

Abstract
The mitochondrial protein sirtuin 3 (SIRT3) can counteract cell damage caused by oxidative stress and inflammation, and contribute to cell survival primarily by improving mitochondrial function. However, the effects of SIRT3 in dopaminergic neuronal cells (DACs) remain unclear. In our previous studies, microglia activation-associated cytotoxicity was observed to promote the apoptosis of DACs, along with the decrease of SIRT3 expression. The aim of the present study was to explore the potential neuroprotective effect of SIRT3 expression against dopaminergic neuron injury caused by microglia activation, and clarify its possible mechanisms. SIRT3 overexpression in DACs reduced the production of intracellular reactive oxygen species (ROS), cell apoptosis rate, mitochondrial membrane potential (ΔΨm) depolarization, opening of mitochondrial permeability transition pore (mPTP) and cyclophilin D (CypD) protein level, and promoted cell cycle progression. However, SIRT3 siRNA-mediated knockdown further aggravated microglia activation-mediated cytotoxicity, including ROS accumulation, increased cell apoptosis and mPTP opening, elevated the CypD level, enhanced mitochondrial ΔΨm depolarization, concomitant to cell cycle arrest at G0/G1 phase. The mechanisms of SIRT3 mitigated microglia activation-induced DAC dysfunction, which included decreased mPTP opening and Bax/Bcl-2 ratio, inhibition of mitochondrial cytochrome c release to the cytoplasm, reduced caspase-3/9 activity, increased LC3II/LC3I and beclin-1 protein expression levels, and decreased nucleotide-binding oligomerization domain, leucine rich repeat and pyrin domain-containing protein 3 (NLRP3), caspase-1, IL-1β and IL-18 protein expression. In conclusion, these results indicated that SIRT3 expression attenuated cell damage caused by microglia activation through the mitochondrial apoptosis pathway in DACs. The mitophagy-NLRP3 inflammasome pathway may also be associated with this neuroprotection. These findings may provide new intervention targets for the survival of dopaminergic neurons and the prevention and treatment of Parkinson's disease.
Keywords: sirtuin 3, dopaminergic neuronal cell, microglia activation, apoptosis, mitophagy
Introduction
The loss of dopaminergic neurons and formation of Lewy bodies in the substantia nigra are the main pathological characteristics of Parkinson's disease (PD) (1). Studies have found that aging, heredity, epigenetic changes, and mitochondrial dysfunction are principal factors promoting the occurrence of PD (2,3). At present, the specific molecular mechanisms of dopaminergic neuron degeneration in the substantia nigra are not yet fully understood. Recent studies have shown that neuroinflammation is involved in the occurrence and development of PD (4) and that neuroinflammation and oxidative stress damage mediated by neuroglial activation play an important role in the degeneration of dopaminergic neurons (5,6). Our previous study revealed that lipopolysaccharide (LPS)-activated microglia could release pro-inflammatory factors, which resulted in inflammatory and oxidative injury in dopaminergic neurons, leading to impaired cell proliferation and increased apoptosis (7). Therefore, the identification of molecular targets to reduce the death of dopaminergic neurons caused by inflammation or oxidative injury may help slow down the progression of PD.
Sirtuin 3 (SIRT3), a mitochondrial protein with deacetylase activity, has a number of biological functions and participates in the regulation of inflammatory damage, oxidative stress, reactive oxygen species (ROS) clearance and mitochondrial dysfunction (8,9). SIRT3 plays a pivotal role in the occurrence and development of a variety of neurodegenerative diseases (10,11). SIRT3 knockdown suppresses the protective effects of dioscin on β-amyloid (Aβ) oligomer-mediated ROS production, cell apoptosis and neurotoxicity in an Alzheimer's disease (AD) model (12). Mitochondrial SIRT3 has been shown to exert neuroprotective effects in Huntington's disease by improving mitochondrial dynamics and oxidative challenges (13). Inflammatory injury and mitochondrial dysfunction can facilitate ROS generation and the irreversible destruction of protein and DNA structure. Neurons are particularly susceptible to mitochondrial dysfunction, and the elevated ROS level promotes neuronal death and protein deposition, thus accelerating the progression of neurodegenerative diseases (14). SIRT3 exerts a neuroprotective effect by eliminating mitochondrial ROS, inhibiting inflammatory cytokine expression and cell apoptosis, regulating energy metabolism and the balance of mitochondrial respiratory electron transport chain, and increasing adenosine triphosphate (ATP) production (15,16). The newly synthesized rhamnoside derivative PL171 can rescue Aβ42 oligomer-associated cell senescence, mitochondrial dysfunction and oxidative stress injury by activating SIRT3 in an AD cell model (17). By improving mitochondrial function, SIRT3 protects parvalbumin and calretinin interneurons against Aβ-mediated degeneration and dysfunction in amyloid precursor protein and presenilin 1 (APP/PS1) double transgenic AD model mice, thus repressing neuronal network hyperactivity (18). Zhang et al (19) proposed that ε-viniferin could enhance SIRT3-mediated forkhead box O3 (FOXO3) deacetylation, increase ATP production, decrease ROS production and alleviate mitochondrial depolarization, thus inhibiting rotenone-induced neuronal cell apoptosis. Oxidative stress injury and mitochondrial dysfunction caused by abnormal aggregation of Aβ, α-synuclein, or Huntington's disease in different target neurons can result in severe neuron loss (20-22), which is the common pathogenesis of different neurodegenerative diseases. SIRT3 promotes the survival of target neurons, such as dopaminergic or cholinergic neurons (23). Thus, SIRT3 could be used as a potential intervention target for dopaminergic neuron survival promotion.
A previous study demonstrated that SIRT3 expression in the brain tissue of PD mice is reduced, and that SIRT3 gene deletion significantly exacerbates the degeneration of dopaminergic neurons in the substantia nigra and decreases the expression of mitochondrial antioxidant enzyme superoxide dismutase 2 (SOD2), suggesting that SIRT3 plays a protective role in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced dopaminergic neurotoxicity by scavenging free radicals in mitochondria (11). SIRT3 also exerts a protective effect on dopaminergic neurons by enhancing the activities of mitochondrial citrate synthase and isocitrate dehydrogenase (24). Theacrine inhibits dopaminergic neuron apoptosis by directly increasing SIRT3 activity, which has been shown to restore mitochondrial functions and reduce ROS accumulation in multiple animal or cell models of PD (25). In addition, SIRT3 expression has also been shown to be reduced in an MPTP-induced PD cell model, with SIRT3 overexpression inhibiting neuronal cell apoptosis (26). Our previous study found that microglia activation-induced oxidative stress could downregulate the protein levels of SIRT3 and SOD2 in dopaminergic neurons (7). The aforementioned results suggested that SIRT3 was associated with the occurrence and development of PD, although the detailed mechanism of action has not been fully explored. It is also unclear whether SIRT3 can inhibit the microglia activation-mediated death of dopaminergic neurons.
A number of studies have revealed that SIRT3 expression can promote mitochondrial autophagy or inhibit nucleotide-binding oligomerization domain, leucine rich repeat and pyrin domain-containing protein 3 (NLRP3) inflammasome activation in various cell types (27-29). Targeting SIRT3 to improve mitophagy and alleviate senescence in bone marrow mesenchymal stem cells may be a potential therapeutic strategy for alleviating senile osteoporosis caused by advanced glycation end products (27). SIRT3 overexpression has been found to prominently facilitate wound healing speed under high glucose conditions by activating mitophagy (28). SIRT3 overexpression can also activate mitochondrial autophagy and reduce mitochondrial damage, as well as cardiomyocyte apoptosis; on the contrary, SIRT3 deficiency can inhibit mitophagy by reducing FOXO3A deacetylation and Parkin expression, and contribute to the development of diabetic cardiomyopathy (29). Zheng et al (30) have demonstrated that SIRT3 upregulation significantly attenuates Ti particle-induced osteogenic suppression by improving osteogenesis and inhibiting the NLRP3 inflammasome in vivo and in vitro. SIRT3 overexpression can markedly weaken the activation of NLRP3 inflammasome in human vascular endothelial cells and delay the progression of vascular inflammation (31). However, whether SIRT3 expression in dopaminergic neurons can alleviate the inflammation and oxidative stress injury caused by microglia activation through the mitophagy-NLRP3 inflammasome pathway remains unknown.
The aim of the present study was to explore whether SIRT3 expression exerts a protective effect against cytotoxicity resulting from microglia activation in dopaminergic neuronal cells (DACs) by improving mitochondrial function. In addition, whether the mitophagy-NLRP3 inflammasome pathway was involved in this SIRT3 neuroprotection was also investigated.
Materials and methods
Cell culture
The MN9D mouse midbrain dopaminergic cell line was sourced from the American Type Culture Collection and maintained in high-glucose DMEM (Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% (v/v) fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.), 100 µg/ml streptomycin and 100 U/ml penicillin (Invitrogen; Thermo Fisher Scientific, Inc.) in an incubator with an atmosphere of 5% CO2 at 37˚C. The murine microglial cell line BV-2 (cat. no. GDC0311; China Center for Type Culture Collection) was cultured in flasks containing DMEM supplemented with 10% FBS at 37˚C with 5% CO2.
Transwell co-culture system
A total of 2x105 DACs (MN9D cells) were cultured in 6-well plates in the lower chamber of a Transwell co-culture system (cat. no. 3412; Corning, Inc.), with 2x105 microglia (BV-2 cells) in the upper chamber. LPS (1 µg/ml, MilliporeSigma) was added to the microglia layer. Cell migration was observed using an Olympus IX71 microscope (magnification, x100; Olympus Corporation). DACs and microglia shared the same DMEM medium with FBS at 37˚C for 48 h, although direct cell-to-cell interaction was unlikely, as two types of cells were physically separated by a 0.4 µm polycarbonate membrane.
Experimental groups
Experiments were conducted using the following groups: i) Blank, DACs were co-cultured with microglia for 48 h; ii) control, DACs were co-cultured with microglia exposed to 1 µg/ml LPS for 48 h; iii) SIRT3 or Vector, DACs in the chamber were pretreated with SIRT3-adenoviral or empty vector (Vigene Biosciences, Inc.) for 6 h respectively, and DACs were then co-cultured with microglia stimulated with 1 µg/ml LPS for 48 h; iv) siSIRT3 or Scrambled, DACs in the chamber were pretreated with SIRT3 small interfering RNA (siRNA) or negative control siRNA (Santa Cruz Biotechnology, Inc.) for 6 h respectively, then co-cultured with microglia exposed to 1 µg/ml LPS for 48 h.
Adenovirus infection and siRNA transfection
A recombinant adenoviral vector overexpressing SIRT3 (Ad-SIRT3-GFP; cat. no. VH820507) and a non-targeting adenoviral vector (Ad-GFP) were purchased premade from Vigene Biosciences, Inc. SIRT3 siRNA (cat. no. sc-61556) and negative control siRNA (cat. no. sc-37007) were purchased from Santa Cruz Biotechnology, Inc. For overexpression of SIRT3, when DACs reached a ~50% confluence, cells were infected with Ad-SIRT3-GFP (SIRT3 group) or Ad-GFP (Vector group) at a multiplicity of infection of 100, after 6 h, the cells were switched to fresh medium and incubated for an additional 24 h. For SIRT3 knockdown, when DACs reached ~60% confluence, the cells were transfected with 50 nM SIRT3 siRNA (siSIRT3 group) or negative control siRNA (Scrambled group) using Lipofectamine® 2000 (cat. no. 11668019; Invitrogen; Thermo Fisher Scientific, Inc.) at 37˚C for 6 h according to the manufacturer's instructions. After transfection, the serum-free transfection mixture was replaced with DMEM + 10% FBS and cells were cultured for an additional 24 h. SIRT3 and SOD2 gene expression in DACs was evaluated using reverse transcription-quantitative polymerase chain reaction (RT-qPCR) and western blotting. After verifying the overexpression and knockdown of SIRT3, the cells were used for subsequent experiments.
Evaluation of intracellular ROS production
The fluorescent probe dihydroethidium (cat. no. S0063) and the hydrogen peroxide assay kit (cat. no. S0038) were obtained from Beyotime Institute of Biotechnology and used to assess the intracellular accumulation of superoxide anion and hydrogen peroxide, respectively, according to the manufacturer's instructions. The fluorescence intensity was measured using a plate reader (Thermo Fisher Scientific, Inc.). These experiments were carried out as previously described (7).
RT-qPCR
TRIzol® (Thermo Fisher Scientific, Inc.) was used to extract total RNA from DACs according to the manufacturer's protocol. RNA concentration was detected using a NanoDrop™ 2000 spectrophotometer (Thermo Fisher Scientific, Inc.). cDNA synthesis was performed from 1.5 µg of RNA using HiScript 1st Strand cDNA Synthesis Kit (cat. no. R111-02; Vazyme Biotech Co., Ltd.) according to the manufacturer's protocol. RT-qPCR was performed for target genes using SYBR Green PCR kit (cat. no. Q111-02; Vazyme Biotech Co., Ltd.), as per the manufacturer's instructions. The thermocycling conditions were as follows: Initial denaturation at 95˚C for 5 min; followed by 40 cycles of denaturation at 95˚C for 10 sec, annealing at 60˚C for 30 sec and extension at 72˚C for 10 sec. GAPDH served as the internal reference gene and the relative mRNA expression was measured using the 2-ΔΔCq method (32). The primer sequences used were as follows: SIRT3 forward, 5'-ATGCCTGAAGACAGCTCCAACAC-3' and reverse, 5'-AGACATCCCTGGTCAGCCTTTCC-3'; SOD2 forward, 5'-TAAGGAGAAGCTGACAGCCGTGT-3' and reverse, 5'-AGAGCAGGCAGCAATCTGTAAGC-3'; LC3 forward, 5'-CAGCCACACCCTTTCACT-3' and reverse, 5'-GTCAGCAACCCCTGGAC-3'; Parkin forward, 5'-AAACAAGCAACCCTCACCT-3' and reverse, 5'-GGCACTCACCACTCATCC-3'; p62 forward, 5'-CAGCACAGGCACAGAAGA-3' and reverse, 5'-GTCCCACCGACTCCAAG-3'; cytochrome c oxidase IV (COX-IV) forward, 5'-CTACCCCTTGCCTGATGTG-3' and reverse, 5'-TGGATGCGGTACAACTGAA-3'; and GAPDH forward, 5'-TGTTTCCTCGTCCCGTAGA-3' and reverse, 5'-ATCTCCACTTTGCCACTGC-3'.
Cell apoptosis
A total of 2x105 DACs were collected by centrifugation at 168 x g for 5 min at room temperature, and Annexin V-FITC binding solution was used to resuspend the cell pellet gently. Next, 20 µg/ml Annexin V-FITC and 20 µg/ml propidium iodide (PI) were added to the suspension in sequence and mixed gently, incubated for 20 min at room temperature (20-25˚C) in the dark, then placed in icy water for 5 min. Detection was finished within 1 h from staining and was carried out using a flow cytometer (FACSCalibur™; BD Biosciences). CellQuest Pro software (version 5.1.0; BD Biosciences) was used for analysis. The Annexin V-FITC/PI assay kit (cat. no. 556547) was purchased from BD Bioscience. Experimental procedures were performed as previously described (7).
Cell cycle distribution analysis
A total of 2x105 DACs were collected by centrifugation at 168 x g for 5 min at room temperature, washed with ice-cold PBS, then fixed with ice-cold 70% ethanol (cat. no. 459836; MilliporeSigma) overnight in a freezer at 4˚C. The fixed cells were centrifuged at 1,000 x g for 5 min to collect the pellet, washed with ice-cold PBS and resuspended with staining buffer containing 50 µg/ml PI, 100 µg/ml RNase A, and 0.2% Triton X-100. The cell suspension was incubated at 37˚C for 30 min in the dark. Flow cytometry (FACSCalibur; BD Biosciences) was performed to evaluate cell cycle distribution within 24 h from staining. CellQuest Pro software (version 5.1.0; BD Biosciences) was used for analysis. The cell cycle analysis kit (cat. no. C1052) was purchased from Beyotime Institute of Biotechnology.
Mitochondrial membrane potential (ΔΨm) measurement
Tetraethylbenzimidazolylcarbocyanine iodide (JC-1, 10 µg/ml; cat. no. C2006; Beyotime Institute of Biotechnology) was added to the 6-well plates with 2x105 DACs and incubated at 37˚C for 20 min. Next, the cells were collected by centrifugation at 168 x g for 5 min at room temperature, the supernatant was discarded, and the cells were washed with JC-1 staining buffer. The cell suspension was centrifuged at 168 x g for 5 min at room temperature and pellets were resuspended with JC-1 staining buffer again. The fluorescence intensity was detected promptly at dual excitation wavelengths (shift from 490 to 525 nm) and dual emission wavelengths (shift from 530 to 590 nm) using an Olympus IX71 fluorescence microscope (Olympus Corporation; magnification, x100). There is an inverse relationship between the degree of mitochondrial depolarization and the ratio value of red/green fluorescence intensity (7).
Mitochondrial permeability transition pore (mPTP) opening assay
A total of 2x105 DACs were incubated with 5 µM calcein-AM (cat. no. sc-203865; Santa Cruz Biotechnology, Inc.) and 5 µM CoCl2 (cat. no. 60818; MilliporeSigma) simultaneously for 15 min at 37˚C. Cells were centrifuged at 168 x g for 5 min at room temperature and washed, and cell pellets were resuspended in 0.4 ml PBS. Flow cytometry (FACSCalibur; BD Biosciences) was then performed to detect the mitochondrial calcein-AM fluorescence. CellQuest Pro software (version 5.1.0; BD Biosciences) was used for analysis. The maximum excitation and emission wavelengths of calcein-AM are 494/517 nm. There is an inverse correlation between the number of mPTP opening and the calcein-AM fluorescence intensity (7).
Mitochondrial and cytosolic protein extraction
Mitochondrial and cytosolic protein extractions were performed using Cell Mitochondria Isolation Kit (cat. no. C3601; Beyotime Institute of Biotechnology). DACs were collected and resuspended in mitochondrial separation reagent containing 1 mM PMSF. After incubation on ice for 15 min, the cell suspension was homogenized, then centrifuged at 1,000 x g for 10 min at 4˚C. The supernatant was then centrifuged at 11,000 x g for 10 min at 4˚C. The precipitation was blended with mitochondrial lysate solution to obtain mitochondrial proteins. The supernatant was centrifuged at 12,000 x g for 20 min at 4˚C to obtain cytosolic proteins.
Western blotting
The treated cells were harvested and completely lysed with RIPA lysis buffer (cat. no. P0013B), and a BCA protein assay kit (cat. no. P0012; Beyotime Institute of Biotechnology) was used to determine the total protein level in the sample. Cell protein extracts (20 µg) were separated using 10-15% SDS-PAGE and transferred to PVDF membranes. After blocking with 5% non-fat milk powder (cat. no. P0216; Beyotime Institute of Biotechnology) at room temperature for 1.5 h, the following primary antibodies were added and incubated overnight at 4˚C: Anti-SIRT3 (cat. no. 5490), anti-cytochrome c (Cyt c, cat. no. 4272), anti-Bax (cat. no. 2772), anti-Bcl-2 (cat. no. 3498), anti-cleaved caspase-3 (cat. no. 9664), anti-COX-IV (cat. no. 4844) and anti-caspase-1 (cat. no. 24232) (all from Cell Signaling Technology, Inc. and diluted 1:1,000); anti-cyclophilin D (CypD, cat. no. ab167513), anti-LC3 (cat. no. ab192890), anti-Beclin-1 (cat. no. ab207612) and anti-NLPR3 (cat. no. ab270449) (all from Abcam and diluted 1,1,000); anti-SOD2 (cat. no. 24127-1-AP; 1:1,000) and anti-GAPDH (cat. no. 10494-1-AP; 1:2,000) (both from ProteinTech Group, Inc.). Subsequently, HRP-conjugated goat anti-rabbit IgG (H+L) secondary antibody (cat. no. SA00001-2; 1:5,000; ProteinTech Group) was added at room temperature for 2 h. The bands were visualized using an enhanced chemiluminescence reagent kit (cat. no. P0018M; Beyotime Institute of Biotechnology). The intensity of the optical bands was quantified using AlphaEaseFC software (version 4.0.0; Alpha Innotech Corporation).
Detection of caspase-3 and caspase-9 enzyme activities
The spectrophotometric method (at a wavelength of 405 nm) was used to measure the caspase-3 and caspase-9 enzyme activities in the cell lysate. The caspase-3 and caspase-9 testing kits (cat. nos. C1116 and C1158) were purchased from Beyotime Institute of Biotechnology.
Detection of IL-1β and IL-18 levels
IL-1β and IL-18 levels in the cell lysate were determined using the spectrophotometric method (at a wavelength of 450 nm), according to the manufacturer's instructions. The IL-1β and IL-18 ELISA kits (cat. nos. PI301 and PI553) were purchased from Beyotime Institute of Biotechnology.
Statistical analysis
SPSS software (version 20.0; IBM Corporation) was used to analyze the data. The minimal number of independent experiments performed for the different assays was four. Statistical significance of the differences between groups was determined using one-way ANOVA followed by the least significant difference (three groups) or Tukey's post hoc test (four groups). P<0.05 was considered to indicate a statistically significant difference.
Results
SIRT3 overexpression mitigates microglia activation-mediated cytotoxicity in DACs
Our previous studies confirmed that inflammatory factors released from activated microglia could promote apoptosis through the mitochondrial pathway and markedly reduce SIRT3 gene expression in DACs (7,16). In the present study, whether SIRT3 expression played a neuroprotective role in DACs was further examined.
As shown in Fig. 1A, the mRNA levels of SIRT3 and SOD2 in DACs transfected with SIRT3 plasmid (SIRT3 group) increased ~7-fold, compared with those in the vector or control groups. Moreover, the protein levels of SIRT3 and SOD2 in the SIRT3 group were significantly higher than those in other two groups (Fig. 1B).
Figure 1.

SIRT3 overexpression mitigates microglia-mediated cytotoxicity in DACs. DACs were incubated with SIRT3 adenoviral vector or non-targeting empty vector, then co-cultured with activated-microglia for 48 h. The microglia were stimulated with 1 µg/ml lipopolysaccharide. (A) The mRNA expression levels of SIRT3 and SOD2 in DACs were measured using reverse transcription-quantitative PCR. (B) The protein expression of SIRT3 and SOD2 in DACs was determined using western blotting. (C) The fluorescent probe dihydroethidium was used to detect intracellular superoxide anion levels, and a hydrogen peroxide assay kit was used to detect intracellular hydrogen peroxide levels in DACs. (D) Cell apoptosis rate and (E) cell cycle distribution of DACs were measured using flow cytometry. (F) The fluorescence probe JC-1 was used to detect the loss of mitochondrial membrane potential. Scale bar, 100 µm. (G) Mitochondrial calcein-AM green fluorescence intensity was examined using flow cytometry to evaluate the number of the membrane permeability transport pores opening in DACs. MitoTracker (red) staining indicates the localization of mitochondria. Scale bar, 50 µm. (H) Western blotting was performed to detect the protein levels of mitochondrial cyclophilin D in DACs. Data in A, B, C, F, G and H are shown as fold change over the Control group. Data are presented as mean ± S.D. n=4. *P<0.05 vs. Control; #P<0.05 vs. Vector. SIRT3, sirtuin 3; DAC, dopaminergic neuronal cell; SOD2, superoxide dismutase 2; JC-1, tetraethylbenzimidazolylcarbocyanine iodide.
SIRT3 overexpression also inhibited superoxide anion and hydrogen peroxide production in DACs induced by microglia activation, compared with the control and vector groups (Fig. 1C). The results of flow cytometry showed that SIRT3 overexpression in DACs could significantly decrease their apoptotic rate (Fig. 1D), increase the frequency of S and G2/M phase cells, and reduce the number of G0/G1 phase cells (Fig. 1E). JC-1 and calcein-AM fluorescent probe assays indicated that SIRT3 overexpression in DACs reduced ΔΨm depolarization (Fig. 1F) and microglia activation-induced mPTP opening (Fig. 1G). SIRT3 overexpression also significantly inhibited the protein expression of CypD in the SIRT3 group (Fig. 1H). These results revealed that SIRT3 overexpression in DACs could relieve microglia activation-induced neuron injury.
Microglia activation-induced cytotoxicity is increased following SIRT3 knockdown
The next experiments aimed to determine whether SIRT3 knockdown could exacerbate microglia activation-induced cytotoxicity in DACs. As shown in Fig. 2A, the mRNA levels of SIRT3 and SOD2 in DACs transfected with SIRT3 siRNA (siSIRT3 group) were decreased by ~70% compared with those in the scrambled (transfected with negative control siRNA) or control groups. The protein levels of SIRT3 and SOD2 in the siSIRT3 group were also significantly lower than those in the other two groups (Fig. 2B).
Figure 2.

SIRT3 knockdown by siRNA transfection impairs DAC function. DACs were treated with SIRT3 siRNA (siSIRT3 group) or scrambled siRNA, then co-cultured with activated-microglia for 48 h. Microglia were exposed to 1 µg/ml lipopolysaccharide. (A) Reverse transcription-quantitative PCR was performed to measure the mRNA expression of SIRT3 and SOD2 in DACs. (B) The protein levels of SIRT3 and SOD2 in DACs were determined using western blotting. (C) The fluorescent probe dihydroethidium was used to detect intracellular superoxide anion level, and a hydrogen peroxide assay kit was used to detect intracellular hydrogen peroxide level in DACs. (D) Cell apoptosis rate and (E) cell cycle distribution of DACs in the Control, Scrambled and siSIRT3 groups were measured using flow cytometry. (F) The fluorescence probe JC-1 was used to detect the loss of mitochondrial membrane potential. Scale bar, 100 µm. (G) Mitochondrial calcein-AM green fluorescence intensity was examined using flow cytometry to evaluate the number of the membrane permeability transport pores opening in DACs. MitoTracker (red) staining indicates the localization of mitochondria. Scale bar, 50 µm. (H) Protein level of mitochondrial cyclophilin D in DACs was determined using western blotting. Data in A, B, C, F, G and H are shown as fold change over Control group. Data are presented as mean ± S.D. n=4. *P<0.05 vs. Control; #P<0.05 vs. Scrambled. SIRT3, sirtuin 3; DAC, dopaminergic neuronal cell; si/siRNA, small interfering RNA; SOD2, superoxide dismutase 2; JC-1, tetraethylbenzimidazolylcarbocyanine iodide.
SIRT3 silencing further promoted intracellular microglia activation-induced ROS production in the siSIRT3 group (Fig. 2C). The flow cytometry results demonstrated that SIRT3 knockdown in DACs significantly increased their apoptotic rate (Fig. 2D), decreased the frequency of cells in the S and G2 phases, and increased the number of cells in the G0/G1 phase (Fig. 2E). JC-1 and calcein-AM fluorescent probe assays indicated that SIRT3 knockdown in DACs further enhanced microglia activation-induced ΔΨm depolarization (Fig. 2F) and mPTP opening (Fig. 2G). siSIRT3 transfection also promoted the protein expression of CypD in the siSIRT3 group (Fig. 2H). In conclusion, these data provided evidence that SIRT3 knockdown in DACs exacerbated microglia activation-induced cell damage.
SIRT3-induced neuroprotection in DACs is associated with the mitochondrial apoptotic pathway
In order to study the molecular mechanisms underlying SIRT3-induced neuroprotection in DACs against microglia activation-mediated cytotoxicity, ELISA and western blotting were performed to measure the levels of several key proteins in the mitochondrial apoptotic signaling pathway. Compared with the vector or control group, SIRT3 overexpression significantly decreased the protein levels of cytoplasmic Cyt C, Bax and total cleaved caspase-3 in SIRT3-overexpressing DACs, and increased the protein levels of mitochondrial Cyt C and total Bcl-2 (Fig. 3A). By contrast, compared with the control or scrambled group, SIRT3 knockdown significantly increased the expression of cytoplasmic Cyt C, Bax and total cleaved caspase-3 in siSIRT3 group DACs, and decreased the expression of mitochondrial Cyt C and total Bcl-2 (Fig. 3B).
Figure 3.

SIRT3 in DACs contributes to neuroprotection via mitochondrial apoptotic pathway. (A) Protein levels of Bax, Bcl-2, cleaved caspase-3, Mito-Cyt C and Cyto-Cyt C in DACs from the SIRT3 group, Vector group, and Control group were measured using western blotting. (B) Protein levels of Bax, Bcl-2, cleaved caspase-3, Mito-Cyt C and Cyto-Cyt C in DACs from the siSIRT3 group, Scrambled group, and Control group were determined using western blotting. Representative western blot (left panel) and quantification (right panel) of Bax, Bcl-2, Cyt C and cleaved caspase-3 protein expression in DACs transfected with scrambled siRNA or siSIRT3 for 48 h. The spectrophotometric method was used to detect caspase-3 and caspase-9 enzyme activities in DACs transfected with SIRT3 plasmid or empty vector (C), and scrambled siRNA or SIRT3 siRNA (D) for 48 h. Results are normalized to those of the Control group. Data are presented as mean ± S.D. n=4. *P<0.05 vs. Control; #P<0.05 vs. Vector or Scrambled. SIRT3, sirtuin 3; DAC, dopaminergic neuronal cell; si/siRNA, small interfering RNA; Mito-Cyt C, mitochondrial cytochrome c; Cyto-Cyt C, cytoplasmic cytochrome c.
In addition, SIRT3 overexpression significantly reduced the enzymatic activities of caspase-3 and -9 (Fig. 3C), while SIRT3 knockdown significantly increased them (Fig. 3D). These results demonstrated that SIRT3 in DACs attenuated microglia activation-induced cell damage through the mitochondrial signaling pathway, which included a decreased Bax protein level and caspase-3/9 enzymatic activity, decreased mitochondrial Cyt C release into the cytoplasm and increased Bcl-2 protein levels.
The mitophagy-NLRP3 pathway is correlated with the neuroprotective effect of SIRT3 in DACs
In order to clarify whether the neuroprotection of SIRT3 was associated with mitochondrial autophagy and NLRP3 inflammasome activation, the protein levels of the key molecules in the mitophagy-NLRP3 inflammasome pathway were detected. As shown in Fig. 4A, the protein levels of LC3II/LC3I and Beclin-1 in the control group were significantly reduced compared with the blank group, and those of NLRP3 and caspase-1 were significantly increased. Compared with the control group, SIRT3 overexpression increased the levels of LC3II/LC3I and Beclin-1, and decreased those of NLRP3 and caspase-1. Conversely, SIRT3 knockdown led to an opposite protein expression trend. The results of RT-qPCR showed that microglia activation significantly decreased the mRNA levels of LC3 and Parkin, and increased the mRNA levels of p62 and mitochondrial marker COX-IV. SIRT3 knockdown further exacerbated the changing trends of these genes expression, except for the COX-IV gene. However, SIRT3 overexpression increased LC3 and Parkin mRNA levels, and decreased those of p62 (Fig. 4B). Furthermore, compared with the blank group, microglia activation significantly elevated the protein levels of IL-1β and IL-18 in the control group. Compared with the control group, SIRT3 knockdown further increased IL-1β and IL-18 levels in siSIRT3-transfected DACs, but decreased them in SIRT3-overexpressing DACs (Fig. 4C).
Figure 4.

SIRT3 overexpression promotes mitochondrial autophagy and inhibits NLRP3 inflammasome activation in DACs. (A) Protein expression of LC3II/LC3I, Beclin-1, NLRP3 and caspase-1 in DACs treated with 1 µg/ml lipopolysaccharide, SIRT3 plasmid or SIRT3 siRNA were analyzed using western blotting. (B) The mRNA expression levels of LC3, Parkin, p62 and COX-IV in DACs from the Blank, Control, SIRT3 and siSIRT3 group were detected using reverse transcription-quantitative PCR. (C) IL-1β and IL-18 protein levels in DACs from the Blank, Control, SIRT3 and siSIRT3 group were measured using ELISA. *P<0.05 vs. blank; #P<0.05 vs. control. SIRT3, sirtuin 3; DACs, dopaminergic neuronal cells; siRNA, small interfering RNA; NLRP3, nucleotide-binding oligomerization domain, leucine-rich repeat and pyrin domain-containing protein 3.
Discussion
In the present study, it was found that SIRT3 overexpression in dopaminergic neurons reduced microglia activation-mediated apoptosis and promoted cell cycle progression, which were conducive to the neuronal survival, whereas SIRT3 knockdown had the opposite effects in DACs upon exposure to LPS-challenged microglia. Pharmacologically increasing the SIRT3 level could counteract α-synuclein-induced mitochondrial dysfunction by decreasing α-synuclein oligomer formation and normalizing mitochondrial bioenergetics in a rodent model of PD (33). Xu et al (34) suggested that rat chondrocytes underwent mitochondrial dysfunction, inflammation, cell apoptosis and degeneration following IL-1β stimulation, which were inhibited by SIRT3 overexpression and were enhanced in chondrocytes following SIRT3 knockdown. Decreased NAD+ levels and SIRT3 activity are involved in the aging process and have been pathologically associated with PD pathogenesis (35). SIRT3 chemical activators and NAD+ precursors can upregulate SIRT3 activity to protect against dopaminergic neuron degeneration in PD models (35). A decrease in SIRT3 expression via the inhibitor nicotinamide or siRNA transfection results in the accumulation of Cyt C oxidase 1 acetylation, increased cell apoptosis and decreased ΔΨm in primary neuronal cells (36). Clinical and experimental studies have demonstrated that the expression of peroxisome proliferator-activated receptor coactivator 1α (PGC-1α) is decreased in the brain tissue of patients with PD and animal models (26). PGC-1α overexpression can inhibit α-synuclein aggregation, and PGC-1α can also bind to the SIRT3 gene promoter to increase SIRT3 transcription, SIRT3 activates SOD2 and ATP synthase to reduce ROS accumulation and ATP consumption, respectively, thereby reducing neurotoxicity and inhibiting the loss of dopaminergic neurons (26).
The results of the present study revealed that the molecular mechanism underlying the protective effect of SIRT3 against inflammatory and oxidative damage in dopaminergic neurons is linked to the mitochondrial apoptosis pathway. Specifically, SIRT3 overexpression in dopaminergic neurons could maintain ΔΨm, prevent mPTP opening and Cyt C release from the mitochondria to the cytoplasm, inhibit caspase-3/9 activity, upregulate Bcl-2 protein expression, and downregulate CypD and Bax protein expression. SIRT3 knockdown resulted in an opposite trend in these parameters. PL171 inhibits ΔΨm reduction and ROS generation induced by Aβ42 oligomers in an SIRT3-dependent manner, and the weakening of SIRT3 activity abolishes the protective actions of PL171 in human neuronal cells (17). The neuroprotection of the sesamin and sesamol compounds in H2O2-treated human neuronal cells can be attributed to the activation of SIRT3-FOXO3A expression, Bax inhibition and Bcl-2 upregulation (37). Ginsenoside Rb1 protects human umbilical vein endothelial cells from high glucose-induced apoptosis through the SIRT3 signaling pathway. Rb1 increases the activities of antioxidant enzymes, decreases Cyt C release from the mitochondria to the cytosol and maintains the ΔΨm; the action of Rb1 against high glucose-induced mitochondria-related apoptosis is inhibited by SIRT3 inhibitor 3-(1H-1,2,3-triazol-4-yl) pyridine (3-TYP) (38). Yang et al (39) reported that ischemic injury resulted in cell apoptosis and mPTP opening by reducing SIRT3, which helped identify a novel target for the treatment of ischemic stroke. The conclusions of this study are in line with those of previous reports.
The present study also determined that LPS-stimulated microglia activation could reduce the levels of key proteins in the mitophagy pathway and increase the expression of important molecules in the NLRP3 inflammasome pathway in DACs. SIRT3 silencing further aggravated mitophagy inhibition and NLRP3 inflammasome activation. Conversely, SIRT3 overexpression could significantly reverse these phenomena. SIRT3 expression in dopaminergic neurons relieved microglial activation-induced cytotoxicity, which was associated with the mitophagy-NLRP3 inflammasome pathway. It has been reported that SIRT3 promotes autophagy through the adenosine monophosphate-activated protein kinase (AMPK)-mTOR signaling pathway, thereby reducing α-synuclein aggregation, inhibiting oxidative stress and alleviating mitochondrial dysfunction in a rotenone-induced PD cell model (40). Moderate oxygen-glucose deprivation can upregulate the expression of SIRT3 in neurons, and SIRT3 also facilitates mitochondrial autophagy through the AMPK-mTOR pathway, thus contributing to neuronal survival (41). SIRT3 overexpression can reverse autophagy inhibition caused by IL-1β in rat chondrocytes (33). Furthermore, SIRT3 can bind to and deacetylate PTEN-induced kinase 1 (PINK1) and Parkin to facilitate mitochondrial autophagy. The level of mitophagy is markedly increased in diabetic corneal epithelial cells when the FOXO3A/PINK1-Parkin pathway is activated via SIRT3 overexpression (28). SIRT3 knockdown results in the downregulation of Beclin-1 and LC3II, suggesting that SIRT3 is an important regulator of autophagy in Aβ1-42 oligomer-treated HT22 cells (12). Mitochonic acid 5 effectively alleviates neuroinflammatory damage by inhibiting mitochondrial dysfunction and enhancing cell survival. The protective effect of mitochonic acid 5 on neuronal damage can be attributed to the activation of AMPK-SIRT3 pathway and Parkin-related mitophagy (42). Metformin suppresses IL-1β-mediated oxidative and osteoarthritis-like inflammatory damage by activating the SIRT3/PINK1/Parkin signaling pathway, and the SIRT3 inhibitor 3-TYP effectively inhibits mitophagy initiation and reduces the LC3II/LC3I ratio (43). Cyperone increases SIRT3 expression and decreased ROS production in the hippocampus of depressed mice, NLRP3 inflammasome-related proteins including NLRP3, caspase-1, IL-1β and IL-18 were downregulated (44). The aforementioned results and those of the present study are consistent.
Stilbene glycoside treatment facilitates mitophagy and cell survival and reduces apoptosis; however, SIRT3 knockdown reduces ΔΨm, cell viability, and LC3II/I and Bcl-2 levels, while markedly increasing apoptosis, and Bax and caspase 3 protein expression in ischemic PC12 cells (45). SIRT3-knockout macrophages exhibit autophagy dysfunction, as well as enhanced NLRP3 inflammasome expression and vascular inflammation, whereas SIRT3 overexpression induces autophagosome maturation and inhibits NLRP3 inflammasome activation (46). AEDC, a cycloartane triterpenoid isolated from Actaea vaginata, increased SIRT3 expression to reverse mitochondrial dysfunction and activate AMPK, which together induced autophagy and in turn suppressed the NLRP3 inflammasome activation in LPS-treated THP-1 macrophages (47). The SIRT3 agonist resveratrol inhibits NLRP3 inflammasome activation by maintaining mitochondrial integrity and amplifying the effect of autophagy (48). Resveratrol also protects neuronal cells against tunicamycin-induced endoplasmic reticulum stress damage by the SIRT3-mediated mitochondrial autophagy pathway (49). The data in the aforementioned reports are also in line with our findings. SIRT3 in dopaminergic neurons plays a role in resisting neuroinflammation through the mitophagy-NLRP3 inflammasome pathway. However, the findings the present study are not sufficient to support this conclusion, which needs to be verified by additional experiments using specific inhibitors and gene silencing in the future. Future experiments should include differentiation of Lund human mesencephalic cells, an immortalized precursor cell line of human dopaminergic neuron, to further validate the results obtained in the present study.
In conclusion, the results of the present study indicated that mitochondrial SIRT3 in dopaminergic neurons can improve mitochondrial dysfunction and decrease ROS generation, thereby reducing the loss of dopaminergic neurons caused by microglia activation, which plays a role in resisting neuroinflammation and oxidative stress injury in PD. The molecular mechanisms underlying this neuroprotection may be associated with increased mitophagy, and reduced NLRP3 inflammasome activation (Fig. 5). The exact association between the mitophagy-NLRP3 pathway and the neuroprotective effect of SIRT3 in DACs needs to be further investigated and verified in the future. The findings of the present study elucidated the mechanism of SIRT3 neuroprotection in PD, and identified new intervention targets for the clinical prevention and treatment of PD and the development of innovative drugs.
Figure 5.
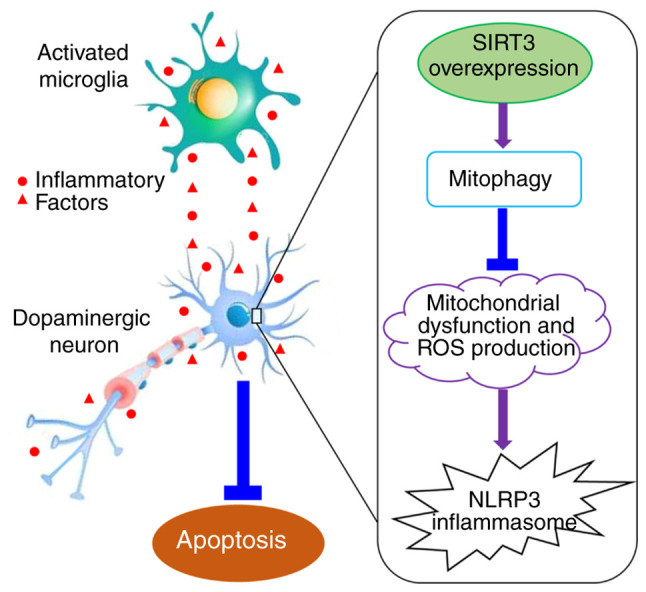
Summary of the results. Lipopolysaccharide-activated microglia release a variety of inflammatory factors that can result in oxidative stress damage and promote the apoptosis of dopaminergic neurons. SIRT3 overexpression in dopaminergic neurons can inhibit apoptosis induced by the excessive microglia activation. The mechanism of this action may be associated with the expression of SIRT3 promoting mitophagy, restoring mitochondrial dysfunction and reducing ROS production, thereby inhibiting NLRP3 inflammasome activation. SIRT3, sirtuin 3; ROS, reactive oxygen species; NLRP3, nucleotide-binding oligomerization domain, leucine rich repeat and pyrin domain-containing protein 3.
Acknowledgements
Not applicable.
Funding Statement
Funding: This study was supported by grants from the Natural Science Foundation of Guangxi Zhuang Autonomous Region of China (grant nos. 2021GXNSFAA220001 and 2018GXNSFAA050002).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
DQJ designed and performed experiments, and drafted manuscript. QMZ designed experiments. LLJ and CSL analyzed the data. SHZ and LCX performed experiments. CSL and LCX reviewed and edited the manuscript. DQJ and QMZ confirm the authenticity of all the raw data. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Martínez-Menárguez JÁ, Martínez-Alonso E, Cara-Esteban M, Tomás M. Focus on the small GTPase Rab1: A key player in the pathogenesis of Parkinson's disease. Int J Mol Sci. 2021;22(12087) doi: 10.3390/ijms222112087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chu YT, Tai CH, Lin CH, Wu RM. Updates on the genetics of Parkinson's disease: Clinical implications and future treatment. Acta Neurol Taiwan. 2021;30:83–93. [PubMed] [Google Scholar]
- 3.Luo Y, Hoffer A, Hoffer B, Qi X. Mitochondria: A therapeutic target for Parkinson's disease? Int J Mol Sci. 2015;16:20704–20730. doi: 10.3390/ijms160920704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rasheed M, Liang J, Wang C, Deng Y, Chen Z. Epigenetic regulation of neuroinflammation in Parkinson's disease. Int J Mol Sci. 2021;22(4956) doi: 10.3390/ijms22094956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Badanjak K, Fixemer S, Smajić S, Skupin A, Grünewald A. The contribution of microglia to neuroinflammation in Parkinson's disease. Int J Mol Sci. 2021;22(4676) doi: 10.3390/ijms22094676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zheng T, Zhang Z. Activated microglia facilitate the transmission of α-synuclein in Parkinson's disease. Neurochem Int. 2021;148(105094) doi: 10.1016/j.neuint.2021.105094. [DOI] [PubMed] [Google Scholar]
- 7.Jiang DQ, Ma YJ, Wang Y, Lu HX, Mao SH, Zhao SH. Microglia activation induces oxidative injury and decreases SIRT3 expression in dopaminergic neuronal cells. J Neural Transm (Vienna) 2019;126:559–568. doi: 10.1007/s00702-019-02005-z. [DOI] [PubMed] [Google Scholar]
- 8.Lin W, Qian X, Yang LK, Zhu J, Wang D, Hang CH, Wang Y, Chen T. Inhibition of miR-134-5p protects against kainic acid-induced excitotoxicity through Sirt3-mediated preservation of mitochondrial function. Epilepsy Res. 2021;176(106722) doi: 10.1016/j.eplepsyres.2021.106722. [DOI] [PubMed] [Google Scholar]
- 9.Yang W, Wang Y, Hao Y, Wang Z, Liu J, Wang J. Piceatannol alleviate ROS-mediated PC-12 cells damage and mitochondrial dysfunction through SIRT3/FOXO3a signaling pathway. J Food Biochem. 2021;46(e13820) doi: 10.1111/jfbc.13820. [DOI] [PubMed] [Google Scholar]
- 10.Anamika Khanna A, Acharjee P, Acharjee A, Trigun SK. Mitochondrial SIRT3 and neurodegenerative brain disorders. J Chem Neuroanat. 2019;95:43–53. doi: 10.1016/j.jchemneu.2017.11.009. [DOI] [PubMed] [Google Scholar]
- 11.Liu L, Peritore C, Ginsberg J, Kayhan M, Donmez G. SIRT3 attenuates MPTP-induced nigrostriatal degeneration via enhancing mitochondrial antioxidant capacity. Neurochem Res. 2015;40:600–608. doi: 10.1007/s11064-014-1507-8. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Z, Han K, Wang C, Sun C, Jia N. Dioscin protects against Aβ1-42 oligomers-induced neurotoxicity via the function of SIRT3 and autophagy. Chem Pharm Bull (Tokyo) 2020;68:717–725. doi: 10.1248/cpb.c20-00046. [DOI] [PubMed] [Google Scholar]
- 13.Naia L, Carmo C, Campesan S, Fão L, Cotton VE, Valero J, Lopes C, Rosenstock TR, Giorgini F, Rego AC. Mitochondrial SIRT3 confers neuroprotection in Huntington's disease by regulation of oxidative challenges and mitochondrial dynamics. Free Radic Biol Med. 2021;163:163–179. doi: 10.1016/j.freeradbiomed.2020.11.031. [DOI] [PubMed] [Google Scholar]
- 14.Nissanka N, Moraes CT. Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett. 2018;592:728–742. doi: 10.1002/1873-3468.12956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen Y, Wu Q, Shi J, Zhou S. Regulation of SIRT3 on mitochondrial functions and oxidative stress in Parkinson's disease. Biomed Pharmacother. 2020;132(110928) doi: 10.1016/j.biopha.2020.110928. [DOI] [PubMed] [Google Scholar]
- 16.Jiang DQ, Wang Y, Li MX, Ma YJ, Wang Y. SIRT3 in neural stem cells attenuates microglia activation-induced oxidative stress injury through mitochondrial pathway. Front Cell Neurosci. 2017;11(7) doi: 10.3389/fncel.2017.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y, Lu J, Cao X, Zhao H, Gao L, Xia P, Pei G. A newly synthesized rhamnoside derivative alleviates Alzheimer's amyloid-β-induced oxidative stress, mitochondrial dysfunction, and cell senescence through upregulating SIRT3. Oxid Med Cell Longev. 2020;2020(7698560) doi: 10.1155/2020/7698560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng A, Wang J, Ghena N, Zhao Q, Perone I, King TM, Veech RL, Gorospe M, Wan R, Mattson MP. SIRT3 Haploinsufficiency aggravates loss of GABAergic interneurons and neuronal network hyperexcitability in an Alzheimer's disease model. J Neurosci. 2020;40:694–709. doi: 10.1523/JNEUROSCI.1446-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang S, Ma Y, Feng J. Neuroprotective mechanisms of ε-viniferin in a rotenone-induced cell model of Parkinson's disease: Significance of SIRT3-mediated FOXO3 deacetylation. Neural Regen Res. 2020;15:2143–2153. doi: 10.4103/1673-5374.282264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hong C, Seo H, Kwak M, Jeon J, Jang J, Jeong EM, Myeong J, Hwang YJ, Ha K, Kang MJ, et al. Increased TRPC5 glutathionylation contributes to striatal neuron loss in Huntington's disease. Brain. 2015;138:3030–3047. doi: 10.1093/brain/awv188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karuppagounder SS, Xu H, Shi Q, Chen LH, Pedrini S, Pechman D, Baker H, Beal MF, Gandy SE, Gibson GE. Thiamine deficiency induces oxidative stress and exacerbates the plaque pathology in Alzheimer's mouse model. Neurobiol Aging. 2009;30:1587–1600. doi: 10.1016/j.neurobiolaging.2007.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Y, Li H, Li Y, Yang M, Wang X, Peng Y. Velvet antler methanol extracts ameliorate Parkinson's disease by inhibiting oxidative stress and neuroinflammation: From C. elegans to mice. Oxid Med Cell Longev. 2021;2021(8864395) doi: 10.1155/2021/8864395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee S, Jeon YM, Jo M, Kim HJ. Overexpression of SIRT3 suppresses oxidative stress-induced neurotoxicity and mitochondrial dysfunction in dopaminergic neuronal cells. Exp Neurobiol. 2021;30:341–355. doi: 10.5607/en21021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cui XX, Li X, Dong SY, Guo YJ, Liu T, Wu YC. SIRT3 deacetylated and increased citrate synthase activity in PD model. Biochem Biophys Res Commun. 2017;484:767–773. doi: 10.1016/j.bbrc.2017.01.163. [DOI] [PubMed] [Google Scholar]
- 25.Duan WJ, Liang L, Pan MH, Lu DH, Wang TM, Li SB, Zhong HB, Yang XJ, Cheng Y, Liu B, et al. Theacrine, a purine alkaloid from kucha, protects against Parkinson's disease through SIRT3 activation. Phytomedicine. 2020;77(153281) doi: 10.1016/j.phymed.2020.153281. [DOI] [PubMed] [Google Scholar]
- 26.Zhang X, Ren X, Zhang Q, Li Z, Ma S, Bao J, Li Z, Bai X, Zheng L, Zhang Z, et al. PGC-1α/ERRα-Sirt3 pathway regulates DAergic neuronal death by directly deacetylating SOD2 and ATP synthase β. Antioxid Redox Signal. 2016;24:312–328. doi: 10.1089/ars.2015.6403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo Y, Jia X, Cui Y, Song Y, Wang S, Geng Y, Li R, Gao W, Fu D. Sirt3-mediated mitophagy regulates AGEs-induced BMSCs senescence and senile osteoporosis. Redox Biol. 2021;41(101915) doi: 10.1016/j.redox.2021.101915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu J, Kan T, Hu X. Sirt3 regulates mitophagy level to promote diabetic corneal epithelial wound healing. Exp Eye Res. 2019;181:223–231. doi: 10.1016/j.exer.2019.02.011. [DOI] [PubMed] [Google Scholar]
- 29.Yu W, Gao B, Li N, Wang J, Qiu C, Zhang G, Liu M, Zhang R, Li C, Ji G, Zhang Y. Sirt3 deficiency exacerbates diabetic cardiac dysfunction: Role of Foxo3A-Parkin-mediated mitophagy. Biochim Biophys Acta Mol Basis Dis. 2017;1863:1973–1983. doi: 10.1016/j.bbadis.2016.10.021. [DOI] [PubMed] [Google Scholar]
- 30.Zheng K, Bai J, Li N, Li M, Sun H, Zhang W, Ge G, Liang X, Tao H, Xue Y, et al. Protective effects of sirtuin 3 on titanium particle-induced osteogenic inhibition by regulating the NLRP3 inflammasome via the GSK-3β/β-catenin signalling pathway. Bioact Mater. 2021;6:3343–3357. doi: 10.1016/j.bioactmat.2021.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen ML, Zhu XH, Ran L, Lang HD, Yi L, Mi MT. Trimethylamine-N-oxide induces vascular inflammation by activating the NLRP3 inflammasome through the SIRT3-SOD2-mtROS signaling pathway. J Am Heart Assoc. 2017;6(e006347) doi: 10.1161/JAHA.117.006347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta DeltaC(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 33.Park JH, Burgess JD, Faroqi AH, DeMeo NN, Fiesel FC, Springer W, Delenclos M, McLean PJ. Alpha-synuclein-induced mitochondrial dysfunction is mediated via a sirtuin 3-dependent pathway. Mol Neurodegener. 2020;15(5) doi: 10.1186/s13024-019-0349-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu K, He Y, Moqbel SAA, Zhou X, Wu L, Bao J. SIRT3 ameliorates osteoarthritis via regulating chondrocyte autophagy and apoptosis through the PI3K/Akt/mTOR pathway. Int J Biol Macromol. 2021;175:351–360. doi: 10.1016/j.ijbiomac.2021.02.029. [DOI] [PubMed] [Google Scholar]
- 35.Zhou ZD, Tan EK. Oxidized nicotinamide adenine dinucleotide-dependent mitochondrial deacetylase sirtuin-3 as a potential therapeutic target of Parkinson's disease. Ageing Res Rev. 2020;62(101107) doi: 10.1016/j.arr.2020.101107. [DOI] [PubMed] [Google Scholar]
- 36.Tu LF, Cao LF, Zhang YH, Guo YL, Zhou YF, Lu WQ, Zhang TZ, Zhang T, Zhang GX, Kurihara H, et al. Sirt3-dependent deacetylation of COX-1 counteracts oxidative stress-induced cell apoptosis. FASEB J. 2019;33:14118–14128. doi: 10.1096/fj.201900708R. [DOI] [PubMed] [Google Scholar]
- 37.Ruankham W, Suwanjang W, Wongchitrat P, Prachayasittikul V, Prachayasittikul S, Phopin K. Sesamin and sesamol attenuate H2O2-induced oxidative stress on human neuronal cells via the SIRT1-SIRT3-FOXO3a signaling pathway. Nutr Neurosci. 2021;24:90–101. doi: 10.1080/1028415X.2019.1596613. [DOI] [PubMed] [Google Scholar]
- 38.Ke SY, Yu SJ, Liu DH, Shi GY, Wang M, Zhou B, Wu L, Song ZM, Zhu JM, Wu CD, Qian XX. Ginsenoside Rb1 protects human umbilical vein endothelial cells against high glucose-induced mitochondria-related apoptosis through activating SIRT3 signalling pathway. Chin J Integr Med. 2021;27:336–344. doi: 10.1007/s11655-020-3478-8. [DOI] [PubMed] [Google Scholar]
- 39.Yang Y, Tian Y, Guo X, Li S, Wang W, Shi J. Ischemia Injury induces mPTP opening by reducing Sirt3. Neuroscience. 2021;468:68–74. doi: 10.1016/j.neuroscience.2021.06.003. [DOI] [PubMed] [Google Scholar]
- 40.Zhang M, Deng YN, Zhang JY, Liu J, Li YB, Su H, Qu QM. SIRT3 protects rotenone-induced injury in SH-SY5Y cells by promoting autophagy through the LKB1-AMPK-mTOR pathway. Aging Dis. 2018;9:273–286. doi: 10.14336/AD.2017.0517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dai SH, Chen T, Li X, Yue KY, Luo P, Yang LK, Zhu J, Wang YH, Fei Z, Jiang XF. Sirt3 confers protection against neuronal ischemia by inducing autophagy: Involvement of the AMPK-mTOR pathway. Free Radic Biol Med. 2017;108:345–353. doi: 10.1016/j.freeradbiomed.2017.04.005. [DOI] [PubMed] [Google Scholar]
- 42.Huang D, Liu M, Jiang Y. Mitochonic acid-5 attenuates TNF-α-mediated neuronal inflammation via activating Parkin-related mitophagy and augmenting the AMPK-Sirt3 pathways. J Cell Physiol. 2019;234:22172–22182. doi: 10.1002/jcp.28783. [DOI] [PubMed] [Google Scholar]
- 43.Wang C, Yang Y, Zhang Y, Liu J, Yao Z, Zhang C. Protective effects of metformin against osteoarthritis through upregulation of SIRT3-mediated PINK1/Parkin-dependent mitophagy in primary chondrocytes. Biosci Trends. 2019;12:605–612. doi: 10.5582/bst.2018.01263. [DOI] [PubMed] [Google Scholar]
- 44.Xia B, Tong Y, Xia C, Chen C, Shan X. α-Cyperone confers antidepressant-like effects in mice via neuroplasticity enhancement by SIRT3/ROS mediated NLRP3 inflammasome deactivation. Front Pharmacol. 2020;11(577062) doi: 10.3389/fphar.2020.577062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li Y, Hu K, Liang M, Yan Q, Huang M, Jin L, Chen Y, Yang X, Li X. Stilbene glycoside upregulates SIRT3/AMPK to promotes neuronal mitochondrial autophagy and inhibit apoptosis in ischemic stroke. Adv Clin Exp Med. 2021;30:139–146. doi: 10.17219/acem/130608. [DOI] [PubMed] [Google Scholar]
- 46.Liu P, Huang G, Wei T, Gao J, Huang C, Sun M, Zhu L, Shen W. Sirtuin 3-induced macrophage autophagy in regulating NLRP3 inflammasome activation. Biochim Biophys Acta Mol Basis Dis. 2018;1864:764–777. doi: 10.1016/j.bbadis.2017.12.027. [DOI] [PubMed] [Google Scholar]
- 47.Zhang T, Fang Z, Linghu KG, Liu J, Gan L, Lin L. Small molecule-driven SIRT3-autophagy-mediated NLRP3 inflammasome inhibition ameliorates inflammatory crosstalk between macrophages and adipocytes. Br J Pharmacol. 2020;177:4645–4665. doi: 10.1111/bph.15215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chang YP, Ka SM, Hsu WH, Chen A, Chao LK, Lin CC, Hsieh CC, Chen MC, Chiu HW, Ho CL, et al. Resveratrol inhibits NLRP3 inflammasome activation by preserving mitochondrial integrity and augmenting autophagy. J Cell Physiol. 2015;230:1567–1579. doi: 10.1002/jcp.24903. [DOI] [PubMed] [Google Scholar]
- 49.Yan WJ, Liu RB, Wang LK, Ma YB, Ding SL, Deng F, Hu ZY, Wang DB. Sirt3-mediated autophagy contributes to resveratrol-induced protection against ER stress in HT22 cells. Front Neurosci. 2018;12(116) doi: 10.3389/fnins.2018.00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.