

Abstract
Objectives
How inflammatory signaling contributes to OA susceptibility is undetermined. An allele encoding a hyperactive form of the RIPK2 proinflammatory signaling intermediate has been associated with familial OA. To test whether altered NOD/RIPK2 pathway activity causes heightened OA susceptibility, we investigated whether variants affecting additional pathway components are associated with familial OA. To determine whether the Ripk2104Asp disease allele is sufficient to account for the familial phenotype, we determined the effect of the allele on mice.
Methods
Genomic analysis of 150 independent families with dominant inheritance of OA affecting diverse joints was used to identify coding variants that segregated strictly with occurrence of OA. Genome editing was used to introduce the OA-associated RIPK2 (p.Asn104Asp) allele into the genome of inbred mice. The consequences of the Ripk2104Asp disease allele on physiology and OA susceptibility in mice were measured by histology, immunohistochemistry, serum cytokine levels, and gene expression.
Results
We identified six novel variants affecting components of the NOD/RIPK2 inflammatory signaling pathway that are associated with familial OA affecting the hand, shoulder, or foot. The Ripk2104Asp allele acts dominantly to alter basal physiology and response to trauma in the mouse knee. Whereas the knees of uninjured Ripk2Asp104 mice appear normal histologically, the joints exhibit a set of marked gene expression changes reminiscent of overt OA. Although the Ripk2104Asp mice lack evidence of chronically elevated systemic inflammation, they do exhibit significantly increased susceptibility to PTOA.
Conclusions
Two types of data support the hypothesis that altered NOD/RIPK2 signaling confers susceptibility to OA.
Keywords: osteoarthritis genetics, osteoarthritis risk factors, osteoarthritis gene, RIPK2, NOD1, NOD2, RIP2, interphalangeal joint osteoarthritis, 1st MTP joint osteoarthritis, shoulder osteoarthritis
INTRODUCTION
The molecular pathways that are rate-limiting in the onset and progression of osteoarthritis (OA) are unknown, consistent with the complete lack of disease-modifying drugs currently available1–4. Knowledge of these pathways is required for identifying individuals at risk for disease, for understanding mechanisms that trigger or amplify disease processes, and for development of effective therapies. One proven approach toward identifying pathways and biological processes whose normal functions limit disease has been to identify gene variants responsible for highly penetrant familial forms of the disease. Increasing evidence demonstrates there are no/few differences between the genes contributing to “monogenic” disease and those contributing to complex disease5–10. Pathways that can be mutated to have determinate effects promoting OA will also be vulnerable to the modest genetic or environmental perturbations that underlie common spontaneous forms of OA5. Despite its promise, to date there have been relatively few studies of non-syndromic familial OA11–14. We have used a unique medical genetics resource, the Utah Population Database, to identify a large number of multigenerational families with dominantly inherited OA15. Here we employ genomic analyses of these families and functional analyses in mice to test the hypothesis that perturbation of the NOD/RIPK2 proinflammatory pathway is sufficient to significantly elevate susceptibility to OA.
In previous work, we identified a rare allele of the Receptor Interacting Protein Kinase 2 (RIPK2) gene (p.Asn104Asp) that was associated with dominant inheritance of early-onset OA of the first metatarsophalangeal (1st MTP) joint in a single family11. The NOD/RIPK2 signaling pathway is a key arm of the innate immunity system, playing critical roles both in clearing bacterial infections and maintaining immune homeostasis16. The intracellular nucleotide-binding oligomerization domain (NOD) receptors are activated by bacterial cell wall breakdown products and additional damage-associated molecular patterns16, 17. Activated NOD receptors signal through the Receptor Interacting Protein Kinase 2 (RIPK2), stimulating the MAPK and NF-κB pathways to elicit tissue-specific responses, most notably inflammatory responses18–21. NOD/RIPK2 signaling is tightly regulated, as mutations that either abrogate or elevate signaling are associated with chronic inflammatory diseases, including Crohn‟s, Blau syndrome, early-onset sarcoidosis, and Behcet‟s disease16, 22, 23. Although chronic inflammatory diseases are often associated with arthritis, no single inflammatory pathway has yet been linked to classic non-syndromic forms of OA4, 24.
Here we investigate the hypothesis that function of the NOD/RIPK2 signaling pathway limits susceptibility to OA. We find that individual gene variants affecting any of several components of the pathway appear sufficient to confer heightened susceptibility to OA in families. Significantly, variants affecting the NOD/RIPK2 signaling pathway are associated with divergent forms of familial OA. Last, we demonstrate that an OA-associated RIPK2 gene variant encoding a single amino acid change in the kinase domain is sufficient to alter basal gene expression patterns in primary chondrocytes and bone marrow macrophages and confer increased susceptibility to post-traumatic OA when introduced into the mouse genome. Our data provide strong support for the hypothesis that modulation of the NOD/RIPK2 signaling pathway is a significant risk factor for OA.
METHODS
Study approval
Written informed consent was obtained under the guidance of the Institutional Review Boards of the University of Utah and Intermountain Healthcare, and the Resource for Genetic and Epidemiologic Research approved this study. Mice were maintained in accordance with approved institutional protocols at the University of Utah.
Identification of families with a dominant pattern of OA inheritance
Our study utilizes data drawn from the Utah Population Database (UPDB) (https://uofuhealth.utah.edu/huntsman/utah-population-database/). The UPDB provides person-based interlinked records documenting genealogy, medical records, and vital statistics for over 11 million individuals from the late 18th century to the present. Medical records derive from the two largest healthcare providers in Utah (Intermountain Healthcare and University of Utah Health), Medicare claims, and the Utah and Idaho Cancer Registries. Vital records include statewide birth, death, and marriage certificates, as well as drivers‟ licenses. UPDB data are available for approved research projects. Privacy of individuals whose data is available through UPDB is strictly protected through the Utah Resource for Genetic and Epidemiological Research (https://rge.utah.edu), established by executive order of the Governor of Utah. We identified individuals with OA between 1996 and 2021 in the UPDB using the following diagnosis (ICD) and related procedure (CPT) codes: 1st MTP joint OA – ICD-9 735.2 or CPT 28289 and 28750; distal and proximal interphalangeal joint OA – ICD-9 715.14, ICD-10 M19.04x and CPT 26862, 26863, 26860, 26861, 26535, or 26536; and glenohumeral osteoarthritis OA – ICD-9 715.11, ICD-10 M19.011, M19.012, M19.019, Z96.611, Z96.612, or M19.0x and CPT 23472. Individuals with any of the following codes were excluded: ICD-9 714.0, 714.2, or 714.3 and ICD-10 M05.xxx, M06.xx, or M08.xxx. A detailed description of the ICD and CPT codes are provide in the online supplementary methods. Manual chart review was performed on affected individuals to verify our coding strategy to identify OA cases and determine if individuals had OA in additional joints. To determine if there was excess familial clustering of OA in each pedigree, we utilized the Familial Standardized Incidence Ratio (FSIR), with a threshold of ≥ 2.0. FSIR allows for the quantification of familial risk of a disease by comparing the incidence of a disease in a family to its expected incidence in the general population. See Kazmers, 2021 and Kazmers, 2020 for detailed methods25, 26. Pedigrees segregating a dominant pattern of OA inheritance were selected for genomic analysis.
Statistical Analyses
Statistical analysis was performed using GraphPad Prism software. Tests performed and statistical significance are indicated in the figure legends. P-values <0.05 were considered statistically significant.
RESULTS
Rare alleles of NOD-RIPK2 pathway genes are associated with multiple types of familial OA
We took an unbiased genetic approach to determine if the NOD/RIPK2 signaling pathway has a strong effect on OA susceptibility. We assembled a cohort of 150 independent OA families with a dominant inheritance pattern of OA. Each family is characterized by disease that primarily affects a distinct subset of joints: distal and proximal interphalangeal OA27, glenohumeral OA28, or 1st MTP joint OA29, 30. While all affected individuals have OA in the primary joint used for identification, two families contain a subset of individuals with OA in additional joints. The proband in MTP25 was diagnosed with triscaphe and thumb OA and had a total knee and hip arthroplasty. His daughter was also diagnosed with spine OA. The proband‟s sister in SA735 had surgery for thumb OA and bilateral total knee and hip arthroplasty (online supplemental table 1). Whole exome sequence (WES) analysis was performed on informative members of families and coding variants that invariably segregated with OA were identified11. Variants were prioritized using the pedigree Variant Annotation, Analysis & Search Tool (pVAAST)31, which identifies the most likely causal variants in a pedigree based on gene tolerance to mutation, variant frequency, phylogenetic conservation and biological function. We next used the PHEnotype-driven Variant Ontological Re-ranking (PHEVOR)32 tool with the Human Phenotype Ontology (HPO) search term ‘Osteoarthritis’ to identify high-priority candidate genes in each family.
In addition to the previously identified OA-associated RIPK2 allele, six novel alleles affecting five NOD/RIPK2 pathway genes were found associated with OA in the cohort of families (table 1 and online supplemental figure 1). Consistent with a dominant pattern of inheritance with strong penetrance, the variants are rare in human populations. Three variants affected the NOD1 (FIJ744 - pVAAST: P-value = 0.00809; LOD = 0.6; PHEVOR: score 3.26, final rank = 33) and NOD2 genes (UUHR2 - pVAAST: P -value = 0.0592; LOD = 7.195; PHEVOR: score 3.42, final rank = 6 and FIJ7 - pVAAST: P -value = 0.00333; LOD = 15.556; PHEVOR: score 4.7, final rank = 1), which encode intracellular receptors that function as upstream activators of RIPK2. The amino acid substitutions encoded by two of the variants reside within the autoinhibitory domain of the respective NOD protein33. Candidate variants in three additional families affected genes known to modify activity of the NOD/RIPK2 pathway, CARD9 (FIJ9 - pVAAST: P -value = 0.00106; LOD = 16.838; PHEVOR: score 3.08, final rank = 19), CHUK (MTP25 - pVAAST: P -value = 0.00102; LOD = 12.55; PHEVOR: score 4.55, final rank = 3), and IKBKB (SA735 - pVAAST: P -value = 0.000743; LOD = 16.15; PHEVOR: score 4.23, final rank = 10)34–37. The family studies indicate a striking correlation between inheritance of variants that alter conserved sites within proteins of the NOD/RIPK2 signaling pathway and the occurrence of disease within families exhibiting OA of the hand, 1st MTP joint, or shoulder.
Table 1.
NOD/RIPK2 Pathway Variants Identified in Independent Osteoarthritis Families
| Gene | OA Phenotype (Family) | Variant | Minor Allele Frequency | Protein Domain Affected by Variant |
|---|---|---|---|---|
| NOD1 | Finger Interphalangeal Joint OA (FIJ744) | c.G2114A:p.R705Q | 0.0008 | Leucine Rich Repeat Domain |
| NOD2 | 1st MTP Joint OA (UUHR2) | c.C2465T:p.A822V | 0.00007 | Leucine Rich Repeat Domain |
| NOD2 | Finger Interphalangeal Joint OA (FIJ7) | c.G247A:p.A83T | 0.00008 | Caspase Activation and Recruitment Domain |
| IKBKB | Glenohumeral OA (SA735) | c.G1663A:p.G555R | 0.00008 | Scaffold Dimerization Domain |
| CARD9 | Finger Interphalangeal Joint OA (FIJ9) | c.G722A:p.R241Q | 0.00005 | Structural Maintenance of Chromosomes |
| CHUK | 1st MTP Joint OA (MTP25) | c.A376T:p.S126C | 0.0008 | Kinase Domain |
| RIPK2* | 1st MTP Joint OA (UUHR1) | c.A310G:pN104D | 0.0004 | Kinase Domain |
- Previously described in Jurynec, 201811.
Generation of the Ripk2104Asp mouse
Nod1/2 and Ripk2 are expressed in uninjured joints of mice, and the pathway is activated following injury (online supplemental figure 2). To determine whether altered pathway signaling is sufficient to confer increased susceptibility to OA, we used precise genome editing to introduce the human RIPK2104Asp variant into the C57BL/6J inbred strain of mice, thereby creating an isogenic pair of mouse lines: the parental strain, which encodes the mammalian lineage-conserved Asn at position 104 (WT), and a derived line whose Ripk2 allele encodes Asp at position 104 (Ripk2104Asp) (figure 1A). The modified allele is expressed at WT levels (figure 1B and online supplemental figure 3). Homozygous and heterozygous mice carrying the Ripk2104Asp allele are viable and display no overt phenotypes. To recapitulate the dominant human phenotype11, heterozygous Ripk2104Asp mice were used for all subsequent analyses.
Figure 1.
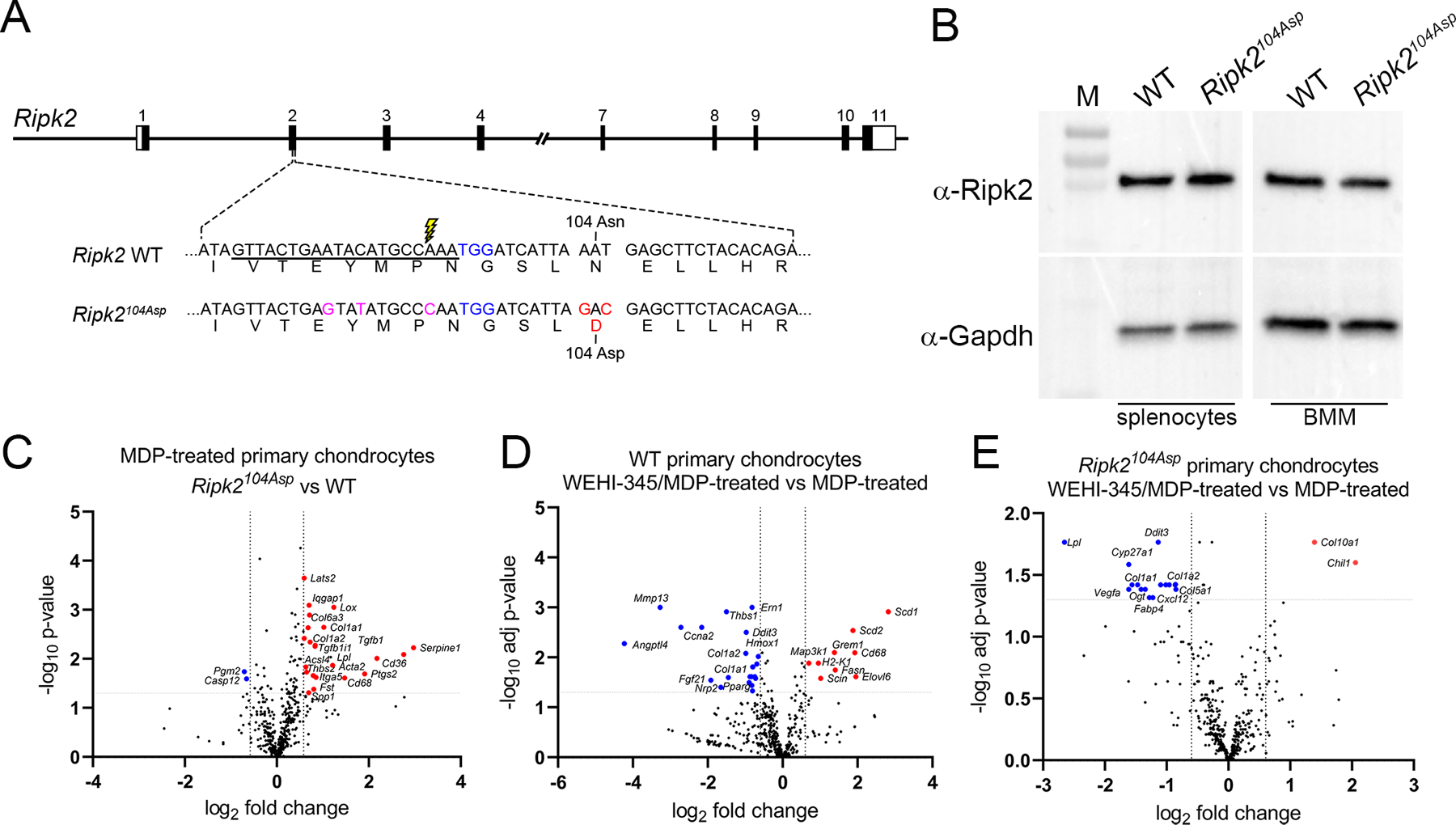
Generation and validation of the Ripk2104Asp mouse. (A) Schematic illustration of the mouse Ripk2 locus and detailed view of exon 2. CRISPR/Cas9-stimulated homology directed repair was used to edit sequences of C57Bl/6J mice (WT) encoding the Ripk2104Asn protein to generate an isogenic line that expressed the OA-associated Ripk2104Asp protein from the native locus. The guide RNA target sequence is underlined, the PAM site is highlighted in blue, and the Cas9 cleavage site is denoted with lightning bolt. An oligonucleotide donor was used as a template to create the mutations to generate Asp 104 (red) as well as silent mutations (magenta) to prevent targeting of the modified locus. (B) Immunoblot analysis indicates similar Ripk2 protein present in WT and Ripk2104Asp splenocytes or bone marrow derived macrophages (BMM). Gapdh is used as a loading control. M = protein mass standards in kDa. (C) A single copy of Ripk2104Asp is sufficient to alter the gene expression response of primary chondrocytes to MDP treatment. Volcano plots indicate genes significantly upregulated (red) or downregulated (blue) in MDP-treated Ripk2104Asp as compared to MDP-treated WT primary chondrocytes. (D and E) Increased gene expression in response to MDP stimulation is dependent on Ripk2 activity. D) WT or E) Ripk2104Asp primary chondrocytes were stimulated with MDP in the presence or absence of the Ripk2 inhibitor, WEHI-345. Volcano plots indicate genes significantly upregulated (red) or downregulated (blue) upon MDP-stimulation in the presence of the inhibitor.
A single copy of the Ripk2104Asp allele is sufficient to alter gene expression in primary chondrocytes
To determine if the Ripk2104Asp allele perturbed NOD/RIPK2 signaling, cultured primary chondrocytes38 were stimulated with the Nod2 agonist, muramyldipeptide (MDP). Both WT and Ripk2104Asp chondrocytes responded to MDP by upregulating genes associated with proinflammatory signaling (online supplemental figure 4). However, the transcriptional response of Ripk2104Asp chondrocytes was significantly amplified as compared with that of WT controls, consistent with previous functional assays of the Ripk2104Asp allele11 (figure 1C). MDP-stimulated gene expression was indeed dependent on Ripk2 activity as co-incubation of chondrocytes with the Ripk2 inhibitor WEHI-34539 significantly reduced expression of many genes, including those whose expression is directly associated with OA (Mmp13, Col1a1, Col1a2, and Ccna2) (figure 1D and E). These data indicate the Ripk2104Asp allele confers heightened gene expression upon activation of the NOD2 receptor.
The Ripk2104Asp allele acts dominantly and is sufficient to confer increased susceptibility to OA
The joints of mature Ripk2104Asp animals appear structurally similar to those of WT mice with no histological evidence of joint degeneration (figure 2 A-D, I and J). Nevertheless, Ripk2104Asp mice displayed increased sensitivity to experimentally induced OA, initiated by destabilization of the medial meniscus (DMM) of the stifle (knee) joint40. Eight weeks after DMM surgery Ripk2104Asp mice exhibited a significant increase in the extent and severity of cartilage damage on the medial (both femoral condyle and tibial plateau) and lateral (femoral condyle) faces of the knee as compared to operated WT controls (figure 2A-J and online supplemental figure 5). Blinded histological scoring of the entire joint or individual quadrants revealed highly significant differences in the average and maximal OARSI scores40 of DMM-treated WT and Ripk2104Asp mice (figure 2I and J). In contrast, no significant difference was evident in the degree of synovitis observed in the operated joints of the two groups of mice (figure 2K). As a non-invasive alternate method for inducing PTOA in 16-week-old mice, we employed mechanical rupture of the anterior cruciate ligament (ACL)41. Again, Ripk2104Asp mice showed a significant increase in cartilage damage compared to WT controls when examined 5 weeks post-rupture (online supplemental figure 6).
Figure 2.
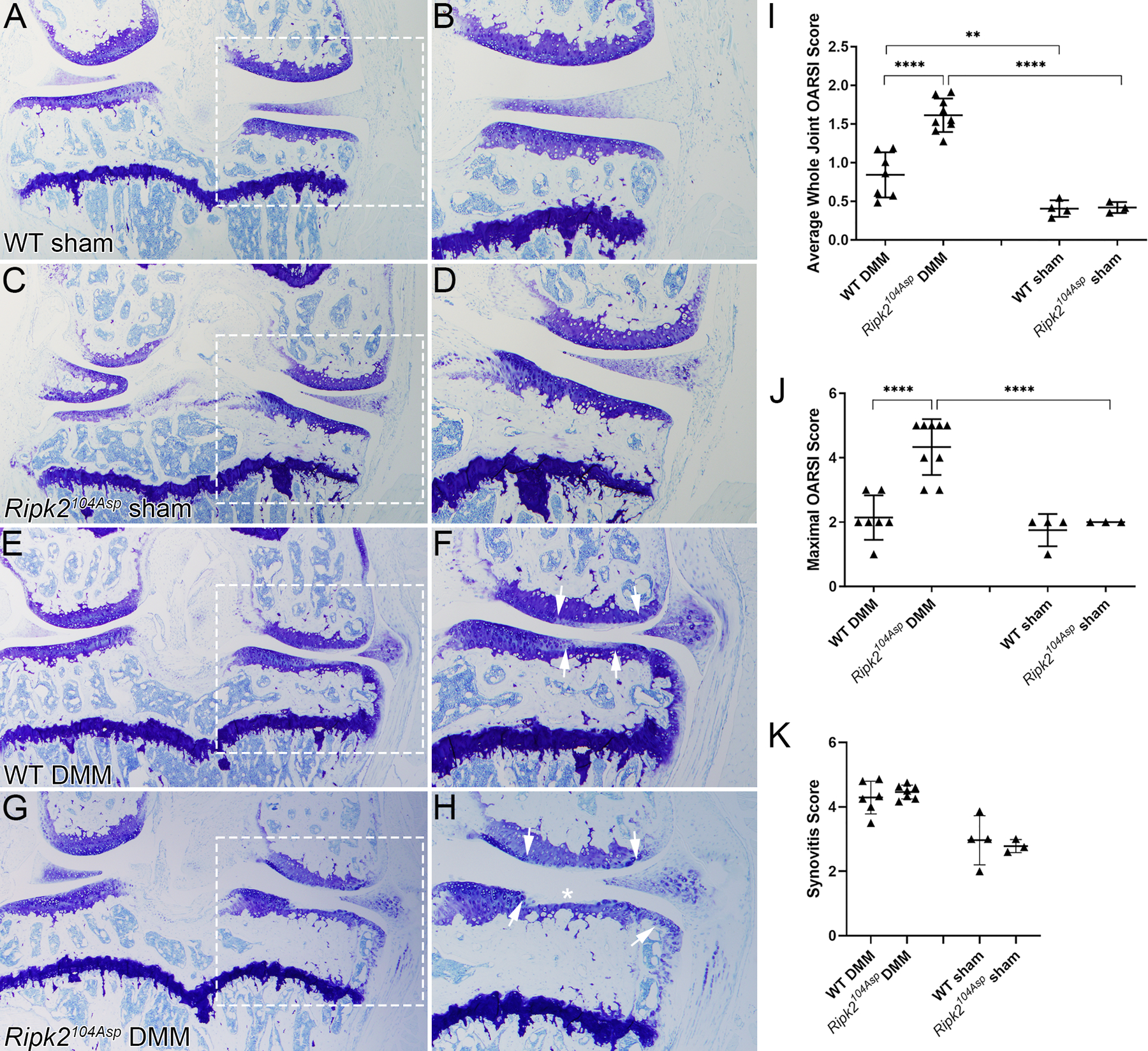
The Ripk2104Asp allele acts dominantly and is sufficient to confer increased susceptibility to post-traumatic osteoarthritis. (A-D) Knee joints of WT and Ripk2104Asp mice that underwent sham surgery are similar histologically, with no indication of an OA phenotype. (E, F) Following DMM surgery, WT knees displayed mild/moderate loss of proteoglycan content in the articular cartilage on the medial side of the knee (indicated by loss of toluidine blue staining). The extent of the damage is indicated by the arrows in F, G, H. Following DMM surgery, joints of Ripk2104Asp mice displayed moderate/severe loss of proteoglycan content (arrows in H), cartilage fibrillation, and complete loss of articular cartilage in the medial tibial plateau (asterisk in H). (I) Average whole joint and (J) maximal OARSI scores of sham-operated and DMM-operated knee joints. (K) There was no difference in the degree of synovitis between different genotypes. A, C, G, E are images of the entire knee joint; dashed boxes were magnified in B, D, F, H to focus on degradation on the medial side of the joint. Femur is up and medial is to the right in all images. WT sham (n=4), Ripk2104Asp sham (n=3), WT DMM (n=7), Ripk2104Asp DMM (n=9). All animals were analyzed 8 weeks post-surgery. Error bars represent ±SD and statistically significant differences of P ≤ 0.01 (**), P ≤ 0.001 (***), and P ≤ 0.0001 (****) were determined by two-way ANOVA with Tukey‟s multiple comparisons test.
Injured knee joints of Ripk2104Asp mice exhibited gene expression signatures associated with classical OA, but indicative of a more advanced disease state as compared with WT. Genes upregulated in the injured Ripk2104Asp knee joints soon after ACL rupture are involved in both the innate and adaptive immune response (Prg2, Trpc6, Bank1, Siglecf, Clca3a1, Isg15, Ighg1, Ighg2b, and Rsad2) and include genes linked to OA (B2m, A2m, Il17r, Ctsk, Angpt4, Aldh1a2, and Alox15) (figure 3A). Conversely, many ECM genes whose depletion is associated with OA pathogenesis were significantly downregulated in Ripk2104Asp joints, including Acan, Col2a1, Col3a1, Comp, Prg4, Fn1, Hspg2, Matn2, and Cspg4 (figure 3A). Thus, whereas the transcriptional response of Ripk2104Asp mice to injury closely parallels that of WT, as indicated by the altered expression of OA-associated genes, it is exaggerated, with increased expression of inflammatory and catabolic factors and increased down-regulation of many ECM genes.
Figure 3.
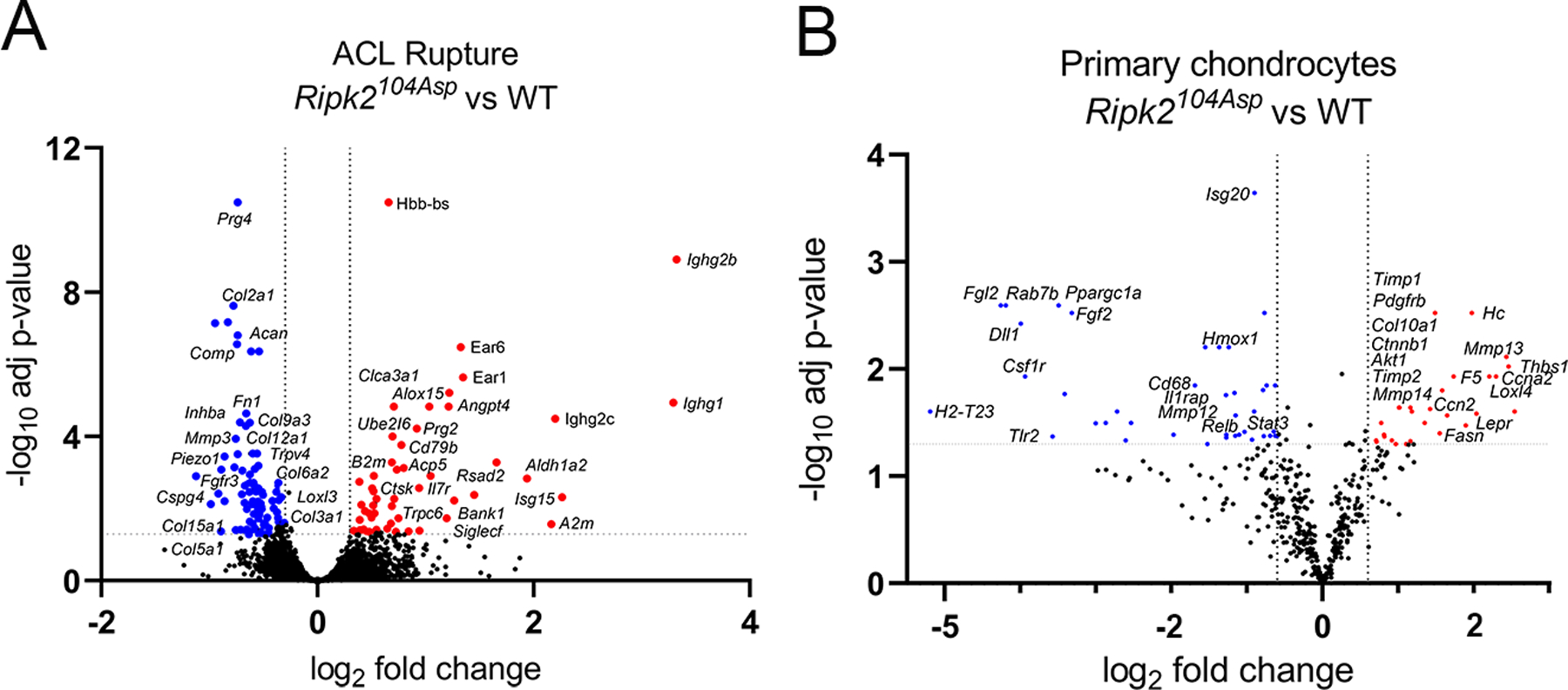
Ripk2104Asp enhances expression of OA-associated markers in the surgically injured joint as well as primary chondrocytes. (A) Comparative analysis of RNA-seq performed on whole joints isolated from WT or Ripk2104Asp mice 10 days post ACL rupture. The volcano plot indicates genes significantly upregulated (red) or downregulated (blue) in Ripk2104Asp as compared to WT joints. (B) The nCounter Fibrosis panel was used to measure gene expression in AC cultured from WT or Ripk2104Asp mice. Volcano plot indicates genes significantly upregulated (red) or downregulated (blue) in Ripk2104Asp primary chondrocytes compared to WT primary chondrocytes.
The Ripk2104Asp allele alters the basal physiology of knees joints and the response to PTOA
In the absence of evident tissue remodeling that might indicate emergent OA in the knee joints of Ripk2104Asp mice (figure 2A-D, I and J), we asked whether the joints exhibited altered signs of gene expression and/or inflammatory state. Analysis of primary chondrocytes revealed that gene expression was significantly altered in Ripk2104Asp cells as compared with those isolated from WT mice (figure 3B). Genes upregulated included well-known markers of OA, including those associated with hypertrophic chondrocytes and ECM remodeling (Mmp13, Mmp14, Timp1, Timp2, Loxl4, and Col10a1), growth factor signaling (Ctnnb1, Ckap4, and Pdgfrb), leptin signaling (Lepr), PI3K/Akt/mTOR signaling (Akt1), as well as genes involved in inflammatory signaling (Lpcat1, Rbx1, Ccn2, Fasn, Cfhr2, F5, Elovl6, Hc, and Thbs1) (figure 3B).
The striking linkage between the altered gene expression profile of cultured Ripk2104Asp chondrocytes and markers of mature OA led us to investigate if altered marker expression presaged the response to injury in the whole joint. The joints of both sham-operated and DMM mice revealed substantive effects of the Ripk2104Asp allele on Nod/Ripk2 activity, matrix components, and markers of inflammation (figure 4 and online supplemental figure 7). Although the Ripk2 protein is present at low levels in tibial chondrocytes of WT and Ripk2104Asp animals subjected to sham surgery (figure 4A), pathway activity appears elevated in the Ripk2104Asp joint, as reflected in higher levels of activated phospho-NF-κB (pNF-κB) compared with that of WT sham controls (figure 4B and online supplemental figure 7). Following DMM surgery, pathway activity differences are enhanced, as both the level of Ripk2 expression and the number of chondrocytes expressing NF-κB are considerably elevated in the operated joints of Ripk2104Asp mice relative to WT (figure 4A, B, C, and online supplemental figure 7). Similarly, expression of matrix markers is altered in sham-operated Ripk2104Asp joints in a manner that parallels the gene expression changes associated with overt OA. As compared with WT sham controls, knee joints of sham surgery Ripk2104Asp mice express elevated levels and have increased numbers of chondrocytes expressing Mmp13, a metalloproteinase that targets collagen for degradation (figure 4C and E). Mmp13 expression in the Ripk2104Asp mice extends beyond the superficial layer of cartilage into deeper layers relative to WT controls (brackets in figure 4C and online supplemental figure 7). Consistent with this finding, collagen deposition scored by the presence of Col2 appears relatively deficient in the joints of sham-operated Ripk2104Asp mice (figure 4D, C, and online supplemental figure 7). The differences between WT and Ripk2104Asp mice in the levels of expression and numbers of chondrocytes expressing Mmp13 and Col2 between WT and Ripk2104Asp mice become even more exaggerated following DMM surgery (figure 4C, D, E and online supplemental figure 7). Altogether these data indicate the Ripk2104Asp allele alters the basal physiological state of the joint so that it expresses features normally associated with overt OA.
Figure 4.
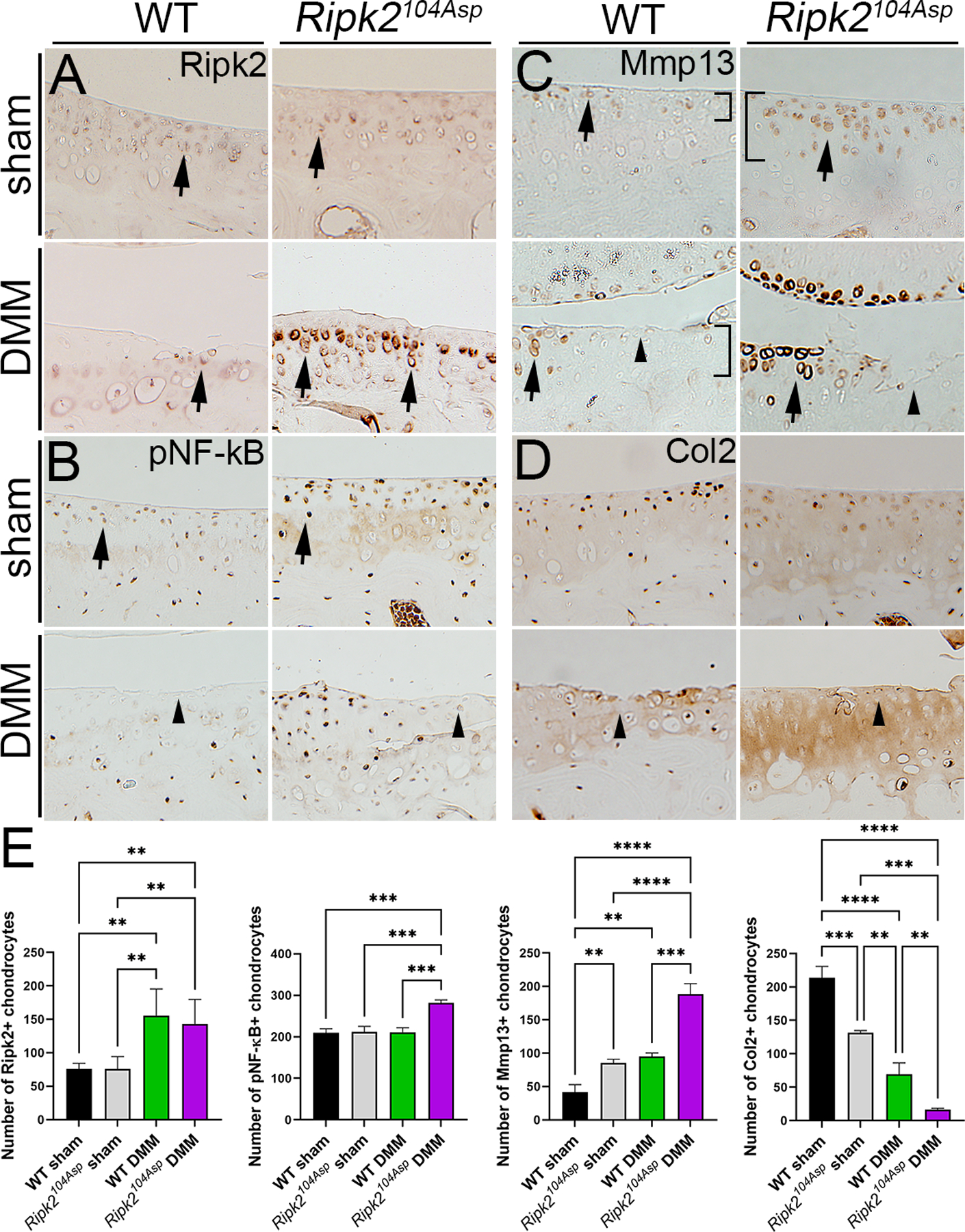
Ripk2104Asp enhances NOD/RIPK2 signaling as well as expression of OA-associated markers of matrix remodeling in uninjured joints and joints with PTOA. Immunohistochemical detection of (A) Ripk2, (B) pNF-κB, (C) Mmp13, or (D) Col2 in WT and Ripk2104Asp mice 8 weeks following sham or DMM surgery. (A) Ripk2 is expressed at low levels in chondrocytes (arrows) of WT sham-, WT DMM-, and Ripk2104Asp sham-operated joints. In contrast, Ripk2 expression is highly elevated in chondrocytes in Ripk2104Asp knees following DMM surgery (arrows). (B) Activated NF-κB (pNF-κB) expression levels are higher in Ripk2104Asp sham-operated joints as compared to WT controls (arrows). The relatively increased Ripk2 expression is maintained in Ripk2104Asp joints following DMM surgery. (C) Mmp13 expression is elevated in chondrocytes of sham-operated Ripk2104Asp mice as compared to WT controls (arrows); the relatively increased expression Ripk2 is maintained in Ripk2104Asp mice following DMM surgery (arrows). Furthermore, in the unoperated Ripk2104Asp joint, Mmp13 expression extends into deeper layers of cartilage, an expression domain normally only seen in WT joints following DMM surgery (brackets in C). (D) Col2 expression is reduced in Ripk2104Asp sham surgery mice relative to WT controls, and this loss is further exacerbated by DMM surgery (arrows). In regions with severe cartilage damage (arrowheads), pNF-κB, Mmp13, and Col2 expression is low in both WT and Ripk2104Asp DMM-operated joints. Images are of selected regions of the medial tibial condyle (see online supplemental figure 7). (E) Quantification of the number of Ripk2, pNF-κB, Mmp13, and Col2 positive chondrocytes in the medial knee joint of WT sham, Ripk2Asp104 sham, WT DMM, and Ripk2Asp104 DMM mice. n = 3 independent animals for each experimental condition. Error bars represent ±SD and statistically significant differences of P ≤ 0.01 (**), P ≤ 0.001 (***), and P ≤ 0.0001 (****) were determined by two-way ANOVA with Tukey‟s multiple comparisons test.
As Ripk2104Asp chondrocytes have elevated expression of proinflammatory genes, and as local inflammation can sensitize joints to PTOA42, knees were examined in situ for the expression of inflammatory markers. iNos is a major inflammatory mediator that is increased in OA. It is expressed in many tissues of the joint, including chondrocytes and macrophages43, both of which contribute directly to homeostatic maintenance of the joint44, 45. Whereas sham-operated WT mice have uniformly low levels of iNos expression throughout the joint, following DMM surgery iNos is highly induced in the joints, especially in the meniscus and osteophyte (figure 5A). In contrast, even sham-operated Ripk2104Asp mice exhibit a striking elevation of proinflammatory iNos signal in cartilage, meniscus, and synovial tissue (figure 5A). Following DMM surgery, elevated iNos expression is also evident in the cartilage, meniscus, and osteophytes (figure 5A and B).
Figure 5.
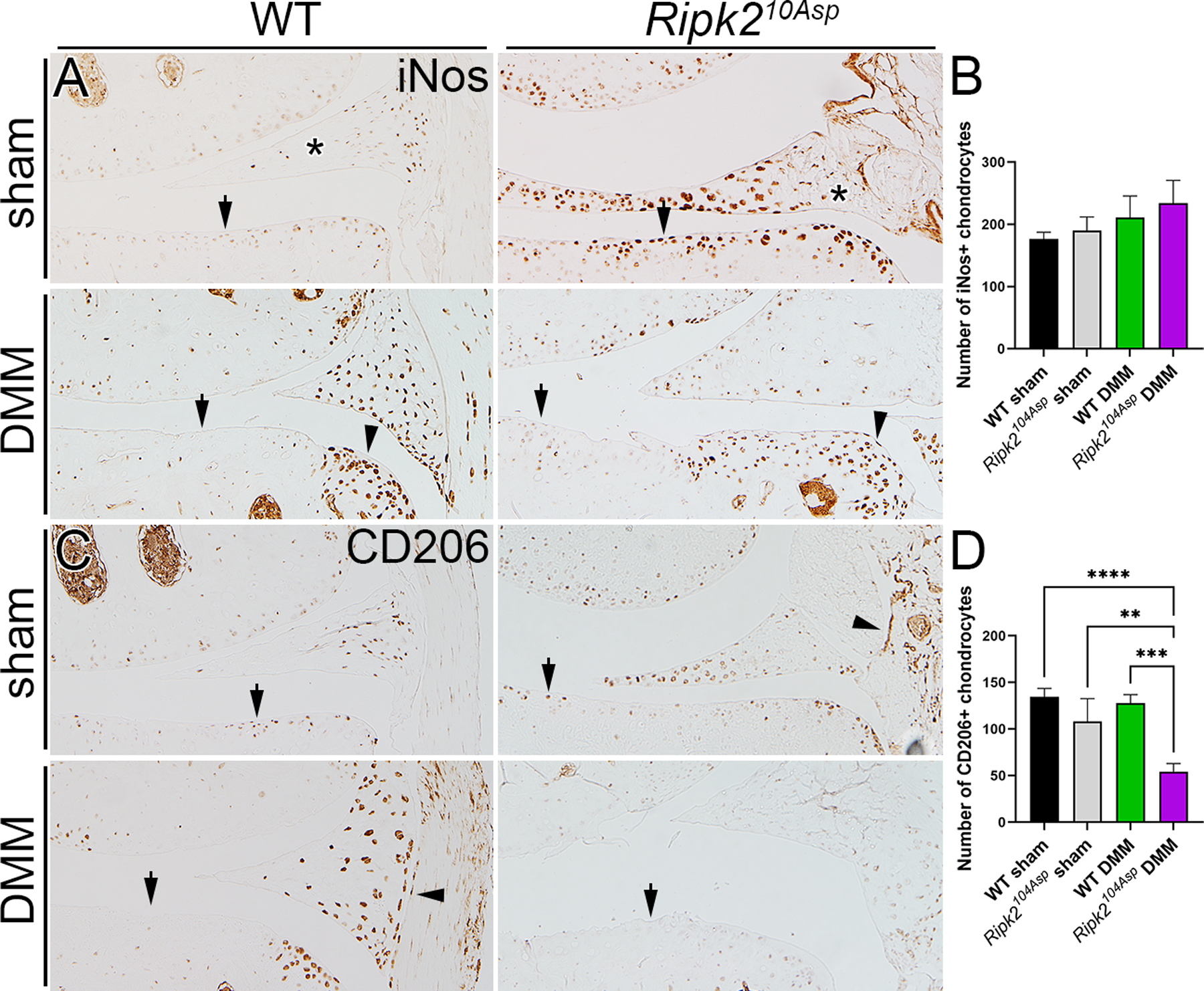
Ripk2104Asp joints have elevated expression of proinflammatory markers. (A) In WT mice, the proinflammatory marker iNos is normally expressed at low levels in the joint, and is markedly elevated following DMM surgery. In contrast, knee joints of Ripk2104Asp have chronically high levels of iNos expression, independent of injury. In A arrows indicate chondrocytes, asterisks mark the meniscus, and arrowheads indicate osteophytes in DMM-operated joints. (B) There is no difference in expression of the anti-inflammatory marker, CD206, between sham-operated surgery WT and Ripk2104Asp joints. Following DMM surgery, CD206 is prominently expressed in WT joints whereas it is almost absent in the joints of Ripk2104Asp mice. In B arrows indicate chondrocytes and arrowheads indicate synovium. All joints are 8 weeks post-surgery. C) Quantification of the number of iNos and CD206 positive chondrocytes in the medial knee joint of WT sham, Ripk2Asp104 sham, WT DMM, and Ripk2Asp104 DMM mice. n = 3 independent animals for each experimental condition. Error bars represent ±SD and statistically significant differences of P ≤ 0.01 (**), P ≤ 0.001 (***), and P ≤ 0.0001 (****) were determined by two-way ANOVA with Tukey‟s multiple comparisons test.
In the normal response to inflammation, CD206+ macrophages accrue in joints and are involved in resolving inflammation and tissue repair. Despite elevated iNos expression in the knees of sham-operated Ripk2104Asp mice, they did not have increased numbers of CD206+ cells relative to WT joints (figure 5C and D). Moreover, Ripk2104Asp mice had a clear deficit in the recruitment of anti-inflammatory CD206+ cells into the cartilage, meniscus, and synovium of operated joints as compared with joints of WT mice (figure 5C and D). In sum, prior to overt injury, knee joints of Ripk2104Asp mice exhibit higher than normal levels of inflammatory gene expression; following injury, Ripk2104Asp mice are relatively poor at recruiting factors to resolve acute inflammation.
The Ripk2104Asp mice do not exhibit an elevated inflammatory phenotype
The NOD/RIPK2 signaling pathway operates broadly and the heightened expression of OA-associated markers in the knee joints of Ripk2104Asp mice may reflect widespread elevation of inflammation. We examined the ability of cultured bone marrow-derived macrophages (BMM) from WT and Ripk2104Asp mice to respond to MDP. In contrast, to the effects of the Ripk2104Asp allele on the expression profile of chondrocytes, relatively few genes were differentially expressed upon stimulation of the BMM cells (online supplemental figure 8). To assess systemic differences in the inflammatory states of the mice, serum cytokine levels pre- or post-DMM surgery were measured. There was no difference in the serum concentration of any of 13 inflammatory cytokines sampled from 16-week old pre-surgery WT and Ripk2104Asp mice (figure 6), indicating unoperated Ripk2104Asp mice do not have a measurably elevated systemic phenotype. In contrast, in response to localized joint injury, Ripk2104Asp mice mount a highly augmented systemic response. At 4 weeks following DMM surgery both WT and Ripk2104Asp responded with raised serum levels of the proinflammatory cytokines IL1β, INFβ, and the anti-inflammatory IL10, but the degree of increase and the levels of these cytokines were significantly elevated in the Ripk2104Asp mice (figure 6). The differences in serum cytokine response are transient, as they are resolved by 8 weeks post-surgery (figure 6).
Figure 6.

DMM surgery induces an acute systemic inflammatory response in Ripk2104Asp mice. Quantification of serum A) IL1β, B) IFNβ, and C) IL10 levels from 16-week-old WT and Ripk2104Asp mice just prior to DMM surgery (black diamonds and triangles) and at 4, 8, and 12 weeks post-surgery (magenta triangles and red circles). Error bars represent ±SD and statistically significant differences of P ≤ 0.01 (**) and P ≤ 0.001 (***) were determined by one-way ANOVA with Tukey‟s multiple comparisons test (4-week post-DMM group) and a two-tailed unpaired t-test (8 and 12 week groups).
DISCUSSION
Animals carrying the single amino acid change encoded by the Ripk2104Asp variant have a magnified response to joint injury that leads to a predisposition to develop OA. The allele creates a chronically hyperactive inflammatory state in the joint with early signs of defective joint maintenance, as evidenced by gene expression in chondrocytes isolated from young mice and altered expression of pNF-κB, iNos, Mmp13, and Col2 in mature animals. Nevertheless, the elevated activity of the NOD/RIPK2/NF-κB pathway caused by the variant allele has a very modest effect on tissue remodeling under normal laboratory conditions. The Ripk2104Asp allele does not alter which signaling pathways and genes are activated in response to joint injury. Rather, consistent with the elevated signaling activity of the variant protein11, it simply leads mice to mount an accelerated and elevated response to injury, characterized locally in the joint by amplified Ripk2 and pNF-κB expression, exaggerated changes in the expression of genes indicative of OA progression, including Mmp13 and Col2, and histologically recognizable deterioration. Both local as well as systemic inflammatory responses are amplified following acute injury, seen by the deficit of CD206+ cells in the joint, altered gene expression in BMM, and the transient rise in serum cytokines.
Multiple sources of inflammation have been proposed as potential drivers of OA, including mechanosensory signaling and responses of chondrocytes or other joint resident cells to damage-associated molecular patterns (DAMPs)4, 46–48. Our work helps identify which of the potential inflammatory signaling pathways functions to limit or promote the occurrence of OA and which cells are essential for promoting the inflammatory response that leads to the tissue remodeling seen in OA24. Here we demonstrate the NOD/RIPK2 is an important inflammatory pathway in the development of OA. Mice that constitutively express the Ripk2Asp104 allele exhibit a set of marked changes in their knee joints without chronic systemic inflammation, reflecting alteration in normal local homeostatic mechanisms in the joint. Although NOD/RIPK2 signaling is known to activate multiple downstream pathways (e.g., NF-κB, p38, etc.), we do not yet know which of the targets of the activated Ripk2Asp104 protein are particularly significant for OA susceptibility. Finally, even though our results suggest Ripk2Asp104 activity functions in the knee joint, we have not defined the specific cells and tissues of the joint that require Ripk2Asp104 activity for OA development. Conditional spatial and temporal activation of Ripk2Asp104 will allow us to test the necessity of the NOD/RIPK2 pathway in OA development.
Understanding the ligands that activate the NOD/RIPK2 pathway is important for determining risk factors. The NODs were initially described as activated by bacterial peptidoglycan fragments49, 50, yet there is increasing evidence they can be activated by non-pathogen associated molecules, including DAMPs51. One possible DAMP activator of NODs in the synovial joint is the pro-catabolic Fibronectin fragment (FN-f), which is produced by cleavage of the Fibronectin protein in response to injury52, 53. NOD1 and NOD2 expression is upregulated in human chondrocytes treated with the 29-kDa amino-terminal FN-f, and activation NF-κB pathway is dependent on NOD2/RIPK2 activity17. Thus, local hyperactivity of the NOD/RIPK2 pathway may augment the response to FN-f release after injury to the joint and lead to increased OA susceptibility. Interestingly, two of the OA-associated NOD variants we have identified affect the Leucine Rich Repeat (LRR) domain, which functions in autoinhibition of NOD-mediated signaling activity and ligand binding. In the absence of ligand, the LLR domain interacts with the NOD domain to keep NOD1/2 in an inactive state and prevent NOD:NOD protein interactions that promote signaling18, 33, 54. Thus, the NOD variant proteins may have heightened basal activity or may be hyperresponsive to ligand binding. Functional analyses of the NOD variants will allow us determine if the variants alter signaling activity and if the altered activity is dependent on FN-f or other ligands.
Our study is focused on the NOD/RIPK2 pathway in OA of the 1st MTP joint, hand, and shoulder. We selected these joints to minimize the phenotypic heterogeneity to increase our power of identifying causal variants in these families. We have excluded families with knee and hip OA from our study due to confounding factors often present in affected individuals (e.g., traumatic injury, ligament tear, developmental dysplasia of the hip, Perthes disease, and avascular necrosis of the hip). Given that we have identified NOD/RIPK2 pathway variants in multiple joints, we predict that alteration of the pathway is also a risk factor for knee and hip OA. It is possible the NOD/RIPK2 pathway is not a major risk factor for these joints as no NOD/RIPK2 pathway genes have been identified in GWAS studies to date55. Identification and analysis of knee and hip OA families free from confounding factors will allow us to test this hypothesis. Further, we only address the role of Ripk2104Asp in PTOA and do not test if the allele leads to increased OA onset or severity in aged animals. Given the Ripk2104Asp allele creates a chronically hyperactive inflammatory state in the joint, the most parsimonious hypothesis is that aged animals carrying the disease allele may have increased prevalence of OA. Alternatively, in the absence of traumatic injury to the joint, aged Ripk2104Asp mice may not have elevated OA susceptibility.
In sum, we propose modulation of the NOD/RIPK2 signaling pathway is a general vulnerability factor for OA. Supporting our hypothesis, we have found rare variants altering conserved positions in NOD/RIPK2 pathway proteins in 7 of the 151 (5%) families we have examined with non-syndromic OA. Consistent with a causal connection between pathway activity and OA, hyperactivity of the NOD/RIPK2 pathway is associated with several human inflammatory syndromes that include arthritis as a comorbidity22, 23, 56. Our data indicate that modification of the NOD/RIPK2 pathway can render multiple joints (both weight and non-weight bearing) susceptible to OA. While the initiating factor (injury, repetitive use, diabetes, obesity, etc.) and activating ligand may differ between joints and individuals, our work has shown that elevated NOD/RIPK2 signaling is a predictive indicator of susceptibility to OA. Further pursuit of this signaling pathway and the spatiotemporal requirement for its activity may lead to assays for detection of early stages of disease and have therapeutic potential.
Supplementary Material
Key messages.
What is already known about this subject?
A coding variant that elevates the proinflammatory activity of the RIPK2 signal transducer has been associated with familial early-onset OA, raising the possibility that perturbations of NOD/RIPK2 signaling may confer susceptibility to OA.
What does this study add?
We discover alleles affecting several components of the NOD/RIPK2 signaling pathway are associated with multiple forms of familial OA, supporting the novel hypothesis that the pathway is a common vulnerability factor for OA.
Introduction of the OA-associated hyperactive Ripk2104Asp allele into the mouse genome causes changes in the basal physiological status of the joint in ways that presage a definitive OA state.
Although Ripk2104Asp mice display no evidence of systemic inflammation or histological evidence of joint degeneration, they displayed significantly increased sensitivity to experimentally induced OA.
How might this impact on clinical practice or future developments?
By showing that altered NOD/RIPK2 signaling is predictive of susceptibility to multiple forms of OA, the work brings new focus to the functions of the signaling pathway in maintaining joint homeostasis, may guide development of assays to detect early stages of OA, and may indicate new therapeutic approaches to disease intervention.
Acknowledgments
We would like to thank Charles L. Saltzman, Jerry Kaplan, and the members of the Department of Orthopaedics Osteoarthritis Discovery and Treatment Initiative advisory board for their support and critical feedback. We thank the families that participated in this study. We would also like to thank the anonymous reviewers for their critique and suggestions to improve our manuscript.
Funding
This work was funded by the Skaggs Foundation for Research (MJJ and NHK), the Utah Genome Project (MJJ, NHK, DJG) the Arthritis National Research Foundation (MJJ – 707634), and the National Institute on Aging (MJJ and DJG – R21AG063534–01A1).
Footnotes
Competing Interests
None.
Data Availability Statement
Data are available upon request.
Patient and Public Involvement
None.
Research Ethics Approval
The Institutional Review Boards of the University of Utah (IRB # 79442) and Intermountain Healthcare (IRB # 1050554), and the Resource for Genetic and Epidemiologic Research approved this study.
REFERENCES
- 1.Aury-Landas J, Marcelli C, Leclercq S, Boumediene K, Bauge C. Genetic Determinism of Primary Early-Onset Osteoarthritis. Trends in molecular medicine 2016. Jan; 22(1):38–52. [DOI] [PubMed] [Google Scholar]
- 2.Cibrian Uhalte E, Wilkinson JM, Southam L, Zeggini E. Pathways to understanding the genomic aetiology of osteoarthritis. Hum Mol Genet 2017. Oct 01; 26(R2):R193–R201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ramos YF, Meulenbelt I. Implementation of Functional Genomics for Bench-to-Bedside Transition in Osteoarthritis. Curr Rheumatol Rep 2015. Aug; 17(8):53. [DOI] [PubMed] [Google Scholar]
- 4.Vincent TL. Of mice and men: converging on a common molecular understanding of osteoarthritis. Lancet Rheumatol 2020. Oct; 2(10):e633–e645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Freund MK, Burch KS, Shi H, Mancuso N, Kichaev G, Garske KM, et al. Phenotype-Specific Enrichment of Mendelian Disorder Genes near GWAS Regions across 62 Complex Traits. Am J Hum Genet 2018. Oct 4; 103(4):535–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Claussnitzer M, Cho JH, Collins R, Cox NJ, Dermitzakis ET, Hurles ME, et al. A brief history of human disease genetics. Nature 2020. Jan; 577(7789):179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun BB, Kurki MI, Foley CN, Mechakra A, Chen CY, Marshall E, et al. Genetic associations of protein-coding variants in human disease. Nature 2022. Feb 23. [DOI] [PMC free article] [PubMed]
- 8.Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell 1991. Aug 09; 66(3):589–600. [DOI] [PubMed] [Google Scholar]
- 9.Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, et al. APC mutations occur early during colorectal tumorigenesis. Nature 1992. Sep 17; 359(6392):235–237. [DOI] [PubMed] [Google Scholar]
- 10.Ji W, Foo JN, O’Roak BJ, Zhao H, Larson MG, Simon DB, et al. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat Genet 2008. May; 40(5):592–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jurynec MJ, Sawitzke AD, Beals TC, Redd MJ, Stevens J, Otterud B, et al. A hyperactivating proinflammatory RIPK2 allele associated with early-onset osteoarthritis. Hum Mol Genet 2018. Jul 1; 27(13):2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ramos YF, Bos SD, van der Breggen R, Kloppenburg M, Ye K, Lameijer EW, et al. A gain of function mutation in TNFRSF11B encoding osteoprotegerin causes osteoarthritis with chondrocalcinosis. Ann Rheum Dis 2015. Sep; 74(9):1756–1762. [DOI] [PubMed] [Google Scholar]
- 13.Ruault V, Yauy K, Fabre A, Fradin M, Van-Gils J, Angelini C, et al. Clinical and Molecular Spectrum of Nonsyndromic Early-Onset Osteoarthritis. Arthritis Rheumatol 2020. Jun 8. [DOI] [PubMed]
- 14.Sliz E, Taipale M, Welling M, Skarp S, Alaraudanjoki V, Ignatius J, et al. TUFT1, a novel candidate gene for metatarsophalangeal osteoarthritis, plays a role in chondrogenesis on a calcium-related pathway. PloS one 2017; 12(4):e0175474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kazmers NH, Meeks HD, Novak KA, Yu Z, Fulde GL, Thomas JL, et al. Familial clustering of erosive hand osteoarthritis in a large statewide cohort. Arthritis Rheumatol 2020. Sep 17. [DOI] [PMC free article] [PubMed]
- 16.Philpott DJ, Sorbara MT, Robertson SJ, Croitoru K, Girardin SE. NOD proteins: regulators of inflammation in health and disease. Nat Rev Immunol 2014. Jan; 14(1):9–23. [DOI] [PubMed] [Google Scholar]
- 17.Hwang HS, Lee MH, Choi MH, Kim HA. NOD2 signaling pathway is involved in fibronectin fragment-induced pro-catabolic factor expressions in human articular chondrocytes. BMB Rep 2019. Feb 14. [DOI] [PMC free article] [PubMed]
- 18.Caruso R, Warner N, Inohara N, Nunez G. NOD1 and NOD2: signaling, host defense, and inflammatory disease. Immunity 2014. Dec 18; 41(6):898–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goncharov T, Hedayati S, Mulvihill MM, Izrael-Tomasevic A, Zobel K, Jeet S, et al. Disruption of XIAP-RIP2 Association Blocks NOD2-Mediated Inflammatory Signaling. Molecular cell 2018. Feb 15; 69(4):551–565 e557. [DOI] [PubMed] [Google Scholar]
- 20.Jeong YJ, Kang MJ, Lee SJ, Kim CH, Kim JC, Kim TH, et al. Nod2 and Rip2 contribute to innate immune responses in mouse neutrophils. Immunology 2014. Oct; 143(2):269–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jun JC, Cominelli F, Abbott DW. RIP2 activity in inflammatory disease and implications for novel therapeutics. Journal of leukocyte biology 2013. Nov; 94(5):927–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takeuchi M, Mizuki N, Meguro A, Ombrello MJ, Kirino Y, Satorius C, et al. Dense genotyping of immune-related loci implicates host responses to microbial exposure in Behcet’s disease susceptibility. Nat Genet 2017. Mar; 49(3):438–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou Q, Wang H, Schwartz DM, Stoffels M, Park YH, Zhang Y, et al. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat Genet 2016. Jan; 48(1):67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Griffin TM, Lories RJ. Cracking the code on the innate immune program in OA. Osteoarthritis and Cartilage 2020; 28(5):529–531. [DOI] [PubMed] [Google Scholar]
- 25.Kazmers NH, Meeks HD, Novak KA, Yu Z, Fulde GL, Thomas JL, et al. Familial Clustering of Erosive Hand Osteoarthritis in a Large Statewide Cohort. Arthritis Rheumatol 2021. Mar; 73(3):440–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kazmers NH, Yu Z, Barker T, Abraham T, Romero R, Jurynec MJ. Evaluation for Kienbock Disease Familial Clustering: A Population-Based Cohort Study. J Hand Surg Am 2020. Jan; 45(1):1–8 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaufmann RA, Logters TT, Verbruggen G, Windolf J, Goitz RJ. Osteoarthritis of the distal interphalangeal joint. J Hand Surg Am 2010. Dec; 35(12):2117–2125. [DOI] [PubMed] [Google Scholar]
- 28.Ibounig T, Simons T, Launonen A, Paavola M. Glenohumeral Osteoarthritis: An Overview of Etiology and Diagnostics. Scand J Surg 2020. Jul 14:1457496920935018. [DOI] [PubMed] [Google Scholar]
- 29.Coughlin MJ, Shurnas PS. Hallux rigidus: demographics, etiology, and radiographic assessment. Foot Ankle Int 2003. Oct; 24(10):731–743. [DOI] [PubMed] [Google Scholar]
- 30.Shurnas PS. Hallux rigidus: etiology, biomechanics, and nonoperative treatment. Foot Ankle Clin 2009. Mar; 14(1):1–8. [DOI] [PubMed] [Google Scholar]
- 31.Hu H, Roach JC, Coon H, Guthery SL, Voelkerding KV, Margraf RL, et al. A unified test of linkage analysis and rare-variant association for analysis of pedigree sequence data. Nat Biotechnol 2014. Jul; 32(7):663–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Singleton MV, Guthery SL, Voelkerding KV, Chen K, Kennedy B, Margraf RL, et al. Phevor combines multiple biomedical ontologies for accurate identification of disease-causing alleles in single individuals and small nuclear families. Am J Hum Genet 2014. Apr 03; 94(4):599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maekawa S, Ohto U, Shibata T, Miyake K, Shimizu T. Crystal structure of NOD2 and its implications in human disease. Nat Commun 2016. Jun 10; 7:11813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Larabi A, Barnich N, Nguyen HTT. New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy 2020. Jan; 16(1):38–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moreira LO, Zamboni DS. NOD1 and NOD2 Signaling in Infection and Inflammation. Front Immunol 2012; 3:328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Negroni A, Pierdomenico M, Cucchiara S, Stronati L. NOD2 and inflammation: current insights. J Inflamm Res 2018; 11:49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rothwarf DM, Zandi E, Natoli G, Karin M. IKK-gamma is an essential regulatory subunit of the IkappaB kinase complex. Nature 1998. Sep 17; 395(6699):297–300. [DOI] [PubMed] [Google Scholar]
- 38.Jonason JH, Hoak D, O’Keefe RJ. Primary murine growth plate and articular chondrocyte isolation and cell culture. Methods Mol Biol 2015; 1226:11–18. [DOI] [PubMed] [Google Scholar]
- 39.Nachbur U, Stafford CA, Bankovacki A, Zhan Y, Lindqvist LM, Fiil BK, et al. A RIPK2 inhibitor delays NOD signalling events yet prevents inflammatory cytokine production. Nat Commun 2015. Mar 17; 6:6442. [DOI] [PubMed] [Google Scholar]
- 40.Glasson SS, Blanchet TJ, Morris EA. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthritis Cartilage 2007. Sep; 15(9):1061–1069. [DOI] [PubMed] [Google Scholar]
- 41.Christiansen BA, Anderson MJ, Lee CA, Williams JC, Yik JH, Haudenschild DR. Musculoskeletal changes following non-invasive knee injury using a novel mouse model of post-traumatic osteoarthritis. Osteoarthritis Cartilage 2012. Jul; 20(7):773–782. [DOI] [PubMed] [Google Scholar]
- 42.Mendez ME, Sebastian A, Murugesh DK, Hum NR, McCool JL, Hsia AW, et al. LPS-Induced Inflammation Prior to Injury Exacerbates the Development of Post-Traumatic Osteoarthritis in Mice. J Bone Miner Res 2020. Nov; 35(11):2229–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ahmad N, Ansari MY, Haqqi TM. Role of iNOS in osteoarthritis: Pathological and therapeutic aspects. J Cell Physiol 2020. Oct; 235(10):6366–6376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Griffin TM, Scanzello CR. Innate inflammation and synovial macrophages in osteoarthritis pathophysiology. Clin Exp Rheumatol 2019. Sep-Oct; 37 Suppl 120(5):57–63. [PMC free article] [PubMed] [Google Scholar]
- 45.Takahata Y, Murakami T, Hata K, Nishimura R. Molecular Mechanisms Involved in the Progression and Protection of Osteoarthritis. Curr Mol Pharmacol 2021; 14(2):165–169. [DOI] [PubMed] [Google Scholar]
- 46.Houard X, Goldring MB, Berenbaum F. Homeostatic mechanisms in articular cartilage and role of inflammation in osteoarthritis. Curr Rheumatol Rep 2013. Nov; 15(11):375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Loeser RF, Goldring SR, Scanzello CR, Goldring MB. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum 2012. Jun; 64(6):1697–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van den Bosch MHJ, van Lent PLEM, van der Kraan PM. Identifying effector molecules, cells, and cytokines of innate immunity in OA. Osteoarthritis and Cartilage 2020; 28(5):532–543. [DOI] [PubMed] [Google Scholar]
- 49.Girardin SE, Boneca IG, Carneiro LA, Antignac A, Jehanno M, Viala J, et al. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science 2003. Jun 6; 300(5625):1584–1587. [DOI] [PubMed] [Google Scholar]
- 50.Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem 2003. Mar 14; 278(11):8869–8872. [DOI] [PubMed] [Google Scholar]
- 51.Kuss-Duerkop SK, Keestra-Gounder AM. NOD1 and NOD2 Activation by Diverse Stimuli: a Possible Role for Sensing Pathogen-Induced Endoplasmic Reticulum Stress. Infect Immun 2020. Jun 22; 88(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ding L, Guo D, Homandberg GA, Buckwalter JA, Martin JA. A single blunt impact on cartilage promotes fibronectin fragmentation and upregulates cartilage degrading stromelysin-1/matrix metalloproteinase-3 in a bovine ex vivo model. J Orthop Res 2014. Jun; 32(6):811–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Homandberg GA, Meyers R, Xie DL. Fibronectin fragments cause chondrolysis of bovine articular cartilage slices in culture. J Biol Chem 1992. Feb 25; 267(6):3597–3604. [PubMed] [Google Scholar]
- 54.Boyle JP, Parkhouse R, Monie TP. Insights into the molecular basis of the NOD2 signalling pathway. Open biology 2014. Dec; 4(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boer CG, Hatzikotoulas K, Southam L, Stefansdottir L, Zhang Y, Coutinho de Almeida R, et al. Deciphering osteoarthritis genetics across 826,690 individuals from 9 populations. Cell 2021. Sep 2; 184(18):4784–4818 e4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kanazawa N, Okafuji I, Kambe N, Nishikomori R, Nakata-Hizume M, Nagai S, et al. Early-onset sarcoidosis and CARD15 mutations with constitutive nuclear factor-kappaB activation: common genetic etiology with Blau syndrome. Blood 2005. Feb 1; 105(3):1195–1197. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.