

Abstract
Introduction: The search for effective inhibitors to multiple infectious agents including influenza, smallpox and hemorrhagic fever viruses is an area of active research as many of these agents pose dramatic health and economic challenges to the human population. Many of these infectious agents are not only endemic threats in different parts of the globe, but are also considered to have the potential of being used as bioterrorism agents.
Areas covered: This review focuses on inhibitors that are currently in use in the research community against specific emerging infectious agents and those that have bioterrorism potential. The paper provides information about the availability of FDA approved drugs, whenever applicable, and insights into the specific aspect of the agent life cycle that is affected by drug treatment, when known.
Expert opinion: The key message that is conveyed in this review is that a combination of pathogen and host-based inhibitors may have to be used for successful control of viral replication to limit the development of drug resistance.
Keywords: hemorrhagic fever viruses, host response-based inhibitors, influenza, inhibitors, pathogen-based inhibitors, smallpox
1. Introduction
There are a plethora of bacterial and viral pathogens that are constantly emerging and evolving for which we lack effective and approved therapeutics. Although many of these infectious agents have come under scrutiny as bioterrorism agents, these are also pathogens that cause natural infections and pandemics in various parts of the globe (Table 1). This review is an attempt to provide a compilation of some of the recently described therapeutics against influenza virus, smallpox virus and hemorrhagic fever viruses.
Table 1.
Characteristics of discussed viruses.
| Family | Virus | Vector/reservoir | Disease | Endemic region |
|---|---|---|---|---|
| Orthomyoviridae | Influenza A | Wild birds | Flu | |
| Influenza B | Flu | |||
| Influenza C | Flu | |||
| Poxviridae | Variola | None | Smallpox | Eradicated |
| Filoviradae | Ebolavirus | Unknown | Ebola hemorrhagic fever | Sub-Saharan Africa |
| Marburgvirus | Unknown | Marburg hemorrhagic fever | Sub-Saharan Africa | |
| Arenavirdae | Lassa virus | Rodents | Lassa fever | West Africa |
| Junin virus | Rodents | Argentine hemorrhagic fever | South America | |
| Machupo virus | Rodents | Bolivian hemorrhagic fever | South America | |
| Guanarito virus | Rodents | Venezuelan hemorrhagic fever | South America | |
| Sabia virus | Rodents | Hemorrhagic fever | South America | |
| Bunyaviradae | Crimean Congo hemorrhagic fever virus | Ticks | Crimean Congo hemorrhagic fever | Africa, China, Middle East |
| Hantavirus | Rodents | Hemorrhagic fever with renal syndrome, human pulmonary syndrome | Americas, Asia, and Europe | |
| Rift valley fever virus | Mosquitoes | Rift valley fever | Sub-Saharan Africa, Egypt, Saudi Arabia, Yemen |
2. Influenza
Influenza virus is a negative strand, 8-segmented, single-stranded RNA virus belonging to the family orthomyxoviridae [1]. Evolution of new strains with pandemic potential due to antigenic drift or shift is entirely arbitrary which underscores the urgent need for broad spectrum, high efficacy inhibitors that can work across strain barriers. Current drugs against influenza include zanamivir (Relenza), oseltamivir (Tamiflu), amantadine and rimantadine (Figure 1). There are several candidate anti-influenza antivirals that are described in the literature and the review by De Clercq provides a comprehensive picture of the newer developments [2].
Figure 1.
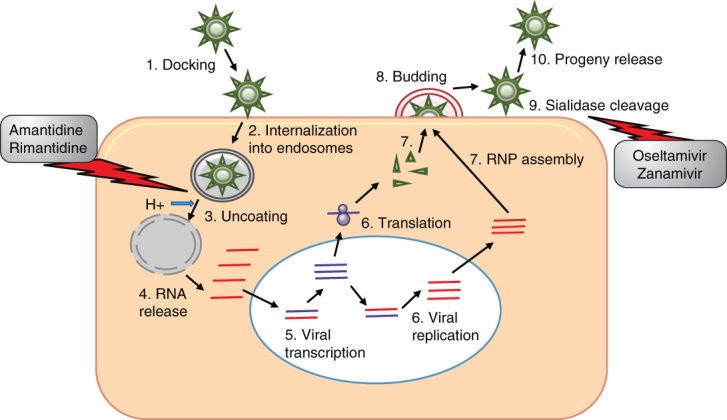
Life cycle of influenza virus is indicated. Following docking and internalization in endosomes, pH alterations cause release of viral RNA into the cytoplasm. Following transcription and translation, the viral RNP is assembled and the virus buds out of the cell on sialidase cleavage. The point of action of the FDA approved M2 ion channel blockers and neuraminidase inhibitors is specified.
M2: Matrix 2; RNP: Ribonucleoprotein.
2.1. Ion channel blockers
Matrix 2 (M2) ion channels are found in the surface of the virion. After host cell entry and uptake into endosomes, the acidic pH of the endosome enables entry of H+ ions into the interior of the viral particle. This process is necessary for viral uncoating and release of viral genetic material into the host cytoplasm. Ion channel inhibitors block the interior channel thus preventing H+ ion influx.
2.1.1. Amantadine and rimantadine
Amantadine was the first synthetic compound that was demonstrated to inhibit influenza virus replication [3]. Amantadine and rimantadine are M2 ion channel blockers and activity of both drugs in prophylaxis is similar with 70 – 90% efficacy, which could be higher in the presence of previous antibodies. An important differentiating factor between the two is that they are metabolized differently. Prescription of adamantine to those with renal insufficiency and the elderly has to be done with care as the active metabolites are retained within the body for much longer [4]. Rimantadine is preferred if adamantanes have to be prescribed. The major pitfall with adamantanes is the high emergence of drug resistant variants and the variants are as ‘fit’ as the wild type in their transmissibility. A specific change, S31N, was associated with > 98% of adamantine resistance observed in H3N2 resistant variants isolated in recent years [2]. Derivatives of amantadine and rimantadine exist and are effective against H2N2 and H3N2. However, potency, safety and emergence of resistance still need to be evaluated.
2.2. Neuraminidase inhibitors
Influenza neuraminidase (NA) is a surface glycoprotein (GP) that is found on the viral envelope which cleaves sialic acid residues that bridge the virus with the host. Cleavage of this residue is essential for the newly formed virus to be released from the host cell. Treatment with NA inhibitors results in virions accumulating on the surface of the host cell without being able to infect other cells and hence viral spread is prevented. The first NA inhibitors were sialic acid analogs, 2-deoxy-2,3-didehydro-N-acetylneuraminic acid and 2-deoxy-2,3-dehydro-N-trifluoroacetylneuraminic acid, which were the lead compounds for the development of zanamivir and oseltamivir.
2.2.1. Oseltamivir
Oseltamivir is a potent selective inhibitor of NA [5]. Tumpey et al. have reported that recombinant virus possessing the 1918 NA is inhibited by oseltamivir in cultured cells and in mice [6] and, therefore, may be effective against a reemerging 1918 or 1918-like virus. Oseltamivir is also regularly used as a drug against seasonal influenza. When administered early, it reduces the amount of shed virus, duration of shedding and duration of symptoms/complications such as bronchitis and sinusitis.
The high desirability of oseltamivir was due to the low reports of resistant viruses until a few years ago. The resistant viruses were ‘unfit’ and were not as infectious as the parental version. Specific mutations associated with oseltamivir resistance include E119K, R292K, H274Y and R152K [7]. E119K and R292K mutants can be easily explained by the fact that oseltamivir makes contacts with arginine in position 292 and glutamic acid in position 119 [8]. The E119K mutant is more worrisome as it retains growth and transmissibility of the wild-type virus. The H274Y mutation has been observed in H5N1 infected influenza patients [9] and in some cases associated with death. However, of great concern is the emergence of oseltamivir resistant versions of H1N1 in the 2007 – 2008 seasons [10]. Additionally, person to person transmission of oseltamivir resistant versions of influenza A and B viruses has also been documented [11]. H5N1 also shows resistance to oseltamivir [12].
2.2.2. Zanamivir
Zanamivir is a second NA inhibitor that is an inhaled powder and its effect is localized to the respiratory tract. Zanamivir may be associated with side effects especially in the context of pre-existing pulmonary disease [13]. There are reports of resistance to zanamivir and the mutations associated with resistance are E119G/A/D, R152K and R292K. Using zanamivir as the lead compound, dimeric NA inhibitors were synthesized that displayed good efficacies in in vitro and in vivo studies [14].
2.2.3. Peramivir
Peramivir is one of several cytopentane derivatives that is also considered an NA inhibitor [15]. Peramivir is effective against H1N1 and H5N1 in mice. However, its strong efficacy did not extend to humans due to low bioavailability. The FDA has authorized emergency administration of peramivir as an investigational new drug (IND) for 2009 H1N1 hospitalized adult and pediatric patients.
2.3. Entry inhibitors
Hemagglutinin is an influenza virus membrane protein that recognizes sialylglycoconjugate receptors on the host cell surface. Sialic acid-containing lipids [16] and polymers [17] are used as entry blockers. Dendritic sialic polymeric inhibitors are effective against H3N2 and H2N2 in vitro [18]. Sialyloligosaccharides containing poly l-glutamic acid backbones [19] and sialyllactose-carrying polystyrene [20] are also effective inhibitors of viral multiplication in vitro. Sialic acid-mimic peptides have been developed as alternative inhibitors that are effective against H1N1 and H3N2 in vitro [21]. A recombinant fusion protein containing the sialidase catalytic domain from Actinomyces viscosus fused with a cell surface anchoring sequence conferred broad spectrum protection against influenza in cell culture and in animal models [22].
2.4. IFN (inducers)/NS1 inhibitors
NS1 is an IFN antagonist [23] and is essential for efficient viral replication. Natural mutations and deletions occurring in NS1 impair its anti-IFN activity and cause attenuation of virus [24]. Hence, chemical inhibitors of NS1 or IFN inducers could function in an antiviral capacity. A screen for NS1 inhibitors using 2000 compounds from the National Cancer Institute Diversity Set library identified four compounds (NSC109834, NSC128164, NSC95676, NSC125044) that inhibited influenza replication in cultured cells [25]. The manuscript demonstrated that NSC109834 and NSC128164 strongly affected viral protein synthesis and hypothesizes that the decrease may be due to inhibition of viral re-infection and spread. Their data support a significant increase in IFN mRNA production in the presence of the drugs and inhibition of NS1 function, which in turn contributed to the low titers. Interestingly, they also demonstrated that the effects are specific to influenza, as respiratory syncytial virus (RSV) is not susceptible.
2.5. RNA polymerase inhibitors
Influenza viruses use a heterotrimeric viral polymerase complex consisting of three subunits PB1, PB2 and PA for replication and transcription. The fact that the formation of this heterotrimeric complex is essential and that PB1 is highly conserved lend the polymerase structural association and function as an attractive target for design of novel inhibitors.
T-705 or favipiravir is an IND that has been demonstrated to have antiviral activity against influenza A, B and C viruses in vitro and in vivo [26]. It affects viral RNA synthesis and is effective against oseltamivir resistant and sensitive H5N1 pathogenic viruses as well as in a mouse model [27]. T-705 is also effective against a wide range of RNA viruses including poliovirus, rhinovirus, RSV, West Nile Virus and arenaviruses as is indicated in other sections of this review.
A novel fusion peptide (a PB1 derived peptide of influenza A with a single influenza B specific amino-acid substitution) that can bind to both influenza A and B PB1 subunit (PB11 – 25AT6Y) has been shown to inhibit H1N1 and H5N1 viral spread by blocking polymerase activity in cell culture [28].
Three siRNAs targeting the polymerase A gene (ps-PA496, ps-PA1116 and ps-PA1473) of H5N1 can effectively inhibit viral replication in cell culture [29].
Influenza is dependent on the host mRNAs for obtaining the 5′ cap structures for its own messages. A series of mesoionic heterocycles with 5,6-fused ring systems analogous to the N(7m)G component of mRNA cap structures were synthesized and examined for the ability to inhibit the cap-binding activity of the RNA polymerase complex [30]. None of the compounds tested were able to inhibit binding and endonucleolytic cleavage of a synthetic radiolabeled capped mRNA in vitro. However, compounds analogous to the mesoionic N(7m)G component of mRNA cap structures offer promise as potential inhibitors of the polymerase. Similar to cap analogs, compounds called 4-substituted 2,4-dioxobutanoic acids were able to inhibit cap-dependent endonuclease activity of the polymerase complex and inhibit viral replication in cell culture and animal models [31]. This was demonstrated more than a decade ago and to our knowledge, no publications about these compounds have been seen in the recent years raising the question if these compounds should be revisited.
2.6. Host-based inhibitors
2.6.1. Ribavarin
Ribavarin has long been recognized for its broad spectrum antiviral properties and lack of resistant mutants. Ribavarin has a range of effects in the host cell including targeting the host cell inosine-5′-monophosphate dehydrogenase (IMP dehydrogenase) enzyme, acting as a nucleoside mimic and influencing mRNA translation. Ribavarin is effective against human and avian influenza viruses [32]. A triple combination of ribavarin, oseltamivir and amantadine was demonstrated to be highly synergistic in inhibition of influenza viruses (H1N1, H3N2 and H5N1) and has a 2- to 13-fold greater inhibitory effect than any double combinations in cell culture studies [33]. Viramidine, a 3-carboxamidine derivative of ribavarin, is effective against H1N1, H3N2 and H5N1 viruses in vitro and in mice [32].
2.6.2. Signal transduction inhibitors
A recent focus for the development of antivirals against highly mutable viruses such as influenza is on cellular factors that influence viral replication. Intracellular signaling cascades and their associated inhibitors are promising candidates. Inhibition of the mitogenic Raf/MEK/ERK kinase cascade and blocking the activation of NF-κB impair influenza viral production in vitro and in vivo [34]. As proof of concept, U0126, a MEK inhibitor, reduced viral titers in the lungs of infected mice. Similarly, inhibition of NF-κB in cell culture using BAY11-7085, BAY11-7082 or Ly294002 was effective in increasing resistance to influenza virus infection [35]. Acetylsalicylic acid (ASA), also known as aspirin, is an effective and selective inhibitor of IKK2, the NF-κB activating kinase. Treatment of cells with ASA blocked replication of influenza viruses including H5N1 [35]. Greatly encouraging is the fact that when ASA was administered in aerosol form, it was protective in mice, reducing viral titers in the lung and promoting survival [35].
2.6.3. mAbs
Recent reports demonstrate the in vitro and in vivo efficacy of using mAbs (A06) against isolates of the 2009 H1N1 influenza virus [36]. A06 was capable of neutralizing H5N1, seasonal H1N1 and 2009 Swine influenza with similar potency in vitro, and could be used to treat infected mice highlighting the potential of mAbs as therapeutics. Another example of a mAb used as a therapeutic in case of influenza is MAb 9F4, which was shown to be efficacious against influenza in murine models [37]. While we have listed only a couple of examples, this is a promising strategy that is currently being explored as an effective treatment option against influenza.
3. Smallpox
Variola major, the causative agent of smallpox belongs to the poxviridae family. The successful vaccination program carried out by the WHO had eradicated smallpox by the 1980s. Since then, vaccinations have been stopped and this has created a large population of humans who are vulnerable to infections by poxviruses. The recent outbreak of monkeypox in the US in 2003 underscores the serious threat. Also, smallpox is considered a biothreat agent as it can be transmitted in an aerosol form which has initiated a search for therapeutics against smallpox and monkeypox infections.
3.1. Viral DNA synthesis inhibitors
Inhibitors that interfere with poxvirus DNA synthesis are nucleoside analogs that include IMP dehydrogenase inhibitors, S-adenosylhomocysteine (SAH) hydrolase inhibitors, orotidine 5′-monophosphate (OMP) decarboxylase/CTP synthetase inhibitors, thymidylate synthase inhibitors and acyclic nucleoside phosphonates [38].
Ribavarin, a broad spectrum antimicrobial that has been discussed in other parts of this review, is also an inhibitor of poxviruses. Ribavarin efficacy as a topical in vaccinia virus-induced keratitis has been demonstrated in rabbits [39]. There are structurally related analogs with similar antiviral activity and the most promising of these is EICAR (5-ethynyl-1-d-ribofuranosylimidazole-4-carboxamide) [40].
SAH hydrolase is an enzyme that breaks down SAH into adenosine and homocysteine. The enzymatic activity is an essential component in the metabolic pathways of sulfur-containing amino acids and, so, inhibitors are expected to be useful as antimicrobials [38]. Some of the SAH inhibitors that have antiviral activity against vaccinia virus in cell culture include dihydroypropyl adenine [41], carbocyclic 3-deaza-adenosine [42] and neplanocins A and C [43].
Both OMP decarboxylase and CTP synthetase inhibitors affect RNA synthesis by preventing conversion of OMP to UMP and UTP to CTP, respectively. Pyrazofurin is the prototype OMP decarboxylase inhibitor that inhibits vaccinia virus without apparently toxicity to the host cell [38]. Carbodine, cyclopentyl cytosine (C-Cyd) and cyclopentenyl cytosine (Ce-Cyd) are examples of CTP synthetase inhibitors.
5-substituted deoxyuridines have been recognized as strong antiviral agents [38]. Many of these compounds, particularly thymidylate synthase inhibitors, are also known for their antitumor activity [44]. They exert their antiviral and antitumor function by interfering with the step that converts deoxy UMP to deoxy TMP.
Adenine arabinoside A (Ara-A) is a nucleoside analog that targets viral DNA synthesis [45]. By competing with dATP, Ara-A exerts a negative effect on the viral DNA polymerase.
Cidofovir or CDV-Vistide (an acyclic nucleoside phosphonate) is the only antiviral that is currently FDA approved for use against orthopoxviruses [46]. It is also licensed for treatment of retinitis caused by cytomegalovirus (CMV) [47] in AIDS patients. It was recently demonstrated that cidofovir inhibited genome encapsidation and affected morphogenesis in vaccinia infection [48]. In vivo efficacy was demonstrated in mice by showing lowered viral replication in different organs and by preventing vaccinia induced death. However, there are two main problems associated with the use of cidofovir. First, there is strong renal toxicity associated with the drug and second, there is low oral bioavailability. The recently developed hexadecyloxypropyl ester of cidofovir (CMX001) shows improved oral bioavailability and displays activity against a number of viruses that encode their own DNA polymerase [49,50].
3.2. Viral maturation inhibitors
The search for antivirals against poxviruses began when it was demonstrated > 50 years ago that thiosemicarbazones that were used as anti-tuberculosis agents were active against vaccinia virus infections. The thiosemicarbazone derivative methisazone was effective in smallpox prophylaxis [51]. The mechanism of action is thought to be interference with viral maturation downstream of viral protein synthesis from late viral mRNAs.
Rifampin is another anti-tuberculosis drug that is effective in curbing vaccinia multiplication. Rifampin is thought to block virus assembly by interacting with a 65 kDa polypeptide that is a product of the D13 gene [52]. However, the drug needed to be administered at high concentrations, which did not bode well for clinical use.
Mitoxantrone is an antineoplastic drug which inhibits DNA replication and DNA-dependent RNA synthesis. It also functions as a topoisomerase II poison and causes double-stranded DNA breaks [53]. It is a synthetic anthraquinone derivative that is used for treatment of acute myeloid leukemia [54], prostate cancer [55] and MS [56]. It was found to inhibit vaccinia replication at a step subsequent to viral early gene expression by a mechanism involving a late stage block to virus assembly [57]. Even though the drug did not perform well in animal models, the strong inhibitory effect in cell culture warrants further studies.
3.3. Egress inhibitors
Tecovirimat, also called ST-246 [46,58,59], is an inhibitor of orthopoxvirus egress from infected cells by targeting viral p37 protein orthologs (Figure 2). The drug is to be administered orally. It is an inhibitor of the core protein cysteine proteinase encoded by the poxvirus I7L gene. It blocks the ability of the virus to spread to neighboring cells and hence prevents disease. ST-246 has been shown to be effective in small animal and non-human primate studies. A Phase II clinical trial is currently in progress [59].
Figure 2.
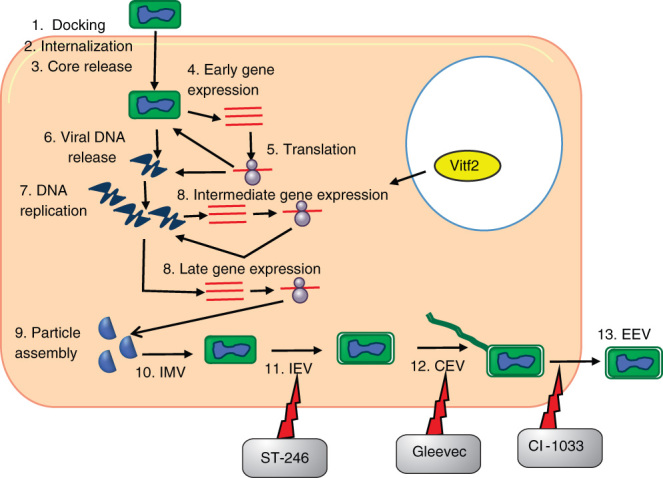
The complex life cycle of smallpox virus is depicted. After docking on the host cell surface, the virus releases its core into the cytoplasm. The gene expression pattern follows an early, intermediate and late gene expression time line after which the viral particles are assembled. The complex particle assembly culminates in the enveloped extracellular virion being released from the infected cell. Points of action by the morphogenesis inhibitors are specified.
3.4. Host-based inhibitors
3.4.1. Gleevec (STI-571)
Poxvirus cell associated enveloped virions (CEVs) use abl and src family tyrosine kinases for actin mobility. Release of CEV from the host cell requires abl family tyrosine kinases and is blocked by STI-571 or gleevec (imatinib mesylate (Figure 2)), an abl family kinase inhibitor used to treat chronic myeloid leukemia in humans [60]. Reduced infectivity in the presence of gleevec was demonstrated in cell culture and a mouse model [61]. Of great interest is the fact that gleevec is only one of three kinase inhibitors that has been cleared by the FDA for use in humans as a cancer drug.
3.4.2. CI-1033
EGF-like growth factors carried by poxviruses play important roles in viral pathogenesis. Smallpox growth factor has an EGF-like domain that targets ErbB-1 kinases and induces tyrosine phosphorylation of host substrates, facilitating viral replication. CI-1033 is a 4-anilinoquinazoline with strong specificity to ErbB kinases that has been used as an anticancer therapeutic [62]. CI-1033 had a prominent effect on comet formation in vaccinia infected cells. Reduction in comet size indicates a delay in formation of extracellular enveloped virus (EEV) and net reduction in EEV numbers. This suggests the interference of CI-1033 in viral morphogenesis is a way for preventing viral egress and spreading to neighboring cells (Figure 2). While this was effectively demonstrated in vitro, in animals the drug was most effective when used in conjunction with the anti-L1R mAb [63].
4. Hemorrhagic fever viruses
Hemorrhagic fever viruses (HFV) are a group of diverse viruses that have the ability to cause viral hemorrhagic fever (VHF). VHF is caused by multiple viral families, including filoviruses, arenaviruses, bunyaviruses and flaviviruses [64]. The majority of HFV are considered to pose serious risk as biological weapons and have, therefore, been classified as Category A agents [64]. Here, we limit our discussion to the filoviruses (Figure 3), arenaviruses (Figure 4) and bunyaviruses (Figure 5).
Figure 3.
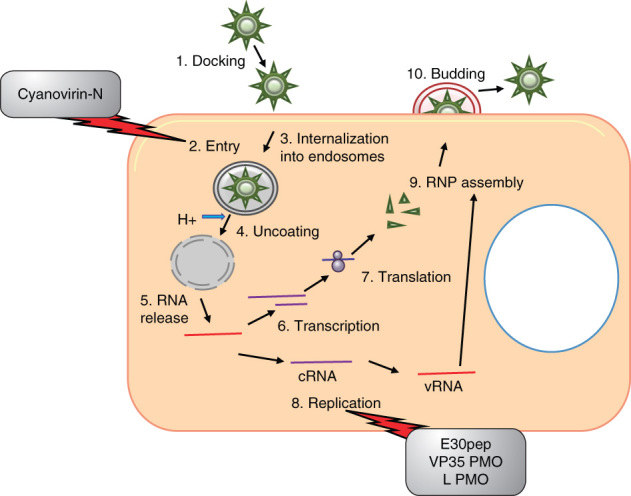
Inhibitors of filoviruses in relation to the viral life cycle. Filoviruses replicate entirely in the cytoplasm. Filoviruses bind to target cells and undergo fusion and internalization into endosomes, resulting in the uncoating and release of RNPs into the cytoplasm. Transcription occurs from the 3′ to 5′ end beginning at the leader sequence and the RdRp recognizes distinct genes through the start and stop signals flanking the viral genes. Structural proteins and RNPs are transported to the plasma membrane where assembly and budding occur. The entry inhibitor Cyanovirin-N and PMOs that inhibit viral replication are shown.
PMO: Phosphorodiamidate morpholino oligomer; RNP: Ribonucleoprotein.
Figure 4.
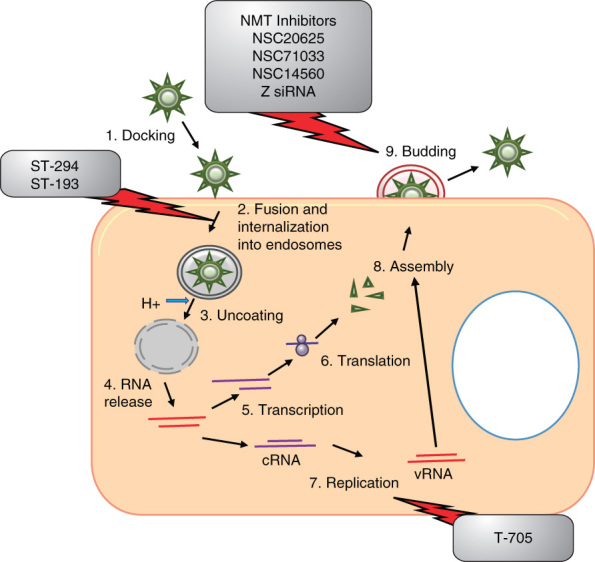
Inhibitors of arenaviruses in relation to the viral life cycle. Arenaviruses are cytoplasmic viruses. Arenaviruses bind to target cells and undergo fusion and internalization into endosomes, resulting in the uncoating and release of RNPs into the cytoplasm. Arenaviruses use an ambisense coding strategy for transcription and replication. Following translation, structural proteins and RNPs are transported to the plasma membrane where assembly and budding occur. The fusion inhibitors ST-294 and ST-193, replication inhibitor T705 and multiple budding inhibitors are shown.
RNP: Ribonucleoprotein.
Figure 5.
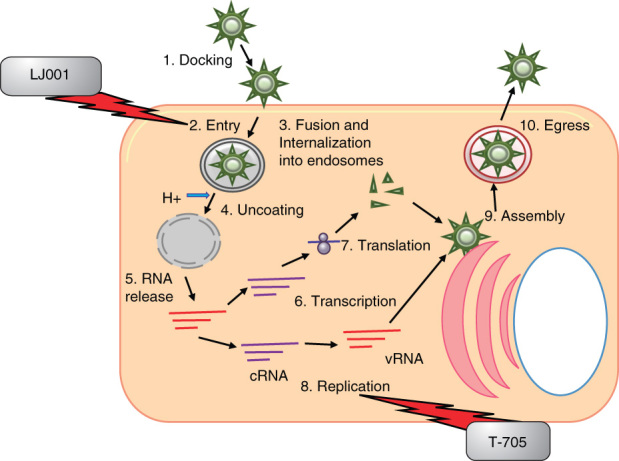
Inhibitors of bunyaviruses in relation to the viral life cycle. Bunyaviruses have an entirely cytoplasmic life cycle. They enter cells via receptor-mediated endocytosis, RNPs are released into the cytoplasm, and transcription and translation occur with glycoproteins being translated and maturing in the ER. Replication of the viral genome occurs and the viral RNP and glycoproteins are transported to the Golgi where budding takes place. Large secretory vesicles filled with numerous viruses are transported to the plasma membrane and virus release through exocytosis. The entry inhibitor LJ001 and the RdRp inhibitor T705 are depicted.
ER: Endoplasmic reticulum; RNP: Ribonucleoprotein.
4.1. Filoviruses
Ebolavirus (EBOV) and marburgvirus (MARV) were identified during deadly outbreaks of hemorrhagic fever in 1976 and 1967, respectively [65]. Filoviruses are negative-stranded RNA viruses of 18 – 19 kb in size. There are several vaccine candidates in development for filoviruses which show promising results in non-human primates [65], including virus-like particle vaccines, replication-defective adenovirus serotype 5, Venezuelan equine encephalitis virus (VEEV) replicon vaccine and a recombinant vesicular stomatitis virus vaccine [66]. As there are no FDA approved vaccines or antivirals, the scientific community is focusing heavily of the identification of novel antivirals for treatment.
4.1.1. Entry inhibitors
The entry process of filoviruses is still an evolving field of research, but there are some similarities between filovirus and HIV entry. Cyanovirin-N (CV-N) is a cyanobacterial lectin that has demonstrated antiviral activity against numerous enveloped viruses, including HIV and EBOV [67]. CV-N recognizes N-linked high-mannose oligosaccharides, Man-8 and Man-9. Zaire EBOV GP and MARV GP-pseudotyped viruses can be inhibited by treatment with CV-N [68]. Treatment with CV-N in tissue culture resulted in inhibition of cytopathic effects and mice infected with EBOV displayed delayed time to death [69]. In addition, CV-N binds to GP1 and GP2 with similar affinity as it binds to HIV gp120 [69]. While the filovirus receptor(s) is currently unknown, CV-N may prevent interactions between the viral GPs and the cellular receptors inhibiting viral entry and fusion. The crystal structure of the trimeric EBOV GP in complex with neutralizing antibody fragment has recently been described [70,71] and should provide important information to aid in rational antibody and small molecule compound design for entry inhibition.
4.1.2. Transcription inhibitors
The RNA-dependent RNA polymerase L is the main driving factor for filovirus transcription. To date, there are no known EBOV L polymerase inhibitors described in the literature. The nucleocapsid protein NP and the IFN antagonist VP35 are also critical for filovirus transcription [65]. In addition, VP30 is an essential EBOV-specific transcription factor that must form oligomers to allow EBOV transcription [72]. Hartlieb et al. identified a 25-mer peptide (E30pep) that binds to VP30, preventing its oligomerization, resulting in inhibition of viral replication in tissue culture [72]. These novel results are the first demonstration of inhibiting EBOV replication through targeting the replication complex.
4.1.3. Oligonucleotide antivirals
In the filovirus field there has been a push for the use of oligonucleotides, siRNAs and nucleic acid analogs, phosphorodiamidate morpholino oligomers (PMOs), as antiviral therapeutics [73]. Warfield et al. utilized antisense PMOs targeting VP24, VP35 and L for the treatment of EBOV infection [74]. Mice treated with PMOs either pre- or post-EBOV exposure were 100% protected when a combination of all three PMOs were utilized. Treatment with VP24 or VP35 PMOs alone also provided nearly complete protection, whereas the L PMO treatment resulted in only 30% survival. Combination of all three viral PMOs protected 75% of rhesus macaques from a lethal EBOV challenge. To further enhance PMOs, arginine-rich peptides can be conjugated to the backbone, allowing greater uptake into the cells, and are referred to as p-PMOs. A VP35 p-PMO has been shown to be effective both pre- and post-infection in a lethal EBOV mouse model [75]. P-PMOs that had two to four positive charges on the backbone demonstrated enhanced activity in vitro as well as in mice infected with EBOV [76]. In addition, PMOplus molecules have been developed, which contain piperazine linkages within the PMO backbone [76]. A 2010 study indicated that both pre- and post-exposure treatment with a combination of VP35 and VP24 PMOplus molecules (designated AVI-6002) resulted in substantial protection from Zaire EBOV challenge in both mice (greater than 90%) and guinea-pigs (83%) [77]. In addition, treatment initiated 30 – 60 min post-exposure results in > 60% protection in rhesus macaques [77]. A combination of NP and VP24 PMOplus molecules for Lake Victoria MARV (designated AVI-6003) demonstrated 100% protection of cynomolgus monkeys [77]. Based on these results, IND applications for both AVI-6002 and AVI-6003 have been submitted to the FDA [77].
siRNAs are a related strategy that have shown promise for both EBOV and MARV infections. siRNAs against MARV NP, VP35 and VP30 decreased the levels of viral proteins and viral release [78]. Likewise, Groseth et al. screened Zaire EBOV siRNA candidates through the use of a minigenome system and found siRNA targeting NP (ZNP1) to be the most effective at reducing viral titers in vitro [79]. Collectively, these studies indicated that oligonucleotide-based therapies are a viable option for the treatment of filovirus infection.
4.1.4. Host-based inhibitors
As stated above, SAH hydrolase is a cellular enzyme that catalyzes the hydrolysis of SAH to adenosine and homocysteine. A number of nucleoside analog inhibitors of SAH hydrolase inhibit EBOV replication, including carbocyclic 3-deazaadenosine and 3-deazaneplancin A [80-82]. A dramatic induction of IFN-α production was observed in EBOV infected mice treated with 3-deazaneplancin A, suggesting that IFN stimulation contributes to the observed protection [83]. While this class of compounds is able to inhibit EBOV infection in a lethal mouse model, limitations include toxicity issues and lack of post-infection efficacy.
There has been an intense effort to identify novel antivirals through high-throughput screening (HTS). FGI-103 (2-(2-(5-(amino(imino)methyl)-1-benzofuran-2-yl)vinyl)-1H-benzimidazole-5-carboximidamide) is a small molecule compound that was selected through in vitro screening of a GFP-expressing Zaire EBOV [84]. Further testing of FGI-103 indicated that it also inhibits wild-type Zaire EBOV, Sudan EBOV, MARV Ci67 and MARV Ravn. Treatment with FGI-103 24 h post-infection resulted in completed protection of mice against both EBOV- and MARV-induced death. While the mechanism of FGI-103 is not established, treatment resulted in delayed pro-inflammatory cytokine responses and decreased viral titers in the kidney, liver and spleen. Likewise, FGI-104 was identified through a screen aimed at identifying host-based inhibitors [85]. Importantly, FGI-104 demonstrated broad spectrum activity against HCV, HBV, HIV, EBOV, VEEV, Cowpox virus and porcine reproductive and respiratory syndrome virus infection in vitro. In vivo studies indicated that FGI-104 prevents lethality from EBOV. TSG-101 has been suggested as the molecular target of FGI-104, but studies are underway to confirm this hypothesis. Finally, a third antiviral, FGI-106, was identified through HTS. FGI-106 displayed broad spectrum activity with the ability to inhibit EBOV, Rift valley fever virus (RVFV) and dengue [86]. In vivo, FGI-106 has shown great promise, protecting mice from lethal EBOV infection.
Through a proteomic and RNAi approach, Spurgers et al. have recently identified host proteins that are specifically incorporated in EBOV and MARV virions [87]. The identified proteins fell into eight different categories, PI3K pathway, integrin signaling pathway, cell structure and motility, small GTPase, annexin, protein targeting and localization, stress response and chaperone proteins. siRNA-mediated knockdown of heat-shock 70 kDa protein 5 (HSPA5), ribosomal protein L18 (RPL18), ribosomal protein L5 (RPL5) and ubiquitin C (UBC) significantly inhibited EBOV replication without inducing cellular toxicity. HSPA5, RPL18 and UBC knockdown had a similar effect on MARV replication, while RPL5 resulted in an increase in MARV replication. These results suggest that inhibition of the host proteins can efficiently inhibit filovirus replication.
4.2. Arenaviruses
The family Arenaviridae contains 25 viruses, which can be subdivided into two major groups, the New World and the Old World arenaviruses [88]. Of these, Junin (JUNV), Machupo, Guanarito, Sabia, Lujo, Chapare and Lassa viruses (LASV) can cause hemorrhagic fever [88-90]. Arenaviruses are enveloped viruses with a bi-segmented negative ssRNA genome. There are no FDA approved vaccines or treatment options for arenaviruses. However, Candid #1 is a live attenuated vaccine against JUNV that was developed at USAMRIID [91], which has proven to be safe and effective in protecting agricultural workers during a Phase III clinical trial in South America [92]. Candid #1 is also an IND vaccine in the US [93]. Other therapeutic options for JUNV and other arenaviruses include transfusion of immune plasma and ribavirin. However, ribavirin is only partially effective and is associated with negative side effects [94]. Here, we discuss novel therapies that show potential for arenavirus treatment.
4.2.1. Z inhibitors
The Z protein is the main driving force for arenavirus budding. The Z protein is myristoylated at glycine position 2 [95] and this post-translation modification has become a target for antiviral design. N-myristoyl transferase (NMT) catalyzes the addition of myristate to N-terminal glycines. NMT inhibitors have been demonstrated to inhibit LASV and JUNV [96,97]. More recently, the zinc finger reactive compound NSC20625 has been shown to induce unfolding and oligomerization of Z as well as disrupt the interaction of Z protein with PML [98-100]. The aromatic disulfides NSC4492 and NSC71033 and the thiuram disulfide NSC14560 have antiviral activity against JUNV and two other arenaviruses [101]. It has been suggested that the viral inhibition is due to the loss of membrane localization of the Z protein.
siRNA-based antivirals targeting the Z protein have also been tested. Four Z-specific siRNAs (Z1- to Z4-siRNAs) were tested and Z2-siRNA, which targets nucleotides 179 – 197 of the Z gene, proved most effective [102]. Z2-siRNA displayed 92.8% JUNV yield reduction in plaque assays and a 91% reduction in expression of Z-EGFP fusion protein [102]. These results highlight the feasibility of Z inhibitor-based therapies.
4.2.2. GP2 inhibitors
The hemorrhagic fever causing arenaviruses must be studied at BSL-4; thus, often times, a related model virus is chosen for initial studies. Tacaribe virus is a BSL-2 arenavirus that can be used as a model for New World arenaviruses, such as JUNV. HTS of 40,000 small molecule compounds against Tacaribe virus identified ST-294 as a lead compound for drug development [103]. ST-294 was also capable of inhibiting JUNV, Machupo and Guanarito as well as protecting against Tacaribe in a mouse challenge model. Additional studies have indicated that ST-294 interferes with the GP precursor (GPC)-mediated membrane fusion [104]. Another small molecule compound, ST-193, which is a benzimidazole derivative, was also identified through a HTS assay for arenavirus entry inhibitors [105]. ST-193 can inhibit a broad range of arenaviruses including LASV, JUNV, Machupo and Guanarito and has been suggested to have a similar mechanism of action as ST-294 [104,105]. Both ST-294 and ST-193 prevented pH-mediated dissociation of the G1-receptor binding subunit from GPC, which is critical for fusion to occur. The inhibition of viral fusion is a promising strategy; however, the efficacy of these inhibitors in vivo has yet to be demonstrated.
4.2.3. Host-based inhibitors
T-705 is a novel pyrazine derivative that has been shown to successfully treat numerous RNA virus infections including arenaviruses (Tacaribe, JUNV and Pichinde virus (PICV)), without the toxicity observed with ribavirin [106]. T-705 has been discussed earlier in this review in relation to influenza. T-705 was efficacious in vivo in the PICV hamster infection model of arenaviral hemorrhagic fever [107]. Recently, Gowen et al. have tested T-705 in the late stages of PICV infection, which is critical for the successful clinical use of T-705. When treatment was initiated 4 or 5 days post-infection, 95% of the infected hamsters survived, but hamsters treated at day 6 showed only 50% survival [107]. Surprisingly, there was no added benefit of combination ribavirin and T-705 therapy. Collectively, these studies indicate that T-705 is a strong candidate for arenaviral therapy.
The GPC must be processed via the cellular proprotein convertase site 1 protease (S1P), also referred to as subtilisin-kexin-isozyme 1. To target this critical step for antiviral therapy, Rojek et al. used a peptide-based S1P inhibitor, decanoyl (dec)-RRLL-chloromethylketone (CMK). They found that dec-RRLL-CMK was able to block LCMV viral production and spread [108]. Treatment in combination with ribavirin resulted in more potent inhibition.
PI3K/AKT signaling is altered in many viral infections [109]. JUNV infection also results in induction of this signaling pathway, potentially through viral internalization [110]. Importantly, treatment of JUNV infected cells with the PI3K inhibitor, Ly294002, decreased viral protein synthesis and viral titers. Interestingly, in LY294002 treated cells, transferring cell-receptor recycling is blocked, suggesting reduced viral binding to the cell surface as a possible mechanism of action [110]. Along these same lines, treatment of the PICV infected cells with the tyrosine kinase inhibitor genistein inhibited infection [111]. PICV infection results in phosphorylation of the transcription factors ATF-2 and CREB, which can both be inhibited following treatment with genistein. Overall, these results indicate that tyrosine kinase activity is needed for arenavirus infection, but the particular kinase responsible has yet to be identified.
4.3. Bunyaviruses
Bunyaviridae is a large family of viruses, containing 5 genera and > 300 species. Within the Bunyaviridae family, RVFV, Crimean Congo hemorrhagic fever virus (CCHFV) and Hantaviruses can cause hemorrhagic fever. The main focus of research for CCHFV has been on the use and efficacy of ribavirin [112]. Disappointingly, one recent study indicated that treatment with ribavirin did not make any significant contribution to patient outcome [113]. Due to limited studies on additional antivirals for the treatment of CCHF, only RVFV and Hantaviruses are discussed below.
4.3.1. Rift valley fever virus
RVFV is a zoonotic virus that infects a number of wild and domestic animals and humans. Rift valley fever is a rapidly emerging disease that could have implications for the US population. There are two IND vaccines currently for RVFV. One is a formalin-inactivated vaccine and the other is a live attenuated vaccine, MP-12, which is made available to at-risk laboratory workers and troops [114-116].
4.3.1.1. Entry inhibitors
LJ001 is a novel broad spectrum inhibitor that is able to intercalate into viral membranes, but not cellular membranes [117]. It has no effect on non-enveloped viruses, but has been shown to inhibit influenza A, filoviruses, poxviruses, arenaviruses, bunyaviruses, paramyxoviruses, flaviviruses and HIV. LJ001 is a rhodanine derivative that can irreversibly inhibit viral entry after viral binding but before virus–cell fusion.
4.3.1.2. Host-based inhibitors
Viral infection stimulates the IFN response, which many viruses have evolved mechanisms to evade. RVFV encodes an IFN antagonist, NSs, which potently and rapidly inhibits IFN stimulation through altering transcription and chromatin remodeling factors [118]. Based on this known virulence factor, studies have been performed to test the effect of IFN treatment. IFN alfacon-1 (trade name Infergen, Three Rivers Pharmaceuticals, Cranberry Township, PA, USA) was tested in both cell culture models of Punta Toro virus (PTV) and RVFV infection. Results indicated that pretreatment of cells with IFN alfacon-1 reduced viral titers [119]. Importantly, when IFN alfacon-1 was tested in a PTV hamster model, complete protection to a lethal challenge was observed at up to 36 h post-infection.
Like many of the RNA viruses discussed, T-705 is also a promising antiviral for RVFV infection. T-705 was shown to be efficacious in treating PTV infection in both mouse and hamster models, allowing a higher percent survival than treatment with ribavirin alone [106]. More recently, a related pyrazine, T-1106, was tested for its antiviral activity against PTV and found to be a promising therapeutic both in in vitro and in vivo model systems [120].
4.3.2. Hantaviruses
Hantaviruses can be divided into Old and New World Hantaviruses. Old World Hantaviruses are the causative agent of hemorrhagic fever with renal syndrome (HFRS) and New World Hantaviruses cause human pulmonary syndrome [121].
There are limited studies on therapeutics against Hantaviruses. However, one particular area of focus has been on preventing Hantavirus entry through targeting the integrin receptor. Antibodies against β3 integrin and its ligand, vitronectin, were able to prevent NY-1 and Sin Nombre infection, but not Prospect Hill infection [122]. Instead, Prospect Hill virus infection was inhibited by fibronectin and β1-specific antibodies. To take advantage of the importance of β3 integrin for Sin Nombre entry, Larson et al. developed a cyclic nonapeptide, cyclo-[CPFVKTQLC], which inhibited Sin Nombre virus entry [123]. This inhibition was comparable to inhibition displayed by ReoPro, a humanized mAb against the β3 subunit. ReoPro is an FDA approved antithrombotic, making it a perfect candidate for treatment of human pulmonary syndrome through drug repositioning. This peptide has been further refined for optimal viral inhibition and the resulting peptide is cyclo-[CPFVC] [124].
For HFRS, two antibody therapies show promise. One is a murine mAb against Hantaan virus (designated AHM) that has recently undergone a Phase I clinical trial [125]. The trial showed that AHM had a relatively long bioavailability and was well tolerated in volunteers as evidenced by no severe adverse effects. The second antibody, scFv3G1, which targets Hantaan virus GP binds to Hantaan GP and is internalized in Hantaan virus infected cells [126]. While scFv3G1 is clearly at a much more preliminary stage of development, it shows potential for treatment of Hantaan virus infection.
5. Conclusion
Continuing and unpredictable onslaughts by multiple emerging pathogens pose serious economic and health burden all over the globe. Additionally, the threat of engineered bioterrorism agents highlights our extreme unpreparedness to deal with a manmade or a natural outbreak. Long-term use of many antivirals has led to emergence of drug resistance in many instances. Renewed efforts have focused on understanding the pathogens and their relationship with the host to highlight pathogen vulnerabilities that can be exploited. This review only takes into consideration a handful of examples. The pathogen list is much longer and growing thus bringing into stark focus the immediate need for some broad spectrum antimicrobials.
6. Expert opinion
For many years, antimicrobials have centered on targeting the pathogen for inhibition of synthesis, activity or egress of the pathogen. The repeated lesson from many highly mutable viruses such as influenza and HIV is that viruses find a way around the inhibitor. A second consideration is that the pathology is also a result of host response gone awry. Many host responses are set in motion early in the infectious process that trigger a domino effect with multiple cascades feeding into one another. Researchers have now recognized and acknowledged the host response as being an important component that requires attention. An apparent advantage in controlling adverse host responses is the ability to ‘buy the host more time’ to stabilize the host before the administered antiviral can control viral multiplication.
Viruses are dependent on the host for successful replication. Many viruses, with some exceptions such as the smallpox virus, have limited genomes and hijack host components to complete their life cycle and it is this dependence that can be exploited. As has been shown for many pathogens, inhibitors to multiple key signal transduction components have proven to be effective in inhibiting viral multiplication and infectivity. Utilizing the host machinery to design drugs against viruses has two main advantages. First, it is virtually impossible for the virus to evolve drug resistance. Second, as many of these pathways intercept at multiple nodes and have common components as upstream activators and downstream targets broad spectrum inhibitors can be developed. However, it should not be forgotten that targeting conserved host pathways may prove to be deleterious to the host itself. This necessitates stringent toxicity control studies before a host target can be therapeutically utilized. The concept of combinatorial therapy is also gaining much attention these days. Combinatorial therapy has to be looked at beyond targeting multiple components of the virus at once. In the case of influenza, combinations of oseltamivir, peramivir and ribavarin effectively targeted multiple components of the viral life cycle while also placing a check on the host. An important consideration during development of combinatorial therapies, however, is that obtaining regulatory approval for usage of the product can be more difficult and expensive as multiple criteria for all the components in the combinatorial strategy have to be satisfied.
‘Drug repositioning’ is a catch phrase that would appeal to drug makers, the public and the scientific community. Repositioned drugs are those that have been designed, FDA approved and are in the market for a completely unrelated disease, but work effectively as antivirals. Now, many drugs that are in the market for treatment of cancerous conditions are tested for their ability to function as antimicrobials. This is a useful avenue to pursue not only because we minimize time and effort in designing additional drugs, but we also bypass the expenses involved in optimization, clinical trials, FDA approvals and so on. Current tools utilized to identify candidates that can be repositioned, however, utilize mathematical models, computerized prediction tools and such methods that often make certain assumptions and may exclude relevant information. Such efforts often focus on the obvious benefit, but attention should also be focused on the risk-side of the equation such as adverse effect profile for the repositioned candidates.
There are many tools available to conquer these pathogens. The pathogen–host interaction is an ever-evolving dance where for every push there is a counter push. Newer technologies that are constantly emerging provide greater sensitivity in detection and avenues for development of potent drugs. The same technologies also reveal how far behind the scientific community is in their ability to effectively inhibit many infectious agents.
Article highlights.
Influenza is a disease that has affected the human population for many years and attempts to find an effective therapeutic continue. The article highlights the currently approved drugs for use as anti-influenza therapeutics while also emphasizing the problem of drug resistance.
Smallpox vaccinations have been discontinued due to its status of being ‘eradicated’. This has created an entire population of individuals who are susceptible to infection should there be any reason for an exposure in the future. The scientific community is addressing the issue of novel therapeutics against smallpox. Drug repositioning appears to be providing some solutions to the problem.
Hemorrhagic fever viruses pose a great threat to the population due to the lack of FDA approved drugs against many of these viruses. Extensive research is ongoing in this arena and multiple candidate therapeutics against the different families of hemorrhagic fever viruses has been indicated.
The article considers the exciting possibility of using host responses for the design of novel therapeutics. Host response-based therapeutics has been shown to be effective by many researchers against influenza, smallpox and hemorrhagic fever virus infections.
This box summarizes key points contained in the article.
Declaration of interest
This manuscript was supported by DOE funding (DOE Award #DE-SC0001599) for C Bailey.
Bibliography
- 1.Taubenberger JK, Morens DM. Influenza: the once and future pandemic. Public Health Rep 2010;125(Suppl 3):16-26 [PMC free article] [PubMed] [Google Scholar]
- 2.De Clercq E. Antiviral agents active against influenza A viruses. Nat Rev Drug Discov 2006;5(12):1015-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davies WL, Grunert RR, Haff RF,. Antiviral activity of 1-adamantanamine (Amantadine). Science 1964;144:862-3 [DOI] [PubMed] [Google Scholar]
- 4.Nakai K, Takeda K, Kimura H,. Obstructive acute renal failure related to amantadine intoxication. Am J Emerg Med 2009;27(3):371 e5-71 e7 [DOI] [PubMed] [Google Scholar]
- 5.Smith JR. Oseltamivir in human avian influenza infection. J Antimicrob Chemother 2010;65(Suppl 2):ii25-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tumpey TM, Garcia-Sastre A, Mikulasova A,. Existing antivirals are effective against influenza viruses with genes from the 1918 pandemic virus. Proc Natl Acad Sci USA 2002;99(21):13849-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferraris O, Lina B. Mutations of neuraminidase implicated in neuraminidase inhibitors resistance. J Clin Virol 2008;41(1):13-9 [DOI] [PubMed] [Google Scholar]
- 8.Yen HL, Herlocher LM, Hoffmann E,. Neuraminidase inhibitor-resistant influenza viruses may differ substantially in fitness and transmissibility. Antimicrob Agents Chemother 2005;49(10):4075-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hurt AC, Holien JK, Parker MW, Barr IG. Oseltamivir resistance and the H274Y neuraminidase mutation in seasonal, pandemic and highly pathogenic influenza viruses. Drugs 2009;69(18):2523-31 [DOI] [PubMed] [Google Scholar]
- 10.Hauge SH, Dudman S, Borgen K,. Oseltamivir-resistant influenza viruses A (H1N1), Norway, 2007-08. Emerg Infect Dis 2009;15(2):155-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duwe S, Heider A, Braun C,. Person-to-person transmission of oseltamivir-resistant influenza A/H1N1 viruses in two households; Germany 2007/08. J Clin Virol 2009;46(3):295-7 [DOI] [PubMed] [Google Scholar]
- 12.Hurt AC, Lowther S, Middleton D, Barr IG. Assessing the development of oseltamivir and zanamivir resistance in A(H5N1) influenza viruses using a ferret model. Antiviral Res 2010;87(3):361-6 [DOI] [PubMed] [Google Scholar]
- 13.Williamson JC, Pegram PS. Neuraminidase inhibitors in patients with underlying airways disease. Am J Respir Med 2002;1(2):85-90 [DOI] [PubMed] [Google Scholar]
- 14.Masuda T, Shibuya S, Arai M,. Synthesis and anti-influenza evaluation of orally active bicyclic ether derivatives related to zanamivir. Bioorg Med Chem Lett 2003;13(4):669-73 [DOI] [PubMed] [Google Scholar]
- 15.Castillo R, Holland LE, Boltz DA. Peramivir and its use in H1N1 influenza. Drug Today 2010;46(6):399-408 [DOI] [PubMed] [Google Scholar]
- 16.Guo CT, Sun XL, Kanie O,. An O-glycoside of sialic acid derivative that inhibits both hemagglutinin and sialidase activities of influenza viruses. Glycobiology 2002;12(3):183-90 [DOI] [PubMed] [Google Scholar]
- 17.Matrosovich M, Klenk HD. Natural and synthetic sialic acid-containing inhibitors of influenza virus receptor binding. Rev Med Virol 2003;13(2):85-97 [DOI] [PubMed] [Google Scholar]
- 18.Landers JJ, Cao Z, Lee I,. Prevention of influenza pneumonitis by sialic Acid-conjugated dendritic polymers. J Infect Dis 2002;186(9):1222-30 [DOI] [PubMed] [Google Scholar]
- 19.Totani K, Kubota T, Kuroda T,. Chemoenzymatic synthesis and application of glycopolymers containing multivalent sialyloligosaccharides with a poly(L-glutamic acid) backbone for inhibition of infection by influenza viruses. Glycobiology 2003;13(5):315-26 [DOI] [PubMed] [Google Scholar]
- 20.Tsuchida A, Kobayashi K, Matsubara N,. Simple synthesis of sialyllactose-carrying polystyrene and its binding with influenza virus. Glycoconj J 1998;15(11):1047-54 [DOI] [PubMed] [Google Scholar]
- 21.Matsubara T, Onishi A, Saito T,. Sialic acid-mimic peptides as hemagglutinin inhibitors for anti-influenza therapy. J Med Chem 2010;53(11):4441-9 [DOI] [PubMed] [Google Scholar]
- 22.Malakhov MP, Aschenbrenner LM, Smee DF,. Sialidase fusion protein as a novel broad-spectrum inhibitor of influenza virus infection. Antimicrob Agents Chemother 2006;50(4):1470-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ehrhardt C, Seyer R, Hrincius ER,. Interplay between influenza A virus and the innate immune signaling. Microbes Infect 2010;12(1):81-7 [DOI] [PubMed] [Google Scholar]
- 24.Solorzano A, Webby RJ, Lager KM,. Mutations in the NS1 protein of swine influenza virus impair anti-interferon activity and confer attenuation in pigs. J Virol 2005;79(12):7535-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Basu D, Walkiewicz MP, Frieman M,. Novel influenza virus NS1 antagonists block replication and restore innate immune function. J Virol 2009;83(4):1881-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smee DF, Hurst BL, Egawa H,. Intracellular metabolism of favipiravir (T-705) in uninfected and influenza A (H5N1) virus-infected cells. J Antimicrob Chemoth 2009;64(4):741-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kiso M, Takahashi K, Sakai-Tagawa Y,. T-705 (favipiravir) activity against lethal H5N1 influenza A viruses. Proc Natl Acad Sci USA 2010;107(2):882-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wunderlich K, Mayer D, Ranadheera C,. Identification of a PA-binding peptide with inhibitory activity against Influenza A and B virus replication. PLoS ONE 2009;4(10):e7517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang W, Wang CY, Yang ST,. Inhibition of highly pathogenic avian influenza virus H5N1 replication by the small interfering RNA targeting polymerase A gene. Biochem Biophys Res Commun 2009;390(3):421-6 [DOI] [PubMed] [Google Scholar]
- 30.Mickleburgh I, Geng F, Tiley L. Mesoionic heterocyclic compounds as candidate messenger RNA cap analogue inhibitors of the influenza virus RNA polymerase cap-binding activity. Antivir Chem Chemother 2009;19(5):213-18 [DOI] [PubMed] [Google Scholar]
- 31.Hastings JC, Selnick H, Wolanski B, Tomassini JE. Anti-influenza virus activities of 4-substituted 2,4-dioxobutanoic acid inhibitors. Antimicrob Agents Chemother 1996;40(5):1304-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sidwell RW, Bailey KW, Wong MH,. In vitro and in vivo influenza virus-inhibitory effects of viramidine. Antiviral Res 2005;68(1):10-17 [DOI] [PubMed] [Google Scholar]
- 33.Nguyen JT, Hoopes JD, Le MH,. Triple combination of amantadine, ribavirin, and oseltamivir is highly active and synergistic against drug resistant influenza virus strains in vitro. PLoS ONE 2010;5(2):e9332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ludwig S. Targeting cell signalling pathways to fight the flu: towards a paradigm change in anti-influenza therapy. J Antimicrob Chemother 2009;64(1):1-4 [DOI] [PubMed] [Google Scholar]
- 35.Nimmerjahn F, Dudziak D, Dirmeier U,. Active NF-kappaB signalling is a prerequisite for influenza virus infection. J Gen Virol 2004;85(Pt 8):2347-56 [DOI] [PubMed] [Google Scholar]
- 36.Kashyap AK, Steel J, Rubrum A,. Protection from the 2009 H1N1 pandemic influenza by an antibody from combinatorial survivor-based libraries. PLoS Pathog 2010;6(7):e1000990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oh HL, Akerstrom S, Shen S,. An antibody against a novel and conserved epitope in the hemagglutinin 1 subunit neutralizes numerous H5N1 influenza viruses. J Virol 2010;84(16):8275-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.De Clercq E. Vaccinia virus inhibitors as a paradigm for the chemotherapy of poxvirus infections. Clin Microbiol Rev 2001;14(2):382-97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sidwell RW, Allen LB, Khare GP,. Effect of 1-beta-D-ribofuranosyl1-1,2,4-triazole-3-carboxamide (virazole, ICN 1229) on herpes and vaccinia keratitis and encephalitis in laboratory animals. Antimicrob Agents Chemother 1973;3(2):242-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.De Clercq E, Cools M, Balzarini J,. Antiviral activities of 5-ethynyl-1-beta-D-ribofuranosylimidazole-4-carboxamide and related compounds. Antimicrob Agents Chemother 1991;35(4):679-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Clercq E, Descamps J, De Somer P, Holy A. (S)-9-(2,3-Dihydroxypropyl)adenine: an aliphatic nucleoside analog with broad-spectrum antiviral activity. Science 1978;200(4341):UNKNOWN [PubMed] [Google Scholar]
- 42.de Clercq E, Montgomery JA. Broad-spectrum antiviral activity of the carbocyclic analog of 3-deazaadenosine. Antiviral Res 1983;3(1):17-24 [DOI] [PubMed] [Google Scholar]
- 43.Borchardt RT, Keller BT, Patel-Thombre U. Neplanocin A. A potent inhibitor of S-adenosylhomocysteine hydrolase and of vaccinia virus multiplication in mouse L929 cells. J Biol Chem 1984;259(7):4353-8 [PubMed] [Google Scholar]
- 44.De Clercq E. Antiviral and antitumor activities of 5-substituted 2′-deoxyuridines. Methods Find Exp Clin Pharmacol 1980;2(5):253-67 [PubMed] [Google Scholar]
- 45.Hasobe M, McKee JG, Borcherding DR, Borchardt RT. 9-(trans-2′,trans-3′-Dihydroxycyclopent-4′-enyl)-adenine and -3-deazaadenine: analogs of neplanocin A which retain potent antiviral activity but exhibit reduced cytotoxicity. Antimicrob Agents Chemother 1987;31(11):1849-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smee DF. Progress in the discovery of compounds inhibiting orthopoxviruses in animal models. Antivir Chem Chemother 2008;19(3):115-24 [DOI] [PubMed] [Google Scholar]
- 47.De Clercq E. The acyclic nucleoside phosphonates from inception to clinical use: historical perspective. Antiviral Res 2007;75(1):1-13 [DOI] [PubMed] [Google Scholar]
- 48.Jesus DM, Costa LT, Goncalves DL,. Cidofovir inhibits genome encapsidation and affects morphogenesis during the replication of vaccinia virus. J Virol 2009;83(22):11477-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Buller RM, Owens G, Schriewer J,. Efficacy of oral active ether lipid analogs of cidofovir in a lethal mousepox model. Virology 2004;318(2):474-81 [DOI] [PubMed] [Google Scholar]
- 50.Parker S, Siddiqui AM, Oberle C,. Mousepox in the C57BL/6 strain provides an improved model for evaluating anti-poxvirus therapies. Virology 2009;385(1):11-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Quenelle DC, Keith KA, Kern ER. In vitro and in vivo evaluation of isatin-beta-thiosemicarbazone and marboran against vaccinia and cowpox virus infections. Antiviral Res 2006;71(1):24-30 [DOI] [PubMed] [Google Scholar]
- 52.Charity JC, Katz E, Moss B. Amino acid substitutions at multiple sites within the vaccinia virus D13 scaffold protein confer resistance to rifampicin. Virology 2007;359(1):227-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seiter K. Toxicity of the topoisomerase II inhibitors. Expert Opin Drug Saf 2005;4(2):219-34 [DOI] [PubMed] [Google Scholar]
- 54.Tallman MS, Gilliland DG, Rowe JM. Drug therapy for acute myeloid leukemia. Blood 2005;106(4):1154-63 [DOI] [PubMed] [Google Scholar]
- 55.Mike S, Harrison C, Coles B,. Chemotherapy for hormone-refractory prostate cancer. Cochrane Database Syst Rev 2006;(4):CD005247. [DOI] [PubMed] [Google Scholar]
- 56.Martinelli V, Radaelli M, Straffi L,. Mitoxantrone: benefits and risks in multiple sclerosis patients. Neurol Sci 2009;30(Suppl 2):S167-70 [DOI] [PubMed] [Google Scholar]
- 57.Deng L, Dai P, Ciro A,. Identification of novel antipoxviral agents: mitoxantrone inhibits vaccinia virus replication by blocking virion assembly. J Virol 2007;81(24):13392-402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.De Clercq E. Emerging antiviral drugs. Expert Opin Emerg Drugs 2008;13(3):393-416 [DOI] [PubMed] [Google Scholar]
- 59.Bolken TC, Hruby DE. Tecovirimat for smallpox infections. Drugs Today (Barc) 2010;46(2):109-17 [DOI] [PubMed] [Google Scholar]
- 60.Chen Y, Peng C, Sullivan C,. Novel therapeutic agents against cancer stem cells of chronic myeloid leukemia. Anticancer Agents Med Chem 2010;10(2):111-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Reeves PM, Bommarius B, Lebeis S,. Disabling poxvirus pathogenesis by inhibition of Abl-family tyrosine kinases. Nat Med 2005;11(7):731-9 [DOI] [PubMed] [Google Scholar]
- 62.Pytel D, Sliwinski T, Poplawski T,. Tyrosine kinase blockers: new hope for successful cancer therapy. Anticancer Agents Med Chem 2009;9(1):66-76 [DOI] [PubMed] [Google Scholar]
- 63.Yang HL, Kim SK, Kim M,. Antiviral chemotherapy facilitates control of poxvirus infections through inhibition of cellular signal transduction. J Clin Invest 2005;115(2):379-87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Borio L, Inglesby T, Peters CJ,. Hemorrhagic fever viruses as biological weapons: medical and public health management. JAMA 2002;287(18):2391-405 [DOI] [PubMed] [Google Scholar]
- 65.Ascenzi P, Bocedi A, Heptonstall J,. Ebolavirus and marburgvirus: insight the filoviridae family. Mol Aspects Med 2008;29(3):151-85 [DOI] [PubMed] [Google Scholar]
- 66.Geisbert TW, Bausch DG, Feldmann H. Prospects for immunisation against Marburg and Ebola viruses. Rev Med Virol 2010;20(6):344-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Barrientos LG, Gronenborn AM. The highly specific carbohydrate-binding protein cyanovirin-N: structure, anti-HIV/Ebola activity and possibilities for therapy. Mini Rev Med Chem 2005;5(1):21-31 [DOI] [PubMed] [Google Scholar]
- 68.Barrientos LG, Lasala F, Otero JR,. In vitro evaluation of cyanovirin-N antiviral activity, by use of lentiviral vectors pseudotyped with filovirus envelope glycoproteins. J Infect Dis 2004;189(8):1440-3 [DOI] [PubMed] [Google Scholar]
- 69.Barrientos LG, O'Keefe BR, Bray M,. Cyanovirin-N binds to the viral surface glycoprotein, GP1,2 and inhibits infectivity of Ebola virus. Antiviral Res 2003;58(1):47-56 [DOI] [PubMed] [Google Scholar]
- 70.Lee JE, Saphire EO. Neutralizing ebolavirus: structural insights into the envelope glycoprotein and antibodies targeted against it. Curr Opin Struct Biol 2009;19(4):408-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee JE, Saphire EO. Ebolavirus glycoprotein structure and mechanism of entry. Future Virol 2009;4(6):621-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hartlieb B, Modrof J, Muhlberger E,. Oligomerization of Ebola virus VP30 is essential for viral transcription and can be inhibited by a synthetic peptide. J Biol Chem 2003;278(43):41830-6 [DOI] [PubMed] [Google Scholar]
- 73.Spurgers KB, Sharkey CM, Warfield KL, Bavari S. Oligonucleotide antiviral therapeutics: antisense and RNA interference for highly pathogenic RNA viruses. Antiviral Res 2008;78(1):26-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Warfield KL, Swenson DL, Olinger GG,. Gene-specific countermeasures against Ebola virus based on antisense phosphorodiamidate morpholino oligomers. PLoS Pathog 2006;2(1):e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Enterlein S, Warfield KL, Swenson DL,. VP35 knockdown inhibits Ebola virus amplification and protects against lethal infection in mice. Antimicrob Agents Chemother 2006;50(3):984-93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Swenson DL, Warfield KL, Warren TK,. Chemical modifications of antisense morpholino oligomers enhance their efficacy against Ebola virus infection. Antimicrob Agents Chemother 2009;53(5):2089-99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Warren TK, Warfield KL, Wells J,. Advanced antisense therapies for postexposure protection against lethal filovirus infections. Nat Med 2010;16(9):991-4 [DOI] [PubMed] [Google Scholar]
- 78.Fowler T, Bamberg S, Moller P,. Inhibition of Marburg virus protein expression and viral release by RNA interference. J Gen Virol 2005;86(Pt 4):1181-8 [DOI] [PubMed] [Google Scholar]
- 79.Groseth A, Hoenen T, Alimonti JB,. In vitro evaluation of antisense RNA efficacy against filovirus infection, by use of reverse genetics. J Infect Dis 2007;196(Suppl 2):S382-9 [DOI] [PubMed] [Google Scholar]
- 80.Bray M, Driscoll J, Huggins JW. Treatment of lethal Ebola virus infection in mice with a single dose of an S-adenosyl-L-homocysteine hydrolase inhibitor. Antiviral Res 2000;45(2):135-47 [DOI] [PubMed] [Google Scholar]
- 81.Huggins J, Zhang ZX, Bray M. Antiviral drug therapy of filovirus infections: S-adenosylhomocysteine hydrolase inhibitors inhibit Ebola virus in vitro and in a lethal mouse model. J Infect Dis 1999;179(Suppl 1):S240-7 [DOI] [PubMed] [Google Scholar]
- 82.Vittori S, Dal Ben D, Lambertucci C,. Antiviral properties of deazaadenine nucleoside derivatives. Curr Med Chem 2006;13(29):3529-52 [DOI] [PubMed] [Google Scholar]
- 83.Bray M, Raymond JL, Geisbert T, Baker RO. 3-deazaneplanocin A induces massively increased interferon-alpha production in Ebola virus-infected mice. Antiviral Res 2002;55(1):151-9 [DOI] [PubMed] [Google Scholar]
- 84.Warren TK, Warfield KL, Wells J,. Antiviral activity of a small-molecule inhibitor of filovirus infection. Antimicrob Agents Chemother 2010;54(5):2152-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kinch MS, Yunus AS, Lear C,. FGI-104: a broad-spectrum small molecule inhibitor of viral infection. Am J Transl Res 2009;1(1):87-98 [PMC free article] [PubMed] [Google Scholar]
- 86.Aman MJ, Kinch MS, Warfield K,. Development of a broad-spectrum antiviral with activity against Ebola virus. Antiviral Res 2009;83(3):245-51 [DOI] [PubMed] [Google Scholar]
- 87.Spurgers KB, Alefantis T, Peyser BD,. Identification of essential filovirion-associated host factors by serial proteomic analysis and RNAi screen. Mol Cell Proteomics 2010;9(12):2690-703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Charrel RN, de Lamballerie X. Arenaviruses other than Lassa virus. Antiviral Res 2003;57(1-2):89-100 [DOI] [PubMed] [Google Scholar]
- 89.Briese T, Paweska JT, McMullan LK,. Genetic detection and characterization of Lujo virus, a new hemorrhagic fever-associated arenavirus from southern Africa. PLoS Pathog 2009;5(5):e1000455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Delgado S, Erickson BR, Agudo R,. Chapare virus, a newly discovered arenavirus isolated from a fatal hemorrhagic fever case in Bolivia. PLoS Pathog 2008;4(4):e1000047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.McKee KT Jr, Oro JG, Kuehne AI,. Candid No. 1 Argentine hemorrhagic fever vaccine protects against lethal Junin virus challenge in rhesus macaques. Intervirology 1992;34(3):154-63 [DOI] [PubMed] [Google Scholar]
- 92.Maiztegui JI, McKee KT Jr, Barrera Oro JG,. Protective efficacy of a live attenuated vaccine against Argentine hemorrhagic fever. AHF Study Group. J Infect Dis 1998;177(2):277-83 [DOI] [PubMed] [Google Scholar]
- 93.Geisbert TW, Jahrling PB. Exotic emerging viral diseases: progress and challenges. Nat Med 2004;10(12 Suppl):S110-21 [DOI] [PubMed] [Google Scholar]
- 94.McKee KT Jr, Huggins JW, Trahan CJ, Mahlandt BG. Ribavirin prophylaxis and therapy for experimental argentine hemorrhagic fever. Antimicrob Agents Chemother 1988;32(9):1304-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Perez M, Greenwald DL, de la Torre JC. Myristoylation of the RING finger Z protein is essential for arenavirus budding. J Virol 2004;78(20):11443-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cordo SM, Candurra NA, Damonte EB. Myristic acid analogs are inhibitors of Junin virus replication. Microbes Infect 1999;1(8):609-14 [DOI] [PubMed] [Google Scholar]
- 97.Strecker T, Maisa A, Daffis S,. The role of myristoylation in the membrane association of the Lassa virus matrix protein Z. Virol J 2006;3:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Garcia CC, Topisirovic I, Djavani M,. An antiviral disulfide compound blocks interaction between arenavirus Z protein and cellular promyelocytic leukemia protein. Biochem Biophys Res Commun 2010;393(4):625-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Garcia CC, Ellenberg PC, Artuso MC,. Characterization of Junin virus particles inactivated by a zinc finger-reactive compound. Virus Res 2009;143(1):106-13 [DOI] [PubMed] [Google Scholar]
- 100.Garcia CC, Djavani M, Topisirovic I,. Arenavirus Z protein as an antiviral target: virus inactivation and protein oligomerization by zinc finger-reactive compounds. J Gen Virol 2006;87(Pt 5):1217-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sepulveda CS, Garcia CC, Damonte EB. Inhibition of arenavirus infection by thiuram and aromatic disulfides. Antiviral Res 2010;87(3):329-37 [DOI] [PubMed] [Google Scholar]
- 102.Artuso MC, Ellenberg PC, Scolaro LA,. Inhibition of Junin virus replication by small interfering RNAs. Antiviral Res 2009;84(1):31-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bolken TC, Laquerre S, Zhang Y,. Identification and characterization of potent small molecule inhibitor of hemorrhagic fever New World arenaviruses. Antiviral Res 2006;69(2):86-97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.York J, Dai D, Amberg SM, Nunberg JH. pH-induced activation of arenavirus membrane fusion is antagonized by small-molecule inhibitors. J Virol 2008;82(21):10932-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Larson RA, Dai D, Hosack VT,. Identification of a broad-spectrum arenavirus entry inhibitor. J Virol 2008;82(21):10768-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gowen BB, Wong MH, Jung KH,. In vitro and in vivo activities of T-705 against arenavirus and bunyavirus infections. Antimicrob Agents Chemother 2007;51(9):3168-76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gowen BB, Smee DF, Wong MH,. Treatment of late stage disease in a model of arenaviral hemorrhagic fever: T-705 efficacy and reduced toxicity suggests an alternative to ribavirin. PLoS ONE 2008;3(11):e3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rojek JM, Pasqual G, Sanchez AB,. Targeting the proteolytic processing of the viral glycoprotein precursor is a promising novel antiviral strategy against arenaviruses. J Virol 2010;84(1):573-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ji WT, Liu HJ. PI3K-Akt signaling and viral infection. Recent Pat Biotechnol 2008;2(3):218-26 [DOI] [PubMed] [Google Scholar]
- 110.Linero FN, Scolaro LA. Participation of the phosphatidylinositol 3-kinase/Akt pathway in Junin virus replication in vitro. Virus Res 2009;145(1):166-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Vela EM, Bowick GC, Herzog NK, Aronson JF. Genistein treatment of cells inhibits arenavirus infection. Antiviral Res 2008;77(2):153-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ergonul O. Treatment of Crimean-Congo hemorrhagic fever. Antiviral Res 2008;78(1):125-31 [DOI] [PubMed] [Google Scholar]
- 113.Koksal I, Yilmaz G, Aksoy F,. The efficacy of ribavirin in the treatment of Crimean-Congo hemorrhagic fever in Eastern Black Sea region in Turkey. J Clin Virol 2010;47(1):65-8 [DOI] [PubMed] [Google Scholar]
- 114.Pittman PR, Liu CT, Cannon TL,. Immunogenicity of an inactivated Rift Valley fever vaccine in humans: a 12-year experience. Vaccine 1999;18(1-2):181-9 [DOI] [PubMed] [Google Scholar]
- 115.Niklasson B, Peters CJ, Bengtsson E, Norrby E. Rift Valley fever virus vaccine trial: study of neutralizing antibody response in humans. Vaccine 1985;3(2):123-7 [DOI] [PubMed] [Google Scholar]
- 116.Ikegami T, Makino S. Rift valley fever vaccines. Vaccine 2009;27(Suppl 4):D69-72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wolf MC, Freiberg AN, Zhang T,. A broad-spectrum antiviral targeting entry of enveloped viruses. Proc Natl Acad Sci USA 2010;107(7):3157-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bouloy M, Weber F. Molecular biology of rift valley Fever virus. Open Virol J 2010;4:8-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gowen BB, Wong MH, Jung KH,. Prophylactic and therapeutic intervention of Punta Toro virus (Phlebovirus, Bunyaviridae) infection in hamsters with interferon alfacon-1. Antiviral Res 2008;77(3):215-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gowen BB, Wong MH, Jung KH,. Efficacy of favipiravir (T-705) and T-1106 pyrazine derivatives in phlebovirus disease models. Antiviral Res 2010;86(2):121-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Muranyi W, Bahr U, Zeier M, van der Woude FJ. Hantavirus infection. J Am Soc Nephrol 2005;16(12):3669-79 [DOI] [PubMed] [Google Scholar]
- 122.Gavrilovskaya IN, Shepley M, Shaw R,. Beta3 Integrins mediate the cellular entry of hantaviruses that cause respiratory failure. Proc Natl Acad Sci USA 1998;95(12):7074-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Larson RS, Brown DC, Ye C, Hjelle B. Peptide antagonists that inhibit Sin Nombre virus and hantaan virus entry through the beta3-integrin receptor. J Virol 2005;79(12):7319-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hall PR, Malone L, Sillerud LO,. Characterization and NMR solution structure of a novel cyclic pentapeptide inhibitor of pathogenic hantaviruses. Chem Biol Drug Des 2007;69(3):180-90 [DOI] [PubMed] [Google Scholar]
- 125.Xu R, Yang XY, Yang DF,. Phase I evaluation of the safety and pharmacokinetics of a single-dose intravenous injection of a murine monoclonal antibody against Hantaan virus in healthy volunteers. Antimicrob Agents Chemother 2009;53(12):5055-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yang J, Chen R, Wei J,. Production and characterization of a recombinant single-chain antibody against Hantaan virus envelop glycoprotein. Appl Microbiol Biotechnol 2010;86(4):1067-75 [DOI] [PMC free article] [PubMed] [Google Scholar]