

Abstract
Background and Purpose:
β-agonists relieve airflow obstruction by activating β2-adrenergic receptors (β2ARs), which are G protein-coupled receptors (GPCRs) expressed on human airway smooth muscle (HASM) cells. The currently available β-agonists are balanced agonists, however, and signal through both the stimulatory G protein (Gs)- and β-arrestin-mediated pathways. While Gs signalling is beneficial and promotes HASM relaxation, β-arrestin activation is associated with reduced Gs efficacy. In this context, biased ligands that selectively promote β2AR coupling to Gs signalling represent a promising strategy to treat asthma. Here we examined several β-agonists to identify Gs-biased ligands devoid of β-arrestin-mediated effects.
Experimental approach:
Gs-biased ligands for the β2AR were identified by high-throughput screening and then evaluated for Gs interaction, Gi interaction, cAMP production, β-arrestin interaction, GPCR kinase (GRK) phosphorylation of the receptor, receptor trafficking, ERK activation, and functional desensitization of the β2AR.
Key results:
We identified ractopamine, dobutamine and higenamine as Gs-biased agonists that activate the Gs/cAMP pathway upon β2AR stimulation while showing minimal Gi or β-arrestin interaction. Furthermore, these compounds didn’t induce any receptor trafficking and had reduced GRK5-mediated phosphorylation of the β2AR. Finally, we observed minimal physiological desensitization of the β2AR in primary HASM cells upon treatment with the biased agonists.
Conclusion and Implications:
Our work demonstrates that Gs-biased signaling through the β2AR may prove to be an effective strategy to promote HASM relaxation in the treatment of asthma. Such biased compounds may also be useful in identifying the molecular mechanisms that determine biased signaling and in the design of safer drugs.
Keywords: asthma, β2-adrenergic receptor, airway smooth muscle, biased signaling, β-arrestins, desensitization, G protein, G protein-coupled receptor
1. INTRODUCTION
Asthma is a complex heterogenous disease with a prevalence in the United States estimated at 8% of adults and 7% of children (according to the Centers for Disease Control and Prevention, 2020). Asthma is mainly characterized by chronic airway inflammation and bronchial obstruction due to airway smooth muscle (ASM) contraction. As a cornerstone in the management of asthma or chronic obstructive pulmonary disease (COPD), bronchodilators such as β-agonists (short and long acting) reverse or prevent airflow obstruction by targeting the β2-adrenergic receptor (β2AR) (Wendell et al., 2020).
The β2AR is a well-studied G protein-coupled receptor (GPCR) that, upon binding to endogenous catecholamines or exogenous agonists, couples to the heterotrimeric stimulatory G protein Gs to activate adenylyl cyclase that increases cyclic adenosine monophosphate (cAMP) production (Pierce et al., 2002). This signalling cascade is responsible for mediating ASM tone, heart rhythm, and other processes of the sympathetic nervous system (Insel, 1996). β2AR signalling is primarily terminated via receptor phosphorylation by GPCR kinases (GRKs) and subsequent recruitment of β-arrestins through a process called desensitization (Krupnick and Benovic, 1998). The role of β-arrestins as a negative regulator of β2AR signalling and as a scaffold to promote receptor internalization has been expanded to include signal transduction through G protein independent pathways (DeWire et al., 2007). Moreover, in addition to cAMP production through the Gs pathway, the β2AR can couple to the inhibitory G protein (Gi), promote ERK1/2 activation, and possibly engage other signalling pathways (Benovic, 2002; Azzi et al., 2003; Shenoy et al., 2006). Among all signalling pathways induced by the β2AR, several studies identified the Gs/cAMP/PKA pathway as the primary mechanism behind the beneficial effects of β2AR-specific agonist (β2-agonist) administration in treating asthma, mainly by inducing ASM relaxation (Penn and Benovic, 2008; Walker et al., 2011). For this reason, β2-agonists have been used efficaciously and extensively in the clinical treatment of mild to severe asthma (Matera et al., 2020). Despite this, β2-agonists have also shown unwanted side effects that include increased risk of asthma morbidity/mortality in patients chronically treated with β2-agonists (Castle et al., 1993; Nelson et al., 2006; Salpeter et al., 2006), leading the US Food and Drug Administration to add a ‘black box warning’. Due to such increasing concern of β2-agonist safety, identifying the molecular mechanisms behind such unwanted side effects has been a major research goal in the development of newer and safer drugs.
Previous studies aimed at understanding the basis for β2-agonist-mediated side effects have suggested that β-arrestins may contribute to this process. For example, β2AR signalling was increased in cultured ASM cells with β-arrestin-knockdown, and β-arrestin2 knockout mice showed greater relaxant tone in response to β-agonist administration (Deshpande et al., 2008; Penn and Benovic, 2008). Thus, one potential mechanism involves chronic stimulation by β-agonists leading to β-arrestin-mediated desensitization of the β2AR with a loss of drug effectiveness in ASM cells (Walker et al., 2011) and consequently a loss of bronchoprotective action in asthmatic patients (Cheung et al., 1992; Bhagat et al., 1995; Grove and Lipworth, 1995). In addition, a role for β-arrestins in promoting an inflammatory response has also been implicated (Walker et al., 2003; Hollingsworth et al., 2010; Walker and DeFea, 2014; Chen et al., 2015).
The β2-agonists currently used in clinical practice are balanced agonists, meaning that upon interaction with the β2AR, they are able to induce both the Gs and β-arrestin mediated pathways (Matera et al., 2020). Such activation of the β2AR might contribute to the side effects encountered with chronic administration of β2-agonists in asthmatic patients. In this scenario, the unique properties of biased ligands to selectively activate a subset of the normal signaling repertoire could be extremely helpful (Smith et al., 2018). In the case of the β2AR, a Gs-biased agonist would promote Gs-mediated bronchodilation while eliminating β-arrestin-induced desensitization of Gs signaling. To date, few biased ligands have been identified for the β2AR and their development is challenging (Ippolito and Benovic, 2021). Indeed, the molecular basis of GPCR-mediated biased signaling remains vague since linking ligand binding to dynamic conformational changes in a receptor is difficult to study. Here, we used high-throughput screening to identify and pharmacologically characterize a series of Gs-biased agonists for the β2AR. These findings may represent a starting point to better understand biased signaling at the β2AR and to develop safer drugs for treating airway disease.
2. MATERIALS AND METHODS
2.1. Materials
All β-agonists were purchased from Sigma-Aldrich (St. Louis, MO, USA) except for higenamine which was from ChemSpace (Monmouth Junction, NJ, USA). Chemical structures of these compounds are provided in Table 1. Agonists were dissolved in H2O with 1 mM ascorbic acid (Sigma-Aldrich) or in dimethyl sulfoxide (DMSO, Sigma-Aldrich), stored as stock solutions at −20 °C, and then diluted in DPBS (Corning Inc, Corning, NY, USA) for analysis. Ninety-six-well white microplates were from PerkinElmer (Waltham, MA, USA). Deep blue coelenterazine (DBC) was from NanoLight Technology (Pinetop, AZ, USA) and coelenterazine H was from Cayman Chemical Company (Ann Arbor, MI, USA).
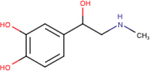
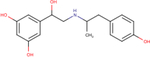
Table 1 – Name, chemical structure and pIC50 of β-agonists.
Name and associated chemical structure of the β-agonists used in this study are given. pIC50 ± SEM, n = 3, extrapolated from radiolabeled competition binding experiments with [3H]DHA (Fig. S1) is also provided. For direct comparison, pEC50 ± SEM, n = 6 for cAMP production and β-arrestin recruitment extrapolated from curve-fitting in Fig. 1C and D is also provided.
| Agonist | Chemical structure | Binding pIC50 ± SEM (n = 3) | cAMP production EC50 ± SEM (n = 6) | β-arrestin recruitment pEC50 ± SEM (n = 6) |
|---|---|---|---|---|
| (−)-Isoproterenol |
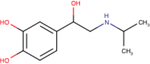
|
6.2 ± 0.1 | 7.8 ± 0.1 | 7.3 ± 0.1 |
| (−)-Epinephrine |
|
5.4 ± 0.2 | 6.8 ± 0.1 | 6.6 ± 0.1 |
| (−)-Norepinephrine |
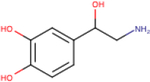
|
3.5 ± 0.3 | 5.4 ± 0.2 | 5.2 ± 0.1 |
| Fenoterol |
|
5.5 ± 0.2 | 6.1 ± 0.1 | 6.9 ± 0.1 |
| Ractopamine |
|
6.6 ± 0.1 | 7.8 ± 0.5 | 4.4 ± 0.6 |
| Buphenine |
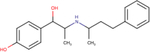
|
6.6 ± 0.1 | 8.2 ± 0.8 | 4.5 ± 0.7 |
| Ritodrine |
|
5.9 ± 0.7 | 6.1 ± 0.7 | 4.7 ± 0.5 |
| Dobutamine |
|
5.3 ± 0.1 | 6.3 ± 1.0 | nd |
| Bamethane |
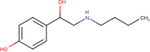
|
4.5 ± 0.1 | 5.4 ± 0.6 | nd |
| Salbutamol |
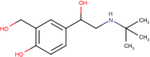
|
5.6 ± 0.1 | 6.3 ± 0.1 | 6.3 ± 0.2 |
| Salmeterol |
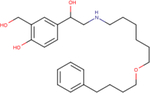
|
6.8 ± 0.0 | 8.2 ± 0.5 | 7.6 ± 0.4 |
| Higenamine |
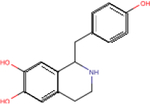
|
5.3 ± 0.1 | 5.7 ± 0.3 | 5.1 ± 0.3 |
2.2. Cell culture and transfection
HEK293 (RRID:CVCL_0045) cells were maintained in Dulbecco’s Modified Eagles Medium (DMEM) containing L-glutamine (Corning) and supplemented with 10% fetal bovine serum (FBS) (Corning), 100 U/ml penicillin and 0.1 mg/ml streptomycin (Gibco, Amarillo, TX, USA). HEK293 cells with stable overexpression of the β2AR (β2AR stable cells) were grown in the presence of G418 sulfonate (Sigma-Aldrich). HASM cells were cultured in Ham’s F12 Medium containing L-glutamine (Corning) supplemented with 10% FBS, 100 U/ml penicillin and 0.1 mg/ml streptomycin. Cells were incubated at 37 °C in a humidified incubator with 5% CO2. Transient transfections were performed in suspension in a 96-well plate using Metafectene PRO (Biontex, München, Germany) following the manufacturer’s protocol. Briefly, two solutions were prepared and incubated separately for 5 min. One solution contained plasmid DNAs encoding for BRET acceptor and BRET donor diluted in 25 μl/well of DMEM without phenol red and FBS while the other solution contained 0.5 μl/well of Metafectene Pro diluted in 25 μl/well of DMEM without phenol red and FBS. After the first incubation, the solutions were mixed, incubated for 20 min, added to 100,000 cells/well diluted in a final volume of 200 μl of DMEM without phenol red, and dispensed in ninety-six-well white microplates.
2.3. High-throughput assays
For cAMP production, 6,250 GloSensor™ cells in 25 μL of culture medium were plated per well in sterile, white, tissue-culture treated 384-well plates (Greiner Bio-One, Kremsmunster, Austria) and allowed to attach overnight at 37 °C in 5% CO2 atmosphere. Plates were removed from the incubator, 25 μL of CO2-independent medium (Thermo-Fisher, Waltham, MA, USA) plus 10% FBS and 4% GloSensor™ cAMP reagent (Promega, Madison, WI, USA) was added to each well and plates were allowed to equilibrate to room temperature for two hours. 0.25 μL of orthogonally compressed libraries from the LIMR Chemical Genomics Center (LCGC, Wynnewood, PA, USA) were transferred into plates using a high-density replication tool and allowed to incubate with cells at 37 °C. Luminescence was determined in kinetic mode at room temperature (23–25 °C) on a PolarStar Optima plate reader (BMG Labtech, Ortenberg, Germany). Response was analyzed relative to forskolin (7 μM) and alprenolol (1 μM) as positive and negative controls, respectively.
For β-arrestin recruitment to β2AR, 5,000 PathHunter™ ADRB2 cells in 20 μL of DiscoveRx cell plating medium (Eurofins, Luxembourg, Grand Duchy of Luxembourg) were plated per well in sterile, white, tissue-culture treated 384-well plates and allowed to attach overnight at 37 °C in 5% CO2 atmosphere. 0.25 μL of orthogonally compressed libraries from the LCGC were transferred into plates using a high-density replication tool and allowed to incubate with cells for 30 minutes at 37 °C. Five μL of cell plating media was then added to each well and plates were incubated for 1 hour at 37 °C. Plates were removed from the incubator, and 12.5 μL of DiscoveRx PathHunter™ detection solution was added to each well. Plates were incubated in the dark at room temperature for one hour. Luminescence was determined at room temperature (23–25 °C) on a PolarStar Optima plate reader. Response was analyzed relative to isoproterenol (1 μM) and alprenolol (1 μM) as positive and negative controls, respectively.
In both cAMP production and β-arrestin recruitment screening, negative control signal was subtracted from all data and responses were normalized relative to full response (positive control minus negative control). Relative assay bias was calculated from the ratio of normalized cAMP and β-arrestin recruitment responses. Wells from orthogonally-compressed libraries displaying Gs-bias were deconvoluted to identify compounds for follow-up analysis (Donover et al., 2013). Candidate compounds were titrated from 10 μM to 10 nM concentration in half-logarithmic steps in GloSensor™ and PathHunter™ assays in order to determine IC50 values for cAMP production and β-arrestin recruitment. Compounds displaying confirmed Gs-bias were progressed to secondary screening.
2.4. Radioligand binding assays
β2AR stable cells were lysed on ice in 50 mM Tris-HCl, pH 7.7 for 10 minutes, then scraped and pelleted by centrifugation at 1,000 rpm for five minutes. The pellet was washed with 50 mM Tris-HCl, pH 7.7 and passed through a 22-gauge needle five times. Lysed cells were spun at 27,000 × g for 10 minutes and the pellet was resuspended in buffer containing 15 mM Tris-HCl, pH 7.4, 120 mM sodium chloride, 5.4 mM potassium chloride, 1.8 mM calcium chloride, and 5 mM glucose. Total membrane protein concentration was determined by Bradford assay (Bio-Rad, Hercules, CA, USA).
Thirty micrograms of β2AR stable cell membrane protein were incubated with 3 nM [3H]dihydroalprenolol ([3H]DHA, PerkinElmer) in the presence of increasing concentrations of agonist (10−10 to 10−3 M) in a final volume of 1 ml. Non-specific binding was determined with 10 μM alprenolol. The mixture was incubated at 25 °C for 2 hours and then filtered over 25 mm Whatman GF/C glass microfiber filters (GE Healthcare, Chicago, IL, USA) soaked in 0.05% polyethyleneimine. The filters were washed 3 times with ice cold Tris buffered saline. Bound radioactivity was measured with a TriCarb 4910 TR liquid scintillation analyzer (PerkinElmer) and expressed as % of maximum [3H]DHA specific binding.
2.5. BRET sensors
The bioluminescence resonance energy transfer (BRET) assay permits the evaluation of protein-protein co-localization by virtue of the energy transfer between a donor (Renilla luciferase) and acceptor (fluorescent protein). To evaluate Gαs and Gαi recruitment, cells were transiently transfected with β1AR or β2AR C-terminally fused with donor RlucII (β1AR-RlucII and β2AR-RlucII) along with acceptor NES-Venus-mGs or NES-Venus-mGsi (Wan et al., 2018). In order to measure real-time cAMP response, HEK293 cells with endogenous β2AR were transfected with the BRET-based intramolecular cAMP sensor CAMYEL (both the donor and the acceptor are fused to the cAMP binding domain EPAC) (Jiang et al., 2007) that upon cAMP binding undergoes a conformational change resulting in a change in the BRET signal. For the assessment of β-arrestin2 recruitment, HEK293 cells were transiently co-transfected with plasmids for β-arrestin2-GFP10 along with β1AR-RlucII or β2AR-RlucII. To measure receptor internalization and endosomal localization, HEK293 cells were transiently transfected with β2AR-RlucII and either Venus-KRas as a membrane marker or Venus-Rab5 as an early endosome marker (Lan et al., 2011).
2.6. Cell stimulation and BRET measurement
Forty-eight hours after transfection, media was removed and cells were stimulated for 20 min with increasing concentrations of β-agonist (10−10 to 10−4 M) in the presence of 5 μM DBC or coelenterazine H diluted with PBS to a final volume of 50 μl/well. Signals at 395 nm (donor) and 530 or 510 nm (acceptor) were recorded in an Infinite F500 plate reader (Tecan, Männedorf, Switzerland).
2.7. Multivariate analysis
Transduction coefficients (LogR, R = τ/KA) for Gαs recruitment, Gαi recruitment, cAMP production and β-arrestin recruitment were obtained after statistical fitting of the operational model for each concentration-activity curve using the normalized data from Figs. 1A–D in GraphPad Prism v.9.2.0 (GraphPad Software, San Diego, CA, USA, RRID:SCR_002798) (Kenakin et al., 2012). We performed global fitting to identify LogR and found that constraining the following parameters provided a better estimation: n (Hill slope) = 1; basal (signal in the absence of ligand) = 0; Emax (maximum possible signal) = 100. In some cases, due to poor concentration-activity curve fitting, the model was unable to determine transduction coefficients and was reported as not determined (nd). To better visualize and compare activity of β-agonists across the different signalling pathways, we represented the transduction coefficients and Emax in a radial graph generated in Microsoft Excel v. 16.54 (Microsoft, Redmond, WA, USA, RRID:SCR_016137).
Figure 1 – β-agonist promoted transducer-coupling profile.
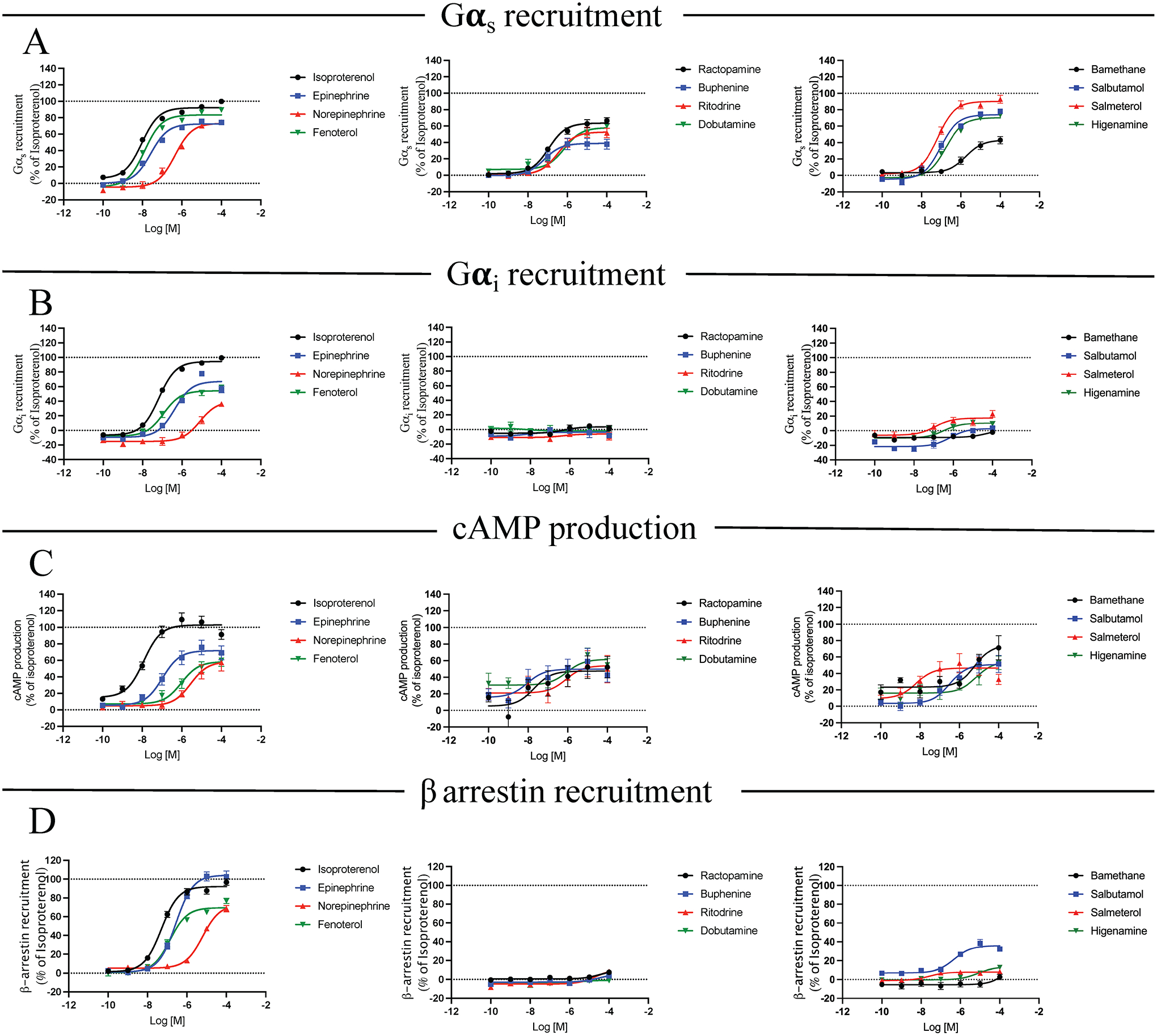
Gαs recruitment (A), Gαi recruitment (B), cAMP production (C) and β-arrestin recruitment (D) were analyzed. HEK293 cells were transiently co-transfected with BRET donor β2AR-Rluc and BRET acceptor NES-venus-mGs, NES-venus-mGi or GFP-β-arrestin 2 depending on the read-out. For evaluation of cAMP production, cells were transiently transfected with the intramolecular BRET sensor (donor and acceptor are on the same sensor) CAMYEL. After 48 h, cells were stimulated with increasing concentrations of compound (10−10 to 10−4 M) for 20 min. Signal generated by each condition was plotted and fitted using the three parameters non-linear regression curve fitting in GraphPad Prism to generate concentration/activity curves. Signal was normalized to the average maximal response of ISO as 100%. Data are mean ± SEM, n = 6.
2.8. In vitro β2AR phosphorylation
A time course of GRK5-mediated phosphorylation of the β2AR was assayed by incubating purified C-terminally strep-tagged GRK5 (100 nM) with purified β2AR (1.5 μM) reconstituted into bicelles with PIP2 in reaction buffer (20 mM Tris-HCl, pH 7.4, 5 mM MgCl2, 30 mM NaCl, 0.5 mM EDTA, 0.8 mM CaCl2, and 100 μM [γ32P]ATP (1000–2000 cpm/pmol)). Isoproterenol, ractopamine, dobutamine, or higenamine was added in reaction buffer at 50 μM concentration. Reactions proceeded for 1, 3, 10, 20 and 40 minutes at 30 °C and were quenched with SDS sample buffer, and samples were separated by SDS-PAGE. Gels were stained with Coomassie blue (Sigma-Aldrich), dried, exposed to autoradiography film and 32P-labeled proteins were excised and counted to determine the amount of phosphate transferred. Reaction rates were normalized to β2AR phosphorylation in the presence of isoproterenol at 40 min of incubation.
2.9. Detection of ERK phosphorylation
The experimental details provided below for Western blotting experiments complies with BJP’s Guidelines for immunoblotting and immunohistochemistry (Alexander et al., 2018). HEK293 cells with endogenous β2AR were stimulated with 10 μM β-agonist for 0, 2, 5, 10, 15, 20, 25, or 30 minutes. The cells were lysed in Laemmli buffer, and equal amounts of protein were analyzed by SDS PAGE and Western blot. The membranes were incubated overnight at 4 °C with anti-phospho-ERK 1/2 (P-p44/42 MAPK; rabbit mAB; Ref:4370S; Lot 31; 1:5000; Cell Signaling Technologies, Danvers, MA, USA) and anti-total ERK (ERK1/2 (C-9); sc-514302; Lot #I19.3; mouse monoclonal IgG2B; 1:10000; Santa Cruz Biotechnology, Dallas, TX, USA) followed by IRDye conjugates (IRDye800CW, goat anti-Mouse, 926–32210, Lot C91210–09/ IRDye680RD, goat-anti-rabbit 926–68071, Lot D00115–06; 1:300; Li-Cor Biosciences, Lincoln, NE, USA) as secondary antibodies. Protein bands were detected with a LI-Cor Odyssey CLX far-infrared scanner (Li-Cor Biosciences, Lincoln, NE, USA) and the bands were quantified by densitometry with ImageJ v. 1.53 (RRID:SCR_003070) (Schneider et al., 2012).
2.10. Magnetic twisting cytometry
Dynamic changes in cell stiffness were measured in isolated HASM cells using forced motions of functionalized beads anchored to the cytoskeleton through cell surface integrin receptors, as previously described (An et al., 2016). The increase or decrease in stiffness is considered an index of single-cell smooth muscle contraction and relaxation, respectively. For these studies, serum-deprived, post-confluent cultured HASM cells were plated at 30,000 cells/cm2 on plastic wells (96-well Removawell, Immulon II, Dynetech Laboratories Inc, Chantilly, VA, USA) previously coated with type I collagen (VitroCol; Advanced BioMatrix, Inc., Carlsbad, CA, USA) at 500 ng/cm2, and maintained in serum-free media for 24 hours at 37 °C in humidified air containing 5% CO2. These conditions have been optimized for seeding cultured cells on collagen matrix and for assessing their mechanical properties (An et al., 2016). For each individual cell, baseline stiffness was measured for the first 60 seconds and then measured continuously for the next 240 seconds after β-agonist addition. To evaluate desensitization, HASM cells were pretreated with or without 10 μM agonist for 0.5, 4, or 24 hours, washed, and then stiffness was measured after addition of 10 μM isoproterenol as described above.
2.11. Data and statistical analysis
This manuscript complies with BJP’s recommendations and requirements on experimental design and analysis (Curtis et al., 2015). Studies were designed to generate groups of equal size using randomization and blinded analysis where possible. Statistical analysis was undertaken as described below only for studies where each group size was at least n=5.
Results from concentration/activity curves are shown as mean ± SEM from six independent experiments. For normalization, we first subtracted the basal signal (wells stimulated with PBS in the absence of ligand) from each well stimulated with β-agonist, and the values of all replicates were then divided by the mean of isoproterenol-induced maximal responses and multiplied by 100 for any given read-out. To obtain values for Emax and EC50, Gαs recruitment, Gαi recruitment, cAMP production, β-arrestin recruitment, internalization and endosomal localization data from Fig. 1 and Fig. 3C and D were fit using the function log(agonist) vs response (three parameters) of the non-linear curve fitting in GraphPad Prism. In those cases where the model reported an “ambiguous” result due to poor curve fitting and large uncertainty, values for Emax and EC50 were reported as “not determined”. After assessing variances are equal among the different datasets, statistical significance of multiple comparisons between ISO-induced Emax and EC50 (as reference agonist) and β-agonist-induced Emax and EC50 was assessed by one-way ANOVA with Dunnet’s post-test. P-values were considered significant when < 0.05. Statistical comparison between values for Emax and EC50 induced by β1AR versus β2AR for Gαs recruitment and β-arrestin recruitment from Fig. 2C and D was assessed by t-test with Welch’s correction, P-values were considered significant when < 0.05. T-test with Welch’s correction was also used to assess statistical significance for internalization and endosomal localization related values for Emax and EC50 from Fig. 3C and D, and P-values were considered significant when < 0.05. To obtain values for TC, Gαs recruitment, Gαi recruitment, cAMP production, β-arrestin recruitment, internalization and endosomal localization data from Fig. 1 were fit using the operational model of agonism in GraphPad Prism. In those cases where the model reported an “ambiguous” result due to poor curve fitting and large uncertainty, the values for TC were reported as “not determined”. After assessing variances are equal, statistical significance of multiple comparisons between ISO-induced values for TC (as reference agonist) and β-agonist-induced values for TC was assessed by one-way ANOVA with Dunnet’s post-test. P-values were considered significant when < 0.05. Agonist-induced relaxation in HASM cells in Fig. 5 was analyzed by one-way ANOVA, followed by Dunnett’s multiple comparisons test.
Figure 3 – Analysis of β-agonist induced GRK5 phosphorylation and β-arrestin-mediated trafficking of the β2AR.
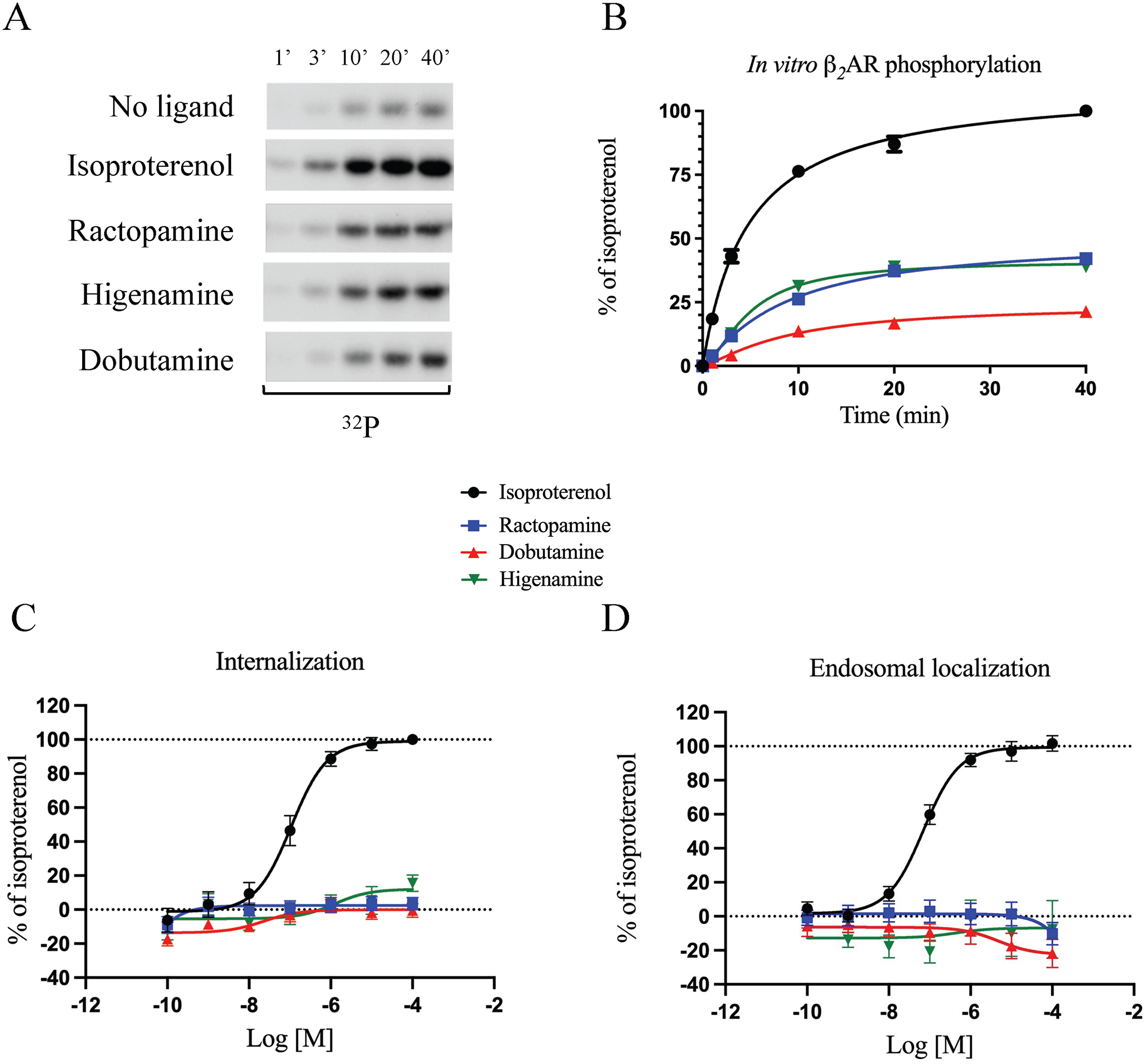
Kinetics of in vitro GRK5-mediated phosphorylation of β2AR in the presence of 50 μM ISO, RAC, DOB and HIG are shown as (A) a representative autoradiograph of 32P incorporation into β2AR and (B) time course of β2AR phosphorylation compared to ISO. Reaction rates were normalized to β2AR phosphorylation in the presence of ISO at 40 min of incubation and plotted as kinetic curves using GraphPad Prism. Data are mean ± SEM, n = 3. Internalization (C) and endosomal localization (D) of β2AR was assessed by BRET. β2AR-mediated internalization and endosomal localization were assessed by transfecting HEK293 cells with BRET donor β2AR-Rluc and acceptors Venus-Kras as membrane marker or Venus-Rab5 as early endosome marker. After 48 h, cells were stimulated with increasing concentrations of ISO, RAC, DOB or HIG (10−10 to 10−4 M) for 20 min. Signal generated by each condition was plotted and fitted using the three parameters non-linear regression curve fitting in GraphPad Prism to generate concentration/activity curves. Signal was normalized to the average maximal response of ISO as 100%. Data are mean ± SEM, n = 6.
Figure 2 – Selection of the best Gs-biased candidates.
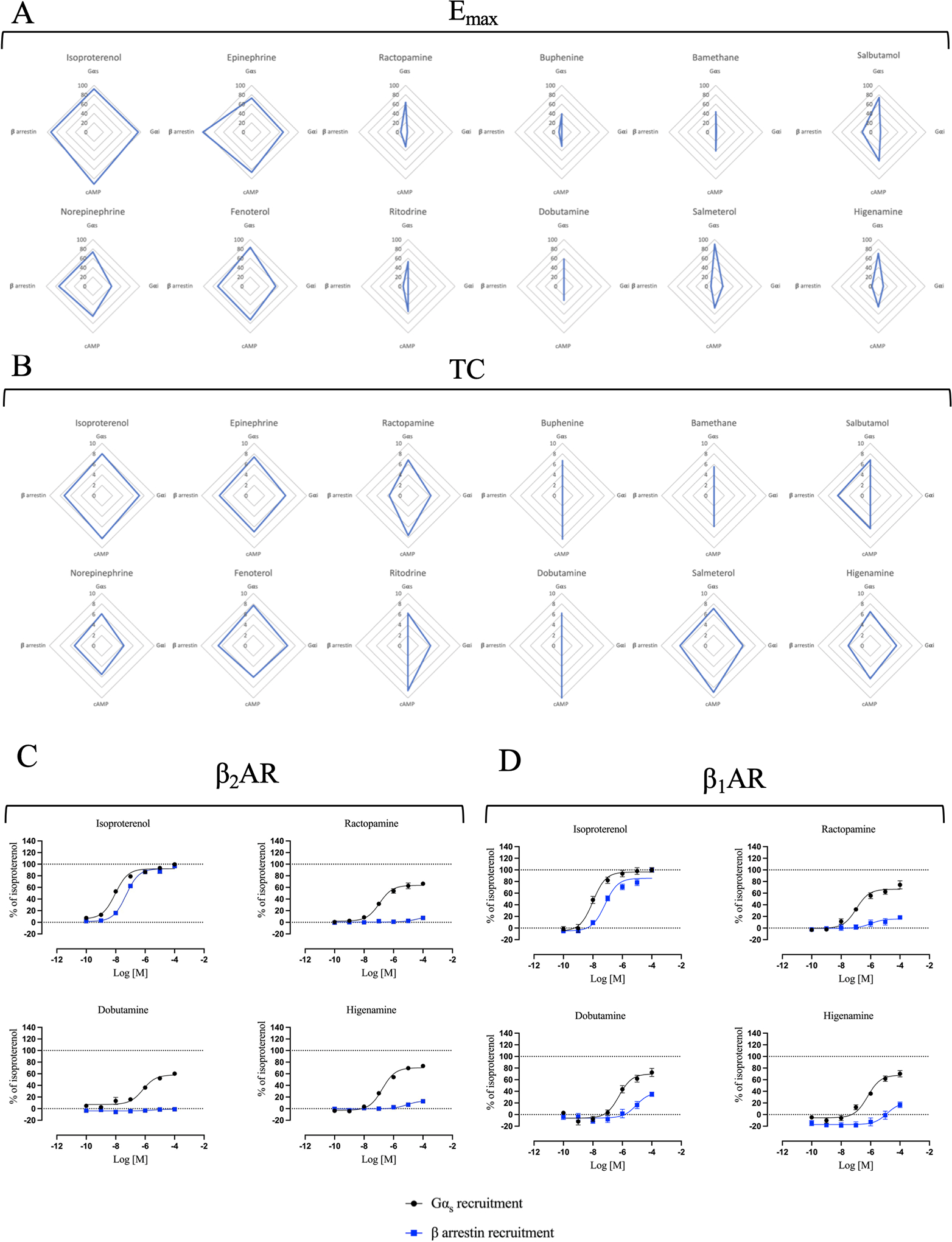
Multivariate analysis of (A) maximal efficacies (Emax) extrapolated from curve fitting of the concentration/activity curves from Fig. 1 and Table S1 and (B) transduction coefficients (TC) obtained after statistical fitting of the operational model of agonism to data extrapolated from Fig. 1, were represented in radial plots. Emax range is 0 – 100 while TC range is 0 – 10. Note that values that were very low and could not be accurately determined were given a value of zero in these plots. Direct comparison of concentration/activity curves for Gαs recruitment and β-arrestin recruitment induced by ISO, RAC, DOB and HIG at the β2AR (C) and the β1AR (D). Data for β2AR comparison were taken from Fig. 1A and B. For β1AR comparison, HEK293 cells were transiently transected with BRET donor β1AR-Rluc and BRET acceptor NES-venus-mGs or GFP-β-arrestin 2 depending on the read-out. After 48 h, cells were stimulated with increased concentration of ISO, RAC, DOB or HIG (10−10 to 10−4 M) for 20 min. Signal generated by each condition was plotted and fitted using the three parameters non-linear regression curve fitting in GraphPad Prism to generate concentration/activity curves. Signal was normalized to the average maximal response of ISO as 100%. Data are mean ± SEM, n = 6.
Figure 5 – Activity of ISO and the Gs-biased agonists on HASM cell relaxation.
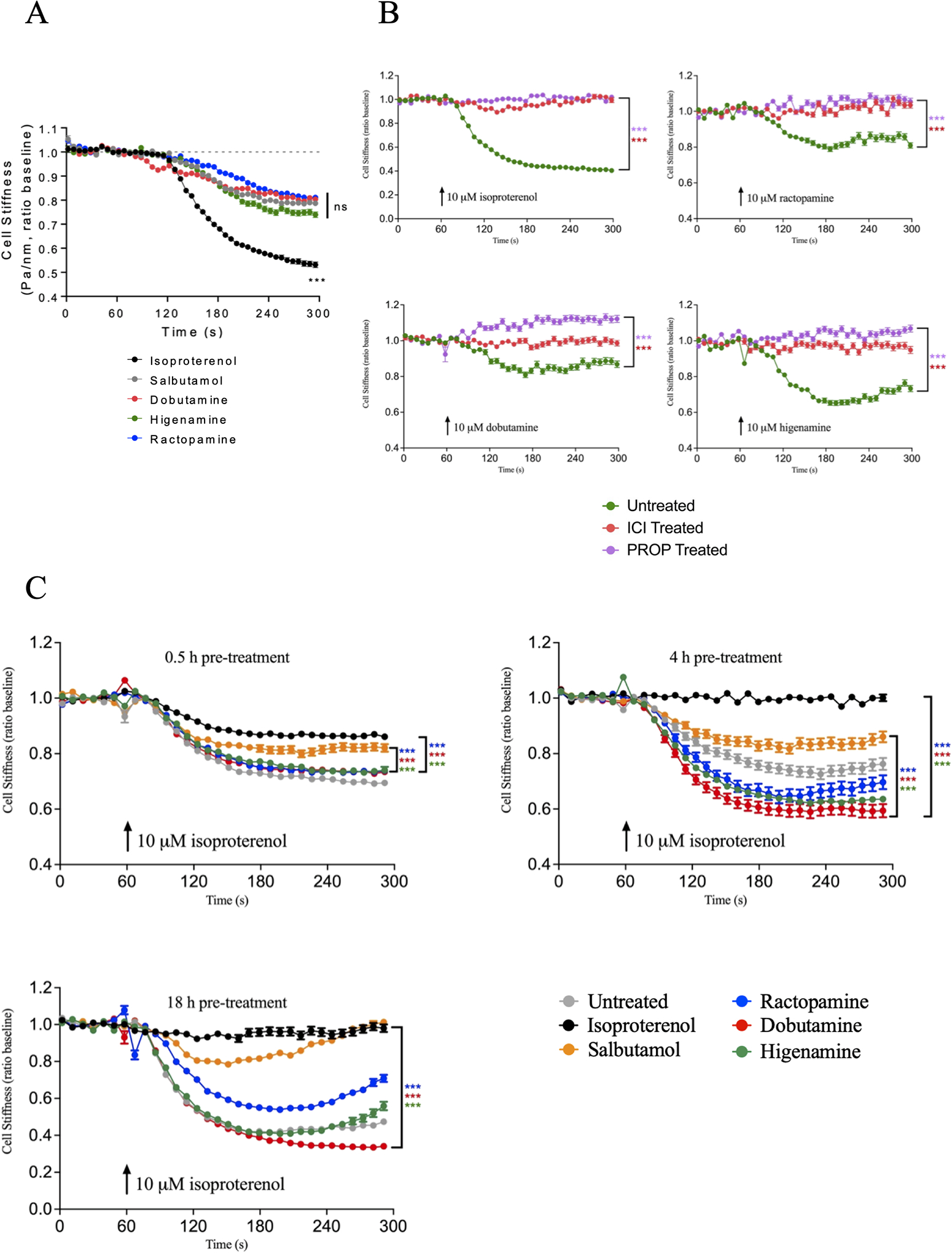
(A) Time course of HASM cell relaxation induced by 10 μM ISO, SALB, RAC, DOB, or HIG. Data are mean ± SEM from 3 different HASM cell lines: ISO (n=342), SALB (n=257), DOB (n=215), HIG (n=214), RAC (n=218) where n is the number of cells per condition. Agonist-induced relaxation at 300 s was analyzed by one-way ANOVA, followed by Dunnett’s multiple comparisons test (comparing all data to SALB). (B) Inhibition of 10 μM ISO, RAC, DOB, or HIG-induced HASM cell relaxation by the β2AR-specific antagonist ICI-118551 (10 μM ICI treated) or the β-antagonist propranolol (10 μM PROP treated). Data are mean ± SEM (n for untreated, ICI-treated and PROP-treated, respectively: ISO (n=258, 162, 156), RAC (n=152, 178, 167), DOB (n=122, 161, 192), HIG (n=204, 128, 150). Agonist-induced relaxation at 300 s was analyzed by one-way ANOVA, followed by Dunnett’s multiple comparisons test (comparing ISO, RAC, DOB and HIG in the presence or absence of ICI or PROP). (C) HASM cell desensitization induced by pretreating cells with 10 μM ISO, SALB, RAC, DOB, or HIG for 0.5, 4 or 18 h, followed by relaxation induced by 10 μM ISO. Data are mean ± SEM from 0.5, 4 and 18 h pretreatments, respectively: untreated (n=380, 262, 254), ISO (n=338, 278, 107), SALB (n=211, 189, 251), RAC (n=355, 227, 323), DOB (n=331, 209, 252) and HIG (n=229, 224, 172). Agonist-induced relaxation at 300 s was analyzed by one-way ANOVA, followed by Dunnett’s multiple comparisons test (comparing RAC, DOB and HIG to either SALB or ISO). ***p < 0.001
2.12. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, and are permanently archived in the Concise Guide to PHARMACOLOGY 2021/22 (Alexander et al., 2021).
3. RESULTS
3.1. Identification and characterization of Gs-biased agonists for the β2AR
In an effort to identify small-molecule Gs-biased agonists for the β2AR, an orthogonally compressed library containing 152,000 compounds was screened in primary and secondary assays for cAMP production and β-arrestin2 recruitment to the β2AR. In primary screens, cells expressing β2AR were stimulated in the presence or absence of 10 μM test compounds and evaluated for cAMP production using the GloSensor™ assay and β2AR/β-arrestin2 association using the PathHunter™ ADRB2 assay. Hits from primary screening were then titrated in GloSensor and PathHunter assays to identify compounds capable of promoting cAMP production in a dose-dependent manner with minimal effect on β-arrestin2 association. The most potent compounds from the primary screens were further characterized using bioluminescence resonance energy transfer (BRET) assays to analyze compound-induced signalling pathways. The orthogonal nature of the detection methods used for primary and secondary screening allowed us to confirm compound-induced phenotypes and rule out false positives arising from assay artifacts. A total of 12 compounds (chemical structures can be found in Table 1) were studied in depth including the endogenous catecholamines epinephrine (EPI) and norepinephrine (NE), the full agonist isoproterenol (ISO) (used as a reference ligand throughout the study), the partial agonists salbutamol (SALB) and salmeterol (SALM) (both used clinically), and the potential Gs-biased agonists identified in the screening: fenoterol (FEN), ractopamine (RAC), buphenine (BUP), ritodrine (RIT), dobutamine (DOB), bamethane (BAM), and higenamine (HIG).
To evaluate the potential orthosteric nature of the agonists, radioligand competition assays were performed. In these studies, increasing concentrations (10−10 to 10−3 M) of each compound were incubated with isolated cell membranes harvested from cells expressing β2AR in the presence of a 3 nM concentration of the tritiated β-antagonist DHA (Fig. S1). Non-specific binding was assessed by competition with 10 μM of unlabeled alprenolol while specific binding was calculated by subtracting the non-specific binding and is represented as a percentage of the maximal DHA specific binding. Each compound tested was able to compete for [3H]DHA binding to the β2AR indicating a receptor specific interaction most likely at the orthosteric site of the receptor with IC50s ranging from high nM to low μM (Table 1). These values are in agreement with previous reports for some of the compounds (Toews et al., 1983) and demonstrate comparable affinities for newly characterized compounds. While these studies help to establish the compound affinities for the β2AR, this does not preclude the possibility of effects at other receptors or potential allosteric effects for the β2AR.
3.2. Profiling of agonist-induced signalling at the β2AR
To assess efficacy at downstream signalling pathways of the β2AR, a panel of Bioluminescent Resonance Energy Transfer (BRET) biosensors was used to generate a transducer interaction profile for each compound. This technique allows monitoring protein-protein interaction or co-localization by virtue of changes occurring in the energy transfer between a donor (Renilla luciferase) and acceptor (fluorescent protein) when both are close enough (~10–100 angstroms) to generate a signal. For this purpose, HEK293 cells were transfected with β2AR C-terminally fused with donor RlucII along with acceptor NES-Venus-mGs to measure Gs interaction or β-arrestin-GFP to measure β-arrestin interaction. We also assessed real-time cAMP response by using the intramolecular cAMP sensor CAMYEL which carries both donor and acceptor fused to the cAMP biding domain EPAC (Fig. 1A–1D). Cells were stimulated with increasing concentrations of the compounds and the BRET signal was plotted in a concentration/activity sigmoid as a percentage of the ISO maximal response for the different read-outs. The profiled compounds demonstrated a variety of pharmacological behaviors with differences in efficacies and potencies for the different outputs, ranging from full agonist to partial agonist to no effect in some assays (Table S1A and B). As a confirmation of the initial high-throughput screening, all compounds showed efficacy in recruiting Gαs to the receptor in a concentration dependent manner upon β2AR stimulation (Fig. 1A). ISO, FEN and SALM were full agonists with comparable efficacies and potencies while the other compounds behaved as partial agonists. Among these, EPI, NE, RAC, DOB, SALB and HIG were able to induce ~60% or more of Gαs recruitment compared to ISO. When assessing compound-induced cAMP production, a similar pattern of pharmacological behaviors to Gαs was found (Fig. 1C). Despite this, it is worth noting that for some compounds the efficacy and potency for cAMP production did not fully match those obtained for Gαs recruitment (Table S1A and B). This discrepancy may be due to a difference in receptor concentrations since we did not overexpress the β2AR in the cAMP assay compared to the Gαs assay, although there are many factors that ultimately contribute to agonist-promoted cAMP levels (e.g., phosphodiesterase expression). We also noticed that basal cAMP levels were higher for some β-agonists at very low concentrations, although there were no statistically significant differences between the compounds and ISO (Fig. 1C).
The β2AR has been reported to also bind to Gi (Daaka et al., 1997; Ma et al., 2020). To evaluate if the selected agonists were able to promote Gi binding, we measured Gαi recruitment to the receptor upon β2AR stimulation using a similar approach as for Gαs (Fig. 1B). Specifically, we used a combination of BRET sensors that included the β2AR fused with donor RlucII along with acceptor NES-Venus-mGsi. ISO, EPI, NE and FEN were able to recruit Gαi to the β2AR with efficacies generally similar to Gαs recruitment while the potencies were reduced approximately 10-fold (Table S1A and B). Interestingly, the other compounds including SALM and SALB displayed weak or no Gαi recruitment suggesting a possible G protein-bias towards Gαs vs Gαi association with the β2AR. The distinction between G protein-bias vs. Gs-bias phenotypes at the β2AR has not been previously evaluated in depth, and this may provide insight into differential physiological responses to drugs classically defined as β-agonists.
Finally, we analyzed β-arrestin recruitment to the β2AR upon stimulation with the compounds (Fig. 1D). Again, ISO, EPI, NE and FEN were the most efficacious in recruiting β-arrestin with potencies comparable to Gαi binding, highlighting their balanced pharmacology. SALB behaved as a partial agonist being less than 40% efficacious compared to ISO while the other compounds showed weak or no β-arrestin recruitment suggesting they are G-protein-biased.
3.3. Multivariate analysis and selection of Gs-biased candidates
To visualize the pharmacological contribution of each compound to the different signaling pathways, we used radial plots to represent the maximal efficacies (Fig. 2A) obtained from the concentration-activity curve fitting (Fig. 1 and Table S1A) and the transduction coefficients (TC) (Fig. 2B) extrapolated from the curve fitting of the operational model of biased agonism (Table S2A). TC is a pharmacological parameter representing the agonist activity of a ligand on a specific signalling pathway (Kenakin et al., 2012). In some cases, the model was not able to identify the Emax or the TC due to poor curve fitting. Therefore, the shapes of some radial graphs are stretched because of the lack of such values. The analysis confirmed that ISO, EPI, NE and FEN are balanced agonists that are able to induce robust efficacies and TC in all pathways. We identified RAC, DOB and HIG as the most promising compounds displaying Gs-biased properties based on the maximal efficacies and transduction coefficients for Gs and β-arrestin. Indeed, direct comparison of the concentration-activity curves for these two pathways induced by these compounds showed a clear signaling bias in favor of Gs recruitment, being over 55% efficacious for Gαs recruitment compared to ISO while showing very weak or no efficacy for β-arrestin recruitment (Fig. 2C, Table S1A and B). Additional compounds including ritodrine, buphenine and bamethane also appeared to be Gs-biased although their efficacy for Gαs recruitment was lower and thus were not studied in more detail.
Because the β2AR shares high structural similarity with the closely related β1-adrenergic receptor (β1AR), the majority of β-agonists can activate both receptors (Baker, 2010). Therefore, we also evaluated compound-mediated Gαs and β-arrestin recruitment to the β1AR by transfecting HEK293 cells with BRET donor β1AR fused with RlucII along with acceptor NES-Venus-mGs or β arrestin-GFP to measure Gs and β-arrestin interaction at the β1AR, respectively. Cells were stimulated with increasing concentrations of ISO, RAC, DOB or HIG and the BRET signal was plotted in a concentration/activity sigmoid as percentage of the ISO maximal response in the two read-outs considered.
ISO produced concentration-activity curves for both pathways comparable to those obtained for the β2AR, confirming its balanced behavior (Table S2B and C). Similarly, RAC and HIG induced β1AR-mediated Gαs recruitment analogous to the β2AR while DOB was slightly more efficacious for β1AR (Table S2B and C). Of note, we also observe higher efficacy for β-arrestin recruitment by RAC, DOB, and HIG at the β1AR compared to the β2AR (Table S2B and C), suggesting a degree of Gs-biased selectivity at the β2AR over the β1AR.
3.4. Compound activity on β-arrestin integrated signaling
GRK phosphorylation of the β2AR C-terminal tail is a requisite step for β-arrestin interaction with the receptor, and thus plays a role in orchestrating biased agonism (Choi et al., 2018). To evaluate the ability of the compounds to promote GRK phosphorylation of the β2AR, an in vitro-kinase assay was performed. For these experiments, purified GRK5 was incubated with purified β2AR reconstituted into bicelles in the presence of phosphatidylinositol 4,5-bisphosphate (Komolov et al., 2017). 50 μM of ISO, RAC, DOB or HIG were added to the mixture and reactions were stopped after 1, 3, 10 and 40 minutes. ISO induced robust phosphorylation of the β2AR while the compounds that displayed Gs-biased signalling profiles (RAC, DOB, and HIG) showed greatly reduced phosphorylation, indicating that they were partial agonists for GRK5-mediated phosphorylation (Fig. 3A and B). While we did not assess the effects of the Gs-biased compounds on β2AR phosphorylation in intact cells, the reduced β2AR phosphorylation induced by RAC, DOB and HIG in vitro may contribute to the reduced β-arrestin binding in cells.
β-arrestin binding to the β2AR is also essential for agonist-promoted internalization of the receptor (Tian et al., 2014). To evaluate the ability of the compounds to modulate β2AR internalization, we used two BRET based sensors: the membrane marker Venus-Kras and the early endosome marker Venus-Rab5 (Lan et al., 2011). Such sensors in combination with the RlucII-tagged receptor allow monitoring of β2AR trafficking upon stimulation. As expected, ISO evoked concentration-dependent internalization (Fig. 3C) and endosomal localization (Fig. 3D) of the β2AR. In contrast, the Gs-biased RAC, DOB and HIG induced minimal internalization or endosomal localization of the β2AR, even at high concentrations, confirming their inability to promote β-arrestin-dependent pathways (Fig. 3C and D, Table S3). Of note, DOB appeared to behave as a weak inverse agonist for the endosomal localization of the receptor (Fig. 3D).
Taken together, the inability of RAC, DOB, and HIG to promote internalization or endosomal localization of the β2AR further corroborates their inability to promote β-arrestin binding to the receptor.
3.5. Analysis of ERK1/2 activation
ERK1/2 is a central downstream kinase that can be phosphorylated by GPCR-mediated pathways to promote a series of biological effects (Wortzel and Seger, 2011). Activation and regulation of ERK1/2 is complex, involving different molecular players including G protein- and β-arrestin-mediated pathways. It has been shown that the initial GPCR-mediated phosphorylation of ERK1/2 is mainly dependent on G protein-activation that results in a transient peak while more sustained ERK1/2 phosphorylation is largely dependent upon β-arrestin activation (Shenoy et al., 2006). To evaluate the ability of the Gs-biased compounds to promote ERK1/2 activation, pERK1/2 was measured by western blot. To minimize potential effects of β2AR overexpression, we used untransfected HEK293 cells for these studies. Cells were stimulated with 10 μM compound for up to 30 minutes and pERK1/2 bands were quantified and normalized to total ERK (Fig. 4A and B). ISO induced an approximately 4-fold increase in pERK1/2 at 5 minutes with some sustained activation through 30 minutes. In contrast, RAC, DOB and HIG induced a similar early peak, but lacked the sustained phosphorylation induced by ISO. These results agree with previous studies and suggest that β-arrestin recruitment plays a role in sustained agonist promoted ERK phosphorylation while Gs activation through the β2AR likely initiates this response.
Figure 4 – Activity of β-agonists on ERK phosphorylation.
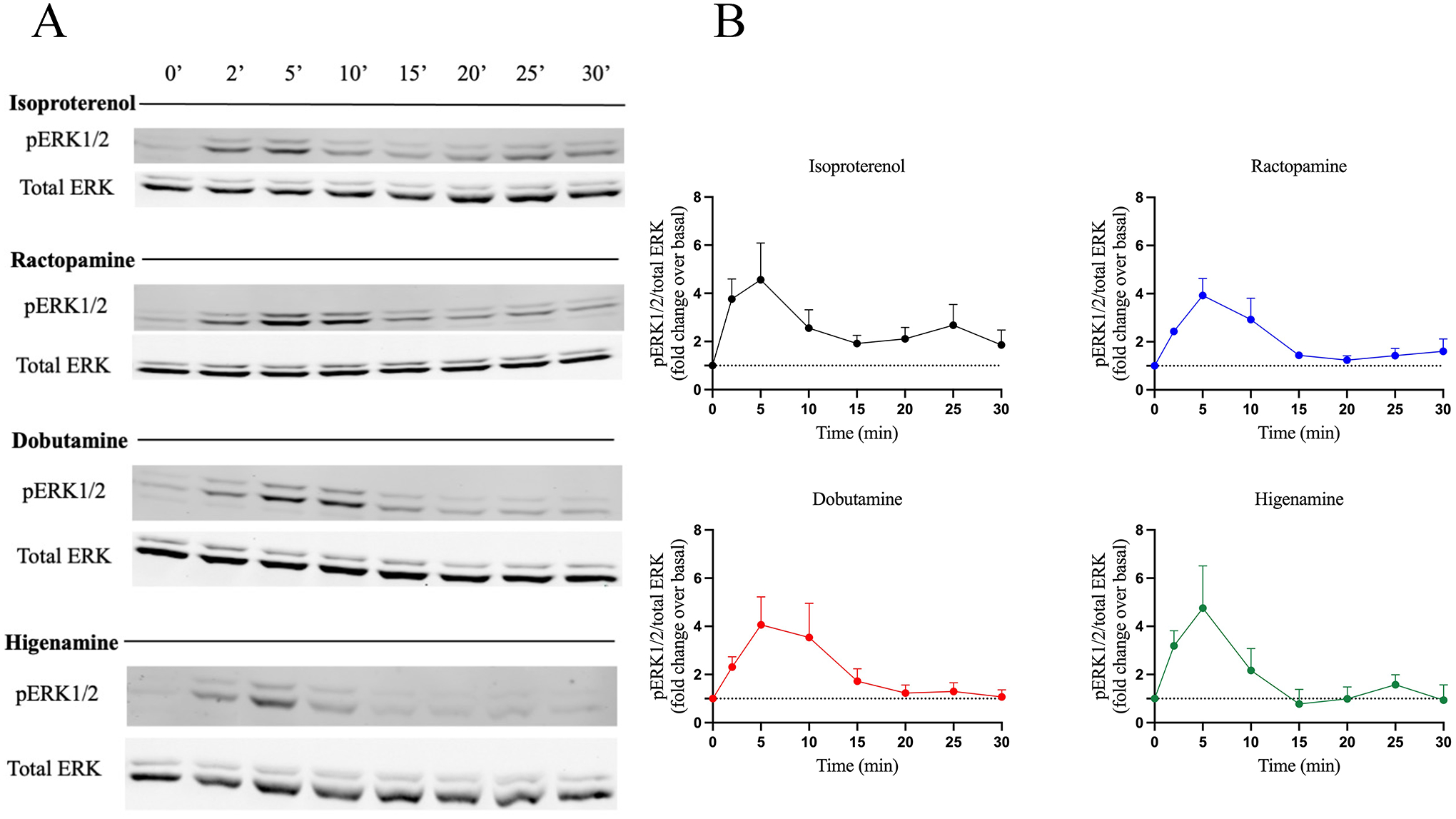
ERK phosphorylation was assessed by western blotting and results are shown as (A) a representative western blot from three independent experiments and (B) normalized data after quantification using ImageJ. HEK293 cells endogenously expressing the β2AR were stimulated with 10 μM ISO, RAC, DOB or HIG for 0, 2, 5, 10, 15, 20, 25 and 30 min. Cells were lysed and equal amounts of protein were analyzed by western blot to detect ERK1/2 phosphorylation. Phopho-ERK1/2 (pERK1/2) bands were quantified and normalized to total ERK1/2 (pERK/total ERK) and are represented as fold change over basal. Data are mean ± SEM, n = 3.
3.6. Evaluation of agonist-induced HASM relaxation and desensitization
A major limitation to many studies investigating biased signalling phenotypes is the lack of recapitulation of observed effects in physiological systems (Michel and Charlton, 2018). To try to address this limitation, agonist-induced desensitization of response to β-agonists was studied in primary HASM cells using magnetic twisting cytometry (MTC), a technique that monitors dynamic changes in cell stiffness. The endpoint of agonist-induced desensitization was chosen due to its role in the loss of therapeutic response to treatment and its potential role in severe adverse effects in asthmatic patients (Cheung et al., 1992; Bhagat et al., 1995; Grove and Lipworth, 1995). Indeed, several studies have implicated β-arrestins as contributing to the adverse effects of β-agonists in murine models of asthma (Walker et al., 2003; Thanawala et al., 2015; Forkuo et al., 2016), and strategies to bias β2AR signalling towards the Gs pathway may attenuate the harmful effects of β-agonists while maintaining therapeutic response (Dror et al., 2011; Christopoulos, 2014; Thanawala et al., 2015).
Our initial studies compared the ability of 10 μM ISO, SALB, RAC, DOB and HIG to promote relaxation of HASM cells using MTC. As shown in Fig. 5A, SALB, RAC, DOB and HIG all promoted relaxation achieving 40–55% of that observed for ISO treatment after 4 min. These effects were β2AR-specific since agonist-induced relaxation was effectively inhibited by the β-antagonist propranolol as well as the β2AR-selective inverse agonist ICI-118551 (Fig. 5B). We next evaluated whether these compounds induced any desensitization of the β2AR. HASM cells were initially pretreated with 10 μM ISO, RAC, DOB, HIG or SALB for either 0.5, 4 or 18 hours. The cells were then washed, and cell relaxation induced by 10 μM ISO was measured. The responsiveness of the cells was effectively desensitized by pretreatment with ISO, while DOB and HIG pretreatment did not induce any desensitization and RAC induced partial desensitization only with 18-hour pretreatment (Fig. 5C). Interestingly, at the 4-hour time point, the Gs-biased compounds appeared to be protective since they were even more responsive to ISO-induced relaxation than cells that were not pretreated. To gauge whether these effects might be due to the Gs-biased compounds being partial agonists, we also evaluated pretreatment with the partial agonist SALB. SALB induced significant desensitization particularly at the longer time points, although the loss of responsiveness was not as great as that observed for ISO pretreatment (Fig. 5C). Thus, the Gs-biased compounds RAC, DOB and HIG were able to effectively promote HASM relaxation through activation of β2AR without promoting significant functional desensitization.
4. DISCUSSION
Identifying biased ligands is considered a promising strategy to attenuate side effects that some balanced agonists show in their action. Indeed, the ability to selectively promote a therapeutically relevant signalling pathway while avoiding potentially detrimental pathways is a feasible goal in targeting GPCRs. Therefore, it is becoming increasingly apparent that classifying receptors by their ability to modulate a single transducer is incomplete. Thorough profiling of receptor-transducer interactions coupled to regulatory and physiological responses provides an opportunity to discover ligands that promote therapeutic responses while minimizing interactions that may be harmful.
Biased agonism at the β2AR came into focus in the early 2000s with an initial distinction between G protein and β-arrestin signalling (Drake et al., 2008; Casella et al., 2011). Specifically, the β2AR has several important physiological roles and most of the drugs targeting this receptor show side effects including the β-agonists used in the treatment of asthma. These compounds are generally classified as short acting β-agonists (SABA) or long-acting β-agonists (LABA) based on their duration of action and both have shown severe side effects including exacerbation of an asthmatic attack and an increase in mortality (Matera et al., 2020; Wendell et al., 2020). Several in vitro and in vivo studies identified differential contributions to the therapeutic and adverse effects in the Gs vs β-arrestin pathways, respectively, highlighting the importance of developing Gs-biased ligands. In the present work, we have used high-throughput screening to search for β-agonists with the desired characteristics. A panel of 12 small-molecule β-agonists were profiled and among these ractopamine, dobutamine and higenamine, showed potent Gs-biased signalling. We believe that the approach described herein can facilitate a more detailed evaluation of agonist promoted interactions at the β2AR that could be applied more broadly to drug discovery efforts to reveal more finely tuned signalling phenotypes.
Our work shows that the β2AR can be effectively biased towards Gs by small molecules. It is worth noting that previous studies reported Gs-biased behaviors of β-agonists such as salmeterol, ritodrine and dobutamine (Ippolito and Benovic, 2021). Specifically, salmeterol is the most prescribed LABA for the treatment of asthma and various studies reported a 5 to 20-fold bias towards Gs vs. β-arrestin (Cazzola and Donner, 2000; Cazzola et al., 2013; van der Westhuizen et al., 2014; Gimenez et al., 2015). While this Gs-biased behavior is confirmed in our analysis, salmeterol has shown severe side effects in clinical practice (Nelson et al., 2006) suggesting that even low efficacies for β-arrestins may contribute to unwanted side effects. Ritodrine and dobutamine were previously reported to be Gs-biased in a study evaluating the ability of β-agonists to differentially promote complexes with Gs and β-arrestin upon β1AR or β2AR activation (Casella et al., 2011). Both compounds exhibited good efficacy for Gs with poor efficacy for β-arrestin, similar to what we found. Interestingly, a Gs bias preference for the β2AR over the β1AR by dobutamine was also observed (Casella et al., 2011). Comparable to our study, dobutamine induced β-arrestin binding to the β1AR but not the β2AR, showing a certain degree of bias “selectivity”. Perhaps this contributes to the adverse effects of dobutamine on cardiac muscle when administered for the treatment of heart failure (Maack et al., 2019). We also noticed such disparity of action between β2AR and β1AR for the Gs-bias preference for ractopamine and higenamine. Such phenomenon underscores the importance of evaluating cross-reactivity and possible receptor bias between structurally-related receptors when performing bias analysis. This should give a broader understanding of the potential mechanisms and help to predict potential off-target effects.
Ractopamine was also identified as Gs-biased in our study; and, it is a well-known β-agonist with a very similar chemical structure to dobutamine (Table 1) (Colbert et al., 2011). Ractopamine is used as an additive in animal feed (Liu et al., 2014), although this use is controversial and banned in some countries. It is worth noting that the ractopamine that we used is a racemic mixture of RR, RS, SR and SS and previous studies have shown that the RR stereoisomer is a full agonist, SR is a partial agonist, and SR and SS have no agonist activity for β2AR-mediated cAMP production (Mills et al., 2003). This characteristic of ractopamine stereoisomers may be useful in structure-function studies aimed at determining the molecular features of biased signalling at the β2AR.
The other Gs-biased agonist identified in our study is higenamine, a plant alkaloid used in traditional Chinese medicine (Hudzik et al., 2021). Higenamine was previously identified as a β-agonist in studies using CHO cells overexpressing the β2AR (Bai et al., 2008) and it was also found to induce relaxant effects in smooth muscle cells from isolated tracheae (Tsukiyama et al., 2009; Ueki et al., 2011). Our study corroborates that higenamine is likely an orthosteric β-agonist and classify this compound as a Gs-biased agonist for the β2AR. While the Gs-bias of higenamine from our studies is clear, a recent study reported higenamine as a β-arrestin biased β-agonist (Zhang et al., 2021). While some of this discrepancy between the studies may be explained by cell type differences, the pharmacological characterization in the previous study was limited to higenamine modulation of ERK1/2 phosphorylation. Since ERK1/2 can be activated by G protein and β-arrestin pathways upon β2AR activation (Shenoy et al., 2006), it is difficult to know whether the effects of higenamine involve activation of the Gs pathway, the β-arrestin pathway, or a combination of the two.
To try to better understand whether the Gs-biased effects seen in the initial pharmacological characterization could be translated to a more physiological model of asthma, we tested the compounds in primary human airway smooth muscle cell relaxation and desensitization. For these experiments, we compared the Gs-biased compounds with the partial agonist SALB in addition to our reference agonist ISO. While SALB is a short acting β2AR-selective agonist that is used to acutely relax ASM during an asthma attack, it can have unwanted side effects when administered chronically. Our studies show that while the Gs-biased compounds and SALB have a comparable ability to promote HASM cell relaxation, RAC, HIG and DOB promote significantly less desensitization compared to SALB (Fig. 5). Such observations help to corroborate the hypothesis that the beneficial effects of the currently used β-agonists in asthma are mostly induced by the Gs pathway while the efficacy of SALB in activating β-arrestin pathways may be responsible for desensitizing the β2AR contributing to the clinical side-effects. We propose that identifying β-agonists that selectively activate Gs signaling may have a similar beneficial effect on asthma patients as SALB but with less desensitization and possibly fewer side effects. Interestingly, a very recent study that screened a large scaffold ranking library also identified a Gs-biased agonist for the β2AR with low μM affinity and reduced physiological desensitization in HASM cells (Kim et al., 2021).
Another interesting observation of our study is the ability of some agonists to distinguish between Gs and Gi interaction with the β2AR. To our knowledge, this is the first report of G protein-biased signalling at the β2AR. It is likely that some molecules described as biased in the literature may be amenable to further sub-classification with an expanded analysis of their activity at the β2AR. We focused on HASM cells as a model system for airway physiology, however, the β2AR is expressed in multiple cell types in the airways and each may display distinct phenotypes based on the subcategory of biased agonist used. While our study focused on an expanded set of protein-protein interactions and regulatory outcomes compared to some prior efforts, there may be additional value in extending the analysis further to include additional non-canonical G protein coupling partners and signalling assays to create a more comprehensive signalling profile for a given ligand. Such an expanded approach has been extensively used by the Bouvier laboratory (Costa-Neto et al., 2016). While this introduces increased complexity to drug discovery applications, it may ultimately lead to reduced late-stage failures during the translation of new lead molecules.
As GPCR pharmacology becomes more complex, characterization and analysis of molecules targeting GPCRs must also follow suit. Advances in biosensors, biophysical techniques, and high throughput cell biology will help to streamline this process and generate new types of highly informative data. As shown here, multiple protein-protein interaction endpoints, signalling outcomes, and regulatory responses were able to be adapted to scalable microplate assays. Incorporating these types of studies into drug discovery efforts will be critical so that identification of molecules with improved therapeutic utility will not be a matter of chance. Increasing the dimensionality of information collected about ligand promoted pathway engagement and physiological responses will mitigate risk in the translational process and provide a foundation for therapeutic development. Additionally, coupling pathway studies to physiological models should help overcome the limitations of screening in overexpression systems that can lead to spurious results. Moreover, examining the ligand-promoted interactome holds value for even historically well-studied GPCRs like the β2AR and will allow for a better understanding of the potential therapeutic utility of novel GPCR drug targets.
Although the compounds identified in this study may not be suitable for clinical translation due to their lack of β2AR subtype selectivity, further structural studies with the Gs-biased ligands may provide insight into the molecular mechanisms of G protein discrimination at the β2AR. This structure-activity profiling would provide a solid framework for rational drug design targeting specific signalling phenotypes. Additionally, tying these ligand classes to physiological systems will help determine which signalling axes promote therapeutically desirable effects and which promote deleterious side effects. Taken together, these types of studies will greatly inform next generation drug development for given drug targets and indications.
Supplementary Material
What is already known:
Current treatments for asthma use balanced agonists for the β2AR that can induce side effects.
Gs signalling is beneficial in treating asthma while β-arrestins inhibit Gs signalling.
What this study adds:
Identification of ractopamine, dobutamine and higenamine as Gs-biased agonists for the β2AR.
Gs-biased agonists for the β2AR constitute an additional strategy to consider for treating asthma.
Clinical significance:
These compounds can be used to design safer drugs to treat asthma.
Acknowledgments:
The authors thank Dr. Mark von Zastrow for providing HEK293 cells stably expressing the β2AR, Dr. Brian Kobilka for providing purified β2AR, Dr. Michel Bouvier for providing β1AR-RlucII, β2AR-RlucII and β-arrestin2-GFP10, Dr. Nevin Lambert for providing NES-Venus-mGs, NES-Venus-mGsi, Venus-KRas and Venus-Rab5, Dr. Lily I. Jiang for providing CAMYEL, Dr. Melvin Reichman for providing access to the LIMR small molecule library, and Dr. Reynold Panettieri for helpful comments.
Funding statement:
Research reported in this publication was supported by National Institutes of Health awards R35GM122541 (JLB), R01HL136219 (JLB), P01HL114471 (RSA, SSA, CPS, JLB), T32GM100836 (MI) and F31HL139104 (MI), the New Jersey Alliance for Clinical and Translational Science (UL1TR0030117, SSA), and utilized the MetaOmics Shared Resource at the Sidney Kimmel Cancer Center at Jefferson Health supported by NIH award P30CA056036. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Declaration of Transparency and Scientific Rigour:
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design and Analysis, and Immunoblotting and Immunochemistry, and as recommended by funding agencies, publishers and other organizations engaged with supporting research.
Abbreviations:
- β1AR
β1-adrenergic receptor
- β2AR
β2-adrenergic receptor
- BAM
bamethane
- BRET
bioluminescence resonance energy transfer
- BUP
buphenine
- cAMP
3’,5’-cyclic adenosine monophosphate
- COPD
chronic obstructive pulmonary disease
- DBC
deep blue coelenterazine
- DHA
dihydroalprenolol
- DMSO
dimethyl sulfoxide
- DOB
dobutamine
- DMEM
Dulbecco’s modified Eagle’s medium
- EPI
epinephrine
- FBS
fetal bovine serum
- FEN
fenoterol
- GPCR
G protein-coupled receptor
- GRK
G protein-coupled receptor kinase
- HASM
human airway smooth muscle
- HIG
higenamine
- pIC50
negative log of the half-maximal inhibitory concentration
- ISO
isoproterenol
- LABA
long acting β-agonist
- nd
not determined
- NE
norepinephrine
- PBS
phosphate buffered saline
- RAC
ractopamine
- RIT
ritodrine
- SABA
short acting β-agonist
- SALB
salbutamol
- SALM
salmeterol
- TC
transduction coefficient
Footnotes
Conflict of interest disclosure: The authors declare no conflicts of interest.
Data availability statement:
All relevant data are included in the figures and tables. Additional primary data sources supporting the study are available upon reasonable request from the corresponding author.
References
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Mathie A, Peters JA, et al. (2021). THE CONCISE GUIDE TO PHARMACOLOGY 2021/22: G protein‐coupled receptors. British J Pharmacology 178:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Roberts RE, Broughton BRS, Sobey CG, George CH, Stanford SC, et al. (2018). Goals and practicalities of immunoblotting and immunohistochemistry: A guide for submission to the British Journal of Pharmacology. Br J Pharmacol 175: 407–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An SS, Mitzner W, Tang W-Y, Ahn K, Yoon A-R, Huang J, et al. (2016). An inflammation-independent contraction mechanophenotype of airway smooth muscle in asthma. Journal of Allergy and Clinical Immunology 138: 294–297.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzi M, Charest PG, Angers S, Rousseau G, Kohout T, Bouvier M, et al. (2003). β-arrestin-mediated activation of MAPK by inverse agonists reveals distinct active conformations for G protein-coupled receptors. Proceedings of the National Academy of Sciences of the United States of America 100: 11406–11411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai G, Yang Y, Shi Q, Liu Z, Zhang Q, and Zhu Y (2008). Identification of higenamine in Radix Aconiti Lateralis Preparata as a beta2-adrenergic receptor agonist 1. Acta Pharmacologica Sinica 29: 1187–1194. [DOI] [PubMed] [Google Scholar]
- Baker JG (2010). The selectivity of β-adrenoceptor agonists at human β1-, β2- and β3-adrenoceptors: β-Adrenoceptor agonist selectivity. British Journal of Pharmacology 160: 1048–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benovic JL (2002). Novel beta2-adrenergic receptor signaling pathways. J Allergy Clin Immunol 110: S229–235. [DOI] [PubMed] [Google Scholar]
- Bhagat R, Kalra S, Swystun VA, and Cockcroft DW (1995). Rapid onset of tolerance to the bronchoprotective effect of salmeterol. Chest 108: 1235–1239. [DOI] [PubMed] [Google Scholar]
- Casella I, Ambrosio C, Grò MC, Molinari P, and Costa T (2011). Divergent agonist selectivity in activating β 1- and β 2-adrenoceptors for G-protein and arrestin coupling. Biochemical Journal 438: 191–202. [DOI] [PubMed] [Google Scholar]
- Castle W, Fuller R, Hall J, and Palmer J (1993). Serevent nationwide surveillance study: comparison of salmeterol with salbutamol in asthmatic patients who require regular bronchodilator treatment. BMJ 306: 1034–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazzola M, and Donner CF (2000). Long-Acting ??2 Agonists in the Management of Stable Chronic Obstructive Pulmonary Disease: Drugs 60: 307–320. [DOI] [PubMed] [Google Scholar]
- Cazzola M, Page CP, Rogliani P, and Matera MG (2013). β 2 -Agonist Therapy in Lung Disease. Am J Respir Crit Care Med 187: 690–696. [DOI] [PubMed] [Google Scholar]
- Chen M, Hegde A, Choi YH, Theriot BS, Premont RT, Chen W, et al. (2015). Genetic Deletion of β-Arrestin-2 and the Mitigation of Established Airway Hyperresponsiveness in a Murine Asthma Model. Am J Respir Cell Mol Biol 53: 346–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung D, Timmers MC, Zwinderman AH, Bel EH, Dijkman JH, and Sterk PJ (1992). Long-term effects of a long-acting beta 2-adrenoceptor agonist, salmeterol, on airway hyperresponsiveness in patients with mild asthma. N. Engl. J. Med 327: 1198–1203. [DOI] [PubMed] [Google Scholar]
- Choi M, Staus D, Wingler L, Ahn S, \ldots, B.P.-S., and undefined 2018. G protein–coupled receptor kinases (GRKs) orchestrate biased agonism at the β2-adrenergic receptor. Stke.Sciencemag.Org. [DOI] [PubMed] [Google Scholar]
- Christopoulos A (2014). Advances in GPCR Allostery: From Function to Structure. Molecular Pharmacology 463–478. [DOI] [PubMed] [Google Scholar]
- Colbert WE, Williams PD, and Williams GD (2011). β-Adrenoceptor Profile of Ractopamine HCl in Isolated Smooth and Cardiac Muscle Tissues of Rat and Guinea-pig. Journal of Pharmacy and Pharmacology 43: 844–847. [DOI] [PubMed] [Google Scholar]
- Costa-Neto CM, Parreiras-e-Silva LT, and Bouvier M (2016). A Pluridimensional View of Biased Agonism. Mol Pharmacol 90: 587–595. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SPA, Giembycz MA, et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daaka Y, Luttrell LM, and Lefkowitz RJ (1997). Switching of the coupling of the β2-adrenergic receptor to different G proteins by protein kinase A. Nature 390: 88–91. [DOI] [PubMed] [Google Scholar]
- Deshpande DA, Theriot BS, Penn RB, and Walker JKL (2008). β‐Arrestins specifically constrain β2‐adrenergic receptor signaling and function in airway smooth muscle. FASEB j 22: 2134–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWire SM, Ahn S, Lefkowitz RJ, and Shenoy SK (2007). Beta-arrestins and cell signaling. Annu. Rev. Physiol 69: 483–510. [DOI] [PubMed] [Google Scholar]
- Donover PS, Yohn M, Sim M, Wright A, Gowda S, Allee C, et al. (2013). New Informatics and Automated Infrastructure to Accelerate New Leads Discovery by High Throughput Screening (HTS; ). [DOI] [PubMed] [Google Scholar]
- Drake MT, Violin JD, Whalen EJ, Wisler JW, Shenoy SK, and Lefkowitz RJ (2008). β-arrestin-biased agonism at the β2-adrenergic receptor. Journal of Biological Chemistry 283: 5669–5676. [DOI] [PubMed] [Google Scholar]
- Dror RO, Arlow DH, Maragakis P, Mildorf TJ, Pan AC, Xu H, et al. (2011). Activation mechanism of the β2 -adrenergic receptor. Proceedings of the National Academy of Sciences of the USA 108: 18684–18689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forkuo GS, Kim H, Thanawala VJ, Al-Sawalha N, Valdez D, Joshi R, et al. (2016). Phosphodiesterase 4 inhibitors attenuate the asthma phenotype produced by β2-adrenoceptor agonists in phenylethanolamine N-methyltransferase-knockout mice. American Journal of Respiratory Cell and Molecular Biology 55: 234–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimenez LE, Baameur F, Vayttaden SJ, and Clark RB (2015). Salmeterol Efficacy and Bias in the Activation and Kinase-Mediated Desensitization of β 2-Adrenergic Receptors. Mol Pharmacol 87: 954–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grove A, and Lipworth B (1995). Tolerance with beta 2-adrenoceptor agonists: time for reappraisal. British Journal of Clinical Pharmacology 39: 109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingsworth JW, Theriot BS, Li Z, Lawson BL, Sunday M, Schwartz DA, et al. (2010). Both hematopoietic-derived and non-hematopoietic-derived {beta}-arrestin-2 regulates murine allergic airway disease. Am. J. Respir. Cell Mol. Biol 43: 269–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudzik TJ, Patel M, and Brown A (2021). β 2 ‐Adrenoceptor agonist activity of higenamine. Drug Test Anal 13: 261–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel PA (1996). Adrenergic receptors - Evolving concepts and clinical implications (Massachusetts Medical Society; ). [DOI] [PubMed] [Google Scholar]
- Ippolito M, and Benovic JL (2021). Biased agonism at β-adrenergic receptors. Cellular Signalling 80: 109905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang LI, Collins J, Davis R, Lin K-M, DeCamp D, Roach T, et al. (2007). Use of a cAMP BRET Sensor to Characterize a Novel Regulation of cAMP by the Sphingosine 1-Phosphate/G13 Pathway. Journal of Biological Chemistry 282: 10576–10584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T, Watson C, Muniz-Medina V, Christopoulos A, and Novick S (2012). A Simple Method for Quantifying Functional Selectivity and Agonist Bias. ACS Chem. Neurosci 3: 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Tokmakova A, Lujan LK, Strzelinski HR, Kim N, Najari Beidokhti M, et al. (2021). Identification and characterization of an atypical Gαs-biased β 2 AR agonist that fails to evoke airway smooth muscle cell tachyphylaxis. Proc Natl Acad Sci USA 118: e2026668118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupnick JG, and Benovic JL (1998). The role of receptor kinases and arrestins in G protein-coupled receptor regulation. Annu Rev Pharmacol Toxicol 38: 289–319. [DOI] [PubMed] [Google Scholar]
- Lan T-H, Kuravi S, and Lambert NA (2011). Internalization Dissociates β2-Adrenergic Receptors. PLoS ONE 6: e17361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Grandy DK, and Janowsky A (2014). Ractopamine, a Livestock Feed Additive, Is a Full Agonist at Trace Amine–Associated Receptor 1. J Pharmacol Exp Ther 350: 124–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Hu Y, Batebi H, Heng J, Xu J, Liu X, et al. (2020). Analysis of β 2 AR-G s and β 2 AR-G i complex formation by NMR spectroscopy. Proc Natl Acad Sci USA 117: 23096–23105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maack C, Eschenhagen T, Hamdani N, Heinzel FR, Lyon AR, Manstein DJ, et al. (2019). Treatments targeting inotropy. European Heart Journal 40: 3626–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matera MG, Page CP, Calzetta L, Rogliani P, and Cazzola M (2020). Pharmacology and Therapeutics of Bronchodilators Revisited. Pharmacol Rev 72: 218–252. [DOI] [PubMed] [Google Scholar]
- Michel MC, and Charlton SJ (2018). Biased agonism in drug discovery - is it too soon to choose a path? Molecular Pharmacology mol.117.110890. [DOI] [PubMed] [Google Scholar]
- Miller WE, and Lefkowitz RJ (2001). Expanding roles for beta-arrestins as scaffolds and adapters in GPCR signaling and trafficking. Curr Opin Cell Biol 13: 139–145. [DOI] [PubMed] [Google Scholar]
- Mills SE, Kissel J, Bidwell CA, and Smith DJ (2003). Stereoselectivity of porcine β-adrenergic receptors for ractopamine stereoisomers1,2. Journal of Animal Science 81: 122–129. [DOI] [PubMed] [Google Scholar]
- Nelson HS, Weiss ST, Bleecker ER, Yancey SW, and Dorinsky PM (2006). The Salmeterol Multicenter Asthma Research Trial. Chest 129: 15–26. [DOI] [PubMed] [Google Scholar]
- Penn RB, and Benovic JL (2008). Regulation of Heterotrimeric G Protein Signaling in Airway Smooth Muscle. Proceedings of the American Thoracic Society 5: 47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce KL, Premont RT, and Lefkowitz RJ (2002). Seven-transmembrane receptors (Nature Publishing Group; ). [DOI] [PubMed] [Google Scholar]
- Salpeter SR, Buckley NS, Ormiston TM, and Salpeter EE (2006). Meta-analysis: effect of long-acting beta-agonists on severe asthma exacerbations and asthma-related deaths. Ann. Intern. Med 144: 904–912. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, et al. (2006). beta-arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor. J. Biol. Chem 281: 1261–1273. [DOI] [PubMed] [Google Scholar]
- Smith JS, Lefkowitz RJ, and Rajagopal S (2018). Biased signalling: from simple switches to allosteric microprocessors. Nature Reviews Drug Discovery 17: 243–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanawala VJ, Valdez DJ, Joshi R, Forkuo GS, Parra S, Knoll BJ, et al. (2015). β-Blockers have differential effects on the murine asthma phenotype. British Journal of Pharmacology 172: 4833–4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X, Kang DS, and Benovic JL (2014). β-Arrestins and G Protein-Coupled Receptor Trafficking. In Arrestins - Pharmacology and Therapeutic Potential, Gurevich VV, ed. (Berlin, Heidelberg: Springer Berlin Heidelberg; ), pp 173–186. [Google Scholar]
- Toews ML, Harden TK, and Perkins JP (1983). High-affinity binding of agonists to beta-adrenergic receptors on intact cells. Proceedings of the National Academy of Sciences 80: 3553–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukiyama M, Ueki T, Yasuda Y, Kikuchi H, Akaishi T, Okumura H, et al. (2009). β 2 -Adrenoceptor-Mediated Tracheal Relaxation Induced by Higenamine from Nandina domestica Thunberg. Planta Med 75: 1393–1399. [DOI] [PubMed] [Google Scholar]
- Ueki T, Akaishi T, Okumura H, Morioka T, and Abe K (2011). Biphasic Tracheal Relaxation Induced by Higenamine and Nantenine From Nandina domestica THUNBERG. J Pharmacol Sci 115: 254–257. [DOI] [PubMed] [Google Scholar]
- Walker J, Penn R, Hanania N, Dickey B, and Bond R (2011). New perspectives regarding β2-adrenoceptor ligands in the treatment of asthma: New paradigms for β2-adrenoceptor ligands in asthma. British Journal of Pharmacology 163: 18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JKL, and DeFea KA (2014). Role for β-arrestin in mediating paradoxical β2AR and PAR2 signaling in asthma. Curr Opin Pharmacol 16: 142–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JKL, Fong AM, Lawson BL, Savov JD, Patel DD, Schwartz DA, et al. (2003). Beta-arrestin-2 regulates the development of allergic asthma. J. Clin. Invest 112: 566–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Q, Okashah N, Inoue A, Nehmé R, Carpenter B, Tate CG, et al. (2018). Mini G protein probes for active G protein–coupled receptors (GPCRs) in live cells. Journal of Biological Chemistry 293: 7466–7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendell SG, Fan H, and Zhang C (2020). G Protein–Coupled Receptors in Asthma Therapy: Pharmacology and Drug Action. Pharmacol Rev 72: 1–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westhuizen ET van der, Breton B, Christopoulos A, and Bouvier (2014). Quantification of Ligand Bias for Clinically Relevant β 2 -Adrenergic Receptor Ligands: Implications for Drug Taxonomy. Mol Pharmacol 85: 492–509. [DOI] [PubMed] [Google Scholar]
- Wortzel I, and Seger R (2011). The ERK Cascade: Distinct Functions within Various Subcellular Organelles. Genes & Cancer 2: 195–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Zhu H, Li Z, and Dong E (2021). A novel β2-AR agonist, Higenamine, induces β-arrestin-biased signaling. Sci. China Life Sci [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant data are included in the figures and tables. Additional primary data sources supporting the study are available upon reasonable request from the corresponding author.