

Abstract
While significant advancements have been made in cancer therapeutics and treatments, early disease detection and diagnosis remains critical to ensuring favorable outcomes for patients. To that end, we propose a microfluidic based approach to the sensitive detection of an intriguing cancer biomarker, extracellular vesicles (EVs). Our extracellular vesicle on demand (EVOD) chip utilizes a catalyst-free click chemistry to rapidly and specifically isolate EVs of interest. This specific isolation is followed by subsequent dithiothreitol release of the isolated EVs for downstream functional analysis. This joint isolation and release provide a powerful tool for the screening and quantification of EVs of interest. By incorporating antibodies against cancer associated surface proteins into the click-chemistry, we were able to selectively recover cancer-associated exosomes, allowing for important insights into patient disease. This platform was also tested using non-small cell lung cancer (NSCLC) patient samples, where anti-epidermal growth factor receptor (EGFR) assisted platform were able to selectively isolate and release 76% more exosomes from NSCLC patients than from healthy donors. This matches the previously reported higher EGFR expression commonly found in NSCLC EVs. Through its rapid isolation kinetics and adaptability in marker targeting, the EVOD device provides a highly versatile liquid biopsy platform for clinicians to use in the fight against cancer.
Keywords: Extracellular vesicles, microfluidics, click chemistry, vesicle release, immunoaffinity isolation
Graphical Abstract
1. Introduction
Exosomes are small (50–200nm) extracellular vesicles secreted from most membranous cells in the body. Exosomes are considered to play a role in cell signaling and have recently been implicated in tumor progression and metastasis (Mathivanan et al., 2010; Steinbichler et al., 2017; Tucci et al., 2018). These nano-scale vesicles act as reliable information carriers because of their innate stability in physiological conditions including in harsh tumor microenvironments. While they display common exosomal markers (CD63, CD9), these EVs also express proteins and genetic information specific to their parent cell (Han et al., 2016). The expression of surface proteins on exosomes derived from the parent cell has allowed tumor derived exosomes (TDEs) to emerge as potential markers for the diagnosis and monitoring of disease progression (Soung et al., 2017). Profiling these TDEs can give clinicians minimally invasive insights into treatment responses in patients, allowing for exosomes to potentially populate a liquid biopsy to monitor disease progression. For example, increased levels of exosomal protein and nucleic acids in peripheral blood of cancer patients have been noted (Li et al., 2017; Huang et al., 2019). Exosome concentration in blood has also been suggested as a possible cancer diagnostic marker, with higher EV concentrations found in patients compared to healthy donors (Eldh et al., 2014). In addition to use in diagnostics and prognostics, several novel approaches of exosomes towards therapeutics have been made recently. Certain subsets such as tumor-derived and immunogenic exosomes can be recovered and utilized for therapeutic purposes and vaccine development. For example, our group has recently demonstrated the therapeutic potential of isolated and released immunogenic natural killer cell derived exosomes (unpublished work), harnessing their cytotoxic abilities towards cancerous cells. To enhance the utility of these novel applications, several strategies for the exosome recovery for downstream utilization in analysis and therapeutics have been developed in recent years in order to meet this need.
Exosome isolation strategies take advantage of various characteristics either inherent to all EVs or specific to the vesicles cell of origin. Amongst the most common isolation techniques are ultracentrifugation, sized based, and chemical based capture including immunoaffinity isolation and polymer-aided exosome precipitation (Li et al., 2017). Ultracentrifugation and sized based separation techniques, which target the EVs density and size/shape, respectively, are highly effective in isolating exosomes in large quantities with relatively high specificity. However, each of these strategies contains drawbacks, the most glaring of which is the inability to target specific subsets of exosomes while filtering out others. To get around this issue, recent interest has been shown in immunoaffinity based isolation strategies. In this method, antibody coated surfaces are used to target and bind proteins and receptors displayed on the exosomal membrane allowing for high specificity isolation of EVs of a target origin. Immunoaffinity based capture is versatile in its application, as it can be incorporated into different platforms ranging from immunomagnetic beads (Tauro et. al., 2012), organic hydrogel surfaces (Kang et al, 2017), and microfluidic devices (Yang et. al, 2017). While the drawback of most immunoaffinity-based platforms is low sample throughput compared to other techniques, advances in microfluidics allows for the efficient rapid handling of samples of all sizes with minimal loss and high purity.
The versatility of microfluidics offers efficient isolation of rare cells such as circulating tumor cell (CTCs) and cancer stem cells (Hajba et al., 2014; Zhu et al., 2014; Ashcroft et al., 2012), and has recently been extended to exosome isolation.
Thus far, microfluidic immunoaffinity-based exosome isolation typically incorporates pre-defined interfaces conjugated with specific antibodies against common exosomal surface markers (CD63, CD9), or to markers exclusive to exosome subsets such as those derived from tumors using epithelial cell adhesion molecule (EpCAM) or programmed death-ligand 1(PD-L1). In doing this, various biochemical anchoring strategies have been explored for increased efficacy of these immunoaffinity isolation devices. Amongst them, avidin-biotinylated antibody complexes take advantage of strong biotin/avidin binding affinity to anchor biotinylated antibodies to the device surface (Chen et al., 2010). While this reaction is highly specific and irreversible, the processing time is lengthy. Biotin/Avidin based microfluidic isolation has also been used without capture antibodies, with biotin being directly conjugated to the exosome surface. This method yields benefits in increased non-source defined exosome isolation for multiplexed analysis, with the drawbacks of still being long processing times and low isolation specificity (Lee et al., 2018; Kang, Purcell, and Hadlock et al., 2019; Wan et al., 2017).
Also, as biotin is often taken as a supplement for its health benefits, background and specificity issues may be observed when evaluating samples from patients who have overconsumed biotin containing substances (Luong et al., 2019; Luong & Vashist., 2020). Annexin V functionalized microfluidic surfaces rapidly and specifically bind to phosphatidylserine in the exosome membrane, however there is still the drawback of discerning the origin of the exosome as EVs derived from various types of cancer highly display this phospholipid (Lea et al., 2017; Kang et al., 2019).
A promising isolation technique that contains the same advantages of specificity and capture efficiency, while lacking many of the disadvantages of common aforementioned linker strategies is click chemistry. Specifically, the inverse electron demand Diels-Alder (IEDDA) reaction between trans-cyclooctane and tetrazine, which does not require the cytotoxic copper catalysts necessitated in other click reactions (Takayama et al, 2019). This allows us to take advantage of the benefits of click chemistry-based capture (rapid kinetics and high specificity) while avoiding the downsides such as a lack of catalyst biocompatibility. Of the many possible IEDDA reagents, tetrazine and trans-clyclooctene (TCO) was chosen as its binding kinetics (K2 > 104 M−1 S−1) are rapid enough to meet the suggested threshold needed to make bioorthogonal reactions viable in practical applications (Wang et al. 2014; Devaraj & Weissleder 2011). Tetrazine conjugated antibody (TzAb) cocktails bind to exosome surface markers before rapidly and specifically binding to a TCO functionalized microfluidic surface. This allows users to modify the antibody cocktails to suit the needs of the specific EV of interest without having to alter the functionalized device.
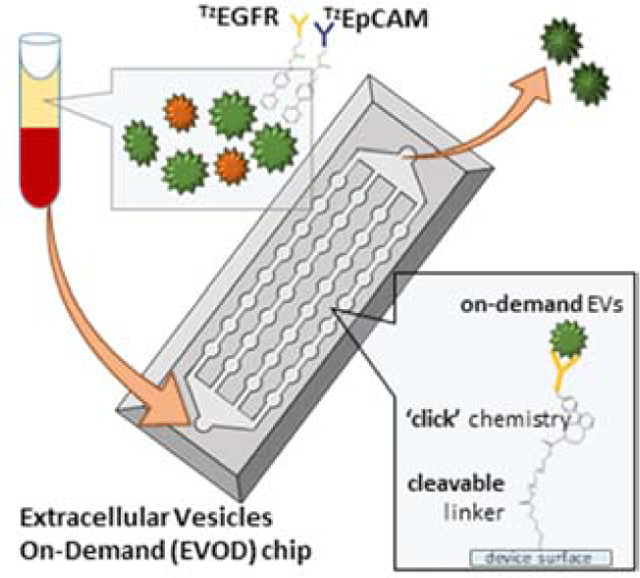
In this paper, we propose the Extracellular Vesicles on Demand (EVOD) device (Fig.1), utilizing IEDDA click chemistry for efficient isolation and analysis of lung cancer derived exosomes. Exosomes derived from different cancers can display specific surface protein markers which make them identifiable. While specific markers have been identified as having heightened expression in TDEs of several cancers, the marker profile of EVs isolated from lung cancer have not been fully determined (Wang et al., 2018). Lung cancer is not only the most common cancer in the world with 2.1 million reported cases, it is also responsible for the most deaths with 1.8 million fatalities in 2018 (Miranda-Filho et al., 2019). Careful analysis of exosomes in the blood of lung cancer patients would provide clinicians with more information in treating the disease.
Figure 1.

Extracellular Vesicle on Demand (EVOD) microfluidic chip for isolation, release, and analysis of tumor derived exosomes from patient plasma samples: (A) working principle of exosome isolation using EVOD chip. Plasma bound exosomes are conjugated with TZantibodies, processed and isolated by TCO bound to the device surface. (B) Photographic image of fabricated EVOD device (Scale bar=500μm)
Along with its capacity for sensitive and rapid EV isolation, the EVOD chip allows for the release of captured exosomes by incorporating a cleavable disulfide bond containing cross linker. Most elution methods, such as temperature (Yoon et al., 2016) and pH-based bond breakage (Ng et al., 2011), can damage the isolated target and lack specificity in what is released into the sample. This can cause serious issues in extracting reliable data from downstream analysis. Disulfide bond crosslinkers provide a platform that is easily and specifically cleavable under mild conditions which do not affect the exosome. We chose 3,3’dithiobis(sulfosuccinimidyl propionate) (DTSSP) as the disulfide bond containing cross linker due to its dual endcap NHS esters, providing the necessary bridge between the free amines of (3-Aminopropyl)triethoxysilane (APTES) and TCO. DTT was chosen as the disulfide reducing agent due to its rapid kinetics which limit any damage caused to the exosomes by the cleaving molecule (Liang et al., 2011; Ren et al., 2011; Kang and Kim et al., 2017; Szajewski & Whitesides., 1980; Smith et al., 2001). Release of isolated exosomes yields additional opportunities for downstream analysis and therapeutics. Furthermore, the biocompatibility of our proposed system in recovering isolated exosomes allows for their extended utilization in functional studies towards various therapeutics and vaccines. To demonstrate the capabilities of this system in clinical settings, we developed a microfluidic device incorporating a TCO conjugated capture surface, enabling the isolation of tetrazine associated exosomes from complex samples, such as plasma. Tetrazine-modified EGFR and EpCAM antibodies were chosen to study exosomes specifically in non-small cell lung cancer (NSCLC), and the results were compared to healthy donors to evaluate clinical value of the system.
2. Experimental section
2.1. EVOD chip fabrication and surface modification for IEDDA click chemistry
EVOD devices were fabricated using standard soft lithography procedures (Kang et al., 2019) with modification for IEDDA click EV capture. The PDMS device top is bonded to a glass slide using O2 plasma treatment (Covance, Femto Science, South Korea). After bonding, each device was heated to 80 ºC for 10 minutes using a hot plate, then allowed to cool to room temperature. A 33% APTES solution in ethanol was applied to each device and allowed to incubate for 30 minutes. This solution was gently washed out with ethanol, with the ethanol then evaporated using a hot plate at 115ºC for 5 minutes. This evaporation step aids in the formation of a monomer layer for uniform application (Howarter & Youngblood., 2006). Each device was then primed with filtered PBS before the application of 10mM DTSSP cross linker solution in PBS, which was incubated for 1 hour. Excess DTSSP was washed out of the devices using filtered PBS, followed by treatment with TCO-NHS-Amine solution. After a 1-hour incubation, unbound TCO-NHS-Ester was removed using filtered PBS, with the devices then ready for us in EV isolation.
2.2. Preparation of Click-antibody-EV conjugation
Two different tetrazine conjugated antibodies were prepared for the purposes of this research. 50mM Tetrazine in DMSO stock solution was prepared using Tetrazine-(PEG)5-NHS. A 1μM Tetrazine-EGFR antibody solution was prepared from the 50mM Tetrazine stock, 10uM antiEGFR antibody stock, and filtered PBS. A 1μM Tetrazine-EPCAM antibody solution was prepared from the 50mM Tetrazine stock, using a 100μg/ml anti-EpCAM antibody solution and filtered PBS. Both solutions could incubate for 1 hour, followed by reaction quenching using 100mM glycine solution in filtered PBS. Excess glycine/tetrazine was removed from the antibodies using desalt spin columns following the manufacturers listed procedure. The purified tetrazine-antibody solutions were now ready for EV surface application.
2.3. Surface functionalization of EVs
EVs were modified with either tetrazine or tetrazine conjugated antibodies (TzAb), depending on the stated purpose. While EV concentration varied by sample, tetrazine-EVs (TzEVs) were defined by the concentration of tetrazine solution in each sample. For tetrazine-EV samples, 10mM solutions of tetrazine (from 50mM stock) in PBS with EV sample (targeting ~10E8 concentration) were prepared and incubated for 1 hour. Unbound tetrazine was removed by ultracentrifugation of 150μl samples at 20psi for 30 minutes, with resulting pellets resuspended in 200μl PBS. For tetrazine-antibody modified EVs, 8μl of previously defined tetrazine-antibody solutions (either anti-EpCAM or anti-EGFR) were diluted in 92μl filtered PBS, with EV samples added targeting ~10E8 EVs/ml. These solutions were incubated for 1 hour, with unbound tetrazine-antibodies being removed by ultracentrifugation at 20psi. Resulting pellets were resuspended in 20μl PBS, and were then ready for application to EVOD devices.
2.4. EV isolation using EVOD microfluidic device
EV isolation by EVOD devices was first characterized using tetrazine modified EVs. Before sample application, 3-inch inlet and outlet tubing was attached to each EVOD device. Devices were then blocked with a 1% bovine serum albumin (BSA) solution to limit non-specific bonding. 100μl of purified tetrazine conjugated EVs were then applied to each device using syringe pumps (Harvard apparatus, USA) at 300μl/hr. Each sample was allowed to incubate for 10 minutes before being washed out with 200μl filtered PBS at 900μl/hr. Both the 100μl of sample and the 200μl of PBS wash were collected in the same “post-capture” vial for analysis. For release of isolated EVs, Dithiothreitol (DTT) solution was prepared to cleave the DTSSP contained disulfide. 50mM DTT solution was prepared in DI water, with the solution then filtered through a 0.2μm filter. 200μl of the 50mM DTT solution was applied to each EVOD device at 900μl/hr and allowed to incubate for 10 minutes upon completion of the injection. DTT solution was then washed out of each device using 100μl of filtered PBS, with both the PBS and DTT solutions collected in the same “post-release” vial for further analysis.
2.5. Performance evaluation using EVOD chip
Ability of the EVOD device to selectively isolate EVs using IEDDA click chemistry was examined using TzEVs. EVOD devices functionalized with TCO as previously described were compared with blank control devices. These control devices consisted of the EVOD PDMS chamber plasma bonded to a glass slide with only PBS applied to the device. For capture and release comparison, both EVOD and blank devices were blocked with 1% BSA, followed by application of 100μl of tetrazine conjugated EV sample. The full EV capture and release procedure previously described was then followed, with nanoparticle tracking analysis used for performance evaluation. The release of EVs isolated on EVOD chip was then further evaluated and its procedures optimized. Following isolation of tetrazine conjugated EV samples on EVOD devices, various DTT concentrations were tested on different devices and examined for release quantity and purity. The flow rate of the applied DTT was also examined at multiple settings on different devices and their results evaluated in section 3.
2.6. EV isolation and release using disulfide bond cleavage
The ability of our EVOD device to isolate and release extracellular vesicles conjugated with tetrazine modified antibodies was evaluated amongst several cell lines. Our tetrazine-EpCAM solution was tested using MCF7, MDA-MB-231, and MRC cell line derived EVs. The tetrazine-EGFR solution was tested using H3255 and MRC5 cell line derived EVs. All of these tetrazine-antibody EV solutions were prepared as previously described. For experiments comparing the viability of these tetrazine modified antibodies to selectively bind to surface antigens present on EV surface and allow for IEDDA isolation, the same processing procedure was used as with direct tetrazine on EV surface modification experiments. 100μl of purified tetrazine modified antibody conjugated vesicles were applied to each device at 300μl/hr with 10-minute incubation followed by 200μl PBS wash. Both the 100μl of sample and the 200μl of PBS wash were collected in the same “post-capture” vial for analysis. For release of isolated EVs, 200μl of the 50mM DTT solution was applied to each EVOD device at 900μl/hr with a 10-minute incubation. Remaining DTT solution was then washed out using 100μl of filtered PBS, with both the PBS and DTT solutions collected in the same “post-release” vial for further analysis.
2.7. EVOD Isolation and Release of Patient Samples
Plasma extracted from blood samples of 3 lung cancer patients and 2 healthy donors were applied to EVOD devices. A CD9 TzAb solution was prepared from a 3.5μM stock of anti-CD9 (CD9 Rabbit mAb, Cell Signaling Tech, United States). The CD9 TzAb solution was prepared by incubating 10μl of the 3.5μM anti-CD9 stock solution with 5μl of Tetrazine-PEG5-NHS Ester in 83μl of PBS for 1 hour. Excess tetrazine was removed using a desalting column, and desalting column particulate removed using ultracentrifugation at 22 psi for 30 minutes. The CD9 TzAb solution was brought to a final concentration of 0.24μM anti-CD9 using PBS.
For the experiment, 15 fully functionalized EVOD devices were prepared as previously described, with 1% BSA applied following the TCO incubation. For each patient and healthy donor sample, 3 100μl aliquots were prepared in individual vials. To these 3 100μl aliquots, one received CD9 TzAb, one received EGFR TzAb, and one received EpCAM TzAb, all at a concentration of 0.02μM. The TzAb solutions in each sample were allowed to incubate for 1 hour, before injection into individual EVOD devices at 300μl/hr with a 10-minute incubation.Effluent from each device’s outlet were collected for NTA, including a 100μl PBS wash step to remove unbound EVs from each sample. A separate vial was then used to collect effluent from the 50mM DTT release solution and subsequent wash step. The 200μl DTT solution was applied at 900μl/r with a 10-minute incubation, and the wash step consisted of 100μl filtered PBS injected at 900μl/hr.
2.8. Nanoparticle Tracking Analysis
EV concentration and size distribution of collected “post-capture” and “post-release” samples were examined by nanoparticle tracking analysis (NTA) using NanoSight NS300 (Marven Instruments, UK). 40μl of each sample was applied to the systems laser and processed through the instrument which analyses the Brownian motion of the particles. Concentration and size distribution measurements were taken in triplicate over 20 second intervals. NTA software prepared raw data was used to calculate post-capture and post-release concentrations in target size ranges, recovery rates, and sample purities. In order to evaluate EV concentration and purity, the concentration of exosome sized particle (30–150 nm) was re-evaluated and considered for the EV concentration and purity. Purity was defined as the concentration of exosome-sized particle (30–150 nm) found in the sample, divided by the total concentration of particles of all sizes as determined by Nanoparticle Tracking Analysis system.
2.9. Total Protein Quantification and Western Blotting
To confirm EV concentration data, protein analysis was performed on EV samples isolated on EVOD device using western blotting. After on-chip EV isolation, RIPA buffer (ThermoFisher, USA) was applied for EV membrane lysis in order to harvest proteins. Total exosomal protein quantity was then determined using a micro BCA kit (ThermoFisher, USA).
2.10. Surface Characterization Using Field Emission Electron Microscope (FE-SEM)
Extracellular vesicle surface conjugation with tetrazine modified anti-EpCAM was examined using field emission electron microscopy. Following on-chip isolation of tetrazine-EpCAM-EV complex as previously described, an SEM sample preparation procedure was performed. A biopsy punch was used to extract pieces of PDMS chamber out of EVOD devices, with the pieces then washed in a PBS bath. Each specimen was then fixed in a 2% glutaraldehyde solution for 1 hour. Glutaraldehyde was washed off with PBS before each specimen was incubated in increasing concentrations of ethanol (50%, 70%, 90%, and 100%). The specimens were then incubated with 50/50 hexamethyldisilazane (Sigma-Aldrich, USA) in ethanol solution, followed by 100% hexamethyldisilazane in a fume hood overnight to dry. Once dehydrated, each PDMS specimen was affixed to an SEM stub using carbon conductive tape, before being coated with a thin layer of gold. The vesicles isolated on the PDMS surface were then examined by FEI Nova 200 anolab Dualbeam FIB scanning electron microscope under low beam energies (2.0–5.0 kV) at the Electron Microscopy Analysis Lab (MC2) at University of Michigan.
2.11. Cell Culture and EV sample preparation
Two human breast cancer cell lines, MCF-7 (highEpCAM expressions) and MDA-MB-231 (lowEpCAM expressions), and one human lung fibroblast cell line, MRC 5 were used as a set for TzEpCAM experiments. One human lung cancer cell line, H3255 (high EGFR mutation and expression) and MRC5 were used as a set for TzEGFR experiments. All cells were cultured in medium for three days and then in serum-free medium for another three days in humidified atmosphere with 5% of CO2 at 37°C. For model sample experiments, all cell supernatants were collected and ultracentrifuged. EV pellets was resuspended into PBS buffer and EV concentrations were measured using NanoSight. Before further experiments, a known number of EVs were spiked into buffer solution.
2.12. Human Blood Sample Preparation
The sample collection and experiments were approved by University of Michigan Institutional Review Board (IRB). Informed consents were obtained from all participants of this clinical study and NSCLC blood samples were obtained after approval of the institutional review board at the University of Michigan. All experiments were performed in accordance with the approved guidelines and regulations by the ethics committee at the University of Michigan. Each blood sample was centrifuged at 4,000xg for 15 minutes to separate plasma layer and the supernatant from the sample was used after a filtration using a 0.22μm filter (Millipore Sigma, USA).
2.13. Immunofluorescence Staining
Direct visualization of EV isolation and release on EVOD device was further examined using immunofluorescence imaging. TzEV samples were applied to EVOD device as previously described. After EV isolation, lipophilic dye (DiO) was used to stain the EVs lipid bilayer. A 1:100 DiO solution was prepared in filtered PBS, with 100μl of this solution applied to EVOD devices after EV isolation. This dye was allowed to incubate for 10 minutes before being washed out with 200μl of filtered PBS. The stained EVOD devices were then examined sing Ti2 microscope (Nikon, Japan). Images were taken in FITC channel at 10x, 20x, and 40x magnifications. EV release was then confirmed by applying 200μl of DTT solution and incubating for 10 minutes, followed by a 100μl PBS wash. Post-release images were taken using the same microscope settings as for post-capture analysis.
3. Results and discussion
3.1. Strategy for recovery of cancer-associated EVs using IEDDA click chemistry
Recovery of extracellular vesicles was to be accomplished using a rapid, catalyst free type of click chemistry known as IEDDA. The strategy incorporates interaction between IEDDA reagents transcyclooctene (TCO) and tetrazine, which bind in a bio-orthogonal reaction. As shown in Figure 2, TCO pre-linked with primary amines was anchored to a silane treated capture surface using a DTSSP crosslinker, containing NHS esters on either end for amine bonding, as well as a cleavable disulfide bond in the middle. The APTES silane solution coats both the glass slide and the PDMS chamber, thus creating a 3-dimensional isolation surface. As for the other half of the IEDDA reaction, tetrazine was affixed to EV surface before application to device. For the purposes of these experiments, two separate tetrazine solutions were used. The first, and more basic method involved using tetrazine linked with NHS ester in order to directly bond to free primary amines displayed on the EV membrane. The other solution contained a tetrazine solution that had been bonded with different antibodies in our lab. These antibodies would then bind to specific proteins on the EV surface. Once EVs were “pre-labeled” with tetrazine, the vesicles were applied to the EVOD device, where the reaction between TCO and the membrane bound tetrazine allowed for rapid and specific isolation, as shown in Figure 1.
Figure 2.

Fabrication and surface modification of EVOD device to isolate and release of exosomes using IEDDA click chemistry with a use of disulfide cross linker.
3.2. Device Fabrication and Verification of IEDDA Click Chemistry
Initial examination of the capacity for IEDDA based isolation within our device was carried out using CY5 conjugated tetrazine. Click ready EVOD devices were applied with the CY5-tetrazine, followed by DTT based disulfide bond cleavage for release. Fluorescent imaging results of the post-capture and post-release states of the device are shown in Figure S1. These results demonstrate the ability of TCO functionalized surfaces to specifically bind with tetrazine modified targets. The capability of DTT to cleave the cross linker and clear the device of CY5 dye is also readily apparent and allows us to confidently translate this system to exosomes.
3.3. Exosome Capture and Release with Direct Tetrazine Surface Conjugation (TzEV)
Extracellular vesicle isolation within the EVOD device was examined using EVs directly modified with tetrazine. Identical samples of tetrazine-exos were applied to both TCO conjugated EVOD devices and blank devices. Fluorescence microscopy utilizing lipophilic dye to stain vesicle membranes was used to demonstrate the general efficacy of our EV isolation and release scheme (Fig 3a). SEM images (Fig. 3b) were also taken to confirm the presence of isolated EVs on the EVOD device’s PDMS surface. Effluent was collected from each device’s outlet and analyzed using NTA for concentration and size measurements. As shown in Figure 3c, blank devices allowed 69% more EVs to pass through without isolation than EVOD devices. This demonstrates the ability of TCO on the surface of the EVOD device to rapidly bond with the tetrazine displayed on the surface of the EVs. Following confirmation of IEDDA capture within EVOD devices, we next examine the ability to release isolated EVs by DTT induced cross linker cleavage. After the previously mentioned EV isolation, DTT solution was applied to each device for cleavage of the disulfide bonds contained in DTSSP. Device effluent was collected, and EV concentrations measured using NTA. Figure 3c shows that EVOD device efficiently captured introduced TzEVs, and successfully released the isolated TzEVs.
Figure 3.

Isolation and release of TZExos by EVOD chip: (A) EVOD chip stained with lipophilic dye (DiO) before capture (left), after isolation of TZExos by click reaction between tetrazine and TCO (center) and after release of exosomes by DTT mediated disulfide bond cleavage (right) (Scale bar=100μm); (B) scanning microscope image of isolated TzExos on the surface of the EVOD chip; (C) nanoparticle tracking analysis of post capture and post release samples.
We then looked to optimize DTT release to maximize the downstream capabilities of our EVOD system. DTT release concentration was examined using 10mM, 50mM, and 100mM solutions of DTT, with results shown in Figure 4a–b. These results show that the 50mM solution was unrivaled in both the amount of EVs it released and in the yielded purity, thus making it the preferred release condition for pure and high yield vesicle recovery. The flow rate of DTT application was also examined, with results in Figure 4c–d showing optimal release quantity and purity obtained at 900μl/hr.
Figure 4.

Performance evaluation and optimization of EVOD chip using exosomes directly conjugated with tetrazine (TzExos): (A-B) concentration (A) and purity (B) of recovered exosomes by EVOD chip depending on DTT release solution concentrations; (C-D) concentration (C) and purity (D) of recovered exosomes by EVOD chip depending on flow rates of DTT solution; (E) recovered total EVs and exosome-sized vesicle amounts compared to blank control devices; (F) relative EV recovery performance before and after performance optimization.
Additional optimization was carried out examining the ideal concentration of APTES to use for the silane application. Several APTES concentrations were tested based on the concentrations used in previous studies (Howarter & Youngblood., 2006; Vashist et al., 2014) with post isolation results found in Figure S2. Based on these results, we saw similar isolation and release performance regarding EV quantity for both the 0.5% and 33% APTES solution, with the 10% solution lagging behind in both metrics. While all three solutions demonstrated high post capture purities, the post release purity was significantly greater when using the 10% and 33% APTES solutions, and thus, due to its combination of high recovery quantity and purity, the 33% concentration was chosen for all experiments. This result was noteworthy, as high APTES concentrations have been previously shown to have issues with uniformity due to formation of multiple binding layers. However, we attribute the success of the high APTES concentration to the immediate heating of the silane treatment in the device at 115°C, which allows for the formation of more stable film-like layers.
The fully optimized system was examined using tetrazine-EVs isolated on both EVOD and blank control devices, followed by release using 50mM DTT solution and a PBS wash. Effluent from both DTT and wash steps were collected together and analyzed by NTA. Concentration of EVs in these released samples are shown in Figure 4e, which show an 86% increase in vesicles released by EVOD chip over blank control devices. Figure 4f demonstrates the efficacy of our fully optimized device and processing procedure compared to the initial trial and control devices. These results outline the EVOD devices’ ability to utilize IEDDA click chemistry for isolation and release of tetrazine-associated EVs. On-chip capture profile and reproducibility of the fully optimized system was also evaluated and shown in Figure S4. These results confirm that isolation takes place at all examined locations, but most of the isolation take place near the inlet of the device due to rapid binding kinetics of the IEDDA click chemistry.
3.4. Exosome Capture and Release Using indirect Tetrazine Modified Antibodies (TzAb-Exo)
Following verification that the IEDDA click system can reliably isolate extracellular vesicles, we then examined the isolation of EVs modified with tetrazine conjugated antibodies specific to a target EV type. EpCAM and EGFR were chosen as the preferred antibodies for testing due to their high antigen expression on breast cancer and long cancer derived EVs, respectively. Antibodies were conjugated with tetrazine-PEG-NHS ester and purified using desalt spin columns. The tetrazine conjugated antibodies were then applied to cell line derived EVs for surface modification, followed by isolation using EVOD device. The ratio of TzAbs to EV containing solutions was optimized, with results shown in Figure 5a. These results show significant increase in vesicle recovery with decrease in TzAb added to samples up to a 1:5 molar ratio, with further reductions in TzAb providing no notable performance gains.
Figure 5.

Performance evaluation of EVOD chip isolation and release using TZAB conjugated exosomes: (A) determination of optimal TZAB:Exosome testing 1:1, 1:5, and 1:10 volumetric ratios of a 10mM TZAB solution with 1010 exo ml−1 solution. (B) relative EV recovery performance comparison based on on-chip total protein quantity analysis without exosome release; (C) Post-release exosome concentration comparison TZEpCAM conjugated exosomes derived from MCF7, MDA, and MRC5 cell lines; (D) Post-release exosome concentration comparison TZEGFR conjugated exosomes derived from H3255 and MRC5 cell lines; (E) clinical application of the EVOD device in quantitative analysis of clinical exosomes using three different tetrazine conjugated antibodies (CD9, EGFR, and EpCAM).
Following this optimization, we first aimed to evaluate capture performance of the device regardless of releasing capabilities. To accomplish this, we used total protein quantity analysis using data collected through BCA analysis. Tetrazine-EpCAM solution was applied to EV samples derived from MCF7, MDA, and MRC5 then processed though the EVOD system. TzEGFR solution was also applied to samples derived from MRC5 and H3255 cell lines, followed by EVOD device isolation. Following isolation, all samples were lysed on-chip using RIPA solution and BCA protein analysis was performed. Relative EV recovery performance was evaluated using total on-chip protein quantity, with results shown in Figure 5b. Compared to EVs derived from MRC5 cell line, EVOD devices isolated 20% more MDA EVs and 35% MCF7 derived EVs. We also see TzEGFR EVs successfully isolated 24% more H3255 derived EVs than those derived from MRC5. While both the TzEpCAM and TzEGFR EV isolation results are in line with reported trends in expression levels of the markers for each cell line tested here, the relative performance differences are lower than expected based on reported marker expression levels (Sterzyńska et al., 2012; Endaya et al., 2017; Kohno et al., 2011).
We further extended our study to NTA-based quantitative analysis of captured and released EVs from the device. These results (Fig 5c) show the EVOD system isolated and released 137% more MCF7 derived EVs (2.25E8 ± 6.29E7) than MRC5 EVs (8.68E7 ± 3.21E7), and 32% more MCF7 EVs than those derived from MDA cell lines (1.39E8 ± 3.24E7). As exosomal surface protein expression typically mirrors that of its parent cell, these results are in line with reported surface expression levels of EpCAM for each of these cell lines. Anti-EGFR conjugated tetrazine was then applied to H3255 and MRC5 cell line derived EVs and processed through the EVOD system for NTA. Figure 5d shows that the EVOD system captured and released 84% more H3255 cell line derived EVs (1.7E8 ± 3.54E7) than MRC5 derived EVs (7.56E7 ± 2.93E7). As with TzEpCAM, these results are in line with reported expression levels of the target antigen on the surface of tested EVs. H3255 cell line derived EVs highly display EGFR on the surface, while MRC5 shows very little EGFR expression. Our NTA-based comparison studies showed better discrimination performance between cancerous EVs and control EVs over BCA analysis. This is likely due to loss of specificity during RIPA-based EV lysis on chip. We lysed all membranous EVs isolated on chip including non-specifically bound EVs, thus yielding overestimated protein amounts for all cases. To obtain a more accurate understanding of the EVODs capabilities, we ran the same experiment using identical samples, released them and analyzed using NTA, which best outlines the overall performance of the system.
Thus, these results show the ability of these tetrazine conjugated antibodies to specifically bind to appropriate antigens on the surface of targeted EVs and allow for isolation by TCO functionalized surfaces. Furthermore, the results from NTA imply that DTT-based release and NTA analysis give better understanding of each EV, maintaining high specificity of the system.
3.5. Pre-Clinical Studies of Exosome Isolation and Release Using EVOD Device
Pre-clinical studies were conducted using three lung cancer patient samples and two healthy donors. These blood samples were separated to obtain plasma from whole blood, filtered through 0.22μm filter, and a 100μl portion of each sample was incubated with either TzCD9, TzEGFR, or TzEpCAM pre-Tzconjugated antibodies. These samples were processed though fully functionalized EVOD devices and released using DTT, with results shown in Figure 5e. These results demonstrate a significant increase in EVs isolated with EGFR TzAbs in all lung cancer patient samples compared to healthy donors. Overall, EVOD devices isolated and released 76% more EVs from patient samples than from healthy donors when using EGFR TzAbs. Furthermore, no healthy donor sample showed a release concentration within 5×108 exos/ml of the lowest concentrated lung cancer sample processed using EGFR TzAbs. As EGFR is a well-known lung cancer marker highly expressed on EVs in NSCLC patients (Yamashita et al., 2013), this result shows the ability of the EVOD device to selectively isolate and release EVs from plasma using the TzAbs. TzEpCAM Abs also showed significant increase in isolation and release of EVs from lung cancer patients compared to healthy donors. However, we found significant variation in this result compared to isolation with TzEGFR Abs. Two of the lung cancer patients had at least double the number of released EVs as either healthy donor, while the third lung cancer patient released fewer vesicles than any other sample tested. While EpCAM is a well-known cancer marker, variations in its expression on NSCLC patient EVs have been found in other studies as well (Sandfeld-Paulsen et al., 2016; Rupp et al., 2011). This large difference in EpCAM TzAb results may be due to the varying mechanisms with which NSCLC develops, many of which we still do not fully understand. Similar to the EpCAM results, CD9 TzAbs showed an increase in released EVs in LC2, and LC3 compared to healthy donors, but nearly equal release quantity between LC1 and the donor samples. As CD9 is a general exosome marker, the smaller difference in release concentrations amongst all samples compared to that found using EGFR and EpCAM is to be expected, as the total quantity of plasma bound EVs is not believed to drastically increase due to cancer. Furthermore, the variation in EV concentration we see in LC2 and LC3 compared to the other tested samples may be due to the usage of only a single general EV marker (CD9). As CD9 expression is not constant amongst all extracellular vesicles and all subjects, additional general EV markers may lead to more normalized results.
4. Conclusion
We proposed the EVOD devices for sensitive, rapid isolation and release of circulating tumor EVs for multiplexed analysis and potential downstream therapeutics. The device functionalized with cleavable DTSSP crosslinkers followed by the click-chemistry TCO compound captured tetrazine functionalized EVs 200% more effectively than control devices. Further TzAb based on-demand EV captures showed that anti-EGFR modified tetrazine sensitively recovered 84% more H3255 cell line derived EVs than MRC5 cell line derived EVs. Tetrazine modified EpCAM was also used to recover MCF7 derived EVs 137% and 32% more effectively than MRC5 and MDA EVs, respectively. Separate tetrazine modified antibodies using anti-CD9, anti-EGFR, and ant-EpCAM were prepared and applied to tumor derived EV samples from lung cancer patients and healthy donors. The EVOD system successfully targeted and recovered 76% more EVs from patients than from healthy donors, in line with high EGFR expression found in lung cancer cells and EVs. Through its rapid isolation kinetics and adaptability in marker targeting, the EVOD device provides a highly versatile liquid biopsy platform for clinicians to use in the fight against cancer.
Supplementary Material
Highlights.
We present a novel platform for on-demand specific extracellular vesicle isolation and release.
Inverse electron demand Diels–Alder (IEDDA) pre-labeling allows for rapid on-chip isolation of circulating cancer exosomes.
Click-chemistry based antibody pre-labeling allows for rapid and specific isolation of cancer exosomes.
Incorporation of a cleavable disulfide bond allows for release of target exosomes for functional study.
Lung cancer patient plasma contains increased quantities of exosomes highly displaying epidermal growth factor receptor (EGFR).
Acknowledgement
The authors thank Dr. Ting-Wen Lo for his assistance with scanning electron microscope analysis; Emma Purcell for the preparation of antibody reagents and clinical sample preparation; Nna-Emeka Onukwugha for his assistance with BCA analysis; Dr. Chitra Subramanian for the preparation of fibroblast cell line, MRC‐ 5, which was used as non-cancer cell line. The authors also acknowledge the Lurie Nanofabrication Facility at the University of Michigan. This work was supported by grants from National Institute of Health (NIH), 5-R33-CA-202867-02 to S.N. and 1-R01-CA-208335-01-A1 to S.N.
Footnotes
Appendix A. Supplementary materials
Supplementary data associated with this article can be found in the online version.
Yoon-Tae Kang: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Supervision, Writing - original draft Thomas Hadlock: Conceptualization, Data curation, Investigation, Methodology, Writing - original draft Shruti Jolly: Resources, Validation, Writing - review & editing Sunitha Nagrath: Conceptualization, Funding acquisition, Project administration, Supervision, Writing - review & editing
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference
- Han C, Sun X, Liu L, Jiang H, Shen Y, Xu X, Li J, Zhang G, Huang J, Lin Z, Xiong N, & Wang T 2016. Stem cells international, 2016, 7653489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soung YH, Ford S, Zhang V, & Chung J 2017. Cancers, 9(1), 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang T, & Deng CX 2019. Int. j. biological sciences, 15(1), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, Zhang T, Zheng M, Liu Y, & Chen Z 2017. Journal of hematology & oncology, 10(1), 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldh M, Olofsson Bagge R, Lässer C, Svanvik J, Sjöstrand M, Mattsson J, Lindnér P, Choi DS, Gho YS, & Lötvall J 2014. BMC cancer, 14, 962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Kaslan M, Lee SH, Yao J, & Gao Z 2017. Theranostics, 7(3), 789–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauro BJ, Greening DW, Mathias RA, Ji H, Mathivanan S, Scott AM, Simpson R 2012. Methods, 56, 293–304 [DOI] [PubMed] [Google Scholar]
- Kang Y-T., Kim YJ., Lee TH., Shim J-E., Cho Y-H. 2017. Proceedings of the American Association for Cancer Research Annual Meeting 2017, Abstract nr 1719, DOI: 10.1158/1538-7445.AM2017-1719 [DOI] [Google Scholar]
- Yang Y, Liao X, Tian Y, Li G 2017. Biotechnol. J. 12, 1600699 [DOI] [PubMed] [Google Scholar]
- Hajba L, Andras Guttman A 2014. TrAC, 59, 9–16 [Google Scholar]
- Chen C, Skog J, Hsu C, Lessard RT, Balaj L, Wurdinger T, Carter BS, Breakefield XO, Toner T, & Irimia D 2010. Lab Chip, 10, 505–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luong JHT, Male KB Glennon JD 2019. Biotech. Adv. 37, 634–641 [DOI] [PubMed] [Google Scholar]
- Luong JHT, & Vashist SK ACS Omega 2020, 5, 1, 10–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lea J, Sharma R, Yang F, Zhu H, Ward ES, & Schroit AJ 2017. Oncotarget, 8(9), 14395–14407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang YT, Purcell E, Palacios‐Rolston C, Lo TW, Ramnath N, Jolly S, Nagrath S 2019. Small, 15(47), 1903600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama Y, Kusamori K, & Nishikawa M 2019. Molecules, 24(1), 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda-Filho A, Piñeros M, Bray F 2019. Salud Publica Mex, 61(3), 219–229. [DOI] [PubMed] [Google Scholar]
- Liang J, & Fernandez JM 2011. J. Am. Chem. Soc. 133, 3528–3534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu B, Nahmias Y, Yarmush ML, Murthy SK 2014. Stem Cells Trans Med, 3(11), 1354–1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft BA, de Sonneville J, Yuana Y, Osanto S, Bertina R, Kuil ME, & Oosterkamp TH 2012. Biomedical microdevices, 14(4), 641–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren C, Song Z, Zheng W, Chen X, Wang L, Kong D, & Yang Z 2011. Chem. Commun, 47, 1619–1621 [DOI] [PubMed] [Google Scholar]
- Kang Y-T, Kim YJ, Bu J, Cho Y-H, Han SW, Moon BI 2017. Nanoscale, 9(36), 13495–13505 [DOI] [PubMed] [Google Scholar]
- Szajewski RP, & Whitesides GM 1980. Journal of the American Chemical Society, 102 (6), 2011–2026 [Google Scholar]
- Smith EA, Wanat MJ, Cheng Y, Barreira SVP, Frutos AG, Corn RM 2001. Langmuir, 17, 2502–2507 [Google Scholar]
- Lee K, Fraser K, Ghaddar B, Yang K, Kim E, Balaj L, Chiocca EA, Breakefield XO, Lee H, & Weissleder R 2018. ACS nano, 12(1), 494–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang YT, Purcell E, Hadlock T, Lo TW, Mutukuri A, Jolly S, & Nagrath S 2019. Analyst, 144, 5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y, Cheng G, Liu X, Hao SJ, Nisic M, Zhu CD, Xia YQ, Li WQ, Wang ZG, Zhang WL, Rice SW, Sebastian A, Albert I, ; Belani CP, & Zheng SY 2017. Nature Biomedical Engineering, 1(4), 0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandfeld-Paulsen B., Aggerholm-Pedersen N., Bæk R., Jakobsen KR., Meldgaard P., Folkersen BH., Rasmussen TR., Varming K., Jørgensen MM., & Sorensen BS. 2016. Molecular Oncology, 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupp AK, Rupp C, Keller S, Brase JC, Ehehalt R, Fogel M, Moldenhauer G, Marmé F, Sültmann H, & Altevogt P 2011. Gynecol. Oncol, 122, 437–446 [DOI] [PubMed] [Google Scholar]
- Steinbichler TB, Dudas J, Riechelmann H, & Skvortsova II 2017. Sem. Cancer Bio. 44, 170–181 [DOI] [PubMed] [Google Scholar]
- Tucci M, Mannavola F, Passarelli A, Stucci LS, Cives M, & Silvestris F 2018. Oncotarget, 9(29), 20826–20837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathivanan S, Ji H, & Simpson RJ 2010. J. Proteomics. 73(10). 1907–1920 [DOI] [PubMed] [Google Scholar]
- Yoon HJ, Shanker A, Wang Y, Kozminsky M, Jin Q, Palanisamy N, Burness ML, Azizi E, Simeone DM, Wicha MS, Kim J, & Nagrath S 2016. Adv Mat. 28(24), 4891–4897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PK, & Snyder MA 2011. J Separation Science. 35(1), 29–35 [DOI] [PubMed] [Google Scholar]
- Sterzyńska K, Kempisty B, Zawierucha P, & Zabel B 2012. Folia Histochem Cytobiol. 50(4):534–541. [DOI] [PubMed] [Google Scholar]
- Kohno M, Horibe T, Haramoto M, Yano Y, Ohara K, Nakajima O, Matsuzaki K, & Kawakami KA 2011. European Journal of Cancer, 47 (5), 773–783. [DOI] [PubMed] [Google Scholar]
- Endaya B, Guan SP, Newman JP, Huynh H, Sia KC, Chong ST, Kok C, Chung A, Liu BB, Hui KM, & Lam P 2017. Oncotarget, 8(33), 54629–54639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T, Kamada H, Kanasaki S, Maeda Y, Nagano K, Abe Y, Inoue M, Yoshioka Y, Tsutsumi Y, Katayama S, Inoue M, & Tsunoda S 2013. Die Pharmazie. 68(12), 969–973(5) [PubMed] [Google Scholar]
- Devaraj Neal K., & Weissleder Ralph. 2011. Accounts of Chemical Research, 44 (9), 816827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Na Z, Fu J, Tan CYJ, Zhang H, Yao SQ, Sun H 2014. Chem. Comm. 80. [DOI] [PubMed] [Google Scholar]
- Howarter JA, Youngblood JP Langmuir 2006, 22, 11142. [DOI] [PubMed] [Google Scholar]
- Vashist SK, Lam E, Hrapovic S, Male KB, & Luong JHT 2014. Chemical Reviews 114 (21), 11083–11130. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.