

Abstract
Adulte interfollicular epidermis (IFE) renewal is likely orchestrated by physiological demands of its complex tissue architecture comprising spatial and cellular heterogeneity. Mouse tail and back skin display two kinds of basal IFE spatial domains that regenerate at different rates. Here, we elucidate the molecular and cellular states of basal IFE domains by marker expression and single‐cell transcriptomics in mouse and human skin. We uncover two paths of basal cell differentiation that in part reflect the IFE spatial domain organization. We unravel previously unrecognized similarities between mouse tail IFE basal domains defined as scales and interscales versus human rete ridges and inter‐ridges, respectively. Furthermore, our basal IFE transcriptomics and gene targeting in mice provide evidence supporting a physiological role of IFE domains in adaptation to differential UV exposure. We identify Sox6 as a novel UV‐induced and interscale/inter‐ridge preferred basal IFE‐domain transcription factor, important for IFE proliferation and survival. The spatial, cellular, and molecular organization of IFE basal domains underscores skin adaptation to environmental exposure and its unusual robustness in adult homeostasis.
Keywords: epidermal domains, human skin, scRNA‐seq, Sox6, UV
Subject Categories: Development, Skin, Stem Cells & Regenerative Medicine
Adult interfollicular epidermis in mouse and human shows unexpected spatial and molecular heterogeneity, suggesting adaption to differential environmental challenges.
Introduction
Adult skin interfollicular epidermis (IFE) makes the essential body barrier to the outside world and must respond to a variety of environmental insults and physiological demands (Blanpain & Fuchs, 2009; Gola & Fuchs, 2021). Homeostatic IFE renewal is likely resulting from local microenvironmental challenges, locally imposed on an intricate tissue architecture that comprises multiple levels of heterogeneity. The vicinity of the hair follicle, body region variations, skin maturation and aging, and stress conditions such as mechanical stretching or wounding pose additional challenges to IFE renewal (Roy et al, 2016, 2020; Aragona et al, 2017, 2020; Park et al, 2017; Ichijo et al, 2021). Development of skin cancers, such as melanoma, is also associated with IFE regional heterogeneity and UV‐exposure (Kohler et al, 2017; Moon et al, 2017). We and others have previously documented that adult mouse IFE presents two spatially distinct domains that renew at different rates during homeostasis. These two domains are found in both mouse tail skin, known as scales and interscales, and in back skin; they can be identified as H2B‐GFP or BrdU non‐label retaining cells (non‐LRCs) domains and LRCs domains (Riquelme et al, 2008; Gomez et al, 2013; Roy et al, 2016; Sada et al, 2016; Sanchez‐Danes et al, 2016; Aragona et al, 2017). Human skin also presents two kinds of spatial domains, known as rete ridges and inter‐ridge (Lawlor & Kaur, 2015), which based on their location in the epidermis have differential exposure to the outside environment. The functional significance of the two spatial IFE domains and their correspondence from mouse to human skin are currently unknown. Recent single‐cell transcriptomics of mouse skin revealed basal layer (BL) cell heterogeneity with multiple cell states (Joost et al, 2018; Aragona et al, 2020; Dekoninck et al, 2020; Haensel et al, 2020; Lin et al, 2020), but how these states are spatially and functionally organized in the IFE is unclear. Recent data from human newborn foreskin suggest that different basal IFE cell states might be spatially organized (Wang et al, 2020). A persistent lack of markers to distinguish spatial and functional basal cellular subsets impedes our current understanding of IFE organization.
Here, we use markers of basal IFE cellular subsets in mouse and human skin to examine the molecular, cellular, and functional organization of two IFE spatial basal domains. We show that the two IFE domains have different gene expression patterns and predominantly different paths of basal layer (BL) cell differentiation (Synopsis). Despite these differences, they both contain comparable mixtures of cell states previously defined as stem, proliferating, and differentiating basal cell types. We find that mouse back skin IFE domain organization stands out as somewhat unique, but we unearth previously unrecognized similarities of basal IFE organization between mouse tail scales and interscales with human rete ridges and inter‐ridges, respectively (Synopsis). Different molecular pathways—notably UV‐response genes—are differentially upregulated in the two IFE domains, corresponding to their differential exposure to the outside environment. Our gene targeting functional studies in mice support the hypothesis that this heterogeneity is physiologically important, enabling the skin's adaptive response to UV exposure (Synopsis). This spatial and molecular organization of the IFE may help explain the remarkable robustness of long‐term skin homeostasis in the face of environmental challenges.
Results
Mouse tail scale/non‐LRC vs. interscale/LRC basal IFE domains express preferred genes at different levels
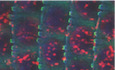
We previously labeled skin cells based on proliferation history and isolated IFE label retaining cells (LRCs) and non‐LRCs from mouse back skin using our tet‐repressible K5tTA x pTRE‐H2BGFP transgenic mice (Tumbar et al, 2004; Sada et al, 2016). The classical stem cell (SC)‐transit amplifying (TA) cell model (Kaur & Potten, 2011) predicted that LRCs would be exclusively SCs. However, we found that both LRCs and non‐LRCs contained long‐lived regenerative (stem) cells, which could be marked with distinct genetic drivers (Dlx1‐CreER and Slc1a3‐CreER, respectively). Moreover, LRCs and non‐LRCs and their corresponding marked lineages segregated preferentially in two spatial domains in mouse back and tail skin (Fig 1A; Sada et al, 2016). In tail skin, LRC domains correspond to interscales, whereas non‐LRC domains correspond to scales (Figs 1A and EV1A and B; Sada et al, 2016) (also see a top view example in Fig EV1A and B). These findings were also reported by two other groups (Gomez et al, 2013; Sada et al, 2016; Sanchez‐Danes et al, 2016; Dekoninck et al, 2020). The interscale includes a subregion that unites all the hair follicles, which we called the “line” (Fig 1A; Sada et al, 2016). Using two inducible systems (K14‐rTA and K5‐tTA), we confirm here that 2‐week chase via K5‐tTA renders easily detectable LRCs in tail skin, as expected (Sada et al, 2016), and they were preferentially in K10+ regions or interscales (Fig EV1C and D). Lack of IFE LRCs after 1‐week chase (Rompolas et al, 2016; Piedrafita et al, 2020) suggests short chase periods may not be sufficient to render differences in proliferation rates in the different domains. In our chases, LRCs were also present in other body regions, such as back, ear, and paw (Fig EV2). We also used tamoxifen (TM) induction experiments and confirmed preferential Slc1a3‐CreER or Dlx1‐CreER marking of non‐LRCs or LRCs spatial clusters in tail (Fig 1B and C). The drivers are also active in back, ear, and paw skin (Fig EV2A–D), except Dlx1‐CreER is highly inefficient in back skin, where we use K14‐CreER as a surrogate LRC control. Next, we tested marking via Aspm‐CreER, another basal non‐LRC gene from the back skin microarray (Sada et al, 2016; Kang et al, 2020), but this was not enriched in scale (non‐LRC) domain (Figs 1B and C, and EV1A). Interestingly, the Dlx1‐CreER marking was enriched along the interscale/scale boundary, and all three drivers marked the interscale “line” substructure (Fig 1A–C; Sada et al, 2016). This data suggest that although the H2B‐GFP non‐LRCs cluster preferentially in scales while the LRCs cluster preferentially in interscales, the LRC/non‐LRC gene expression spatial patterns are more complex.
Figure 1. Basal layer cells express different LRC/non‐LRC preferred‐genes in scales vs. interscales of mouse tail skin.
-
AModel of tail skin organization in IFE domains.
-
B, C(B) Whole mount low magnification images show marking in interscale (K10+) vs. scale regions, quantified in (C). Error bars are SDs. The data shown are average of four different time points (2 week, 3 month, 6 month and 1 year) each consisting of two mice and five images per mice. P‐value is calculated for difference between scale vs. interscale using Student's t‐test (mixed Anova).
-
DTail skin sections immunofluorescence stained for interscale (K10) or scale (K31) supra‐basal markers, and basal markers. Scale bar = 20 μm or 50 μm as indicated on the panels. Asterisks indicate that photoshop was used to erase the autoflourescent cornified layer. Dashed lines indicate ROI in basement membrane.
-
EQuantification of images like those in (D) with n = 3–4 mice and 6–7 images per mouse showing preferred‐gene enrichment. Error bars are SDs. P‐values calculated by Student's t‐test (mixed effect model).
- F
-
GCartoon summarizing spatial distribution of markers in the tail IFE domains.
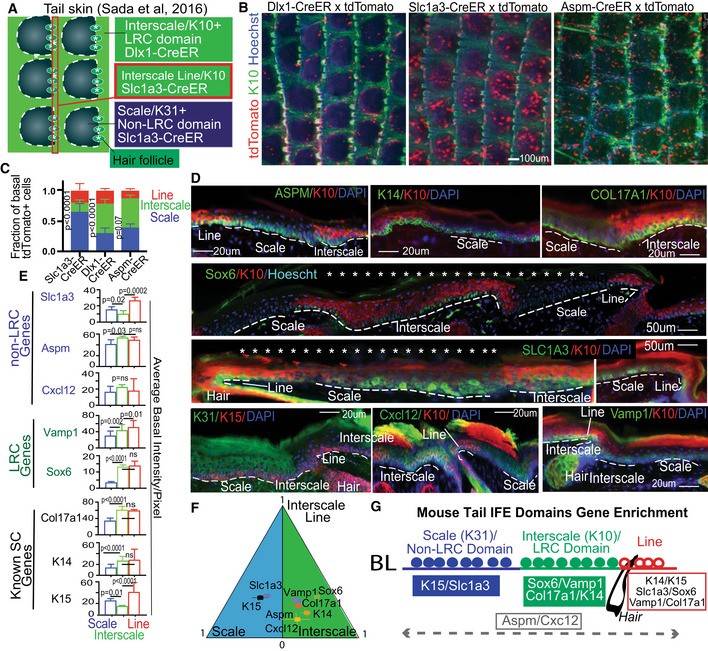
Figure EV1. H2B‐GFP LRC and non‐LRC IFE domains in mouse tail skin from lineage‐traced mice (Associated with Fig 1).
-
AWhole mount view (top) and sections (bottom) of mouse tail skin from Dlx1‐/Slc1a3‐/AspmCreER x K5‐tTA/pTRE‐H2BGFP mice showing H2BGFP label retaining cells (LRCs) after doxycycline (doxy) chases. Scale bar = 50 or 200 μm as indicated on panels. Interscales regions have been previously shown enriched in LRCs (Mascre et al, 2012; Gomez et al, 2013; Sada et al, 2016). Here, scale and interscale regions are tentatively outlined based on the known H2B‐GFP LRC clustering, the 3‐hair follicle grouping patterns and DAPI staining.
-
BCartoon showing the microscopic view for whole mount and sections.
-
CTop Panels: Schematics of tet‐off (left) versus tet‐on (right) inductions used for comparison with (Piedrafita et al, 2020). Bottom Panels: Tail skin sections from K5‐tta x and K14‐rta x pTRE‐H2BGFP mice stained with interscale (K10) marker (as indicated). “Mosaicism” denotes areas of the IFE devoid of H2BGFP due to deficiency in transgene expression. High vs. low exposure images demonstrate enrichment of LRCs in the interscale areas in both mouse models. Better contrast is obtained in the K5‐tTa (our preferred) mouse model compared to the K14‐rTa (Piedrafita et al, 2020) and a minimum of 2‐week chase is required to note differences.
-
DTail skin sections stained for LRC/non‐LRC markers as in Fig 1D, exhibit preferred overlap with H2BGFP LRC or non‐LRCs IFE domains. Dotted lines indicate scale or interscale, depending on which is written within the image.
Source data are available online for this figure.
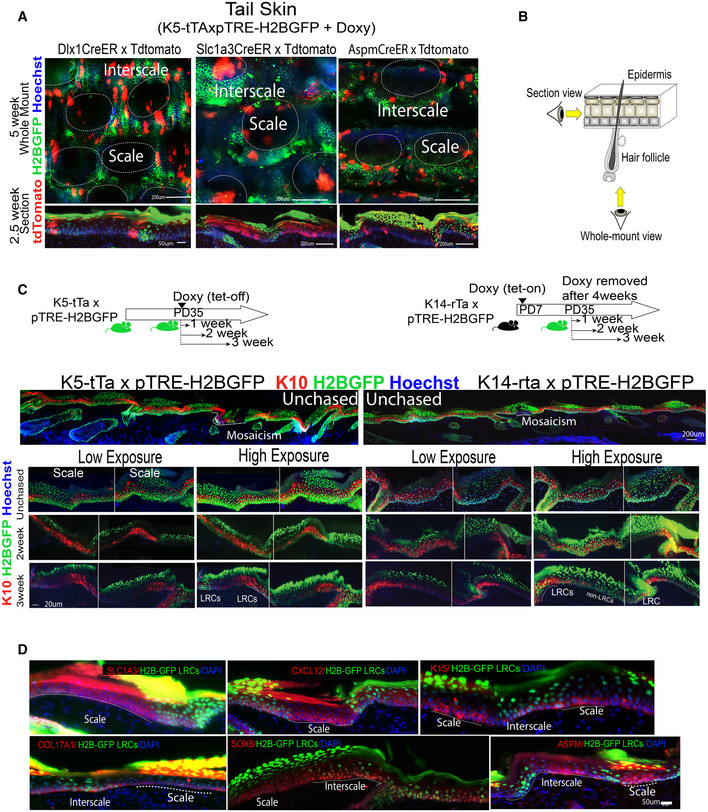
Figure EV2. H2B‐GFP LRC and non‐LRC IFE domains in mouse non‐tail body regions from lineage‐traced mice (Associated with Fig 1).
-
ASchematic of tamoxifen and doxy chase times for various body skin regions.
-
BBack and ear skin sections from unchased K5‐tTA x pTRE‐H2BGFP mice. Mosaicism is due to deficiency in transgene expression in ear skin. Scale bars = 200 μm.
-
C, DWhole mount (C) and section (D) view of back from lineage‐traced mice showing 2‐week chased H2B‐GFP (green) postinjection with high tamoxifen (TM) dose. Scale bars = 200 μm (C) or as indicated (D). Dashed line in (C) indicates non‐LRC patches devoid of H2BGFP signal. Dotted lines in (D) indicated basement membrane.
-
EImages of skin from back, ear and paw immuno‐stained for some markers from Fig 1D. Dotted lines in (E) indicates ROI in basement membrane. Scale bar is as indicated per row.
-
F, G(F) Quantification of images like those in (E) with n = 2–5 mice and 6–7 images per mouse showing marker heterogeneity and (G) overlap with H2BGFP LRCs vs. non‐LRCs domains. P‐values were calculated by two‐tail Student's t‐test.
Source data are available online for this figure.
To examine spatial patterns of protein expression in the IFE basal domains, we used immunofluorescence (IF) staining and microscopy on different body regions of mice at different ages. We tested commercial antibodies to > 10 gene candidates we previously identified by microarrays of back skin IFE LRCs/non‐LRCs (Sada et al, 2016), and obtained working conditions for Sox6 and Vamp1 (enriched in LRCs) and Slc1a3, Cxcl12, and Aspm (enriched in non‐LRCs). We also used antibodies to some previously known SC‐enriched genes—K15 (Webb et al, 2004; Cotsarelis, 2006), Col17a1 (Watanabe et al, 2017; Li et al, 2019), and K14 (Mascre et al, 2012). Most LRC/non‐LRC‐enriched factors, but especially, the Slc1a3 protein, were expressed in distinct basal cell clusters in adult tail (Figs 1D and EV1D). Back, ear, and paw BL also showed heterogeneous expression for some of these genes (Fig EV2D–F). However, overall correlation to LRC/non‐LRCs was generally weaker in non‐tail tissues (Fig EV2E–G). This might reflect more fluidity of the LRCs/non‐LRC cell states and gene expressions in non‐tail tissues. Newborn skin was relatively homogeneous to these expressions when compared with adult skin (Fig EV3A–C). Tail image quantification measured the differential expression of our LRC/non‐LRC‐enriched genes in the scale, interscale, and line basal IFE regions (Fig 1E). Except for Aspm and Cxcl12, the non‐LRC‐enriched genes (e.g., Slc1a3) were preferentially expressed in scale, whereas the LRC‐enriched genes (Sox6 and Vamp1) were preferentially expressed in interscale (Fig 1F). Unexpectedly, previously known SC‐expressed genes also showed preferred expression: K15 was enriched in scale, and K14 and Col17a1 were enriched in interscale (Fig 1E and F).
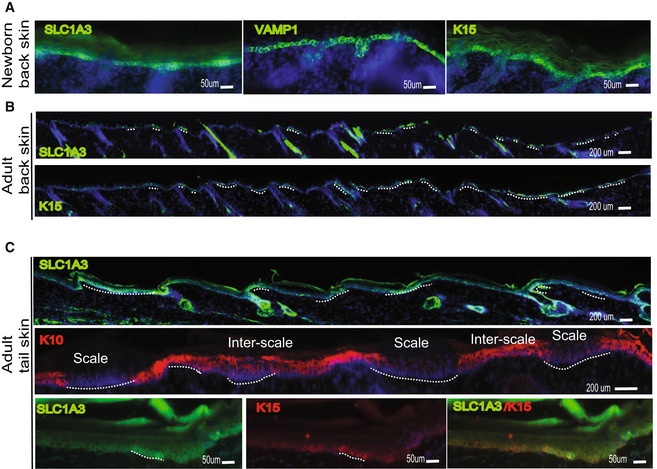
Figure EV3. Expression of select markers in newborn versus adult mouse skin (Associated with Fig 1).
-
A–CImmunostaining for select markers analyzed in Fig 1D shown here in mouse skin from newborn (A) vs. adult back (B) and adult tail (C) skin. Markers show more uniform expression in newborn back skin. Scale = 50 or 200 μm as indicated on panels.
Source data are available online for this figure.
In summary, we identified preferred basal layer (BL) factors that distinguish the scale (K15/Slc1a3), a non‐LRC‐enriched tail domain, from interscale (Vamp1/Sox6/Col17a1/K14), a LRC‐enriched tail domain (Fig 1G). Several of these genes also show heterogeneous expression in BL of other mouse body regions, but the correlation with LRC/non‐LRCs is reduced in non‐tail skin regions.
Human inter‐ridges vs. rete ridges resemble the mouse interscale vs. scale domains
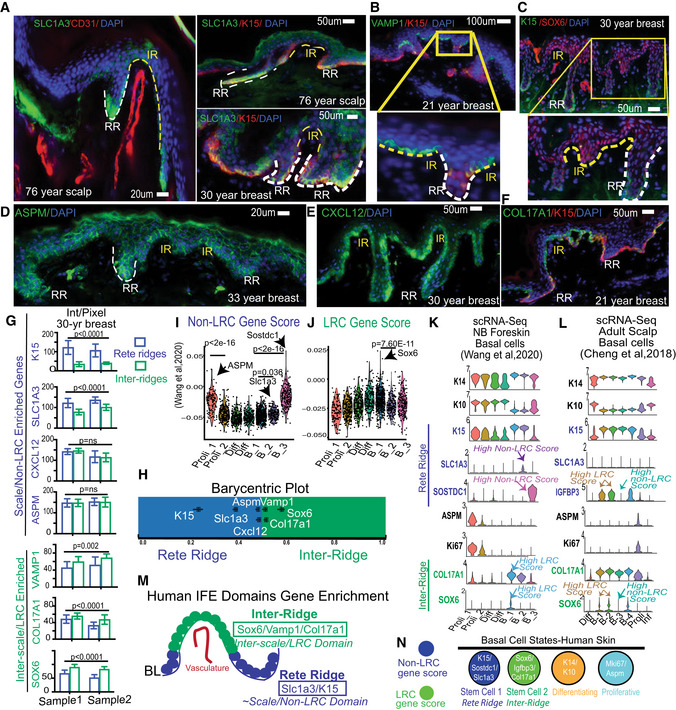
Human IFE is characterized by an undulating pattern of rete ridges (RR), which are spatial domains embedded deep into the dermis, and inter‐ridges (IR), which are more raised and exposed IFE spatial domains (Lawlor & Kaur, 2015). Here, we used human skin samples to probe for expression of our newly identified preferred‐genes from mouse IFE LRC domain (interscale in tail) and non‐LRC domain (scale in tail). Interestingly, we found preferential expression of Slc1a3/K15 in the rete ridges and of Vamp1, Sox6, and Col17a1 in the inter‐ridges (Figs 2A–H and EV4A–D). This correlation is maintained to some extent in human breast, forehead, abdomen, scalp, arm, cheek, ear, and newborn foreskin (ages 21, 30, 45, 52, 76‐year‐old; Fig EV4B and Appendix Table S1). These data reveal previously unrecognized similarities between mouse tail and human IFE organization in basal layer. Specifically, mouse tail interscales/LRC domains correspond to inter‐ridges in human skin, and scales/non‐LRC domains correspond to rete ridges (compare Figs 1G and 2M). Other basal markers such as Cxcl12 and Aspm were found in small basal cell clusters in both IR and RR (Fig 2D and E), reminiscent of their presence in both scales and interscales in mouse tail (Fig 1D–G). We briefly inquired into the proliferative status of RR and IR, but Ki67+ proliferative cells showed no obvious difference (Fig EV4A), as reported (Ruetze et al, 2010). Notably, marker expression heterogeneity in the newborn human foreskin was less pronounced than in aged skin (Fig EV4C and D), reminiscent of the mouse age‐dependent differences in heterogeneity (Fig EV3).
Figure 2. Human IFE shows basal domains and cell clusters enriched in mouse LRC/non‐LRC preferred‐genes.
-
A–FHuman skin immunofluorescence images of mouse basal non‐LRC/LRC preferred‐factor staining in rete ridges (RR) versus inter‐ridges (IR). Representative images from 30 samples of different body regions and ages (see Appendix Table S1). Scale bars are indicated on the panels.
-
GLRC/non‐LRC preferred gene expression in RR and IR samples from two 30‐year‐old breast samples, quantified using background subtraction (Materials and Methods). P‐values from paired Student's t‐test. Error bars are SDs. 8–10 images were quantified each from two females.
-
HRelative RR vs. IR gene expression/area normalized to sum to one, shown as barycentric plot. A gene expressed only in inter‐ridges would be plotted at 1. Error bars are SEMs.
-
I, JGene score analysis in cell clusters from (Wang et al, 2020). P‐values from pairwise Wilcoxon rank‐sum tests with Benjamini‐Hochberg correction.
-
K, LscRNA‐seq identified clusters show specific gene expression of relevant markers.
-
MCartoon summarizing differential RR vs. IR marker distribution.
-
NModel of human IFE basal cell states with LRC/ non‐LRC gene expression distributions.
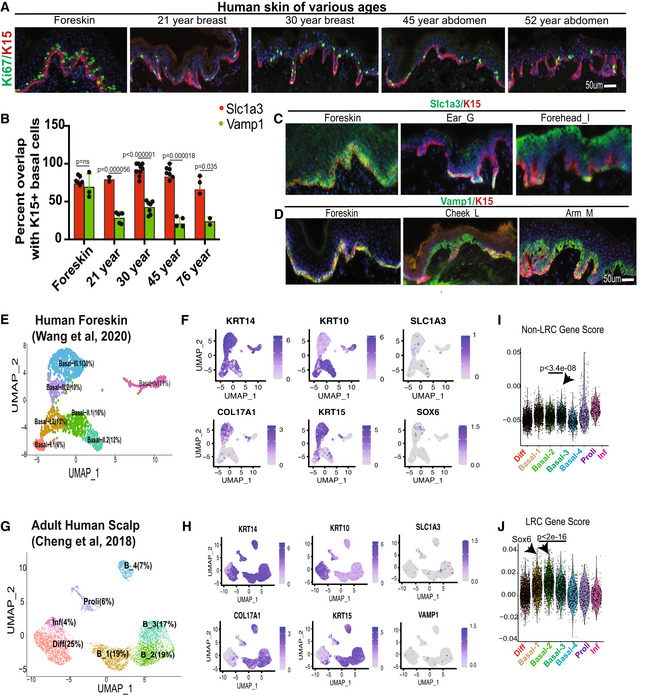
Figure EV4. Human skin LRC/non‐LRC marker staining and scRNA‐seq analysis (Associated with Fig 2).
-
AKi67 staining of human skin at various ages. Scale = 50 μm for all the panels.
-
BQuantification demonstrates preferential colocalization of K15 with Slc1a3 (non‐LRC/scale marker), but not with Vamp1 (LRC/interscale marker) in human skin of all ages, except in newborn foreskin. Error bars are SDs. 2–9 images were quantified from two different samples at every age. P‐values were calculated by two‐tailed Student's t‐test.
-
C, DExamples of staining in newborn foreskin and other adult body regions suggests more pronounced heterogeneity in the adult. Scale = 50 μm for all the panels.
-
E–H(E, G) Seurat generated clusters from scRNA‐seq extracted from indicated studies shown as UMAP plots. (F, H) Feature plots show expression of specific markers in clusters from (E) and (G), respectively.
-
I, JNon‐LRC and LRC gene enrichment score analysis in cell clusters from (Cheng et al, 2018). P‐values were calculated using pairwise Wilcoxon rank‐sum tests with Benjamini‐Hochberg correction.
Source data are available online for this figure.
To further investigate our mouse IFE LRC/non‐LRC preferred‐genes in human skin, we used Seurat analysis (Satija et al, 2015; Butler et al, 2018) with Uniform Manifold Approximation and Projection (UMAP) (preprint: McInnes et al, 2018; Korsunsky et al, 2019) to probe two human single‐cell (sc)RNA‐seq databases recently generated from newborn foreskin (Wang et al, 2020) and adult scalp (Cheng et al, 2018). We identified seven clusters in human basal layer IFE (Fig EV4E and G), and examined marker expression of known SC, proliferation, differentiation, and our mouse LRC/non‐LRC genes (Figs 2I–L and EV4E–J). All clusters expressed K14, indicative of their basal layer identity, but some had low levels of K10, suggesting they are initiating differentiation, as was also reported for mouse basal IFE (Rompolas et al, 2016; Park et al, 2017; Lin et al, 2020; preprint: Cockburn et al, 2021; Fig EV4F and H). Several clusters expressed Ki67, indicative of actively proliferating cells, and interestingly, those clusters also expressed our non‐LRC marker Aspm. The remaining basal clusters that qualify as undifferentiated and non‐proliferative (e.g., putative G0 SC states) expressed preferentially either non‐LRC/scale makers (Slc1a3, Sostdc1, and K15) or LRC/interscale markers (Sox6, Igfbp3, and Col17a1) (Fig 2K and L). Furthermore, these clusters were enriched in either LRC or non‐LRC computed gene scores (see Materials and Methods and Dataset EV1) (Figs 2I and J, and EV4I and J). We conclude that human IFE contains multiple basal cell states: differentiating, proliferative, and several putative SC states (Fig 2N). The SC states are enriched in either non‐LRC or LRC gene, and the expression of preferred‐genes place them in either rete ridges or inter‐ridges (Fig 2N). This analysis adds to work and interpretation from (Wang et al, 2020). Importantly, the non‐LRC vs. LRC gene enrichment in human rete ridges vs. inter‐ridges resembles mouse tail scales vs. interscales, respectively (compare Figs 2M and 1G and see Synopsis). This suggests an unexpected and novel link between the organization of the IFE basal layer in tail skin and human skin.
LRCs/non‐LRCs sorted from mouse back and tail skin harbor a mixture of basic basal cell states
To characterize the cell states present in our IFE LRC/non‐LRC domains, we next used mouse tail and back skin isolated cellular subsets and performed scRNA‐seq analysis (Figs 3A–J and EV5A–N; Appendix Fig S1A–E). We FACS purified basal (Sca1+/α6‐integrin+) IFE cells as H2BGFP LRCs, mid‐LRCs, and non‐LRCs from K5‐tTA × pTRE‐H2BGFP mice (Tumbar et al, 2004; Fig 3A) and used 10X Genomics scRNA‐seq technology (see Materials and Methods). After quality control and filtering, we obtained a total of 13,484 cells from back skin of two mice, or ~ 4,500 cells each for LRC, mid‐LRC and non‐LRC sorted population (Fig EV5A–E). LRC vs. non‐LRC gene scores computed from previous microarrays (Materials and Methods; Sada et al, 2016) confirmed the expected marker gene enrichment in our LRC vs. non‐LRC scRNA‐seq databases (Fig 3B). Some LRC/non‐LRC gene expression differences previously found by microarrays (Sada et al, 2016) were detectable by scRNA‐seq (e.g., Igfbp3, Chit1, and Sostdc1), but many others were not (Fig EV5F and G). Quantitative reverse transcriptase (QRT)–PCR confirmed the microarray results for some of the individually tested genes (Fig EV5H), underscoring detection limitations of scRNA‐seq data.
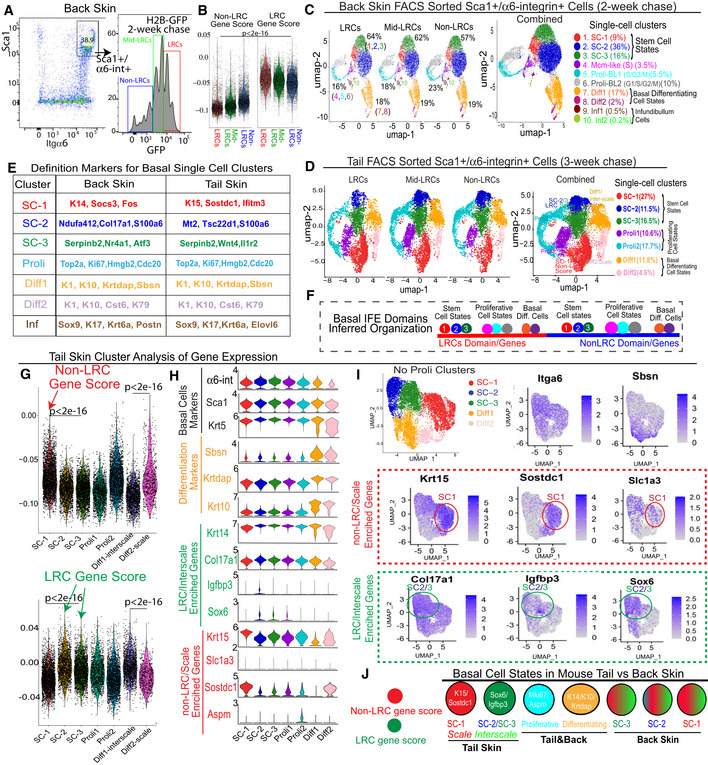
Figure 3. Single‐cell RNA‐seq of basal IFE LRCs/non‐LRCs sorted from mouse back and tail skin.
-
AFACS sorting of basal LRCs, mid‐LRCs and non‐LRCs.
-
BscRNA‐seq data of sorted cells from (A) analyzed for LRC/non‐LRC gene scores computed from microarray data (Sada et al, 2016).
- C, D
-
EBasal cluster definition markers.
-
FModel of basal IFE domain cell states.
-
GLRCs and non‐LRCs computed gene scores are differentially enriched in tail SC clusters. P‐values from pairwise Wilcoxon rank‐sum tests with Benjamini‐Hochberg correction.
-
H, IViolin and feature plots of markers in tail skin without proliferative (Proli) clusters. Note LRC/interscale markers Sox6&Igfbp3 enriched in SC‐2&3 and non‐LRC/scale markers K15/Sostdc1 enriched in SC‐1. Slc1a3 is in SC‐1 (scale); SC‐2 expression is likely due to Slc1a3 expression in interscale line.
-
JModel of LRC/non‐LRC gene scores and marker expression in scRNA‐seq clusters found in tail vs. back skin.
Figure EV5. Mouse back skin scRNA‐seq of sorted basal LRCs, Mid‐LRCs, Non‐LRCs (Associated with Figs 3 and 4).
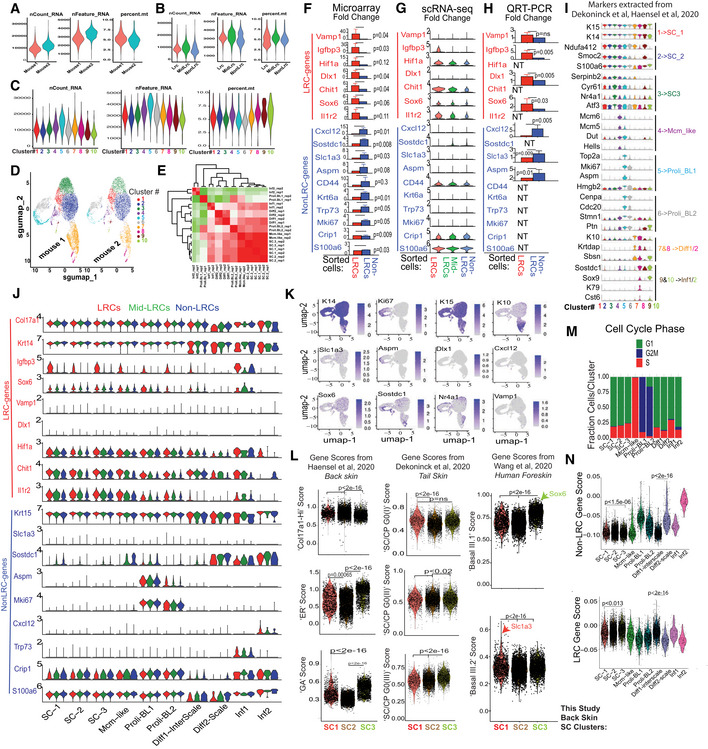
-
A–C(A) mRNA or gene count and percent mitochondrial genes reflecting the quality of selected cells used for scRNA‐seq analysis are shown as violin plots for the (A) two mouse replicates and (B) three sorted IFE cell samples and (C) ten clusters used in our scRNA‐seq experiment.
-
D, EHigh correlation between UMAP plots (D) and clusters in gene expression matrix (E) from the two mouse replicates.
-
F–HGene expression changes in sorted basal cells from microarray (Sada et al, 2016) (F), scRNA‐seq (G) and qRT‐PCR (H); SEM and P‐values are indicated. N = 3 in (F), two mice in (G) and 4–5 mice (NT is not tested) in (H). Note low or undetectable levels of many markers in (G). Two‐tailed Student's t‐test.
- I
-
JViolin plot representation for the LRC/non‐LRC marker expression in the given clusters across the samples showing generally low expression or weak differences in clusters.
-
KFeature plots of cluster‐defining markers and LRC/non‐LRC genes.
-
LScores extracted from previous publication as indicated (Dekoninck et al, 2020; Haensel et al, 2020; Wang et al, 2020) demonstrate correlations among our 3 SC populations with the other databases (Dataset EV1). P‐value was calculated using pairwise Wilcoxon rank‐sum tests with Benjamini‐Hochberg correction.
-
MCell cycle scoring of cells in various clusters as defined by cell‐cycle phase‐specific gene lists using Addmodulescore (see Materials and Methods) function in Seurat.
-
NLRCs and non‐LRCs gene scores from previous microarray of back skin (Sada et al, 2016) detect both gene sets in all single‐cell derived clusters with some enrichment. P‐value was calculated using pairwise Wilcoxon rank‐sum tests with Benjamini‐Hochberg correction.
Source data are available online for this figure.
Despite these limitations, cluster analysis of scRNA‐seq data helped define basic basal cell states present in all sorted LRC, mid‐LRC, non‐LRC IFE populations (Fig 3C). Seurat and Harmony integration (Korsunsky et al, 2019) with quality control parameters and UMAP (preprint: McInnes et al, 2018) reduction method showed the 10 back skin basal cell clusters were well‐correlated in two mouse samples (Fig EV5C–E). The results were similar in cluster analysis of 14,884 (4,402 LRCs, 6,236 mid‐LRCs and 4,245 Non‐LRCs) basal IFE cell fractions sorted from two tail skin replicates (Fig 3D; Appendix Fig S1A and B). To assign cell‐cluster identity, we used previously published cluster‐markers (Figs 3E and EV5I for back and Appendix Fig S1C for tail and Dataset EV1), cell cycle analysis (Fig EV5M), and gene scores (Fig EV5L; Appendix Fig S1D) extracted from other previously defined IFE cellular subsets (Joost et al, 2016; Dekoninck et al, 2020; Haensel et al, 2020). This classified both the back and the tail skin IFE clusters as: Three putative G0 SC states well‐correlated in tail and back skin (Appendix Fig S1E); actively proliferating (Proli) cells; and basal‐differentiating cells (Diff) (Fig 3C–E). Based on these data, we can infer that the two spatial (LRC/non‐LRC) IFE basal domains of mouse back and tail skin harbor mixtures of basic basal cell states that can be defined by scRNA‐seq (Fig 3F).
Next, we examined how the LRC/non‐LRC gene expression signatures defined as gene scores (Materials and Methods) from the back skin microarrays (Sada et al, 2016) may be represented in the scRNA‐seq basal cell clusters. The back skin scRNA‐seq clusters showed mild, if any, differential enrichment in LRC/non‐LRC‐specific marker expression and computed gene scores (Fig EV5J and K, and N). This was reminiscent of IF staining results from back skin that showed poor correlation of preferred‐gene expression with LRCs/non‐LRC (Fig EV2G). In contrast, the tail skin SC clusters showed strong differences in LRC/non‐LRC gene scores (Fig 3G and H). Specifically, the SC‐1 tail cluster was enriched in non‐LRC gene scores and K15&Sostdc1 expressions (Fig 3G and H), which makes it a scale‐preferred cluster. Conversely, SC‐2 and SC‐3 tail clusters were enriched in LRC gene scores and Igfbp3&Sox6 (Fig 3G and H), making them interscale‐preferred. This was also apparent to some extent in feature plots of specific LRC/non‐LRC preferred‐genes (Fig 3I). Aspm marked proliferative clusters of both tail and back skin (Figs 3H and EV5J), as previously seen in human skin (Fig 2K and L). In summary, we propose that several basic basal cell states that include stem, proliferative, and differentiating cells exist in both LRC and non‐LRC spatial domains of mouse back and tail skin. In the mouse back skin, the LRC/non‐LRC gene signatures do not differentiate well the three predicted stem cell states (Fig 3F). However, in tail skin, distinct scRNA‐seq defined stem cell states express preferentially either the LRC or the non‐LRC gene expression signatures (Fig 3J), a situation also seen in human undifferentiated basal cell clusters (Fig 2N). This further underscores the previously unrecognized similarities between mouse tail and human skin.
Two basal cell differentiation paths reflect the IFE spatial domain organization
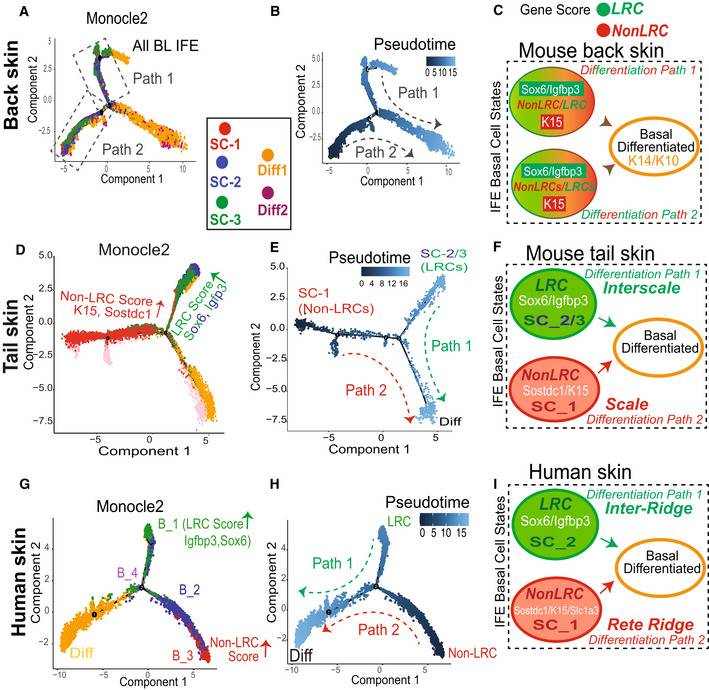
To understand how the IFE basal cell states might relate to each other in transcriptomic lineage trajectory maps, we used Monocle 2 and Pseudotime analysis (Trapnell et al, 2014) from which the proliferative cell clusters were removed to eliminate bias due to cell cycle status. We analyzed all our mouse back and tail databases (Fig 4), along with two databases previously published from human (Cheng et al, 2018; Wang et al, 2020) (Appendix Fig S1F–H) and three from mouse skin (Joost et al, 2016; Dekoninck et al, 2020; Haensel et al, 2020) (Appendix Fig S1I–K). Our mouse sorted basal cells rendered a uniquely rich mouse dataset combining both high sequencing depth and large numbers of basal sorted cells. These combined qualities of our dataset uncovered a previously unidentified bifurcated lineage tree for the basal IFE, with three differentiation branches (Fig 4A and D), not as apparent in other smaller databases from mouse skin (Appendix Fig S1I–K). This 3‐branched lineage tree was also present in a human skin dataset from the adult scalp (Fig 4G). The basal differentiating (K14+/K10+) cells were found on one branch, and the three predicted SC clusters were split on the other two branches (Fig 4A and D, and G).
Figure 4. Transcriptomic lineage trajectory models reveal two basal IFE differentiation paths.
-
A–I(A, D, and G) Monocle 2 lineage trajectory model and associated Pseudotime predictions (B, E, and H) for skin samples indicated with proliferative and infundibulum clusters excluded. (G) is human scalp from (Cheng et al, 2018). Pseudotime places the “Diff” basal cluster branch as most differentiated, as expected based on marker expression. (C, F, and I) Cartoons depicting two differentiation paths that based on LRC/non‐LRC marker expression can be defined as tail scale vs. interscales (F) and human rete ridges inter‐ridges (I); note mixed expressions along the two paths for back skin (C).
Pseudotime analysis assigned the two SC branches as ground states, and they both converged onto the more committed, basal differentiating cells (Diff) (Fig 4B and E, and H). This suggests the existence of two distinct paths of IFE basal cell differentiation in mouse back and tail skin and in human scalp in the samples analyzed. In mouse back skin where LRC/non‐LRC gene scores and expressions were not strongly differentiated in the three SC states (Figs 3J and EV2G), all three SC states intermingled together along each of the two differentiation paths (Fig 4A). In contrast, in mouse tail and human scalp skin where LRC/non‐LRC gene scores and markers were strongly differentiated among the SC clusters (Figs 2N and 3J), the SC states were strongly segregated along the two basal differentiation paths (Fig 4D and G). Specifically, in tail, SC‐1 (K15/Sostdc1‐enriched; non‐LRC score/e.g., of the scale) was on one path, while SC‐2 and SC‐3 (Igfbp3/Sox6‐enriched; LRC score/ e.g., of the interscale) were on the other path (Fig 4D). This distribution was similar in human skin, where a basal cluster highly enriched in LRCs score/Sox6 expression (e.g., of the inter‐ridge) was found on one path, while a different basal cluster enriched in non‐LRC score (e.g., of the rete ridge) was found on the other (Fig 4G). The two differentiation paths seem to reflect at least in part the spatial organization of the IFE in mouse tail scales vs. interscales and in human rete ridges vs. inter‐ridges (Fig 4F and I). Two basal paths of differentiation were detectable in mouse back skin as well, but a possible fluidity of SC states might exist along the two paths in this distinct tissue type (Fig 4C; see also Discussion and Synopsis).
Molecular pathway differences suggest IFE basal domain‐specific adaptation to UV exposure
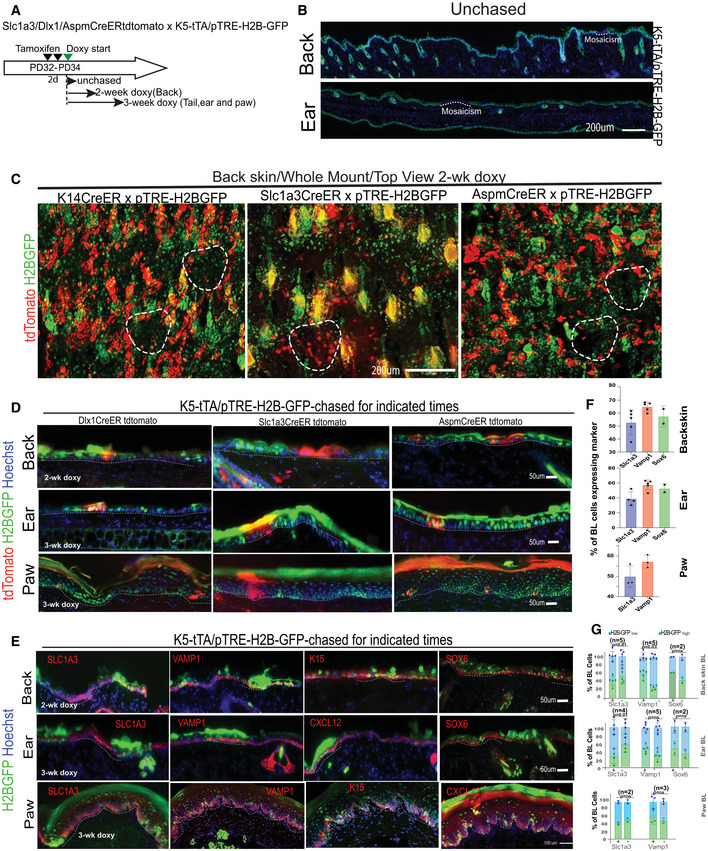
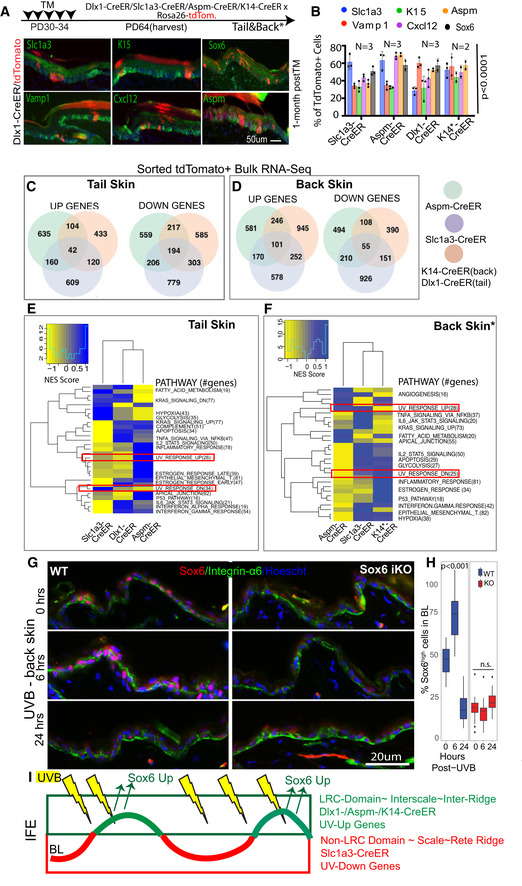
To further examine the cellular and molecular make up of our IFE basal populations enriched in the two spatial domains, we stained the back and tail skin of Slc1a3‐CreER x, Aspm‐CreER x, Dlx1‐CreER x tdTomato mice at 1‐month post‐TM with basal markers (Slc1a3, K15, Aspm, Vamp1, and Cxcl12; Fig 5A and B; Appendix Fig S2A–D). K14‐CreER x tdTomato mice served as a control population previously defined as a broad epidermal stem cell (Mascre et al, 2012). This population shows even distribution of all our five tested basal markers (Fig 5B). The Dlx1‐CreER, Slc1a3‐CreER, and Aspm‐CreER progenies were enriched in their respective “parent” lineage‐marker, and each showed a unique pattern of different marker distributions (Fig 5B). Notably, by 1‐month post‐TM, only a fraction of progenies expressed the parent‐marker, suggesting that some descending cells convert into other basal cell states. This was true in both scales and interscales (Appendix Fig S2B). Therefore, we conclude that progenies of our IFE populations evolve over time, but overall remain different from each other, attesting to their distinct and heterogeneous molecular nature in the two spatial domains.
Figure 5. Molecular differences in IFE populations suggest domain‐specific adaptation to environmental UV exposure.
-
ASchematic showing the tamoxifen injection paradigm for tdTomato‐labeling of all IFE lineages in tail and back. Co‐expression of markers with the lineage‐traced cells at 1‐month post‐tamoxifen. Dlx1‐CrER is shown as example in tail; see also Appendix Fig S2A and C (*Back skin). Scale bar = 50 μm.
-
BQuantification of images like those in (A) and Appendix Fig S2A, showing % tdTomato+ cells expressing indicated markers. Sox6 (black bar) was not analyzed in K14‐Cre/EYFP lineage. See also Appendix Fig S2B for scale vs. interscale split. Error bars are SDs. P‐values calculated by Student's t‐test (mixed effects model) from n = 2–3 mice and 5–6 images per replicate.
-
C–F(C, D) Venn diagram showing bulk RNA‐seq data in the three sorted IFE basal lineages and (E, F) heatmap for their differentially regulated Hallmark_Pathways.
-
GImmunofluorescence images of Sox6 staining in noUVB (0 h) or UVB‐exposed (6 h‐ or 24‐h post) mouse back skin.
-
HBoxplot shows % Sox6 expressing basal cells quantified from images like those in (G). Central band is the mean value, the top of box indicates 25th percentile while the bottom indicates 75th percentile. Whiskers indicate minimum and maximum values in the data, with dots indicating potential outliers. Linear mixed model statistical test was used with data from 18 images from 3 biological replicates.
-
IModel of IFE LRC/non‐LRC domains and marked lineages with distinct degree of UV exposure in normal conditions.
To systematically characterize the molecular differences among these IFE populations and obtain clues on their potential physiological purpose, we used bulk RNA‐seq analysis for each of the individually sorted IFE lineages. For this, we FACS purified > 10,000 tdTomato+/Sca1+/α6‐int+ (basal) cells from both back and tail skin at 2‐week post‐TM induction from each of four CreER mouse lines and normalized them to Sca1+/α6‐int+ sorted from the same mouse. Because the Dlx1‐CreER is extremely inefficient in mouse back skin, we relied on K14‐CreER as a control population in this body region. Hierarchical clustering and principal component analysis revealed hundreds of genes with differential expression in our IFE populations in both back and tail skin (Fig 5C–F; Appendix Table S2). GSEA analysis of Hallmark Pathways (Subramanian et al, 2005) revealed differential upregulation of many pathways including metabolism, inflammation, and hypoxia (Appendix Table S3) in both back and tail (Fig 5E and F). Two pathways that differed among our sorted IFE cells especially caught our attention, as they were related to UV‐response genes (Fig 5E and F). Similarly, in scRNA‐seq analysis, SC2/3 clusters enriched in Sox6 (an interscale/inter‐ridge marker) also showed upregulation of UV‐response genes as defined by Hallmark Analysis in both mouse tail and human scalp (Appendix Fig S2E–G). A second UV‐signature list extracted from previously published work (Dataset EV1; Li et al, 2021; Shen et al, 2019) was also found enriched in the Sox6 expressing scRNA‐seq clusters (e.g., SC3 in tail and B‐1/2/4 in human scalp) (Appendix Fig S2H and I).
That UV‐pathways might be differentially regulated in the distinct IFE population and domains was especially intriguing, given that in human skin, the basal layer in the domains (rete ridges and inter‐ridges) clearly faces unequal UV exposure. Specifically, rete ridges, enriched in Slc1a3 and other non‐LRC genes (Fig 2), are deeply embedded in the human skin and hence more UV‐protected. Conversely, inter‐ridges, enriched in LRC markers Sox6 and Vamp1 (Fig 2), are highly exposed to UV. In mouse tail, scales may be less UV‐exposed than interscales, as they are more than double in thickness due to IFE undulations and very thick cornified envelope that retains nuclei in scales (Appendix Fig S3A–C). The degree of UV exposure of an IFE basal domain might explain the differential regulation of UV‐response pathways detected in these domains.
To directly test whether UV exposure might affect the expression of some of our IFE domain preferred‐genes, we irradiated the mouse back skin with UVB (Moon et al, 2017; Roy et al, 2020), which superficially affects the epidermis without penetrating to the dermal cells. We then performed IF staining for Aspm, Slc1a3, and Sox6 at several time points (Fig 5G; Appendix Fig S3D). Sox6 IF signal was clearly increased in basal cells by 6‐h post‐UVB exposure, and no signal was detected in Sox6 KO epidermis, confirming Sox6 antibody specificity (Fig 5G–H). Moreover, our quantification demonstrates that there is an increase in % Sox6high cells in response to UVB irradiation of the mouse back skin, resulting from Sox6low cells acquiring more Sox6 expression (Fig 5G and H; Appendix Fig S3E and F). Further mining of the databases revealed several other UV‐regulated gene candidates expressed at higher levels in the LRC‐enriched SC clusters located in the more exposed IFE domains (Appendix Fig S4A–C). These results suggest that increased UV exposure of inter‐ridges and interscales might contribute to Sox6 and possibly other LRC‐domain genes upregulation in these regions (Fig 5I) and see Discussion. The upregulation of LRC‐domain genes such as Sox6 in UV‐exposed regions and in response to acute UV irradiation poses the question of what role might these genes play in skin homeostasis and in response to environmental challenges, such as UV exposure.
Sox6 promotes IFE proliferation and survival during homeostasis and in UV exposure
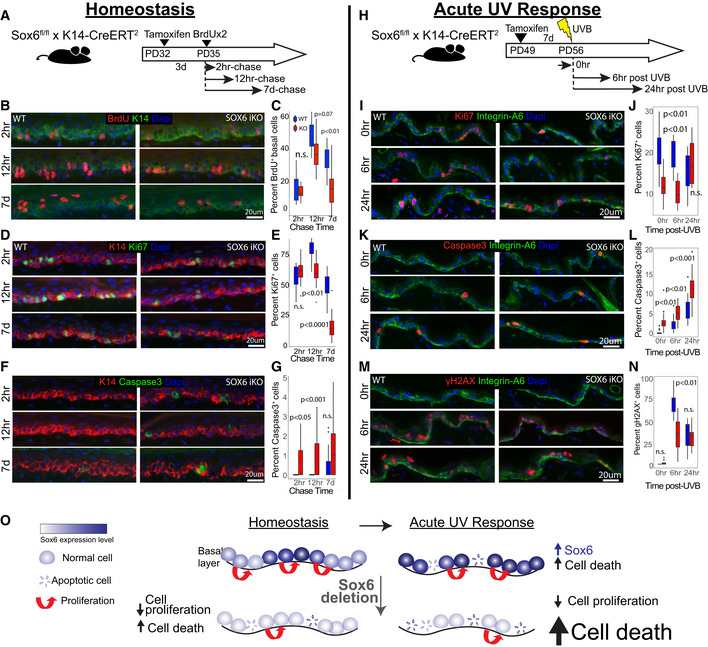
To first examine the role of Sox6 in the IFE during homeostasis, we deleted Sox6 from the skin epithelium and examined basal layer cell proliferation, differentiation, and survival in mouse tail. We used inducible Sox6 knockout (iKO) mice obtained by crossing the K14‐CreERT2 (Indra, 1999) and Sox6fl/fl mice (Dumitriu et al, 2006), and injected TM at PD32 followed by a short BrdU pulse‐chase experiment (Fig 6A). We then sacrificed both wild‐type (WT) and iKO littermates at 2‐h, 12‐h, and 7‐day chase and quantified BrdU+ cells. Initially, the number of BrdU‐labeled basal cells was comparable in WT vs. Sox6 iKO, but by 7‐day chase, they significantly decreased in the iKO (Fig 6B and C; Appendix Fig S5A). The DNA damage (Appendix Fig S5B and C) and the epidermis thickness marked by K10 (Appendix Fig S5D) were not significantly different between two groups. To check whether the BrdU+ cell loss in the iKO IFE during chase is caused by changes in proliferation, cell death, or both, we performed Ki67 and Caspase3 staining and quantified the results. We observed decreased proliferation (Fig 6D and E) and a transient increase in cell death in the iKO IFE compared with CT skin (Fig 6F and G). Importantly, these data indicate that Sox6 promotes cell proliferation and survival in the IFE basal layer. To our surprise, despite Sox6 upregulation in interscale compared with scale, the gene loss affected basal cells in both scale and interscale (Appendix Fig S5E–M). Apparently, even at lower expression levels as those found in the scales, Sox6 actively promotes normal IFE proliferation and survival during skin homeostasis. Differences in proliferation as indicated by Ki67 staining, survival, and DNA damage were subtle, if any, and not detectable in these assays between scale and interscale by IF staining (Appendix Fig S5N–Q). Interestingly, whereas initially Sox6 loss results in increased cell death and decreased proliferation, by 30‐day post‐TM injection, cell death resolves and proliferation appear upregulated in the epidermis (Appendix Fig S5R–U). This is likely due to robust skin compensatory mechanisms to overcome tissue abnormalities in long‐term.
Figure 6. Sox6 role in basal cell proliferation and survival during homeostasis and acute UV response.
-
ASchematic of Sox6 epithelial knockout (iKO) and BrdU pulse‐chase experiment.
-
B–G(B, D, and F) Images of tail skin immune‐stained for markers indicated and (C, E, and G) corresponding box plots.
-
HSox6 role in acute UV‐response was assessed in back skin to avoid compounding effects from the nucleus‐retaining cornified envelope in tail scales.
-
I–N(I, K, and M) Images of skin immune‐stained for markers indicated and (J, L, and N) corresponding box plots.
-
OModel depicting the role of Sox6 in IFE homeostasis and acute UV response.
Data information: In box plots, central band is the mean value, and the top indicates 25th percentile while the bottom indicates 75th percentile. Whiskers indicate minimum and maximum values in the data, with dots indicating potential outliers. Linear mixed model statistical test was used with data from 18 images from three biological replicates.
Since Sox6 was upregulated in the IFE upon acute UVB irradiation (Fig 5G and H), we then asked whether Sox6 plays an additional or additive role in UV response. Thus, we UVB irradiated back skin of WT and Sox6 iKO mice and analyzed them at different time points post‐exposure (Fig 6H). Staining with Ki67+ demonstrated that proliferative cells were fewer in the IFE of iKO mice at 0 and 6‐h post‐UVB, although interestingly and unlike in homeostasis, they recovered by 24 h (Fig 6I and J). In contrast, cell apoptosis was consistently increased in the Sox6 iKO IFE upon UVB treatment (Fig 6K and L). Furthermore, the frequency of DNA‐damaged cells was decreased in the iKO IFE by 6‐h post‐UV (Fig 6M and N). These results together suggest that in acute UVB stress upon Sox6 loss, IFE basal cells may resort more to apoptosis rather than DNA damage repair, compounding the effect this gene loss has on cell survival during homeostasis. Overall, our data demonstrate that the LRC‐factor Sox6, enriched in more UV‐exposed IFE regions (e.g., inter‐ridges in human and interscales in mouse tail), plays a vital role in basal cell proliferation and survival during homeostasis and in acute UV exposure (Fig 6O).
Discussion
Here, we characterize the molecular, cellular, and functional organization of two types of IFE basal spatial domains in both mouse and human skin. The two spatial IFE domains were previously shown in mouse tissue kinetics studies to renew at (~ 2x) different rates in mouse back and tail skin (Mascre et al, 2012; Gomez et al, 2013; Roy et al, 2016; Sada et al, 2016; Sanchez‐Danes et al, 2016), hence harboring preferentially either LRCs or non‐LRCs in pulse‐chase experiments. This relatively small proliferation differences cannot be detected by short chases (Rompolas et al, 2016; Piedrafita et al, 2020) or by less quantitative tissue kinetics studies, such as Ki67 staining. Based on lineage tracing data with different CreER drivers, we previously proposed that the two spatial domains each contain a spatially distinct stem cell population (Sada et al, 2016). Here, we demonstrate by in situ analysis that the two basal IFE domains express LRC/non‐LRC preferred genes and some other known SC markers at different protein levels. The two domains contain mixtures of molecularly discrete basal cell states on their path to differentiation, as defined by single‐cell transcriptomics. Importantly, we propose that the spatial and molecular organization of these two domains reflects in part the IFE adaptation to environmental exposure.
The spatial organization of IFE in domains is reflected in two basal cell differentiating paths, as described by our single‐cell transcriptomic analysis of mouse and human skin. The two differentiation paths harbor distinct stem cell states and, unexpectedly, show strong similarities in human skin and mouse tail but not mouse back skin. Specifically, the tail IFE domains, defined as scales and interscales, show similarities of preferred‐gene expression and lineage differentiation trajectories with the human rete ridges and inter‐ridges, respectively (Synopsis). Furthermore, our IF and single‐cell transcriptomic data suggest that mouse scales and their newly found human counterpart—rete ridges—harbor stem cell states enriched in our non‐LRC gene signatures. Conversely, the mouse interscales and their new human counterpart—the inter‐ridges—harbor SCs enriched in our LRC gene signatures (Synopsis). The mouse back skin, which has so far been considered a prototype of human skin, shows the most distinct behavior among all these skin types. Although the mouse back skin presents two distinct differentiation paths, the SC states are intermingled along the paths and present a mixed identity based on LRC/non‐LRC gene signatures (Synopsis). An important difference between tail and back is that in tail, the LRC/non‐LRC domains remain as permanent fixed structures (e.g., interscale/scales) throughout the animals' life. In contrast, in back skin, an LRC domain may be a temporary structure with a fluid identity. This identity may be influenced by local and changing micro‐environmental conditions, or by the waves of synchronized hair cycle progression specific to mouse back skin (Plikus et al, 2011; Roy et al, 2016), which influence the overall rates of epidermal proliferation. In contrast to back skin, and similar with tail scales/interscales, human skin rete ridges/inter‐ridges also may remain as fixed spatial structures throughout animal's life and thus display strong segregation of stem cell identities along the two basal layer differentiation paths.
An important finding of our study is that cells from the two IFE domains express a set of genes and molecular pathways that distinguish them at the functional level. Many pathways are differentially regulated, and future studies will be needed to decipher the full spectrum of these differences. However, given the spatial organization of IFE domains, one clear functional link that emerges from our study is with UV‐response. This may be explained at least in part by the differential exposure of the two IFE domains to the outside world (e.g., rete ridges and scales are less exposed than inter‐ridges and interscales). A link of UV response with spatial organization was also proposed for smaller epidermal domains, which are strictly linked to the vicinity of the hair follicle (Roy et al, 2020). We propose that the differential pattern of basal gene expressions in the more environment‐exposed regions (e.g., the LRC gene signature) may be in part (although not in total, see below paragraph), a consequence of skin's increased UV stress in these IFE regions (Synopsis). These genes likely underline the adaptation that provides each epidermal domain with equal chances of survival despite differential exposure. This model is supported by our data on the LRC‐factor Sox6, a novel interscale/inter‐ridge enriched gene. Interestingly, Sox6 is also upregulated in skin with atopic dermatitis, a disease in which epidermal barrier defects cause excessive environmental susceptibility (Liew et al, 2020). Our gene targeting data in mice demonstrate that Sox6 promotes basal cell proliferation and survival during homeostasis. In long‐term, the epidermis compensates for its loss by upregulating proliferation. We also find that Sox6 is readily upregulated by basal cells upon UV exposure to combat cellular stress and prevent excessive apoptosis (Synopsis). Although future work is required to substantiate downstream mechanisms, Sox6 emerges here as a novel LRC‐domain gene prototype, which helped us decipher at least one potential physiological meaning of epidermal spatial organization. In future, it will be interesting to examine how exactly is Sox6 performing its role in epidermis survival and proliferation in homeostasis and UV response.
One exciting possibility is that UV‐protection may be a broader characteristic of LRC‐related genes, as several UV‐induced master regulators (Shen et al, 2019; Li et al, 2021) appeared in our analysis preferentially upregulated in the LRC/interscale/inter‐ridge‐enriched population. These observations warrant a more systematic future testing of LRC signature genes implication in UV and other environmental‐stress responses. While exposure to UV and other environmental challenges may contribute to spatial and molecular IFE domain organization, this is likely not the absolute origin of it. The laboratory mouse is not naturally exposed much to UV, and the scale/interscale regions form early on after birth, by ~ PD9 (Gomez et al, 2013; Sada et al, 2016), before extensive exposure were possible. Furthermore, the distinct spatial IFE domains already exist to some extent in newborn human foreskin ([Wang et al, 2020] and this work, suggesting the domains may be established during skin development. We plan to investigate the question of epidermal spatial patterning in the future.
In closing, we propose that the spatial organization of the IFE basal layer in distinct regenerative domains encompasses complex cellular and molecular organization. This organization may reflect the skin adaptation to local environmental challenges, imposed on a non‐uniform 3D tissue structure. The spatial, cellular, and molecular complexity of the basal layer may contribute to the remarkable robustness of epidermis, this extraordinarily resilient and essential tissue that faces constant challenges during adult homeostasis.
Materials and Methods
Mice
All mouse work was carried out according to Cornell University Institutional Animal Care and Use Committee guidelines (protocol number no. 2007–0125). To employ the H2B–GFP tet‐off system, double‐transgenic K5–tTA (FVB) (Diamond et al, 2000)/pTRE–H2B–GFP (CD1) (Tumbar et al, 2004) mice were used. Mice were fed with doxy chow (1 g doxy/1 kg, Bio‐serv) for the indicated chase periods, starting at 1–3 months of age. Chase times are indicated for all the different experiments.
For lineage tracing, K14‐CreER (CD1) (Vasioukhin et al, 1999), Dlx1‐CreER (C57BL6) (Taniguchi et al, 2011) (The Jackson Laboratory, no. 014551) Slc1a3‐CreER (C57BL6) (Nathans, 2010) (The Jackson Laboratory, no. 012586) mice or Aspm‐CreER (Madisen et al, 2010; Marinaro et al, 2011), were crossed with Rosa–tdTomato reporter mice (Madisen et al, 2010; The Jackson Laboratory). CreER/Rosa–tdTomato mice without TM injections were used to examine the leakiness of Cre. Dlx1‐CreER, Slc1a3‐CreER or Aspm‐CreER quadruple‐transgenic mice (CreER/Rosa–tdTomato/K5–tTA/pTRE–H2B–GFP) were obtained after several steps of intercrossing the above lines. Cre‐ER/K5tTa (using Tet860‐1060 primers), Rosa‐EYFP (used for K14‐CreER) and tdTomato mice were genotyped as recommended by the Jackson manufactures primer and protocol.
For short‐term BrdU pulse‐chase and UVB‐irradiation experiment, K14‐CreERT2 mice (Indra, 1999) were crossed with Sox6fl/fl mice (Dumitriu et al, 2006) to generate inducible epithelial‐specific Sox6 knockout (iKO) mice.
Tamoxifen injection
K14‐CreER‐H2BGFP quadruple‐transgenic mice were injected intraperitoneally with a single dose of TM (Sigma) (75 μg g−1 body weight for FACS, 200 μg g−1 in K14‐CreER‐EYFP). For lineage tracing and FACS experiments (using Dlx1‐CreER and Slc1a3‐CreER lines), mice were injected with TM (100 μg g−1 body weight) for five consecutive days at 4–7 weeks of age. Intermediate dose (100 μg g−1 body weight for two consecutive days) was used for Aspm CreER and Slc1a3‐CreER lines in the lineage staining experiments. Mice were euthanized at the indicated times after the last injection.
To induce epithelial‐specific Sox6 deletion for short‐term BrdU pulse‐chase and UVB experiments, K14‐CreERT2 × Sox6fl/fl mice were injected with a single dose of 100 μg g−1 body weight tamoxifen at PD32 or PD49, respectively.
Human samples
Human skin samples were de‐identified except age and sex was recorded. They were obtained as frozen OCT tissue samples, under Cornell Office of Research Integrity and Assurance IRB for Human Participants, Protocol# 0908000777.
Whole‐mount immunostaining in the tail epidermis
Tail skin pieces (5 mm × 5 mm) were incubated in EDTA (20 mM)/PBS on a shaker at 37°C for 2 h to separate the epidermis from the dermis as an intact sheet. Epidermal sheets were fixed in 4% paraformaldehyde (PFA) overnight at 4°C. The skin pieces were washed, incubated in blocking buffer (1% BSA, 2.5% donkey serum, 2.5% goat serum, 0.8% Triton in PBS) for 3 h at room temperature, and incubated with primary antibodies/blocking buffer overnight at room temperature. Samples were washed 4× in PBS with 0.2% Tween for 1 h at room temperature, and were incubated overnight with secondary antibodies at 4°C. After washing, samples were counterstained with Hoechst for 1 h and mounted. For the H2B‐GFP samples, back and tail skin were chased for 2, 3 or 6‐week, respectively. No‐GFP staining was done to enhance the signal.
Primary antibody dilutions: mouse anti‐K10 (1:100, BioLegend 904301), rat anti‐β4‐integrin (1:200, BD bioscience 553745), guinea pig anti‐K31 (1:100, PROGEN Biotechnik GP‐hHa1), rabbit anti‐K14 (1:100, BioLegend 905301). All secondary antibodies (TxR, FITC, Cy5 or Alexa‐594, Jackson ImmunoResearch) were used at a 1:500 dilution. The MOM kit (Vector Laboratories) was used for blocking when staining with mouse primary antibodies.
Preparations were analyzed by confocal microscopy (Zeiss LSM710 or Zeiss LSM880) with Zen 2012 software. All confocal data are shown as projected Z‐stack images viewed from the basal surface.
Prefixation of tdTomato expressing tissues before embedding and immunostaining of mouse and human skin sections
For lineage‐traced mice (both tdTomato or EYFP), back and tail skin (with intact dermis) were prefixed in 4% PFA overnight and passed through sucrose gradient (15 and 30%) before embedding. Non‐CreER or only H2BGFP chased mice back and tail skin was directly embedded in optimal cutting temperature (OCT) compound (Tissue Tek, Sakura). The frozen sections (10 μm) were fixed with 4% PFA for 10 min at room temperature. After blocking in normal serum, sections were incubated with primary antibodies overnight at 4°C. The following day, the sections were washed and incubated for 1 h with secondary antibodies at room temperature. After washing, the sections were counterstained with Hoechst and mounted. For staining with anti‐BrdU antibody, the sections were treated with 2 M HCl for 55 min at 37°C after blocking and stained as described above.
Primary antibody dilutions were as follows: rabbit anti‐K14 (1:1,000, BioLegend 905301), mouse anti‐K10 (1:100, BioLegend 904301), guinea pig anti‐K31 (1:100, PROGEN Biotechnik GP‐hHa1), chicken anti‐K15 (1:150, BD Biosciences 833904), rabbit anti‐Ki67 (1:100, Abcam ab15580), rabbit anti‐Slc1a3 (1:300, abcam 416), Aspm (1:1,000, Proteintech 26223‐1‐AP), Vamp1 (1:500, Proteintech 13115‐1‐AP), Col17a1 (1:20,000, Invitrogen MA5‐31984) and Cxcl12 (1:500, Cell Signaling Technology 97958S). Rabbit anti‐Sox6 (1:4,000, Abcam ab30455), anti‐Caspase3 (1:4,000, R&D Systems AF835), anti‐gamma H2AX (1:4,000, Abcam ab2893), rat anti‐α6 integrin (1:4,000, BD Biosciences 555734), anti‐BrdU (1:400, Abcam ab6326), chicken anti‐K14 (1:40,000, BioLegend 906004), mouse anti‐K10 (1:4,000, Abcam ab9026). All secondary antibodies (TxR, FITC, Cy5 or Alexa‐594, Jackson ImmunoResearch) were used at 1:500 dilution. For mouse primary antibodies, the MOM kit (Vector Laboratories) was used for blocking.
Preparations were examined using a fluorescent microscope (Nikon) and digitally imaged using a CCD (charge‐coupled device) 12‐bit digital camera (Retiga EXi; QImaging) and IP‐Lab software (MVI).
Short‐term BrdU pulse‐chase
For short‐term bromodeoxyuridine (BrdU) pulse‐chase labeling, 35 days old K14‐CreERT2/ Sox6fl/fl mice and their control litter mates (n = 3 per group) were injected with 50 μg/g body weight BrdU intraperitoneally twice at 12‐h intervals. 2 h‐chased mice were harvested 2 h after the first BrdU injection, while 12 h‐ and 7 days‐chased mice were harvested at the indicated times after the last injection.
UVB‐irradiation
UVB irradiation was performed on 56‐day‐old K14‐CreERT2/Sox6fl/fl mice and their control litter mates (n = 3 per group) following the procedure described in a previously published protocol (Moon et al, 2017). Isoflurane was used to anesthetize mice during dorsal skin shaving at PD55 and UVB‐irradiation procedure at PD56, and 180 mJ/cm2 UVB was irradiated on dorsal skin during the irradiation. No UVB (0 h UVB) and UVB‐exposed skin samples (6 h or 24 h post‐UVB) were collected at indicated times after the procedure.
Quantification of microscope images
Relative fluorescence intensity per unit area/pixel or fraction of tdtomato+ cells of the basal layer were quantified by using ImageJ software. The scale/interscale regions are defined based on the retention of nuclei in the cornified layer in the scale region and/or K10 (interscale) or K31(scale) expression. The data were normalized by subtracting background intensity (the region of lowest intensity in the basal layer) per image. For H2B‐GFP overlap analyses, we first examined basal cells that were marker positive vs. negative, qualitatively define cells with low GFP intensity, that was then quantified and set as threshold to further classify cells in GFPlow vs. GFPhigh. Next, we counted both marker positive and negative cells per image and calculated % BL expressing marker irrespective of H2BGFP. For whole mount clonal data, the number of tdTomato+ clones of the tail epidermis was counted on maximal projections Z‐stack confocal images. Clones are defined as clusters of cells that contain at least one basal or suprabasal cell. Quantifications were independently performed on ≥ 2 mice/per time point/per genotype, and ≥ 50 clones/structure were counted per mouse.
FACS sorting
Mouse back/tail skin was incubated in 0.25% trypsin/versene overnight at 4°C and for 30 min at 37°C. Single‐cell suspensions were prepared by scraping off the fat and subcutaneous tissue from the dermal side of the skin followed by enzymatic digestions and subsequent filtering with strainers (70 mm, followed by 40 mm). Cells were stained with the following antibodies for 30 min on ice: FITC rat anti‐Sca1 (1:300, 557405) or PE‐Cy7 rat anti‐Sca1 (1:400, 558162) and Brilliant Violet 421 rat anti‐α6‐integrin (1:200, 313624) from BD Biosciences. Dead cells were excluded by AmCyan Live/Dead dye (1:500, Invitrogen L34966) staining. FACS (FACS Aria, BD Biosciences) analyses were performed in the Cornell Flow Cytometry facility. FACS data were analyzed with the FlowJo software.
RNA isolation and RT–PCR
Total RNAs were isolated from sorted skin cells prepared by TriZol method and used for reverse transcription by Super script III (Invitrogen). The primers used were as follows: Gapdh, 5′‐ACTGCCACCCAGAAGACTGT‐3′ and 5′‐GATGCAGGGATGATGTTCT‐3′; Dlx1, 5′‐ATGCCAGAAAGTCTCAACAGC‐3′ and 5′‐AACAGTGCATGGAGTAGTGCC‐3′; Igfbp3, 5′‐TCTAAGCGGGAGACAGAATACG‐3′ and 5′‐CTCTGGGACTCAGCACATTGA‐3′; Sox6, 5′‐GGTCATGTTTCCCACCCACAA‐3′ and 5′‐TTCAGAGGGGTCCAAATTCCT‐3′; Slc1a3,5′‐ACCAAAAGCAACGGAGAAGAG‐3′ and 5′‐GGCATTCCGAAACAGGTAACTC‐3′; Aspm, 5′‐TGGCTATGAGTGAATGCTCTTCC‐3′ and 5′‐TCGCGTAAAAACAGTGGCAAG 3′; Vamp1,5′‐CAGTGCTGCCAAGCTAAAAA‐3'and 5′‐CCAGTAGCCGTCTCCATACC‐3′; Cxcl12, 5′‐GCGCTCTGCATCAGTGAC‐3'and 5′‐TTTCAGATGCTTGACGTTGG‐3′ K14, 5′‐AAGGTCATGGATGTGCACGAT‐3′ and 5′‐CAGCATGTAGCAGCTTTAGTTCTTG‐3′; K10, 5′‐GGAGGGTAAAATCAAGGAGTGGTA‐3′ and 5′‐TCAATCTGCAGCAGCACGTT‐3′; K15, 5′‐GGAGGTGGAAGCCGAAGTAT‐3′ and 5′‐GAGAGGAGACCACCATCGCC‐3′ qRT–PCR for each gene is normalized to GAPDH. The relative level for each gene is set to 1 in the control population.
Single‐cell capturing, library generation and processing of scRNA‐seq data
FACS collected α6‐integrin+/Sca1+ cells from back skin epidermis were further purified by GFP intensity into LRCs, Mid‐LRCs and Non‐LRCs from two male mice at PD47 (telogen). Single‐cell suspension of each cell type (~ 4,000 live cells) were processed to generate single‐cell 3′ cDNA libraries using individually barcoded 10X Chromium Single‐Cell 3′ gel bead and library Kit v3, according to the manufacturer's recommendations (10x Genomics). RNA from the barcoded cells was reverse transcribed, followed by amplification, shearing 50 adaptor and sample index attachment. The final libraries were quantified using Agilent Bioanalyzer high‐sensitivity DNA chip and sequenced using an Illumina NextSeq‐500. The raw data files were demultiplexed to generate the sample‐specific FASTQ files, which were aligned to the mouse reference genome (mm10‐3.0.0) using the 10X Genomics Cellranger pipeline (v3.1.0) with default parameters. Approximately 270 M reads were generated for the LRCs, Mid‐LRCs and Non‐LRCs libraries from the back skin, with a mean number of 68,000, 56,000 and 42,000 reads per cell. After filtering, 3,674, 4,308 and 5,500 cells were analyzed for back skin, detecting a median of 3,000 genes per cell. For tail skin, we analyzed 4,402, 6,236 and 4,245 cells combined from two replicates for the LRCs, Mid‐LRCs and Non‐LRCs respectively. Total reads per cell and the number of genes detected were comparable to back skin.
Single‐cell RNA‐seq data analysis
The output from the Cellranger from the 10X platform consists of a matrix of raw read counts that was further analyzed in R using the Seurat package version 3.1 (Satija et al, 2015). High‐quality cells that contain at least 200–5,000 genes with a mitochondrial gene percentage under 10% were filtered. Expression value scaling and normalization, PCA and UMAP dimensionality reductions and clustering were performed using the Seurat (Butler et al, 2018) R package (version 3.0.1). LRCs, Mid‐LRCs and Non‐LRCs were integrated using harmony (version 1.0) (Korsunsky et al, 2019) after scaling expression values for each sample independently using Seurat, resulting a total of 13,487 cells by combining two replicates for further analyses, which could be further splitted back into LRCs, Mid‐LRCs and Non‐LRCs for sample‐specific information. To identify cell clusters, principle component analysis (PCA) was first performed and the top 10 PCs with a 0.55 resolution were used to obtain 10 clusters. Differentially expressed genes for all the clusters were acquired using FindAllMarkers function with log2 fold change > 0.25 using the Wilcoxon Ranked Sum test. Multiple resolutions (0.3, 0.5, 0.7, 0.9) were assessed to reveal clusters with biological significance and perform marker gene discovery. Markers were then selected by setting the threshold to all genes with an adjusted P‐value lower than 0.05. After filtering, cells belonging to the infundibulum (Sox9high, Krt17high, and Krt79high), differentiated cells (Krt10high, Krt1high, and Sbsnhigh), putative SC cell types (SC1, SC2, and SC3; Krt14high Krt15 high, and Col17a1high), proliferating cells (enriched in either S‐phase MCMs and/or G2‐M cell‐cycle related genes were remarkably segregated; Mki67, Top2a, and Cdc20) were manually assigned based on their expression of various related genes (Dekoninck et al, 2020; Haensel et al, 2020).
Pseudotime lineage trajectory
Trajectory analysis was performed using Monocle (Trapnell et al, 2014). First, the Seurat object (v3) was converted to a monocle cds (cell data set) and then loaded into Monocle2. We used the top 2,000 differentially expressed genes that were calculated using the standard Seurat workflow to assign pseudotime values to individual cells. The cells that contain lower than 200 transcripts were removed from further analyses. The trajectories were constructed with DDRTree.
LRC/non‐LRC gene score computation and cell cycle analysis
Cell‐cycle phases of different clusters were predicted using Seurat's Cell‐Cycle scoring method Each cell was scored based on the expression of certain G2M and S phase markers. Any cell that did not express either G2M or S phase markers was predicted to be in G0/G1. The AddModuleScore function in the Seurat R package was used to calculate signature scores and determine whether there was any significant difference between the subpopulations. This function was widely used for scoring various gene sets from previous literature for both mice (Dekoninck et al, 2020; Haensel et al, 2020) and human (Wang et al, 2020). Specific genes in each gene set are listed in Dataset EV1. The two‐sided Wilcoxon rank‐sum test (Ji et al, 2020) was used to evaluate whether there are significant differences in the computed signature scores between two groups of cells.
Bulk RNA‐sequencing
RNA‐seq were performed in triplicates (BL Slc1a3/Dlx1/Aspm‐tdTomato positive BL and total BL α6‐integrin+/Sca1+ cells) isolated from three female mice at PD49 (telogen). Total RNA was isolated from sorted cell populations using Trizol (Thermo Fisher) according to the commercial protocol with the following additions: after the first phase separation, additional chloroform extraction step of the aqueous layer in Phase‐lock Gel heavy tubes (Quanta Biosciences); addition of 1ul Glyco‐blue (Thermo Fisher) immediately prior to isopropanol precipitation; two washes of the RNA pellet with 75% ethanol. The RNA sample quality was confirmed using a Qubit3 (RNA HS kit; Thermo Fisher) to determine concentration and with a Fragment Analyzer (Advanced Analytical) to determine RNA integrity. The PolyA+ RNA was enriched with the NEBNext Poly(A) mRNA Magnetic Isolation Module (New England Biolabs).
Illumina library preparation and sequencing
Since our samples resulted in low‐input RNA (< 20 nt total RNA), truSeq‐barcoded RNA‐seq libraries were generated with the Ultra II RNA Library Prep Kit (non‐directional) (New England Biolabs). Each library was quantified with a Qubit 2.0 (dsDNA HS kit; Thermo Fisher), and the size distribution was determined with a Fragment Analyzer (Advanced Analytical) prior to pooling. Libraries were sequenced on an Illumina instrument (Hiseq4000). 2x150 bp HiSeq reads were generated with a depth of 20 M.
Mapping the reads
Preprocessing: reads were trimmed for low quality and adaptor sequences with TrimGalore v0.6.0 (ref 1), a wrapper for cutadapt (ref 2) and fastQC (ref 3). Parameters: ‐j 1 ‐e 0.1 ‐‐nextseq‐trim = 20 ‐O 1 ‐a AGATCGGAAGAGC ‐‐length 50 ‐‐fastqc;unwanted reads were removed with STAR v 2.7.0e (ref 4). Parameters: ‐‐outReadsUnmapped Fastx mapping: reads were mapped to the reference genome/transcriptome (mouse/mm10) using STAR v2.7.0e (ref 4). GeneCounts gene expression analysis: SARTools and DESeq2 v1.26.0 were used to generate normalized counts and statistical analysis of differential gene expression (ref 5,6). Parameters: fitType parametric, cooks Cutoff TRUE, independent Filtering TRUE, alpha 0.05, pAdjustMethod BH, typeTrans VST, locfunc median.
Software references
-
1
TrimGalore: Felix Krueger.
http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/
-
2
cutadapt: Marcel Martin.
https://cutadapt.readthedocs.io/en/stable/
http://journal.embnet.org/index.php/embnetjournal/article/view/200
-
3
fastQC: Simon Andrews.
-
4
STAR: Alexander Dobin.
-
5
SARTools: Hugo Varet.
-
6
DEseq2: Michael Love.
http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html
https://genomebiology.biomedcentral.com/articles/10.1186/s13059‐014‐0550‐8
Quality control analysis
Principal component analysis was performed on the raw counts for the different tdTomato+ sorted populations normalized in both back and tail skin with their respective BL (α6‐integrin+/Sca1+) sorted control and the scores were represented in a three‐dimensional scatter plot. Hierarchical clustering was performed on all the samples to reflect sample grouping between tail and back epidermal populations After normalization, the genes having counts with a signal value < 100 or reported “absent” were excluded. The remaining genes that are ≥ 2‐fold up‐ or downregulated in BL relative to BL tdTomato+ lineages were selected. Signature genes were defined as those that were ≥ 2‐fold upregulated in one lineage over its own BL. To compare the populations gene‐wise, lists of ‘≥ 2‐fold change in Aspm/Slc1a3/Dlx1/K14‐CreER; tdTomato normalized over total BL (~ 400 genes) were used. For pathway comparison, lists of ‘log FC ≥ 1 or < 1 genes without any cut‐off on raw count signal (~ 4,000 genes) was used (Appendix Table S2). The numbers of overlapping genes are shown in the Venn diagram (Fig 6C). Gene Set Enrichment analyses using Hallmark pathways ([using GSEA 4.0 with classic parameters]; Subramanian et al, 2005) were performed on differentially expressed genes (Appendix Table S3).
Statistics and reproducibility
All experiments with or without quantification were independently performed at least twice with different mice and the representative data are shown. All statistical analyses were performed either using the two‐tailed Student's t‐test (mixed Anova) or linear mixed model as indicated in each figure legend; statistical significance was defined as P < 0.05.
The sample size was dictated by experimental considerations and not by a statistical method. The experiments were not randomized. The investigators were not blinded to allocation and outcome assessment during experiments.
Author contributions
Sangeeta Ghuwalewala: Conceptualization; data curation; software; formal analysis; validation; investigation; visualization; methodology; writing – original draft; project administration; writing – review and editing. Seon A Lee: Conceptualization; data curation; software; formal analysis; validation; investigation; visualization; methodology; writing – original draft; project administration; writing – review and editing. Kevin Jiang: Data curation; formal analysis; validation; investigation; visualization. Joydeep Baidya: Formal analysis; validation; visualization. Gopal Chovatiya: Software; methodology; writing – review and editing. Pritinder Kaur: Resources; data curation; formal analysis; writing – review and editing. David Shalloway: Data curation; software; formal analysis; validation; methodology; writing – review and editing. Tudorita Tumbar: Conceptualization; resources; formal analysis; supervision; funding acquisition; investigation; visualization; methodology; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interests.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Source Data for Expanded View
PDF+
Acknowledgments
We thank L. Tesfia and J.E. Mahoney for FACS; C. J. Bayles, R. M. Williams, and J. M. DelaCruz for confocal imaging (BRC Facility) and data processing; J. Grenier, A.E. Tate and F. Ahmed for RNA sequencing (TREX facility); P.A. Schweitzer for 10x genomics scRNA‐seq run; B. Cosgrove for help with scRNA‐seq analysis; T.E. Hall for Vamp1 antibody staining conditions. The Cornell CARE staff for mouse husbandry. The Cornell Biotechnology Research Center and Imaging Facility as supported by NIH Grant 1S10RR025502‐01. The research was supported by grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases (R01AR070157 and R01AR073806) to TT.
The EMBO Journal (2022) 41: e110488.
Data availability
The scRNA‐seq data for both the replicates reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database under accession code GSE205746 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE205746), which is the super Series linked to GSE205745 (12 scRNA‐seq samples; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE205745). Bulk RNA sequencing data that support the findings of this study have been deposited in the GEO under accession code GSE205744 (28 bulk RNA‐seq samples; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE205744). All other data supporting the findings of this study are available from the corresponding author on request.
References
- Aitchison J (1982) The statistical analysis of compositional data. J R Stat Soc B Methodol 44: 139–177 [Google Scholar]
- Aragona M, Dekoninck S, Rulands S, Lenglez S, Mascre G, Simons BD, Blanpain C (2017) Defining stem cell dynamics and migration during wound healing in mouse skin epidermis. Nat Commun 8: 14684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragona M, Sifrim A, Malfait M, Song Y, Van Herck J, Dekoninck S, Gargouri S, Lapouge G, Swedlund B, Dubois C et al (2020) Mechanisms of stretch‐mediated skin expansion at single‐cell resolution. Nature 584: 268–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanpain C, Fuchs E (2009) Epidermal homeostasis: a balancing act of stem cells in the skin. Nat Rev Mol Cell Biol 10: 207–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, Satija R (2018) Integrating single‐cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36: 411–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng JB, Sedgewick AJ, Finnegan AI, Harirchian P, Lee J, Kwon S, Fassett MS, Golovato J, Gray M, Ghadially R et al (2018) Transcriptional programming of normal and inflamed human epidermis at single‐cell resolution. Cell Rep 25: 871–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockburn K, Annusver K, Ganesan S, Mesa KR, Kawaguchi K, Kasper M, Greco V (2021) Gradual differentiation uncoupled from cell cycle exit generates heterogeneity in the epidermal stem cell layer. bioRxiv 10.1101/2021.01.07.425777 [PREPRINT]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotsarelis G (2006) Epithelial stem cells: a folliculocentric view. J Invest Dermatol 126: 1459–1468 [DOI] [PubMed] [Google Scholar]
- Dekoninck S, Hannezo E, Sifrim A, Miroshnikova YA, Aragona M, Malfait M, Gargouri S, de Neunheuser C, Dubois C, Voet T et al (2020) Defining the design principles of skin epidermis postnatal growth. Cell 181: 604–620.e22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond I, Owolabi T, Marco M, Lam C, Glick A (2000) Conditional gene expression in the epidermis of transgenic mice using the tetracycline‐regulated transactivators tTA and rTA linked to the keratin 5 promoter. J Invest Dermatol 115: 788–794 [DOI] [PubMed] [Google Scholar]
- Dumitriu B, Dy P, Smits P, Lefebvre V (2006) Generation of mice harboring a Sox6 conditional null allele. Genesis 44: 219–224 [DOI] [PubMed] [Google Scholar]
- Gola A, Fuchs E (2021) Environmental control of lineage plasticity and stem cell memory. Curr Opin Cell Biol 69: 88–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez C, Chua W, Miremadi A, Quist S, Headon DJ, Watt FM (2013) The interfollicular epidermis of adult mouse tail comprises two distinct cell lineages that are differentially regulated by Wnt, Edaradd, and Lrig1. Stem Cell Reports 1: 19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haensel D, Jin S, Sun P, Cinco R, Dragan M, Nguyen Q, Cang Z, Gong Y, Vu R, MacLean AL et al (2020) Defining epidermal basal cell states during skin homeostasis and wound healing using single‐cell transcriptomics. Cell Rep 30: 3932–3947.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichijo R, Kabata M, Kidoya H, Muramatsu F, Ishibashi R, Abe K, Tsutsui K, Kubo H, Iizuka Y, Kitano S et al (2021) Vasculature‐driven stem cell population coordinates tissue scaling in dynamic organs. Sci Adv 7: eabd2575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indra AK (1999) Temporally‐controlled site‐specific mutagenesis in the basal layer of the epidermis: comparison of the recombinase activity of the tamoxifen‐inducible Cre‐ERT and Cre‐ERT2 recombinases. Nucleic Acids Res 27: 4327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji AL, Rubin AJ, Thrane K, Jiang S, Reynolds DL, Meyers RM, Guo MG, George BM, Mollbrink A, Bergenstrahle J et al (2020) Multimodal analysis of composition and spatial architecture in human squamous cell carcinoma. Cell 182: e422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joost S, Zeisel A, Jacob T, Sun X, La Manno G, Lonnerberg P, Linnarsson S, Kasper M (2016) Single‐cell transcriptomics reveals that differentiation and spatial signatures shape epidermal and hair follicle heterogeneity. Cell Syst 3: 221–237.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joost S, Jacob T, Sun X, Annusver K, La Manno G, Sur I, Kasper M (2018) Single‐cell transcriptomics of traced epidermal and hair follicle stem cells reveals rapid adaptations during wound healing. Cell Rep 25: 585–597.e7 [DOI] [PubMed] [Google Scholar]
- Kang S, Long K, Wang S, Sada A, Tumbar T (2020) Histone H3 K4/9/27 trimethylation levels affect wound healing and stem cell dynamics in adult skin. Stem Cell Reports 14: 34–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur P, Potten CS (2011) The interfollicular epidermal stem cell saga: sensationalism versus reality check. Exp Dermatol 20: 697–702 [DOI] [PubMed] [Google Scholar]
- Kohler C, Nittner D, Rambow F, Radaelli E, Stanchi F, Vandamme N, Baggiolini A, Sommer L, Berx G, van den Oord JJ et al (2017) Mouse cutaneous melanoma induced by mutant BRaf arises from expansion and dedifferentiation of mature pigmented melanocytes. Cell Stem Cell 21: 679–693.e6 [DOI] [PubMed] [Google Scholar]
- Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh PR, Raychaudhuri S (2019) Fast, sensitive and accurate integration of single‐cell data with harmony. Nat Methods 16: 1289–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor KT, Kaur P (2015) Dermal contributions to human interfollicular epidermal architecture and self‐renewal. Int J Mol Sci 16: 28098–28107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li KN, Jain P, He CH, Eun FC, Kang S, Tumbar T (2019) Skin vasculature and hair follicle cross‐talking associated with stem cell activation and tissue homeostasis. Elife 8: e45977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MY, Flora P, Pu H, Bar C, Silva J, Cohen I, Galbo PM Jr, Liu H, Yu X, Jin J et al (2021) UV‐induced reduction in Polycomb repression promotes epidermal pigmentation. Dev Cell 56: e2548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liew WC, Sundaram GM, Quah S, Lum GG, Tan JSL, Ramalingam R, Common JEA, Tang MBY, Lane EB, Thng STG et al (2020) Belinostat resolves skin barrier defects in atopic dermatitis by targeting the dysregulated miR‐335:SOX6 axis. J Allergy Clin Immunol 146: 606–620.e12 [DOI] [PubMed] [Google Scholar]
- Lin Z, Jin S, Chen J, Li Z, Lin Z, Tang L, Nie Q, Andersen B (2020) Murine interfollicular epidermal differentiation is gradualistic with GRHL3 controlling progression from stem to transition cell states. Nat Commun 11: 5434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR et al (2010) A robust and high‐throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13: 133–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinaro C, Butti E, Bergamaschi A, Papale A, Furlan R, Comi G, Martino G, Muzio L (2011) In vivo fate analysis reveals the multipotent and self‐renewal features of embryonic AspM expressing cells. PLoS One 6: e19419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascre G, Dekoninck S, Drogat B, Youssef KK, Brohee S, Sotiropoulou PA, Simons BD, Blanpain C (2012) Distinct contribution of stem and progenitor cells to epidermal maintenance. Nature 489: 257–262 [DOI] [PubMed] [Google Scholar]
- McInnes L, Healy J, Melville J (2018) UMAP: uniform manifold approximation and projection for dimension reduction (Version 3). arXiv 10.48550/ARXIV.1802.03426 [PREPRINT] [DOI] [Google Scholar]
- Moon H, Donahue LR, Choi E, Scumpia PO, Lowry WE, Grenier JK, Zhu J, White AC (2017) Melanocyte stem cell activation and translocation initiate cutaneous melanoma in response to UV exposure. Cell Stem Cell 21: 665–678.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathans J (2010) Generation of an inducible Slc1a3‐cre/ERT transgenic allele. MGI Direct Data Submission [MGI Ref ID J:157151]
- Park S, Gonzalez DG, Guirao B, Boucher JD, Cockburn K, Marsh ED, Mesa KR, Brown S, Rompolas P, Haberman AM et al (2017) Tissue‐scale coordination of cellular behaviour promotes epidermal wound repair in live mice. Nat Cell Biol 19: 155–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piedrafita G, Kostiou V, Wabik A, Colom B, Fernandez‐Antoran D, Herms A, Murai K, Hall BA, Jones PH (2020) A single‐progenitor model as the unifying paradigm of epidermal and esophageal epithelial maintenance in mice. Nat Commun 11: 1429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plikus MV, Baker RE, Chen CC, Fare C, de la Cruz D, Andl T, Maini PK, Millar SE, Widelitz R, Chuong CM (2011) Self‐organizing and stochastic behaviors during the regeneration of hair stem cells. Science 332: 586–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riquelme PA, Drapeau E, Doetsch F (2008) Brain micro‐ecologies: neural stem cell niches in the adult mammalian brain. Philos Trans R Soc Lond B Biol Sci 363: 123–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rompolas P, Mesa KR, Kawaguchi K, Park S, Gonzalez D, Brown S, Boucher J, Klein AM, Greco V (2016) Spatiotemporal coordination of stem cell commitment during epidermal homeostasis. Science 352: 1471–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy E, Neufeld Z, Cerone L, Wong HY, Hodgson S, Livet J, Khosrotehrani K (2016) Bimodal behaviour of interfollicular epidermal progenitors regulated by hair follicle position and cycling. EMBO J 35: 2658–2670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy E, Wong HY, Villani R, Rouille T, Salik B, Sim SL, Murigneux V, Stark MS, Fink JL, Soyer HP et al (2020) Regional variation in epidermal susceptibility to UV‐induced carcinogenesis reflects proliferative activity of epidermal progenitors. Cell Rep 31: 107702 [DOI] [PubMed] [Google Scholar]
- Ruetze M, Gallinat S, Wenck H, Deppert W, Knott A (2010) In situ localization of epidermal stem cells using a novel multi epitope ligand cartography approach. Integr Biol 2: 241 [DOI] [PubMed] [Google Scholar]
- Sada A, Jacob F, Leung E, Wang S, White BS, Shalloway D, Tumbar T (2016) Defining the cellular lineage hierarchy in the interfollicular epidermis of adult skin. Nat Cell Biol 18: 619–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez‐Danes A, Hannezo E, Larsimont JC, Liagre M, Youssef KK, Simons BD, Blanpain C (2016) Defining the clonal dynamics leading to mouse skin tumour initiation. Nature 536: 298–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satija R, Farrell JA, Gennert D, Schier AF, Regev A (2015) Spatial reconstruction of single‐cell gene expression data. Nat Biotechnol 33: 495–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Chan G, Xie M, Zeng W, Liu L (2019) Identification of master regulator genes of UV response and their implications for skin carcinogenesis. Carcinogenesis 40: 687–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES et al (2005) Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci U S A 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi H, He M, Wu P, Kim S, Paik R, Sugino K, Kvitsiani D, Fu Y, Lu J, Lin Y et al (2011) A resource of Cre driver lines for genetic targeting of GABAergic neurons in cerebral cortex. Neuron 71: 995–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL (2014) The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 32: 381–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumbar T, Guasch G, Greco V, Blanpain C, Lowry WE, Rendl M, Fuchs E (2004) Defining the epithelial stem cell niche in skin. Science 303: 359–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasioukhin V, Degenstein L, Wise B, Fuchs E (1999) The magical touch: genome targeting in epidermal stem cells induced by tamoxifen application to mouse skin. Proc Natl Acad Sci U S A 96: 8551–8556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Drummond ML, Guerrero‐Juarez CF, Tarapore E, MacLean AL, Stabell AR, Wu SC, Gutierrez G, That BT, Benavente CA et al (2020) Single cell transcriptomics of human epidermis identifies basal stem cell transition states. Nat Commun 11: 4239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Natsuga K, Nishie W, Kobayashi Y, Donati G, Suzuki S, Fujimura Y, Tsukiyama T, Ujiie H, Shinkuma S et al (2017) Type XVII collagen coordinates proliferation in the interfollicular epidermis. Elife 6: e26635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb A, Li A, Kaur P (2004) Location and phenotype of human adult keratinocyte stem cells of the skin. Differentiation 72: 387–395 [DOI] [PubMed] [Google Scholar]