

Abstract
Recent advances in the manufacturing, modification, purification, and cellular delivery of ribonucleic acid (RNA) have enabled the development of RNA-based therapeutics for a broad array of applications. The approval of two SARS-CoV-2–targeting mRNA-based vaccines has highlighted the advances of this technology. Offering rapid and straightforward manufacturing, clinical safety, and versatility, this paves the way for RNA therapeutics to expand into cancer immunotherapy. Together with ongoing trials on RNA cancer vaccination and cellular therapy, RNA therapeutics could be introduced into clinical practice, possibly stewarding future personalized approaches. In the present review, we discuss recent advances in RNA-based immuno-oncology together with an update on ongoing clinical applications and their current challenges.
Introduction
RNA has become a widely popular tool for vaccination against infectious diseases following the SARS-CoV2 pandemic (1). Potentially, this molecule can be exploited for many more applications, especially in cancer immunotherapy, offering a broad range of RNA-based therapeutic strategies (2, 3). Since its discovery in 1961 by Brenner and colleagues, RNA has evolved from what was initially considered a mere intermediary between DNA and protein to a versatile molecule operating at multiple cellular levels, exploitable for immunotherapeutic applications (4). In vitro transcribed messenger RNA (iVT-mRNA), self-amplifying RNA (SAM), antisense oligonucleotides (ASO), aptamers, small interfering RNA (siRNA), and microRNA (miRNA) are the most studied RNA formats at present, which can be categorized into two main groups: (i) coding RNA (cRNA) translating into protein, including mRNA and SAM, and (ii) noncoding RNA (ncRNA) that does not translate into proteins but rather regulates cell physiology and functions, including among others ASOs, aptamers, siRNA, and miRNA. Based on their role related to cancer biology, ncRNA can function as oncogenes or tumor suppressor genes (Fig. 1; ref. 5) and therefore be implemented for therapeutic use. Initially, RNA was not considered a suitable therapeutic tool due to its unstable nature and susceptibility to rapid degradation by ubiquitous ribonucleases (RNases). Other concerns regarding potential toxicity, unspecific immune activation, and unknown effectiveness also needed further investigation (6). At present, many limitations have been overtaken, including chemical modification of the RNA structure, paralleled with the development of novel technologies for RNA delivery and protection. This allows fast, cost-effective, and versatile generation of mRNA suitable for clinical application in the context of cancer and infectious diseases (4, 7), introducing RNA as a promising pharmaceutical product (Table 1; refs. 8–12).
Figure 1.

Overview of coding and noncoding RNA structures. Left, in vitro transcription (iVT) of messenger RNA (mRNA). mRNA has several conserved features, including a 5′ cap structure, two extended untranslated regions (UTR) at the 5′ and 3′ end of the ORF, and a 3′ poly-A tail. The final iVT product can be mRNA or self-amplifying mRNA. Right, solid-phase synthesis of noncoding and antisense oligonucleotides. Abbreviations: ASO, antisense oligonucleotides; mRNA, messenger RNA; UTR, untranslated region; rNTP, ribonucleoside triphosphates; miRNA, microRNA; siRNA, small interfering RNA. Adapted from an image created with BioRender.com.
Table 1.
Advantages and disadvantages of RNA- compared with DNA- and protein-based therapeutics (data from references 8–12).
| RNA | DNA | Protein | |
|---|---|---|---|
| Synthesis | + | + | − |
| Manufacture timea | +/− | +/− | − |
| Genome integration | − | +/− | − |
| Self-adjuvancy | +/− | +/− | − |
| Storageb | +/− | +/− | + |
| Administration | + | + | − |
aRNA manufacture time is fast but depends on a DNA template.
bStorage requirements depend on the formulation used for the therapeutic product.
In vitro transcribed mRNA and self-amplifying RNA
Mature eukaryotic mRNA consists of highly conserved molecular features, including a cap structure at the 5′ terminus, two extended untranslated regions (UTR) at the 5′ and 3′ end of the open reading frame (ORF), and a poly-A tail at the 3′ terminus (13), all influencing mRNA degradation, stability, immunogenicity, and translatability (14). For the generation of iVT-mRNA, a DNA template is required, commonly derived from a linearized plasmid (15), from which mRNA is transcribed using RNA polymerase enzymes such as T7, SP6, or T3 (16). Other main components of the iVT reaction mix comprise a RNase inhibitor, pyrophosphatases, and a reaction buffer including the four ribonucleoside triphosphates (rNTP) and a capping reagent (ARCA, Clean CapTM). The final concentration of each component, the reaction temperature, and time will determine the final mRNA yield obtained (Fig. 1; refs. 16, 17).
After the iVT reaction, the DNA template, all enzymes used, organic and inorganic contaminants and secondary transcription products, such as incomplete/truncated RNA molecules and double-stranded RNA (dsRNA) have to be removed (18). A DNase digestion is performed to eliminate the DNA template, followed by a salt and ethanol precipitation or more accurately by high-pressure/performance liquid chromatography (HPLC) to obtain pure mRNA (19). The purification process reduces immune activation triggered by contaminants and therefore can improve mRNA translation upon delivery into the cell (19). In addition to manufacturing and purification, thorough quality controls are performed to assess the final RNA integrity, purity, concentration, capping efficiency, sequence, and poly-A tail length (20).
Poor mRNA uptake and in vivo expression are often observed due to extracellular RNase activity (15) and intrinsic mRNA immunogenicity (21), triggering interferon (IFN) pathway activation (7). Packaging mRNA into lipid nanoparticles (LNP) has proven an effective solution to ensure protection and successful RNA delivery into the cell (22). A multitude of nanoparticles have been tested and extensively reviewed by others (23–25). At present, the LNP-mRNA COVID-19 vaccines Tozinameran and Elasomeran, with LNPs composed of ionizable lipids, phospholipids, cholesterol, and PEGylated lipids, have been clinically approved (26). Moreover, RNA chemical modifications such as N1-methyladenosine, 5-methylcytidine, pseudouridine, N6-methyladenosine, 5-methyl deoxycytidine, and inosine pioneered by Kariko and colleagues (27–30) have been exploited to generate stable, nonimmunogenic, and high-translatable RNA molecules, including also modified base analogs as 5`cap structures (31, 32). Other work on the optimization of the poly-A tail length showed enhanced mRNA stability and lowered immunogenic profile (33). Additional optimization of UTRs and codon optimization paired with sequence design of the encoding sequence (34) aimed to improve RNA lifetime stability and expression (35–37).
Noncoding and antisense oligonucleotides
Solid-phase synthesis is mostly used for manufacturing (antisense) oligonucleotides. The first nucleotide of the desired molecule is attached to a solid phase and elongated using a repetition of a four-step cycle, adding one nucleotide per cycle until the oligonucleotide is fully synthesized. The four-step cycle includes detritylation/deblocking, coupling, capping, and oxidation or thiolation. The synthesis is performed in a column reactor under programmed delivery of all reagents (Fig. 1). A 70% to 80% success synthesis rate is achieved of the desired oligonucleotide length, requiring a final HPLC purification step, removing all polymers showing incorrect length (n ± x; ref. 4). Chemical modifications were implemented to this process, aiming for improved pharmacokinetics (38). The most used modifications include a substitution of the phosphodiester bond with a phosphorothioate, proving to increase resistance toward nuclease degradation and reduce binding to plasma proteins, and decreasing renal clearance (39). However, reduced target affinity is also observed, and therefore 2′-methoxyethyl, 2′-deoxy-2′-fluoro, and 2′-O-methyl modifications were introduced on the ribose of the RNA to improve specificity (20).
A similar manufacturing approach is exploited for the synthesis of RNA aptamers. A widely used method to design aptamers is by Systematic Evolution of Ligands by Exponential enrichment (SELEX). In short, a high diversity library of single-stranded RNA (ssRNA) is synthetized by in vitro transcription (40). From the RNA library, a selective target binding ssRNA is isolated through repeated rounds of exposure, binding, selection, and amplification. Once the desired sequence/design of the RNA aptamers is obtained, the aptamers are manufactured using solid-phase synthesis (41).
RNA as a Vaccination Strategy
Therapeutic cancer vaccines aim to generate antigen-specific T-cell responses targeting tumor cells and potentially achieve long-term clinical benefits (42, 43). The approval of two mRNA vaccines for COVID-19 prevention has highlighted the potential of mRNA technology (44). Two main RNA-based approaches have been extensively explored for cancer vaccination: ex vivo mRNA-loaded dendritic cell (DC) vaccines (45, 46) and mRNA-LNP vaccines (ref. 47; Fig. 2). In both strategies, mRNA is used to deliver the tumor-associated antigen (TAA) or tumor-specific antigen to elicit an antitumor immune response. Upon cellular entry followed by translation of the mRNA, the proteasome processes the mRNA-encoded protein into peptides that ultimately will be processed and presented by human leucocyte antigen (HLA) class I molecules to CD8+ T cells (48). CD4+ T-cell stimulation is also recommended to support the CD8+ T-cell response (49). In this regard, coupling an HLA class II sorting signal, such as the signal sequence of the invariant chain, lysosomal-associated membrane protein (LAMP), or DC-LAMP, to the antigen sequence is required to ensure that antigen-derived peptides enter the HLA class II presentation pathway, despite the protein being synthesized in the cytosol of the cell (50, 51).
Figure 2.

Overview of active mRNA-based immunotherapeutic strategies. Left, LNP-antigen mRNA vaccine delivered systemic or locally, followed by antigen expression resulting in T-cell priming and eventually cancer cell killing. Right, monocytes or hematopoietic progenitor cells are isolated from blood, further cultured, and differentiated into DCs. mRNA is then used to load the DCs ex vivo with tumor antigens. The modified DCs are administered to patients, where they will prime T cells, eventually resulting in the killing of cancer cells. Adapted from an image created with BioRender.com.
Cancer vaccines, mostly targeting cancer–testis and differentiation antigens, as monotherapy, have not shown significant activity thus far. Most clinical trials have been ineffective, and the induced immune reactivity was insufficient, limited in time, and narrow. The lack of clinical efficacy from vaccine treatment alone can be attributed to weak antigen delivery modalities that induced low T-cell titers as well as immune checkpoints remaining intact, which ultimately prevented tumor cell killing. It is now thought that neoantigens generated by somatic alterations could be differentially recognized by the immune system as these proteins/peptides would be unique to the tumor and the T-cell repertoire recognizing such neoantigens would not have been subjected to central tolerance mechanisms, as is the case for cancer–testis- and differentiation antigen-specific T cells (8, 52).
mRNA-based dendritic cell vaccines
mRNA-based DC cancer vaccination was introduced more than two decades ago (53). At present, more than 30 clinical trials have been published, recently reviewed by Dorrie and colleagues (46). For DC generation, monocytes or hematopoietic progenitor cells are isolated from blood and further cultured and differentiated into DCs (54). mRNA is then used to load the DCs ex vivo with tumor antigens. The modified DCs are administered to patients (Fig. 2) via intravenous, intradermal, subcutaneous, or intranodal injections (55). DC maturation is most often induced using a cytokine cocktail (56), feasible with both protein and mRNA delivery (57). Examples of mRNA-induced functional manipulation of DCs include TriMix mRNA, a mixture consisting of CD40 Ligand (CD40L), CD70, and constitutively active Toll-like receptor 4 (TLR4) encoding mRNA. Several studies showed that TriMix-DC vaccination induces robust, TAA-specific T-cell responses in the majority of analyzed patients (58–60). Other strategies to improve the activation of T cells by mRNA-modified DC have been studied and show promising results (61–65).
DC-based vaccines have shown to induce adaptive immune responses, and RNA transfection is emerging as an ideal method for antigen-loading and functional manipulation of the applied cells. DC vaccination rarely produces adverse events and has a highly safe profile (46). However, challenges that call for imperative improvements such as the manufacturing process, the optimal choice of DC subset or their in vitro generation, the antigen choice, the route of administration, and vaccination schedule still need to be addressed (66).
mRNA-based vaccines
First clinical results were observed with intradermal or intranodal naked mRNA administration, resulting in mRNA uptake by antigen-presenting cells in the dermis or lymph nodes followed by antigen presentation and T-cell stimulation (67). Even though it has a positive clinical outcome and favorable safety profile, the many limitations, such as limited mRNA uptake, resulting in low bioavailability of antigens and lowered immune responses, hampered its implementation (15).
Recent attempts opted for intramuscular or intravenous administration of mRNA encapsulated by delivery carriers (24). Four recent clinical trials, using mRNA packaged in LNPs, have shown promising clinical and immunologic results in patients with solid tumors. In the Lipo-Merit trial (NCT02410733; Table 2), a vaccine consisting of nonmodified lipoplexed mRNA targeting a variety of TAAs (NY-ESO-1, MAGE-A3, tyrosinase, and TPTE) was administered intravenously to advanced melanoma patients. The adverse events (pyrexia, chills, and flu-like symptoms) were mild to moderate and transient. Expansion and activation of antigen-specific T cells with cytolytic activity against tumor cells could be documented. Continuous vaccination resulted in the persistence of antigen-specific memory T cells. Encouraging clinical responses have been reported (68, 69).
Table 2.
Overview of active and recruiting clinical trials using mRNA-based therapeutics.
| IT type | Cancer | Study | Phase | Target | Formulation | Combination |
|---|---|---|---|---|---|---|
| CAR | Solid tumors | NCT04981691 | Phase I | MESO | EP autologous T cells | NA |
| Vaccine | NSCLC, CRC, PDAC | NCT03948763 | Phase I | KRAS | V941 mRNA | Pembrolizumab |
| siRNA (CAS3/SS3) | B-cell NHL | NCT04995536 | Phase I | TLR9, STAT3 | siRNA linked to CpG oligonucleotide | Radiotherapy |
| Synthetic naked mRNA | BC | NCT03788083 | Phase I | TriMix | NA | NA |
| Vaccine | OC | NCT04163094 | Phase I | 3 OC TAA | LNP | Carboplatin/paclitaxel |
| Vaccine | Esophageal, NSCLC | NCT03908671 | Pilot study | Neoantigen | Undisclosed | NA |
| Vaccine | Melanoma | NCT03897881 | Phase II | Neoantigen | LNP | Pembrolizumab |
| Vaccine | Solid tumors | NCT03313778 | Phase I | Neoantigen | LNP | Pembrolizumab |
| Vaccine | Melanoma | NCT03815058 | Phase II | Neoantigen | RNA-LPX | Pembrolizumab |
| Vaccine | Melanoma | NCT02410733 | Phase I | 4 TAA | RNA-LPX | NA |
| Vaccine | Prostate | NCT04382898 | Phase I/II | 5 PC TAA | LNP | Cemiplimab |
| Vaccine | Esophageal, gastric cancer, CRC, PC | NCT03468244 | Pilot study | Neoantigen | Undisclosed | NA |
| Vaccine | GBM | NCT03688178 | Phase II | CMV pp65 | mRNA-Loaded autologous DC | Varlilumab |
| Vaccine | GBM | NCT02649582 | Phase I/II | WT-1 | mRNA-Loaded autologous DC | Temozolomide |
| Vaccine | NSCLC | NCT03164772 | Phase I/II | 6 NSCLC TAA | BI 1361849 | Durvalumab, tremelimumab |
| Vaccine | Melanoma | NCT01456104 | Phase I | Melanoma TAA | Autologous LC EP with TAA mRNA | NA |
| Vaccine | GBM | NCT03927222 | Phase II | CMV | Pp65-LAMP mRNA-loaded autologous DC | Temozolomide, GM-CSF Tetanus-diphteria, toxoid |
| Vaccine | GMB | NCT02465268 | Phase II | CMV | Pp65-shLAMP mRNA-loaded DC | GM-CSF |
| Vaccine | Brain metastasis | NCT02808416 | Phase II | Neoantigen | mRNA tumor antigen pulsed DC | NA |
| Vaccine | GBM | NCT00639639 | Phase I | CMV | Pp65-LAMP mRNA-loaded autologous DC | Tetanus toxoid |
| Vaccine | PC | NCT01197625 | Phase I/II | Tumor antigen | Tumor mRNA-loaded DC | hTERT Survivin |
| Vaccine | AML | NCT01686334 | Phase I/II | WT1 antigen | mRNA EP autologous DC | (Potentially) low-dose chemotherapy |
| Vaccine | MM | NCT01995708 | Phase I | CT7, MAGE-A3, WT1 | mRNA EP autologous LC | Standard of care |
| Vaccine | AML, HRMS | NCT03083054 | Phase I/II | WT1 antigen | mRNA EP autologous DC | NA |
| Vaccine | NSCLC, GEA, mUC, MSS-CRC | NCT03639714 | Phase I/II | Neoantigen | SAM | ChAd, nivolumab, ipilimumab |
| Vaccine | MSS-CRC, NSCLC, PDAC | NCT03953235 | Phase I/II | Shared neoantigen Kras | SAM | ChAd, nivolumab, ipilimumab |
| Vaccine | CRC | NCT05141721 | Phase II/III | Neoantigen | SAM | ChAd, standard of care atezolizumab, ipilimumab |
| RNAi | PDAC | NCT01676259 | Phase II | KRAS | siG12D-LODER | Gemcitabine, paclitaxel, FOLFIRINOX |
| RNAi | Advanced malignant solid neoplasms | NCT01591356 | Phase I | EPHA2 | DOPC encapsulation | NA |
| Vaccine | Melanoma | NCT02410733 | Phase I | NYESO-1, MAGE A3 tyrosinase, TPTE | Lipo-MERIT | NA |
| Vaccine | Melanoma | NCT03815058 | Phase II | Neoantigen | Lipoplex | Pembrolizumab |
| mAB | Solid tumors | NCT04683939 | Phase I/II | CLDN18.2 | Undisclosed | Paclitaxel, gemcitabine |
| Immune inducers | Solid tumors, lymphoma | NCT03739931 | Phase I/II | NA | OX40L, IL23, IL36y coding mRNA | Durvalumab |
Abbreviations: BC, breast cancer; ChAd, chimpanzee adenovirus; CMV, cytomegalovirus; EP, electroporated; GBM, glioblastoma multiforme; GEA, gastroesophageal adenocarcinoma; HRMS, high-risk myelodysplastic syndrome; IT, immunotherapy; LCs, Langerhans cells; AML, acute myeloid leukemia; MESO, mesothelin; MSS-CRC, microsatellite stable colorectal cancer; mUC, metastatic urothelial carcinoma; NA, not applicable; NHL, non-Hodgkin lymphoma; NSCLC, non–small cell lung carcinoma; OC, ovarian cancer; PC, prostate cancer; PDAC, pancreatic ductal adenocarcinoma; pHGG, pediatric high-grade glioma; STAT3, signal transducer and activator of transcription 3; TAA, tumor-associated antigens; TLR9, Toll-like receptor 3.
In the R07198457-trial (NCT03815058; Table 2), the administration of nonmodified mRNA encoding up to 20 patient-specific neoantigens has been studied as monotherapy and in combination with a PD-L1 inhibitor, in patients with advanced solid tumors (breast cancer, prostate cancer, ovarian cancer, melanoma, non–small cell lung cancer, bladder cancer, and colorectal cancer; refs. 70, 71). Also, in this trial, the adverse events were mostly of grade 1–2 and transient. Neoantigen-specific T-cell responses were observed in most of the patients. Promising clinical results in these often heavily pretreated patients were noted.
In both the KEYNOTE-603 trial (NCT03313778; Table 2) and the KEYNOTE-942 trial (NCT03897881; Table 2; Moderna and Merck), the safety and immunogenicity of intramuscularly administered lipid-protected modified mRNA encoding neoantigens were evaluated, either as monotherapy or in combination with anti–PD-1 monoclonal antibodies (mAb, pembrolizumab) in patients with solid tumors (72–74). This mRNA-based personalized cancer vaccine has an acceptable safety profile along with observed clinical responses in combination with pembrolizumab. Preliminary efficacy analysis from checkpoint inhibition-naïve relapsed/refractory human papillomavirus (HPV) negative head and neck squamous cell carcinoma (cohort suggests activity of this combination (73).
An alternative mRNA-based approach uses SAM, originating from positive ssRNA alphaviruses, consisting of the RNA replication machinery of the alphavirus (self-assembly genes; Fig. 1) and replacing other genetic regions with the gene sequence encoding the antigen(s) of interest. SAM amplifies over time (up to 2 months) and consequently induces more potent and persistent immune responses (75, 76). Clinical applications using SAM have been promising in preventing infectious diseases (77) and are transitioning into the cancer immunotherapy field. Gritstone, a California-based company, is performing clinical studies, where a viral prime and a SAM boost are used to induce immune responses against private or shared neoantigens (78).
RNA in Passive Immunotherapy
Passive immunotherapy is used as an umbrella term to describe any strategy designed to help a patient to fight disease by administration of immune system components that have been generated in the laboratory, including delivery of proinflammatory cytokines, immune-modulatory mAbs, or ex vivo manipulated autologous effector immune cells (79, 80). As with active immunotherapies, passive strategies can also benefit from the implementation of not only iVT-mRNA but also ncRNA, as passive immunotherapy covers a broader range of strategies (Fig. 3).
Figure 3.

Overview of passive immunotherapeutic strategies. Left, systemic administration of TCR and CAR LNP-mRNA causes specific uptake by CD8+ T cells, followed by expression and antigen recognition, resulting in cancer cell death. Top right, ex vivo TCR/CAR LNP-mRNA manipulation of T cells, followed by systemic administration. Bottom right, intratumoral or systemic delivery of LNP-mRNA encoding monoclonal antibodies (mAbs), immune-inducing cytokines, or stimulatory receptors. Abbreviations: TCR, T-cell receptor; CAR, chimeric antigen receptor. Adapted from an image created with BioRender.com.
mRNA for protein therapy in vivo
In vivo delivery of antibody encoding mRNA
Since the development of the hybridoma technique in 1975 by Milstein and Köhler, therapeutic mAbs have been introduced for numerous indications. The mAb-mediated blockade of immune checkpoints, such as programmed cell death-1 (PD-1) and cytotoxic T lymphocyte-associated protein 4 (CTLA-4), has revolutionized cancer treatment (81–83). Besides full-sized mAb, antibody fragments, such as single-chain variable fragments (scFv) and heavy-chain VH domains, have been heavily studied. In parallel, a variety of tumor-associated targets, such as TAAs, vascular, and stromal cells, have been explored as targets (10, 80, 84, 85).
Although mRNA-based antibody therapies have yet to face technical and clinical challenges (e.g., frequency and route of administration; ref. 86), the use of mRNA for in vivo production of therapeutic antibodies remains a promising approach (10).
In 2019, Rybakova and colleagues demonstrated the delivery of mRNA encoding the humanized anti-human epidermal growth factor receptor 2 (HER2) antibody, trastuzumab, via LNP in tumor-bearing mice. The reported serum–antibody concentrations were detectable up to 14 days after LNP injection, demonstrating more favorable pharmacodynamics compared with the recombinant mAbs. In general, the mRNA transcribed antibody retained its cell toxicity properties in vivo, which contributed to a significant delay in HER2-positive tumor growth when administered weekly (87).
The potential of bispecific T-cell–engaging antibodies is high, but their manufacturing is often challenging. Stadler and colleagues tested the in vivo production of bispecific antibodies by treating mice with pharmacologically optimized, nucleoside-modified iVT-mRNA encoding the bispecific antibody. Sustained endogenous synthesis of the bispecific antibody was achieved, eliminating advanced tumors as effectively as the corresponding purified bispecific antibody was achieved. This approach could accelerate the clinical development of novel bispecific antibodies (88).
A clinical trial by BioNTech is investigating the safety and pharmacokinetics of BNT141 (NCT04683939; Table 2) and BNT142, both mRNA encoding antibodies targeting CLDN18.2 and CD3−CLDN6, respectively, in unresectable or metastatic claudin 18.2-positive solid tumors for which no available standard therapy is likely to confer clinical benefits.
mRNA-engineered adoptive cell therapies
Although immune-checkpoint blocking and mAb-based methods have shown promising results in breast cancer (89), melanoma (90), and non–small cell lung carcinoma (91, 92), among others, many patients still develop disease progression after these therapies, requiring additional treatment options (80, 93, 94). This demand was met with the introduction of adoptive cell therapy (ACT), pioneered by Steven A. Rosenberg, who demonstrated the in vivo antitumor activity of tumor-infiltrating lymphocytes (TIL; refs. 80, 88, 94). Over the years, ACT moved beyond the use of TILs. New strategies include ex vivo expansion and modification of tumor-residing or peripheral T cells with TCRs or CARs that convey specificity for the cancer cells (94, 95). Although ACT mainly focuses on the introduction of T cells expressing a TCR or CAR, Exteberria and colleagues showcased the therapeutic effect of the intratumoral administration of T cells transiently expressing IL12 in combination with transient CD137 ligand expression resulting in antitumor toxicity (96).
Successful clinical outcome has been reported with CAR-T therapies in hematologic malignancies. For instance, CAR-T therapy targeting CD19 in chronic lymphocytic and acute lymphoblastic leukemia (97), CD33 and CD123 CAR targeting in acute myeloid leukemia (AML; refs. 97, 98), and anti-BCMA CAR-T cell therapy in multiple myeloma (MM; ref. 99). This success was unmet when translated to solid malignancies as it is among others more challenging to identify the right target molecules.
At present, the clinical implementation of engineered T cells still raises many questions, including cross reactivity, controllability of permanently modified cellular products, and safety assessments (100). Because of these safety concerns, there has been an increasing interest for mRNA-based T-cell manipulation as transient expression is ensured in both TCR and CAR approaches (97, 100). However, despite this major advantage, other disadvantages such as insufficient longevity of mRNA-encoded CAR or TCR expression need to be addressed. This drawback also results in higher T-cell demands as repeated administration would be necessary to compensate the reduced half-life of CAR or TCR expression (97).
In 2020, Parayath and colleagues reported on the use of an injectable nanocarrier to deliver CAR or TCR encoding mRNA directly to circulating T cells, eliminating the need for ex vivo T-cell expansion (101). In this study, using leukemia and prostate cancer mouse models, the nanoparticles were manufactured using poly  -amino ester, which self-assembles into nanocomplexes when interacting with anionic nucleic acids. These nanoparticles specifically targeted CD8+ T cells by incorporation of an anti-CD8–linked polyglutamic acid. For CAR therapy, the nanoparticles were administered weekly, as the CAR expression lasted up to 8 days. A similar duration of TCR expression was achieved. The authors demonstrated that the use of injectable nanocarrier for mRNA delivery was sufficient to bring disease regression (101). More recently, the successful in vivo generation of CAR-T cells by delivery of modified mRNA packaged into T-cell targeted LNPs was reported. Transient expression and functionality of the CAR were observed (102).
-amino ester, which self-assembles into nanocomplexes when interacting with anionic nucleic acids. These nanoparticles specifically targeted CD8+ T cells by incorporation of an anti-CD8–linked polyglutamic acid. For CAR therapy, the nanoparticles were administered weekly, as the CAR expression lasted up to 8 days. A similar duration of TCR expression was achieved. The authors demonstrated that the use of injectable nanocarrier for mRNA delivery was sufficient to bring disease regression (101). More recently, the successful in vivo generation of CAR-T cells by delivery of modified mRNA packaged into T-cell targeted LNPs was reported. Transient expression and functionality of the CAR were observed (102).
Phase I trials in mesothelioma (NCT01355965) and pancreatic cancer (NCT01897415) have been initiated using autologous T cells transfected with mRNA encoding a mesothelin targeting CAR. The use of such mRNA-engineered T cells appeared to be feasible and safe. Early signs of antitumor activity and absence of overt off-tumor on-target toxicity were observed (103). A phase I clinical trial using autologous cMet-redirected T cells administered intratumorally in patients with breast cancer (NCT01837602) has shown that cMet-CAR-T cell injections were well tolerated, as no patients experienced above grade 1 adverse events, whereas tumor necrosis, and a consequential inflammatory response, was present when IHC was performed on tumor resections (104).
mRNA-based modulation of the tumor microenvironment
The intratumoral delivery of therapeutic compounds is an attractive option to increase the in situ bioavailability and, thus, the efficacy of immunotherapies. This applies to compounds targeted to tumor tissue as well as for compounds targeting immune cells that play an important role in immune evasion, such as regulatory T cells, tumor-associated macrophages (TAM), neutrophils (TAN), and immature DCs (105–109). Therefore, delivery of mRNA encoding such compounds can contribute to antitumor immunity, as shown before by delivery of mRNA encoding a fusokine consisting of IFN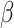 and the ectodomain of the TGF
and the ectodomain of the TGF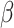 type III receptor (110). In a study reported by Haabeth and colleagues, charge-altering releasable transporters were used for the intratumoral delivery of mRNA encoding immune modulators (111). In this study, a monotherapy with mRNA encoding for IFN
type III receptor (110). In a study reported by Haabeth and colleagues, charge-altering releasable transporters were used for the intratumoral delivery of mRNA encoding immune modulators (111). In this study, a monotherapy with mRNA encoding for IFN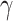 , IL12, CD70, CD80, CD86, and CD40L was investigated, and in particular a significant tumor growth delay was observed for CD40L, CD80, and CD86, as confirmed also by Van Lint and colleagues through intratumoral delivery of TriMix mRNA (112). More recent studies have published similar results with mRNA formulated in saline solution (113). mRNA encoding IFN
, IL12, CD70, CD80, CD86, and CD40L was investigated, and in particular a significant tumor growth delay was observed for CD40L, CD80, and CD86, as confirmed also by Van Lint and colleagues through intratumoral delivery of TriMix mRNA (112). More recent studies have published similar results with mRNA formulated in saline solution (113). mRNA encoding IFN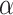 , IL12 single chain, granulocyte–monocyte colony stimulation factor (GM-CSF), and IL15 sushi was administered intratumorally, resulting in an increase of immune cell populations accompanied by intratumoral IFN
, IL12 single chain, granulocyte–monocyte colony stimulation factor (GM-CSF), and IL15 sushi was administered intratumorally, resulting in an increase of immune cell populations accompanied by intratumoral IFN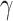 induction, systemic antigen-specific T-cell expansion, increased granzyme B+ T-cell infiltration, and formation of immune memory (113). In another preclinical study, iVT-mRNA encoding IL15 was administered in vivo. This mRNA was complexed using a protamine/liposome system. In both local and systemic administration, the CLLP/IL15 mRNA resulted in significant tumor-inhibitory effects in subcutaneous, abdominal cavity, and pulmonary metastasis models (114).
induction, systemic antigen-specific T-cell expansion, increased granzyme B+ T-cell infiltration, and formation of immune memory (113). In another preclinical study, iVT-mRNA encoding IL15 was administered in vivo. This mRNA was complexed using a protamine/liposome system. In both local and systemic administration, the CLLP/IL15 mRNA resulted in significant tumor-inhibitory effects in subcutaneous, abdominal cavity, and pulmonary metastasis models (114).
Several clinical trials using intratumoral delivery of mRNA are ongoing. A phase I study (NCT03739931; Table 2) is evaluating the intratumoral delivery of LNP-encapsulated mRNA encoding human OX40L, IL23, and IL36γ, either as monotherapy or in combination with immune-checkpoint blockade in patients with advanced malignancies. BioNTech is testing the intratumoral delivery of BNT131 or a mRNA mixture encoding IL12 single chain, IFN-alpha2b, GM-CSF, and IL15 sushi as monotherapy and in combination with PD-1 targeting cemiplimab in advanced solid tumors. Another phase I study by eTheRNA in collaboration with VUB-UZ aims to deliver synthetic naked mRNA encoding TriMix intratumorally in early-stage breast cancer (NCT03788083; Table 2).
ncRNA for the reduction of expression
RNA interference–based therapeutic interventions
All the above-mentioned applications involve the use of mRNA to mediate the expression of immune-boosting proteins. Notably, progressively more studies have focused on RNA as an intermediary, not only for expression but also for the regulation of expression, since RNA interference (RNAi) has been discovered in 1998 (115). The starting feature of RNAi is siRNA, short hairpin RNA (shRNA), or miRNA (116, 117), which after cleavage by the Dicer enzyme and after association with the RISC/ago2 enzyme complex has the capacity to hybridize with a complementary mRNA strand and results in the cleaving of that strand (118–120). This was successfully applied in immuno-oncology by Li and colleagues, where siRNA-PD-L1 (siPD-L1) was codelivered with imatinib in liposomal nanoparticles, resulting in the reduced expression of PD-L1, synergistically causing a tumor delay more significant than the monotherapies (121). Similar results were achieved in a mouse melanoma model by Wang and colleagues (122), indicating that, in combination with chemotherapy, or extracellular targeting, RNAi-mediated cell disruption can significantly promote the antitumor effects of already clinically available cancer treatment (116). Moreover, RNAi can as well be exploited beyond the alleviation of inhibitory pathways, but also for TME remodeling on TAMs, TANs, and immature DCs (123, 124) and in conjugation with mRNA for DC vaccination (61, 125). Although RNAi holds promise, important improvements regarding clinical applications, such as pharmacodynamics and pharmacokinetics, as well as toxicity need to be addressed (120). However, improvements regarding toxicity have already been booked with LNP formulations, as toxicity is often due to unintended and on-target off-tissue RNAi activity.
Beyond RNAi
Besides their role in RNAi, small ncRNA, such as miRNA, could act not only as tumor suppressor miRNA (TS-miR), but also as oncogenes (oncomiR), depending on the target (126). miRNA mimics, classifiable as ASOs can be implemented for anticancer therapy. Around 20–25 bases long, ASOs bind to their miRNA targets, preventing interaction of that miRNA with its target mRNA, and resulting in RNase H–mediated degradation (127). The use of ASOs has already been demonstrated in a preclinical setting for glioma using anti-miR-21 with miR-21 being on oncomiR, suppressing IL12 (126) for anticancer therapy. Around 20–25 bases long, ASOs bind to their miRNA targets, preventing the interaction of that miRNA with its target mRNA and resulting in RNase H mediated degradation (127). The use of ASOs has already been demonstrated in a preclinical setting for glioma using anti-miR-21 with miR-21 being on oncomiR, suppressing IL12 (126). Next to ASOs, aptamers have also entered the scope of RNA-mediated immunotherapy. Aptamers possess a small molecular weight, making them suitable for TME entry. Moreover, their longer shelf-life and low immunogenicity, combined with their possibility of cell-free manufacturing, give them advantageous features for clinical applicability (128). In a study by Gao and colleagues, aptamers targeting PD-L1 were developed and validated (129). Besides this, aptamers targeting CXCL12 (NOX-A12) and CCL-2 (NOX-E36) have been tested in clinical trials (130), from which monotherapy of NOX-A12 showed induction of T helper 1 cytokines and resulted in prolonged time on treatment versus prior therapy in 35% of patients with metastatic microsatellite stable colorectal or pancreatic cancer in combination with pembrolizumab (131). These studies concluded that aptamers can be considered a valid alternative compared with mAbs, as the production costs are significantly lower and similar tumor inhibition and binding affinity as for mAbs was obtained (128).
Conclusion and Perspectives
The SARS-CoV-2 pandemic has unlocked the great potential of mRNA as a therapeutic agent, due to the extreme need for a prompt development of an effective COVID-19 vaccine. This rapid progress was possible only because of the preexisting long-term experience and already developed mRNA technology of the past three decades. Next, the mRNA format's high versatility could push further the implementation of mRNA-based personalized cancer therapies into the clinic, relying on an easily convertible manufacturing process (132). Nevertheless, personalized therapies still require the identification of novel, cancer-specific targets (including neoantigens) for which abundance and immunogenicity studies remain the main challenge. However, improvements in in silico prediction algorithms and next-generation sequencing (are expected to address this implementation; refs. 132, 133).
Despite high versatility, reduced costs, and quick manufacturing of mRNA vaccines, further insights are still required, especially regarding the mechanisms of action and therefore understanding the contribution of the innate immunogenicity of mRNA (134). The two SARS-CoV-2 mRNA-based vaccines, BNT162b2 (Tozinameran) and mRNA-1273 (Elasomeran), showed that mRNA chemical modifications and purity play an important role in reducing intrinsic immunogenicity, and this is key for intramuscular injected prophylactic vaccines (28). Low intrinsic immunogenicity is also necessary for other RNA therapeutics, where the protein level needs to be as high as possible including antibody encoding mRNA and mRNA-based modulation of the TME (28). Furthermore, the use of adjuvants and even the mRNA self-adjuvancy level has not yet been extensively evaluated in terms of potential benefits or adverse effects in mRNA cancer vaccine studies (21, 135).
The prompt optimization of LNP for clinical formulations contributed to the success of RNA as a therapeutic agent. However, many parameters need further investigation, such as biodegradability, tissue and cell tropism, long-term side effects, route of administration and delivery, all having a major effect on the overall cost, efficacy, and safety profile of LNP (23, 136).
Regarding RNAi, hereditary transthyretin amyloidosis and acute hepatic porphyria can already benefit from treatment options (137). Nevertheless, for the treatment of cancer, RNAi and ncRNA formulations have not yet been approved. The main challenge here is the scarce delivery of effector molecules in tumor cells to induce a clinically significant response. Two clinical trials (NCT01676259 and NCT01591356) are ongoing, using RNAi targeting KRAS and EPHA2, respectively. Positive results from this work could further accelerate the implementation of RNAi into the clinic (Table 2).
The use of RNA in passive immunotherapeutic approaches is catalyzed by current results from ongoing clinical trials (Table 2; ref. 2). More preclinical studies are necessary to thoroughly investigate RNA kinetics and dosage (80).
If, on the one hand, RNA therapeutics boast of a high safety profile (138) due to a transient dwelling time, on the other hand, more frequent administrations are required, which in terms of ACT might hamper the manufacturing process, as T cells are limited. In addition to this, injected T cells might also fail to induce a potent response as expression could be lost before reaching the tumor site. Local administrations could work as an efficient alternative, but to date have been unsuccessful in human clinical trials (98).
Altogether, the mentioned developments in the RNA field indicate its potential as an ideal candidate anticancer therapeutic agent, expanding on its current use for antiviral vaccination. Because of its manufacturing benefits, RNA therapeutics could establish a general presence in the drug development industry, going even beyond implementation for cancer immunotherapy.
Acknowledgments
This research was supported by the Belgian Foundation against Cancer [FAF-C/2018/1222 (2018-128) and FAF-F/2018/1223 (2018-089)], Flanders Innovation and Entrepreneurship (VLAIO, HBC.2019.2522 and HBC.2019.2564), and the Research Council of the Vrije Universiteit Brussel (Strategic Research Program 48). W. De Mey is a PhD fellow funded via the Oncology Research Center and Scientific Fund Willy Gepts.
Authors' Disclosures
K. Thielemans reports a patent for WO2009/034172 issued and licensed to eTheRNA and a patent for WO2021/185833 pending. K. Breckpot reports grants from Research Council VUB, Stichting tegen Kanker, and VLAIO and other support from Oncology Research Center and Scientific Fund Willy Gepts during the conduct of the study. No disclosures were reported by the other authors.
References
- 1. Dolgin E. The tangled history of mRNA vaccines. Nature 2021;597:318–24. [DOI] [PubMed] [Google Scholar]
- 2. Pastor F, Berraondo P, Etxeberria I, Frederick J, Sahin U, Gilboa E, et al. An RNA toolbox for cancer immunotherapy. Nat Rev Drug Discov 2018;17:751–67. [DOI] [PubMed] [Google Scholar]
- 3. Van Hoecke L, Verbeke R, Dewitte H, Lentacker I, Vermaelen K, Breckpot K, et al. mRNA in cancer immunotherapy: beyond a source of antigen. Mol Cancer 2021;20:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mollocana-Lara EC, Ni M, Agathos SN, Gonzales-Zubiate FA. The infinite possibilities of RNA therapeutics. J Ind Microbiol Biotechnol 2021;48:kuab063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Damase TR, Sukhovershin R, Boada C, Taraballi F, Pettigrew RI, Cooke JP. The limitless future of RNA therapeutics. Front Bioeng Biotechnol 2021;9:628137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dammes N, Peer D. Paving the road for RNA therapeutics. Trends Pharmacol Sci 2020;41:755–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pardi N, Hogan MJ, Weissman D. Recent advances in mRNA vaccine technology. Curr Opin Immunol 2020;65:14–20. [DOI] [PubMed] [Google Scholar]
- 8. Hollingsworth RE, Jansen K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines 2019;4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stephens AJ, Burgess-Brown NA, Jiang S. Beyond just peptide antigens: the complex world of peptide-based cancer vaccines. Front Immunol 2021;12:696791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schlake T, Thran M, Fiedler K, Heidenreich R, Petsch B, Fotin-Mleczek M. mRNA: a novel avenue to antibody therapy? Mol Ther 2019;27:773–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu MA. A comparison of plasmid DNA and mRNA as vaccine technologies. Vaccines 2019;7:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Diken M, Kranz LM, Kreiter S, Sahin U. mRNA: a versatile molecule for cancer vaccines. Curr Issues Mol Biol 2017;22:113–28. [DOI] [PubMed] [Google Scholar]
- 13. Gallie DR. The cap and poly(A) tail function synergistically to regulate mRNA translational efficiency. Genes Dev 1991;5:2108–16. [DOI] [PubMed] [Google Scholar]
- 14. Holtkamp S, Kreiter S, Selmi A, Simon P, Koslowski M, Huber C, et al. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood 2006;108:4009–17. [DOI] [PubMed] [Google Scholar]
- 15. Pardi N, Hogan MJ, Porter FW, Weissman D. mRNA vaccines - a new era in vaccinology. Nat Rev Drug Discov 2018;17:261–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pardi N, Muramatsu H, Weissman D, Karikó K. In vitro transcription of long RNA containing modified nucleosides. Methods Mol Biol 2013;969:29–42. [DOI] [PubMed] [Google Scholar]
- 17. Weissman D, Pardi N, Muramatsu H, Karikó K. HPLC purification of in vitro transcribed long RNA. Methods Mol Biol 2013;969:43–54. [DOI] [PubMed] [Google Scholar]
- 18. Baiersdörfer M, Boros G, Muramatsu H, Mahiny A, Vlatkovic I, Sahin U, et al. A facile method for the removal of dsRNA contaminant from in vitro-transcribed mRNA. Mol Ther Nucleic Acids 2019;15:26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Karikó K, Muramatsu H, Ludwig J, Weissman D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res 2011;39:e142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Demelenne A, Servais AC, Crommen J, Fillet M. Analytical techniques currently used in the pharmaceutical industry for the quality control of RNA-based therapeutics and ongoing developments. J Chromatogr A 2021;1651:462283. [DOI] [PubMed] [Google Scholar]
- 21. Verbeke R, Lentacker I, De Smedt S, Dewitte H. Three decades of messenger RNA vaccine development. Nano Today 2019;28: 100766. [Google Scholar]
- 22. Kowalski PS, Rudra A, Miao L, Anderson DG. Delivering the messenger: advances in technologies for therapeutic mRNA delivery. Mol Ther 2019;27:710–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hou X, Zaks T, Langer R, Dong Y. Lipid nanoparticles for mRNA delivery. Nat Rev Mater 2021;6:1078–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zeng C, Zhang C, Walker PG, Dong Y. Formulation and delivery technologies for mRNA vaccines. Curr Top Microbiol Immunol 2020;10.1007/82_2020_217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Uchida S, Perche F, Pichon C, Cabral H. Nanomedicine-based approaches for mRNA delivery. Mol Pharm 2020;17:3654–84. [DOI] [PubMed] [Google Scholar]
- 26. Suzuki Y, Ishihara H. Difference in the lipid nanoparticle technology employed in three approved siRNA (Patisiran) and mRNA (COVID-19 vaccine) drugs. Drug Metab Pharmacokinet 2021;41:100424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Karikó K, Buckstein M, Ni H, Weissman D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity 2005;23:165–75. [DOI] [PubMed] [Google Scholar]
- 28. Nelson J, Sorensen EW, Mintri S, Rabideau AE, Zheng W, Besin G, et al. Impact of mRNA chemistry and manufacturing process on innate immune activation. Sci Adv 2020;6:eaaz6893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Karikó K, Muramatsu H, Welsh FA, Ludwig J, Kato H, Akira S, et al. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol Ther 2008;16:1833–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Harcourt EM, Kietrys AM, Kool ET. Chemical and structural effects of base modifications in messenger RNA. Nature 2017;541:339–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Henderson JM, Ujita A, Hill E, Yousif-Rosales S, Smith C, Ko N, et al. Cap 1 messenger RNA synthesis with co-transcriptional CleanCap. Curr Protoc 2021;1:e39. [DOI] [PubMed] [Google Scholar]
- 32. Andreas K, Hiromi M, Katalin K, Stephanie F, Ugur S. 5'-cap-trinucleotide- or higher oligonucleotide compounds and their use in stabilizing RNA, expressing proteins in therapy. Patent application #202047044688; 2021. [Google Scholar]
- 33. Lima SA, Chipman LB, Nicholson AL, Chen YH, Yee BA, Yeo GW, et al. Short poly(A) tails are a conserved feature of highly expressed genes. Nat Struct Mol Biol 2017;24:1057–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Thess A, Grund S, Mui BL, Hope MJ, Baumhof P, Fotin-Mleczek M, et al. Sequence-engineered mRNA without chemical nucleoside modifications enables an effective protein therapy in large animals. Mol Ther 2015;23:1456–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Orlandini von Niessen AG, Poleganov MA, Rechner C, Plaschke A, Kranz LM, Fesser S, et al. Improving mRNA-based therapeutic gene delivery by expression-augmenting 3' UTRs identified by cellular library screening. Mol Ther 2019;27:824–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Asrani KH, Farelli JD, Stahley MR, Miller RL, Cheng CJ, Subramanian RR, et al. Optimization of mRNA untranslated regions for improved expression of therapeutic mRNA. RNA Biol 2018;15:756–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sample PJ, Wang B, Reid DW, Presnyak V, McFadyen IJ, Morris DR, et al. Human 5' UTR design and variant effect prediction from a massively parallel translation assay. Nat Biotechnol 2019;37:803–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Smith CIE, Zain R. Therapeutic oligonucleotides: state of the art. Annu Rev Pharmacol Toxicol 2019;59:605–30. [DOI] [PubMed] [Google Scholar]
- 39. Bennett CF, Swayze EE. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu Rev Pharmacol Toxicol 2010;50:259–93. [DOI] [PubMed] [Google Scholar]
- 40. Liu Q, Zhang W, Chen S, Zhuang Z, Zhang Y, Jiang L, et al. SELEX tool: a novel and convenient gel-based diffusion method for monitoring of aptamer-target binding. J Biol Eng 2020;14:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gold L, Janjic N, Jarvis T, Schneider D, Walker JJ, Wilcox SK, et al. Aptamers and the RNA world, past and present. Cold Spring Harb Perspect Biol 2012;4:a003582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Antonarelli G, Corti C, Tarantino P, Ascione L, Cortes J, Romero P, et al. Therapeutic cancer vaccines revamping: technology advancements and pitfalls. Ann Oncol 2021;32:1537–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Saxena M, van der Burg SH, Melief CJM, Bhardwaj N. Therapeutic cancer vaccines. Nat Rev Cancer 2021;21:360–78. [DOI] [PubMed] [Google Scholar]
- 44. Chakraborty C, Sharma AR, Bhattacharya M, Lee SS. From COVID-19 to cancer mRNA vaccines: moving from bench to clinic in the vaccine landscape. Front Immunol 2021;12:679344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Filin IY, Kitaeva KV, Rutland CS, Rizvanov AA, Solovyeva VV. Recent advances in experimental dendritic cell vaccines for cancer. Front Oncol 2021;11:730824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dörrie J, Schaft N, Schuler G, Schuler-Thurner B. Therapeutic cancer vaccination with ex vivo RNA-transfected dendritic cells-an update. Pharmaceutics 2020;12:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Miao L, Zhang Y, Huang L. mRNA vaccine for cancer immunotherapy. Mol Cancer 2021;20:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vigneron N, Abi Habib J, Van den Eynde BJ. Learning from the proteasome how to fine-tune cancer immunotherapy. Trends Cancer 2017;3:726–41. [DOI] [PubMed] [Google Scholar]
- 49. Galaine J, Borg C, Godet Y, Adotévi O. Interest of tumor-specific CD4 T helper 1 cells for therapeutic anticancer vaccine. Vaccines 2015;3:490–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bonehill A, Heirman C, Tuyaerts S, Michiels A, Breckpot K, Brasseur F, et al. Messenger RNA-electroporated dendritic cells presenting MAGE-A3 simultaneously in HLA class I and class II molecules. J Immunol 2004;172:6649–57. [DOI] [PubMed] [Google Scholar]
- 51. Bonehill A, Heirman C, Tuyaerts S, Michiels A, Zhang Y, van der Bruggen P, et al. Efficient presentation of known HLA class II-restricted MAGE-A3 epitopes by dendritic cells electroporated with messenger RNA encoding an invariant chain with genetic exchange of class II-associated invariant chain peptide. Cancer Res 2003;63:5587–94. [PubMed] [Google Scholar]
- 52. Jou J, Harrington KJ, Zocca MB, Ehrnrooth E, Cohen EEW. The changing landscape of therapeutic cancer vaccines: novel platforms and neoantigen identification. Clin Cancer Res 2021;27:689–703. [DOI] [PubMed] [Google Scholar]
- 53. Hsu FJ, Benike C, Fagnoni F, Liles TM, Czerwinski D, Taidi B, et al. Vaccination of patients with B-cell lymphoma using autologous antigen-pulsed dendritic cells. Nat Med 1996;2:52–8. [DOI] [PubMed] [Google Scholar]
- 54. Perez CR, De Palma M. Engineering dendritic cell vaccines to improve cancer immunotherapy. Nat Commun 2019;10:5408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Benteyn D, Heirman C, Bonehill A, Thielemans K, Breckpot K. mRNA-based dendritic cell vaccines. Expert Rev Vaccines 2015;14:161–76. [DOI] [PubMed] [Google Scholar]
- 56. Massa C, Thomas C, Wang E, Marincola F, Seliger B. Different maturation cocktails provide dendritic cells with different chemoattractive properties. J Transl Med 2015;13:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bonehill A, Tuyaerts S, Van Nuffel AM, Heirman C, Bos TJ, Fostier K, et al. Enhancing the T-cell stimulatory capacity of human dendritic cells by co-electroporation with CD40L, CD70 and constitutively active TLR4 encoding mRNA. Mol Ther 2008;16:1170–80. [DOI] [PubMed] [Google Scholar]
- 58. Wilgenhof S, Van Nuffel AM, Corthals J, Heirman C, Tuyaerts S, Benteyn D, et al. Therapeutic vaccination with an autologous mRNA electroporated dendritic cell vaccine in patients with advanced melanoma. J Immunother 2011;34:448–56. [DOI] [PubMed] [Google Scholar]
- 59. Van Nuffel AM, Benteyn D, Wilgenhof S, Pierret L, Corthals J, Heirman C, et al. Dendritic cells loaded with mRNA encoding full-length tumor antigens prime CD4+ and CD8+ T cells in melanoma patients. Mol Ther 2012;20:1063–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wilgenhof S, Van Nuffel AMT, Benteyn D, Corthals J, Aerts C, Heirman C, et al. A phase IB study on intravenous synthetic mRNA electroporated dendritic cell immunotherapy in pretreated advanced melanoma patients. Ann Oncol 2013;24:2686–93. [DOI] [PubMed] [Google Scholar]
- 61. Aerts-Toegaert C, Heirman C, Tuyaerts S, Corthals J, Aerts JL, Bonehill A, et al. CD83 expression on dendritic cells and T cells: correlation with effective immune responses. Eur J Immunol 2007;37:686–95. [DOI] [PubMed] [Google Scholar]
- 62. Tuyaerts S, Van Meirvenne S, Bonehill A, Heirman C, Corthals J, Waldmann H, et al. Expression of human GITRL on myeloid dendritic cells enhances their immunostimulatory function but does not abrogate the suppressive effect of CD4+CD25+ regulatory T cells. J Leukoc Biol 2007;82:93–105. [DOI] [PubMed] [Google Scholar]
- 63. Pen JJ, Keersmaecker BD, Heirman C, Corthals J, Liechtenstein T, Escors D, et al. Interference with PD-L1/PD-1 co-stimulation during antigen presentation enhances the multifunctionality of antigen-specific T cells. Gene Ther 2014;21:262–71. [DOI] [PubMed] [Google Scholar]
- 64. Willemen Y, Van den Bergh JM, Lion E, Anguille S, Roelandts VA, Van Acker HH, et al. Engineering monocyte-derived dendritic cells to secrete interferon-α enhances their ability to promote adaptive and innate anti-tumor immune effector functions. Cancer Immunol Immunother 2015;64:831–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Van den Bergh JMJ, Smits ELJM, Versteven M, De Reu H, Berneman ZN, Van Tendeloo VFI, et al. Characterization of interleukin-15-transpresenting dendritic cells for clinical use. J Immunol Res 2017;2017:1975902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gu YZ, Zhao X, Song XR. Ex vivo pulsed dendritic cell vaccination against cancer. Acta Pharmacol Sin 2020;41:959–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Faghfuri E, Pourfarzi F, Faghfouri AH, Abdoli Shadbad M, Hajiasgharzadeh K, Baradaran B. Recent developments of RNA-based vaccines in cancer immunotherapy. Expert Opin Biol Ther 2021;21:201–18. [DOI] [PubMed] [Google Scholar]
- 68. Sahin U, Oehm P, Derhovanessian E, Jabulowsky RA, Vormehr M, Gold M, et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature 2020;585:107–12. [DOI] [PubMed] [Google Scholar]
- 69. BioNTech[homepage on the Internet]. mRNA-based BNT111 FixVac melanoma trial in Nature [cited 2022 Jan]. Available from: https://investors.biontech.de/news-releases/news-release-details/biontech-publishes-data-mrna-based-bnt111-fixvac-melanoma-trial/.
- 70. Braiteh F, LoRusso P, Balmanoukian A, Klempner S, Camidge DR, Hellmann M, et al. Abstract CT169: a phase Ia study to evaluate RO7198457, an individualized neoantigen specific immunoTherapy (iNeST), in patients with locally advanced or metastatic solid tumors. Cancer Res 2020;80:CT169. [Google Scholar]
- 71. Lopez JS, Camidge R, Iafolla M, Rottey S, Schuler M, Hellmann M, et al. Abstract CT301: a phase Ib study to evaluate RO7198457, an individualized neoantigen specific immunotherapy (iNeST), in combination with atezolizumab in patients with locally advanced or metastatic solid tumors. Cancer Res 2020;80:CT301. [Google Scholar]
- 72. Burris HA, Patel MR, Cho DC, Clarke JM, Gutierrez M, Zaks TZ, et al. A phase I multicenter study to assess the safety, tolerability, and immunogenicity of mRNA-4157 alone in patients with resected solid tumors and in combination with pembrolizumab in patients with unresectable solid tumors. J Clin Oncol 2019;37:2523. [Google Scholar]
- 73. Bauman J, Burris H, Clarke J, Patel M, Cho D, Gutierrez M, et al. 798 Safety, tolerability, and immunogenicity of mRNA-4157 in combination with pembrolizumab in subjects with unresectable solid tumors (KEYNOTE-603): an update. J ImmunoTher Cancer 2020;8:A477. [Google Scholar]
- 74. Meisel AaPS. mRNA vaccines against infectious diseases and cancer. healthbook TIMES Oncol Hematol 2021;9:24–31. [Google Scholar]
- 75. Lundstrom K. Self-amplifying RNA viruses as RNA vaccines. Int J Mol Sci 2020;21:5130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wang Y, Zhang Z, Luo J, Han X, Wei Y, Wei X. mRNA vaccine: a potential therapeutic strategy. Mol Cancer 2021;20:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bloom K, van den Berg F, Arbuthnot P. Self-amplifying RNA vaccines for infectious diseases. Gene Ther 2021;28:117–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gritstone [homepage on the Internet]. Corporate presentation [cited 2022 Jan]. Available from: https://ir.gritstonebio.com/investors/presentations.
- 79. Cha HR, Lee JH, Ponnazhagan S. Revisiting immunotherapy: a focus on prostate cancer. Cancer Res 2020;80:1615–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Schlake T, Thess A, Thran M, Jordan I. mRNA as novel technology for passive immunotherapy. Cell Mol Life Sci 2019;76:301–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rotte A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J Exp Clin Cancer Res 2019;38:255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Schnell A, Bod L, Madi A, Kuchroo VK. The yin and yang of co-inhibitory receptors: toward anti-tumor immunity without autoimmunity. Cell Res 2020;30:285–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. D'Arrigo P, Tufano M, Rea A, Vigorito V, Novizio N, Russo S, et al. Manipulation of the immune system for cancer defeat: a focus on the T cell inhibitory checkpoint molecules. Curr Med Chem 2020;27:2402–48. [DOI] [PubMed] [Google Scholar]
- 84. Lecocq Q, De Vlaeminck Y, Hanssens H, D'Huyvetter M, Raes G, Goyvaerts C, et al. Theranostics in immuno-oncology using nanobody derivatives. Theranostics 2019;9:7772–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Chen W, Yuan Y, Jiang X. Antibody and antibody fragments for cancer immunotherapy. J Control Release 2020;328:395–406. [DOI] [PubMed] [Google Scholar]
- 86. Deal CE, Carfi A, Plante OJ. Advancements in mRNA encoded antibodies for passive immunotherapy. Vaccines 2021;9:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Rybakova Y, Kowalski PS, Huang Y, Gonzalez JT, Heartlein MW, DeRosa F, et al. mRNA delivery for therapeutic anti-HER2 antibody expression in vivo. Mol Ther 2019;27:1415–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Stadler CR, Bähr-Mahmud H, Celik L, Hebich B, Roth AS, Roth RP, et al. Elimination of large tumors in mice by mRNA-encoded bispecific antibodies. Nat Med 2017;23:815–7. [DOI] [PubMed] [Google Scholar]
- 89. Vranic S, Cyprian FS, Gatalica Z, Palazzo J. PD-L1 status in breast cancer: current view and perspectives. Semin Cancer Biol 2021;72:146–54. [DOI] [PubMed] [Google Scholar]
- 90. Luke JJ, Flaherty KT, Ribas A, Long GV. Targeted agents and immunotherapies: optimizing outcomes in melanoma. Nat Rev Clin Oncol 2017;14:463–82. [DOI] [PubMed] [Google Scholar]
- 91. Mayor M, Yang N, Sterman D, Jones DR, Adusumilli PS. Immunotherapy for non-small cell lung cancer: current concepts and clinical trials. Eur J Cardiothorac Surg 2016;49:1324–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science 2018;359:1350–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Van Hoecke L, Roose K. How mRNA therapeutics are entering the monoclonal antibody field. J Transl Med 2019;17:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Rohaan MW, Wilgenhof S, Haanen JBAG. Adoptive cellular therapies: the current landscape. Virchows Arch 2019;474:449–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Hanssens H, Meeus F, De Veirman K, Breckpot K, Devoogdt N. The antigen-binding moiety in the driver's seat of CARs. Med Res Rev 2022;42:306–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Etxeberria I, Bolaños E, Quetglas JI, Gros A, Villanueva A, Palomero J, et al. Intratumor adoptive transfer of IL-12 mRNA transiently engineered antitumor CD8. Cancer Cell 2019;36:613–29. [DOI] [PubMed] [Google Scholar]
- 97. Soundara Rajan T, Gugliandolo A, Bramanti P, Mazzon E. In vitro-transcribed mRNA chimeric antigen receptor T cell (IVT mRNA CAR T) therapy in hematologic and solid tumor management: a preclinical update. Int J Mol Sci 2020;21:6514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Foster JB, Barrett DM, Karikó K. The emerging role of in vitro-transcribed mRNA in adoptive T cell immunotherapy. Mol Ther 2019;27:747–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Feng D, Sun J. Overview of anti-BCMA CAR-T immunotherapy for multiple myeloma and relapsed/refractory multiple myeloma. Scand J Immunol 2020;92:e12910. [DOI] [PubMed] [Google Scholar]
- 100. Mensali N, Myhre MR, Dillard P, Pollmann S, Gaudernack G, Kvalheim G, et al. Preclinical assessment of transiently TCR redirected T cells for solid tumour immunotherapy. Cancer Immunol Immunother 2019;68:1235–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Parayath NN, Stephan SB, Koehne AL, Nelson PS, Stephan MT. In vitro-transcribed antigen receptor mRNA nanocarriers for transient expression in circulating T cells in vivo. Nat Commun 2020;11:6080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Rurik JG, Tombácz I, Yadegari A, Méndez Fernández PO, Shewale SV, Li L, et al. CAR T cells produced in vivo to treat cardiac injury. Science 2022;375:91–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res 2014;2:112–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Tchou J, Zhao Y, Levine BL, Zhang PJ, Davis MM, Melenhorst JJ, et al. Safety and efficacy of intratumoral injections of chimeric antigen receptor (CAR) T cells in metastatic breast cancer. Cancer Immunol Res 2017;5:1152–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Champiat S, Tselikas L, Farhane S, Raoult T, Texier M, Lanoy E, et al. Intratumoral immunotherapy: from trial design to clinical practice. Clin Cancer Res 2021;27:665–79. [DOI] [PubMed] [Google Scholar]
- 106. Melero I, Castanon E, Alvarez M, Champiat S, Marabelle A. Intratumoural administration and tumour tissue targeting of cancer immunotherapies. Nat Rev Clin Oncol 2021;18:558–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Van der Jeught K, Bialkowski L, Daszkiewicz L, Broos K, Goyvaerts C, Renmans D, et al. Targeting the tumor microenvironment to enhance antitumor immune responses. Oncotarget 2015;6:1359–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Van der Jeught K, Van Lint S, Thielemans K, Breckpot K. Intratumoral delivery of mRNA: overcoming obstacles for effective immunotherapy. Oncoimmunology 2015;4:e1005504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Yuan J, Khilnani A, Brody J, Andtbacka RHI, Hu-Lieskovan S, Luke JJ, et al. Current strategies for intratumoural immunotherapy: beyond immune checkpoint inhibition. Eur J Cancer 2021;157:493–510. [DOI] [PubMed] [Google Scholar]
- 110. Van der Jeught K, Joe PT, Bialkowski L, Heirman C, Daszkiewicz L, Liechtenstein T, et al. Intratumoral administration of mRNA encoding a fusokine consisting of IFN-β and the ectodomain of the TGF-β receptor II potentiates antitumor immunity. Oncotarget 2014;5:10100–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Haabeth OAW, Blake TR, McKinlay CJ, Tveita AA, Sallets A, Waymouth RM, et al. Local delivery of Ox40l, Cd80, and Cd86 mRNA kindles global anticancer immunity. Cancer Res 2019;79:1624–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Van Lint S, Renmans D, Broos K, Goethals L, Maenhout S, Benteyn D, et al. Intratumoral delivery of TriMix mRNA results in T-cell activation by cross-presenting dendritic cells. Cancer Immunol Res 2016;4:146–56. [DOI] [PubMed] [Google Scholar]
- 113. Hotz C, Wagenaar TR, Gieseke F, Bangari DS, Callahan M, Cao H, et al. Local delivery of mRNA-encoded cytokines promotes antitumor immunity and tumor eradication across multiple preclinical tumor models. Sci Transl Med 2021;13:eabc7804. [DOI] [PubMed] [Google Scholar]
- 114. Lei S, Zhang X, Men K, Gao Y, Yang X, Wu S, et al. Efficient colorectal cancer gene therapy with IL-15 mRNA nanoformulation. Mol Pharm 2020;17:3378–91. [DOI] [PubMed] [Google Scholar]
- 115. Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998;391:806–11. [DOI] [PubMed] [Google Scholar]
- 116. Monty MA, Islam MA, Nan X, Tan J, Tuhin IJ, Tang X, et al. Emerging role of RNA interference in immune cells engineering and its therapeutic synergism in immunotherapy. Br J Pharmacol 2021;178:1741–55. [DOI] [PubMed] [Google Scholar]
- 117. Berber B, Aydin C, Kocabas F, Guney-Esken G, Yilancioglu K, Karadag-Alpaslan M, et al. Gene editing and RNAi approaches for COVID-19 diagnostics and therapeutics. Gene Ther 2021;28:290–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Mello CC, Conte D. Revealing the world of RNA interference. Nature 2004;431:338–42. [DOI] [PubMed] [Google Scholar]
- 119. Lin YX, Wang Y, Blake S, Yu M, Mei L, Wang H, et al. RNA nanotechnology-mediated cancer immunotherapy. Theranostics 2020;10:281–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Setten RL, Rossi JJ, Han SP. The current state and future directions of RNAi-based therapeutics. Nat Rev Drug Discov 2019;18:421–46. [DOI] [PubMed] [Google Scholar]
- 121. Li C, Han X. Melanoma cancer immunotherapy using PD-L1 siRNA and imatinib promotes cancer-immunity cycle. Pharm Res 2020;37:109. [DOI] [PubMed] [Google Scholar]
- 122. Wang C, Shi X, Song H, Zhang C, Wang X, Huang P, et al. Polymer-lipid hybrid nanovesicle-enabled combination of immunogenic chemotherapy and RNAi-mediated PD-L1 knockdown elicits antitumor immunity against melanoma. Biomaterials 2021;268:120579. [DOI] [PubMed] [Google Scholar]
- 123. Chen J, Dou Y, Tang Y, Zhang X. Folate receptor-targeted RNAi nanoparticles for silencing STAT3 in tumor-associated macrophages and tumor cells. Nanomedicine 2020;25:102173. [DOI] [PubMed] [Google Scholar]
- 124. Zins K, Abraham D. Cancer immunotherapy: targeting tumor-associated macrophages by gene silencing. Methods Mol Biol 2020;2115:289–325. [DOI] [PubMed] [Google Scholar]
- 125. Breckpot K, Aerts-Toegaert C, Heirman C, Peeters U, Beyaert R, Aerts JL, et al. Attenuated expression of A20 markedly increases the efficacy of double-stranded RNA-activated dendritic cells as an anti-cancer vaccine. J Immunol 2009;182:860–70. [DOI] [PubMed] [Google Scholar]
- 126. Milani R, Brognara E, Fabbri E, Manicardi A, Corradini R, Finotti A, et al. Targeting miR-155-5p and miR-221-3p by peptide nucleic acids induces caspase-3 activation and apoptosis in temozolomide-resistant T98G glioma cells. Int J Oncol 2019;55:59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Bajan S, Hutvagner G. RNA-based therapeutics: from antisense oligonucleotides to miRNAs. Cells 2020;9:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Wei J, Gilboa E, Calin GA, Heimberger AB. Immune modulatory short noncoding RNAs targeting the glioblastoma microenvironment. Front Oncol 2021;11:682129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Gao T, Mao Z, Li W, Pei R. Anti-PD-L1 DNA aptamer antagonizes the interaction of PD-1/PD-L1 with antitumor effect. J Mater Chem B 2021;9:746–56. [DOI] [PubMed] [Google Scholar]
- 130. Shigdar S, Schrand B, Giangrande PH, de Franciscis V. Aptamers: cutting edge of cancer therapies. Mol Ther 2021;29:2396–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Halama N, Williams A, Prüfer U, Frömming A, Beyer D, Eulberg D, et al. Abstract CT117: phase 1/2 study with CXCL12 inhibitor NOX-A12 and pembrolizumab in patients with microsatellite-stable, metastatic colorectal or pancreatic cancer. Cancer Res 2020;80:CT117. [Google Scholar]
- 132. Esprit A, de Mey W, Bahadur Shahi R, Thielemans K, Franceschini L, Breckpot K. Neo-antigen mRNA vaccines. Vaccines 2020;8:776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Lang F, Schrörs B, Löwer M, Türeci Ö, Sahin U. Identification of neoantigens for individualized therapeutic cancer vaccines. Nat Rev Drug Discov 2022;21:261–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Maruggi G, Zhang C, Li J, Ulmer JB, Yu D. mRNA as a transformative technology for vaccine development to control infectious diseases. Mol Ther 2019;27:757–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Linares-Fernández S, Lacroix C, Exposito JY, Verrier B. Tailoring mRNA vaccine to balance innate/adaptive immune response. Trends Mol Med 2020;26:311–23. [DOI] [PubMed] [Google Scholar]
- 136. Chung JY, Thone MN, Kwon YJ. COVID-19 vaccines: the status and perspectives in delivery points of view. Adv Drug Deliv Rev 2021;170:1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Sundara Rajan S, Ludwig KR, Hall KL, Jones TL, Caplen NJ. Cancer biology functional genomics: from small RNAs to big dreams. Mol Carcinog 2020;59:1343–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Beck JD, Reidenbach D, Salomon N, Sahin U, Türeci Ö, Vormehr M, et al. mRNA therapeutics in cancer immunotherapy. Mol Cancer 2021;20:69. [DOI] [PMC free article] [PubMed] [Google Scholar]