucose uptake in the brain,[9,12] and induction of leptin resistance.[13] Prior studies have also shown this model to induce behavioral changes as a result of circadian disruption, such as desynchronization and alterations in locomotor activity[8a,b,10–11], and changes in sleep schedule without causing sleep deprivation.[8c,12] Although each of these individual effects of CD is potentially important contributors, the studies in this manuscript focus on how these effects culminate to impact tumor growth and immune dysregulation.
After 4 weeks of acclimation to the standard lighting (SL) schedule or the CD schedule, mice were inoculated with tumors. We first tested the mouse mammary carcinoma cell line, 4T1, in BALB/c mice. 4T1 was chosen because it has been shown to have nonfunctional α and β adrenergic receptors, thus isolating the potential effects of perturbed adrenergic signaling to nontumor cells within the tumor.[14] In a separate experiment, we inoculated C57BL/6 mice with B16-F10 murine melanoma tumors. Mice in the CD group had significantly larger tumors than those in the SL group in both tumor models (Figure 2). 4T1 tumor-bearing mice were euthanized 26 days after tumor inoculation and B16 tumor-bearing mice were euthanized 19 days after tumor inoculation. Directly following euthanasia, tumors and spleens were processed and stained for flow cytometric analysis of MDSC and T cell populations. Polymorphonuclear-MDSCs (PMN-MDSCs) were characterized as CD11b+Ly6G+Ly6Clo and monocytic-MDSCs (M-MDSCs) were characterized as CD11b+Ly6G−Ly6Chi.
Figure 2.
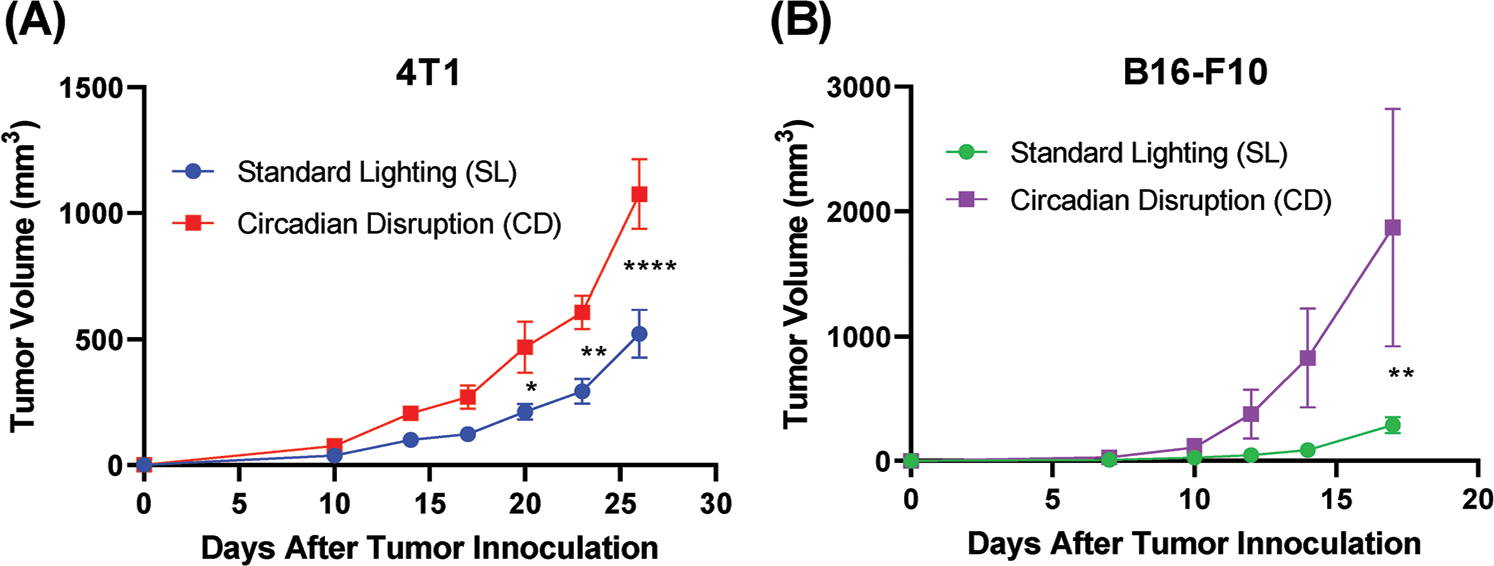
Circadian disruption increases tumor growth. Tumor growth in mice bearing 4T1 (A) or B16-F10 (B) tumors, housed in either SL conditions or CD lighting conditions (n = 5). Data presented as mean ± standard error of the mean (SEM). Two-way ANOVA was used to determine statistical significance. *p < 0.05, **p < 0.01, and ****p < 0.0001.
In both tumor models, a significant difference in the proportion of intratumoral PMN-MDSCs was observed, with more PMN-MDSCs making up the live cell population in CD tumors compared to SL (Figure 3A,C). In the 4T1 tumor model, the M-MDSC population was also found to be increased in the spleen (Figure 3B). Significant differences in the proportion of intratumoral M-MDSCs were not observed in either tumor model (Figure 3B,D). The cellular composition of the tumors is also notable, with around 10% of the live cells within the 4T1 tumors being composed of PMN-MDSCs. This contrasts with the B16 model, where MDSC populations composed less than 1% of the live cells within the tumor.
Figure 3.
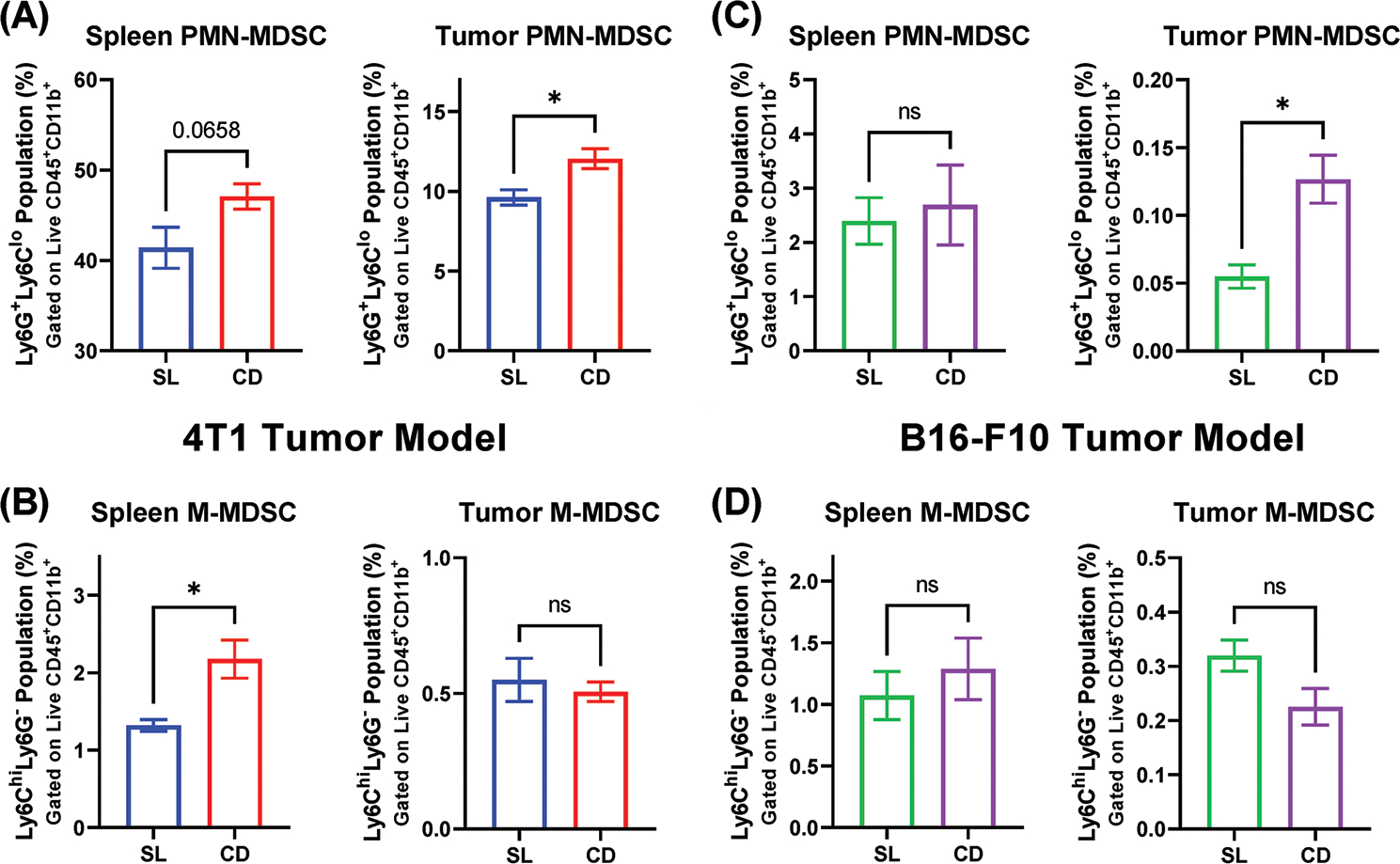
Circadian disruption alters MDSCs accumulation in spleen and tumor. Flow cytometric analysis of MDSC subtypes in the spleen and tumor tissue of mice, as a proportion of the live cell population. A) PMN-MDSC populations in 4T1 tumor-bearing mice. B) M-MDSC populations in 4T1 tumor-bearing mice. C) PMN-MDSC populations in B16-F10 tumor-bearing mice D) M-MDSC populations in B16-F10 bearing mice. (n = 3–5) Data presented as mean ± SEM. A two-tailed unpaired Student’s t-test was used to analyze the statistical significance between the SL and CD groups. * = p < 0.05.
In the spleens and tumors from 4T1 and B16 tumor-bearing mice, CD4+ and CD8+ T cell proportions did not differ significantly between SL and CD lighting conditions (Figure 4). This suggests that T cell recruitment into the tumors was not affected by the circadian disruption, and that suppression of the effector function of T cells within the tumor could contribute to the observed differences in tumor volume.
Figure 4.
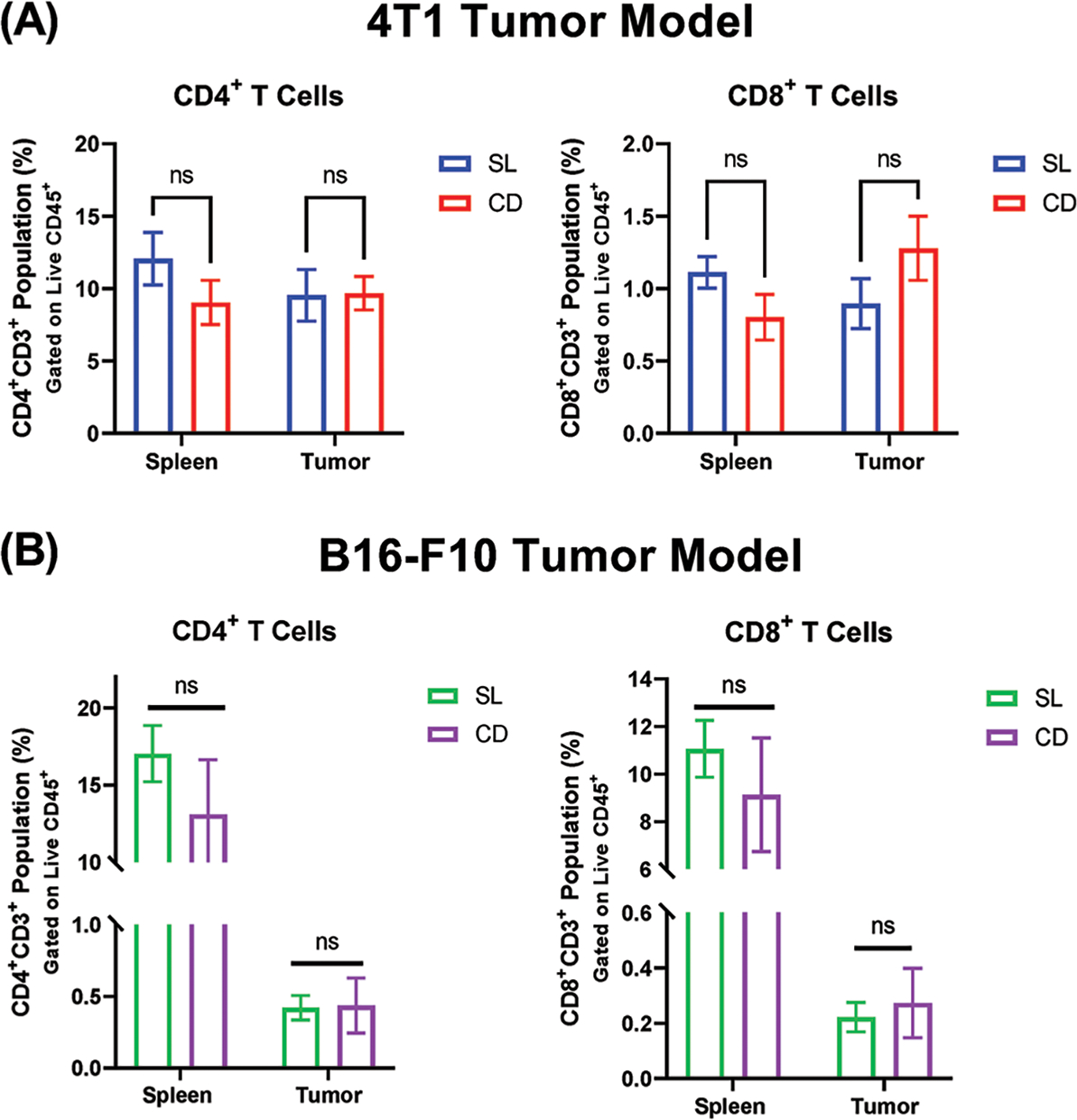
T cell proportions in tumor-bearing mice are not altered by circadian disruption. Flow cytometric analysis of CD4+ and CD8+ T cells in the spleen and tumor tissue of mice, as a proportion of the live cell population. A) T cell populations in 4T1 tumor-bearing mice B) T cell populations in B16-F10 tumor-bearing mice. (n = 4–5) Data presented as mean ± SEM. A two-tailed Student’s t-test was used to analyze statistical significance between the SL and CD groups. p-value less than 0.05 was considered to be statistically significant.
3. Discussion
Our experiments using murine breast and melanoma tumor models showed that mice with a disrupted circadian rhythm experience faster tumor growth compared to those at SL conditions. This is consistent with previous studies using other tumor models.[10,15] From the results of this study and others, it is clear that external derailment of a mouse’s circadian rhythm alone is enough to amplify tumor growth. However, the mechanisms that lead to this enhanced tumor growth are not as clear, and there are likely multiple factors involved. External CD through light manipulation may lead to disruptions of the internal circadian rhythms within cancer cells, potentially amplifying growth and proliferation. Central CD may lead to altered signaling from noncancerous cells within the tumor that could promote tumor growth as well. However, differences in MDSC accumulation suggest a contribution from MDSCs to tumor growth, which would likely be suppression of the tumor-specific immune response.
This is the first known circadian rhythm study to look at the subtypes of MDSCs, PMN-MDSCs, and M-MDSCs, which are phenotypically and morphologically distinct, each having unique functional characteristics.[16] Due to differences in suppressive capabilities and mechanisms of immune suppression between the two subtypes, it is important to quantify each type within the tumor as well as in peripheral immune organs, such as the spleen.
The proportionality of splenic and tumoral MDSC subtypes differed greatly between the two tumor models, indicating that MDSC accumulation differs between tumor models and may be more relevant to specific cancer types. The 4T1 tumors were highly infiltrated by PMN-MDSCs, while M-MDSCs made up only a small portion of the total cells within the tumor. In contrast, the B16 model featured MDSCs as a much lower proportion of the live cell population, and M-MDSCs were more prominent in the tumor compared to PMN-MDSCs, but this is the opposite of what was seen in the spleen. However, in the field of MDSCs, more research is necessary to determine the relationship between MDSC proportionality and subsequent immune suppression. Some studies have shown M-MDSCs to be more suppressive on a per cell basis, so the much larger proportion of PMN-MDSCs compared to M-MDSCs in the 4T1 tumor model does not mean that the PMN-MDSCs are solely responsible for any immune suppression within the tumor.[17]
This study showed increases in tumoral and splenic MDSCs in mice with disrupted circadian rhythms. However, further studies are necessary to determine the underlying causes of these increases in MDSCs. Prior research in our laboratory has shown that adrenergic stress increases the frequency and suppressive function of MDSCs.[7,18] Experiments using the beta blocker, propranolol, or β2-AR−/− mice have shown reductions in MDSCs in tumor-bearing mice compared to untreated or wild-type mice. Since the nervous system is responsible for the detection of zeitgebers and for relaying these signals to peripheral tissues, it is possible that the increased accumulation of MDSCs is due to increased adrenergic signaling caused by circadian disruption.
The SCN of the hypothalamus is the central circadian pacemaker, and is entrained to solar time via signaling from the retina.[19] Circadian signaling from the SCN regulates the systemic production of epinephrine and norepinephrine via neural linkages between the SCN and the paraventricular nucleus, which is the driver of the hypothalamic–pituitary–adrenal (HPA) axis.[20] Systemic catecholamine production is also regulated by the innervation of the adrenal glands via neurons connected to the SCN. The SCN can also send direct signals to tissues through the sympathetic nervous system, inducing the release of norepinephrine at the innervated tissue site.[21] Additionally, circadian control of the HPA axis can alter levels of circulating glucocorticoids in addition to catecholamines, which can modulate the activity of immune cells. Circadian control of the sympathetic nervous system can induce local adrenergic signaling in immune tissues and organs. This type of signaling is responsible for circadian fluctuations in circulating hematopoietic stem cells and their expression of CXCL12.[22] Rhythmic adrenergic signaling through sympathetic innervation of the spleen modulates the cytokine expression and cytolytic function of natural killer (NK) cells.[23] In order to determine the contribution of adrenergic signaling to the tumor-promoting effects of circadian disruption, future studies could utilize β2-adrenergic receptor global knockout mice or treat mice in CD conditions with beta blockers, such as propranolol.
If increased adrenergic signaling due CD is responsible for increased MDSCs, further research is needed to determine the mechanisms that ultimately lead to increased intratumoral and splenic MDSCs. Sympathetic signaling within the bone marrow could directly induce the production of MDSCs. Circadian fluctuations in circulating hematopoietic stem cells have been shown to be controlled by the local noradrenergic release of sympathetic nerves within the bone marrow,[22] so it is possible that MDSC progenitors also undergo sympathetic signaling directly tied to central circadian rhythms. However, further research is needed to determine if fluctuations in tissue levels of MDSCs are also dependent on circadian rhythms. Additionally, chronic stress has been shown to mobilize hematopoietic stem progenitor cells to induce extramedullary myelopoiesis in the spleen, mediated by adrenergic signaling.[24] Increases in myelopoiesis through extramedullary myelopoiesis could result in greater overall numbers of MDSCs in mice in CD conditions.
CD mediated alterations in the production of cytokines and signaling factors could also be responsible for the increased MDSCs in CD mice. The accumulation of MDSCs is dependent on two groups of signals: signals involved in the expansion of immature myeloid cells and signals mediated by inflammatory cytokines and damage-associated molecular patterns.[25] The signals involved in myeloid expansion include granulocyte-macrophage colony-stimulating factor (GM-CSF), granulocyte-CSF (G-CSF), macrophage-CSF (M-CSF), stem cell factor (SCF), and vascular endothelial growth factor (VEGF), and are produced directly by the tumor or are produced within the bone marrow as a result of infection or inflammation. The second group of signals includes inflammatory signals such as IFN-γ, IL-1β, IL-6, and TNFα. Many of these signaling factors are known to fluctuate rhythmically within circulation or are expressed and released from specific cell types in a rhythmic manner under normal or inflammatory conditions,[26] and perturbations in circadian rhythm could alter the signaling to immature myeloid cells and bias them toward an MDSC phenotype. Additionally, alterations in central circadian rhythm could induce tumor cells to produce more of these signaling factors, especially if their transcription is regulated by clock genes in the tumor cells. Knockout of the clock proteins Cry1 and Cry2 have been shown to cause constitutive elevation of proinflammatory cytokines through constitutive NF-κB and protein kinase A (PKA) signaling, showing direct circadian gene regulation of cytokine expression.[27] In future studies, cytokines that contribute to the development of MDSCs should be measured within the tumor and bone marrow to determine if they are present in sufficient concentrations to drive the development of MDSCs.
While greater MDSC accumulation under disrupted circadian rhythm may be due to increased production of MDSCs, it could also be a result of enhanced MDSC trafficking into peripheral tissues and the tumor. Perturbations in the internal circadian rhythm of individual MDSCs could induce alterations in their trafficking patterns, as myeloid-specific deletion of circadian genes has been shown to disrupt diurnal rhythms in chemokine expression and recruitment to tissues of Ly6Chi monocytes.[28] Additionally, adhesion molecules and chemokine expression in endothelial cells have been found to oscillate in a circadian fashion, mediated by local sympathetic innervation, resulting in circadian oscillations in leukocyte adhesion and extravasation.[29]
The importance of circadian rhythms in their regulation of essential biological functions is demonstrated by pathologies that occur when circadian rhythms are disrupted. With regard to CD and human cancers, epidemiological studies have identified higher levels of breast, colorectal, and prostate cancers in shift workers.[30] Outside of more specific cases of circadian disruption, such as shift-work and jet lag, the general population is at risk for CD due to the sleep-wake habits of modern lifestyles and the regular usage of man-made light sources when sunlight is absent. There is some evidence for artificial light at night (ALAN) being correlated with breast cancer incidence,[31] and ALAN has been shown to drive resistance to tamoxifen therapy in rats with breast tumors.[32] ALAN may also contribute to the development of conditions that are associated with the development of cancer, such as obesity.[33] Light pollution in urban environments presents as another source of circadian disruption, as animal studies utilizing dim light at night with similar intensity to urban light pollution have shown CD as a result.[34] Due to the widespread prevalence of ALAN and light pollution in our modern lives, more research is necessary to determine subsequent risks to human health and how these factors influence the development and progression of cancer and the subsequent immune response.
Increases in MDSC accumulation are likely only part of the explanation for increased tumor growth under CD conditions. The immune contribution to this phenomenon likely involves the action of other immunosuppressive cell types as well as disruption of the internal circadian rhythms of effector cells. Increased levels of circulating glucocorticoids and catecholamines likely have a broad range of effects, one being direct suppression of effector cells. Effects outside of the immune system likely contribute to the increased tumor growth as well, as disruption of the central circadian clock puts all individual cells at risk for internal CD and subsequent effects. However, this study and others show that the function of the immune system is controlled by circadian rhythm and the immune system’s response and contribution to tumor progression can be perturbed by external manipulation of circadian rhythm. Modern life contains many sources of external stimuli capable of inducing circadian disruption, and a greater understanding of this phenomena in the context of cancer and immunity is necessary to efforts to improve cancer prevention and treatment efficacy.
3.1. Limitations
In this study, we did not conduct an extensive immunophenotyping of the spleen and tumor immune cells and therefore did not collect data on the proportions of regulatory T cells, NK cells, and tumor-associated macrophages. We also did not collect data on the expression of immune checkpoint molecules on the surface of leukocytes or tumor cells such as programmed cell death protein 1 (PD-1) and programmed death-ligand 1 (PD-L1). The expression of markers of senescence and exhaustion on T cells was also not tested in this study. While CD4+ and CD8+ T cell levels were similar between the experimental groups, those populations could differ greatly in their expression of functional markers and immune checkpoint molecule expression. These limitations demonstrate a need for more extensive immunophenotyping in follow-up studies in order to determine the root cause of immunosuppression in the CD model. Future studies could also utilize T cell intracellular cytokine assays to determine the functional capabilities of intratumoral T cells to determine if their function is suppressed by increased MDSC populations. Future studies should also monitor physiologic changes and behaviors of the mice such as locomotion, food intake, body weight, and chronicity of activities to determine if observed immune perturbations can be attributed in part to these specific factors. The brain and peripheral organs should also be taken at the endpoint and the expression of circadian genes in these tissues should be compared between experimental groups and to a separate group housed with the experimental mice prior to CD acclimation to serve as a baseline. This data would confirm the perturbation of endogenous circadian rhythms from the CD model. Nonsignificant differences in our data could potentially be due to a lack of statistical power. Future studies should conduct power calculations to determine mouse sample sizes to ensure that experiments are not underpowered.
4. Experimental Section
Animals:
4-week-old BALB/c and C57BL/6 mice were purchased from Charles River. All mice used in this study were female. All mice were age maintained in specific-pathogen-free housing, and all mouse studies were reviewed and approved by the Roswell Park Comprehensive Cancer Center IACUC (protocol numbers 757M and 1038M).
Light Schedules and Housing:
Mice were housed in standard cages placed inside chambers equipped with fluorescent lighting that was controlled by a light timer program. The lighting chambers blocked all external light, leaving mice in complete darkness during the dark period. Mice were provided food and water ad libitum and cages were changed weekly. All mice underwent a 4-week acclimation period in the light chambers prior to tumor inoculation to acclimate them to the new housing environment in addition to the lighting conditions. SL schedule consisted of 12 h of light, followed by 12 h of darkness, which would change at the same time every day. CD also featured 12 h of light and dark but would shift by 6 h every 2–3 days (Figure 1). Mice were observed once daily during the scheduled light period, and all interactions with the mice took place during the scheduled light period.
Cell Culture:
The murine mammary carcinoma cell line, 4T1, was used in all experiments involving BALB/c mice, and the murine melanoma cell line, B16-F10, was used in experiments involving C57BL/6 mice. Both cell lines were purchased from the ATCC. Cell lines were cultured in RPMI 1640 supplemented with 10% fetal bovine serum (FBS), 1% L-glutamine, and 1% penicillin/streptomycin. Cell lines were thawed and passaged twice prior to tumor inoculation. 4T1 cells were injected into the mammary fat pad of female BALB/c mice at a concentration of 10 000 cells in 100 μL of phosphate buffered saline (PBS). B16-F10 cells were injected subcutaneously into the lower left abdomen of female C57BL/6 mice at a concentration of 200 000 cells in 100 μL of PBS.
Circadian Rhythm Disruption Experimental Design:
Mice were acclimated to their assigned lighting schedules for 4 weeks prior to tumor injection. Tumors for both light conditions were implanted at 12:00 when the CD group was under the same light conditions as the SL controls. Mice continued living in their specific lighting conditions following tumor implantation. Tumors were allowed to grow for a week and then were measured every 3–4 days using calipers. Tumor measurements were scheduled to take place during the light period for all experimental groups. Tumor volume was estimated using the following equation: , with SD being the shorter dimension of an ovoid tumor and LD being the longer dimension.
Flow Cytometry:
At the experimental endpoint, tumors and spleens were removed for processing. Tumors were chopped into small pieces ≈5 mm3 and were enzymatically dissociated with collagenase/hyaluronidase, then passed through a 70 μm nylon cell strainer. Spleens were mechanically disrupted and passed through a 70 μm cell strainer and red blood cells were lysed with ACK buffer. Tumor and spleen cells were washed with flow buffer and incubated with Fc receptor block for 10 min. For the MDSC panel, cells were stained for CD45, CD11b, Ly6C, Ly6G, and L/D Aqua was used to gate live cells. For the T cell panel, cells were stained for CD45, CD3, CD4, CD8, and L/D Aqua. All data were collected on an LSR Fortessa flow cytometer and analyzed with FlowJo v7 software.
Statistical Analysis:
Exclusions in the flow cytometry data were determined based on abnormally low viability staining compared to the other samples in the group as well as compared to the viability reading of the same tissue sample used in other panels. These outliers are attributed to errors in sample processing and were therefore excluded. All data are presented as mean ± SEM. For each tumor model, five mice were used per experimental group. For single variable comparisons, an unpaired, two-tailed Student’s t-test was used. For comparisons of tumor growth curves, two-way ANOVA (Analysis of Variance) was used with multiple comparisons using the Šidák correction. For all analyses, a p-value less than 0.05 was considered to be statistically significant. Statistical analyses were conducted in GraphPad Prism 9 software.
Acknowledgements
The authors thank Jeanne M. Prendergast and Samuel A. Ministero for technical assistance. The authors also thank the Roswell Park Laboratory Animal Shared Resource and the Flow Cytometry Core Facility for their expertise and support. The Table of Contents graphic and Figure 1 graphic were created using BioRender. This project was supported the Roswell Park Alliance Foundation and by the National Institutes of Health (NIH) grants: R21CA227375 (to M.A. and E.A), P30CA016056, R01CA205246, and R01CA236390 (to E.A.), K99HL155792 and F32CA239356 (to H.M.), and T32CA085183 and F30CA265127 (to C.M.).
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
The ORCID identification number(s) for the author(s) of this article can be found under https://doi.org/10.1002/adbi.202200031.
Contributor Information
Nathan T. Roberts, Department of Immunology, Roswell Park Comprehensive Cancer Center, Buffalo, NY 14203, USA
Cameron R. MacDonald, Department of Immunology, Roswell Park Comprehensive Cancer Center, Buffalo, NY 14203, USA
Hemn Mohammadpour, Department of Cell Stress Biology, Roswell Park Comprehensive Cancer Center, Buffalo, NY 14203, USA.
Marina P. Antoch, Department of Pharmacology and Therapeutics, Roswell Park Comprehensive Cancer Center, 665 Elm St, Buffalo, NY 14203, USA
Elizabeth A. Repasky, Department of Immunology, Roswell Park Comprehensive Cancer Center, Buffalo, NY 14203, USA
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- [1].a) Patke A, Young MW, Axelrod S, Nat. Rev. Mol. Cell Biol. 2020, 21, 67; [DOI] [PubMed] [Google Scholar]; b) Kervezee L, Kosmadopoulos A, Boivin DB, Eur. J. Neurosci. 2020, 51, 396; [DOI] [PubMed] [Google Scholar]; c) Thosar SS, Butler MP, Shea SA, J. Clin. Invest. 2018, 128, 2157; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Karatsoreos IN, Bhagat S, Bloss EB, Morrison JH, McEwen BS, Proc. Natl. Acad. Sci. USA 2011, 108, 1657; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Wulff K, Gatti S, Wettstein JG, Foster RG, Nat. Rev. Neurosci. 2010, 11, 589; [DOI] [PubMed] [Google Scholar]; f) Sulli G, Lam MTY, Panda S, Trends Cancer 2019, 5, 475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Savvidis C, Koutsilieris M, Mol. Med. 2012, 18, 1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Scheiermann C, Kunisaki Y, Frenette PS, Nat. Rev. Immunol. 2013, 13, 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Chen DS, Mellman I, Immunity 2013, 39, 1. [DOI] [PubMed] [Google Scholar]
- [5].Raskov H, Orhan A, Christensen JP, Gögenur I, Br. J. Cancer 2021, 124, 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Marvel D, Gabrilovich DI, J. Clin. Invest. 2015, 125, 3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Mohammadpour H, MacDonald CR, Qiao G, Chen M, Dong B, Hylander BL, McCarthy PL, Abrams SI, Repasky EA, J. Clin. Invest. 2019, 129, 5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Inokawa H, Umemura Y, Shimba A, Kawakami E, Koike N, Tsuchiya Y, Ohashi M, Minami Y, Cui G, Asahi T, Ono R, Sasawaki Y, Konishi E, Yoo S-H, Chen Z, Teramukai S, Ikuta K, Yagita K, Sci. Rep. 2020, 10, 2569; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yamaguchi Y, Suzuki T, Mizoro Y, Kori H, Okada K, Chen Y, Fustin J-M, Yamazaki F, Mizuguchi N, Zhang J, Dong X, Tsujimoto G, Okuno Y, Doi M, Okamura H, Science 2013, 342, 85; [DOI] [PubMed] [Google Scholar]; c) Castanon-Cervantes O, Wu M, Ehlen JC, Paul K, Gamble KL, Johnson RL, Besing RC, Menaker M, Gewirtz AT, Davidson AJ, J. Immunol. 2010, 185, 5796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Khan S, Yong VW, Xue M, Carcinogenesis 2021, 42, 864. [DOI] [PubMed] [Google Scholar]
- [10].Filipski E, Delaunay F, King VM, Wu M-W, Claustrat B, Gréchez-Cassiau A, Guettier C, Hastings MH, Francis L, Cancer Res. 2004, 64, 7879. [DOI] [PubMed] [Google Scholar]
- [11].Casiraghi LP, Alzamendi A, Giovambattista A, Chiesa JJ, Golombek DA, Physiol. Rep. 2016, 4, e12743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gao Q, Khan S, Zhang L, Sci. Data 2020, 7, 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kettner NM, Mayo SA, Hua J, Lee C, Moore DD, Fu L, Cell Metab. 2015, 22, 448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Szpunar MJ, Burke KA, Dawes RP, Brown EB, Madden KS, Cancer Prev. Res. 2013, 6, 1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Aiello I, Fedele MLM, Román F, Marpegan L, Caldart C, Chiesa JJ, Golombek DA, Finkielstein CV, Paladino N, Sci. Adv. 2020, 6, eaaz4530; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Filipski E, King VM, Li X, Granda TG, Mormont M-C, Liu X, Claustrat B, Hastings MH, Lévi F, Natl J. Cancer Inst. 2002, 94, 690; [DOI] [PubMed] [Google Scholar]; c) Filipski E, Lévi F, Integr. Cancer Therapies 2009, 8, 298. [DOI] [PubMed] [Google Scholar]
- [16].Bronte V, Brandau S, Chen S-H, Colombo MP, Frey AB, Greten TF, Mandruzzato S, Murray PJ, Ochoa A, Ostrand-Rosenberg S, Rodriguez PC, Sica A, Umansky V, Vonderheide RH, Gabrilovich DI, Nat. Commun. 2016, 7, 12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gabrilovich DI, Ostrand-Rosenberg S, Bronte V, Nat. Rev. Immunol. 2012, 12, 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].a) MacDonald C, Ministero S, Pandey M, Robinson D, Hong Forti E., Hylander B, McCarthy P, Gordon C, Repasky E, Mohammadpour H, Cell. Immunol. 2021, 361, 104285; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mohammadpour H, MacDonald CR, McCarthy PL, Abrams SI, Repasky EA, Cell Rep. 2021, 37, 109883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hastings MH, Maywood ES, Brancaccio M, Nat. Rev. Neurosci. 2018, 19, 453. [DOI] [PubMed] [Google Scholar]
- [20].Leach S, Suzuki K, Front. Immunol. 2020, 11, 1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Dibner C, Schibler U, Albrecht U, Annu. Rev. Physiol. 2010, 72, 517. [DOI] [PubMed] [Google Scholar]
- [22].Méndez-Ferrer S, Lucas D, Battista M, Frenette PS, Nature 2008, 452, 442. [DOI] [PubMed] [Google Scholar]
- [23].Logan RW, Arjona A, Sarkar DK, Brain, Behav., Immun. 2011, 25, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].McKim DB, Yin W, Wang Y, Cole SW, Godbout JP, Sheridan JF, Cell Rep. 2018, 25, 2552.e2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Veglia F, Perego M, Gabrilovich D, Nat. Immunol. 2018, 19, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].a) Gibbs JE, Blaikley J, Beesley S, Matthews L, Simpson KD, Boyce SH, Farrow SN, Else KJ, Singh D, Ray DW, Loudon ASI, Proc. Natl. Acad. Sci. USA 2012, 109, 582; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Adams KL, Castanon-Cervantes O, Evans JA, Davidson AJ, Biol J. Rhythms 2013, 28, 272; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Born J, Lange T, Hansen K, Mölle M, Fehm HL, Immunol J. 1997, 158, 4454; [PubMed] [Google Scholar]; d) Gibbs J, Ince L, Matthews L, Mei J, Bell T, Yang N, Saer B, Begley N, Poolman T, Pariollaud M, Farrow S, DeMayo F, Hussell T, Worthen GS, Ray D, Loudon A, Nat. Med. 2014, 20, 919; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Keller M, Mazuch J, Abraham U, Eom GD, Herzog ED, Volk H-D, Kramer A, Maier B, Proc. Natl. Acad. Sci. USA 2009, 106, 21407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Narasimamurthy R, Hatori M, Nayak SK, Liu F, Panda S, Verma IM, Proc. Natl. Acad. Sci. USA 2012, 109, 12662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Nguyen KD, Fentress SJ, Qiu Y, Yun K, Cox JS, Chawla A, Science (New York, N.Y.) 2013, 341, 1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Scheiermann C, Kunisaki Y, Lucas D, Chow A, Jang J-E, Zhang D, Hashimoto D, Merad M, Frenette PS, Immunity 2012, 37, 290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Roenneberg T, Merrow M, Curr. Biol. 2016, 26, R432. [DOI] [PubMed] [Google Scholar]
- [31].Stevens RG, Int. J. Epidemiol. 2009, 38, 963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Dauchy RT, Xiang S, Mao L, Brimer S, Wren MA, Yuan L, Anbalagan M, Hauch A, Frasch T, Rowan BG, Blask DE, Hill SM, Cancer Res. 2014, 74, 4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Rybnikova NA, Haim A, Portnov BA, Int. J. Obes 2016, 40, 815. [DOI] [PubMed] [Google Scholar]
- [34].Walker WH, Bumgarner JR, Walton JC, Liu JA, Meléndez-Fernández OH, Nelson RJ, DeVries AC, Int. J. Mol. Sci. 2020, 21, 9360. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.