

Abstract
Hypoxic-ischemic encephalopathy (HIE) is the leading cause of neonatal morbidity and mortality worldwide. Approximately 1 million infants born with HIE each year survive with cerebral palsy (CP) and/or serious cognitive disabilities. While infants born with mild and severe HIE frequently result in predictable outcomes, infants born with moderate HIE exhibit variable outcomes that are highly unpredictable. Here, we describe an umbilical cord occlusion (UCO) model of moderate HIE with a 6-day follow-up. Near term lambs (n=27) were resuscitated after the induction of 5 minutes of asystole. Following recovery, lambs were assessed to define neurodevelopmental outcomes. At the end of this period, lambs were euthanized, and brains harvested for histological analysis. Compared with prior models that typically follow lambs for 3 days, the observation of neurobehavioral outcomes for 6 days enabled identification of animals that recover significant neurological function. Approximately 35 % of lambs exhibited severe motor deficits throughout the entirety of the 6-day course and, in the most severely affected lambs, developed spastic diparesis similar to that observed in infants who survive severe neonatal HIE (severe, UCOs). Importantly, and similar to outcomes in human neonates, while initially developing significant acidosis and encephalopathy, the remainder of the lambs in this model recovered normal motor activity and exhibited normal neurodevelopmental outcomes by 6 days of life (improved, UCOi). The UCOs group exhibited gliosis and inflammation in both white and gray matter, oligodendrocyte loss, and neuronal loss and cellular death in the hippocampus and cingulate cortex. While the UCOi group exhibited more cellular death and gliosis in the parasagittal cortex, they demonstrated more preserved white matter markers, along with reduced markers of inflammation and lower cellular death and neuronal loss in Ca3 of the hippocampus compared with UCOs lambs. Our large animal model of moderate HIE with prolonged follow-up will help further define pathophysiologic drivers of brain injury while enabling identification of predictive biomarkers that correlate with disease outcomes and ultimately help support development of therapeutic approaches to this challenging clinical scenario.
Keywords: ovine model, neonates, brain hypoxia-ischemia, neurodevelopmental outcomes
Introduction
Neonatal HIE remains the leading cause of neonatal morbidity and mortality worldwide (~ 4 million neonates annually), and accounts for nearly 25 % of neonatal deaths (~1 million newborns) [1–4]. Approximately 1 million infants per year survive with CP and/or serious cognitive and other developmental disabilities [5,6]. Furthermore, the global nature of the ischemia associated with HIE is responsible for the impairment of several different organ systems (central nervous system 28 %, cardiovascular system 25 %, kidneys 50 %, and lungs 23 %), resulting in significant additional morbidity [3]. Unfortunately, approximately 47 % of affected infants that undergo therapeutic hypothermia (TH) are still at risk of death or serious disability [7]. While TH results in a reduction in brain injury and mortality following birth asphyxia in moderate to severe HIE [8], it is the only known treatment, the benefits are modest, and its use can be potentially harmful [9,10]. The most commonly used grading system for infants born with HIE remains the Sarnat score, with infants graded as mild, moderate or severe depending on their clinical signs [11]. The approximate breakdown tends to be mild (39%), moderate (39%) and severe (22%) [6]. The management and outcome varies significantly with grade of HIE. While infants born with mild and severe HIE frequently result in predictable outcomes, infants born with moderate HIE exhibit variable outcomes that are highly unpredictable[12]. Infants born with mild HIE are not typically offered TH. The ultimate goal of this study is to develop a large animal model with high translational potential to identify predictive biomarkers and evaluate potential neurotherapeutic agents to improve neurodevelopmental outcomes in the setting of HIE.
Different animal models have their strengths and limitations, but in general they have greatly facilitated our understanding of the pathophysiology of neonatal HIE [13]. An ideal animal model of neonatal HIE is characterized by similarities between both human and animal model in terms of pathophysiology, phenotypic and histopathological characteristics, predictive biomarkers for course or prognosis, response to therapies, suitability for evaluation of drug safety or toxicity, cost effectiveness, and ability to study long-term neurological outcomes [14]. Most commonly used rodent models are inexpensive, and provide excellent insight into basic molecular mechanisms and long-term neurodevelopmental outcomes. However, there is significant variability between animals, as well as strains of species. The most commonly used Rice-Vannucci model leads to unilateral insult, while bilateral common carotid occlusion model has high mortality, with neither fully representing human neonatal HIE. Importantly, the rodent brain significantly differs from the human in both its size and level of cortical gyrification and the small size of rodents does not allow for more precise neurophysiological monitoring and assessment of multi organ function after the insult compared to large animal models [15]. While larger rabbit models of in utero hypoxia-ischemia wherein uterine artery obstruction at 75 % of gestational age reliably produce motor deficits in newborns [16], do not recapitulate injury incurred during the dramatic physiologic changes accompanying the fetal-neonatal transition during birth. Non-human primate models, such as monkeys, or baboons, while highly similar to human, do not fully reproduce the injury seen in human neonates, and their use is restricted by ethical concerns and high experimental costs [15].
Piglet and sheep models have been invaluable in providing critical insights regarding cerebral blood flow and metabolism, delineating the drivers of neurological injury, or assessing responses to hypothermia after hypoxic-ischemic insults in newborns [15]. While existing (141–145 days) term models of umbilical cord occlusion (UCO) in sheep are highly robust in recapitulating the perinatal pathophysiology and neurological impairment observed in the setting of HIE, they typically describe neurological outcomes only up to 72 hours after injury [17–19]. Longer term outcomes are frequently needed in most animal models of brain injury to properly assess therapeutic responses. Along these lines, we predicted that extending the monitoring of neurological outcomes to 6 days in our term model of HIE would enable us to identify potentially differing neurodevelopmental outcomes [20]. Importantly, we have opted not to utilize the near-term (121–123 days) model of bilateral carotid artery occlusion previously used to develop therapeutic hypothermia, as we were primarily interested in defining the mechanisms giving rise to neurological injury as well as the pharmacology of putative therapeutic agents in the setting of the fetal-to-neonatal transition at term as this represents the most likely clinical use scenario globally [21].
Consistent with our prediction, here we describe two groups of animals with similar neurological deficits early after perinatal hypoxic-ischemic injury with significantly differing outcomes by day of life 6. We provide a hemodynamic, histological and neurological comparison of these two groups in our model. Our large animal model of moderate HIE with prolonged follow up will help further define pathophysiologic drivers of brain injury while enabling identification of predictive biomarkers that correlate with disease outcomes and ultimately help support development of therapeutic approaches to this challenging clinical scenario.
Materials and Methods
Animals
All animal research was approved by the University of California Davis Institutional Animal Care and Use Committee and was performed in accordance with the Guide for the Care and Use of Laboratory Animals. Sheep of both sexes were used. Study groups included UCO asphyxiated lambs (UCO), n=16 and the group of sham-surgery lambs (Control), n=11 who were exposed to the same instrumentation as the experimental group but without UCO.
Induction of HIE
HIE was induced via UCO in near term lambs (141–143 days gestation, term ~ 147–150 days; n=11). Time-dated pregnant ewes were fasted for 12–24 hours prior to surgery. The ewes were induced with ketamine and propofol and anesthetized with isoflurane for surgery according to IACUC approved Standard Operating Procedure, SC-20–112, “Sheep Anesthesia: Surgical Research Facility, H-Building at TRACS”. Briefly, a jugular catheter or peripheral venous catheter was placed and the ewe given 4mg/kg slow push IV propofol, and 1–5mg/kg Ketamine. After anesthetic induction and intubation, the ventral abdomen was shaved and cleaned. Immediately prior to surgery, the pregnant ewes underwent ultrasound (US) imaging under general anesthesia to verify pregnancy. After ultrasound, the ewe was then transferred into the operating room where she was placed on a mechanical ventilator. Anesthesia was maintained with 1–5 % isoflurane through the endotracheal tube. The ventral abdomen was then given a standard surgical scrub (using either Betadine or Chlorhexidine and alcohol) and the ewe placed on maintenance IV fluids, usually 5–15ml/kg/hr. Oxygenation of the ewe was monitored with an O2 saturation probe and hemodynamics were monitored with a noninvasive blood pressure cuff. A midline incision along the ventral abdomen (6–10”) was made and the uterus exposed. The ewe was given IV antibiotics (penicillin G potassium 10,000–20,000 units/kg and gentamicin 1–2 mg/kg). After exteriorization of the fetal head the fetus was intubated with an appropriate sized cuffed endotracheal tube (ETT). The lungs were drained of fluid passively by gravity and the ETT was plugged to prevent gas exchange during gasping. Venous and arterial catheters were placed in the jugular vein and carotid artery for hemodynamic monitoring, blood sampling and drug administration. Asphyxia was induced by UCO until the onset of asystole. Lambs used as controls were instrumented in an identical fashion but were delivered and resuscitated immediately following clamping and cutting of the umbilical cord. The umbilical cord was cut, lamb was delivered to a radiant warmer and following 5 minutes of asystole as assessed by invasive hemodynamic monitoring the lambs were resuscitated using neonatal resuscitation program (NRP) guidelines with positive pressure ventilation with a fraction of inspired oxygen FiO2 of 1.0. Resuscitation was not initiated with room air as asystole and the need for chest compressions was universal given the severity of the model and therefore oxygen therapy was clinically indicated. After 30 seconds of ventilation, external chest compressions were initiated. Chest compressions continued for 60-second intervals before reassessing the heart rate and these efforts continued for up to 15 minutes. Epinephrine (0.01 mg/kg) was administered intravenously if inadequate response to oxygen, ventilation and chest compressions was noted after 60 seconds of asystole following initiation of ventilation. Additional boluses of epinephrine were given if animals were unresponsive to initial doses. Saline boluses were not provided as oxygen, epinephrine and chest compressions were typically sufficient to restore adequate perfusion following ROSC. During, as well as following resuscitation, a ventilator provided ongoing mechanical ventilation. Assisted ventilation was decreased and then ceased when the lamb was spontaneously breathing > 50 % of the time and maintained a peripheral oxygen saturation > 85 % at an FiO2 of 0.21. No intravenous fluids were administered during or after resuscitation. After extubation, the lambs were gavage fed for the first 24 to 48 hours. The lambs were assessed over a 6-day period to determine neurodevelopmental outcomes and euthanized on day 6 with an overdose of euthanasia solution (100mg/kg pentobarbitone sodium, Lethabarb™, Virbac Pty. Ltd., Peakhurst, NSW, Australia).
Histology and Immunohistochemistry
Following euthanasia on day 6, brains were flushed with 500 mL of phosphate-buffered saline (PBS) and perfused with 500 mL of 4 % paraformaldehyde in 0.1 M phosphate buffer (pH 7.4). Brains were post-fixed in 4 % paraformaldehyde overnight and transferred to 20 % sucrose for 2 days and 30 % sucrose till they sank (14 days). Brains were then flash frozen in 2-methyl butane on dry ice and stored at −80 °C. Coronal sections were cut on a cryostat (12 μm-thick serial sections). Double immunofluorescence labeling was performed on brain sections that were defrosted and air dried at room temperature for 1 h. Following antigen retrieval in 10 mM citrate buffer (pH 6.0) for 10 min at 80 °C and a PBS wash, sections were incubated in blocking solution (5 % normal donkey serum, 0.4 % Triton X-100 in PBS) for 1 h at RT. Primary antibody incubation was done overnight at 4 °C with rabbit anti-GFAP (GFAP, 1:500, Z0334, Agilent); rabbit anti-CNPase (CNPase, 1:200, C5922, Millipore Sigma); mouse anti-NeuN for neurons (NeuN, 1:200, MAB377, Millipore Sigma), goat anti-Iba1 for microglia (Iba-1, 1:200, NB100–1028, Novus Biologicals), mouse anti-caspase-3 (casp-3, 1:200, NB600–1235, Novus Biologicals), goat anti-oligodendrocyte transcription factor 2 for oligodendrocytes (Olig-2,1:200 AF2418, Novus Biologicals), rat anti-myelin basic protein (MBP, 1:200, NB600–717, Novus Biologicas) and mouse anti-adenomatous polyposis coli protein clone CC-1 (CC-1, 1:100, OP800100UG, Millipore Sigma). After three 5-min PBS washes, sections were incubated for 1 h at RT with appropriate secondary antibodies: donkey anti-goat Alexa Fluor 647 (1:500, A21447, Thermo Fisher), donkey anti-mouse Alexa Fluor 568 (1:500, A10037, Thermo Fisher), donkey anti-rat Alexa Fluor 594 (1:500, A-21209, Thermofischer) and donkey anti-rabbit Alexa Fluor 488 (1:500, A21206, Thermo Fisher). For nuclear staining, sections were stained with 4′,6-diamino-2-phenylindol for 5 min. Slides were then washed and coverslipped with ProLong Gold antifade (P36930, Invitrogen). Images were taken with a Leica TCS SP5 Spectral Confocal Microscope.
Histopathological analysis
To define the anatomical localization of the injury, we grossly evaluated all areas of the brain on sections corresponding to s.640, and 1200 of Sheep Brain Atlas [22] at 5X magnification. For final analysis we chose the anatomical areas where injury was detected and compared these to control animals. Confocal-like Z-stacks (25X oil objective, 10 μm thick, 1 μm Z step) were acquired using a Zeiss microscope equipped with the confocal-like optigrid device and Volocity software (version 6.3, Improvision, Perkin Elmer, Waltham, MA, USA). Matching regions of interest in control animals acquired under the same conditions served as a control. Every brain had a control with no primary antibodies for staining. Image capturing (using Volocity software) and analysis using Imaris software (version 9.6.2. Oxford Instruments America Inc., Pleasanton, CA) to assess NeuN, Olig-2 and CC-1 cell counts and Iba-1, MBP, CNP-ase volumes, Image J software (version 2.0.0.-rc-69/1.52p, National Institutes of Health, Bethesda, MD, USA) to manually count cleaved caspase-3 cells and Ca1/2, Ca3 NeuN- positive cells) was done in a blinded manner. We measured the number of cells that express NeuN, Cleaved caspase-3, Olig-2, CC-1 and total volume of cell bodies and fibers expressing Iba-1, GFAP, MBP and CNPase per field of view (FOV) measuring 1350 × 1050 × 10 μm3 (1.4×107 μm3).
Injury scores
Brain injury scores were determined in coronal sections stained with Fluoro-Jade (FJ) using modified Bjorkman scale of histopathological scoring in a blinded manner [23]. Regions scored included the following: cortex (Ctx), caudate (Caud), putamen (Put), thalamus (Th) and hippocampus (Hc). Scores were assigned for each region as follows: 0 = no injury, 1 = mild manifested by a single layer scattered neurons in cortex or scattered single neurons in CPTHC, 2 = moderate involving 2 layers of neurons in one anatomical area of the CTX, or a single focus of damaged neurons in Caud/Put/Th/Hc, 3 = severe involving > 2 cortical layers or > 2 foci of damaged neurons in Caud/Put/Th/Hc for a total score of 0–15.
Neurological outcomes
Neurological outcomes were evaluated daily for 6 days. Our neurobehavioral assessment (Tab. 1) was based on observations previously conducted in sheep to monitor their well-being following birth [24,25]. We assessed the time (days) taken to reach normal lamb behavioral milestones after birth (head lift and shake; use of front and hind limbs; first use of four legs; standing; feeding proficiency; walking) for a total score of 7 (Tab. 1). Lambs were fed 2 oz every 4 hours by tube the first day. Afterwards, the lambs got fed 2–6 oz by bottle depending on size of lamb 4 times a day. If they were not able to bottle feed, they continued with tube feeding 2–4 oz 4 times a day with bottle attempts every day until they were euthanized. As a result of this assessment, lambs were assigned to two different categories, severe (UCOs), and improved (UCOi), depending on whether their clinical course improved over the course of the 6-day observation period to achieve developmental milestones comparable to controls (N) reflected by full score of 7 (UCOi), or remained severe (< 3) (UCOs).
Table 1:
| Neurological milestone | Scoring |
|---|---|
| activity at rest (head lift,shake) | 0= sleepy; no head lift with stimulation 0.5= wakes up with stimulation, tries to lift the head up 1= lifts the head up, alert active |
| feeding | 0= minimal suckle, tube fed 0.25= requires assistance to find bottle; a few good suckles 0.5= suckling well once finds the bottle 1= able to nurse normally |
| front limbs | 0= no use 0.5= some use, flexes, tries to pull to stand 1= pulls to stand |
| hind limbs | 0= no use or spastic 0.5= some use; unstable 1= normal strength |
| four limbs use | 0= no 1= yes |
| standing | 0= no standing 0.5= unstable standing; or stands with support only 1= stable standing without support |
| coordinated walk | 0= no walk 0.25= walks few steps, poorly coordinated 0.5= longer coordinated walk; tries to run a few steps; some incoordination 0.75= runs with some incoordination 1= normal coordinated walk; runs |
Statistical analysis
Analyses were performed using Prism 7 (GraphPad Software, San Diego, CA). All data are shown as mean ± standard error of measurement. Data was subjected to a normality test. The differences between two groups were assessed by t-tests. Grouped data were analyzed using one-way and two-way analysis of variance and subsequently subjected to Smidak post hoc analyses and differences were considered significant at p < 0.05. The data that did not pass the normality test were analyzed using Kruskal-Wallis test. The hemodynamic data was analyzed using grouped analysis of the individual group’s means for a specific time point. Comparisons were made between the groups with different neurobehavioral outcomes: UCO-subjected with persistent motor deficits and encephalopathy (UCOs), UCO-subjected with transient motor deficits and encephalopathy and complete recovery on day 6 (UCOi) and control group (N); and all injured animals (I) vs control group (N) (Supplementary material).
Results
Neurobehavioral outcomes identified two groups of UCO lambs
Neurodevelopmentally, on day 0, uninjured control animals were able to lift and move their heads, feed and use all four limbs resulting in a stable gait within a few hours of birth (Fig. 1A,B,C). Their walk was brief and not completely coordinated. However, control lambs reached these neurodevelopmental milestones rapidly and exhibited a coordinated walk and run by day 1 after birth (Fig. 1B). A group of UCO lambs (n=3) achieved full neurodevelopmental milestones already on day 0, while the rest of the animals (n=13) suffered from severe disability on days 0 and 1. This included an inability to use the hind limbs (n=13), minimal use of the front limbs (n=6), absent standing or walking (n=13), and minimal head lift (n=9) (Fig. 1B,D–F). However, for some animals a remarkable progress in neurodevelopmental outcomes evolved during our 6 day observation period (Fig. 1A,B). Almost half of the UCO animals (n=7) with poor prior neurological function and total scores ranging from 0 to 2 on day 0 remarkably improved their motor function and achieved close to a full clinical recovery as early as day 2 after the UCO. A subgroup of UCO lambs with poor neurological score on day 0 improved to the full clinical score more slowly than those not as clinically impaired at this early time point, reaching full scores on day 4 (n=1) and 5 (n=1). A final subgroup of UCO animals (UCOs, n=5) remained severely affected with scores ranging from 0–3, never achieving the ability to feed, or use their limbs, and continued to have difficulties with feeding and were minimally active (Fig. 1F). These animals also demonstrated increased motor tone and spasticity of the hindlimbs. These findings of a movement disorder causing persistent activity limitation attributed to disturbances that occurred in the developing fetal brain, often accompanied by disturbances of behavior and secondary musculoskeletal problems are consistent with clinical findings in infants with severe HIE (Fig. 1A–B, D–F) [19]. The distribution of outcomes in this model are thus consistent with the variation of clinical outcomes observed globally. We did not detect significant differences in body weight (Fig. 1G) at birth and explant and brain weight (Fig. 1H) at the end of the 6-day period, nor in brain to body weight ratio (Fig. 1I) among any of the studied groups.
Figure 1: Neurobehavioral outcomes among the studied groups:

The total clinical outcomes differ among UCO and control groups (A), with subset of more severe UCOs animals that never regain their functioning vs less severely injured UCOi animals that reach comparable milestones with controls on day 3 after the UCO. The milestones evaluated (B) included feeding, use of the hind and front limbs, ability to move four limbs, stable standing, walking, and mental status reflected by activity at rest. The images reflect the control animals on day 0 (C) achieving all scored milestones. The UCO animals on day 0 exhibit significant encephalopathy and sleepiness (D), inability to stand (E). The most severe group, UCOs exhibits persistent encephalopathy, spastic diparesis on day 6 after the UCO (F). Plotted are changes in body weight (G), brain weight (H) and brain to body ratio (I). Red-UCOs group, Black-UCOi group, Green- control group. ns-not significant, **p<0.01,***p<0.001, ****p<0.0001.
Hemodynamic parameters
All lambs were hemodynamically monitored via invasive arterial catheters starting in utero prior to UCO. We assessed for changes in hemodynamic parameters continuously for a duration of up to 60 min post-delivery. UCO resulted in hypotension and bradycardia, ultimately resulting in pulseless electrical activity/asystole within approximately 16.18 ± 1.099 minutes (Fig. 2). We did not observe differences in measured hemodynamic parameters (systolic and diastolic blood pressures (SBP, DBP) as well as mean arterial pressure (MAP) between the UCOi and UCOs groups (Fig. 2). The onset of asystole, duration of cardiopulmonary resuscitation (CPR) and time to return of spontaneous circulation (ROSC) were comparable between UCOs and UCOi cohorts (asystole: UCOi 16.64 ±1.65 vs UCOs 15.33 ± 0.84 min; CPR: UCOi 1.81 ± 0.18 vs UCOs 1.66 ±0.33 min; ROSC: UCOi 6.81± 0.18 vs UCOi 6.66 ± 0.33 min). All animals received one dose of epinephrine. Two second doses of epinephrine were necessary to achieve ROSC in UCOi animals (2 of n=11), while one second dose was given to one UCOs animal (1 of n=5). No additional medications were administered.
Figure 2: Hemodynamic characteristics of our UCO model compared to control:

All animals have similar vital signs at the baseline 0 s. UCO leads to profound bradycardia resulting in asystole, and hypotension (A-C). Return of spontaneous circulation is accompanied by transient tachycardia. SBP- systolic blood pressure (A), MAP- mean arterial pressure (B), DBP- diastolic blood pressure (C).
Blood gas and glucose analyses
Blood sampling to assess arterial blood gases (ABG) was undertaken in utero prior to UCO to assess baseline status, at the end of asphyxia just prior to resuscitation (BSN), and post-resuscitation at regular intervals. ABG analysis revealed similar baseline parameters (pH, pCO2, pO2, glucose, lactate, bicarbonate, hemoglobin) for all groups (Fig. 3A–E, Tab. 2, BSN). UCO led to significant acidosis in injured lambs when compared with uninjured controls (Fig. 3A,B,E, Tab. 2, CPR, 10min). Between the 2 clinically different groups of the UCO lambs (UCOs vs UCOi), in the ones with more severe clinical outcome (UCOs) we noticed more significant metabolic acidosis reflected as lower pH (p=0.03), bicarbonate (p=0.02) (data not shown) and more profound base deficit during the CPR (p=0.01). Both groups continued to have significant base deficit during the whole observed post resuscitation period and hyperlactemia at 60 min compared to control (Fig. 3B,E, Tab. 2, 10–60min). While the UCOi animals had significant hypercarbia compared to controls 20min after the UCO (p=0.003), profound hypercarbia developed in the UCOs animals at 30min (p<0.0001) (Fig. 3C, Tab. 2, 20–60min). The UCOs and UCOi animals also developed significant hyperoxia compared to controls (UCOs:p=0.009, UCOi:p=0.04) (Fig. 3D, Tab. 2, 10min). UCO led to significant hypoglycemia in both UCOs and UCOi detected at 10 (UCOs: p=0.006; UCOi: p=0.006) and 20 min (UCOs:p=0.03; UCOi:p=0.01) after the insult (Fig. 3F, Tab. 2 10–30min). The hemoglobin levels were not significantly different in any of the studied groups (data not shown).
Figure 3: Arterial blood gas parameters:

All animals have comparable baseline values. UCO leads to profound respiratory and metabolic acidosis reflected by pH < 7 (A), lower base excess (B), high pCO2 (C), hypoxia (D) and hyperlactatemia (E). UCO also causes glucose variability (F), with profound hypoglycemia immediately following resuscitation, followed by prolonged hyperglycemia. The resuscitation leads to brief period of hyperoxia with high pO2. BSN- baseline, CPR- cardiopulmonary resuscitation, glc- glucose, BE- base excess, lac- lactate.
Table 2:
Blood gas analysis at different timepoints after the UCO.
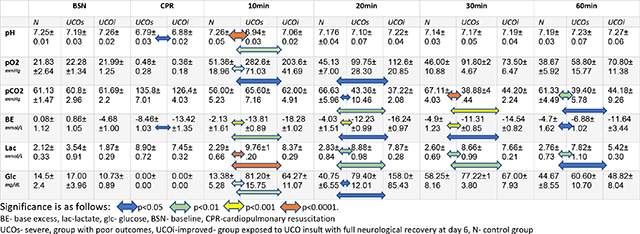
|
Histopathological Injury
Periventricular and subcortical white matter
Given the significant motor deficits observed, we initially evaluated brains for evidence of white matter injury. White matter injury was assessed by measuring the quantity and integrity of the major structural components of the myelin sheath including MBP, CNPase, as well as by the quantity of mature oligodendrocytes stained with CC-1 and total oligodendrocytes labeled by Olig-2. The overall cellularity evaluated by DAPI nuclear counts within periventricular white matter (PVWM), subcortical white matter of the cingulate gyrus (SCWM1) and subcortical white matter of the first parasagittal gyrus (SCWM2) was similar across all groups (data not shown). MBP volume was significantly lower in PVWM and SCWM1 of the UCOs groups compared with control (PVWM: p=0.002; SCWM1: p< 0.0001) (Fig. 4A). HIE-affected lambs in the UCOi group, however, had lower MBP volume vs control only in SCWM1 (p<0.006) (Fig. 4A). The UCOs additionally had lower MBP volumes compared to the UCOi in PVWM (p=0.002) and SCWM2 (p=0.02). The myelin proteins stained by CNPase were unchanged in all groups compared with control (PVWM: UCOi, p=0.75; UCOs, p> 0.99; SCWM1: UCOi, p>0.99; UCOs, p>0.99; SCWM2: UCOi, p=0.91; UCOs, p= 0.54) (Fig. 4B).
Figure 4: Quantitative analysis of white matter markers and markers of inflammation:

The volume of MBP (A), CNPase (B) positive cells and the number of Olig-2 (C) and CC-1 (D) positive cells reflect the changes triggered by UCO in PVWM, SCWM1 and SCWM2. The neuroinflammation was quantified by total volume of Iba-1-positive microglial cells (E) and presence of gliosis (F). The cellular death was quantified by the numbers of caspase-3 positive cells (G). Brackets show significances as follows: *p<0.05, **p<0.01, ***p<0.001, #p<0.0001. UCOs (black)-injured animals with poor outcomes, UCOi (dark gray)- injured animals with improved outcomes on day 6, N (green)- controls, I (light gray)- all injured animals, I=UCOs+UCOi.
Olig-2 staining of oligodendrocyte lineage cells was significantly increased in the UCOi group compared with control only in SCWM2 (p=0.03) (Fig. 4C). Olig-2-positive cells counts differed between UCOs and UCOi groups in SCWM1 (p=0.03) and SCWM2 (p=0.01). The number of mature oligodendrocytes labeled by CC-1 marker did not significantly differ among studied groups (Fig. 4D).
Inflammation of the white matter was investigated by volumetric and morphologic assessment of astroglial (GFAP) and microglial cells (Iba-1). The volume of Iba-1-positive cells was significantly higher in UCOs animals in all areas studied compared to controls (PVWM: p=0.01; SCWM1: p=0.004, SCWM2: p=0.03). UCOi and UCOs brains differed in the Iba-1 volumes in PVWM (p=0.03) and SCWM1 (p=0.003) (Fig. 4E). The astroglial cell volumes did not differ among UCOi, or UCOs compared to controls. Elevated astrogliosis was noted in UCOs compared to UCOi in SCWM2 (p=0.005) (Fig. 4F).
Cell death determined by Casp-3 indicated greater number of Casp-3-positive cells in injured animals compared to controls in all areas studied (PVWM: p=0.05, SCWM1: p=0.01, SCWM2: p=0.007). However, we did not detect differences when subgroups UCOs, UCOi were measured compared to controls in any of the studied groups (Fig. 4G).
Structurally, we observed disrupted myelin structure and myelin loss in PVWM, SCWM1 and SCWM2 when assessed by MBP immunoreactivity (Fig. 5), however no evident changes in CNPase were observed. The astrocytes in injured brains had shorter, thicker, and fewer processes, and larger cell bodies compared to control. Microglia had larger cell bodies, thicker processes and were globally present in higher volumes in injured brains compared to control (Fig. 5).
Figure 5: Histological findings in white matter after UCO:

Representative photomicrographs of MBP, CNPase, Olig-2, Iba-1, GFAP, cleaved caspase-3 and CC-1 in PVWM, SCWM1, SCWM2. MBP shows reduction in the number of myelin fibers and myelin breaks in the injured UCOs brains compared to controls. The Olig-2 is elevated in SCWM2 in UCOi animals. The CNPase does not show significant changes in injured animals compared to the controls. The Iba-1 marker is elevated due to increased number, cell body dimension and thickness of processes in all studied white matter areas in UCOs animals. Similarly, GFAP-stained astrocytes show thicker bodies and shortened processes. The caspase-3 is elevated in injured animals compared to controls. (Scale marker= 100 μm; blue- DAPI nuclear staining; the pathologies are pointed at by an arrowhead). PVWM- periventricular white matter, SCWM1- subcortical white matter of cingulate gyrus, SCWM2- subcortical white mater of the 1st parasagittal gyrus.
Gray matter injury
Histological injury was initially detected by using the FJ marker to identify degenerating neurons in brains on day 6. We observed degenerating neurons primarily in the cingulate gyrus (Ctx1) (Fig. 6A), extending to the parasagittal 1st (Ctx2) and 2nd gyri (Fig. 6B,C). In the cortex, we detected damage to neurons in the II. layer (Fig. 6D,E), more extensive injury involved neurons in the III.-V. layer (Fig. 6F), as well as deeper brain structures, such as caudate (Caud), putamen (Put), hippocampus (Hc) and thalamus (Th) (Fig. 6G–J). The extent of neuronal damage detected by FJ staining did not necessarily correlate with clinical outcomes, as some of the animals showing more extensive injury by FJ were clinically unaffected (Fig. 6K). However, the clinically most severely affected animals all exhibited neurological injury within the Ctx1/Ctx2 (n=5), as well as severe damage to Hc (n=3).
Figure 6: Initial injury evaluation by using FJ marker:

We assigned score 0 for areas with no FJ neurons, 1 for mild injury manifested by a single layer or scattered neurons (A), (D), 2 for moderate injury involving 2 laminar layers of neurons in one anatomical area (B), (E) and 3 for severe injury involving > 2 cortical layers (C), (F) or > 2 foci of damaged neurons in deeper brain structures, such as hippocampus (G), caudate (H), putamen (I) and thalamus for a total score of 0–15. The injury affected predominantly watershed areas of parasagittal cortex and/or basal ganglia (J, yellow marker= injury areas). The FJ score did not correlate with the clinical outcomes (K). Photomicrographs are representative images of FJ staining at 10X (A-C and G-I) and 25X (D-F). Brackets show significances as follows: *p < 0.05.
We assessed the extent of global neuronal loss on day 6 by measuring the total number of NeuN-positive neurons in Ctx1, Ctx2, Caud, Put and hippocampus Ca1/2, Ca3. The total number of NeuN-positive neurons was lower in the cortex of the cingulate gyrus and Ctx1 in UCOs animals compared to controls (p=0.03), as well as the UCOi animals (p=0.002). We did not detect significant changes in cortex of the 1st parasagittal gyrus (Ctx2), nor in caudate or putamen in all groups studied (Fig. 7A). In hippocampus, the neuronal loss was significant in Ca1/2 in the UCOi group (p=0.03), while the Ca3 regon demonstrated fewer NeuN cells in the UCOs group compared to control (p=0.04) as well as compared to the UCOi group (p=0.01)(Fig. 7A). The overall cellularity assessed by DAPI-staining was comparable in all areas studied but Ctx2, where UCOs animals had higher number of DAPI-positive cells compared to controls (p=0.02, data not shown).
Figure 7: Histological changes in gray matter:

Quantitative changes in neuronal counts (NeuN, A), cellular death markers (caspase-3, B) and inflammatory markers of microglial accumulation (Iba-1, C) and gliosis (GFAP, D) in cingulate gyrus cortex (Ctx1), 1st parasagittal gyrus cortex (Ctx2), caudate (caud), putamen (put), Ca1/2 and Ca3 of the hippocampus (ca1/2 and ca3). Brackets show significances as follows: *p < 0.05, **p < 0.01., ***p<0.001. Representative photomicrographs of CA3 area of injured hippocampus (F) show loss of neurons (NeuN-UCO) with more gliosis (GFAP-UCO), accumulation of activated amoeboid microglia (Iba-1-UCO); and significant cellular death (Caspase-3-UCO) yellow arrowheads. The hippocampus in control brain shows normal neuronal structure (NeuN-control), less gliosis (GFAP-control), ramified microglia (Iba-1-control) and lower counts of caspase-3 positive cells (Caspase-3 control); white arrowheads. Scale marker= 100 μm; Blue- DAPI nuclear staining.
We further assessed for apoptotic cell death by counting the total number of Casp-3-positive cells. In the cortex of the parasagittal gyrus, Casp-3-stained cells were measured in UCOi group compared to controls (p=0.01). High Casp-3 counts were found in hippocampus in UCOs animals compared to controls in both, Ca1/2 (p=0.001) and Ca3 (p<0.0001). No increase in Casp-3 positive cells in any group of animals was detected in the cortex of cingulate gyrus, caudate or putamen (Fig. 7B,E).
Iba-1 positive cells were significantly elevated in UCOs animals compared to controls in all areas studied (Ctx1: p= 0.03, Ctx2: p=0.006, caud: p=0.02, put: p= 0.006, ca1/2:p=0.0008, ca33: p=0.002). UCOi animals had elevation in Iba-1 positive cell volume only in the cortical area (ctx1: p=0.03, ctx2: p=0.02) (Fig. 7C).
We did not observe changes in astrogliosis in either Ctx1, Ctx2, caudate or putamen gyrus among all groups studied (Fig. 7D). Increased astrogliosis was noticed in Ca1/2 when we compared all injured animals to control group (p=0.02), however no significant differences were noted between groups UCOs (p=0.06) or UCOi (p=0.94) lambs. In Ca3 of the hippocampus, astrogliosis was observed in the UCOs animals compared to controls (p=0.01) (Fig. 7D). The focal injury site was characterized by shrunken, pyknotic NeuN+cells and neuronal loss (Fig. 7E, Fig. 8). Microglial cells at the injury site had ameboid features with enlarged cell bodies and fewer processes. The injury site exhibited accumulation of GFAP-positive fibers suggestive of gliosis (Fig. 7E, Fig. 8).
Figure 8: Representative photomicrographs of histological changes in gray matter:

Images compare injured (UCO) vs control brains (Control). Arrows point to glial cells with smaller processes and thicker cell bodies (GFAP), loss of NeuN cells with loss of organization of the cortex (NeuN), accumulation of activated microglia with shorter processes, round cell bodies (Iba-1) and apoptotic cells with shrunken cell bodies and pycnotic nuclei suggesting cell death (Caspase-3). Cortex (ctx), caudate (caud) and putamen (put). (Scale marker= 100 μm; blue- DAPI nuclear staining).
Discussion
We have identified two subgroups of animals that differ in their physiologic, histologic, and neurodevelopmental responses after suffering similarly severe metabolic insults observed in the term neonate suffering perinatal asphyxia. The UCO affected lambs developed a severe metabolic acidosis prior to resuscitation similar to infants born with profound HIE, and all lambs exhibited significant encephalopathy typically lasting hours to days. 35 % of lambs exhibited severe motor deficits compromising ambulation throughout the 6-day course and, in the most severely affected lambs, exhibited spastic diparesis similar to that observed in older infants with severe HIE. The UCOs group exhibited gliosis and inflammation in both white and gray matter, oligodendrocyte loss, and neuronal loss and cellular death in the hippocampus and cingulate cortex. The UCOi group showed more cellular death and gliosis in the parasagittal cortex but demonstrated greater preservation of white matter markers, reduced markers of inflammation and lower cellular death and neuronal loss in Ca3 of the hippocampus compared with UCOs lambs.
A common pathophysiologic pathway ultimately resulting in perinatal brain injury in humans is characterized by the sequence of hypotension with or without asystole, leading to primary energy failure due to brain hypoxia and ischemia followed by secondary energy failure due to reperfusion injury [26,27]. Our sheep model met the commonly used clinical criteria that define an acute intrapartum event sufficient to cause HIE, including severe intrapartum acidosis with pH < 7 and base deficit > 12 mmol/L, moderate/severe encephalopathy, spastic di/quadriplegia or dyskinesia, and absence of other factors [28]. Prolonged UCO in the near-term lamb led to a double-insult consisting of profound hypotension and hypoxemia, both needed to create an injury as near-term lambs have more mature cerebral blood flow autoregulation [29]. Utilizing this model, we identified two subsets of animals in the UCO group that significantly differed in their clinical neurological outcomes despite suffering the same insult that led to a comparable change in hemodynamic parameters. The differences in neurological outcomes became evident only 3–6 days after injury. Approximately a third of the animals suffered severe neurological disability resulting in severe encephalopathy and spastic diplegia, while the second subgroup of injured animals improved their mental status and motor outcomes starting on day 3. The neurobehavioral outcomes in the latter group of animals fully matched the uninjured control group at day 6 after the insult. These two subgroups differed, however, in their biochemical parameters, as well as histopathological findings.
Blood gas analysis correlated with neurological outcomes, as the group of animals with more severe neurological injury had more profound acidosis with lower pH and more severe base deficits early during resuscitation. This is similar to near-term ovine UCO studies and human studies where pH, lactate and base deficit belong to the strongest predictors of clinical outcomes [17,30]. These predictors also include the need for resuscitation in babies, however in our study both groups underwent cardiopulmonary resuscitation suggesting presence of other factors impacting the outcomes. Similar to selected human studies, the lactate levels took longer to normalize in the more severely injured animals which could be related to the degree of end-organ dysfunction impacting lactate clearance or seizure activity and may predict outcomes [31,32]. The impact of lactate could be level-dependent, Da Silva observed that plasma lactate concentration greater than 9 mmol/L was associated with moderate or severe encephalopathy with a sensitivity of 84% and a specificity of 67%, while lactate < 5 mmol/L did not lead to significant encephalopathy [33]. The hemodynamic parameters were largely similar between the two injured groups. Notable also is hyperoxia at 10 min in both UCO groups after asystole resulting from the use of 100 % O2 for initial resuscitation. Hyperoxia in the immediate postnatal period has been associated with the increased incidence of HIE in term infants [31]. However, as the hyperoxia is noticed in both groups, we exclude it as a major contributing factor responsible for different clinical neurological outcomes between the UCO groups in our model. The UCO insult was associated with a profound systemic hypoglycemia in injured animals at the time of CPR. Rapid depletion of glucose that occurs during the hypoxic episode is associated with anaerobic metabolism, and rapid rise in lactate in both hemispheres [35] and early hypoglycemia in term infants between 0–6 hours is reported to be associated with worse outcomes [36]. The neonatal brain is especially susceptible to hypoglycemia compared to the adult brain [35,37]. While treatment of the hypoglycemia after hypoxic-ischemic injury may reverse the anoxic vulnerability as shown by Vannucci et al [38], this phase is followed by an episode of hyperglycemia at 10–20 min after the UCO probably as a result of a stress response. Mallard et al [39] observed hyperglycemia as early as 4 min at UCO, however our measurements unfortunately did not include this timepoint. Hypoglycemia, hyperglycemia and high glucose variability are associated with poor neurological outcomes in term neonates [40–43]. However, the changes in glucose were not significantly different between the more and less severe group of UCO animals.
Our histological findings align with 2 major MRI patterns of term neonatal HIE described by Miller et al: watershed-predominant and basal ganglia/thalamus-predominant that are associated with different clinical presentations and neurodevelopmental outcomes [44,45]. Mild to moderate HIE produces parasagittal watershed zone infarcts involving both the cortex and underlying subcortical white matter. Severe HIE results in injury to metabolically active tissues such as thalami, putamina, hippocampi, brainstem, corticospinal tracts, and sensorimotor cortex [46].
Consistent with previous findings of white matter injury in near-term fetal sheep [47–49], our UCO model led to loss and disruption of myelination and increased numbers of microglia in the intragyral and periventricular white matter of the more severely injured animals [50]. While decreased immunoreactivity in MBP with myelin breaks and areas of hypomyelination affected PVWM, SCWM1 and SCWM2 in the more injured animals, the CNPase stained myelin sheaths did not differ among groups. CNPase is considered an index of myelin formation where the amount of immunoreactive CNPase correlates with the thickness of the myelin sheath in the central nervous system [51]. Interestingly, a higher number of Olig2-positive cells was detected in SCWM2 in the less severely injured group. We did not detect significant loss of mature oligodendrocytes labeled by CC-1. Changes in white matter after brain injury are dynamic [52] and the quantitative description of white matter markers at 6 days after UCO likely reflects the process of an ongoing injury, as well as remyelination. We speculate that areas of unchanged oligodendrocyte count, MBP and CNPase in less severe injured animals reflect some degree of remyelination that is less active in the group of animals with worse outcomes, where the MBP remain low, and CNPase and oligodendrocyte counts are unchanged.
Gray matter injury was initially assessed by using Fluoro-Jade staining (FJ) that has been validated as a sensitive and reliable marker of ongoing neuronal degeneration [53,54]. The FJ staining pattern was characterized by injury consistent with a combination of linear necrosis and focal patches. FJ staining revealed areas of specific neuronal susceptibility to the UCO that was located in almost all injured animals predominantly in the areas of the cingulate gyrus and parasagittal cortex. The FJ positive neurons were detected in striatum, hippocampus, and thalamus, although less frequently. This pattern of neuronal susceptibility is similar to other UCO studies in term sheep and term neonates after HIE [29,39,55]. The areas identified by FJ in our model correspond to areas of susceptibility to the regional cerebral blood flow (CBF) changes (parasagittal cortex) [56] and areas of high glucose utilization (cingulate cortex, basal ganglia, and hippocampal regions) in neonates [57,58]. Thus, if the compensatory mechanisms to maintain CBF and glucose levels are exhausted, they lead to a characteristic injury pattern that is responsible for motor and cognitive deficits seen in neonates after HIE [44,46]. While FJ enabled our study to distinguish between the injured and control animals, the overall FJ injury score did not predict clinical outcomes. This could be due to clearance of damaged neurons from the injury site at our chosen timepoint of 6 days. The Iba-1/NeuN/GFAP immunohistochemistries showed global changes, as well as detected focal injury patterns characterized by neuronal loss, microglial activation and glial scar formation, an injury pattern described in non-human primates after severe HIE [59]. Neuronal loss detected by the NeuN marker was predominant in the parasagittal areas.
Interestingly, cellular death at day 6 was more pronounced in the animals with better outcomes in cortex. We speculate that this may be attributed to more active cellular turnover as a part of neuroregeneration. Significant cell death was found in Ca3 areas of hippocampus in the most severely injured animals, together with more profound neuronal loss, presence of microglial cells and more astrogliosis. UCOs animals also demonstrated gliosis and microglial cell accumulation in Ca1/2, however we did not detect significant neuronal loss. Combination of cell death and neuronal loss in Ca1/2 was notable in UCOi animals. This likely suggests more extensive inflammation in hippocampus and cortex in UCOs animals with cellular loss. The neuronal loss more predominant in the CA3 areas is similar to the observation of Mallard et al [60]. Dense activated microglial infiltration notable in hippocampal stratum moleculare and radiatum, suggests possible damage of hippocampal neuronal dendrites. Preserved interaction between pyramidal cells and interneurons in hippocampus is essential for learning and memory formation and for functional recovery after injury [61], thus excessive microglial activation could be one of the factors contributing to delayed hippocampal injury leading to worse neurobehavioral outcomes after HIE.
Our study has several limitations. While the birth weight, initial hemodynamic parameters, and biochemical data were similar among our groups, we cannot exclude other confounders to poor neurological outcomes in one group compared to the other. Defining additional physiological and biochemical factors that worsen outcomes in one group, or improve outcomes in the other animals, such as timing of the cord clamping or depth of chest compressions is crucial in understanding of the pathobiology of neonatal HIE [18], as well as for designing therapies. Because of the cost and maintenance of large animal studies, our study included lower numbers of animals representing individual groups than is typically accomplished with rodent studies. This prevented us from considering well-known effects of sex on response to brain HI and neurological outcomes [62,63]. These factors also limited the duration of follow-up in these lambs. Our hemodynamic parameters did include only the first 60 min after UCO, where the vital signs did not typically return to the baseline preventing us from better defining the hemodynamic alterations. We did not assess subclinical seizure activity by EEG. The variability in severity of neurological injury poses a challenge to measure effectiveness of therapeutic interventions. This variability is also observed in clinical studies and is likely due to variation in intrinsic susceptibilities driven by genetic heterogeneity. Our use of an outbred ovine model thus accurately reflects the heterogeneity observed clinically. Our pathohistological investigation did not include staining for all cell types, regions and processes relevant to HIE, including cerebellum, or different mechanisms of cell death. We plan on addressing these deficiencies in future studies.
It is well known that brain injury following HIE evolves over time, and some potential therapies show benefit even when administered outside of the initial 6-hour window critical for therapeutic hypothermia [26]. Our large animal model of moderate HIE with prolonged follow-up will help further define pathophysiologic drivers of brain injury while enabling identification of predictive biomarkers that correlate with disease outcomes and ultimately help support development of therapeutic approaches to this challenging clinical scenario. Our findings thus suggest that assessing neurodevelopmental outcomes and the effects of neurotherapeutic interventions might need to be extended at last until 6 days after UCO in ovine models.
Supplementary Material
Acknowledgement
We would like to thank to Sandrijn Vanschaik, Jeff Fineman and Division of Pediatric Critical Care and Neonatology at UCSF and UCD for their help and support.
Funding Sources
This work was supported by Bill & Melinda Gates Foundation, NIH grants HD096299 (PV) and HD072929 (SL), R35– 5R35NS097299 (DF), T32GM007546 (YW), K12HD105250 (JKM) and UCSF Pediatric Critical Care Division.
Footnotes
Conflict of Interest Statement
The authors have no conflicts of interest to declare.
Statement of Ethics
All animal research was approved by the University of California San Francisco Institutional Animal Care and Use Committee and was performed in accordance with the Guide for the Care and Use of Laboratory Animals. This study protocol was reviewed and approved by UC Davis IACUC, approval number 20777.
Data Availability Statement
All data generated or analyzed during this study are included in this article [and/or] its supplementary material files. Further enquiries can be directed to the corresponding author.
References
- 1.Lawn JE, Manandhar A, Haws RA, Darmstadt GL. Reducing one million child deaths from birth asphyxia – a survey of health systems gaps and priorities. Heal Res Policy Syst. 2007. Dec;5(1). DOI: 10.1186/1478-4505-5-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lawn JE, Lee AC, Kinney M, Sibley L, Carlo WA, Paul VK, et al. Two million intrapartum-related stillbirths and neonatal deaths: Where, why, and what can be done? Int J Gynecol Obstet. 2009. Oct;107(Supplement). DOI: 10.1016/j.ijgo.2009.07.016 [DOI] [PubMed] [Google Scholar]
- 3.Bhatti A, Kumar P. Systemic Effects of Perinatal Asphyxia. Indian J Pediatr. 2014. Mar;81(3). DOI: 10.1007/s12098-013-1328-9 [DOI] [PubMed] [Google Scholar]
- 4.Edwards AD, Brocklehurst P, Gunn AJ, Halliday H, Juszczak E, Levene M, et al. Neurological outcomes at 18 months of age after moderate hypothermia for perinatal hypoxic ischaemic encephalopathy: synthesis and meta-analysis of trial data. BMJ. 2010. Feb;340(feb09 3). DOI: 10.1136/bmj.c363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hutton JL. Life expectancy in severe cerebral palsy. Arch Dis Child. 2006. Mar;91(3). DOI: 10.1136/adc.2005.075002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee AC, Kozuki N, Blencowe H, Vos T, Bahalim A, Darmstadt GL, et al. Intrapartum-related neonatal encephalopathy incidence and impairment at regional and global levels for 2010 with trends from 1990. Pediatr Res. 2013. Dec;74(S1). DOI: 10.1038/pr.2013.206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jacobs SE, Berg M, Hunt R, Tarnow-Mordi WO, Inder TE DP. Cooling for newborns with hypoxic ischaemic encephalopathy. [Internet]. Cochrane Database Syst Rev. 2013. DOI: 10.1002/14651858.CD003311.pub3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bach AM, Fang AY, Bonifacio S, Rogers EE, Scheffler A, Partridge JC, et al. Early Magnetic Resonance Imaging Predicts 30-Month Outcomes after Therapeutic Hypothermia for Neonatal Encephalopathy. J Pediatr. 2021. Jul DOI: 10.1016/j.jpeds.2021.07.003 [DOI] [PubMed] [Google Scholar]
- 9.Krishnan V, Kumar V, Shankaran S, Thayyil S. Rise and Fall of Therapeutic Hypothermia in Low-Resource Settings: Lessons from the HELIX Trial. Indian J Pediatr. 2021. Jul DOI: 10.1007/s12098-021-03861-y [DOI] [PubMed] [Google Scholar]
- 10.Thayyil S, Pant S, Montaldo P, Shukla D, Oliveira V, Ivain P, et al. Hypothermia for moderate or severe neonatal encephalopathy in low-income and middle-income countries (HELIX): a randomised controlled trial in India, Sri Lanka, and Bangladesh. Lancet Glob Heal. 2021. Sep;9(9). DOI: 10.1016/S2214-109X(21)00264-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sarnat HB, Sarnat MS. Neonatal encephalopathy following fetal distress. A clinical and electroencephalographic study. Arch Neurol. 1976. Oct;33(10):696–705. [DOI] [PubMed] [Google Scholar]
- 12.Ahearne CE, Boylan GB, Murray DM. Short and long term prognosis in perinatal asphyxia: An update. World J Clin Pediatr. 2016. Feb;5(1):67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mota-Rojas D, Villanueva-García D, Solimano A, Muns R, Ibarra-Ríos D, Mota-Reyes A. Pathophysiology of Perinatal Asphyxia in Humans and Animal Models. Biomedicines. 2022. Feb;10(2):347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mergenthaler P, Meisel A. Animal models: value and translational potency. Principles of Translational Science in Medicine. Elsevier; 2021; pp 95–103. [Google Scholar]
- 15.Tetorou K, Sisa C, Iqbal A, Dhillon K, Hristova M. Current Therapies for Neonatal Hypoxic–Ischaemic and Infection-Sensitised Hypoxic–Ischaemic Brain Damage. Front Synaptic Neurosci. 2021. Aug;13. DOI: 10.3389/fnsyn.2021.709301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Derrick M, Drobyshevsky A, Ji X, Tan S. A Model of Cerebral Palsy From Fetal Hypoxia-Ischemia. Stroke. 2007. Feb;38(2). DOI: 10.1161/01.STR.0000251445.94697.64 [DOI] [PubMed] [Google Scholar]
- 17.Aridas JDS, Yawno T, Sutherland AE, Nitsos I, Ditchfield M, Wong FY, et al. Detecting brain injury in neonatal hypoxic ischemic encephalopathy: closing the gap between experimental and clinical research. Exp Neurol. 2014. Nov;261:281–90. [DOI] [PubMed] [Google Scholar]
- 18.Polglase GR, Schmölzer GM, Roberts CT, Blank DA, Badurdeen S, Crossley KJ, et al. Cardiopulmonary Resuscitation of Asystolic Newborn Lambs Prior to Umbilical Cord Clamping; the Timing of Cord Clamping Matters! Front Physiol. 2020;11:902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aridas JDS, Yawno T, Sutherland AE, Nitsos I, Ditchfield M, Wong FY, et al. Systemic and transdermal melatonin administration prevents neuropathology in response to perinatal asphyxia in newborn lambs. J Pineal Res. 2018. May;64(4):e12479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yawno T, Castillo-Melendez M, Jenkin G, Wallace EM, Walker DW, Miller SL. Mechanisms of Melatonin-Induced Protection in the Brain of Late Gestation Fetal Sheep in Response to Hypoxia. Dev Neurosci. 2012;34(6). DOI: 10.1159/000346323 [DOI] [PubMed] [Google Scholar]
- 21.Gunn AJ, Gunn TR, de Haan HH, Williams CE, Gluckman PD. Dramatic neuronal rescue with prolonged selective head cooling after ischemia in fetal lambs. J Clin Invest. 1997. Jan;99(2):248–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson John I., Sudheimer Keith D., Davis Kristina K., Kerndt Garrett M. and BMW. The Sheep Brain Atlas [Internet]. Available from https//brains.anatomy.msu.edu/brains/sheep/index.html [Google Scholar]
- 23.Björkman ST, Foster KA, O’driscoll SM, Healy GN, Lingwood BE, Burke C, et al. Hypoxic/Ischemic models in newborn piglet: comparison of constant FiO2 versus variable FiO2 delivery. Brain Res. 2006. Jul;1100(1):110–7. [DOI] [PubMed] [Google Scholar]
- 24.Dwyer CM, Lawrence AB, Brown HE, Simm G. Effect of ewe and lamb genotype on gestation length, lambing ease and neonatal behaviour of lambs. Reprod Fertil Dev. 1996;8(8). DOI: 10.1071/RD9961123 [DOI] [PubMed] [Google Scholar]
- 25.Castillo-Melendez M, Baburamani AA, Cabalag C, Yawno T, Witjaksono A, Miller SL, et al. Experimental modelling of the consequences of brief late gestation asphyxia on newborn lamb behaviour and brain structure. PLoS One. 2013;8(11). DOI: 10.1371/journal.pone.0077377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Larpthaveesarp A, Pathipati P, Ostrin S, Rajah A, Ferriero D, Gonzalez FF. Enhanced Mesenchymal Stromal Cells or Erythropoietin Provide Long-Term Functional Benefit After Neonatal Stroke. Stroke. 2021. Jan;52(1). DOI: 10.1161/STROKEAHA.120.031191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Allen KA, Brandon DH. Hypoxic Ischemic Encephalopathy: Pathophysiology and Experimental Treatments. Newborn Infant Nurs Rev. 2011. Sep;11(3):125–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neonatal Encephalopathy and Cerebral Palsy: Executive Summary*. Obstet Gynecol. 2004. Apr;103(4). DOI: 10.1097/01.AOG.0000120142.83093.30 [DOI] [PubMed] [Google Scholar]
- 29.Koehler RC, Yang Z-J, Lee JK, Martin LJ. Perinatal hypoxic-ischemic brain injury in large animal models: Relevance to human neonatal encephalopathy. J Cereb Blood Flow Metab. 2018. Dec;38(12). DOI: 10.1177/0271678X18797328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mooney C, O’Boyle D, Finder M, Hallberg B, Walsh BH, Henshall DC, et al. Predictive modelling of hypoxic ischaemic encephalopathy risk following perinatal asphyxia. Heliyon. 2021. Jul;7(7):e07411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu T-W, Tamrazi B, Hsu K-H, Ho E, Reitman AJ, Borzage M, et al. Cerebral Lactate Concentration in Neonatal Hypoxic-Ischemic Encephalopathy: In Relation to Time, Characteristic of Injury, and Serum Lactate Concentration. Front Neurol. 2018. May;9. DOI: 10.3389/fneur.2018.00293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shah S, Tracy M, Smyth J. Postnatal Lactate as an Early Predictor of Short-Term Outcome after Intrapartum Asphyxia. J Perinatol. 2004. Jan;24(1). DOI: 10.1038/sj.jp.7211023 [DOI] [PubMed] [Google Scholar]
- 33.da Silva S, Hennebert N, Denis R, Wayenberg JL. Clinical value of a single postnatal lactate measurement after intrapartum asphyxia. Acta Paediatr. 2000. Mar;89(3). [PubMed] [Google Scholar]
- 34.Kapadia VS, Chalak LF, DuPont TL, Rollins NK, Brion LP, Wyckoff MH. Perinatal Asphyxia with Hyperoxemia within the First Hour of Life Is Associated with Moderate to Severe Hypoxic-Ischemic Encephalopathy. J Pediatr. 2013. Oct;163(4). DOI: 10.1016/j.jpeds.2013.04.043 [DOI] [PubMed] [Google Scholar]
- 35.Mikrogeorgiou A, Xu D, Ferriero DM, Vannucci SJ. Assessing Cerebral Metabolism in the Immature Rodent: From Extracts to Real-Time Assessments. Developmental Neuroscience. S. Karger AG; 2019; pp 463–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nadeem M, Murray DM, Boylan GB, Dempsey EM, Ryan CA. Early blood glucose profile and neurodevelopmental outcome at two years in neonatal hypoxic-ischaemic encephalopathy. BMC Pediatr. 2011. Dec;11(1). DOI: 10.1186/1471-2431-11-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vannucci RC. Experimental Biology of Cerebral Hypoxia-Ischemia: Relation to Perinatal Brain Damage. Pediatr Res. 1990. Apr;27(4). DOI: 10.1203/00006450-199004000-00001 [DOI] [PubMed] [Google Scholar]
- 38.Vannucci RC, Vannucci SJ. Cerebral carbohydrate metabolism during hypoglycemia and anoxia in newborn rats. Ann Neurol. 1978. Jul;4(1). DOI: 10.1002/ana.410040114 [DOI] [PubMed] [Google Scholar]
- 39.Mallard EC, Williams CE, Johnston BM, Gluckman PD. Increased vulnerability to neuronal damage after umbilical cord occlusion in fetal sheep with advancing gestation. Am J Obstet Gynecol. 1994. Jan;170(1). DOI: 10.1016/S0002-9378(94)70409-0 [DOI] [PubMed] [Google Scholar]
- 40.Pinchefsky EF, Hahn CD, Kamino D, Chau V, Brant R, Moore AM, et al. Hyperglycemia and Glucose Variability Are Associated with Worse Brain Function and Seizures in Neonatal Encephalopathy: A Prospective Cohort Study. J Pediatr. 2019. Jun;209. DOI: 10.1016/j.jpeds.2019.02.027 [DOI] [PubMed] [Google Scholar]
- 41.Al Shafouri N, Narvey M, Srinivasan G, Vallance J, Hansen G. High glucose variability is associated with poor neurodevelopmental outcomes in neonatal hypoxic ischemic encephalopathy. J Neonatal Perinatal Med. 2015. Jul;8(2). DOI: 10.3233/NPM-15814107 [DOI] [PubMed] [Google Scholar]
- 42.Chouthai NS, Sobczak H, Khan R, Subramanian D, Raman S, Rao R. Hyperglycemia is associated with poor outcome in newborn infants undergoing therapeutic hypothermia for hypoxic ischemic encephalopathy. J Neonatal Perinatal Med. 2015. Jul;8(2). DOI: 10.3233/NPM-15814075 [DOI] [PubMed] [Google Scholar]
- 43.Basu SK, Salemi JL, Gunn AJ, Kaiser JR. Hyperglycaemia in infants with hypoxic–ischaemic encephalopathy is associated with improved outcomes after therapeutic hypothermia: a post hoc analysis of the CoolCap Study. Arch Dis Child - Fetal Neonatal Ed. 2017. Jul;102(4). DOI: 10.1136/archdischild-2016-311385 [DOI] [PubMed] [Google Scholar]
- 44.Miller SP, Ramaswamy V, Michelson D, Barkovich AJ, Holshouser B, Wycliffe N, et al. Patterns of brain injury in term neonatal encephalopathy. J Pediatr. 2005. Apr;146(4):453–60. [DOI] [PubMed] [Google Scholar]
- 45.Mercuri E, Guzzetta A, Haataja L, Cowan F, Rutherford M, Counsell S, et al. Neonatal Neurological Examination in Infants with Hypoxic Ischaemic Encephalopathy: Correlation with MRI Findings. Neuropediatrics. 1999. Apr;30(02). DOI: 10.1055/s-2007-973465 [DOI] [PubMed] [Google Scholar]
- 46.Bano S, Chaudhary V, Garga UC. Neonatal hypoxic-ischemic encephalopathy: A radiological review. J Pediatr Neurosci. 2017. Jan;12(1):1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guan J, Bennet L, George S, Wu D, Waldvogel HJ, Gluckman PD, et al. Insulin-Like Growth Factor-1 Reduces Postischemic White Matter Injury in Fetal Sheep. J Cereb Blood Flow Metab. 2001. May;21(5). DOI: 10.1097/00004647-200105000-00003 [DOI] [PubMed] [Google Scholar]
- 48.Roelfsema V, Bennet L, George S, Wu D, Guan J, Veerman M, et al. Window of Opportunity of Cerebral Hypothermia for Postischemic White Matter Injury in the Near-Term Fetal Sheep. J Cereb Blood Flow Metab. 2004. Aug;24(8). DOI: 10.1097/01.WCB.0000123904.17746.92 [DOI] [PubMed] [Google Scholar]
- 49.Davidson JO, Yuill CA, Zhang FG, Wassink G, Bennet L, Gunn AJ. Extending the duration of hypothermia does not further improve white matter protection after ischemia in term-equivalent fetal sheep. Sci Rep. 2016. Apr;6(1). DOI: 10.1038/srep25178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Draghi V, Wassink G, Zhou KQ, Bennet L, Gunn AJ, Davidson JO. Differential effects of slow rewarming after cerebral hypothermia on white matter recovery after global cerebral ischemia in near-term fetal sheep. Sci Rep. 2019. Dec;9(1). DOI: 10.1038/s41598-019-46505-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Naureen I, Waheed KAi, Rathore AW, Victor S, Mallucci C, Goodden JR, et al. Fingerprint changes in CSF composition associated with different aetiologies in human neonatal hydrocephalus: glial proteins associated with cell damage and loss. Fluids Barriers CNS. 2013. Dec;10(1). DOI: 10.1186/2045-8118-10-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Michalski D, Keck AL, Grosche J, Martens H, Härtig W. Immunosignals of Oligodendrocyte Markers and Myelin-Associated Proteins Are Critically Affected after Experimental Stroke in Wild-Type and Alzheimer Modeling Mice of Different Ages. Front Cell Neurosci. 2018. Feb;12. DOI: 10.3389/fncel.2018.00023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schmued LC, Albertson C, Slikker W. Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res. 1997. Mar;751(1):37–46. [DOI] [PubMed] [Google Scholar]
- 54.Schmued LC, Hopkins KJ. Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000. Aug;874(2):123–30. [DOI] [PubMed] [Google Scholar]
- 55.Gunn AJ, Parer JT, Mallard EC, Williams CE, Gluckman PD. Cerebral Histologic and Electrocorticographic Changes after Asphyxia in Fetal Sheep. 1992. [DOI] [PubMed] [Google Scholar]
- 56.Volpe JJ, Herscovitch P, Perlman JM, Kreusser KL, Raichle ME. Positron emmission tomography in the asphyxiated term newborn: Parasagittal impairment of cerebral blood flow. Ann Neurol. 1985. Mar;17(3):287–96. [DOI] [PubMed] [Google Scholar]
- 57.Proisy M, Mitra S, Uria-Avellana C, Sokolska M, Robertson NJ, Le Jeune F, et al. Brain Perfusion Imaging in Neonates: An Overview. Am J Neuroradiol. 2016. Oct;37(10). DOI: 10.3174/ajnr.A4778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chugani HT. A Critical Period of Brain Development: Studies of Cerebral Glucose Utilization with PET. Prev Med (Baltim). 1998. Mar;27(2). DOI: 10.1006/pmed.1998.0274 [DOI] [PubMed] [Google Scholar]
- 59.McAdams RM, McPherson RJ, Kapur RP, Juul SE. Focal Brain Injury Associated with a Model of Severe Hypoxic-Ischemic Encephalopathy in Nonhuman Primates. Dev Neurosci. 2017;39(1–4). DOI: 10.1159/000456658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mallard EC, Gunn AJ, Williams CE, Johnston BM, Gluckman PD. Transient umbilical cord occlusion causes hippocampal damage in the fetal sheep. Am J Obstet Gynecol. 1992. Nov;167(5):1423–30. [DOI] [PubMed] [Google Scholar]
- 61.Chavez-Valdez R, Emerson P, Goffigan-Holmes J, Kirkwood A, Martin LJ, Northington FJ. Delayed injury of hippocampal interneurons after neonatal hypoxia-ischemia and therapeutic hypothermia in a murine model. Hippocampus. 2018. Aug;28(8). DOI: 10.1002/hipo.22965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.MURDEN S, BORBÉLYOVÁ V, LAŠTŮVKA Z, MYSLIVEČEK J, OTÁHAL J, RILJAK V. Gender Differences Involved in the Pathophysiology of the Perinatal Hypoxic-Ischemic Damage. Physiol Res. 2019. Dec DOI: 10.33549/physiolres.934356 [DOI] [PubMed] [Google Scholar]
- 63.Rosenkrantz TS, Hussain Z, Fitch RH. Sex Differences in Brain Injury and Repair in Newborn Infants: Clinical Evidence and Biological Mechanisms. Front Pediatr. 2019. Jun;7. DOI: 10.3389/fped.2019.00211 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this article [and/or] its supplementary material files. Further enquiries can be directed to the corresponding author.