

Abstract
Bacteria utilize small extracellular molecules to communicate in order to collectively coordinate their behaviors in response to the population density. Autoinducer‐2 (AI‐2), a universal molecule for both intra‐ and inter‐species communication, is involved in the regulation of biofilm formation, virulence, motility, chemotaxis, and antibiotic resistance. While many studies have been devoted to understanding the biosynthesis and sensing of AI‐2, very little information is available on its export. The protein TqsA from Escherichia coli, which belongs to the AI‐2 exporter superfamily, has been shown to export AI‐2. Here, we report the cryogenic electron microscopic structures of two AI‐2 exporters (TqsA and YdiK) from E. coli at 3.35 Å and 2.80 Å resolutions, respectively. Our structures suggest that the AI‐2 exporter exists as a homo‐pentameric complex. In silico molecular docking and native mass spectrometry experiments were employed to demonstrate the interaction between AI‐2 and TqsA, and the results highlight the functional importance of two helical hairpins in substrate binding. We propose that each monomer works as an independent functional unit utilizing an elevator‐type transport mechanism.
Keywords: autoinducer‐2, pentamer, quorum sensing, TqsA, YdiK
Subject Categories: Structural Biology
Cryo‐EM structures of two autoinducer‐2 exporters give important insights into how bacteria communicate with each other.

Introduction
Quorum sensing (QS) is a cell‐to‐cell communication mechanism that confers bacteria with the ability to sense and decipher their surroundings in terms of not only their own cell density but also other species cohabitating with them (Papenfort & Bassler, 2016; Whiteley et al, 2017). This phenomenon was first described for the marine organism Vibrio fischeri that emits bioluminescence in response to a high population density (Bassler et al, 1993). Later research revealed that the role of QS extends beyond bioluminescence to many other density‐dependent cellular processes like the formation of biofilms, production of virulence factors and antibiotics, sporulation, conjugation, and chemotaxis (Sperandio et al, 1999, 2001, 2002; Antunes et al, 2010; De Araujo et al, 2010; Whiteley et al, 2017). QS specifically modulates those cellular functions and processes, the effect of which will be maximum when all cells act in unison.
In QS, intercellular communication is mediated by small chemical molecules known as autoinducers (Bassler & Losick, 2006). They are synthesized intracellularly and could be passively or actively released into the extracellular milieu. The extracellular concentration of the autoinducer increases as a function of cell number. On reaching a critical concentration threshold, the autoinducers are perceived by their cognate membrane receptors. The bacteria eventually respond to this environmental stimulus by activating signal transduction cascades that ultimately regulate the expression of certain genes. So far, a number of different autoinducers have been identified in various bacteria and they can be classified into three main categories. Acyl‐homoserine lactones (AHLs, autoinducer‐1) are frequently used by Gram‐negative bacteria, while autoinducing peptides (AIPs) are the key autoinducers in Gram‐positive bacteria (Rutherford & Bassler, 2012). These signaling molecules display a large structural diversity and are often specific to each bacterial species. There is a third category of autoinducers, termed autoinducer‐2 (AI‐2), which can be produced and sensed by a majority of bacterial and eukaryotic species and hence is considered as a universal signal molecule, promoting both intra‐ and inter‐species as well as inter‐kingdom communication (Pereira et al, 2013; Ismail et al, 2016; Valastyan et al, 2021).
Bacterial AI‐2 molecules are small cyclic furanone compounds that are converted from their precursor, 4,5‐ dihydroxy‐2,3‐pentanedione (DPD) (Xavier & Bassler, 2005). DPD is synthesized in three enzymatic steps from S‐adenosyl methionine (SAM), a metabolite involved in many metabolic pathways, including transmethylation, transsulfuration, and aminopropylation. In the first step, SAM is converted to S‐adenosyl homocysteine (SAH) by methyltransferases. Subsequently, the SAH nucleosidase hydrolyzes SAH to form S‐ribosylhomocysteine and adenine. Finally, SRH is converted to DPD and homocysteine by the S‐ribosylhomocysteinase (LuxS) enzyme (Pereira et al, 2013). DPD is quite unstable and can spontaneously cyclize to form different cyclic furanone compounds. Among them, a furanosyl borate diester, called S‐2‐methyl‐2,3,3,4‐tetrahydroxy tetrahydrofuran‐borate (S‐THMF‐borate), was reported to be the AI‐2 signal molecule responsible for QS in Vibrio species, whereas a non‐borated enantiomer of DPD, R‐2‐methyl‐2,3,3,4‐tetrahydroxy tetrahydrofuran (R‐THMF), represents the AI‐2 signal in a range of other bacteria, including Salmonella Typhimurium and Escherichia coli (Chen et al, 2002; Miller et al, 2004). AI‐2 is considered to be a universal communication signal because LuxS, the key enzyme in AI‐2 biosynthesis, was found to be widespread in both Gram‐negative and Gram‐positive bacteria (Xavier & Bassler, 2003). This enzyme is also a part of the essential activated methyl cycle, raising skepticism about the idea that AI‐2 is just a metabolic byproduct. Subsequent studies revealed that AI‐2 is involved in regulation of biofilm formation, pathogenesis, chemotaxis, and antibiotic resistance (Pereira et al, 2013; Li et al, 2015, 2017; Laganenka et al, 2016; Xue et al, 2016; Laganenka & Sourjik, 2018).
AI‐2 or DPD is a small hydrophilic compound and is considered to be membrane impermeable (Kamaraju et al, 2011). It is proposed to be an uncharged molecule under physiological conditions, but it hosts four hydroxyl (‐OH) groups in its structure enabling it to form hydrogen bonds (Fig 1). Cell signaling via AI‐2 involves both secretion and sensing that require the transmission of signals across the membrane in both directions, as illustrated in Fig 1. In enteric bacteria, the active import of AI‐2 into the cell is carried out by an ABC‐type transporter, whose encoding genes are part of the lsr operon consisting of eight genes, lsrKRACDBFG, that encodes proteins involved in uptake, phosphorylation, and further processing of AI‐2 (Xavier & Bassler, 2005). However, to the best of our knowledge, the mechanism of export of AI‐2 still remains undetermined. The first protein suggested to be an exporter for AI‐2 was the YdgG protein (later renamed TqsA for transporter of quorum‐sensing signal AI‐2) from E. coli (Herzberg et al, 2006). Previous studies showed that a deletion of tqsA leads to increased biomass production, motility, biofilm formation, intracellular concentrations of AI‐2, and surged lsr operon expression (Herzberg et al, 2006). TqsA is a 38 kDa transmembrane protein, which belongs to a large and underexplored family of putative membrane transporters, presently known as the AI‐2 exporter superfamily. In E. coli, three more proteins (YhhT, YdiK, and PerM) were assigned to this superfamily (Rettner & Saier, 2010). The expression of two of them (PerM and YdiK) was shown to be regulated by the PurR transcription factor that regulates purine metabolism and transport (Cho et al, 2011). The regulation of PurR on the expression of AI‐2 exporters at the cellular level is surely noteworthy. The rationale behind this could either be the production of adenine as a by‐product during the AI‐2 biosynthesis or could indicate the existence of an additional set of substrates (purines) for this exporter family (Rettner & Saier, 2010).
Figure 1. Schematic overview of AI‐2‐mediated quorum sensing in Escherichia coli .
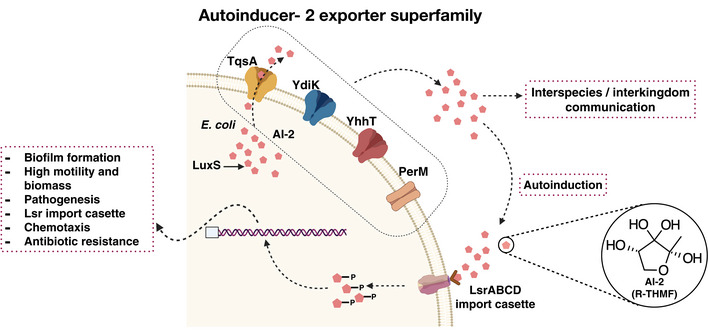
Autoinducer‐2 is a quorum sensing signal molecule that mediates interspecies and inter‐kingdom cell‐to‐cell communication. The four predicted AI‐2 exporters from E. coli are shown embedded in the inner membrane in different shades (TqsA in yellow, YdiK in blue, YhhT in red, and PerM in brown), and AI‐2 is represented as small red pentagonal moieties. LuxS produces DPD, the precursor of AI‐2. The ABC transporter complex LsrABCD is involved in the active import of AI‐2. Phosphorylated AI‐2 inactivates the transcriptional repressor and thereby regulates the expression of quorum sensing‐related genes.
Besides the AI‐2 exporter TqsA from E. coli, recently another member of the AI‐2 exporter superfamily from Halobacillus andaensis (Upf0118) was functionally characterized (Dong et al, 2017; Wang et al, 2020). Unexpectedly, it was observed to function as a Na+/H+ antiporter, and in a subsequent study it was suggested to belong to an independent subgroup in the AI‐2 exporter superfamily (Dong et al, 2017; Wang et al, 2020). So far, only two members (TqsA and Upf0118) of this family have been investigated and they both are proposed to possess divergent functions (Dong et al, 2017; Wang et al, 2020).
In this study, all four AI‐2 exporters from E. coli (TqsA, YhhT, YdiK, and PerM) were purified and studied in an attempt to characterize them both biochemically and structurally. We determined the cryogenic electron microscopy (cryo‐EM) structures of TqsA and YdiK at 3.35 Å and 2.80 Å resolutions, respectively. Our results show that these AI‐2 exporters are homo‐pentameric complexes, and structural analyses in combination with molecular docking studies reveal the importance of two helix–turn–helix motifs for substrate binding. This study provides major insights into this novel and unexplored superfamily of membrane transporters and will serve as the basis for future studies aimed at understanding the mechanisms of AI‐2 secretion.
Results
AI‐2 exporters from Escherichia coli
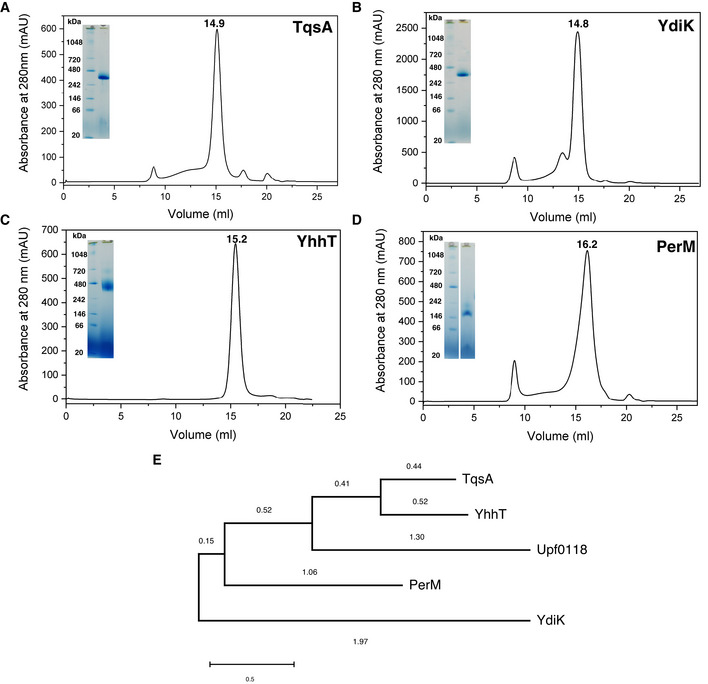
The proteins of the AI‐2 exporter superfamily are approximately 40 kDa in size with a few exceptions (Rettner & Saier, 2010). The results of multiple sequence alignment of the four predicted AI‐2 exporters from E. coli (TqsA, YdiK, YhhT, and PerM) showed that each protein is distantly related to each other with a relatively low overall sequence similarity (Fig EV1E). The highest sequence identity of 46% was observed between TqsA and YhhT, indicating a closer phylogenetic relationship. No clear phylogenetic correlation can be drawn among TqsA, YdiK, and PerM since pairwise sequence identities fall below ≈24%, which may suggest different cellular functions or substrate specificities of the proteins.
Figure EV1. Purification and phylogenetic analyses of AI‐2 exporters from Escherichia coli .
-
A–DSize exclusion chromatography (SEC) and BN–PAGE profiles for the four AI‐2 exporters from E. coli, TqsA (A), YdiK (B), YhhT (C), and PerM (D). The retention volumes for each of the proteins are included. Superose 6 increase 10/300 GE column was used for the final size exclusion step.
-
EPhylogenetic analysis of the four AI‐2 exporters and Upf0118 family using the maximum‐likelihood method. MEGAX was utilized for the analysis and the branch distances are mentioned on the respective branches of the tree.
As mentioned earlier, another member of the AI‐2E family, Upf0118, was recently reported to show Na+/H+ antiport activity. A comparison between TqsA and Upf0118 revealed a low sequence similarity of 24%. Nevertheless, we investigated if any of the four selected AI‐2 exporters from E. coli can function in a similar manner as Upf0118. Results of our growth complementation assay showed that the E. coli Na+‐sensitive strain (Knabc strain) expressing the AI‐2 exporters was unable to grow in the presence of sodium ions, which suggests that they do not act as Na+/H+ antiporters (Appendix Fig S1A and B). In addition, the assay of antiport activity using everted vesicles further confirmed that TqsA, unlike Upf0118, does not transport Na+/Li+ in exchange for H+ (Appendix Fig S1C and D). These results emphasize that the proteins TqsA and Upf0118 represent two diverse groups within the AI‐2 exporter family.
Purification and characterization of four AI‐2 exporters
In this work, four AI‐2 exporters were produced in E. coli top 10 cells. Initially, the detergent GDN was used for the purification. All four proteins could be purified to homogeneity at yields of sub‐milligram proteins per liter of culture using a combination of affinity and size exclusion chromatography (Fig EV1A–D). Gel filtration profiles showed that three AI‐2 exporters (TqsA, YdiK, and YhhT) eluted as single sharp peaks at similar retention volumes of ~15 ml corresponding to a size of about 400 kDa (Fig EV1A–C), while PerM was recovered later from the column as a broader peak at 16.2 ml (Fig EV1D), suggesting the presence of different oligomeric species with the majority possessing apparently smaller hydrodynamic radii.
All purified proteins were analyzed on BN–PAGE gels to investigate their oligomeric state (Fig EV1A–D). Three proteins (TqsA: 37.5 kDa; YdiK: 39.8 kDa; and YhhT: 38.5 kDa) migrated as single dominant bands with apparent molecular masses of about 320 kDa (Fig EV1A–C), which corresponds to around 180 kDa using a correction factor of 1.8 to account for the mass contribution of bound Coomassie dye and detergent molecules (Heuberger et al, 2002). The deduced mass suggests that all three proteins may exist as homo‐oligomeric complexes consisting of four or five protomers. By comparison, a single dominant band migrating at approximately 155 kDa was observed for PerM (39.2 kDa), yielding a mass of ~86 kDa after correction, which may indicate the presence of a dimer.
To investigate the influence of detergents on the oligomerization of AI‐2 proteins, several non‐ionic detergents (α‐DDM, β‐DDM, DM, and LMNG) were also used for purification. After solubilization with β‐DDM, detergent exchange was directly performed on the affinity chromatography column. The oligomeric states of the purified proteins were further assessed by size exclusion chromatography and BN–PAGE analysis. All proteins displayed similar behavior to that described in GDN (Fig EV1A–D), indicating that the assembly of higher oligomeric complex was not affected by the choice of these detergents.
Single‐particle cryo‐EM analyses of AI‐2 exporters
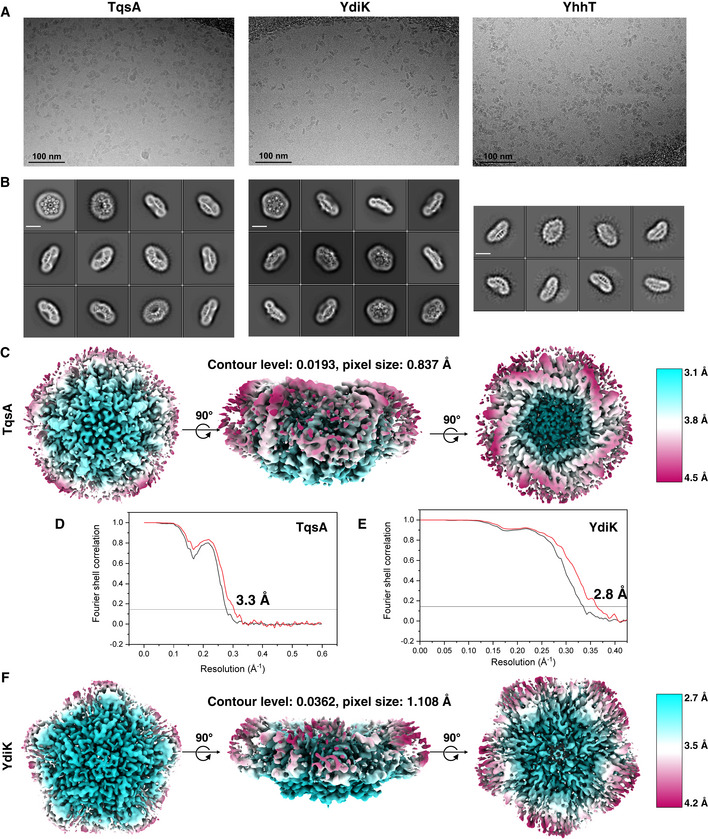
As already mentioned, monomers of AI‐2 exporter proteins are relatively small, and the size is close to the theoretical lower protein size limit for the visualization in cryo‐EM (Henderson, 1995). Nevertheless, the existence of higher oligomeric states of AI‐2 exporters allowed us to successfully execute electron microscopic high‐resolution structure determinations without the need for additional fusion proteins or binding proteins. For the cryo‐EM experiments, we attempted protein preparation in the two commonly used detergents LMNG and GDN. After data processing, we compared different datasets, and we found that GDN was more suitable for the AI‐2 exporters than LMNG, yielding a better quality of reconstructed 3D density maps. A total of 5,452 and 7,372 movie stacks were collected for GDN‐purified TqsA and YdiK, respectively (Fig EV2A). The two‐dimensional (2D) class averages showed that TqsA adopts a characteristic pentameric structure resembling the shape of a “flower” with five petals when viewed from the top and displays a bowl‐shaped architecture equipped with a central cavity when viewed from the side (Fig EV2B). YdiK also forms a similar pentameric arrangement, however, the central cavity is not that pronounced in the 2D class averages (Fig EV2B). The final three‐dimensional (3D) reconstructions yielded maps with 3.3 Å and 2.8 Å resolutions for the overall structures of TqsA and YdiK, respectively (Fig 2A and C). In both EM maps, the bottom and the interior of the bowl are the best resolved portions of the structure (~3.0 Å for TqsA and ~2.7 Å for YdiK), while the rim of the bowl is not that well resolved, especially in the EM map of YdiK, probably due to the high flexibility of this region (Fig EV2C–F). Finally, we built the atomic models de novo for both AI‐2 exporters (Fig 2B and D). Our final model for each TqsA monomer contains 340 of a total of 344 residues, spanning Ala2 to Leu341 (Figs 3A and EV3A). For YdiK, some portions of the protein could not be modeled due to lack of density, and the final model includes three polypeptides, Pro7‐Ile80, Phe163‐Gly230, and Thr279‐Gln354, comprising ~59% of the total amino acid residues (Figs 3C and EV3B).
Figure EV2. Cryo‐EM on AI‐2 exporters.
-
ARaw micrographs for the three GDN‐purified AI‐2 exporters TqsA, YdiK, and YhhT from Titan Krios TEM at 300 kV.
-
BReference‐free 2D class averages for the proteins (scale bar—10 nm).
-
CLocal resolution EM map for TqsA.
-
D, EFourier shell correlation (FSC) curves for the AI‐2 exporters TqsA and YdiK. The profiles for masked and unmasked maps are shown in red and black, respectively.
-
FLocal resolution EM map for YdiK.
Figure 2. Cryo‐EM structures of the pentameric complex of the two AI‐2 exporters.
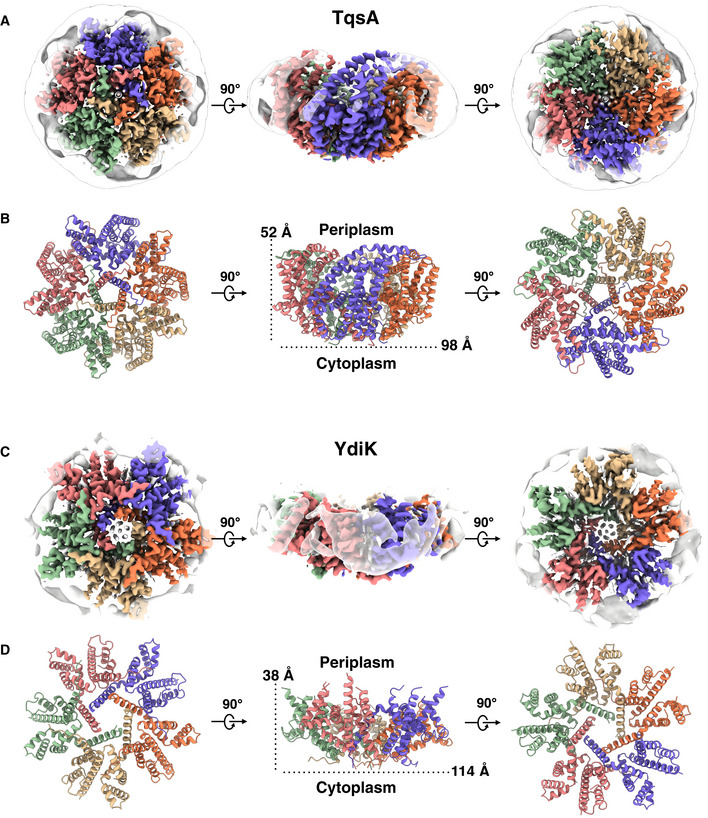
- Cytoplasmic (left), side (middle), and periplasmic (right) view of a cryo‐EM 3D reconstruction map of TqsA at 3.35 Å resolution colored by protomers. The detergent belt surrounding the transmembrane helix region of the protein is shown.
- Corresponding structural model of the TqsA pentamer. Protomers are colored as in A.
- The 2.80 Å cryo‐EM density map of YdiK showing the pentameric organization of YdiK. Each protomer is presented in a different color.
- Corresponding structural model of YdiK pentamer. Protomers are colored as in C.
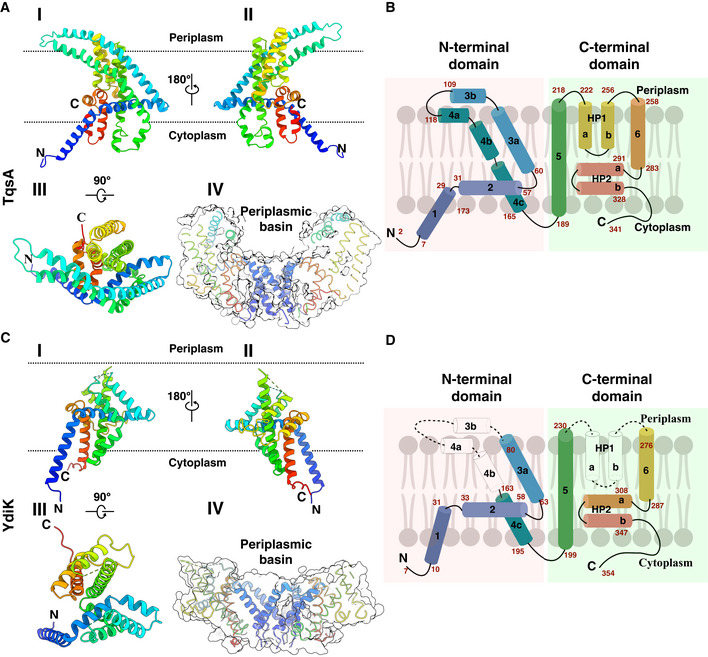
Figure 3. Atomic structures of the two monomeric AI‐2 exporters.
-
ARibbon representation of the TqsA monomer, viewed from the membrane using a rainbow color gradient from the N‐ (blue) to C‐terminus (red). I, II, and III show different orientational views of the monomer. IV represents the sliced surface view of TqsA, highlighting the positioning of H1 and the periplasmic basin.
-
BSchematic representation of the transmembrane topology of TqsA. α‐Helices are shown as cylinders; sequence numbers of amino acids of segments are marked in red. The N‐ and C‐terminal domains are shaded in red and green, respectively.
-
C, DStructure of the YdiK protomer as shown for TqsA in (A) and (B). The hydrophilic part of TM3 and TM4 as well as HP1 are not included in the atomic model due to the weak density for this part in the cryo‐EM map. The missing loops are represented as dashed connecting lines and missing helices are shown as white cylinder in the topology map.
Figure EV3. Model fitting of the AI‐2 exporters.
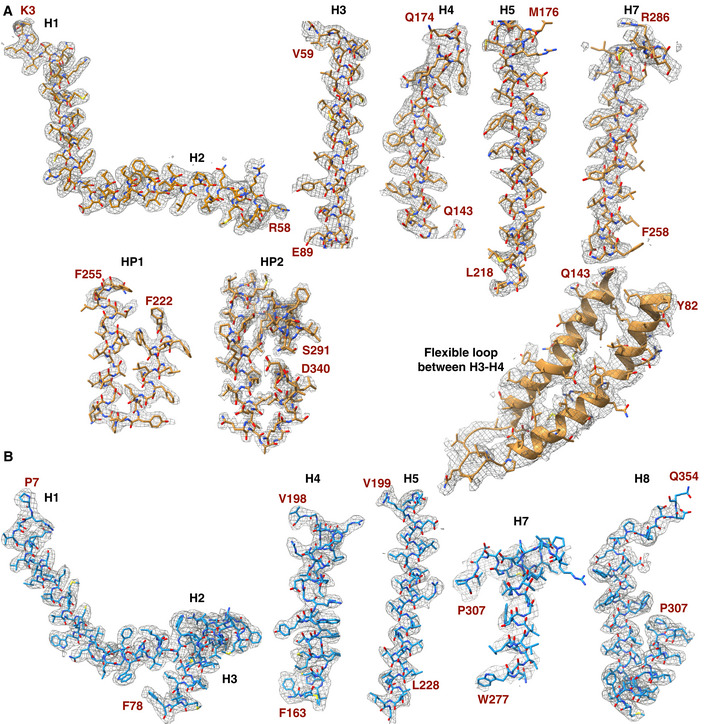
- Model fitting of TqsA in the EM density map for different regions of the monomer structure. The residues constituting a particular structural region are indicated in red.
- Model fitting of YdiK in the EM density map for different regions of the monomer structure is shown with the similar nomenclature as that used for TqsA in A.
In addition, a small set of 989 movies were collected for the GDN‐purified YhhT (Fig EV2A). This dataset exhibited a pronounced preferred orientation, presenting mainly side views of the particles in 2D class averages. Due to the absence of top views, direct visualization of the oligomeric state of YhhT was not possible (Fig EV2B). Nevertheless, YhhT exhibits very similar side views as observed for TqsA and YdiK. The particle size of all three proteins has similar dimensions of 135–145 Å in diameter, thereby providing sufficient evidence for a pentameric nature of YhhT (Fig EV2B).
Additionally, we also purified the AI‐2 exporter Aq_740 (~40.2 kDa), from the hyperthermophilic bacterium Aquifex aeolicus which is the only family member present in its genome. A dataset of 7,254 movies was collected and our preliminary data show that Aq_740 also forms a pentamer (Fig EV4). The 6.4 Å low‐resolution 3D reconstruction showed that it adopts a similar helical arrangement as TqsA and YdiK (Fig EV4).
Figure EV4. Cryo‐EM studies on Aq_740 from Aquifex aeolicus .
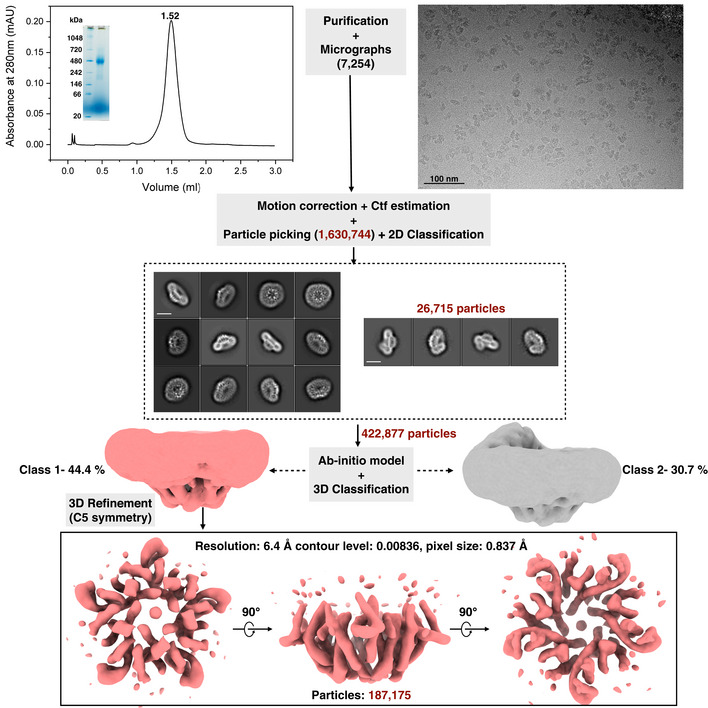
A flowchart for the entire workflow adopted for Aq_740 is represented in the figure. Analytical SEC and BN–PAGE profile for Aq_740 are shown in the beginning on the top of the figure. Superose 6 3.2/300 GE column analytical column was used for the final size exclusion step. An illustrative raw micrograph for GDN purified AI‐2 exporter Aq_740 from Titan Krios TEM at 300 kV with 0.837 Å pixel size is shown on the right. A total of 7,254 gain‐corrected movies were collected. Reference‐free 2D class averages for Aq_740 (scale bar—10 nm) were obtained from RELION 3.1 with an initial particle stack of around 1,630,744 particles. Later, two different and almost equally populated 3D classes were obtained as shown. One of them depicted in light coral shade was further refined with imposed C5 symmetry. It yielded a low‐resolution 3D reconstruction of 6.40 Å which is shown in different angular views at the bottom of the figure upon anti‐clockwise rotation along the X‐axis. The final particle stack of the map is around 187,175 particles. The processing was performed using C1 symmetry. C5 symmetry was only imposed on the later refinement stage of Class 1.
Pentameric architecture and oligomeric interface
The cryo‐EM structures of TqsA and YdiK revealed that both AI‐2 exporters form a pentamer with the fivefold symmetry axis perpendicular to the membrane (Fig 2B and D). The structure of the pentameric complex features a bowl‐shaped architecture with a concave aqueous basin facing the periplasmic space and a tightened base located in the cytoplasm. The TqsA pentamer is about 98 Å in diameter and 52 Å in height (Fig 2B). On the periplasmic side, the rim of the bowl is shaped like a pentagon with each side approximately 39 Å in length. The basin has a depth of around 30 Å and penetrates the membrane bilayer from the periplasm about two‐thirds. The concave surface is mainly hydrophilic which may allow the bulk aqueous solution to access the membrane interior to a considerable depth (Appendix Fig S2A). Between the two TqsA protomers within the pentamer, the interface has an area of 1,717 Å2 per monomer and is mainly formed by hydrophobic interactions of residues present in the N‐terminal regions from the two adjacent monomers (Appendix Fig S3D). In addition, the interface is further strengthened by a few H‐bonds, e.g., between Ser145 (protomer A) and Tyr127 (protomer B) on the periplasmic side as well as between Lys171 (protomer A) and Leu337/Ser339 (protomer B) on the cytoplasmic side.
The overall structure of the YdiK pentamer, which is 114 Å in diameter, is similar to that of the TqsA protein (Fig 2D and Appendix Fig S2B). However, the height of the pentameric YdiK within the membrane bilayer and the depth of its basin cannot be accurately estimated since the resolved structure was only partially modeled and the majority of the missing density lines the periplasmic basin. Nevertheless, one interesting structural feature can be recognized at the cytoplasmic bottom of the pentamer in both proteins. There is a pentagonal central opening connecting the cytoplasm with the periplasmic aqueous basin (Fig 4A and B). The pentagonal opening at the cytoplasmic side has a width of 17 Å with each side of the pentagon of 11 Å for TqsA and a width of 20 Å with each side of the pentagon of 11.6 Å for YdiK (Fig 4A and B). It spans vertically into the membrane to a depth of 22 Å for TqsA and 17 Å for YdiK, and subsequently, it opens up in the basin facing the periplasm. The overall shape of this opening is dissimilar in both AI‐2 exporters. It resembles a cylinder in the case of TqsA, while it appears more like a funnel opening up in the periplasmic basin in YdiK (Fig 4A and B). In both proteins, it is constituted by Helix1 (H1) and this helix from each protomer entangles against the neighboring one. This helix mostly comprises hydrophobic residues resulting in an overall hydrophobic environment in this central area. (Fig 4C and D).
Figure 4. Structural features of the AI‐2 exporters TqsA and YdiK.
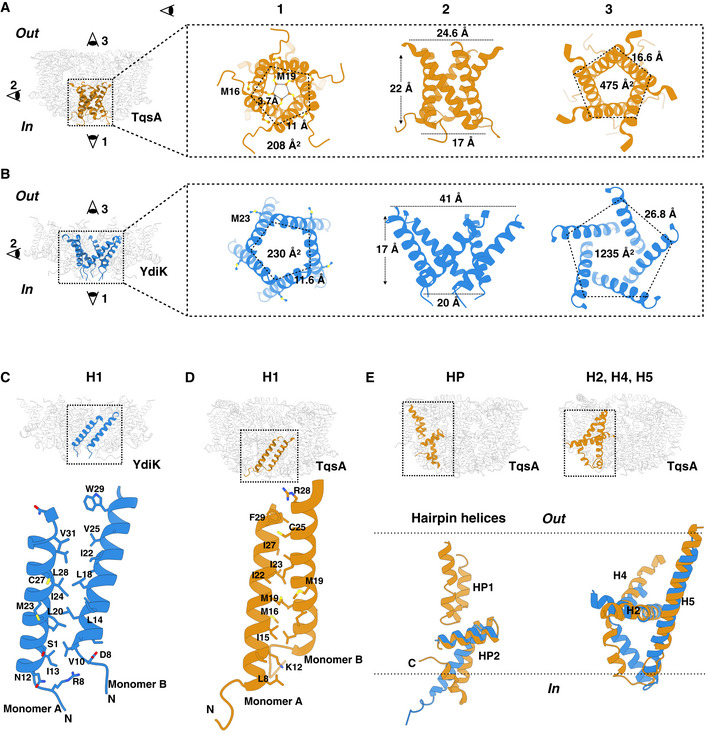
-
A, BThe figures focus on the cytoplasmic central opening of the proteins TqsA (A, brown) and YdiK (B, blue). Images on the left in both figures show the overall positioning of the opening in the pentameric complexes. The illustrations on the right represent distinct views of this region from three different directions.
-
C, DClose views showing the hydrophobic interactions between two adjacent H1 helices, which are vital for maintaining the pentameric architecture of the AI‐2 exporters.
-
EStructural superposition of TqsA and YdiK in specific regions of the proteins in order to highlight their structural deviations. Helical hairpins are shown on the left while H2, H4, and H5 on the right.
We could observe long non‐protein densities in the central cytoplasmic opening of YdiK and around it in the case of TqsA, suggesting the presence of either lipids or detergent molecules. In TqsA, there is a methionine residue (Met19TqsA) facing the interior of the opening from each protomer as shown in Fig 4A. The distance between the adjacent residues is ~3.7 Å and the arrangement of five methionine side chains appears to be like a hydrophobic filter or block in the cavity. Methionine residues, Met16TqsA and Met23YdiK, are also present in the central region, but they face the exterior of this area unlike Met19TqsA (Fig 4A and B). The presence of this methionine hydrophobic filter in TqsA and lipids/detergents in the case of YdiK appears to prevent any possible leakage of ions or water molecules across the membrane through such wide openings in both proteins.
Overall structure of TqsA and YdiK protomer
The TqsA protomers are mainly composed of α‐helical segments with both N‐ and C‐terminus exposed to the cytoplasmic side of the membrane (Fig 3A). Viewed parallel to the membrane plane, the protein has the shape of a “half‐moon”. Each protomer can be clearly divided into an N‐terminal domain formed by four α‐helices (H1‐4) and a C‐terminal domain consisting of two transmembrane α‐helices (TMH5 and 6) as well as two helical hairpin motifs (HP1 and HP2) as shown in Fig 3B.
The fold of the N‐terminal domain is less compact than that of the C‐terminal half of TqsA and displays several unusual structural features, which are not commonly observed in other membrane transporters. H1 (Thr7‐Phe29) is inclined to the membrane normal by about 40° and penetrates only partially into the lipid monolayer (Fig 3A). H1 from all five protomers entwine obliquely around the fivefold axis, forming the major contact point on the cytoplasmic face of the pentamer (Fig 4A). This N‐terminal helix unravels at Ala30, followed by the membrane‐embedded horizontal helix H2. The 42‐Å‐long H2 (Ala31‐Trp57) is oriented nearly parallel to the membrane plane and is localized predominantly in the inner monolayer. This horizontal helix H2 forms an angle of about 105° with H1 and an angle of 60° with H3. The two long helices H3 (Arg61‐Glu109) and H4 (Asp118‐Glu164), which form an anti‐parallel helical bundle, constitute another unusual structural element in the N‐terminal domain of TqsA. Each helix is broken into segments that result in a highly curved overall shape (Fig 3A and B). Both helices can be divided into two approximately equal parts with the hydrophobic portion spanning the membrane and a hydrophilic region exposed to the periplasm. Within the membrane, this two‐helix bundle is tilted from the membrane normal by approximately 45° (Fig 3A and B). In the TM domain of H3 and H4, mainly van der Waals interactions are responsible for the helix–helix interactions. Interestingly, several leucine residues are spaced regularly at three or four amino acid intervals, and this leucine zipper‐like dimerization motif may further stabilize the bent conformation of this two‐helix bundle (Appendix Fig S3B). At the membrane–water interface, H3 and H4 are tilted significantly away from the membrane normal and finally, end almost parallel to the membrane surface at the periplasmic side. The connecting loop between H3 and H4 forms one of the major contact points between two adjacent protomers on the boundary of the periplasmic basin (Appendix Fig S3D). The density of this loop was quite feeble, suggesting flexibility of this region (Fig EV3A).
The C‐terminal domain of TqsA is compactly assembled into a cylinder‐shaped unit. TMH5 (Ala189‐Leu218) is oriented perpendicular to the membrane plane. The loop connecting H4 and TMH5 crosses over H2 and is also one of the flexible loops in the structure (Fig 4E). TMH5 together with H2 form an uncommon cross‐shaped structure, lining the interface between the N‐ and C‐halves, and this feature may provide an anchor for possible conformational rearrangements during the transport cycle (Appendix Fig S3A). Following H5, two helical hairpins HP1 (Phe222‐Asn256) and HP2 (Ser291‐Thr328) constitute a functionally critical element in the C‐half of TqsA (Fig 3A and B). Both HP1 and HP2 are helix–turn–helix supersecondary structures mainly composed of hydrophobic residues. HP1 begins on the periplasmic side of the membrane and penetrates vertically about halfway across the membrane. This helical hairpin is sandwiched between H3 and H4 from the N‐half and TMH5 and TMH6 from the C‐half of TqsA. Both HP1 and HP2 are connected by the 36‐Å‐long helix TMH6 (Phe258‐Ile283) followed by a seven‐residue loop (Met284‐Leu290). Unlike HP1, HP2 is oriented nearly parallel to the membrane plane, with its turn region exposed to the periplasmic basin. HP2 is followed by the C‐terminal region of around 13 residues returning toward the membrane yet exposed in the cytoplasm (Fig 3A and B).
Similar to TqsA, each YdiK protomer comprises six α‐helical segments and two helical hairpins (Fig 3C and D). Due to the lack of density, some portions of the protein could not be modeled, including the hydrophilic region of H3 and H4 and the first helical hairpin (Figs 3C and D, and EV3B). Nevertheless, YdiK shows a similar helical arrangement to that observed in TqsK (Fig 4E). All uncommon structural features that we observed in the TqsA structure, including the membrane‐embedded horizontal helix H2, the cross‐shaped arrangement between H2 and H5, as well as the placement of HP2, can also be discerned in YdiK (Fig 4E).
Interaction with AI‐2
We investigated the interaction between TqsA and its putative substrate AI‐2 (R‐THMF) with in silico substrate docking. Our docking results show that the AI‐2 compound fits well into the cavity formed by HP1, H5, and H6 in the protomer (Fig 5A and C). According to this binding mode, the AI‐2 molecule may interact with several surrounding residues, including Tyr196, Lys200, Pro237, Asn238, Glu280, and Met284 (Fig 5A). Asn238 and the strictly conserved Pro237 are located at the tip of the HP1 (Fig 5B and Appendix Fig S10). Due to the presence of four hydroxyl groups, the AI‐2 molecule is capable to form hydrogen bonds with the neighboring residues as observed in the AI‐2 bound structures of its receptors LsrB (Miller et al, 2004; Kavanaugh et al, 2011; Torcato et al, 2019). Knowing the structure of TqsA with bound AI‐2 will be further beneficial to shed more light on the nature of the binding mechanism of AI‐2 to the AI‐2 exporters.
Figure 5. Molecular docking of AI‐2 into the TqsA protomer.
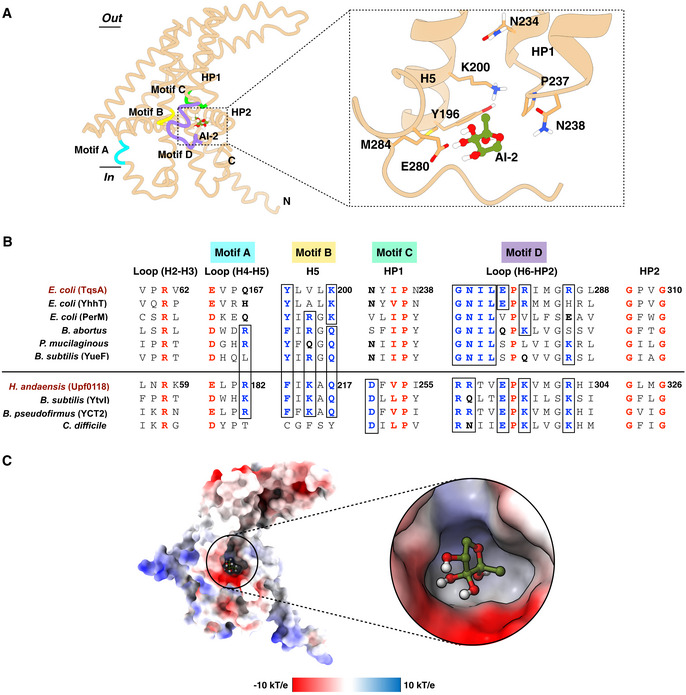
- The AI‐2 molecule is depicted in an olive‐green color. A closer look on the residues lining the docked site is shown on the right. The predicted binding site is formed by the residues from H5, the tip of HP1, and the loop connecting H6 to HP2. Motifs which were previously recognized as critical for Upf0118 function were positioned on the TqsA monomer structure based on a sequence alignment. Motifs in the structure are shaded in distinct colors; Motif A: sky blue, Motif B: yellow, Motif C: green, and Motif D: purple.
- Multiple‐sequence alignment highlighting the relation between AI‐2 exporters and Upf0118. Highly conserved residues are colored in red, and partially conserved ones in blue. The alignment is sectioned based on conserved residues and Upf0118 motifs. Homologs considered for the AI‐2 exporter group include AI‐2 exporters from E. coli (TqsA, YhhT, and PerM), Brucella abortus (UniProt: A0A7U8JIH7), Paenibacillus mucilaginosus (UniProt: F8FLY5), and Bacillus subtilis (YueF, UniProt: O32095). Similarly, homologs considered for Upf0118 group include proteins from H. andaensis (Upf0118, UniProt: A0A1W5X0D5), B. subtilis (YtvI, UniProt O50485), Bacillus pseudofirmus (YCT2, UniProt: Q04454), and Clostridium difficile (UniProt: Q188A3). The choice of the homologs considered for multiple‐sequence alignment is based on the sub‐groups within the AI‐2 exporter family mentioned in the Upf0118 study (Wang et al, 2020). YdiK from E. coli is not included in the alignment due to low sequence similarity. It constitutes a distinct sub‐group in the family.
- Electrostatic potential surface representation of the proposed interaction site of AI‐2 within TqsA, focusing on the charge distribution around the site. Blue represents the presence of positive charged residues while red shows the presence of negative charge residues.
Recently, studies on Upf0118 from H. andaensis have shown that this Na+/H+ antiporter, together with its homologs, shares five functionally critical motifs (Motif A‐E) (Wang et al, 2020). Based on the sequence alignment, we explored the positioning of those motifs in the TqsA protomer structure (Fig 5A and B). Our results show that the potential substrate‐binding site in TqsA is surrounded by the motifs B, C, and D, which are located on H5, the tip of HP1, and the loop connecting H6 to HP2, respectively. The residues observed critical in these motifs (B, C, and D) for Upf0118 were compared with that of TqsA and are briefly shown in Appendix Fig S4. Most of the functionally important residues are not conserved in TqsA and the other AI‐2 exporter subgroup (Fig 5B and Appendix Fig S4A). In TqsA, the motif C forms the short turn region of HP1 and is defined by a pattern of one small hydrophobic residue (Ile236TqsA) followed by one absolutely conserved proline residue (Pro237TqsA) as shown in Fig 5B. Notably, a short E‐P‐R/K tripeptide can be found in the motif D of many AI‐2 exporters as well as in the Upf0118 family proteins (Fig 5B and Appendix Fig S10). Glu296Upf0118 was shown to be vital for the function of Upf0118, and our docking studies also suggested the involvement of an equivalent residue in TqsA (Glu280TqsA) in substrate binding. In addition to the above‐mentioned motifs, two other motifs (A and E) are identified in Upf0118. There are some highly conserved residues among the family including Glu164TqsA present in motif A as shown in Fig 5B, which are located rather far from the putative substrate interaction site and their functional roles require further analyses. Motif E was not considered to be essential for the Na+/H+ antiporter activity of Upf0118 protein, and it is not present in other AI‐2E family proteins as well. Taken together, the canonical AI‐2 exporters, in particular, TqsA from E. coli, share similar yet distinct motifs with the Upf0118 family proteins. The potential substrate‐binding site of TqsA lacks several conserved charged or polar residues that are important for the Na+/H+ antiporter activity seen in Upf0118 family proteins, providing key evidence for the two proteins to belong to the same protein family but with different functions.
In addition to the docking studies, native mass spectrometry (native MS) was also employed to investigate the binding of AI‐2 to TqsA. The mass spectrum of TqsA in 0.05% n‐dodecyl‐N, N‐dimethylamine‐N‐oxide (LDAO) in the absence of AI‐2 substrate display a charge‐state distribution corresponding to pentameric TqsA, which further confirms the oligomeric state that we observed in the structure (Appendix Fig S5A). Additionally, the spectrum also displayed a range of endogenous lipids co‐purified with the protein, whose masses are ~1,388 Da, ~2,963 Da, and 4,416 Da. Further lipidomic analysis on the TqsA sample showed the presence of the common E. coli lipids PE (phosphatidylethanolamine), PG (phosphatidylglycerol), and CDL (cardiolipin) (Appendix Fig S1D). We then added 60 µM DPD/AI‐2 to 4 µM TqsA and recorded the data. The resultant spectra display a clear shift (~140 kDa) in the peak positions for pentameric TqsA (Appendix Fig S5A), indicating the binding of this non‐borate form of AI‐2 to the TqsA complex. To further confirm the binding of AI‐2 to TqsA, we analyzed the samples in the delipidating detergent tetraethylene glycol monooctyl ether (C8E4), where the majority of the protein was found to be in the monomeric form. When AI‐2 was added, the mass spectra clearly show additional peaks which correspond to the binding of AI‐2 to the TqsA monomer (Appendix Fig S5B). A similar methodology was also adopted for YdiK and we could observe AI‐2 interaction with the YdiK monomer as well (Appendix Fig S5C).
Furthermore, the measurements were also performed using several variants of TqsA, which included single‐point mutations of the residues lining our proposed binding site (K200, N234, N238, P237, and E280) and two highly conserved residues R61 and E164 (Appendix Fig S4B and Fig 5A and B). During purification, all variants behaved similarly to the wild‐type TqsA according to the size exclusion and the Blue native PAGE analyses, indicating the complete assembly of the pentamers. A minor effect was observed for K200L, P237A, N238L, and E280Q. The three variants R61L, E164Q, and N234L showed a mild decrease in DPD binding (Appendix Fig S4B). In addition, a quadruple variant (N234L/P237A/N238L/E280Q) was also investigated to test the potential synergistic effect of the mutations. In comparison to the single mutants, the quadruple variant showed a significant effect (Appendix Fig S4B). It should be noted that these results with the TqsA variants are preliminary and further biochemical support is required. Taken together, we could observe the interaction of AI‐2 with TqsA both in silico and in vitro. However, further studies are needed to elucidate the functional importance of the interacting residues.
Discussion
Members of the AI‐2 exporter superfamily are putative secondary active transporters that are widely distributed in prokaryotes and archaea (Rettner & Saier, 2010). This protein family (2.A.86) was renamed (formerly, PerM family, 9.B.22) subsequent to the characterization of its first member TqsA from E. coli as an exporter of the quorum sensing signaling molecule autoinducer‐2 (Saier, 2000). In this work, we could show that three AI‐2 exporters from E. coli (TqsA, YdiK, and YhhT) assemble into homo‐pentameric complexes (Figs 2 and EV2). A pentameric assembly is a rare occurrence among membrane transporters. To the best of our knowledge, the only other transporter family with homo‐pentameric members is the formate/nitrite transporter (FNT) family (Lü et al, 2013).
In this study, using cryo‐EM, we have determined the first structures of two AI‐2 exporters from E. coli, TqsA and YdiK at 3.35 Å and 2.80 Å overall resolutions, respectively (Fig 2A and C and Movie EV1). Both protein complexes comprise a central hydrophobic central opening facing the cytoplasm, which extends around one‐third into the membrane and shows an overall bowl‐shaped appearance, opening up in the periplasmic side (Fig 4A and B). The presence of this opening connecting the periplasmic basin to the interior of the cell may insinuate that this region of the protein can potentially serve as a channel. However, this region appears to be sealed by the hydrophobic filter in the case of TqsA and the presence of lipids in YdiK (Fig 4A).
Very recently, DeepMind has developed the artificial intelligence (AI) deep learning system AlphaFold for the prediction of protein structures (Senior et al, 2020). During the preparation of this manuscript, the AlphaFold protein structure database was launched (Jumper et al, 2021), which also included the predicted structures of the four AI‐2 exporters from E. coli (Appendix Figs S6 and S7). We compared our cryo‐EM structure of TqsA with the one predicted by AlphaFold. The superimposition of both structures showed a surprisingly high similarity with a root mean square deviation (RMSD) of 3.8 Å (Appendix Fig S6A). The main differences are within the connecting loops and the region that is believed to undergo structural rearrangements necessary for transport. In the case of YdiK, our experimentally determined partial structure also shares a remarkable similarity with the corresponding portion of the AlphaFold structure as indicated by an RMSD of 4.6 Å (Appendix Fig S6B). Importantly, the predicted structure helped to fill in the missing parts of the experimental structure. It should be mentioned that the missing region of H3 and H4 was predicted with low confidence, which suggests a high flexibility and conformational variability and may explain why the density here cannot be well resolved. In addition, both YhhT and PerM were predicted to comprise around six helical segments and two helical hairpins and to share similar fold characteristics with TqsA (Appendix Fig S7). Taken together, AlphaFold’s computational predictions appear to be quite good, at least for the four AI‐2 exporters investigated here, although the prediction of the oligomeric state and topology currently remains not possible.
Overall, the AI‐2 exporter TqsA comprises several interesting structural elements. To begin with, the helix arrangement of H1 and H2 is absolutely peculiar (Fig 4A and B). It gives a perception of a “camera shutter” capable of widening and narrowing during the transport activity. Another captivating feature in each protomer is the presence of two “helical hairpins” which is a characteristic and functionally critical structural element reported in many other transporters, such as the glutamate transporter SLC1, the nucleoside transporter CNT, and the citrate transporter NaCT (Yernool et al, 2004; Hirschi et al, 2017; Yu et al, 2019; Sauer et al, 2021; Fig EV5B). However, both hairpins in the AI‐2 exporter are arranged distinctively relative to the other families (Fig EV5B). In most of the other transporters, the tips of the helical hairpins face each other in the overall structure. In AI‐2 exporter, HP1 is perpendicular to the membrane plane while HP2 is curved and bends almost parallel to the membrane plane facing the interior toward the periplasmic basin (Fig EV5B). Both helical hairpins form an L‐shaped structure with the tips pointing in different directions (Fig 4E).
Figure EV5. Comparison between AI‐2 exporter and other transporter families adopting elevator mechanism.
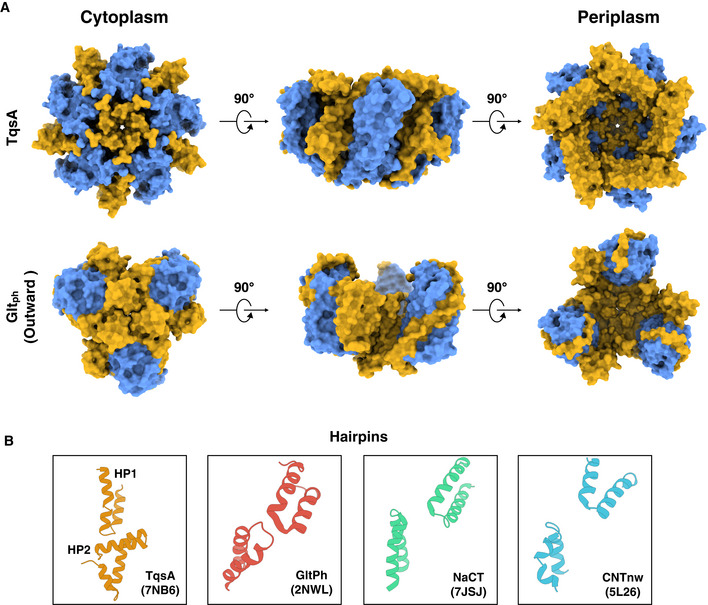
- Contrast between AI‐2 exporter TqsA (PDB: 7NB6) and glutamate transporter Gltph (PDB: 2NWL) is shown. The scaffold domain in both the surface representative models is shown in yellow and the transport domain is illustrated in blue.
- The figure focuses on the differences in the helical hairpins in four different transporter families (AI‐2 exporter (TqsA); 7NB6, glutamate transporter; 2NWL, sodium citrate transporter; 7JSJ, and nucleoside transporter; 5L26).
All above‐mentioned transporters possessing two helical hairpins employ the elevator‐type mechanism for transport of their respective substrates (Yernool et al, 2004; Boudker et al, 2007; Hirschi et al, 2017; Garaeva & Slotboom, 2020; Sauer et al, 2021). Several common features among such transporter families have been highlighted in previous reports like the presence of two distinct domains, one rigid scaffold domain and one mobile transport domain normally segregated by a tilted helix supporting the major conformational changes during the transport cycle (Reyes et al, 2009; Garaeva & Slotboom, 2020; Chen et al, 2021). Our structures also reveal the presence of two such domains that are separated by the long and extremely bent helix H4 (Fig 3B and D, and Fig EV5A). In addition, most elevator‐type transporters exist as either homodimers or homotrimers, in which the subunit contacts are mainly mediated by the scaffold domains (Reyes et al, 2009; Garaeva & Slotboom, 2020). Although AI‐2 exporters assemble in an even higher oligomeric state, still the major contact points between the protomers are formed by the scaffold domains (Fig EV5A). Moreover, a membrane topology with inverted repeats is another commonality of the elevator‐type transporters. In TqsA, internal pseudosymmetry can be only observed in the C‐terminal domain (Fig 3B).
The topology of the C‐terminal transport domain of TqsA shows a remarkable similarity with that of sodium‐/potassium‐dependent glutamate and amino acid transporters, e.g., Gltph from Pyrococcus horikoshii and the human ASCT2 (Yernool et al, 2004; Reyes et al, 2009; Ruan et al, 2017; Yu et al, 2019) (Fig 6A). Previous structural studies of glutamate transporters revealed that this protein forms a bowl‐shaped trimer (Yernool et al, 2004; Reyes et al, 2009). In both the proteins Gltph and TqsA, the transport domain comprises a transmembrane helix followed by two helical hairpins and a connecting helix between the hairpins (Fig 6A). Although the aqueous basin in both proteins faces the same direction, the overall topology of the transport domain and the transmembrane helix connecting N‐ and C‐terminal halves is inverted. This inversion event might be attributed to the fact that the scaffold domain of ΤqsA has one α‐helix less compared to Gltph (Fig 3B).
Figure 6. Proposed transport mechanism for AI‐2 export.
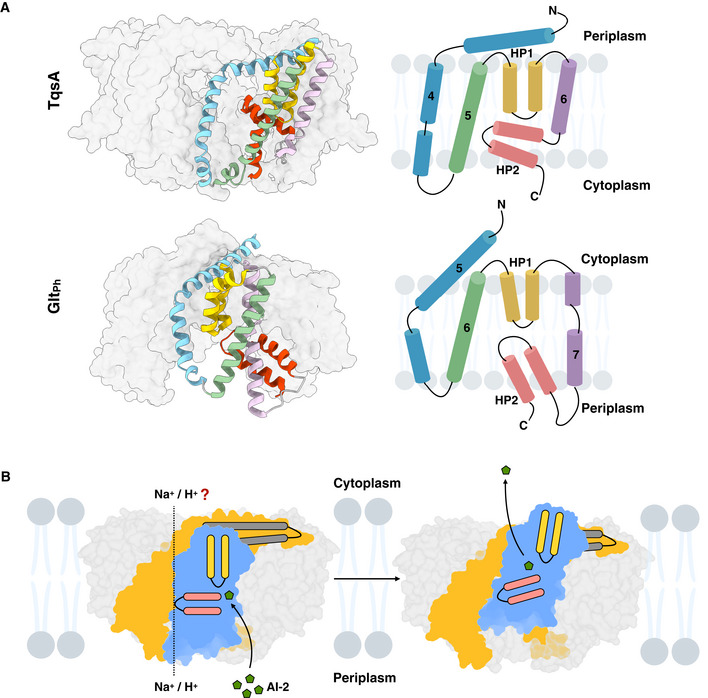
- Similarities between the transport domain of AI‐2 exporter TqsA (PDB: 7NB6) and glutamate transporter (Gltph) from Pyrococcus horikoshii (PDB: 2NWL) are shown. Individual segments of the transport domain of TqsA are shaded in different colors (H4 in blue, H5 in green, HP1 in yellow, H6 in purple, and HP2 in red). Similarly, corresponding structural segments in Gltph are colored in a similar manner. The topology map of the transport domains of both proteins is included on the right of the figure for a better comprehension.
- Proposed transport mechanism of TqsA. AI‐2 is depicted in an olive‐green color in the figure, and homo‐pentameric AI‐2 exporter TqsA is shown embedded in the membrane bilayer as viewed from the membrane plane. The lipids surrounding TqsA are depicted in faint blue. The scaffold and transport domains of the monomer are shown in yellow and blue, respectively. HP1 and HP2 are depicted in yellow and red, respectively, and the long anti‐parallel tilted helix bundle (H3 and H4) is represented in dark gray. The likely outwards facing conformation via elevator motion of TqsA is suggested in the figure on the right. The coupling ion for the transport is kept as an unresolved factor in the figure.
The observation of the structural similarity between the transport domain of the AI‐2 exporter TqsA and that of glutamate transporter yields a strong clue for anticipating the transport mechanism of the AI‐2 exporter. Gltph utilizes an elevator‐type mechanism for providing alternating access of the binding site to either side of the membrane (Yernool et al, 2004; Reyes et al, 2009; Chen et al, 2021). Each protomer of Gltph works as an independent transport unit. The binding site of glutamate in Gltph is observed at the interface of the two helical hairpins, also involving residues from the surrounding helices (TM7 and TM8) and loops in the transport domain. As shown earlier, a similar binding site was predicted for AI‐2 in TqsA from our molecular docking studies involving residues from HP1, H5, and the loop connecting H6 to HP2 (Fig 5A). In Gltph, HP1 is reported to be involved in substrate binding while HP2 acts as a gate, alternatively sealing and opening the binding site during the transport cycle (Zhang & Kanner, 1999; Yernool et al, 2004). The two highly tilted helices in the glutamate transport along which its transport domain slides determine the distance the binding site can travel across the membrane during the transport cycle (fixed barrier) (Garaeva & Slotboom, 2020). Similarly, in TqsA, the two long tilted helices (H3 and H4) and the loop connecting H4 to H5 on the cytoplasmic side may act as a determinant or fixed barrier, deciding the distance which the transport domain can progress (Appendix Fig S3C). The flexible loop connecting H6 to HP2 also seems to be a participant in this barrier, but this speculation requires support from further studies (Appendix Fig S3C). Additionally, the large aqueous basin formed in the glutamate transporter is considered to be beneficial for allowing rapid access of the transmitter to the binding site during synapses (Yernool et al, 2004). A similar bowl‐shaped architecture in TqsA might allow rapid diffusion of the pheromone AI‐2 into the environment through the aqueous periplasmic bowl (Appendix Fig S2).
Taken together, we suggest a similar transport mechanism for AI‐2 exporters as exercised by glutamate transporters (Fig 6B). Each protomer can work as an independent transport unit adopting an elevator‐type movement (Movie EV1). In addition, we could observe a peculiar protein conformational change for Aq_740 in both 2D and 3D class averages during the single‐particle cryo‐EM data processing (Fig EV4). One protomer in the pentameric complex was seen to be risen toward the periplasmic side. This observation supports our suggestion about the elevator‐type mechanism for AI‐2 exporters and allows us to postulate that our apo structures are in inward‐facing conformations. However, further studies are required for a better understanding of the transport mechanism of this family.
In summary, many questions remain open, requiring further studies to elucidate the transport mechanism of the AI‐2 exporters. These include the movement and gating mechanism of the hairpins, sodium being the coupling ion for the transport, the possibility of the presence of any ion channel, and the identification of other potential substrates. Whereas proteins such as TqsA comprise a short α‐helix at the C‐termini, larger homologs in AI‐2 exporter family contain a C‐terminal ATP/GTP binding domain, a sensor GAF domains, or a Tpr‐like ligand‐binding domain (Rettner & Saier, 2010). In search of other potential substrates, such as cyclic AMP, cyclic GMP, nucleosides, and even other quorum‐sensing signaling molecules including AI‐3, an in vitro transport assay system has to be established (Kim et al, 2020). So far, we were unable to demonstrate the transport activity of TqsA using a number of methods, but the attempts are still ongoing. In this study we successfully characterized several members of the Autoinducer‐2 exporter family and highlighted the structural uniqueness. Based on the structural analysis and comparisons, we suggest a transport mechanism for this transporter family. We would still like to accentuate the importance of further studies on the interaction or transport of the suspected substrate AI‐2 which still remains a probable substrate for this family. We are open to the possibility of other substrates as mentioned earlier. However, this study on the pentameric AI‐2 exporters will undoubtedly prove to provide benefits for future studies in the fields of quorum sensing, biofilm formation, and antibiotic resistance.
Materials and Methods
Cloning, expression, and purification of autoinducer‐2 exporters
Primers were designed based on the full‐length gene sequences for each of the four selected AI‐2 exporter family proteins from E. coli K‐12 (TqsA: [UniProt ID: P0AFS5], YdiK: [UniProt ID: P0AFS7], YhhT: [UniProt ID: P0AGM0], and PerM: [UniProt ID: P0AFI9]) and for the A. aeolicus AI‐2 exporter Aq_740 [UniProt ID: O66948]. All primers are listed in Appendix Table S1. Genomic DNA from the E. coli strain DH5α was isolated using the QuickExtract DNA extraction solution (Lucigen). The coding sequences of the four autoinducer‐2 exporter genes were amplified using the Phusion DNA polymerase (Thermo Fisher Scientific). The resulting DNA fragments were gel extracted and cloned individually into the pBADC3 plasmid using InFusion EcoDry cloning kit (TaKaRa). All genes were fused with a C‐terminal StrepII‐tag for affinity purification. The TqsA mutants were prepared with the QuikChange Lightning site‐directed mutagenesis kit (Thermo Fisher Scientific) according to the manufacturer’s instructions.
For protein overexpression, E. coli top 10 cells transformed with the expression vectors were grown in lysogeny broth (LB) medium supplemented with 100 μg/ml carbenicillin at 37°C to an optical density at 600 nm (OD600) of 0.5–0.6. Production of autoinducer‐2 exporters was induced by the addition of 0.02% (w/v) L‐arabinose, and incubation was continued for 4 h. Cells were harvested by centrifugation and stored at −80°C.
For protein purification, cells were resuspended in the lysis buffer (20 mM Tris–HCl, pH 7.5, 250 mM sucrose, and 150 mM choline chloride) supplemented with protease inhibitors (1 mM phenylmethylsulfonyl fluoride, 1 mM benzamidine hydrochloride, and 1 mM 6‐aminocaproic acid), DNase I (approximately 50 μg/ml of buffer), 2.5 mM MgCl2, and 1 mM dithiothreitol (DTT). Cell disruption was conducted by passing through a microfluidizer at 8,000 psi and 4°C for six passes. The cell debris was removed by centrifugation at 4°C and 12,000 × g for 45 min using a GSA rotor. Subsequently, the supernatant containing the membranes was centrifuged at 4°C and 43,000 × g for 2.5 h. The pelleted membranes were resuspended in the lysis buffer without any additives to a concentration of around 20 mg total protein per ml of buffer. Aliquots of the membranes were flash frozen in liquid nitrogen and stored at −80°C. The total protein concentration was determined using the Bradford protein assay.
All purification steps were performed at 4°C. The membrane proteins (100 mg total protein) were solubilized in the solubilization buffer (100 mM Tris–HCl, pH 7.5, 25% glycerol, 300 mM NaCl, and 2% [w/v] n‐dodecyl‐ß‐D maltoside [DDM]) with gentle stirring at 4°C for 1.5 h. Following solubilization, the insoluble membrane fraction was removed by ultracentrifugation at 55,000 × g for 1 h. The supernatant containing the solubilized proteins was supplemented with 40 μg/ml avidin and subsequently loaded onto a 5‐ml StrepTrap HP column (GE Healthcare), which was pre‐equilibrated with binding buffer (50 mM Tris–HCl, pH 8, 100 mM NaCl, 1 mM EDTA, and 0.03% [w/v] DDM), with a peristaltic pump. Following loading, the column was washed with 20 column volumes (CV) of washing buffer (50 mM Tris–HCl, pH 8, 100 mM NaCl, 1 mM EDTA, and 0.1% [w/v] glycol‐diosgenin [GDN]). The StrepII‐tagged proteins were eluted with 6 CV of the washing buffer supplemented with 5 mM desthiobiotin. The protein was further purified via gel filtration using Superose 6 Increase 10/300 GL column (GE Healthcare) pre‐equilibrated with the purification buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, and 0.006% [w/v] GDN). The proteins were concentrated with 50 kDa MWCO Amicon filters (Merck) to around 3–4 mg/ml for subsequent structural studies.
SDS–PAGE and BN–PAGE
The purified proteins were analyzed by sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS–PAGE) using 4–12% Bis‐Tris NuPAGE gels (Invitrogen). Prior to SDS–PAGE analysis, protein samples were heated in the presence of 10 mM DTT at 90°C for 10 min. For Western blot analysis, proteins from SDS–PAGE gels were transferred onto a nitrocellulose membrane using iBlot blotting system (Thermo Scientific). Membranes were blocked for 1 h in TBST (10 mM Tris–HCl, 150 mM NaCl, and 0.05% Tween 20, pH 8) containing 2% (w/v) biotin‐free bovine serum albumin (BSA). The Strep‐II‐tagged proteins were immunodetected using the alkaline phosphatase‐conjugated Strep‐Tactin (IBA) and the color reaction was developed with 5‐bromo‐4‐chloro‐3‐indolyl phosphate (BCIP) and nitroblue tetrazolium (NBT). The Blue native PAGE was performed using the Novex 4–16% Bis‐Tris gels (Invitrogen) according to the manufacturer's instructions.
Sample preparation and cryo‐EM data collection
Before proceeding to cryo‐EM data collection, protein samples (0.01 mg/ml) were negatively stained with uranyl formate and analyzed by negative staining electron microscopy using a Tecnai Spirit transmission electron microscope (TEM) operated at 120 kV. For cryo‐EM, an aliquot of 4 μl of purified proteins (TqsA, YdiK, and YhhT) at around 3–5 mg/ml concentration was applied to freshly glow‐discharged C‐flat holey carbon grids (R1.2/1.3, Cu, 300 mesh). The grids were blotted at blotting force 20 for 4 s at 4°C and 100% humidity and plunged into liquid ethane using a Vitrobot (Thermo Scientific).
High‐resolution cryo‐EM imaging was performed using a 300 kV Titan Krios G3i microscope (Thermo Scientific) equipped with a BioQuantum imaging filter (Gatan) and a K3 direct electron detector (Gatan). Cryo‐EM data were collected in electron counting super‐resolution mode at a nominal magnification of 105,000×. For TqsA, a total of 5,452 movies were collected with a total dose of 100 e−/Å2 (100 frames) using the aberration‐free image‐shift (AFIS) correction in EPU. For YdiK, a total of 7,378 movies were collected with the same exposure settings. In addition, 989 movies were collected for YhhT with a total dose of 43 e−/Å2 (40 frames). The defocus range used for data collection was from −1.1 to −2.1 μm, and cryoSPARC live was employed for constantly monitoring the quality of the incoming movies and for the on‐the‐fly data processing (Punjani et al, 2017).
Single‐particle data processing
For TqsA, dose‐fractionated and gain‐corrected movies were subjected to beam‐induced motion correction using MotionCorr2 (Zheng et al, 2017). Thereafter, motion‐corrected micrographs were contrast transfer function (CTF) estimated using CTFFIND4 (Rohou & Grigorieff, 2015). A total of 3,314 micrographs with CtfMaxResolution values better than 4 A˚ were selected for further processing. Particle picking was done with CrYOLO1.5 using a general model for low‐pass filtering (Wagner et al, 2019). Around 1.2 million particles were picked from selected micrographs and imported in Relion3.1 (Zivanov et al, 2018). The particles were extracted with a down‐sampled pixel size of 3.348 Å and a box size of 80 pixels. These particles were subjected to multiple rounds of reference‐free 2D classification, which resulted in 427,253 particles. Particles from several best 2D classes were used for the initial model building and a 3D classification was applied. A total of 133,640 particles from the best 3D classes were extracted with the original pixel size of 0.837 Å per pixel and a box size of 320 pixels. For the 3D refinement, C5 symmetry was imposed because the pentameric organization was clearly visible in the 2D class averages as well as in 3D maps as shown in the figures, Fig EV2B, and Appendix Fig S8. Refinement was also performed separately with C1 symmetry as well. After the first refinement step, C5 and C1 symmetry EM maps were generated at 3.72 Å and 4.25 Å resolution, respectively (Appendix Fig S8). Iterative rounds of particle polishing and CTF refinement were performed to improve the quality of both maps. Finally, C5 and C1 symmetry EM maps of global resolutions of 3.35 Å and 3.99 Å were obtained, respectively, according to the gold standard FSC = 0.143 criterion. The polishing step was carried out with the first 70 fractions, filtering out the later dose‐damaged fractions. Local resolution maps were generated using Relion3.1, as shown in Fig EV2C and D. The data processing workflow is illustrated in the Appendix Fig S8.
For the YdiK dataset, a similar strategy was applied, and a detailed flowchart is shown in Appendix Fig S9, and local resolution maps are shown in Fig EV2E and F. Finally, a C5 symmetry map at 2.80 Å global resolution was obtained from 619,311 particles.
Model building and validation
For both TqsA and YdiK, initial partial models were obtained using a combination of ARP/wARP 8.0 and Buccaneer in the CCP‐EM software suite (Chojnowski et al, 2018; Hoh et al, 2020). These models were corrected and extended de novo into the EM density maps using COOT (Emsley et al, 2010). A combination of phenix.real_space_refine (Afonine et al, 2018) and Refmac (Brown et al, 2015) was used for the restrained refinement for improving the fitting of the models into the EM maps (Adams et al, 2010; Brown et al, 2015). Map to model and cross validation was carried out using Phenix suite and MolProbity (Rosenthal & Henderson, 2003; Chen et al, 2010). Refinement and validation statistics for both models are summarized in Table 1.
Table 1.
Cryo‐EM data collection, refinement, and validation statistics.
| Specifications | TqsA (EMDB: 12256) (PDB:7NB6) | YdiK (EMDB: 13057) (PDB:7OT9) |
|---|---|---|
| Data collection and processing | ||
| Magnification | 105,000 | 105,000 |
| Voltage (kV) | 300 | 300 |
| Electron exposure (e−/Å2) | 80 | 80 |
| Defocus range (μm) | −1.1 to −2.1 | −1.1 to −2.1 |
| Pixel size (Å) | 0.837 | 0.831 |
| Symmetry imposed | C5 | C5 |
| Initial particle images (no.) | 1,218,552 | 1,222,739 |
| Final particle images (no.) | 134,300 | 619,311 |
| Map resolution (Å) | 3.35 | 2.80 |
| FSC threshold | 0.143 | 0.143 |
| Map resolution range (Å) | 3.0–7.2 | 2.6–6.5 |
| Refinement | ||
| Initial model used | – | – |
| Model resolution (Å) | 2.9 | 2.5 |
| FSC threshold | 0.143 | 0.143 |
| Map sharpening B factor (Å2) | −30 | −10 |
| Model composition | ||
| Non‐hydrogen atoms | 13,020 | 8,410 |
| Protein/nucleotide residues | 1,700 | 1,105 |
| Ligands | – | – |
| B factors (Å2) (min/max/mean) | ||
| Protein | 18.22/197.18/109.91 | 71.51/175.24/100.68 |
| Ligands | – | – |
| R.m.s deviations | ||
| Bond lengths (Å) | 0.010 | 0.006 |
| Bond angles (°) | 1.282 | 1.002 |
| Validation | ||
| MolProbity score | 2.13 | 1.90 |
| Clashscore | 13.42 | 9.94 |
| Poor rotamers (%) | 0.49 | 0 |
| CaBLAM outliers (%) | 2.38 | 2.39 |
| Ramachandran Plot | ||
| Favored (%) | 91.72 | 94.42 |
| Allowed (%) | 8.28 | 5.58 |
| Disallowed (%) | 0 | 0 |
Native mass spectrometry
Prior to MS analysis, the sample buffer was exchanged to 200 mM ammonium acetate, pH 8.0, and two times the critical micelle concentration (CMC) of various detergents (namely LDAO and C8E4) using a Biospin‐6 column (BioRad), and introduced directly into the mass spectrometer using gold‐coated capillary needles (prepared in‐house). Data were collected on a Q‐Exactive UHMR mass spectrometer (Thermo Scientific). The instrument parameters were as follows: capillary voltage 1.1 kV, quadrupole selection from 1,000 to 20,000 m/z range, S‐lens RF 100%, collisional activation in the HCD cell 200 V, trapping gas pressure setting at 7.5, temperature 200°C, in‐source trapping 300 V, and resolution of the instrument 6250. The noise level was set at 3 rather than the default value of 4.64. Data were analyzed using Xcalibur 4.2 (Thermo Scientific). AI‐2/ DPD was provided by Dr. Rita Ventura’s group, ITQB Portugal (Ascenso et al, 2011).
Substrate docking
Substrate docking of AI‐2 to the TqsA structure was performed with Glide (Friesner et al, 2004). The initial coordinates of AI‐2 were taken in the cyclic form from the LsrB structure (PDB ID 6DSP) and an ensemble of conformations was generated using Ligprep (Schrödinger Release 2020‐1: LigPrep, Schrödinger, LLC, New York, NY, 2020). Docking was carried out over a search space of the entire protein volume with 75 runs. In each run, a box size of 15 × 15 × 15 Å for the inner box and 45 × 45 × 45 Å for the outer box were used. All the binding poses obtained from all docking runs (n = 3,950) were pooled together and then clustered using the cluster module of Gromacs program (v2020.2) with the Jarvis–Patrick method and a cut‐off of 4 Å (Abraham et al, 2015). Clustering resulted in 23 clusters and the most populated cluster comprises of more than 25% of all the binding poses (n = 1,091). This cluster is describing the binding modes near HP1, H5, and H6 in the protomer and has been discussed in the text and the representative binding pose is shown in Fig 5A. The second (n = 635) and third (n = 553) most populated clusters are located near the loop between H6 and HP2 and the space H6 and HP1, respectively (not shown in the manuscript).
Sodium ion/proton antiporter assays
Two methods for investigating the functional role of AI‐2 exporters as Na+/H+ antiporters were conducted as described previously (Rosen, 1986; Herz et al, 2003; Dong et al, 2017). One of them, the growth complementation assay, was performed using the E. coli Knabc strain (Herz et al, 2003), which was generously provided by Prof. Etana Padan, (Hebrew University, Israel). Briefly, the cells were transformed individually with the expression vectors containing the genes encoding AI‐2 exporters. Overexpression of the proteins was induced by the addition of 0.02% (w/v) L‐arabinose for 3 h and the growth of the cells was monitored in both liquid and solid media containing 200 mM NaCl. The expression levels of the proteins were determined by Western blot analysis. All cultures were carried out in duplicate and the study was done multiple times at different pH values to confirm the results.
Another assay inspects the Na+/H+ antiporter activity in everted membrane vesicles using the pH‐dependent fluorescent dye acridine orange (Goldberg et al, 1987). Briefly, membrane vesicles of TqsA, NhaB, and empty pBADC3 plasmid from Knabc transformants were prepared in a similar way as the membranes were prepared for purification of the proteins except that the cells were disrupted at lower pressures ~3,000 psi. Acridine orange was used as a fluorescent probe for detection of sodium ion‐induced pH variations. Fluorescence was measured using a Hitachi F4500 fluorimeter at excitation and emission wavelengths of 495 nm and 530 nm, respectively. Everted vesicles (approximately 100 μg of total protein) were added to the buffer containing 10 mM MES‐Tris pH 8, 145 mM choline chloride, 5 mM MgCl2, and 2 μM acridine orange. The vesicles were acidified by addition of 2.5 mM Tris‐D‐lactate and after stabilization of the fluorescence, dequenching was induced with 50 mM NaCl.
Author contributions
Radhika Khera: Conceptualization; Formal analysis; Investigation; Methodology; Writing—original draft; Writing—review and editing. Ahmad R Mehdipour: Validation; Investigation. Jani R Bolla: Investigation. Joerg Kahnt: Investigation. Sonja Welsch: Resources. Ulrich Ermler: Validation. Cornelia Muenke: Investigation. Carol V Robinson: Resources; Writing—review and editing. Gerhard Hummer: Resources; Writing—review and editing. Hao Xie: Supervision; Investigation; Writing—original draft; Writing—review and editing. Hartmut Michel: Resources; Supervision; Project administration; Writing—review and editing.
In addition to the CRediT author contributions listed above, the contributions in detail are:
RK designed the experiments, purified proteins, acquired, and processed cryo‐EM data, built atomic models, and analyzed data. ARM performed molecular docking studies. JRB carried out native MS measurements. JK performed mass spectrometry analyses. SW provided technical assistance for high‐resolution cryo‐EM data collection. UE assisted in building atomic models. CM provided technical assistance in the laboratory. CVR and GH provided the work setup. HX supervised the research and provided guidance for data interpretation; RK and HX prepared figures and wrote the manuscript. All authors discussed the results, read and approved the manuscript. HM supervised this work.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Movie EV1
PDF+
Acknowledgements
We would like to thank the staff at the EM facility at the Max Planck Institute of Biophysics, Simone Prinz, Dr. Susann Kaltwasser, and Mark Linder for their support, training, and assistance for microscopy data collection; Hannelore Mueller for technical assistance in the lab; Jiangfeng Zhao and Philipp Valina Allo for help in cloning and purification of Aq_740; Dr. Schara Safarian for initial training and assistance at the microscope; Dr. Yongchan Lee for guidance and useful insights for cryo‐EM data processing; Prof. Etana Padan for sending the E. coli Knabc strain; Dr. Rita Ventura’s group, ITQB, Portugal, for providing synthetic AI‐2; Prof. Victor Sourjik and Dr. Karina Xavier from MPI for Terrestrial Microbiology and Instituto Gulbenkian de Ciência, respectively, for all their help and swift replies. This work was supported by the Max Planck Society. Open Access funding enabled and organized by ProjektDEAL.
The EMBO Journal (2022) 41: e109990.
See also: S Wang et al (September 2022)
Contributor Information
Hao Xie, Email: hao.xie@biophys.mpg.de.
Hartmut Michel, Email: hartmut.michel@biophys.mpg.de.
Data availability
The atomic coordinates for TqsA and YdiK have been deposited to PDB under the accession numbers: 7NB6 (TqsA, http://www.rcsb.org/pdb/explore/explore.do?structureId=7NB6) and 7OT9 (YdiK, http://www.rcsb.org/pdb/explore/explore.do?structureId=7OT9). The EM density maps including the C1 maps and masks used for final processing for both TqsA and YdiK have been deposited to EMDB under the accession numbers: EMD‐12256 (TqsA, https://www.ebi.ac.uk/emdb/EMD‐12256) and EMD‐13057 (YdiK, https://www.ebi.ac.uk/emdb/EMD‐13057). Further information can be acquired from the corresponding author upon request.
References
- Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E (2015) GROMACS: High performance molecular simulations through multi‐level parallelism from laptops to supercomputers. SoftwareX 1‐2: 19–25. 10.1016/j.softx.2015.06.001 [DOI] [Google Scholar]
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L‐W, Kapral GJ, Grosse‐Kunstleve RW et al (2010) PHENIX: a comprehensive Python‐based system for macromolecular structure solution. Acta Crystallogr. Sect. D . Biol Crystallogr 66: 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine PV, Poon BK, Read RJ, Sobolev OV, Terwilliger TC, Urzhumtsev A, Adams PD (2018) Real‐space refinement in PHENIX for cryo‐EM and crystallography. Acta Crystallogr D Struct Biol 74: 531–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antunes LCM, Ferreira RBR, Buckner MMC, Finlay BB (2010) Quorum sensing in bacterial virulence. Microbiology 156: 2271–2282 [DOI] [PubMed] [Google Scholar]
- Ascenso OS, Marques JC, Santos AR, Xavier KB, Ventura MR, Maycock CD (2011) An efficient synthesis of the precursor of AI‐2, the signalling molecule for inter‐species quorum sensing. Bioorg Med Chem 19: 1236–1241 [DOI] [PubMed] [Google Scholar]
- Bassler BL, Losick R (2006) Bacterially speaking. Cell 125: 237–246 [DOI] [PubMed] [Google Scholar]
- Bassler BL, Wright M, Showalter RE, Silverman MR (1993) Intercellular signalling in Vibrio harveyi: sequence and function of genes regulating expression of luminescence. Mol Microbiol 9: 773–786 [DOI] [PubMed] [Google Scholar]
- Boudker O, Ryan RM, Yernool D, Shimamoto K, Gouaux E (2007) Coupling substrate and ion binding to extracellular gate of a sodium‐dependent aspartate transporter. Nature 445: 387–393 [DOI] [PubMed] [Google Scholar]
- Brown A, Long F, Nicholls RA, Toots J, Emsley P, Murshudov G (2015) Tools for macromolecular model building and refinement into electron cryo‐microscopy reconstructions. Acta Crystallogr. Sect. D . Biol Crystallogr 71: 136–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen I, Pant S, Wu Q, Cater RJ, Sobti M, Vandenberg RJ, Stewart AG, Tajkhorshid E, Font J, Ryan RM (2021) Glutamate transporters have a chloride channel with two hydrophobic gates. Nature 591: 327–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC (2010) MolProbity: all‐atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D . Biol Crystallogr 66: 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Schauder S, Potier N, Van Dorsselaer A, Pelczer I, Bassler BL, Hughson FM (2002) Structural identification of a bacterial quorum‐sensing signal containing boron. Nature 415: 545–549 [DOI] [PubMed] [Google Scholar]
- Cho B‐K, Federowicz SA, Embree M, Park Y‐S, Kim D, Palsson BØ (2011) The PurR regulon in Escherichia coli K‐12 MG1655. Nucleic Acids Res 39: 6456–6464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chojnowski G, Heuser P, Pereira J, Lamzin V (2018) Building atomic models into electron microscopy maps with ARP/wARP version 8.0. Acta Crystallogr A Found Adv 74: e151 [Google Scholar]
- De Araujo C, Balestrino D, Roth L, Charbonnel N, Forestier C (2010) Quorum sensing affects biofilm formation through lipopolysaccharide synthesis in Klebsiella pneumoniae . Res Microbiol 161: 595–603 [DOI] [PubMed] [Google Scholar]
- Dong P, Wang L, Song NA, Yang L, Chen J, Yan M, Chen H, Zhang R, Li J, Abdel‐motaal H et al (2017) A UPF0118 family protein with uncharacterized function from the moderate halophile Halobacillus andaensis represents a novel class of Na+(Li+)/H+ antiporter. Sci Rep 7: 45936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Crystallogr Sect D Biol Crystallogr 66: 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK et al (2004) Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 47: 1739–1749 [DOI] [PubMed] [Google Scholar]
- Garaeva AA, Slotboom DJ (2020) Elevator‐type mechanisms of membrane transport. Biochem Soc Trans 48: 1227–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg EB, Arbel T, Chen J, Karpel R, Mackie GA, Schuldiner S, Padan E (1987) Characterization of a Na+/H+ antiporter gene of Escherichia coli . Proc Natl Acad Sci USA 84: 2615–2619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson R (1995) The potential and limitations of neutrons, electrons and X‐rays for atomic resolution microscopy of unstained biological molecules. Q Rev Biophys 28: 171–193 [DOI] [PubMed] [Google Scholar]
- Herz K, Vimont S, Padan E, Berche P (2003) Roles of NhaA, NhaB, and NhaD Na+/H+ antiporters in survival of Vibrio cholerae in a saline environment. J Bacteriol 185: 1236–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzberg M, Kaye IK, Peti W, Wood TK (2006) YdgG (TqsA) controls biofilm formation in Escherichia coli K‐12 through autoinducer 2 transport. J Bacteriol 188: 587–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuberger EHML, Veenhoff LM, Duurkens RH, Friesen RHE, Poolman B (2002) Oligomeric state of membrane transport proteins analyzed with blue native electrophoresis and analytical ultracentrifugation. J Mol Biol 317: 591–600 [DOI] [PubMed] [Google Scholar]
- Hirschi M, Johnson ZL, Lee S‐Y (2017) Visualizing multistep elevator‐like transitions of a nucleoside transporter. Nature 545: 66–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoh SW, Burnley T, Cowtan K (2020) Current approaches for automated model building into cryo‐EM maps using Buccaneer with CCP‐EM. Acta Crystallogr D Struct Biol 76: 531–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismail AS, Valastyan JS, Bassler BL (2016) A host‐produced autoinducer‐2 mimic activates bacterial quorum sensing. Cell Host Microbe 19: 470–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A et al (2021) Highly accurate protein structure prediction with AlphaFold. Nature 596: 583–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamaraju K, Smith J, Wang J, Roy V, Sintim HO, Bentley WE, Sukharev S (2011) Effects on membrane lateral pressure suggest permeation mechanisms for bacterial quorum signaling molecules. Biochemistry 50: 6983–6993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavanaugh JS, Gakhar L, Horswill AR (2011) The structure of LsrB from Yersinia pestis complexed with autoinducer‐2. Acta Crystallogr Sect F Struct Biol Cryst Commun 67: 1501–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CS, Gatsios A, Cuesta S, Lam YC, Wei Z, Chen H, Russell RM, Shine EE, Wang R, Wyche TP et al (2020) Characterization of Autoinducer‐3 structure and biosynthesis in E. coli . ACS Cent Sci 6: 197–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laganenka L, Colin R, Sourjik V (2016) Chemotaxis towards autoinducer 2 mediates autoaggregation in Escherichia coli . Nat Commun 7: 12984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laganenka L, Sourjik V (2018) Autoinducer 2‐dependent Escherichia coli biofilm formation is enhanced in a dual‐species coculture. Appl Environ Microbiol 84: e02638‐17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Li X, Wang Z, Fu Y, Ai Q, Dong Y, Yu J (2015) Autoinducer‐2 regulates Pseudomonas aeruginosa PAO1 biofilm formation and virulence production in a dose‐dependent manner. BMC Microbiol 15: 192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Li X, Song C, Zhang Y, Wang Z, Liu Z, Wei H, Yu J (2017) Autoinducer‐2 facilitates Pseudomonas aeruginosa PAO1 pathogenicity in vitro and in vivo . Front Microbiol 8: 1944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lü W, Du J, Schwarzer NJ, Wacker T, Andrade SLA, Einsle O (2013) The formate/nitrite transporter family of anion channels. Biol Chem 394: 715–727 [DOI] [PubMed] [Google Scholar]
- Miller ST, Xavier KB, Campagna SR, Taga ME, Semmelhack MF, Bassler BL, Hughson FM (2004) Salmonella typhimurium recognizes a chemically distinct form of the bacterial quorum‐sensing signal AI‐2. Mol Cell 15: 677–687 [DOI] [PubMed] [Google Scholar]
- Papenfort K, Bassler BL (2016) Quorum sensing signal‐response systems in Gram‐negative bacteria. Nat Rev Microbiol 14: 576–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira CS, Thompson JA, Xavier KB (2013) AI‐2‐mediated signalling in bacteria. FEMS Microbiol Rev 37: 156–181 [DOI] [PubMed] [Google Scholar]
- Punjani A, Rubinstein JL, Fleet DJ, Brubaker MA (2017) cryoSPARC: algorithms for rapid unsupervised cryo‐EM structure determination. Nat Methods 14: 290–296 [DOI] [PubMed] [Google Scholar]
- Rettner RE, Saier MH (2010) The autoinducer‐2 exporter superfamily. J Mol Microbiol Biotechnol 18: 195–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes N, Ginter C, Boudker O (2009) Transport mechanism of a bacterial homologue of glutamate transporters. Nature 462: 880–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohou A, Grigorieff N (2015) CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol 192: 216–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen BP (1986) Ion extrusion systems in Escherichia coli . Methods Enzymol 125: 328–336. [DOI] [PubMed] [Google Scholar]
- Rosenthal PB, Henderson R (2003) Optimal determination of particle orientation, absolute hand, and contrast loss in single‐particle electron cryomicroscopy. J Mol Biol 333: 721–745 [DOI] [PubMed] [Google Scholar]
- Ruan Y, Miyagi A, Wang X, Chami M, Boudker O, Scheuring S (2017) Direct visualization of glutamate transporter elevator mechanism by high‐speed AFM. Proc Natl Acad Sci USA 114: 1584–1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford ST, Bassler BL (2012) Bacterial quorum sensing: its role in virulence and possibilities for its control. Cold Spring Harb Perspect Med 2: a012427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saier MH (2000) A functional‐phylogenetic classification system for transmembrane solute transporters. Microbiol Mol Biol Rev 64: 354–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer DB, Song J, Wang B, Hilton JK, Karpowich NK, Mindell JA, Rice WJ, Wang D‐N (2021) Structure and inhibition mechanism of the human citrate transporter NaCT. Nature 591: 157–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senior AW, Evans R, Jumper J, Kirkpatrick J, Sifre L, Green T, Qin C, Žídek A, Nelson AWR, Bridgland A et al (2020) Improved protein structure prediction using potentials from deep learning. Nature 577: 706–710 [DOI] [PubMed] [Google Scholar]
- Sperandio V, Mellies JL, Nguyen W, Shin S, Kaper JB (1999) Quorum sensing controls expression of the type III secretion gene transcription and protein secretion in enterohemorrhagic and enteropathogenic Escherichia coli . Proc Natl Acad Sci USA 96: 15196–15201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperandio V, Torres AG, Girón JA, Kaper JB (2001) Quorum sensing is a global regulatory mechanism in enterohemorrhagic Escherichia coli O157:H7. J Bacteriol 183: 5187–5197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperandio V, Torres AG, Kaper JB (2002) Quorum sensing Escherichia coli regulators B and C (QseBC): a novel two‐component regulatory system involved in the regulation of flagella and motility by quorum sensing in E. coli . Mol Microbiol 43: 809–821 [DOI] [PubMed] [Google Scholar]
- Torcato IM, Kasal MR, Brito PH, Miller ST, Xavier KB (2019) Identification of novel autoinducer‐2 receptors in Clostridia reveals plasticity in the binding site of the LsrB receptor family. J Biol Chem 294: 4450–4463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valastyan JS, Kraml CM, Pelczer I, Ferrante T, Bassler BL (2021) Saccharomyces cerevisiae requires CFF1 to produce 4‐Hydroxy‐5‐Methylfuran‐3(2H)‐One, a mimic of the bacterial quorum‐sensing Autoinducer AI‐2. MBio 12: e03303‐20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner T, Merino F, Stabrin M, Moriya T, Antoni C, Apelbaum A, Hagel P, Sitsel O, Raisch T, Prumbaum D et al (2019) SPHIRE‐crYOLO is a fast and accurate fully automated particle picker for cryo‐EM. Commun Biol 2: 218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Zou Q, Yan M, Wang Y, Guo S, Zhang R, Song Y, Li X, Chen H, Shao LI et al (2020) Polar or charged residues located in four highly conserved motifs play a vital role in the function or pH response of a UPF0118 family Na+(Li+)/H+ antiporter. Front Microbiol 11: 841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteley M, Diggle SP, Greenberg EP (2017) Progress in and promise of bacterial quorum sensing research. Nature 551: 313–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xavier KB, Bassler BL (2003) LuxS quorum sensing: more than just a numbers game. Curr Opin Microbiol 6: 191–197 [DOI] [PubMed] [Google Scholar]
- Xavier KB, Bassler BL (2005) Regulation of uptake and processing of the quorum‐sensing autoinducer AI‐2 in Escherichia coli . J Bacteriol 187: 238–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue T, Yu L, Shang F, Li W, Zhang M, Ni J, Chen X (2016) Short communication: The role of autoinducer 2 (AI‐2) on antibiotic resistance regulation in an Escherichia coli strain isolated from a dairy cow with mastitis. J Dairy Sci 99: 4693–4698 [DOI] [PubMed] [Google Scholar]
- Yernool D, Boudker O, Jin Y, Gouaux E (2004) Structure of a glutamate transporter homologue from Pyrococcus horikoshii . Nature 431: 811–818 [DOI] [PubMed] [Google Scholar]
- Yu X, Plotnikova O, Bonin PD, Subashi TA, McLellan TJ, Dumlao D, Che YE, Dong YY, Carpenter EP, West GM et al (2019) Cryo‐EM structures of the human glutamine transporter SLC1A5 (ASCT2) in the outward‐facing conformation. Elife 8: e48120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Kanner BI (1999) Two serine residues of the glutamate transporter GLT‐1 are crucial for coupling the fluxes of sodium and the neurotransmitter. Proc Natl Acad Sci USA 96: 1710–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache J‐P, Verba KA, Cheng Y, Agard DA (2017) MotionCor2: anisotropic correction of beam‐induced motion for improved cryo‐electron microscopy. Nat Methods 14: 331–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, Scheres SH (2018) New tools for automated high‐resolution cryo‐EM structure determination in RELION‐3. Elife 7: e42166 [DOI] [PMC free article] [PubMed] [Google Scholar]