

Abstract
Acute myeloid leukemia (AML) is an aggressive, heterogenous malignancy characterized by clonal expansion of bone marrow-derived myeloid progenitor cells. While our current understanding of the molecular and genomic landscape of AML has evolved dramatically and opened avenues for molecularly targeted therapeutics to improve upon standard intensive induction chemotherapy, curative treatments are elusive, particularly in older patients. Responses to current AML treatments are transient and incomplete, necessitating the development of novel treatment strategies to improve outcomes. To this end, harnessing the power of bioactive sphingolipids to treat cancer shows great promise. Sphingolipids are involved in many hallmarks of cancer of paramount importance in AML. Leukemic blast survival is influenced by cellular levels of ceramide, a bona fide pro-death molecule, and its conversion to signaling molecules such as sphingosine-1-phosphate and glycosphingolipids. Preclinical studies demonstrate the efficacy of therapeutics that target dysregulated sphingolipid metabolism as well as their combinatorial synergy with clinically-relevant therapeutics. Thus, increased understanding of sphingolipid dysregulation may be exploited to improve AML patient care and outcomes. This review summarizes the current knowledge of dysregulated sphingolipid metabolism in AML, evaluates how pro-survival sphingolipids promote AML pathogenesis, and discusses the therapeutic potential of targeting these dysregulated sphingolipid pathways.
Keywords: Sphingolipid dysregulation, Bcl-2, Mcl-1, ceramide, sphingosine-1-phosphate, therapeutics
1. Introduction
Acute myeloid leukemia (AML) is the most common acute leukemia in adults and comprises a heterogeneous group of hematological malignancies characterized by abnormal proliferation of immature hematopoietic progenitor cells of the myeloid lineage [1]. An uncommon disease, AML accounts for 1.1% of all cancers and has a 5-year survival rate of 29.5% following diagnosis. In the U.S., an estimated 20,240 new AML cases will be diagnosed and approximately 11,400 people will die due to AML in 2021, resulting in 1.9% of all cancer deaths [1]. The disease incidence increases while survival rates decline with ageing [1–3]. Moreover, the incidence of AML appears to be increasing in recent years due to the overall aging population, toxic exposures, and prior use of chemotherapy and/or radiation for the treatment of other malignancies [1, 4].
Risk classification for AML in adults at the time of diagnosis is defined by cytogenetic and mutation events [4]. Further risk assessment can be determined using the detection of measurable residual disease [5–7]. Additional classifications (e.g., somatic events, epigenetic, and gene expression signatures) have also been proposed, but are not in routine clinical use [8–14]. However, this hematopoietic cell malignancy is heterogeneous in cell phenotypes and molecular events [2, 15]. Cell phenotypes (e.g., cytogenetics, somatic variants, cell surface markers) are utilized for diagnostic and prognostic purposes as well as measurable residual disease monitoring [6, 7]. In recent years, genomic studies of AML patient specimens have significantly expanded the understanding of disease heterogeneity [16–19]. These studies have defined patient groups harboring distinct molecular alterations, such as cytogenetic events and somatic mutations in genes involved in epigenetic regulation, cell signaling, tumor suppression, and transcriptional network modulation. Furthermore, disease heterogeneity has expanded to include epigenetic landscapes where mutation-associated and independent DNA methylation patterning associates with distinct disease biology and clinical outcomes [9, 10, 18, 20].
For many decades the main treatment option for AML patients at the time of diagnosis was intensive 7+3 induction chemotherapy consisting of daily infusions of cytarabine (Ara-C) supplemented with an anthracycline on the first three days [15]. Most patients respond to this therapeutic approach and the 5-year survival rate is 40–50% for younger patients with de novo AML [21–25]. Exceptions are patients with acute promyelocytic leukemia, which has a cure rate upwards of 90% when using differentiating agents such as all-trans-retinoic acid and arsenic trioxide [15]. Ultimately, if patients respond to treatment and achieve remission, further treatment with allogeneic hematopoietic stem cell transplantation can be considered for all eligible patients. If patients achieve remission after induction chemotherapy, intensive post-remission consolidation therapy with Ara-C can also be considered [26–29]. Unfortunately, most patients will relapse following induction therapy, and age-related toxicities preclude older patients from receiving intensive chemotherapy, highlighting the need for alternative therapeutic approaches [30].
Therapeutic options have evolved to target molecular and epigenetic characteristics of the disease due to the availability of novel agents and a better understanding of disease pathogenesis [15, 23, 24, 31]. Venetoclax, an orally available selective Bcl-2 inhibitor, is now routinely combined with the traditional single-agent treatments of hypomethylating agents (azacitidine or decitabine) or low-dose Ara-C (LDAC) for frontline AML treatment [32–34]. Glasdegib, an oral hedgehog pathway inhibitor, is also approved combined with LDAC in the same treatment setting [35, 36]. Patients who have specific mutations or molecular aberrations can be considered for other newly approved drugs. These targetable aberrations include AML expressing: CD33 (gemtuzumab ozogamicin [31]); FMS-related tyrosine kinase 3 gene (FLT3) internal tandem duplication mutation (ITD subtype) or point mutation in the tyrosine kinase domain (TKD subtype) (midostaurin [37] and gilteritinib [38]); and isocitrate dehydrogenase mutations (IDH1 (ivosidenib [39]) and IDH2 (enasidenib [40])). These agents can be considered in the frontline and/or relapsed settings. Although there are many recent drug approvals for AML, relapse rates remain high [41, 42].
Current treatments need to be adapted and combined with novel therapeutic regimens to improve clinical outcomes. Bioactive sphingolipids (SLs), once overlooked as just a structural component of membranes, control many cellular processes of paramount importance in cancer progression and represent promising therapeutic targets in cancer. Hence, there is an opportunity to develop SL therapeutics in AML, especially for non-responsive and relapsed patients. In this review, we summarize the current knowledge of SL dysregulation in AML, highlight SL alterations downstream of current AML therapeutics, and discuss the prospects of harnessing SL-based treatments to improve AML patient care.
2. Sphingolipids: Metabolism, clinical relevance, and therapeutic potential
SLs are a unique class of bioactive lipids that regulate cellular processes such as growth, proliferation, apoptosis, autophagy, chemotherapeutic response, stemness, and immune cell activation. They accomplish these functions both directly, as messenger molecules, and indirectly, through lipid raft formation and receptor component stabilization within membranes [43–48]. To effectively regulate the potent bioactive effects of SL signaling, cells utilize a complex network of enzymes that catalyze the generation, breakdown, and interconversion of SL species (depicted as subway systems in Figure 1). The complexity of SL metabolism and biology is further compounded by families of enzymes with distinct as well as redundant functional specificity (e.g., ceramide synthases, ceramidases), varying biological effects of SL species based upon fatty acyl chain length (e.g., C16 to C24) as well as trafficking and sequestration of SL within distinct cellular compartments. Considering the size of the SL network and the numerous signaling pathways it regulates, it is not surprising that dysregulation of this network can be catastrophic to cells, often causing or exacerbating diseases such as lipid-storage disorders and cancer [49]. Fortunately, an improved understanding of these pathways has led to clinical advancements for treating patients with lipid disorders, and efforts to advance SL-based cancer treatments into the clinic are ongoing. In this section, we briefly summarize key SL metabolic pathways and highlight noteworthy findings relevant to cancer biology and therapeutic intervention. For a complete summary of SL biochemistry, metabolism, and signaling, refer to these comprehensive reviews: [43, 45, 47, 50, 51].
Figure 1. Transit map of sphingolipid metabolism.
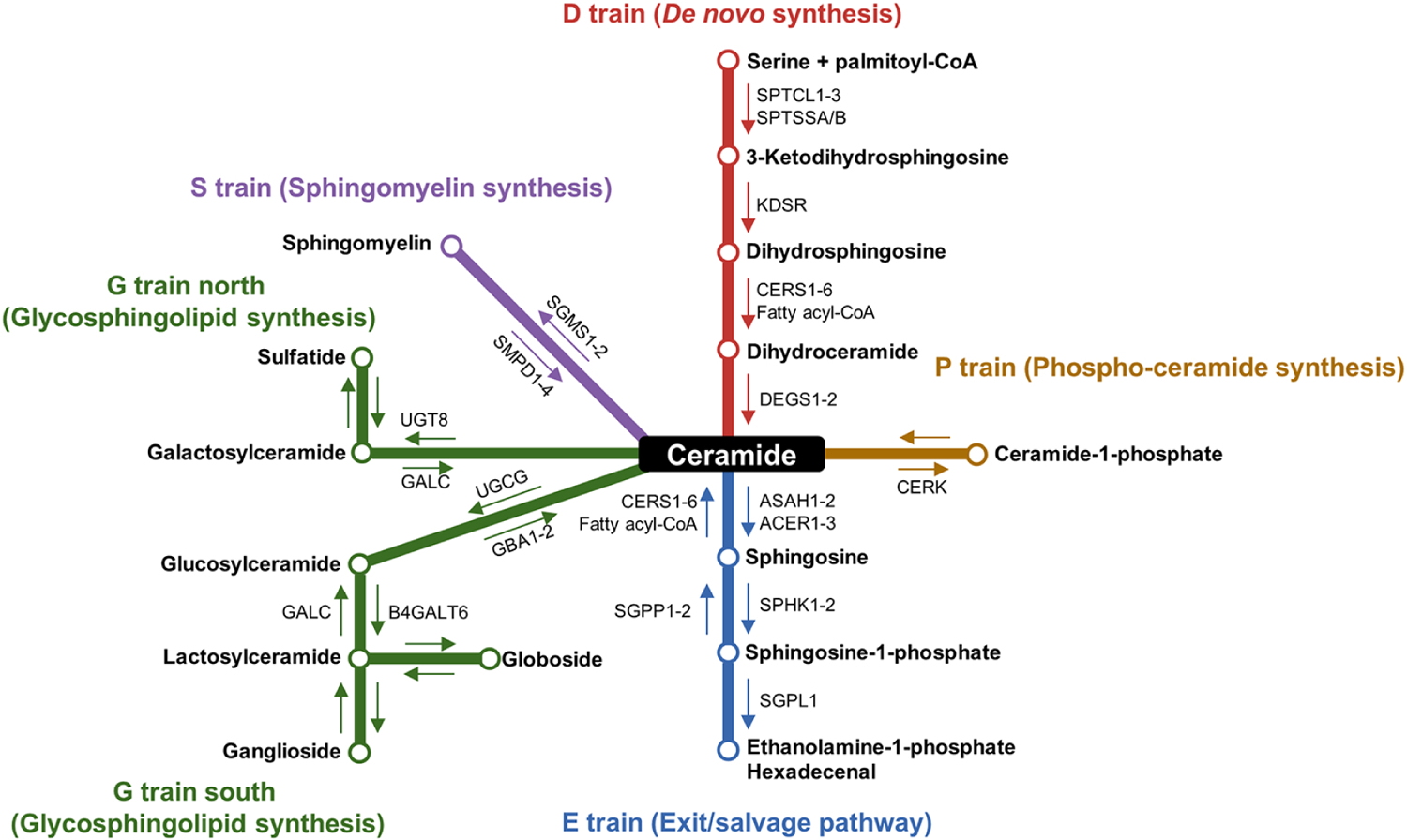
Ceramides reside at the canonical center of the sphingolipid metabolism transit map and serve as the central hub for the other sphingolipids. De novo synthesis begins with the condensation of serine and palmitoyl-CoA by the serine palmitoyltransferase (SPT) complex and further processing by 3-ketodihydrosphingosine reductase (KDSR), dihydroceramide synthases (CERS1–6), and dihydroceramide desaturases (DEGS1–2) to generate ceramide (D train). Other methods of generating ceramides include sphingomyelin hydrolysis (S train) by sphingomyelin phosphodiesterases (SMPD1–4) or CERS activity as part of the exit/salvage pathway (E train). Conversely, there are multiple routes to decrease cellular ceramide content. The addition of a phosphocholine head group by the sphingomyelin synthase enzymes (SGMS1–2) generates sphingomyelin (S train). Ceramidases (ASAH1–2 and ACER1–3) hydrolyze ceramide into sphingosine and free fatty acids, and sphingosine kinases (SPHK1–2) phosphorylate sphingosine into sphingosine-1-phosphate (E train). S1P breakdown by sphingosine-1-phosphate lyase (SGPL1) generates phosphoethanolamine and hexadecenal. Ceramide kinase (CERK) phosphorylates ceramide to generate ceramide-1-phosphate (P train). The synthesis of complex glycosphingolipids begins with the action of galactosylceramide synthase (UGT8) to generate galactosylceramides (G train north) or glucosylceramide synthase (UGCG) to generate glucosylceramides (G train south).
2.1. De novo sphingolipid synthesis and generation of ceramide
The de novo SL synthesis pathway begins with the serine palmitoyl transferase-mediated condensation of serine and palmitoyl-CoA to form 3-ketodihydrosphingosine, which contains the sphingoid backbone common to all SLs. 3-ketodihydrosphingosine reductase (KDSR protein and gene abbreviation) then generates dihydrosphingosine. De novo synthesis culminates with the addition of a fatty acid moiety to the sphingoid backbone by one of six ceramide synthases (CERS1–6 protein and gene abbreviation) and subsequent desaturation of the sphingoid backbone by the desaturase enzymes (DEGS1–2 gene, DES1–2 protein abbreviation) to generate ceramide [43, 45]. While DES1-mediated ceramide generation from dihydroceramide is well characterized, the function of DES2 is less clear and has been implicated in phytoceramide synthesis from dihydroceramides [52]. However, the tissue specificity of DES2 expression is highly restricted and likely excludes hematopoietic tissue (proteinatlas.org), which limits its relevance in leukemia [53]. The six different CERS isoforms preferentially add distinct fatty acids to the sphingoid backbone (Fig. 1, D train) [54–56]. The resulting ceramides sit at the canonical center of the SL metabolic network and can be modified and/or broken down to form nearly every other SL species.
2.2. Ceramide catabolism
Ceramidase enzymes are lipid hydrolases that initiate the breakdown of ceramide via the “exit/salvage pathway.” Although roles for neutral (ASAH2 gene) and alkaline (ACER1–3 gene) ceramidases have been described in cancer, the most studied ceramidase enzyme in cancer is acid ceramidase (ASAH1 gene, AC protein abbreviation) [45]. Working at an acidic pH primarily within the lysosome, AC hydrolyzes ceramides into sphingosine and free fatty acids that can then be utilized as nutrients for the cell or recycled to form other complex lipids [45]. Sphingosine generated from this reaction can either be re-acylated via CERS to regenerate ceramides or phosphorylated by sphingosine kinases (SPHK1–2 protein and gene abbreviation) to generate sphingosine-1-phosphate (S1P) (Fig. 1, E train). S1P is often viewed as antithetical to ceramide and exerts pro-survival effects, in part, by directly binding S1P G protein-coupled receptors (S1PR1–5 protein and gene abbreviation) to regulate autophagy, T-cell migration, and cell survival [57–60]. Additional regulatory enzymes include S1P phosphatase (SGPP1–2 gene), which dephosphorylates S1P to re-generate sphingosine, and S1P lyase (SGPL1 gene), which catabolizes S1P into hexadecenal and ethanolamine-phosphate to exit the SL pathway [45]. Of clinical relevance, several immunomodulating S1PR targeting drugs are clinically approved to target dysregulated T-cell activation and inflammatory signaling (fingolimod and ozanimod) [45, 61, 62].
2.3. Ceramide modifications and conversion into higher-order sphingolipids
Apart from ceramide catabolism, three additional primary pathways deplete ceramides and/or use them as scaffolds to build higher-order SLs: phosphorylation, glycosylation, and sphingomyelin synthesis. Phosphorylation by ceramide kinase (CERK protein and gene abbreviation) generates ceramide-1-phosphate (C1P) (Fig. 1, P train). C1P is thought to induce both cell proliferation and migration, but receptors are poorly defined [45, 58]. One or more sugars may be added to the hydroxyl group of ceramides to generate glycosphingolipids. Galactosylceramide synthase (UGT8 gene) regulates the synthesis of galactosylceramides (Fig. 1, G train north). In contrast, the “gatekeeper” enzyme, glucosylceramide synthase (UGCG gene, GCS protein abbreviation), adds a glucose moiety to form glucosylceramides, which can be further processed to increasingly complex glycosphingolipids (e.g., gangliosides and globosides) (Fig. 1, G train south) [63]. The enzymes GBA1–2 and GALC remove sugar groups from glucosylceramides and galactosylceramides, respectively, to re-generate ceramides [63]. Many lysosomal storage disorders such as Tay-Sachs and Gaucher disease result from a build-up of glycosphingolipids, highlighting the clinical significance of these lipids and their regulating enzymes [64]. Recent studies have elucidated structural roles for various glycosphingolipids in other pathologies as well. Specifically, glucosylceramides stabilize membranes in response to bacterial infection and prevent viral uptake [65, 66]. Others have also linked galactosylceramides and lactosylceramides as secondary messengers to inflammatory pathways [67].
Cellular ceramide levels can also be reduced via the sphingomyelin synthesis pathway. Sphingomyelin synthases (SGMS1–2 gene, SMS1–2 protein abbreviation) transfer a phosphocholine headgroup to ceramide, generating sphingomyelin and diacylglycerol as a byproduct [43]. In some instances, phosphoethanolamine can substitute for phosphocholine [45]. Meanwhile, sphingomyelinase enzymes (SMPD1–4 gene, SMase protein abbreviation) catabolize sphingomyelin back into ceramide (Fig. 1, S train) [43, 45]. Sphingomyelins are of immense structural and signaling importance. Sphingomyelins, along with glycosphingolipids, stabilize cholesterol-dependent raft domains that dictate the thickness and curvature of the membrane, bring together receptors and other components to facilitate signal transduction, and alter interactions with nearby cells and extracellular pathogens [48]. Similar to the lysosomal storage diseases caused by glycosphingolipid dysregulation, mutations in the lysosomal acid SMase (SMPD1 gene) cause Niemann-Pick Disease through the accumulation of sphingomyelin [43]. Moreover, sphingomyelin metabolism is highly relevant to cancer biology, as radiotherapy and a multitude of chemotherapies induce sphingomyelin breakdown into pro-death ceramides to promote cancer cell death [43].
Although the central dogma of dysregulated SL metabolism in cancer states that decreased ceramides and increased S1P promote cell survival, SL signaling is complex and requires further investigation [68]. Indeed, many aspects of the regulation of the complex SL network described above and the specific function of each lipid species therein remain incompletely defined. However, the immense impact of these bioactive molecules on health and disease is exemplified by their linkage with nearly every major metabolic pathway and illness. For this reason, a better understanding of these metabolic pathways, enabled by sphingolipidomics, may redefine the 21st century approach to medicine as the SL field matures from mere membrane components to bona fide prognostic disease biomarkers and/or promising therapeutic targets. The clinical and pathological significance of SLs in AML are best viewed through their roles in many critical aspects of cell biology, as described below.
3. Sphingolipid dysregulation in AML
SL dysregulation plays a major role in cancer through the aberrant expression and localization of SL enzymes, metabolites, and receptors, which results in increased pro-survival lipids and/or blockade of death pathways [43, 46, 47]. The characterization of SL dysregulation in AML has gained traction within the last five to ten years. In general, ceramide-catabolizing enzymes, S1P-synthesizing enzymes, and S1PRs are aberrantly expressed in AML and promote proliferation, survival, drug resistance, and leukemogenesis [69–78]. One notable example showed that increased S1P signaling through elevated S1PR3 expression induced AML in mice [78]. Overall, cumulative evidence strongly supports a higher pro-survival/pro-death lipid ratio in AML and thus, presents a relatively untapped source of both prognostic and diagnostic information [43, 78–80].
Given their key role in cancer progression, many approaches seek to target SLs and their dysregulation for clinical benefit. Strategies aim to either inhibit the generation of pro-survival S1P or promote the formation and accumulation of pro-death ceramides [81]. Noteworthy examples include studies that target aberrant SL players to significantly reduce leukemic burden and increase survival in leukemic mice [70, 73, 74, 82], as well as examples demonstrating improved drug efficiency by combining SL-based therapies with current AML treatments [69, 74]. Importantly, chemotherapy and radiotherapy increase endogenous ceramide levels in cancer cells, which is necessary for their therapeutic effects [83, 84]. SL dysregulation also plays a vital role in drug resistance [69, 85] and leukemic stem cell renewal [86]. Taken together, the SL pathway is a promising target for therapeutic intervention [43, 79–81, 87, 88]. A summary of dysregulated SL signaling in AML is depicted in Figure 2. We also refer readers to important work by Tan et al. [79] and Lewis et al. [80, 88], which have previously reviewed specific aspects of SL metabolism and targeting in AML. The following sections provide the first comprehensive review of the role of SLs in AML survival, drug resistance, and leukemic stem cells, as well as a discussion of SL enzymes and metabolites as predictive biomarkers.
Figure 2. Summary of dysregulated sphingolipid signaling in AML.
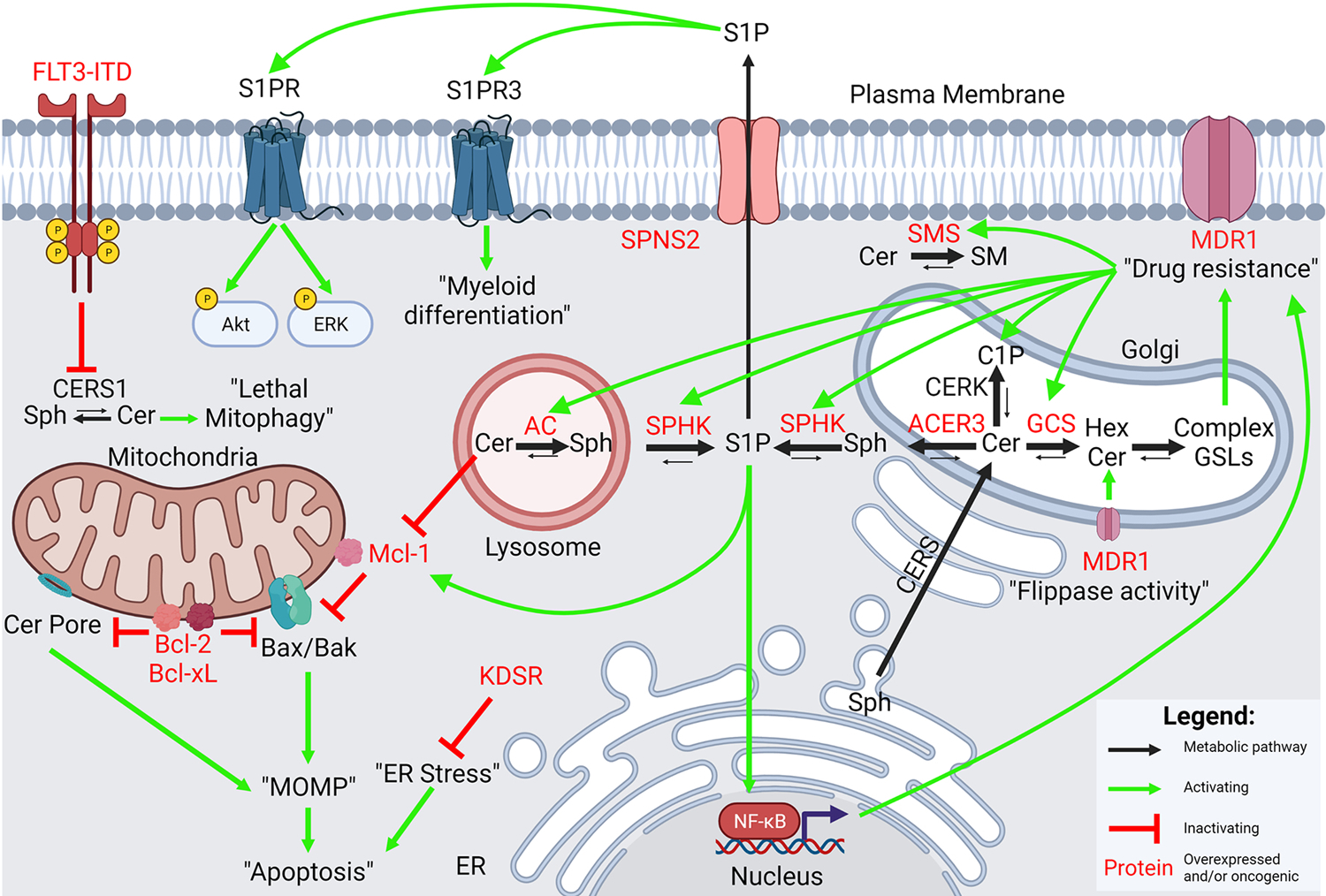
The intersections between dysregulated sphingolipid metabolism and oncogenic signaling in AML are many. Multiple ceramide catabolizing enzymes (AC, SPHK, ACER3) are elevated in de novo AML. Multidrug resistance also upregulates the metabolite C1P and the enzymes AC, SPHK, GCS, and SMS. Overexpression of AC, SPHK, or GCS is sufficient to induce upregulation of MDR1, likely through glycosphingolipids or S1P-mediated NF-κB signaling. Oncogenic FLT3-signaling inhibits ceramide generation by antagonizing CERS1. Overexpression of AC, ACER3, SPHK, and the S1P transporter SPNS2, correlate with poorer overall survival. Bcl-2 and Bcl-xL inhibit the formation of ceramide pores. Ceramides and S1P signaling regulate levels of the pro-survival Bcl-2 family protein, Mcl-1. In addition, S1P-S1PR signaling upregulates levels of phospho-Akt and phospho-ERK. Signaling through S1PR3 promotes myeloid differentiation. KDSR, involved in de novo SL synthesis, prevents ER stress to promote leukemic maintenance. Black arrows = metabolic pathway; green arrows = activating; red inhibitory arrow = inactivating; red text = overexpressed and/or oncogenic proteins.
3.1. Sphingolipid metabolism and AML mutations/chromosomal abnormalities
AML is highly heterogeneous in disease presentation, mutational burden, and prognosis [8, 16, 17, 89]. Although efforts to link aberrant SLs and specific AML molecular alterations are just beginning, a few examples have emerged. In a lipidomic analysis of bone marrow aspirates from AML patients with translocation t(8;21), inversion (inv16), or normal karyotype, Stefanko et al. found that t(8;21) patients have decreased sphingomyelin content and increased membrane fluidity through the shift to polyunsaturated fatty acids. Compared to normal karyotype, both t(8;21) and inv16 AML samples had higher levels of glucosylceramides and decreased core binding factor expression, which regulates several genes in SL metabolism [90].
FLT3
FLT3 is a frequently mutated gene in AML encoding a receptor tyrosine kinase that canonically promotes proper hematopoietic stem cell (HSC) and progenitor cell development. Extracellular FLT3 ligand binds to and activates FLT3 receptor to promote proliferation and differentiation via PI3K, RAS, and STAT5 signaling. FLT3 mutations lead to constitutively activated signaling to promote leukemogenesis and cell survival [91]. Dany et al. published that aberrant FLT3-ITD signaling prevented the formation of endogenous pro-death C18-ceramide by antagonizing CERS1 [92].
IDH1
IDH1 is another frequently mutated gene in AML and the mutant enzyme mediates the conversion of α-ketoglutarate to D-2-hydroxyglutarate. This oncometabolite inhibits α-ketoglutarate-dependent enzymes that regulate epigenetic and signaling pathways [93]. Stuani et al. showed that IDH1R132H mutant expression in the HL-60 AML cell line changed the global SL profile, including upregulation of sphingosines and ceramides [94]. In glioma cells, IDH1R132H expression was associated with decreased S1P, increased ceramides, and increased N,N-dimethyl sphingosine, an endogenous SPHK inhibitor [95]. Interestingly, supplementing IDH1-mutant glioma cells, but not IDH1 wild-type cells, with N,N-dimethyl sphingosine and sphingosine C17 decreased cell growth. These findings highlight a SL-based therapeutic vulnerability that is selectively detected in IDH1-mutant glioma cells. This suggests that the presence of the IDH1R132H mutation in glioma induces a growth disadvantage compared to IDH1 wild-type cells due to the decrease in mitogenic S1P and increase in ceramides. It remains to be seen whether this vulnerability exists in AML. Further studies are needed to define the mechanism and extent of SL changes associated with common AML somatic mutations and their contribution to disease pathology. Moreover, the IDH1 mutation-specific vulnerability to a specific SL inhibitor identified in glioma may represent a new paradigm for SL targeted therapeutics in molecularly defined AML subtypes.
3.2. Sphingolipid dysregulation controls mechanisms of cell death
Dysregulation of bioactive SLs influences cellular signaling and survival pathways, and targeting this dysregulation has revealed novel mechanisms underlying AML survival (summarized in Figure 2 and detailed in the following sections). Here, we discuss the current knowledge and unresolved questions of SLs regulating cell death.
3.2.1. Apoptotic cell death
Leukemic development and progression relies on evasion of apoptotic stimuli triggered by tumor suppressor activation or chemotherapy treatment, which typically culminate in the activation of executioner caspases and apoptotic cell death [96]. Intrinsic apoptosis is primarily regulated at the mitochondria where a delicate balance of SLs and pro-apoptotic (Bim, Bax, Bak, etc.) vs anti-apoptotic (Bcl-2, Mcl-1, Bcl-xL, etc.) Bcl-2 family proteins regulate mitochondrial outer-membrane permeabilization (MOMP) and the release of pro-apoptotic effectors [46, 51]. The ability to resist apoptosis is a classic hallmark of cancer that is regulated in part by SL metabolism. The roles of ceramide as an important signaling molecule and pro-death lipid were initially described by Kolesnick et al. and Obeid et al. in their seminal manuscripts published in 1992 and 1993, respectively [97, 98]. Since then, the role of SLs in intrinsic apoptosis has been evaluated further and key examples of this relationship include: (1) ceramides increase following chemotherapy and are necessary for cell death [83, 84, 99, 100]; (2) inhibition of anti-apoptotic Bcl-2 family proteins increases ceramides [44]; (3) ceramides putatively form “water-filled” channels that permeabilize the mitochondrial outer membrane [101]; (4) SLs are required for proper Bax/Bak function to permeabilize the mitochondrial outer membrane [44]; and (5) ceramide conversion into sphingomyelin, glycosphingolipids, and S1P typically promotes cell survival [43].
Apoptosis resistance is a major mechanism of relapse in AML, which is mediated in part by the overexpression of anti-apoptotic Bcl-2 family proteins [102]. Venetoclax is a selective Bcl-2 antagonist that indirectly activates pro-apoptotic effector proteins (Bim, Bax, Bak) by preventing their association with Bcl-2 [15, 103]. The recent approval of venetoclax in combination with azacitidine or LDAC for elderly AML patients and AML patients unable to undergo intensive chemotherapy highlights the importance of targeting Bcl-2 family proteins [32, 33]. Of additional importance, Mcl-1 is a potent survival factor and mediator of venetoclax resistance in AML due to its shared ability to sequester pro-apoptotic effectors [104, 105]. We have shown that ceramide metabolism modulates expression of the anti-apoptotic Bcl-2 family protein, Mcl-1. Tan et al. demonstrated that AC gene expression is elevated in TCGA AML samples, and validated this finding via gene expression micro-array [70]. AC knockdown in AML cell lines, including drug-resistant variants, reduced total Mcl-1 levels and decreased cell viability [70, 82]. This finding was corroborated with pharmacological inhibition of AC with the lysosomotropic B-13 inhibitor analog, LCL-204. Exogenous addition of ceramide was sufficient to reduce Mcl-1 levels. Conversely, overexpression of AC increased Mcl-1 levels [70]. This study details important links between Mcl-1 and SL biology although the molecular mechanism by which AC and ceramide levels modulate Mcl-1 is unknown. Of note, the alkaline ceramidase, ACER3, is also elevated in AML and its knockdown suppresses cell viability and colony-forming potential in multiple AML cell lines through mechanisms yet to be determined [77].
Ceramidases catabolize ceramides to sphingosine, which can then by phosphorylated by sphingosine kinases SPHK1 and SPHK2 to generate S1P. Pro-survival properties of SPHKs and their product, S1P, have also been identified in AML. SPHK1 likely promotes cell survival through ERK1/2 and Akt signaling since inhibition of SPHK1 with SK1-I significantly downregulated phospho-ERK1/2 and phospho-Akt [106]. A newly developed dual Akt and SPHK1 inhibitor, A-674563, also demonstrated anti-leukemic efficacy [107]. Powell et al. recently demonstrated that pharmacological and molecular inhibition of SPHK1 by MP-A08 and shRNA, respectively, depleted Mcl-1 levels. Signaling through S1P is mediated in part by binding the S1PR G-protein coupled receptors. Treating cells with the putative S1PR2 antagonist, JTE-013, phenocopied the MP-A08-mediated depletion of Mcl-1, suggesting that signaling through S1PR2 modulates Mcl-1 protein levels [74]. However, S1PR2 knockdown did not alter AML cell survival, and additional mechanistic studies of JTE-013 suggest off-target inhibition of DES1 and SPHK1/2, which may be contributory [108]. Taken together, the multiple studies demonstrating that SL signaling modulates levels of Mcl-1, a known mediator of venetoclax resistance [105], suggest that co-targeting AC or SPHK1 may improve venetoclax efficacy in AML.
Endoplasmic reticulum (ER) stress is frequently upregulated in leukemias and results from the accumulation of misfolded proteins as a consequence of rapid proliferation [109]. Cumulative evidence suggests that the unfolded protein response (UPR) is upregulated in AML, restores ER homeostasis, and maintains leukemic survival [109, 110]. Recent work by Liu et al. demonstrated that dysregulated SL metabolism promotes UPR in AML to maintain cell survival [111]. CRISPR knockout of KDSR, the enzyme responsible for the reduction of 3-ketodihydrosphingosine during de novo SL synthesis, results in apoptotic cell death and cell cycle arrest in multiple AML cell lines. Mechanistically, KDSR loss increased 3-ketodihydrosphingosine levels, which downregulated UPR checkpoint proteins, disrupted ER structure, and elicited cell death [111].
Protein phosphatase 2 (PP2A) is a cellular serine-threonine phosphatase that negatively regulates oncogenic signaling, apoptotic signaling, and cell cycle progression. The enzyme is recurrently inactivated in AML via endogenous PP2A inhibitory proteins such as CIP2A, I1PP2A, and I2PP2A (SET) [112, 113]. PP2A regulation is also dependent on SL metabolism [114–116] and ceramides were described as an activator of PP2A [117]. Interestingly, ceramides are also direct ligands of SET, which is a putative inhibitor of PP2A, and thus loss of ceramide correlated with PP2A inactivation [114]. Chen et al. demonstrated that treating Kasumi-1 AML cells with the S1PR-modulating drug and SPHK1 inhibitor, fingolimod (FTY720), induced caspase-dependent cell death coinciding with upregulation of ceramide and PP2A activity. Knockdown of PP2A activity partially inhibited cell death [118]. FTY720 was also recently described as a ligand of SET that can activate PP2A by directly blocking the SET-PP2A interaction [119], which would complement ceramide-mediated SET inactivation.
3.2.2. Other mechanisms of cell death
Autophagy is a process by which cellular materials are sequestered in double-layer membranes that contain microtubule-associated proteins 1A/1B light chain 3B (LC3B) and are termed autophagosomes [120]. Autophagosomes traffic to the lysosome and are degraded as a form of cellular recycling. Mitophagy is a specialized form of autophagy that involves the recycling of dysfunctional or damaged mitochondria to preserve energy metabolism [121].
The role of autophagy in AML appears to be multifaceted and variable based on the context. Numerous studies have shown that pharmacological, molecular, and genetic manipulation of normal autophagic pathways promote leukemia [122]. Intriguingly, modulating these same autophagic pathways appears to enhance certain AML therapeutics – especially FLT3-targeted therapies such as quizartinib and crenolanib. Thus, it can be appreciated that manipulation of the autophagic pathway holds therapeutic promise [122].
It is well established that various SLs and SL analogs can induce both autophagy and mitophagy via a myriad of mechanisms including increased expression of autophagic proteins (LC3B, Beclin 1, and BNIP3), downregulation of nutrient transporters, or direct binding to LC3B-II, which promotes the formation of autophagosomes [43, 46, 120]. Importantly, just as autophagy itself can be beneficial or detrimental in AML, these SL effects can be either protective or lethal depending on cell type, with growing evidence for the latter in AML.
Previous AML data showed that ceramide and ceramide analogs directly bind LC3B-II and target autophagosomes to the mitochondria [123]. Further studies highlighted that the FLT3 inhibitor crenolanib increased ceramide synthesis, mitophagy, and cell death in MV4–11 and MOLM-14 AML cell lines [92]. Also in this study, the positively-charged mitochondria-targeting pyridinium-ceramide analog, LCL-461, induced autophagy and mitophagy. Finally, C6-ceramide co-treatment with tamoxifen increased autophagic flux and synergistic cell death in multiple AML cell lines in a FLT3-ITD-independent manner [124]. Importantly, inhibition of autophagy via knockdown of LC3B and ATG5 reduced cell death from LCL-461 and ceramide plus tamoxifen, respectively. Therefore, ceramide-induced autophagy and mitophagy appear lethal in AML.
In summary, the links between SLs and cell death in AML are many. The recurring theme is that ceramide accumulation is blunted by overexpression of ceramide-catabolizing ceramidases and downregulation of ceramide synthases. This, accompanied with overexpression of SPHKs, promotes a pro-survival phenotype in AML. Indeed, pharmacological and genetic inhibition of these enzymes provide therapeutic benefit in preclinical models. The link between SL metabolism and Mcl-1 levels provides rationale for SL-targeting therapies in combination with venetoclax. However, much remains unknown regarding SLs and AML pathogenicity. For instance, the consequence of less-studied SL biology (e.g., ceramide-1-phosphate, dihydrosphingolipids, sphingoid backbone chain lengths, fatty acyl chain lengths) in AML is not known. While more studies are needed to address these and other uncertainties, the current links between SLs and AML cell death encourage future therapeutic development to target this vulnerability.
3.3. Drug resistance
Drug resistance arising in biological systems is ever-present and widespread. Owing to evolutionary processes and/or sudden selection pressures, nature has provided defenses, some beneficial, some not. These defenses protect from toxins of all kinds, protection that unfortunately extends to chemotherapy drugs. In AML, despite advances in cure rate, a considerable number of patients die from the disease due to the occurrence of multidrug resistance. Drug efflux driven by membrane transport proteins such as multidrug resistance protein 1 (MDR1, also known as P-glycoprotein) [125–129] and multidrug resistance-associated protein 1 (MRP1) [130–132] have been established as the most significant multidrug resistance mechanisms found in AML. Of clinical relevance, a negative correlation between MDR1 and/or MRP1 overexpression, remission rate, and disease-free survival has been demonstrated in patients with AML [130, 133–136]. Even with the known clinical relevance of these pervasive, hallmark proteins, targeting drug transporters has proven elusive [137, 138]. For example, the novel MDR1 antagonist, zosuquidar, in combination with conventional induction chemotherapy, failed to improve AML patient outcomes in a randomized, placebo-controlled trial for newly diagnosed AML [139].
Recent studies in chemotherapy selection pressure have revealed a curious link between MDR1 and SL metabolism. Specifically, wild-type, drug-sensitive AML variants challenged with heterocyclic anticancer agents (daunorubicin, vincristine) generated drug-resistant cells that demonstrated synchronous upregulation of MDR1 as well as several key enzymes of SL metabolism: GCS, AC, SPHK1, and SMS [85, 140–143]. Therefore, selection pressure upregulates MDR1 [128, 144, 145], but additionally enhances expression of enzymes that decrease ceramide, steps that cooperatively contribute to anti-apoptotic, mitogenic behavior that sabotages ceramide-driven cancer cell death. This is significant because mechanisms of cytotoxicity of heterocyclic anti-cancer drugs are, in part, driven by their capacity to generate cytotoxic levels of ceramides [146]. This unique association of SL metabolism with drug resistance has been supported in various works [147–149]. Interestingly, we and others have shown that forced overexpression of GCS, AC, and SPHK1 is sufficient to confer multidrug resistance through direct modulation of MDR1 expression and activity. Importantly, inhibition of these enzymes attenuated multidrug resistance and, in some cases, improved drug retention in AML [69, 71, 150]. In addition, we recently interrogated the consequence of multidrug resistance on mitochondrial bioenergetics [151]. While drug-resistant AML cell lines show increased basal and maximal respiration, they demonstrate a significantly lower fractional oxidative phosphorylation attributed to impaired respiratory complex I compared to wild-type cells. Highlighting the therapeutic implications of this finding are the superb synergistic killing effects observed when SL-modulating agents and the respiratory complex I inhibitor, phenformin, are combined [151].
In addition to its canonical function as a drug exporter at the plasma membrane, MDR1 can act as a “flippase”, transporting glucosylceramides into the Golgi lumen [152]. This raises questions regarding its possible role in ceramide metabolism. Studies in AML have shown that MDR1-expressing cells are resistant to ceramide-induced apoptosis. However, this resistance was averted by exposure to either elacridar or cyclosporin A, MDR1 antagonists [153]. Work of Morad et al. [154] in multidrug resistance AML strongly substantiates the cooperation of MDR1 in the metabolism of ceramide by showing that zosuquidar, a specific MDR1 antagonist, enhanced ceramide-based therapeutics via blockade of ceramide glycosylation. Intriguingly, these works propose that MDR1 contributes to multidrug resistance by acting as an intracellular “ceramide neutralizer”, and prescribe novel indications for MDR1 antagonists in the enhancement of ceramide-centric treatment of AML [43, 154–156]. For example, therapeutic MDR1 approaches were shown to decrease mitochondrial membrane potential, inhibit complex I respiration, and generate ROS in a model of drug-resistant AML [76]. Appropriately, as leukemic stem cells and chemotherapy resistant leukemic cells are thought to play a pivotal role in relapse and exhibit high multidrug resistance transporter expression [157] as well as high oxidative phosphorylation status [158], it is tempting to speculate that these self-renewing and drug-resistant cells could be prime targets for agents that enhance ceramide-driven apoptosis via MDR1 modulation and oxidative stress.
3.4. Leukemic stem cells
Leukemic stem cells (LSCs) are a hallmark of AML and a source of relapse in AML patients [159–161]. LSCs arise from hematopoietic stem cells (HSCs) after one or more mutation events and are generally found within the CD34+CD38− subpopulation, though not exclusively [162–164]. LSCs, like HSCs, have self-renewal capabilities and inherent drug-resistant properties [163]. Targeting LSCs has been challenging due to their poor response to treatment and heterogeneity, especially in relapsed/refractory patients after exposure to chemotherapy [165, 166]. Newer therapeutic strategies seek to exploit LSCs’ unique immunophenotypes, which include CD123 (IL3RA), CD96, C-type lectin-like molecule-1 (CLL-1), and TIM-3 [162, 167], as well as unique metabolic profiles, epigenetic properties [168] and transcriptional regulation [169]. In addition, combinatorial strategies involving LSC targeting with conventional chemotherapy appears beneficial [165]. Hence, exploring and exploiting LSCs’ unique SL profile could provide new approaches to LSC-targeted therapy.
To this point, different subpopulations of progenitor, mature, and HSCs were shown to have distinct SL profiles and these unique SL signatures, which center around the dihydroceramide to ceramide pathway, regulate stem cell renewal and lineage commitment [170]. Importantly, dihydroceramide desaturase 1 (DEGS1 gene, DES1 protein abbreviation) was significantly increased in long-term-HSCs, short-term HSCs, and granulocyte-monocyte progenitors after ex vivo cytokine stimulation. Inhibiting DES1 with fenretinide (4-HPR), a synthetic retinoid, and genetic knockdown induced cellular-stress signaling such as the autophagy pathway and the unfolded protein response to maintain HSC self-renewal [170].
In addition, S1P-S1PR signaling is dysregulated in AML and S1PR3 has been identified as the key player in regulating TNFα-NF-κB signaling in both HSCs and LSCs [86]. Treatment of leukemic mice with the S1PR antagonist, FTY720, decreased leukemic burden and reduced LSC frequency [86, 171]. More importantly, an eight-gene SL signature was identified as significantly associated with overall survival of AML patients, thereby providing an avenue to identify and target LSCs in AML patients. Three of the genes in the SL signature were involved in S1P signaling (SPHK1, S1PR3, and S1PR5). In addition, SPHK1 inhibition in CD34+/CD38−/CD123+ leukemic stem and progenitor cells synergized with Ara-C treatment to potently decrease cell survival over single agents [74]. Collectively, these works showed that targeting S1P signaling in LSCs has therapeutic benefits. Additional studies on LSC-SL relationships are warranted, such as utilizing the eight-gene SL signature to inform studies combining SL therapeutics and LSC-targeting therapies.
3.5. Utility of dysregulated sphingolipid enzymes and metabolites as predictive biomarkers
Investigations into the landscape of SL alterations in AML have revealed a complexity that has yet to reach a consensus. In general, malignancy is associated with increased S1P at the expense of ceramides, which leads to a pro-survival phenotype. Indeed, we and others have demonstrated dysregulated expression of pro-survival SL enzymes in AML and that these changes predict poorer patient outcomes. AC expression and activity are elevated in AML and patients with high AC activity exhibit significantly lower overall survival [70]. IFN regulatory factor 8 (IRF8), a potent tumor suppressor and important factor of proper myeloid cell differentiation, also functions as a negative regulator of AC expression. IRF8 expression is often impaired in AML, which may contribute to elevated AC [172]. High mRNA expression of the alkaline ceramidase, ACER3, also correlated with poor overall survival [77]. This high ceramidase activity produces more substrate for SPHKs to generate S1P. Sobue et al. observed significantly elevated SPHK1 mRNA, but not SPHK2 mRNA, in acute leukemia patient samples [173]. Specific upregulation of SPHK1 in AML was confirmed by Powell et al., who described elevated SPHK1 mRNA and protein levels, but not SPHK2, in AML patient samples of various cytogenetic subgroups [74]. In contrast, Ghazaly et al. reported no alterations in SPHK1 or SPHK2 mRNA within AML specimens [174]. The contradictory results may be due to differences between sample types as Ghazaly et al. primarily utilized unenriched peripheral blood cells with variable blast percentages [174]. The studies describing high SPHK1 levels and activity in acute leukemias utilized bone marrow aspirates [173] or lineage-depleted HSCs [74]. In addition, Sobue et al. demonstrated that increased SPHK1 mRNA and activity from 16 leukemia cell lines strongly correlated with decreasing sensitivity to the AML chemotherapeutic, daunorubicin [175]. Thus, the majority of literature supports the hypothesis that elevated SPHK1 promotes survival in AML and represents a promising therapeutic target [80, 176, 177]. High expression of the S1P transporter, spinster homolog 2 (SPNS2 gene), also correlated with poorer event-free survival and overall survival [178]. This could suggest increased export of cellular S1P in AML. One study indicated lower plasma S1P levels in AML than normal controls [179], which highlights variations in SL content depending on the tissue type being measured (e.g., AML blasts vs plasma).
Primary AML cells exhibit reduced cellular ceramides [174]. This is supported by the study of FLT3-ITD positive AML specimens, as mentioned above [92]. This group reported reduced CERS1 mRNA with FLT3-ITD mutation [92], though this would not fully account for the global reduction in ceramides that was observed. Reduced mRNA expression for the ceramide-generating neutral SMase 2 (SMPD3 gene) may also be contributory [173]. Our group recently showed that AML with myelodysplastic syndrome-related changes (AML-MRC) preferentially metabolizes ceramides into pro-death SL metabolites compared to de novo AML [180]. Thus, single agent ceramide-based therapies seem better suited for AML-MRC while ceramide-based therapies may need to be used in combination with other AML treatments for de novo AML. In contrast to cellular ceramides, circulating ceramides were reported to be elevated in adult AML [179] and female pediatric AML cases [181], though one group reported a decrease in the ceramide ratios as defined as C18:1/C24:1 in adult AML [182].
Others have shown that sphingomyelins were predictors of AML in pediatric males [181]. In adult AML, several circulating sphingomyelin species were reportedly decreased [182], and another report showed lower serum levels of odd-chain sphingomyelins, but not others [183]. Moreover, acid sphingomyelinase-like phosphodiesterase 3b mRNA was upregulated in human AML samples, correlated to cytogenetic risk and karyotypes, and associated with poor overall survival [184].
Metabolism of ceramides to and from glycosphingolipids has also been investigated in AML. One group reported that CD34+ AML myeloblasts had significantly increased levels of the ganglioside, monosialodihexosylganglioside (GM3) [185]. This increase in GM3 was supported by a separate group showing increased expression of the glycosphingolipids: GM3, lactotriaosylceramide, neolactotetraosylceramide, and reduced globosides in AML specimens [186]. Lastly, circulating lactosylceramide may be diminished in pediatric AML [181].
Together, these studies have begun to define SL dysregulation in AML, although some inconsistencies remain. In general, SL profiling suggests decreased ceramides and increased glycosphingolipids and sphingomyelin in AML. The majority of studies found that ceramide-depleting (AC, ACER3) and S1P-generating (SPHK1) enzymes are elevated and correlated with poorer survival in de novo AML while few studies have described decreased SPHK1 levels. These discrepancies likely arise from the population sizes, detection methodologies, sample types, model systems, and the heterogeneity of AML. SL metabolism may be differentially regulated by specific chromosomal rearrangements [90] and specific driver mutations [94]. Increased understanding requires standardized and comprehensive SL measuring techniques to characterize different sample types (e.g., AML blasts vs plasma) and AML subtypes (e.g., mutational profile, cytogenic profile, age). The specific distribution of SL subspecies, their role in leukemic development, and their utility as diagnostic or prognostic biomarkers represent areas of future study and opportunity.
4. Role of sphingolipids in standard AML treatments
Standard AML 7+3 intensive induction chemotherapy has evolved to also include more specific approaches such as epigenetic-modulating regimens and Bcl-2, CD33, FLT3, and IDH-targeted therapies, as described above. SLs control many facets of cell signaling and are important signaling molecules that facilitate cell death following cancer therapy. Here, we summarize what is known regarding the relationship between SLs and AML treatments.
4.1. Induction chemotherapy
Induction chemotherapy, consisting of Ara-C and topoisomerase II inhibition with an anthracycline like daunorubicin, has been the mainstay treatment for newly diagnosed AML for decades. Ceramide generation is important for apoptosis induced by both Ara-C and daunorubicin. Ceramide is upregulated due to increased SMase activity following Ara-C treatment of U-937 cells. This increase in SMase activity was dependent on the formation of reactive oxygen species and the Src-family kinase p53/p56 Lyn. Mechanistically, Ara-C and daunorubicin induce oxidative stress, which activates p53/p56 Lyn and leads to the recruitment and activation of neutral SMase to sphingomyelin-enriched membrane rafts [83, 84].
As discussed previously, AML cells resistant to Ara-C or daunorubicin demonstrated significantly decreased levels of total pro-death ceramides as a consequence of elevated activities of the ceramide catabolizing enzymes AC, GCS, SMS, and SPHK1 [85, 143]. Increases in these enzymes dampen the cytotoxic ceramide induction following SMase activation and contribute to drug resistance. As previously mentioned, increased SPHK1 mRNA and activity correlated strongly with decreased daunorubicin sensitivity in 16 leukemia cell lines. Pharmacological and genetic inhibition of SPHK1 increased sensitivity of K-562 cells to daunorubicin [175]. Interestingly, resistance to Ara-C and daunorubicin did not significantly alter the toxicity of multiple SL enzyme targeting agents (AC, GCS, and SPHK inhibitors), which suggests that these agents may be used to combat drug resistance following induction chemotherapy [85]. Taken altogether, these studies highlight the dichotomy between ceramide-generating processes and mitogenic S1P signaling in the context of AML induction chemotherapy.
4.2. Bcl-2 family-targeted therapeutics
Bcl-2 family proteins are key survival mediators and important therapeutic targets in AML. Several studies have linked SLs and Bcl-2 family proteins. As mentioned previously, AC and SPHK1 inhibition as well as ceramide induction negatively regulate Mcl-1 levels [70, 74]. Moreover, Bcl-2 and Bcl-xL directly inhibit the formation of “water-filled” ceramide pores within the mitochondria [187]. Treating leukemia cell lines U-937, K-562, and MV4–11 with navitoclax, a pan inhibitor targeting Bcl-2, Bcl-xL, and Bcl-w, resulted in cell death and the accumulation of pro-apoptotic C16-ceramide. Intriguingly, knockout of the pore-forming protein BAK prevented ceramide accumulation following navitoclax treatment [188]. SL metabolism also interfaces with the pore-forming proteins, BAK and BAX, to regulate apoptosis. BAK, but not BAX, is required for CERS-dependent long-chain ceramide generation during chemotherapy-induced apoptosis [189]. Hexadecenal is generated from the breakdown of S1P by S1P lyase, and BAK- and BAX-induced mitochondrial outer membrane permeabilization requires a specific lipid composition involving S1P and hexadecenal, respectively [190]. These studies collectively demonstrate key interactions between SL and apoptosis regulation by the Bcl-2 family.
4.3. FLT3-targeted therapy
Twenty-five percent of AML cases harbor internal tandem duplication in the juxtamembrane domain of the FLT3 receptor tyrosine kinase. However, its targeting has yielded limited clinical success due to acquired drug resistance [91]. Dany et al. provided evidence that the FLT3-ITD mutation suppresses ceramide levels and significantly downregulates CERS1 mRNA in CD34+ bone marrow from FLT3-mutated AML [92]. CERS1 protein levels were also reduced in TF-1 cells overexpressing FLT3-ITD. Knockdown of FLT3 in two human AML cell lines (MV4–11 and MOLM-14) harboring the FLT3-ITD mutation significantly rescued CERS1 levels. Pharmacological inhibition of FLT3 with the first-generation FLT3 inhibitor, sorafenib, and the second-generation FLT3 inhibitors, AC220 and crenolanib, significantly increased CERS1 mRNA, CERS1 protein, and C18-ceramide levels. Knockdown of CERS1 protected cells from crenolanib while reconstitution of wild-type CERS1, but not a catalytically inactive CERS1 mutant, sensitized cells to crenolanib [92]. Further studies are warranted to characterize the role of CERS and other SL enzymes in AML with additional molecular drivers.
4.4. Chemotherapy-free AML treatments
Acute promyelocytic leukemia (APL) accounts for 5–10% of AML cases [15] and is characterized by the t(15;17) translocation, which encodes the PML-RARα fusion oncogene. Chemotherapy-free treatment with all-trans-retinoic acid (ATRA) and arsenic trioxide (ATO) result in APL cure rates upwards of 90% [15]. ATRA and ATO increase endogenous ceramides primarily through enhanced activation of acid SMase activity and CERS activity, respectively [191, 192]. ATRA and ATO treatment also significantly decreased GCS activity.
To summarize, preclinical studies suggest that the efficacy of intensive chemotherapy, molecularly-targeted AML therapies, and differentiating agents are all influenced by SL metabolism. Specifically, ceramide accumulation is important for the cytotoxic effects of these treatments, and mechanisms that deplete ceramide promote drug resistance. An intimate understanding of these relationships is necessary to improve therapy responses and prevent drug resistance.
5. Therapeutic agents targeting sphingolipid metabolism
The importance of ceramide and ceramide-metabolizing enzymes in cell survival and drug resistance, as described above, justifies substantial effort to develop small-molecule inhibitors targeting SL enzymes. Below, we summarize the current state of SL enzyme-targeting drugs in AML, and Table 1 provides a representative list of those in preclinical development. Recent studies have identified FDA-approved drugs (tamoxifen [124, 193–195], desipramine [196], dacarbazine [197], and fluphenazine [198]) that have off-target effects on SL metabolism. A representative list of clinically-relevant SL-targeting agents is summarized in Table 2 and mapped onto the SL metabolic pathway in Figure 3. The following subsections focus on drugs that directly target SL metabolism. Please refer to these excellent articles for a comprehensive review of SL-targeting agents: [87, 199].
Table 1.
Summary of sphingolipid-targeting agents in preclinical development for AML. Compounds listed were used as anti-neoplastic agents or tool compounds in AML.
| Target(s) | Sphingolipid Drug | References |
|---|---|---|
| Ceramide analogs and ceramide inducing compounds | ||
| Acid ceramidase | LCL-204 | [70] |
| Acid ceramidase | SACLAC | [82, 151] |
| Glucosylceramide synthase | PDMP | [150, 151, 208, 222] |
| Glucosylceramide synthase | PPMP | [141, 208] |
| Mitochondria | Pyridinium-ceramide (LCL-461) | [92] |
| Antagonists of S1P synthesis and signaling | ||
| Sphingosine kinase 1 Akt | A-674563 | [107] |
| Sphingosine kinase 1 | SK1-I | [106, 151] |
| Sphingosine kinase 1 & 2 | MP-A08 | [74] |
| Sphingosine kinase 1 & 2 | SKI-II | [200, 222] |
| Sphingosine kinase 1 & 2 | SKI-178 | [72, 73] |
| Sphingosine kinase 1 & 2 | SKI-349 | [201] |
| S1PR 2 & 4 DES1 Sphingosine kinase 1 & 2 | JTE-013 | [74, 108] |
Table 2.
Summary of clinically relevant sphingolipid-modulating drugs.
| Target(s) | Sphingolipid Drug | Developmental Stage | References |
|---|---|---|---|
| Ceramide analogs and ceramide inducing compounds | |||
| Acid ceramidase | Dacarbazine | Approved to treat melanoma and Hodgkin’s lymphoma | [197] |
| Acid ceramidase | Desipramine | Approved to treat depression | [196] |
| Acid sphingomyelinase | Fluphenazine | Approved to treat schizophrenia | [198] |
| Dihydroceramide desaturase 1 | Fenretinide (4-HPR) | Phase I clinical trials | [211–215, 225, 226] |
| Estrogen receptor Acid ceramidase Glucosylceramide synthase | Tamoxifen | Approved for hormone receptor+ breast cancer | [193–195] |
| Exogenous ceramide | C6 ceramide nanoliposome | Phase I clinical trials | [151, 180, 209] |
| Glucosylceramide synthase | Eliglustat | Approved for Gaucher disease | [49, 85, 227] |
| Antagonists of S1P Synthesis and Signaling | |||
| Protein kinase C Sphingosine kinase 1 & 2 | Safingol | Phase I clinical trials | [87, 203] |
| Sphingosine kinase 2 Dihydroceramide desaturase 1 | ABC294640 | Phase I/II clinical trials | [88, 202, 224] |
| S1PR modulator Sphingosine kinase 1 S1P lyase | Fingolimod (FTY720) | Approved to treat multiple sclerosis Phase I clinical trials | [118, 119, 218, 219] |
Figure 3. Dysregulated sphingolipid metabolism in AML and potential therapeutic intervention.
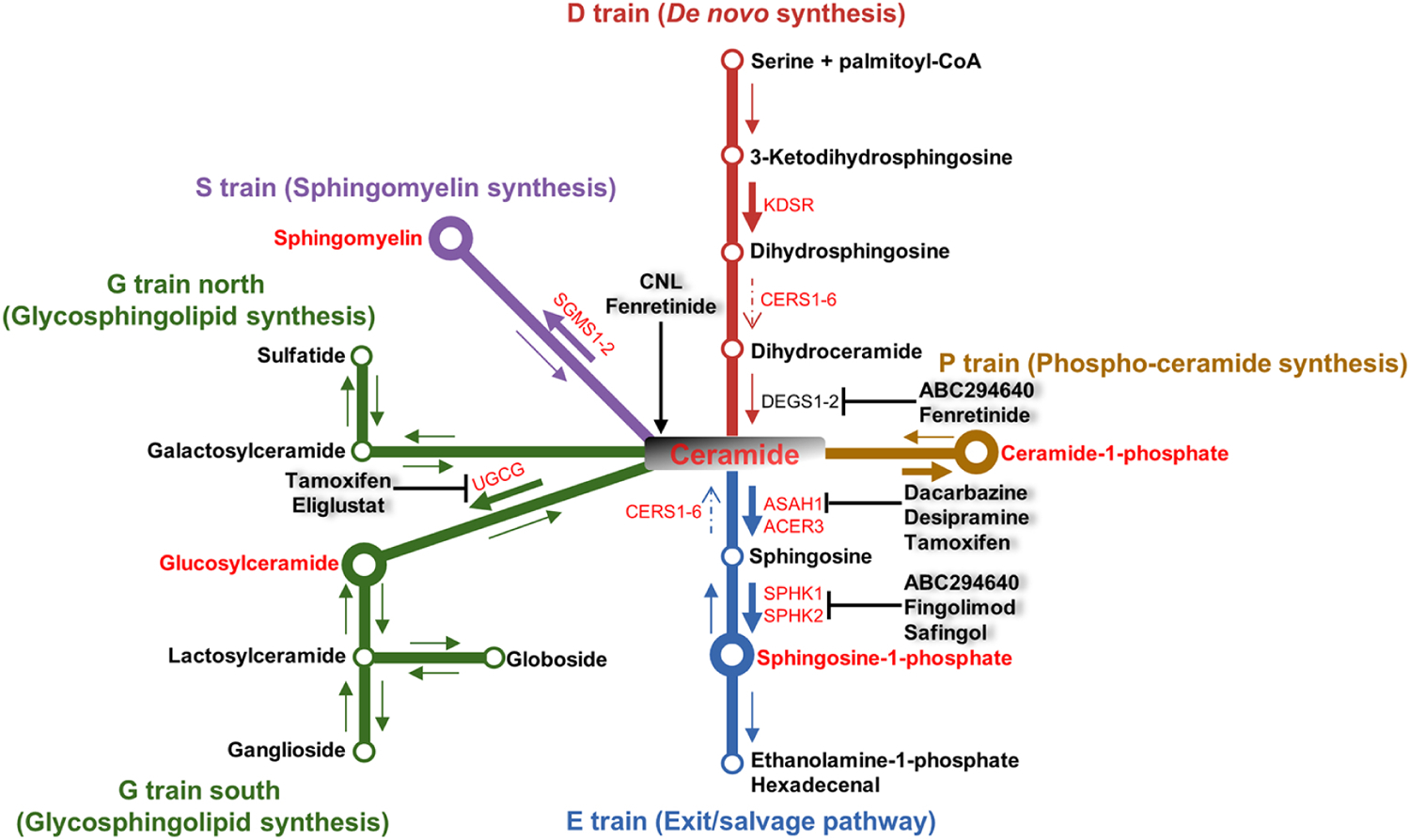
Refer to Figure 2 for a detailed summary of dysregulated sphingolipid metabolism in AML and Table 2 for a representative list of sphingolipid-modulating drugs that are FDA-approved or in clinical trials. In summary, multiple mechanisms contribute to ceramide depletion and S1P accumulation in AML, which are targeted by the indicated therapeutics. Dysregulated sphingolipid metabolites and enzymes are labeled in red. A larger node indicates upregulation of a particular metabolite. A large arrow indicates increased flux while a dashed arrow indicates decreased flux through a pathway. Clinically relevant compounds and their targets are labeled.
5.1. Sphingosine kinase (SPHK) inhibitors
Ceramide breakdown generates the substrate for S1P synthesis by the sphingosine kinases SPHK1 and SPHK2. SPHKs are attractive targets and several inhibitors targeting one or both isoforms have been evaluated in AML [75, 177]. The SPHK1 inhibitor, SK1-I, induced caspase-mediated apoptosis in U-937 cells, which is accompanied by decreased S1P and increased ceramides. Furthermore, SK1-I decreased phospho-ERK1/2 and phospho-Akt signaling and suppressed tumor growth in a U-937 xenograft model [106]. Corroborative findings were reported for the dual SPHK1 and SPHK2 inhibitors SKI-II [200], SKI-178 [72], and SKI-349 [201] in additional AML cell lines. Interestingly, SKI-178 and SKI-349 also target microtubule polymerization, which contributes to their cytotoxic effects [72, 201]. SKI-II was more potent at reducing cell viability and inducing apoptosis than SK1-I in vitro, which may indicate beneficial effects of targeting both SPHK isoforms [200]. An additional pan SPHK inhibitor, MP-A08, promoted caspase-dependent apoptosis in AML cell lines and patient samples. As discussed previously, MP-A08 treatment also induced pro-apoptotic Bcl-2 family proteins while depleting Mcl-1 [74]. These SPHK inhibitors have yet to progress to cancer-related clinical trials due to poor potency and a lack of enzyme specificity [176]. Two S1P-modulating therapies, ABC294640 and safingol, have progressed to clinical trials in advanced solid tumors [202–204]. It remains to be seen if these agents exhibit efficacy in AML.
5.2. Acid ceramidase (AC) inhibitors
We demonstrated that AC is elevated in AML and promotes a pro-survival state by depleting endogenous ceramides, promoting S1P synthesis, and upregulating Mcl-1 [79]. Several small-molecule inhibitors of AC are being explored in AML. We first characterized the effects of LCL-204, which elicited potent caspase-dependent cell death in multiple AML cell lines, patient samples, and a syngeneic mouse model. AC and Mcl-1 levels both decreased following LCL-204 treatment. The ceramide analog SACLAC is an irreversible inhibitor that forms a covalent adduct with Cys143 in the catalytic site of AC [70, 205]. SACLAC treatment also suppressed S1P, upregulated ceramides, led to caspase-dependent apoptosis in AML cells, and reduced leukemic burden in a human AML cell line xenograft model. SACLAC treatment induced alternative splicing of the anti-apoptotic Mcl-1 transcript via ceramide-mediated modulation of the levels of RNA splicing factor SF3B1. Alternative splicing of Mcl-1 resulted in the formation of the Mcl-1-short (Mcl-1S) isoform [82]. Intriguingly, while the full-length Mcl-1 is an anti-apoptotic protein capable of binding and inhibiting pore-forming Bcl-2 family proteins, Mcl-1S lacks the BH1 and BH2 domains, which are necessary to heterodimerize and inhibit pro-apoptotic proteins. Consequently, Mcl-1S is a pro-apoptotic BH3-only protein capable of binding full-length Mcl-1 exclusively [206]. Translation of these AC inhibitors to the clinic is precluded by their micromolar potency as well as poor solubility, pharmacokinetics, and bioavailability, which are likely attributed to their lipophilic nature.
5.3. Glucosylceramide synthase (GCS) inhibitors
The formation of glycosphingolipids promotes chemotherapeutic resistance in cancer [43, 147, 207]. Thus, GCS inhibitors may provide therapeutic benefit since drug resistance is a major factor in treatment failure and relapse in AML. To that end, Plo et al. treated leukemia cell lines with acquired daunorubicin resistance and high levels of the multidrug resistance protein, MDR1, with the GCS inhibitor 1-phenyl-2-decanoylamino-3-morpholino-1-propanolol (PDMP). Treatment improved daunorubicin retention and resensitized cells to daunorubicin-induced cell death while depleting glucosylceramides and other glycosylated SL species [150]. While several studies have characterized GCS in the context of drug resistance in cancer, few have identified the effects of GCS inhibition in drug-sensitive cells. Surprisingly, GCS inhibition with PDMP or 1-phenyl-2-palmitoylamino-3-morpholino-1-propanol (PPMP), in the drug-sensitive leukemic cell lines U-937 and HL-60 protected cells from daunorubicin-induced apoptosis. These protective effects were attributed to the induction of galactosylceramides instead of pro-apoptotic ceramides [208]. These divergent findings highlight the complexity of targeting SL metabolism in cancer. Since GCS inhibition sensitizes drug-resistant cells to chemotherapy while protecting drug-sensitive cells, glucosylceramide inhibition may be best utilized in relapsed/refractory AML. Of note, eliglustat is a potent GCS inhibitor in clinic for Gaucher disease as an effective lipid-reducing therapy. The therapeutic potential of eliglustat has not been extensively evaluated in AML, with one report citing its effectiveness in reducing cell viability in chemotherapy-sensitive and resistant HL-60 cells [85].
5.4. Ceramide nanoliposomes
Ceramides are canonically considered the center of SL signaling and are the most characterized SLs for their anti-cancer properties (Fig. 1). Therapies based on direct administration of exogenous ceramides are being evaluated in a variety of cancers including AML [209], but this approach is limited by poor delivery and solubility. The development of short-chain C6-ceramide nanoliposomes (CNL) was a key enabling technology to overcome delivery challenges of lipid-based drugs [209]. Our group compared the effects CNL as a single agent in de novo AML (DN-AML) versus AML with myelodysplastic-related changes (AML-MRC) [180]. As mentioned above, CNL preferentially exerts pro-apoptotic effects against AML-MRC over DN-AML patient samples. The colony-forming potential of AML-MRC patient samples was particularly sensitive to CNL compared to DN-AML. These findings were corroborated using transgenic mouse models representing AML-MRC versus DN-AML subtypes. The differential sensitivity to CNL between subtypes was attributed to their differences in SL metabolism following CNL treatment. AML patient samples with MDS-related mutations showed elevated pro-death SLs and decreased S1P compared to DN-AML samples, which showed increased pro-survival metabolites such as S1P and C6-hexosylceramides upon CNL treatment [180]. These results exemplify the need to characterize the uniformity of therapeutic responses to SL targeting agents across the heterogenous subtypes of AML. CNL demonstrated modest single agent effects on leukemic burden in immunodeficient mice engrafted with primary human AML cells [210]. Remarkably, CNL exhibited strong synergistic potency and “cures” in approximately 66% of treated leukemic mice when combined with the microtubule targeting agent vinblastine [180]. Our data suggest that this synergy is due, in part, to lysosomal disruption induced by vinblastine and resultant ceramide-induced autophagic cell death.
CNL has recently completed a phase I clinical trial for patients with advanced solid tumors (NCT02834611), exceeding all pharmacokinetic endpoints and showing evidence of efficacy (as defined by stable disease) in nearly 40% of the patients treated (manuscript in preparation). CNL is scheduled to enter a clinical trial for relapsed/refractory AML in combination with LDAC/venetoclax (NCT04716452).
5.5. “Ceramide generator” fenretinide
Fenretinide (4-HPR) is a synthetic retinoid being evaluated in several cancer types including AML. Treating HL-60 AML cells with 4-HPR induced apoptosis and ceramide accumulation. CERS inhibition suppressed ceramide synthesis and attenuated 4-HPR-induced apoptosis [211]. Jiang et al. confirmed these findings in two additional cell lines (NB-4 and U-937) and showed that vitamin C abrogated the apoptotic effects of 4-HPR, suggesting that oxidative stress is important for its anti-proliferative effects [212]. Our group confirmed that de novo ceramide synthesis and oxidative stress are necessary and demonstrated that sphingomyelin-derived ceramides contribute to 4-HPR cytotoxic effects [213]. Importantly, 4-HPR preferentially killed leukemic stem cells over normal stem cells, which was attributed to the induction of oxidative stress and downregulation of NF-κB and Wnt signaling [214]. Zhao et al. showed that AML blasts harboring FLT3 mutations were significantly more sensitive to 4-HPR than FLT3 wild-type AML. These findings were also confirmed using FLT3-mutant AML cell lines [215]. Interestingly, 4-HPR was also reported to inhibit DES1, which inserts a 4,5-trans double bond to the sphingoid backbone of dihydroceramides to generate ceramides [216]. These findings are consistent with 4-HPR treatment-induced ceramide accumulation via CERS and SMase activation, independent of de novo ceramide synthesis. Nanoformulations of 4-HPR are currently in clinical trials for relapsed/refractory T-cell non-Hodgkin lymphoma (NCT04234048) and represent intriguing options for development in AML.
5.6. Fingolimod
Fingolimod (FTY720) is an immunosuppressive drug that is FDA-approved for refractory multiple sclerosis. FTY720 antagonizes the sphingosine-1-phosphate receptor, S1PR1, to sequester T cells within lymph nodes [217]. At higher concentrations, FTY720 also has promising anti-cancer effects attributed to its ability to target and downregulate S1PR1 and SPHK1 [218]. FTY720 increases endogenous ceramide levels, decreases S1P levels, and elicits caspase-dependent apoptosis in Kasumi-1 AML cells, which was partially attributed to PP2A activation [118]. In addition, FTY720 activates PP2A directly via SET antagonism. A detailed mechanistic study of FTY720 in AML found that cytotoxicity was not mediated primarily by caspase activity, necroptosis, ferroptosis, ROS, or autophagy, but rather a form of non-canonical phosphatidylserine exposure that is PP2A dependent and requires cellular vacuolization [219]. It is likely that the therapeutic effects of FTY720 vary depending on the cellular SL metabolic state and are mediated by the combination of ceramide induction, SPHK1 antagonism, and direct SET interaction.
5.7. Novel sphingolipid drug combinations
SLs regulate many facets of cell survival. By defining these functions, synergistic combinatorial therapies can be rationally designed to target specific aspects of SL metabolism in conjunction with other anti-cancer agents. Our group demonstrated that exogenous C6-ceramide in combination with tamoxifen elicited synergistic killing in several AML cell lines that was independent of tamoxifen’s anti-estrogenic function [76]. The efficacy was associated with elevated reactive oxygen species, inhibited complex I respiration, and reduced mitochondrial membrane potential [76]. These effects may also be attributed to tamoxifen’s off-target inhibition of GCS, AC, and SPHK1 [124, 193, 195]. The combination of ceramide and tamoxifen also downregulated the inhibitors of apoptosis proteins survivin and XIAP [76]. Exogenous C6 ceramide and tamoxifen-driven cell death also coincided with upregulated autophagy in AML cell lines [124]. In addition, the combination of nanoliposomal safingol and CNL synergistically killed AML cell lines and patient samples [220]. As described above, we also showed respiratory complex I was impaired in drug-resistant AML and that further ablation of complex I function with phenformin, in combination with ceramide-promoting regimens (CNL treatment or GCS/AC/SPHK1 inhibition), resulted in impressive synergistic killing in multiple drug-sensitive and drug-resistant AML cell lines [151]. Moreover, the combination of CNL and vinblastine demonstrated potent synergy in in vitro and in vivo models of AML. Specifically, the combination synergistically killed primary AML patient samples and dramatically reduced leukemic burden and improved overall survival in multiple mouse models of AML [180]. Efforts are focused on improving CNL activity in combination with Ara-C, low dose liposomal vinblastine, and other agents including an aldehyde dehydrogenase inhibitor developed by our group [221].
Bcl-2 family proteins are important therapeutic targets due to their direct regulation of cellular apoptosis. Combinatorial studies with SL-modulating agents and Bcl-2 family inhibitors show promise in leukemia. Casson et al. demonstrated that inhibiting ceramide detoxification with the GCS inhibitor, PDMP, or the dual SPHK1 and SPHK2 inhibitor, SKI-II in combination with navitoclax resulted in increased ceramides and synergistic killing of U-937 and K-562 leukemia cell lines. Unexpectedly, the combination of PDMP and navitoclax also increased S1P level, which highlights the complexity of SL responses following treatment with anti-cancer therapeutics [222]. Synergistic effects were also observed between SPHK inhibition with MP-A08 and navitoclax treatment in MV4–11 AML cells [74]. Unfortunately, on-target thrombocytopenia induced by navitoclax precludes its use in AML [223]. Thus, studies exploring the therapeutic efficacy of SL-targeting agents in combination with the Bcl-2 inhibitor venetoclax are necessary, but none have been published to date in AML. However, this novel combination has been explored in other leukemias. For instance, the SPHK2 inhibitor, ABC294640, synergizes with venetoclax to reduce cell viability and increase apoptosis in myeloma cell line models. The combined treatment downregulated Mcl-1 and Bcl-xL, which likely contributed to the observed synergy [224]. Ongoing studies within our group are exploring the synergistic potential of CNL or AC inhibitors in combination with venetoclax-containing regimens.
6. Conclusions and future considerations
AML is a highly heterogeneous malignant disorder that continues to require urgent advances in therapeutic strategy and efficacy. In younger patients with newly diagnosed, de novo AML, the 5-year survival rate is only 50%, a discouraging statistic that is nonetheless far superior to the dismal 5-year survival rates seen in older patients or following relapse. Novel approaches based on immunogenic properties, mutated proteins, apoptotic signaling, and epigenetic dysregulation are raising the bar for “bench-to-bedside” AML therapy. However, the rise of relapse cases, the large population of patients unable to tolerate intensive induction chemotherapy, and the poor survival outcomes for the vulnerable older AML patient population collectively dictate that other avenues should be explored and exploited. SLs have emerged as key regulators of numerous aspects of AML biology. Many questions on the precise roles of SLs will need to be addressed to fully exploit the potential of SL modifying drugs to treat AML. Nonetheless, targeting SL dysregulation induces cell death mechanisms and reduces leukemic burden in human AML cell lines and murine models. SL inhibitors in conjunction with AML therapeutic agents have also shown considerable combinatorial synergy in preclinical studies.
Considering the impressive response to the combination of CNL with vinblastine in preclinical in vivo studies, current clinical studies offering CNL alone and in various combinations for relapsed and refractory AML patients are awaited with great interest. It will be of importance to determine whether AML patients exhibit similar promising therapeutic responses to combined cytotoxic and CNL therapy seen in animal models. Of additional importance will be the assessment of relative sensitivity of AML phenotypic and molecular subsets to SL directed therapeutics, including in combination with emerging AML therapeutic strategies including Bcl-2 inhibition, targeting mutant epigenetic/signaling effectors, and modalities which target cell surface molecules in AML. In addition, CNL and other SL directed agents should be studied in relapsed and refractory AML to define dose and toxicity of these agents. Subsequently, their use in combinations in newly diagnosed, poor prognosis patient subsets will also be of great interest. Several FDA-approved drugs have off-target SL modulating effects (tamoxifen, dacarbazine, FTY720) that may allow for repurposing to treat AML. Unfortunately, better SL therapeutic agents are needed to enable widespread clinical utilization. Some of the issues facing the current SL drugs include poor bioavailability, potential toxicity, high micromolar dosing, and inefficient delivery due to lipophilic properties. Further drug development will help resolve these issues to allow combinatorial studies to move forward. Supplementing current AML clinical treatments with SL-centric therapeutics presents a promising opportunity to meet the current challenges of treating AML.
7. Practice points
Preclinical studies demonstrate synergistic efficacies when combining AML treatments with sphingolipid therapeutics.
Sphingolipids and their regulatory enzymes are dysregulated in AML, and the use of sphingolipids as biomarkers is currently under investigation and shows promise.
In vitro studies suggest that ceramide induction is necessary for chemotherapeutic effectiveness.
Enzymes that decrease ceramides and/or increase S1P, glycosphingolipids, and sphingomyelins are frequently upregulated in multidrug-resistant AML.
Sphingolipids modulate Mcl-1 levels in vitro, which should be exploited to improve current venetoclax standard-of-care regimens.
8. Research agenda
Define additional relationships between common AML mutations and sphingolipid metabolism.
Identify AML subtypes with differential sphingolipid metabolism, profiles, flux, and/or enzyme dependencies.
Determine whether AML subtypes, as defined by sphingolipid profiles, exhibit differential sensitivity to AML therapeutics and/or sphingolipid targeting agents.
Demonstrate the utility of sphingolipid enzymes and metabolites as predictive or therapeutic biomarkers in AML.
Define the subcellular localization and flux of sphingolipid metabolites with mass spec-based whole-cell and organelle lipidomics.
Uncover contributions to AML pathogenicity of less-studied sphingolipid biology such as ceramide-1-phosphate, dihydrosphingolipids, sphingoid backbone chain lengths, and fatty acyl chain lengths.
Advance sphingolipid drug development and utilize nanotechnology to improve drug specificity, potency, and solubility.
Evaluate and predict the efficacy of combinatorial drug studies in molecularly defined AML patient samples using bioinformatics and systems biology approaches.
Acknowledgements
We apologize to any investigators whose important work was not included due to space limitations. The authors gratefully acknowledge the collaborative support and discussions of all members of the projects and cores within our funded Program Project (P01) “Targeted Sphingolipid Metabolism for Treatment of AML” (P01CA171983). Figure 2 was created with BioRender.com.
Funding support
This work was supported by the National Institutes of Health under the National Cancer Institute Award Number P01CA171983 (to TPL and MK), under the National Cancer Institute Research Training Award Number 5T32CA009109-45 (to JU), and under the National Cancer Institute Individual Predoctoral to Postdoctoral Fellow Transition Award Number 5K00CA245802 (to PCP). DC is supported by National Cancer Institute Award Number U10CA180820. Studies supported by MSK core facilities were supported in part by MSKCC Support Grant/Core Grant P30 CA008748 and the Marie-Josée and Henry R. Kravis Center for Molecular Oncology. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflicts of interest
Ceramide nanoliposomal technology (CNL) has been licensed by Penn State Research Foundation to Keystone Nano, Inc. (PA, USA) and MK is Chief Technical Officer and co-founder of Keystone Nano and TPL is a member of the Scientific Advisory Board of Keystone Nano. MK has co-founded other technology companies (Oraceu, TX, PA, USA; AgroSpheres Inc., VA, USA; Transfoam, Inc., VA, USA) that do not have access to any Kester sphingolipid-based technologies. TPL is on the Scientific Advisory Board and has stock options for Bioniz Therapeutics and Dren Bio, Inc. TPL and DJF received honoraria from Kymera Therapeutics. DJF has research funding from AstraZeneca. DC is participating in clinical studies of leukemia therapeutics funded by Aptose Biosciences, Astellas Pharma, AbbVie, Daiichi-Sankyo. RLL is on the supervisory board of Qiagen and is a scientific advisor to Imago, Mission Bio, BAKX Therapeutics, Zentalis Pharmaceuticals, Ajax, Auron Therapeutics, Prelude Therapeutics, C4 Therapeutics and IsoPlexis. RLL has received research support from AbbVie, Constellation, Ajax, Zentalis Pharmaceuticals, and Prelude Therapeutics. RLL has received research support from and consulted for Celgene and Roche and has consulted for Syndax Pharmaceuticals, Incyte Corporation, Janssen Pharmaceuticals, Astellas Pharma, MorphoSys AG, and Novartis. RLL has received honoraria from AstraZeneca and Novartis for invited lectures and from Gilead Sciences and Novartis for grant reviews. There are no conflicts of interest with the work presented in this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].National Cancer Institute. Cancer Stat Facts: Leukemia-Acute Myeloid Leukemia (AML).
- [2].De Kouchkovsky I, Abdul-Hay M. ‘Acute myeloid leukemia: a comprehensive review and 2016 update’. Blood Cancer J. 2016;6:e441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Shah A, Andersson TM, Rachet B, Bjorkholm M, Lambert PC. Survival and cure of acute myeloid leukaemia in England, 1971–2006: a population-based study. Br J Haematol. 2013;162:509–16. [DOI] [PubMed] [Google Scholar]
- [4].Dohner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Buchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129:424–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Heuser M, Freeman SD, Ossenkoppele GJ, Buccisano F, Hourigan CS, Ngai LL, et al. 2021 Update Measurable Residual Disease in Acute Myeloid Leukemia: European LeukemiaNet Working Party Consensus Document. Blood. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Schuurhuis GJ, Heuser M, Freeman S, Bene MC, Buccisano F, Cloos J, et al. Minimal/measurable residual disease in AML: a consensus document from the European LeukemiaNet MRD Working Party. Blood. 2018;131:1275–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Costa AFO, Menezes DL, Pinheiro LHS, Sandes AF, Nunes MAP, Lyra Junior DP, et al. Role of new Immunophenotypic Markers on Prognostic and Overall Survival of Acute Myeloid Leukemia: a Systematic Review and Meta-Analysis. Sci Rep. 2017;7:4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. The New England journal of medicine. 2012;366:1079–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Figueroa ME, Lugthart S, Li Y, Erpelinck-Verschueren C, Deng X, Christos PJ, et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell. 2010;17:13–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Li S, Garrett-Bakelman FE, Chung SS, Sanders MA, Hricik T, Rapaport F, et al. Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nat Med. 2016;22:792–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Mosquera Orgueira A, Peleteiro Raindo A, Cid Lopez M, Diaz Arias JA, Gonzalez Perez MS, Antelo Rodriguez B, et al. Personalized Survival Prediction of Patients With Acute Myeloblastic Leukemia Using Gene Expression Profiling. Front Oncol. 2021;11:657191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bullinger L, Dohner K, Bair E, Frohling S, Schlenk RF, Tibshirani R, et al. Use of gene-expression profiling to identify prognostic subclasses in adult acute myeloid leukemia. The New England journal of medicine. 2004;350:1605–16. [DOI] [PubMed] [Google Scholar]
- [13].Verhaak RG, Wouters BJ, Erpelinck CA, Abbas S, Beverloo HB, Lugthart S, et al. Prediction of molecular subtypes in acute myeloid leukemia based on gene expression profiling. Haematologica. 2009;94:131–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bullinger L, Valk PJ. Gene expression profiling in acute myeloid leukemia. J Clin Oncol. 2005;23:6296–305. [DOI] [PubMed] [Google Scholar]
- [15].Kantarjian H, Kadia T, DiNardo C, Daver N, Borthakur G, Jabbour E, et al. Acute myeloid leukemia: current progress and future directions. Blood Cancer J. 2021;11:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. The New England journal of medicine. 2016;374:2209–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Tyner JW, Tognon CE, Bottomly D, Wilmot B, Kurtz SE, Savage SL, et al. Functional genomic landscape of acute myeloid leukaemia. Nature. 2018;562:526–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Cancer Genome Atlas Research N, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. The New England journal of medicine. 2013;368:2059–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19]. https://leucegene.ca/
- [20].Giacopelli B, Wang M, Cleary A, Wu YZ, Schultz AR, Schmutz M, et al. DNA methylation epitypes highlight underlying developmental and disease pathways in acute myeloid leukemia. Genome Res. 2021;31:747–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Dombret H, Itzykson R. How and when to decide between epigenetic therapy and chemotherapy in patients with AML. Hematology Am Soc Hematol Educ Program. 2017;2017:45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Dohner H, Weisdorf DJ, Bloomfield CD. Acute Myeloid Leukemia. The New England journal of medicine. 2015;373:1136–52. [DOI] [PubMed] [Google Scholar]
- [23].Dombret H, Seymour JF, Butrym A, Wierzbowska A, Selleslag D, Jang JH, et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood. 2015;126:291–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sekeres MA, Guyatt G, Abel G, Alibhai S, Altman JK, Buckstein R, et al. American Society of Hematology 2020 guidelines for treating newly diagnosed acute myeloid leukemia in older adults. Blood Adv. 2020;4:3528–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Dohner H, Lubbert M, Fiedler W, Fouillard L, Haaland A, Brandwein JM, et al. Randomized, phase 2 trial of low-dose cytarabine with or without volasertib in AML patients not suitable for induction therapy. Blood. 2014;124:1426–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Schlenk RF, Jaramillo S, Muller-Tidow C. What’s new in consolidation therapy in AML? Semin Hematol. 2019;56:96–101. [DOI] [PubMed] [Google Scholar]
- [27].Dombret H, Gardin C. An update of current treatments for adult acute myeloid leukemia. Blood. 2016;127:53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Schwarer AP, Butler J, Jackson K, Beligaswatte A, Martin L, Kennedy G, et al. A Comparison of High-Dose Cytarabine During Induction Versus Consolidation Therapy in Newly Diagnosed AML. Hemasphere. 2018;2:e158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Li W, Gong X, Sun M, Zhao X, Gong B, Wei H, et al. High-dose cytarabine in acute myeloid leukemia treatment: a systematic review and meta-analysis. PLoS One. 2014;9:e110153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Recher C, Rollig C, Berard E, Bertoli S, Dumas PY, Tavitian S, et al. Long-term survival after intensive chemotherapy or hypomethylating agents in AML patients aged 70 years and older: a large patient data set study from European registries. Leukemia. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Amadori S, Suciu S, Selleslag D, Aversa F, Gaidano G, Musso M, et al. Gemtuzumab Ozogamicin Versus Best Supportive Care in Older Patients With Newly Diagnosed Acute Myeloid Leukemia Unsuitable for Intensive Chemotherapy: Results of the Randomized Phase III EORTC-GIMEMA AML-19 Trial. J Clin Oncol. 2016;34:972–9. [DOI] [PubMed] [Google Scholar]
- [32].DiNardo CD, Pratz K, Pullarkat V, Jonas BA, Arellano M, Becker PS, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. 2019;133:7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wei AH, Strickland SA, Hou JZ, Fiedler W, Lin TL, Walter RB, et al. Venetoclax combined with low-dose cytarabine for previously untreated patients with acute myeloid leukemia: Results from a phase Ib/II study. Journal of Clinical Oncology. 2019;37:1277–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wei AH, Montesinos P, Ivanov V, DiNardo CD, Novak J, Laribi K, et al. Venetoclax plus LDAC for newly diagnosed AML ineligible for intensive chemotherapy: a phase 3 randomized placebo-controlled trial. Blood. 2020;135:2137–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Cortes JE, Heidel FH, Hellmann A, Fiedler W, Smith BD, Robak T, et al. Randomized comparison of low dose cytarabine with or without glasdegib in patients with newly diagnosed acute myeloid leukemia or high-risk myelodysplastic syndrome. Leukemia. 2019;33:379–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Savona MR, Pollyea DA, Stock W, Oehler VG, Schroeder MA, Lancet J, et al. Phase Ib Study of Glasdegib, a Hedgehog Pathway Inhibitor, in Combination with Standard Chemotherapy in Patients with AML or High-Risk MDS. Clin Cancer Res. 2018;24:2294–303. [DOI] [PubMed] [Google Scholar]
- [37].Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. The New England journal of medicine. 2017;377:454–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Perl AE, Martinelli G, Cortes JE, Neubauer A, Berman E, Paolini S, et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. The New England journal of medicine. 2019;381:1728–40. [DOI] [PubMed] [Google Scholar]
- [39].DiNardo CD, Stein EM, de Botton S, Roboz GJ, Altman JK, Mims AS, et al. Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. The New England journal of medicine. 2018;378:2386–98. [DOI] [PubMed] [Google Scholar]
- [40].Stein EM, DiNardo CD, Pollyea DA, Fathi AT, Roboz GJ, Altman JK, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130:722–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Thol F, Ganser A. Treatment of Relapsed Acute Myeloid Leukemia. Curr Treat Options Oncol. 2020;21:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Walter RB, Estey EH. Management of older or unfit patients with acute myeloid leukemia. Leukemia. 2015;29:770–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ogretmen B. Sphingolipid metabolism in cancer signalling and therapy. Nat Rev Cancer. 2018;18:33–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Patwardhan GA, Beverly LJ, Siskind LJ. Sphingolipids and mitochondrial apoptosis. Journal of Bioenergetics and Biomembranes. 2016;48:153–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hannun YA, Obeid LM. Sphingolipids and their metabolism in physiology and disease. Nature Reviews Molecular Cell Biology. 2018;19:175–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Young MM, Kester M, Wang HG. Sphingolipids: Regulators of crosstalk between apoptosis and autophagy. Journal of Lipid Research. 2013;54:5–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Shaw J, Costa-Pinheiro P, Patterson L, Drews K, Spiegel S, Kester M. Novel Sphingolipid-Based Cancer Therapeutics in the Personalized Medicine Era: Elsevier Ltd; 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Codini M, Garcia-Gil M, Albi E. Cholesterol and Sphingolipid Enriched Lipid Rafts as Therapeutic Targets in Cancer. Int J Mol Sci. 2021;22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Barth BM, Shanmugavelandy SS, Tacelosky DM, Kester M, Morad SAF, et al. Gaucher’s Disease and Cancer a Sphingolipid Perspective. Crit Rev Oncog. 2013;18:221–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Gault CR, Obeid LM, Hannun YA. An overview of sphingolipid metabolism: From synthesis to breakdown. Advances in Experimental Medicine and Biology. 2010;688:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Morad SAF, Cabot MC. Ceramide-orchestrated signalling in cancer cells. Nature Reviews Cancer. 2013;13:51–65. [DOI] [PubMed] [Google Scholar]
- [52].Omae F, Miyazaki M, Enomoto A, Suzuki M, Suzuki Y, Suzuki A. DES2 protein is responsible for phytoceramide biosynthesis in the mouse small intestine. Biochemical Journal. 2004;379:687–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Uhlén M, Fagerberg L, Hallström Björn M, Lindskog C, Oksvold P, Mardinoglu A, et al. Tissue-based map of the human proteome. Science. 2015;347:1260419. [DOI] [PubMed] [Google Scholar]
- [54].Cingolani F, Futerman AH, Casas J. Ceramide synthases in biomedical research. Chemistry and Physics of Lipids. 2016;197:25–32. [DOI] [PubMed] [Google Scholar]
- [55].Levy M, Futerman AH. Mammalian ceramide synthases. IUBMB Life. 2010;62:347–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Park JW, Park WJ, Futerman AH. Ceramide synthases as potential targets for therapeutic intervention in human diseases. Biochimica et Biophysica Acta - Molecular and Cell Biology of Lipids. 2014;1841:671–81. [DOI] [PubMed] [Google Scholar]
- [57].Green CD, Maceyka M, Cowart LA, Spiegel S. Sphingolipids in metabolic disease: The good, the bad, and the unknown. Cell Metab. 2021;33:1293–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Hait NC, Maiti A. The Role of Sphingosine-1-Phosphate and Ceramide-1-Phosphate in Inflammation and Cancer. Mediators Inflamm. 2017;2017:4806541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Kunkel GT, Maceyka M, Milstien S, Spiegel S. Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nat Rev Drug Discov. 2013;12:688–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Pyne NJ, El Buri A, Adams DR, Pyne S. Sphingosine 1-phosphate and cancer. Adv Biol Regul. 2018;68:97–106. [DOI] [PubMed] [Google Scholar]
- [61].Baer A, Colon-Moran W, Bhattarai N. Characterization of the effects of immunomodulatory drug fingolimod (FTY720) on human T cell receptor signaling pathways. Sci Rep. 2018;8:10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Sandborn WJ, Feagan BG, Wolf DC, D’Haens G, Vermeire S, Hanauer SB, et al. Ozanimod Induction and Maintenance Treatment for Ulcerative Colitis. The New England journal of medicine. 2016;374:1754–62. [DOI] [PubMed] [Google Scholar]
- [63].Morad SAF, Cabot MC. The Onus of Sphingolipid Enzymes in Cancer Drug Resistance. Adv Cancer Res. 2018;140:235–63. [DOI] [PubMed] [Google Scholar]
- [64].Dunn TM, Tifft CJ, Proia RL. A perilous path: the inborn errors of sphingolipid metabolism. J Lipid Res. 2019;60:475–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Drews K, Calgi MP, Harrison WC, Drews CM, Costa-Pinheiro P, Shaw JJP, et al. Glucosylceramide synthase maintains influenza virus entry and infection. PLoS One. 2020;15:e0228735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Drews K, Calgi MP, Harrison WC, Drews CM, Costa-Pinheiro P, Shaw JJP, et al. Glucosylceramidase Maintains Influenza Virus Infection by Regulating Endocytosis. Journal of Virology. 2019;93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Patterson L, Allen J, Posey I, Shaw JJP, Costa-Pinheiro P, Walker SJ, et al. Glucosylceramide production maintains colon integrity in response to Bacteroides fragilis toxin-induced colon epithelial cell signaling. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2020;34:15922–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Newton J, Lima S, Maceyka M, Spiegel S. Revisiting the sphingolipid rheostat: Evolving concepts in cancer therapy. Experimental Cell Research. 2015;333:195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Tan SF, Dunton W, Liu X, Fox TE, Morad SAF, Desai D, et al. Acid ceramidase promotes drug resistance in acute myeloid leukemia through NF-kappaB-dependent P-glycoprotein upregulation. J Lipid Res. 2019;60:1078–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Tan SF, Liu X, Fox TE, Barth BM, Sharma A, Turner SD, et al. Acid ceramidase is upregulated in AML and represents a novel therapeutic target. Oncotarget. 2016;7:83208–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Bonhoure E, Pchejetski D, Aouali N, Morjani H, Levade T, Kohama T, et al. Overcoming MDR-associated chemoresistance in HL-60 acute myeloid leukemia cells by targeting sphingosine kinase-1. Leukemia. 2006;20:95–102. [DOI] [PubMed] [Google Scholar]
- [72].Dick TE, Hengst JA, Fox TE, Colledge AL, Kale VP, Sung SS, et al. The apoptotic mechanism of action of the sphingosine kinase 1 selective inhibitor SKI-178 in human acute myeloid leukemia cell lines. Journal of Pharmacology and Experimental Therapeutics. 2015;352:494–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Hengst JA, Dick TE, Sharma A, Doi K, Hegde S, Tan SF, et al. SKI-178: A Multitargeted Inhibitor of Sphingosine Kinase and Microtubule Dynamics Demonstrating Therapeutic Efficacy in Acute Myeloid Leukemia Models. Cancer Transl Med. 2017;3:109–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Powell JA, Lewis AC, Zhu W, Toubia J, Pitman MR, Wallington-Beddoe CT, et al. Targeting sphingosine kinase 1 induces MCL1-dependent cell death in acute myeloid leukemia. Blood. 2017;129:771–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Wallington-Beddoe CT, Bradstock KF, Bendall LJ. Oncogenic properties of sphingosine kinases in haematological malignancies. British Journal of Haematology. 2013;161:623–38. [DOI] [PubMed] [Google Scholar]
- [76].Morad SA, Ryan TE, Neufer PD, Zeczycki TN, Davis TS, MacDougall MR, et al. Ceramide-tamoxifen regimen targets bioenergetic elements in acute myelogenous leukemia. J Lipid Res. 2016;57:1231–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Chen C, Yin Y, Li C, Chen J, Xie J, Lu Z, et al. ACER3 supports development of acute myeloid leukemia. Biochem Biophys Res Commun. 2016;478:33–8. [DOI] [PubMed] [Google Scholar]
- [78].Vorbach S, Grunder A, Zhou F, Koellerer C, Jutzi JS, Simoni M, et al. Enhanced expression of the sphingosine-1-phosphate-receptor-3 causes acute myelogenous leukemia in mice. Leukemia. 2020;34:721–34. [DOI] [PubMed] [Google Scholar]
- [79].Tan SF, Pearson JM, Feith DJ, Loughran TP Jr., The emergence of acid ceramidase as a therapeutic target for acute myeloid leukemia. Expert Opin Ther Targets. 2017;21:583–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Lewis AC, Wallington-Beddoe CT, Powell JA, Pitson SM. Targeting sphingolipid metabolism as an approach for combination therapies in haematological malignancies. Cell Death Discovery. 2018;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Liu J, Beckman BS, Foroozesh M. A review of ceramide analogs as potential anticancer agents. Future Med Chem. 2013;5:1405–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Pearson JM, Tan SF, Sharma A, Annageldiyev C, Fox TE, Abad JL, et al. Ceramide Analogue SACLAC Modulates Sphingolipid Levels and MCL-1 Splicing to Induce Apoptosis in Acute Myeloid Leukemia. Mol Cancer Res. 2020;18:352–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Bezombes C, Plo I, Mas VM-D, Quillet-Mary A, Nègre-Salvayre A, Laurent G, et al. Oxidative stress‐induced activation of Lyn recruits sphingomyelinase and is requisite for its stimulation by Ara‐C. The FASEB Journal. 2001;15:1583–5. [DOI] [PubMed] [Google Scholar]
- [84].Grazide S, Maestre N, Veldman RJ, Bezombes C, Maddens S, Levade T, et al. Ara-C- and daunorubicin-induced recruitment of Lyn in sphingomyelinase-enriched membrane rafts. The FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2002;16:1685–7. [DOI] [PubMed] [Google Scholar]
- [85].Kao LP, Morad SAF, Davis TS, MacDougall MR, Kassai M, Abdelmageed N, et al. Chemotherapy selection pressure alters sphingolipid composition and mitochondrial bioenergetics in resistant HL-60 cells. Journal of Lipid Research. 2019;60:1590–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Xie SZ, Kaufmann KB, Wang W, Chan-Seng-Yue M, Gan OI, Laurenti E, et al. Sphingosine-1-phosphate receptor 3 potentiates inflammatory programs in normal and leukemia stem cells to promote differentiation. Blood Cancer Discov. 2021;2:32–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Companioni O, Mir C, Garcia-Mayea Y, Lleonart ME. Targeting Sphingolipids for Cancer Therapy. Frontiers in Oncology. 2021;11:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Lewis CS, Voelkel-Johnson C, Smith CD. Targeting Sphingosine Kinases for the Treatment of Cancer: Elsevier Ltd; 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Weinstein JN, Collisson EA, Mills GB, Shaw KRM, Ozenberger BA, Ellrott K, et al. The cancer genome atlas pan-cancer analysis project. Nature Genetics. 2013;45:1113–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Stefanko A, Thiede C, Ehninger G, Simons K, Grzybek M. Lipidomic approach for stratification of acute myeloid leukemia patients. PLoS One. 2017;12:e0168781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Daver N, Schlenk RF, Russell NH, Levis MJ. Targeting FLT3 mutations in AML: review of current knowledge and evidence. Leukemia. 2019;33:299–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Dany M, Gencer S, Nganga R, Thomas RJ, Oleinik N, Baron KD, et al. Targeting FLT3-ITD signaling mediates ceramide-dependent mitophagy and attenuates drug resistance in AML. Blood. 2016;128:1944–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Cairns RA, Mak TW. Oncogenic isocitrate dehydrogenase mutations: mechanisms, models, and clinical opportunities. Cancer discovery. 2013;3:730–41. [DOI] [PubMed] [Google Scholar]
- [94].Stuani L, Riols F, Millard P, Sabatier M, Batut A, Saland E, et al. Stable Isotope Labeling Highlights Enhanced Fatty Acid and Lipid Metabolism in Human Acute Myeloid Leukemia. Int J Mol Sci. 2018;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Dowdy T, Zhang L, Celiku O, Movva S, Lita A, Ruiz-Rodado V, et al. Sphingolipid Pathway as a Source of Vulnerability in IDH1(mut) Glioma. Cancers (Basel). 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Taylor RC, Cullen SP, Martin SJ. Apoptosis: Controlled demolition at the cellular level. Nature Reviews Molecular Cell Biology. 2008;9:231–41. [DOI] [PubMed] [Google Scholar]
- [97].Obeid LM, Linardic CM, Hannun YA. Programmed cell death induced by ceramide. Science. 1993;259:1769–71. [DOI] [PubMed] [Google Scholar]
- [98].Dressler Kenneth A, Mathias S, Kolesnick Richard N. Tumor Necrosis Factor-α Activates the Sphingomyelin Signal Transduction Pathway in a Cell-Free System. Science. 1992;255:1715–8. [DOI] [PubMed] [Google Scholar]
- [99].Moro K, Nagahashi M, Gabriel E, Takabe K, Wakai T. Clinical Application of Ceramide in Cancer Treatment Breast Cancer. 2019;26:407–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].De Palma C, Perrotta C. Ceramide as a target of chemotherapy: Its role in apoptosis and autophagy. Clinical Lipidology. 2012;7:111–9. [Google Scholar]
- [101].Colombini M. Ceramide channels and their role in mitochondria-mediated apoptosis. Biochimica et Biophysica Acta - Bioenergetics. 2010;1797:1239–44. [DOI] [PubMed] [Google Scholar]
- [102].Molica M, Breccia M, Foa R, Jabbour E, Kadia TM. Maintenance therapy in AML: The past, the present and the future. American Journal of Hematology. 2019;94:1254–65. [DOI] [PubMed] [Google Scholar]
- [103].Kale J, Osterlund EJ, Andrews DW. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death and Differentiation. 2018;25:65–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Kadia TM, Kantarjian HM, Konopleva M. Myeloid cell leukemia-1 dependence in acute myeloid leukemia: A novel approach to patient therapy. Oncotarget. 2019;10:1250–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Konopleva M, Letai A. BCL-2 inhibition in AML: An unexpected bonus? Blood. 2018;132:1007–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Paugh SW, Paugh BS, Rahmani M, Kapitonov D, Almenara JA, Kordula T, et al. A selective sphingosine kinase 1 inhibitor integrates multiple molecular therapeutic targets in human leukemia. Blood. 2008;112:1382–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Xu L, Zhang Y, Gao M, Wang G, Fu Y. Concurrent targeting Akt and sphingosine kinase 1 by A-674563 in acute myeloid leukemia cells. Biochemical and biophysical research communications. 2016;472:662–8. [DOI] [PubMed] [Google Scholar]
- [108].Pitman MR, Lewis AC, Davies LT, Moretti PAB, Anderson D, Creek DJ, et al. The sphingosine 1-phosphate receptor 2/4 antagonist JTE-013 elicits off-target effects on sphingolipid metabolism. Scientific Reports. 2022;12:454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Féral K, Jaud M, Philippe C, Di Bella D, Pyronnet S, Rouault-Pierre K, et al. ER Stress and Unfolded Protein Response in Leukemia: Friend, Foe, or Both? Biomolecules. 2021;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Martelli AM, Paganelli F, Chiarini F, Evangelisti C, McCubrey JA. The Unfolded Protein Response: A Novel Therapeutic Target in Acute Leukemias. Cancers. 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Liu Q, Chan AKN, Chang W-H, Yang L, Pokharel SP, Miyashita K, et al. 3-Ketodihydrosphingosine reductase maintains ER homeostasis and unfolded protein response in leukemia. Leukemia. 2022;36:100–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Cristóbal I, Garcia-Orti L, Cirauqui C, Alonso MM, Calasanz MJ, Odero MD. PP2A impaired activity is a common event in acute myeloid leukemia and its activation by forskolin has a potent anti-leukemic effect. Leukemia. 2011;25:606–14. [DOI] [PubMed] [Google Scholar]
- [113].Perrotti D, Neviani P. Protein phosphatase 2A: A target for anticancer therapy. The Lancet Oncology. 2013;14:e229–e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Oaks J, Ogretmen B. Regulation of PP2A by sphingolipid metabolism and signaling. Frontiers in Oncology. 2015;4:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Chalfant CE, Kishikawal K, Mumby MC, Kamibayashi C, Bielawska A, Hannun YA. Long chain ceramides activate protein phosphatase-1 and protein phosphatase-2A. Journal of Biological Chemistry. 1999;274:20313–7. [DOI] [PubMed] [Google Scholar]
- [116].Chalfant CE, Szulc Z, Roddy P, Bielawska A, Hannun YA. The structural requirements for ceramide activation of serine-threonine protein phosphatases. Journal of Lipid Research. 2004;45:496–506. [DOI] [PubMed] [Google Scholar]
- [117].Dobrowsky RT, Kamibayashi C, Mumby MC, Hannun YA. Ceramide activates heterotrimeric protein phosphatase 2A. Journal of Biological Chemistry. 1993;268:15523–30. [PubMed] [Google Scholar]
- [118].Chen L, Luo LF, Lu J, Li A, Liu YF, Wang J, et al. FTY720 induces apoptosis of M2 subtype Acute Myeloid Leukemia cells by targeting sphingolipid metabolism and increasing endogenous ceramide levels. PLoS ONE. 2014;9:12–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Pippa R, Dominguez A, Christensen DJ, Moreno-Miralles I, Blanco-Prieto MJ, Vitek MP, et al. Effect of FTY720 on the SET-PP2A complex in acute myeloid leukemia; SET binding drugs have antagonistic activity. Leukemia. 2014;28:1915–8. [DOI] [PubMed] [Google Scholar]
- [120].Sheridan M, Ogretmen B. The Role of Ceramide Metabolism and Signaling in the Regulation of Mitophagy and Cancer Therapy. Cancers (Basel). 2021;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Palikaras K, Lionaki E, Tavernarakis N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nature Cell Biology. 2018;20:1013–22. [DOI] [PubMed] [Google Scholar]
- [122].Du W, Xu A, Huang Y, Cao J, Zhu H, Yang B, et al. The role of autophagy in targeted therapy for acute myeloid leukemia. Autophagy. 2021;17:2665–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Sentelle RD, Senkal CE, Jiang W, Ponnusamy S, Gencer S, Selvam SP, et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat Chem Biol. 2012;8:831–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Morad SAF, MacDougall MR, Abdelmageed N, Kao LP, Feith DJ, Tan SF, et al. Pivotal role of mitophagy in response of acute myelogenous leukemia to a ceramide-tamoxifen-containing drug regimen. Exp Cell Res. 2019;381:256–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Gottesman MM, Lavi O, Hall MD, Gillet J-P. Toward a Better Understanding of the Complexity of Cancer Drug Resistance. Annual Review of Pharmacology and Toxicology. 2016;56:85–102. [DOI] [PubMed] [Google Scholar]
- [126].Housman G, Byler S, Heerboth S, Lapinska K, Longacre M, Snyder N, et al. Drug resistance in cancer: an overview. Cancers (Basel). 2014;6:1769–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Bradley G, Ling V. P-glycoprotein, multidrug resistance and tumor progression. Cancer and Metastasis Reviews. 1994;13:223–33. [DOI] [PubMed] [Google Scholar]
- [128].Gottesman MM. Mechanisms of cancer drug resistance. Annual review of medicine. 2002;53:615–27. [DOI] [PubMed] [Google Scholar]
- [129].Sun YL, Patel A, Kumar P, Chen ZS. Role of ABC transporters in cancer chemotherapy. Chinese journal of cancer. 2012;31:51–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].da Silveira Júnior LS, Soares VL, Jardim da Silva AS, Gil EA, Pereira de Araújo MDG, Merces Gonçalves CA, et al. P-glycoprotein and multidrug resistance-associated protein-1 expression in acute myeloid leukemia: Biological and prognosis implications. International journal of laboratory hematology. 2020;42:594–603. [DOI] [PubMed] [Google Scholar]
- [131].Leith CP, Kopecky KJ, Chen IM, Eijdems L, Slovak ML, McConnell TS, et al. Frequency and clinical significance of the expression of the multidrug resistance proteins MDR1/P-glycoprotein, MRP1, and LRP in acute myeloid leukemia: a Southwest Oncology Group Study. Blood. 1999;94:1086–99. [PubMed] [Google Scholar]
- [132].Legrand O, Simonin G, Beauchamp-Nicoud A, Zittoun R, Marie JP. Simultaneous activity of MRP1 and Pgp is correlated with in vitro resistance to daunorubicin and with in vivo resistance in adult acute myeloid leukemia. Blood. 1999;94:1046–56. [PubMed] [Google Scholar]
- [133].de Moraes AC, Maranho CK, Rauber GS, Santos-Silva MC. Importance of detecting multidrug resistance proteins in acute leukemia prognosis and therapy. Journal of clinical laboratory analysis. 2013;27:62–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Chauhan PS, Bhushan B, Singh LC, Mishra AK, Saluja S, Mittal V, et al. Expression of genes related to multiple drug resistance and apoptosis in acute leukemia: response to induction chemotherapy. Experimental and molecular pathology. 2012;92:44–9. [DOI] [PubMed] [Google Scholar]
- [135].Nasilowska-Adamska B, Solarska I, Paluszewska M, Malinowska I, Jedrzejczak WW, Warzocha K. FLT3-ITD and MLL-PTD influence the expression of MDR-1, MRP-1, and BCRP mRNA but not LRP mRNA assessed with RQ-PCR method in adult acute myeloid leukemia. Annals of hematology. 2014;93:577–93. [DOI] [PubMed] [Google Scholar]
- [136].Legrand O, Zompi S, Perrot JY, Faussat AM, Benderra Z, Chaoui D, et al. P-glycoprotein and multidrug resistance associated protein-1 activity in 132 acute myeloid leukemias according to FAB subtypes and cytogenetics risk groups. Haematologica. 2004;89:34–41. [PubMed] [Google Scholar]
- [137].Shaffer BC, Gillet JP, Patel C, Baer MR, Bates SE, Gottesman MM. Drug resistance: still a daunting challenge to the successful treatment of AML. Drug resistance updates : reviews and commentaries in antimicrobial and anticancer chemotherapy. 2012;15:62–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Robey RW, Pluchino KM, Hall MD, Fojo AT, Bates SE, Gottesman MM. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat Rev Cancer. 2018;18:452–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Cripe LD, Uno H, Paietta EM, Litzow MR, Ketterling RP, Bennett JM, et al. Zosuquidar, a novel modulator of P-glycoprotein, does not improve the outcome of older patients with newly diagnosed acute myeloid leukemia: A randomized, placebo-controlled trial of the Eastern Cooperative Oncology Group 3999. Blood. 2010;116:4077–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Gouazé V, Yu JY, Bleicher RJ, Han TY, Liu YY, Wang H, et al. Overexpression of glucosylceramide synthase and P-glycoprotein in cancer cells selected for resistance to natural product chemotherapy. Molecular cancer therapeutics. 2004;3:633–9. [PubMed] [Google Scholar]
- [141].Xie P, Yun-Fen S, Shi Y-P, Ge S-M, Gu Z-H, Wang J, et al. Overexpression of glucosylceramide synthase in associated with multidrug resistance of leukemia cells. Leukemia Research. 2008;32:475–80. [DOI] [PubMed] [Google Scholar]
- [142].Lavie Y, Cao H, Volner A, Lucci A, Han TY, Geffen V, et al. Agents that reverse multidrug resistance, tamoxifen, verapamil, and cyclosporin A, block glycosphingolipid metabolism by inhibiting ceramide glycosylation in human cancer cells. J Biol Chem. 1997;272:1682–7. [DOI] [PubMed] [Google Scholar]
- [143].Itoh M, Kitano T, Watanabe M, Kondo T, Yabu T, Taguchi Y, et al. Possible role of ceramide as an indicator of chemoresistance: Decrease of the ceramide content via activation of glucosylceramide synthase and sphingomyelin synthase in chemoresistant leukemia. Clinical Cancer Research. 2003;9:415–23. [PubMed] [Google Scholar]
- [144].Chaudhary PM, Roninson IB. Induction of multidrug resistance in human cells by transient exposure to different chemotherapeutic drugs. Journal of the National Cancer Institute. 1993;85:632–9. [DOI] [PubMed] [Google Scholar]
- [145].Kohno K, Sato S, Takano H, Matsuo K, Kuwano M. The direct activation of human multidrug resistance gene (MDR1) by anticancer agents. Biochemical and biophysical research communications. 1989;165:1415–21. [DOI] [PubMed] [Google Scholar]
- [146].Senchenkov A, Litvak DA, Cabot MC. Targeting ceramide metabolism--a strategy for overcoming drug resistance. Journal of the National Cancer Institute. 2001;93:347–57. [DOI] [PubMed] [Google Scholar]
- [147].Gouaze-Andersson V, Cabot MC. Glycosphingolipids and drug resistance. Biochimica et Biophysica Acta - Biomembranes. 2006;1758:2096–103. [DOI] [PubMed] [Google Scholar]
- [148].Hajj C, Becker-Flegler KA, Haimovitz-Friedman A. Novel mechanisms of action of classical chemotherapeutic agents on sphingolipid pathways. Biological chemistry. 2015;396:669–79. [DOI] [PubMed] [Google Scholar]
- [149].Sietsma H, Veldman RJ, Kok JW. The involvement of sphingolipids in multidrug resistance. The Journal of membrane biology. 2001;181:153–62. [DOI] [PubMed] [Google Scholar]
- [150].Plo I, Lehne G, Beckstrøm KJ, Maestre N, Bettaïeb A, Laurent G, et al. Influence of ceramide metabolism on P-glycoprotein function in immature acute myeloid leukemia KG1a cells. Molecular Pharmacology. 2002;62:304–12. [DOI] [PubMed] [Google Scholar]
- [151].Fisher-Wellman KH, Hagen JT, Kassai M, Kao L-P, Nelson M, McLaughlin K, et al. Alterations in sphingolipid composition and mitochondrial bioenergetics represent synergistic therapeutic vulnerabilities linked to multidrug resistance in leukemia. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2021:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].van Helvoort A, Smith AJ, Sprong H, Fritzsche I, Schinkel AH, Borst P, et al. MDR1 P-glycoprotein is a lipid translocase of broad specificity, while MDR3 P-glycoprotein specifically translocates phosphatidylcholine. Cell. 1996;87:507–17. [DOI] [PubMed] [Google Scholar]
- [153].Turzanski J, Grundy M, Shang S, Russell N, Pallis M. P-glycoprotein is implicated in the inhibition of ceramide-induced apoptosis in TF-1 acute myeloid leukemia cells by modulation of the glucosylceramide synthase pathway. Exp Hematol. 2005;33:62–72. [DOI] [PubMed] [Google Scholar]
- [154].Morad SAF, Davis TS, MacDougall MR, Tan SF, Feith DJ, Desai DH, et al. Role of P-glycoprotein inhibitors in ceramide-based therapeutics for treatment of cancer. Biochemical pharmacology. 2017;130:21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Barth BM, Cabot MC, Kester M. Ceramide-based therapeutics for the treatment of cancer. Anti-cancer agents in medicinal chemistry. 2011;11:911–9. [DOI] [PubMed] [Google Scholar]
- [156].Canals D, Perry DM, Jenkins RW, Hannun YA. Drug targeting of sphingolipid metabolism: sphingomyelinases and ceramidases. British journal of pharmacology. 2011;163:694–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].de Figueiredo-Pontes LL, Pintão MC, Oliveira LC, Dalmazzo LF, Jácomo RH, Garcia AB, et al. Determination of P-glycoprotein, MDR-related protein 1, breast cancer resistance protein, and lung-resistance protein expression in leukemic stem cells of acute myeloid leukemia. Cytometry Part B, Clinical cytometry. 2008;74:163–8. [DOI] [PubMed] [Google Scholar]
- [158].Farge T, Saland E, de Toni F, Aroua N, Hosseini M, Perry R, et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer discovery. 2017;7:716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Dick JE. Acute myeloid leukemia stem cells. Ann N Y Acad Sci. 2005;1044:1–5. [DOI] [PubMed] [Google Scholar]
- [160].Ye H, Adane B, Khan N, Sullivan T, Minhajuddin M, Gasparetto M, et al. Leukemic Stem Cells Evade Chemotherapy by Metabolic Adaptation to an Adipose Tissue Niche. Cell Stem Cell. 2016;19:23–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Shlush LI, Mitchell A, Heisler L, Abelson S, Ng SWK, Trotman-Grant A, et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature. 2017;547:104–8. [DOI] [PubMed] [Google Scholar]
- [162].Chopra M, Bohlander SK. The cell of origin and the leukemia stem cell in acute myeloid leukemia. Genes Chromosomes Cancer. 2019;58:850–8. [DOI] [PubMed] [Google Scholar]
- [163].Hanekamp D, Cloos J, Schuurhuis GJ. Leukemic stem cells: identification and clinical application. International journal of hematology. 2017;105:549–57. [DOI] [PubMed] [Google Scholar]
- [164].Ng SW, Mitchell A, Kennedy JA, Chen WC, McLeod J, Ibrahimova N, et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature. 2016;540:433–7. [DOI] [PubMed] [Google Scholar]
- [165].Pollyea DA, Jordan CT. Therapeutic targeting of acute myeloid leukemia stem cells. Blood. 2017;129:1627–35. [DOI] [PubMed] [Google Scholar]
- [166].Hills RK, Castaigne S, Appelbaum FR, Delaunay J, Petersdorf S, Othus M, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15:986–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Haubner S, Perna F, Kohnke T, Schmidt C, Berman S, Augsberger C, et al. Coexpression profile of leukemic stem cell markers for combinatorial targeted therapy in AML. Leukemia. 2019;33:64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011;20:66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Minuesa G, Albanese SK, Xie W, Kazansky Y, Worroll D, Chow A, et al. Small-molecule targeting of MUSASHI RNA-binding activity in acute myeloid leukemia. Nat Commun. 2019;10:2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Xie SZ, Garcia-Prat L, Voisin V, Ferrari R, Gan OI, Wagenblast E, et al. Sphingolipid Modulation Activates Proteostasis Programs to Govern Human Hematopoietic Stem Cell Self-Renewal. Cell Stem Cell. 2019;25:639–53 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Yang C, Yamashita M, Suda T. A Novel Function of Sphingolipid Signaling via S1PR3 in Hematopoietic and Leukemic Stem Cells. Blood Cancer Discov. 2021;2:3–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Hu X, Yang D, Zimmerman M, Liu F, Yang J, Kannan S, et al. IRF8 regulates acid ceramidase expression to mediate apoptosis and suppresses myelogeneous leukemia. Cancer Research. 2011;71:2882–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Sobue S, Iwasaki T, Sugisaki C, Nagata K, Kikuchi R, Murakami M, et al. Quantitative RT-PCR analysis of sphingolipid metabolic enzymes in acute leukemia and myelodysplastic syndromes. Leukemia. 2006;20:2042–6. [DOI] [PubMed] [Google Scholar]
- [174].Ghazaly EA, Miraki-Moud F, Smith P, Gnanaranjan C, Koniali L, Oke A, et al. Repression of sphingosine kinase (SK)-interacting protein (SKIP) in acute myeloid leukemia diminishes SK activity and its re-expression restores SK function. J Biol Chem. 2020;295:5496–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Sobue S, Nemoto S, Murakami M, Ito H, Kimura A, Gao S, et al. Implications of sphingosine kinase 1 expression level for the cellular sphingolipid rheostat: Relevance as a marker for daunorubicin sensitivity of leukemia cells. International journal of hematology. 2008;87:266–75. [DOI] [PubMed] [Google Scholar]
- [176].Powell JA, Wallington-Beddoe CT, Pitson SM. Targeting sphingosine kinase 1 in acute myeloid leukemia: translation to clinic. International Journal of Hematologic Oncology. 2017;6:31–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Evangelisti C, Evangelisti C, Buontempo F, Lonetti A, Orsini E, Chiarini F, et al. Therapeutic potential of targeting sphingosine kinases and sphingosine 1-phosphate in hematological malignancies. Leukemia. 2016;30:2142–51. [DOI] [PubMed] [Google Scholar]
- [178].Huang W, Qian T, Cheng Z, Zeng T, Si C, Liu C, et al. Prognostic significance of Spinster homolog gene family in acute myeloid leukemia. J Cancer. 2020;11:4581–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Watek M, Durnas B, Wollny T, Pasiarski M, Gozdz S, Marzec M, et al. Unexpected profile of sphingolipid contents in blood and bone marrow plasma collected from patients diagnosed with acute myeloid leukemia. Lipids Health Dis. 2017;16:235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Barth BM, Wang W, Toran PT, Fox TE, Annageldiyev C, Ondrasik RM, et al. Sphingolipid metabolism determines the therapeutic efficacy of nanoliposomal ceramide in acute myeloid leukemia. Blood Advances. 2019;3:2598–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Petrick L, Imani P, Perttula K, Yano Y, Whitehead T, Metayer C, et al. Untargeted metabolomics of newborn dried blood spots reveals sex-specific associations with pediatric acute myeloid leukemia. Leuk Res. 2021;106:106585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Pabst T, Kortz L, Fiedler GM, Ceglarek U, Idle JR, Beyoglu D. The plasma lipidome in acute myeloid leukemia at diagnosis in relation to clinical disease features. BBA Clin. 2017;7:105–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Hori A, Ishida F, Nakazawa H, Yamaura M, Morita S, Uehara T, et al. Serum sphingomyelin species profile is altered in hematologic malignancies. Clin Chim Acta. 2021;514:29–33. [DOI] [PubMed] [Google Scholar]
- [184].Qu H, Zhu Y. SMPDL3B Predicts Poor Prognosis and Contributes to Development of Acute Myeloid Leukemia. Front Mol Biosci. 2021;8:695601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Smolenska-Sym G, Spychalska J, Zdebska E, Wozniak J, Traczyk Z, Pszenna E, et al. Ceramides and glycosphingolipids in maturation process: leukemic cells as an experimental model. Blood Cells Mol Dis. 2004;33:68–76. [DOI] [PubMed] [Google Scholar]
- [186].Wang Z, Wen L, Ma X, Chen Z, Yu Y, Zhu J, et al. High expression of lactotriaosylceramide, a differentiation-associated glycosphingolipid, in the bone marrow of acute myeloid leukemia patients. Glycobiology. 2012;22:930–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Siskind LJ, Feinstein L, Yu T, Davis JS, Jones D, Choi J, et al. Anti-apoptotic Bcl-2 family proteins disassemble ceramide channels. Journal of Biological Chemistry. 2008;283:6622–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Beverly LJ, Howell LA, Hernandez-Corbacho M, Casson L, Chipuk JE, Siskind LJ. BAK activation is necessary and sufficient to drive ceramide synthase-dependent ceramide accumulation following inhibition of BCL2-like proteins. Biochemical Journal. 2013;452:111–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Siskind LJ, Mullen TD, Rosales KR, Clarke CJ, Hernandez-Corbacho MJ, Edinger AL, et al. The BCL-2 protein BAK is required for long-chain ceramide generation during apoptosis. Journal of Biological Chemistry. 2010;285:11818–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Chipuk JE, McStay GP, Bharti A, Kuwana T, Clarke CJ, Siskind LJ, et al. Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell. 2012;148:988–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Murate T, Suzuki M, Hattori M, Takagi A, Kojima T, Tanizawa T, et al. Up-regulation of acid sphingomyelinase during retinoic acid-induced myeloid differentiation of NB4, a human acute promyelocytic leukemia cell line. Journal of Biological Chemistry. 2002;277:9936–43. [DOI] [PubMed] [Google Scholar]
- [192].Dbaibo GS, Kfoury Y, Darwiche N, Panjarian S, Kozhaya L, Nasr R, et al. Arsenic trioxide induces accumulation of cytotoxic levels of ceramide in acute promyelocyte leukemia and adult T-cell leukemia/lymphoma cells through de novo ceramide synthesis and inhibition of glucosylceramide synthase activity. Haematologica. 2007;92:753–62. [DOI] [PubMed] [Google Scholar]
- [193].Morad SAF, Levin JC, Tan SF, Fox TE, Feith DJ, Cabot MC. Novel off-target effect of tamoxifen - Inhibition of acid ceramidase activity in cancer cells. Biochimica et Biophysica Acta - Molecular and Cell Biology of Lipids. 2013;1831:1657–64. [DOI] [PubMed] [Google Scholar]
- [194].White-Gilbertson S, Lu P, Jones CM, Chiodini S, Hurley D, Das A, et al. Tamoxifen is a candidate first-in-class inhibitor of acid ceramidase that reduces amitotic division in polyploid giant cancer cells—Unrecognized players in tumorigenesis. Cancer Medicine. 2020;9:3142–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Morad SAF, Cabot MC. Tamoxifen regulation of sphingolipid metabolism - Therapeutic implications. Biochimica et Biophysica Acta - Molecular and Cell Biology of Lipids. 2015;1851:1134–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Elojeimy S, Holman DH, Liu X, El-Zawahry A, Villani M, Cheng JC, et al. New insights on the use of desipramine as an inhibitor for acid ceramidase. FEBS Letters. 2006;580:4751–6. [DOI] [PubMed] [Google Scholar]
- [197].Bedia C, Casas J, Andrieu-Abadie N, Fabriàs G, Levade T. Acid ceramidase expression modulates the sensitivity of A375 melanoma cells to dacarbazine. Journal of Biological Chemistry. 2011;286:28200–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Klutzny S, Lesche R, Keck M, Kaulfuss S, Schlicker A, Christian S, et al. Functional inhibition of acid sphingomyelinase by Fluphenazine triggers hypoxia-specific tumor cell death. Cell Death and Disease. 2017;8:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Skácel J, Slusher BS, Tsukamoto T. Small Molecule Inhibitors Targeting Biosynthesis of Ceramide, the Central Hub of the Sphingolipid Network. Journal of Medicinal Chemistry. 2021;64:279–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Yang L, Weng W, Sun ZX, Fu XJ, Ma J, Zhuang WF. SphK1 inhibitor II (SKI-II) inhibits acute myelogenous leukemia cell growth in vitro and in vivo. Biochemical and biophysical research communications. 2015;460:903–8. [DOI] [PubMed] [Google Scholar]
- [201].Hengst JA, Hegde S, Paulson RF, Yun JK. Development of SKI-349, a dual-targeted inhibitor of sphingosine kinase and microtubule polymerization. Bioorganic and Medicinal Chemistry Letters. 2020;30:127453–. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Britten CD, Garrett-Mayer E, Chin SH, Shirai K, Ogretmen B, Bentz TA, et al. A phase I study of ABC294640, a first-in-class sphingosine kinase-2 inhibitor, in patients with advanced solid tumors. Clinical Cancer Research. 2017;23:4642–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Dickson MA, Carvajal RD, Merrill AH, Gonen M, Cane LM, Schwartz GK. A phase I clinical trial of safingol in combination with cisplatin in advanced solid tumors. Clinical Cancer Research. 2011;17:2484–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Coward J, Ambrosini G, Musi E, Truman JP, Haimovitz-Friedman A, Allegood JC, et al. Safingol (L-threo-sphinganine) induces autophagy in solid tumor cells through inhibition of PKC and the PI3-kinase pathway. Autophagy. 2009;5:184–93. [DOI] [PubMed] [Google Scholar]
- [205].Ordóñez YF, Abad JL, Aseeri M, Casas J, Garcia V, Casasampere M, et al. Activity-Based Imaging of Acid Ceramidase in Living Cells. Journal of the American Chemical Society. 2019;141:7736–42. [DOI] [PubMed] [Google Scholar]
- [206].Bae J, Leo CP, Sheau Yu H, Hsueh AJW. MCL-1S, a splicing variant of the antiapoptotic BCL-2 family member MCL-1, encodes a proapoptotic protein possessing only the BH3 domain. Journal of Biological Chemistry. 2000;275:25255–61. [DOI] [PubMed] [Google Scholar]
- [207].Liu Y-Y, Li Y-T. Ceramide Glycosylation Catalyzed by Glucosylceramide Synthase and Cancer Drug Resistance. Advances in Cancer Research. 2013:59–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Grazide S, Terrisse AD, Lerouge S, Laurent G, Jaffrézou JP. Cytoprotective Effect of Glucosylceramide Synthase Inhibition against Daunorubicin-induced Apoptosis in Human Leukemic Cell Lines. Journal of Biological Chemistry. 2004;279:18256–61. [DOI] [PubMed] [Google Scholar]
- [209].Kester M, Bassler J, Fox TE, Carter CJ, Davidson JA, Parette MR. Preclinical development of a C6-ceramide NanoLiposome, a novel sphingolipid therapeutic. Biological chemistry. 2015;396:737–47. [DOI] [PubMed] [Google Scholar]
- [210].Barth BM, Keasey NR, Wang X, Shanmugavelandy SS, Rampal R, Hricik T, et al. Engraftment of Human Primary Acute Myeloid Leukemia Defined by Integrated Genetic Profiling in NOD/SCID/IL2rγnull Mice for Preclinical Ceramide-Based Therapeutic Evaluation. Physiology & behavior. 2014;2:146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].DiPietrantonio AM, Hsieh TC, Olson SC, Joseph MWU. Regulation of G1/S transition and induction of apoptosis in HL-60 leukemia cells by fenretinide (4HPR). International Journal of Cancer. 1998;78:53–61. [DOI] [PubMed] [Google Scholar]
- [212].Jiang L, Pan X, Chen Y, Wang K, Du Y, Zhang J. Preferential involvement of both ROS and ceramide in fenretinide-induced apoptosis of HL60 rather than NB4 and U937 cells. Biochemical and biophysical research communications. 2011;405:314–8. [DOI] [PubMed] [Google Scholar]
- [213].Morad SAF, Davis TS, Kester M, Loughran TP, Cabot MC. Dynamics of ceramide generation and metabolism in response to fenretinide - Diversity within and among leukemia. Leukemia Research. 2015;39:1071–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Zhang H, Mi JQ, Fang H, Wang Z, Wang C, Wu L, et al. Preferential eradication of acute myelogenous leukemia stem cells by fenretinide. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:5606–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Zhao X-y, Zhang R-r, Ye Q, Qiu F, Xu H-y, Wei F-g, et al. 4-Hydroxyphenyl Retinamide Preferentially Targets FLT3 Mutated Acute Myeloid Leukemia via ROS Induction and NF-κB Inhibition. Current Medical Science. 2020;40:810–6. [DOI] [PubMed] [Google Scholar]
- [216].Rahmaniyan M, Curley RW, Obeid LM, Hannun YA, Kraveka JM. Identification of dihydroceramide desaturase as a direct in vitro target for fenretinide. Journal of Biological Chemistry. 2011;286:24754–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Chun J, Hartung HP. Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clinical Neuropharmacology. 2010;33:91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].White C, Alshaker H, Cooper C, Winkler M, Pchejetski D. The emerging role of FTY720 (Fingolimod) in cancer treatment. Oncotarget. 2016;7:23106–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Young MM, Bui V, Chen C, Wang HG. FTY720 induces non-canonical phosphatidylserine externalization and cell death in acute myeloid leukemia. Cell Death and Disease. 2019;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Brown TJ. Therapeutic Combination of Nanoliposomal Safingol and Nanoliposomal Ceramide for Acute Myeloid Leukemia. Journal of Leukemia. 2013;01:1–8. [Google Scholar]
- [221].Charyguly A, Krishne G, Trupti P, Priyanjali B, Su-Fern T, Soumya I, et al. The novel Isatin analog KS99 targets stemness markers in acute myeloid leukemia. Haematologica. 2020;105:687–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Casson L, Howell L, Mathews LA, Ferrer M, Southall N, Guha R, et al. Inhibition of Ceramide Metabolism Sensitizes Human Leukemia Cells to Inhibition of BCL2-Like Proteins. PLoS One. 2013;8:e54525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Pollyea DA, Amaya M, Strati P, Konopleva MY. Venetoclax for AML: Changing the treatment paradigm. Blood Advances. 2019;3:4326–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Sundaramoorthy P, Gasparetto C, Kang Y. The combination of a sphingosine kinase 2 inhibitor (ABC294640) and a Bcl-2 inhibitor (ABT-199) displays synergistic anti-myeloma effects in myeloma cells without a t(11;14) translocation. Cancer Medicine. 2018;7:3257–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Cooper JP, Reynolds CP, Cho H, Kang MH. Clinical development of fenretinide as an antineoplastic drug: Pharmacology perspectives. Experimental Biology and Medicine. 2017;242:1178–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Mohrbacher AM, Yang AS, Groshen S, Kummar S, Gutierrez ME, Kang MH, et al. Phase I study of fenretinide delivered intravenously in patients with relapsed or refractory hematologic malignancies: A California cancer consortium trial. Clinical Cancer Research. 2017;23:4550–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Stirnemann J, Belmatoug N, Camou F, Serratrice C, Froissart R, Caillaud C, et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. International journal of molecular sciences. 2017;18:441. [DOI] [PMC free article] [PubMed] [Google Scholar]