

Abstract
The tumor microenvironment (TME) is a complex network of non‐malignant cells and stroma that perform a wide array of vital roles in tumor growth, immune evasion, metastasis, and therapeutic resistance. These highly diverse roles have been shown to be critically important to the progression of cancers and have already shown potential as therapeutic targets. Therefore, there has been a tremendous push to elucidate the pathways that underlie these roles and to develop new TME‐directed therapies for cancer treatment. Unfortunately, TME‐focused research has been limited by a lack of translational in vitro culture platforms that can model this highly complex niche and can support the integrated analysis of cell biology and function. In the current study, we investigate whether an independently developed reconfigurable microfluidic platform, known as Stacks, can address the critical need for translational multi‐cellular tumor models and integrated analytics in TME research. We present data on multi‐cellular culture of primary human cells in Stacks as well as the orthogonal analysis of cellular polarization, differentiation, migration, and cytotoxicity in this reconfigurable system. These expanded capabilities of Stacks are highly relevant to the cancer research community with the potential to enhance clinical translation of pre‐clinical TME studies and to yield novel biological insight into TME crosstalk, metastasis, and responses to novel drug combinations or immune therapies.
Keywords: high throughput, microfluidics, multi‐culture, multiplex, organoid, primary cells, tumor microenvironment
Abbreviations
- ECM
extracellular matrix
- MDMs
monocyte‐derived macrophages
- MSCs
mesenchymal stem cells
- NF
nuclease‐free
- PDCO
patient‐derived cancer organoid
- PMPs
paramagnetic particles
- TME
tumor microenvironment
1. INTRODUCTION
During cancer progression, tumor cells encounter a wide range of imposing obstacles including limited oxygen and nutrient resources, dense stroma that limits migration, inhospitable metastatic sites, and the constant threat of destruction by immune cells or iatrogenic therapies. 1 , 2 Although each of these obstacles pose a major barrier to cancer progression, tumor cells are ultimately able to overcome them through the support of a diverse and specialized group of non‐malignant cells collectively known as the tumor microenvironment (TME). 3 The cell populations that comprise the TME, which include fibroblasts, endothelial cells, macrophages, T cells, B cells, NK cells, and dendritic cells, perform a wide range of functions that help tumor cells overcome the barriers to progression. These functions include vascular recruitment, stromal remodeling, establishing the pre‐metastatic niche, suppressing anti‐tumor immune responses, and limiting the efficacy of cancer therapies. 4 , 5 , 6 , 7 As a result of this orchestrated support, tumor cells are able to adapt, steadily grow, evade immunity, metastasize to distant sites, and survive as cancer progression occurs. 8
With such a broad range of essential roles in tumor progression, the TME, and the pathways that underlie TME‐mediated progression are valuable therapeutic targets for cancer treatment. 9 While there has been considerable progress in the development of TME‐targeted cancer therapies as evidenced by immune checkpoint inhibitors and anti‐angiogenic therapies, there remains high potential to expand the indications for these therapies and to develop more effective treatment strategies by targeting additional cell types in the TME such as tumor‐associated macrophages and cancer‐associated stromal cells. 9 Further research is therefore needed to gain more insight into TME‐mediated pathways of tumor promotion. Unfortunately, this research poses an array of challenges for traditional in vitro culture platforms, which are typically limited to the simultaneous culture of one or two cell populations in a single, two‐dimensional system. Such platforms are unable to model the diverse cellular networks of the TME and do not incorporate the combined effects of these cell populations. Furthermore, the size and scale of traditional platforms require a reliance on cell lines, which do not reflect patient heterogeneity and can carry important biologic differences from primary cells. 10
To directly address the limitations of current preclinical tools, we have recently developed a platform, known as Stacks, which utilizes open and suspended microfluidic principles to enable culture and analysis of multiple cell types across a wide range of functional and molecular readouts. This platform is a non‐sealed microfluidic device that is composed of polystyrene plates, which contain microwells that are open to the atmosphere. There are 24 microwells on each plate, arranged in a 6‐column by 4‐row configuration (Supporting Information 1, Figure S1). The size and shape of the microwells (surface area of 3.14 mm2 and a thickness of 1.2 mm) are designed to leverage the principles of open and suspended microfluidics so that surface tension prevents the loss of liquid from the wells by creating pinning regions on air‐liquid interfaces. The plates can therefore be stacked on top of each other to create a single fluid system among vertically aligned wells without leakage of fluid between layers or loss of pinning at the bottom (Figure S1). While secreted factors can diffuse freely through each vertically aligned system (with predictable diffusion dynamics), there is no physical connection between the hydrogels or cells in any of the layers (Figure S1B).
Notable advantages of the Stacks platform over conventional culture systems include the ability to multi‐culture up to six patient‐derived cell populations within a single system as well as the option to fully reconfigure layer orientation at any timepoint during culture. Due to the microscale dimensions of the well, input requirements are low (4.5 μl of biomatrix; 10 μl of cells/media), supporting more efficient use of valuable biospecimens and lower experimental costs. Furthermore, the microwells of the Stacks platform are designed to accommodate multi‐channel pipettes, enabling high throughput experimentation and multiplexed analysis of cellular phenotype, gene expression, secretion profiles, cell signaling, cell‐to‐cell interaction, migration, and matrix remodeling (Figure 1). 11 , 12 , 13
FIGURE 1.
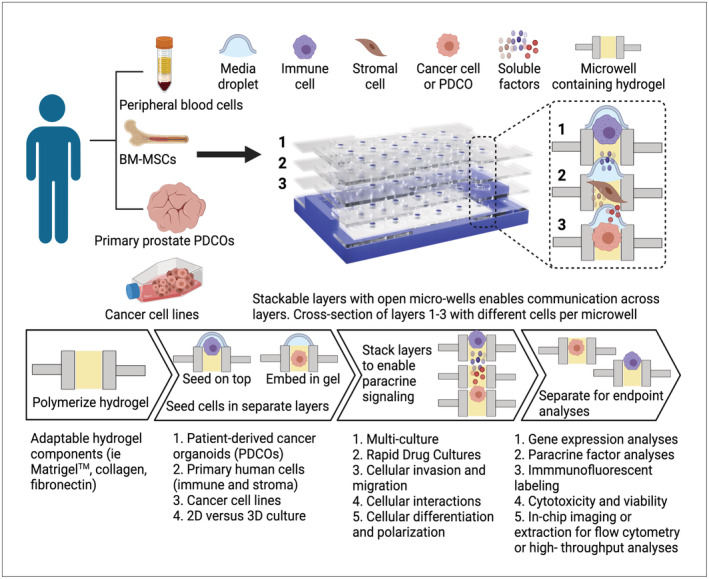
Stacks platform. The Stacks platform is composed of separate plates/layers, each of which contains a series of open microwells. Cells from a variety of different sources, including cell lines, peripheral blood cells, bone marrow‐derived cells, and primary tissue‐derived cells can be cultured in the microwells in both 2D and 3D configurations. Up to six plates can be combined (stacked) to allow chemical or physical communication between wells in vertical alignment at desired intervals. As needed, individual mono‐culture or multi‐culture replicates can be treated (drugs, cytokines, etc.) or left untreated. Layers can be separated at desired timepoints for cell‐specific analysis of mRNA and protein expression, immunophenotype, viability, and migration as well as an array of additional endpoints.
In our initial publication on Stacks, we discussed the microfluidic principles of the platform and demonstrated that mRNA analysis and fluorescence microscopy could be utilized to investigate the morphology and gene expression of cell lines in mono‐ and co‐culture systems. 13 In this manuscript, we expand the application of this technology to enable comprehensive TME‐directed research using primary cells in Stacks. We describe protocols for analyzing an array of orthogonal endpoints in Stacks, including gene expression, phenotypic characterization, cytotoxicity, and cell migration. We provide the analytic data to support each of these protocols and further demonstrate how Stacks can be leveraged to efficiently utilize human biospecimens to perform multiplexed, high throughput investigation of the TME.
2. MATERIALS AND METHODS
2.1. Stack devices
Stacks were sourced from Protolabs, Maple Plain, MN, US (1121–5161‐007). Prior to use, the Stacks plates were prepared by sonication in 100% isopropanol for 60 min and washed in deionized water. All plates, 3D holders, Nunc™Omnitrays™ (Thermo Fisher Scientific, Waltham, MA, USA), and 245 mm square non‐tissue culture‐treated BioAssay dish (Corning Inc., Corning, NY, USA) were sterilized by exposure to UV light for 20 min on each side in a biosafety cabinet. Cells were plated in the Stacks device by adding 10 ul of cells suspended in media to each well. Cells were then allowed to adhere or migrate through the matrix as applicable. When stacking plates, media was removed from the top and bottom of each well leaving a small amount of residual media on each side to prevent the formation of a gas bubble during stacking. Plates were then stacked on top of each other using a holder for alignment. Each well was inspected for gas bubbles, which is a rare occurrence (<1%) that can be detected visually (Supporting Information, Figure S2B). Affected wells were discarded from the analysis. To prevent dehydration, a three‐layer humidifying chamber was assembled as illustrated in the Supporting Information (Figure S1C). Specifically, Stacks devices in 3D holders were placed adjacent to a sterile sponge in a Nunc™Omnitray™ (Thermo Fisher Scientific, Waltham, MA, USA). Sterile ddH20 was applied to the sponge. The omnitray was placed square bioassay dish containing sterile petri dishes filled with 25–30 ml sterile ddH20.
2.2. Isolation of primary cells
Blood specimens were collected in vacutainer tubes (BD Biosciences, Franklin Lake, NJ, USA) with EDTA anticoagulant. All patients provided written, informed consent under an Institutional Review Board (IRB) approved protocol. Whole blood was diluted 1:1 with Hank's balanced salt solution (HBSS, Lonza Group, Basel, Switzerland) before being underlaid with 10 ml of Ficoll‐Paque PLUS (GE Healthcare, Chicago, IL, USA) per 50 ml conical tube. Monocytes were enriched from peripheral blood mononuclear cells (PBMCs) using LS MACS columns following incubation with anti‐CD14 magnetic beads (both from Miltenyi Biotec, Bergisch Gladbach, North Rhine‐Westphalia, Germany). T cells were isolated from the CD14− cell fraction remaining after monocyte enrichment via a Pan T cell isolation kit (Miltenyi Biotec, Germany) and LS MACS columns. Bone marrow‐derived mesenchymal stem cells were isolated from leftover bone marrow filters collected from normal healthy bone marrow donors, based on an IRB‐exempt protocol of the University of Wisconsin Bone Marrow Transplant Program, and bone marrow MSCs were cultured according to standard conditions. 11 , 14 Human prostate tissues were obtained at the University of Wisconsin‐Madison from patients with prostate cancer undergoing radical prostatectomy who had received no prior treatments. The University of Wisconsin IRB approved the utilization of all the tissue samples in this study and written and informed consent was obtained from all patients.
2.3. Cell culture
DU145, 22Rv1, primary T cells, and primary macrophages were cultured in Stacks by seeding as a monolayer on top of a collagen‐fibronectin matrix, consisting of collagen I (Advanced BioMatrix, Carlsbad, CA, USA #5005), fibronectin (Sigma‐Aldrich, Millipore Sigma, Burlington, MA, USA #F1141) prepared according to the manufacturer guidelines. Moisture was retained by storing Stacks inside of a humidifying chamber. DU145 and 22Rv1 cells (acquired from ATCC) were cultured in RPMI1640 media with L‐Glutamine (Corning™ Thermo Fisher Scientific, Waltham, MA, USA), 10% FBS (Gibco™, Thermo Fisher Scientific, Waltham, MA, USA) and 2% penicillin/streptomycin (Hyclone™, VWR, Radnor, PA, USA) and were seeded at a concentration of 300,000 cells/ml. Patient‐derived monocytes and T‐cells were grown in RPMI1640 media with L‐Glutamine, 10% FBS, 5% Glutamax (Gibco™, Thermo Fisher Scientific, Waltham, MA, USA), 2% penicillin/streptomycin. Monocytes were differentiated into macrophages using 50 ng/mL M‐CSF (Tonbo Biosciences, Cytek, San Diego, CA USA) as previously described. 12 T cells were activated with Dynabeads™ Human T‐activator CD3/CD28 beads (Thermo Fisher Scientific, Waltham, MA, USA) and were grown in suspension in 24‐well plates. For migration experiments, they were seeded in Stacks at a concentration of 500,000 cells/ml and allowed to migrate for 24 h.
2.4. Nucleic acid extraction and quantitative RT‐PCR
Monocyte‐derived macrophages were removed from Stacks matrix via 1 mg/mL collagenase I (Sigma‐Aldrich, Millipore Sigma, Burlington, MA, USA) digestion prior to mRNA extraction. mRNA isolation was performed using Invitrogen™Dynabeads™ mRNA DIRECT Kit (Thermo Fisher Scientific, Waltham, MA, USA). Cells were lysed within the culture wells using 10 μl of supplied lysis/binding buffer. Lysate was transferred to tubes containing an additional 30 μl of lysis/binding buffer. Culture wells were washed with an additional 10 μl of lysis/binding buffer, which was added to the lysate. 10 μl of washed beads were added to each sample lysate was washed with 200 μl Buffer A × 2 and 200 μl Buffer B × 1.
The mRNA elution sample containing paramagnetic particles (PMPs) was reverse transcribed using a High‐Capacity cDNA Reverse Transcriptase kit (Applied Biosystems™, Thermo Fisher Scientific, Waltham, MA, USA), according to the manufacturer's directions using Bio‐Rad C1000 Touch™ thermocycler (Bio‐Rad Laboratories, Hercules, CA, USA). The RT reaction (12.5 μl) was then amplified for 10 cycles using TaqMan™ PreAmp (Applied Biosystems™, Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer's directions and diluted 1:3 in 1× TE (10 mM Tris–HCL pH 8, 1 mM EDTA). For TaqMan™ assays, 5 μl of diluted cDNA template was mixed with 10 μl iTaq® master mix (Bio‐Rad, USA), 1 μl TaqMan™ Gene Expression Assay CCL2 (HS00234140_m1), CCL5 (HS00982282_m1), CCL18 (Hs00268113_m1), CCL22 (Hs01574247_m1), CXCL9 (HS00171065_m1), CXCL10 (Hs01124252_g1), CXCL11 (Hs04187682_g1), CXCL12 (Hs03676656_mH), IL‐10 (Hs00961622_m1), and MRC‐1 (Hs00267207_m1) (Life Technologies, USA) and 4 μl nuclease‐free (NF) water. Each reaction was amplified for 45 cycles (denatured at 95°C for 15 s followed by annealing at 60°C for 1 min) using a CFX Connect® Real‐Time PCR System (Bio‐Rad Laboratories, Hercules, CA, USA).
2.5. T cell migration analysis
T cell migration was evaluated by stacking a T cell containing Stacks plate on top of a plate containing 22Rv1 tumor cells in each well. T cells were allowed to migrate through the matrix for 24 h before the Stacks layers were separated. T cells were then fixed at their location within the collagen‐fibronectin matrix with a Foxp3 Fixation/Permeabilization kit (eBioscience, Thermo Fisher Scientific, Waltham, MA, USA), washed, and then stored at 4°C. Staining was performed by adding a solution of 20 mM Hoechst at a 1:250 dilution, anti‐CD4 (BD Biosciences, Franklin Lake, NJ, USA, #564419) at 1:50, and anti‐CD8 (BD Biosciences, USA #555367) at 1:100 to each well followed by overnight incubation at 4°C (reagents listed in Table S1). Each well was imaged on a Yokogawa CSU‐W1 Nikon Spinning Disk Confocal Microscope (Tokyo, Japan). Images were analyzed with NIS‐Elements software (RRID:SCR_014329; Nikon Instruments, Melville, NY, USA). CD4+ or CD8+ populations were identified by thresholding for signal intensities in the FITC and PE channels, respectively. Total migration distance for each cell population was quantified by calculating the distance that each cell had moved from the top of the well. Average migration was calculated by dividing the total migration distance by the total number of cells detected and correcting for the step size of the microscope during imaging.
2.6. Flow cytometry analysis
Stacks culture wells were rinsed with PBS and stained with Fc blocker, CD14, CD80 (BD Biosciences, Franklin Lake, NJ, USA), CD11b, CD200R (Biolegend, San Diego, CA, USA), Ghost Dye™ Violet 510 Live/Dead (Tonbo Biosciences, Cytek, Sand Diego, CA, USA) (listed in Table S1) stain for 30 min at 37°C followed by washing three times with BD Staining Buffer with BSA (#554657) in chip. Cells were then collagenase treated for 45 min at 37°C, flushed into tubes with 120 μl BD Stain buffer, and transferred to ice. 1% PFA (BD Cytofix™ Fixation buffer, BD Biosciences) was added for fixation. Cells were acquired on a BD LSRII instrument (BD Biosciences, Franklin Lake, NJ, USA). Data were analyzed with FlowJo v10.7.1 (FlowJo LLC, by BD Biosciences, Ashland, OR, USA).
2.7. Cytokine analysis
Supernatant media was collected from the microwells of macrophages following differentiation/polarization. Media from wells of an individual condition was combined to a volume of 20 μl and diluted in 40 μl of media prior to centrifugation at 300 g for 3 min. 53 μl of the supernatant was removed, snap frozen, and stored at −80°C until analysis. Samples were analyzed using a custom multiplexed Quantikine® kit according to the manufacturer's protocol (R&D Systems, Minneapolis, MN, USA) using a MAGPIX system. Samples, standards, and blanks were assayed in duplicate. Samples were diluted 1:2 in the assay buffer provided in the kit. Cytokine concentration was quantified using the standard curve generated by the 5‐PL curve fit analysis tool in Prism (GraphPad Software, San Diego, CA, USA). The average concentration value from the blank was subtracted from the concentration of each sample, for each analyte.
2.8. Viability and apoptosis assays
22Rv1 prostate cancer cells were seeded onto a collagen‐fibronectin matrix in Stacks and treated with vehicle or docetaxel for 48 h. Cells were then stained with Calcein AM (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA #C1430) and Hoechst33342 (Thermo Fisher Scientific, Waltham, MA, USA) for 1 h followed by 1xPBS washes. Cells were extracted from each well by collagenase I (Sigma‐Aldrich, USA) digestion and transferred into a 384‐well low‐volume microplate. The fluorescence intensities of Calcein AM and Hoechst33342 staining were detected using a CLARIOstar Plate reader (BMG Labtech, Cary, NC, USA) at the Small Molecule Screening Facility, UW‐Madison, and relative viability was established by comparing the Calcein signal in each condition to the control condition.
Caspase‐3 activity was analyzed by fluorescence microscopy using NucView® 488 Caspase‐3 Substrate (Biotium, Fremont, CA, USA). Cells were imaged on a glass coverslip using a Nikon Ti‐E Eclipse inverted fluorescent microscope. Quantification of caspase‐3 activity was accomplished using NIS‐Elements software. Spot detection function was used to identify individual cells and generate statistics on signal intensity values in each cell and for the sample as a whole. Reagents are listed in Table S1.
2.9. Cytotoxicity analysis
The H358 lung cancer cell line expressing the KRAS G12C alteration was seeded on a collagen‐fibronectin matrix in Stacks and treated with 100 nM of adagrasib, a KRAS G12C small molecule inhibitor (Selleck Chemicals, Houston, TX, USA) or with DMSO for 48 h in serum‐free RPMI media. Cells were stained under live conditions with EpCAM Alexa Fluor 647 at 1:50 dilution, Image‐iT® Dead™ Green at 1:10 000 dilution, and Hoechst33342 for 60 min. Wells were rinsed with PBS, fixed with 1% PFA, washed with PBS, and mounted as described above. Cells were imaged directly in‐chip using a Nikon Yokogawa CSU‐W1 Spinning Disk Confocal Microscope equipped with Hamamatsu Orca Flash cameras, and the percentage of Image‐iT® Dead™‐positive cells were quantified using NIS‐Elements software. Spot detection function was used to identify individual cells and generate statistics on signal intensity values in each cell and for the sample as a whole. This experiment was performed twice with three technical replicates per experiment.
2.10. Prostate patient‐derived cancer organoid drug culture and confocal microscopy analysis
Primary patient‐derived cancer organoids (PDCOs) were derived from radical prostatectomy specimen from patients with high‐risk localized prostate cancer as described before. Biopsy specimen was partially digested and propagated in hanging 50% Matrigel™ droplets (GFR Basement Membrane Matrix, BD Biosciences, Franklin Lake, NJ, USA) followed by transferring to Stacks wells in 50% Matrigel with PrEGM media (Lonza, Basel, Switzerland) supplemented with 0.1 μg EGF (Gibco, Thermo Fisher Scientific, Waltham, MA, USA), and 10 μM Y‐27632 (Sigma‐Aldrich, Millipore Sigma, USA). PDCOs were treated with DMSO, 10 nM Docetaxel, or 100 μM Bicalutamide (Selleck Chemicals, Houston, TX, USA) for 72 h in PrEGM media supplemented with 10 μM Y‐27632 (Selleck Chemicals, Houston, TX, USA). Wells were rinsed three times with 1xPBS prior to staining with Image‐IT Dead™ Green, EpCAM Alexa Fluor 647, and Hoechst33342 (Supporting Information 2, Table S1) for 30 min at 37°C. Wells were then rinsed three times with PBS, fixed with 1% PFA (Cytofix, BD Biosciences) for 15 min followed by three washes with PBS. Wells were then mounted with SlowFade™ Gold Antifade Mounting Media (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) and imaged on a Nikon Yokogawa CSU‐W1 Spinning Disk Confocal Microscope equipped with Hamamatsu Orca Flash cameras. 20x images were then analyzed with NIS‐Elements software.
2.11. Primary human bone marrow mesenchymal stem cell differentiation
Human bone marrow‐derived mesenchymal stem cells were isolated from left‐over bone marrow filters, collected from normal healthy bone marrow donors, based on an IRB‐approved exempt protocol of the University of Wisconsin 15 and cultured as previously described. 16 MSCs were grown in monolayer at 37°C and 5% CO2 and expanded in media consisting of alpha‐MEM containing 1% Glutamax, 15% human platelet lysate (StemCell Technologies, Vancouver, BC, Canada), and 1% penicillin/streptomycin. After initial expansion, MSCs were maintained in media consisting of alpha‐MEM containing 1% Glutamax, 5% human platelet lysate, and 1% penicillin/streptomycin. MSCs were passaged before reaching confluency, and only MSCs up to passage 8 were used for differentiation experiments. MSCs from two separate donors were seeded on a collagen‐fibronectin matrix in Stacks and differentiated toward adipogenic lineage using Adipocyte Differentiation Media (StemCell Technologies, Vancouver, BC, Canada). Cells were incubated in base media for the control condition. Media was changed every 3–4 days, and sterile water was re‐added to the Nunc™Omnitray™ humidifying chamber weekly. After 14 days of differentiation in Stacks, lipid droplets were detected using LipidSpot™ Lipid Droplet Stain (Biotium, Fremont, CA, USA #700‐69 T). After live staining with LipidSpot and Hoechst33342 for 60 min, wells were washed with PBS, fixed, and mounted with SlowFade Gold Antifade mounting media. Wells were imaged in‐chip on a Nikon Yokogawa CSU‐W1 Spinning Disk Confocal Microscope and images were analyzed by Nikon NIS‐Elements software. All experiments were repeated at least three times unless otherwise indicated. Data are reported as means+SEM Differences among treatment groups were determined by t‐test. p < .05 was considered significant. Statistical analysis was performed with the Prism software (GraphPad Software, San Diego, CA, USA).
3. RESULTS
3.1. Orthogonal analysis of cellular polarization by evaluation of gene expression and cellular phenotype integrating qPCR, flow cytometry, and multiplexed bead array assays in Stacks
To establish the utility of Stacks as a platform to polarize primary immune cell subsets as well as analyze cellular features with a comprehensive, multi‐analyte approach, we chose primary monocyte‐derived macrophages (MDMs) as our main experimental model. Primary macrophages were selected for this purpose because they are a TME cell type that is easily derived from liquid specimens. Additionally, they have high plasticity, which enables straightforward manipulation of their phenotype for expression analysis. To derive macrophages, primary CD14+ monocytes were isolated from peripheral blood specimens followed by differentiation in Stacks using M‐CSF as previously described. 12 Cells were cultured as a monolayer on top of a 3D collagen I and fibronectin matrix, which was inserted into each well prior to cell seeding. On Day eight, MDMs were polarized toward an M1‐like or M2‐like phenotype, respectively, or left unpolarized. M1 and M2 are broad phenotypic categories for macrophages that are based on factors such as gene expression, immunophenotype, morphology, and function. An array of protocols allow polarization into these phenotypes in vitro, or the macrophages can alternatively be left in an unpolarized state (M0). Each of these states are associated with well‐defined phenotypic characteristics that can be analyzed through gene expression and functional assays. For our experiments, we utilized IFN‐γ to induce M1 polarization and IL‐4 for M2 polarization, as previously described. 12 M0 macrophages were maintained with M‐CSF. Following polarization, we performed mRNA, protein, and secretory factor analysis of the cells with the goal of identifying whether primary cell phenotype could be controlled and effectively assessed by orthogonal endpoints in Stacks (Figure 2A).
FIGURE 2.
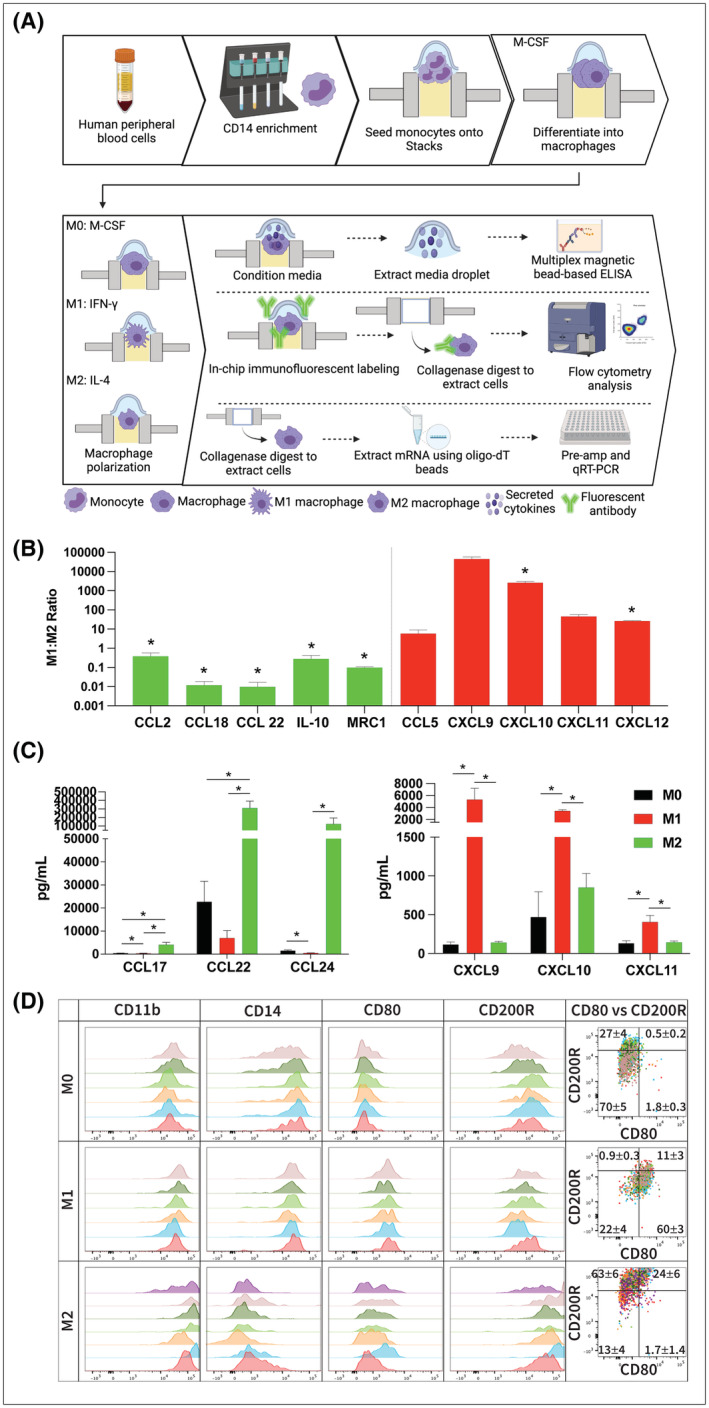
Analysis of gene expression in Stacks. (A) Workflow for isolation, culture, and analysis of gene expression in primary MDMs. (B) mRNA expression of primary MDMs expressed as the M1:M2 ratio (relative expression of each gene in M1 macrophages divided by the relative expression in M2 macrophages). Levels >1 indicates higher expression in M1 macrophages and <1 indicate higher expression in M2 macrophages. Red bars indicate M1‐associated genes and green bars indicate M2‐associated genes (n = 3). (C) Concentration of cytokines in the supernatant media of polarized (M1 and M2) and unpolarized (M0) macrophages as detected by multi‐analyte immunoassay. Left graph is M2‐associated cytokines and right graph is M1‐associated cytokines (n = 3). (D) Flow cytometry analysis of polarized (M1 and M2) and unpolarized (M0) macrophages. Histogram overlays represent CD11b expression on live/single/cells (in first column) and biomarker expression of CD11b+/live/single/cells (in second, third, fourth histogram columns and in dot plots). Individual colors represent individual samples. Data in dot plot overlay quadrants represent the frequency of the parent gate as Mean ± SEM for all samples within the group; *p < .05.
For mRNA analysis, cells were extracted from the microwells by digesting the matrix with collagenase I. mRNA isolation was then performed by magnetic bead separation followed by reverse transcription into cDNA as described in Methods. The cDNA in each sample was pre‐amplified for 10 cycles in a thermocycler prior to performing qPCR analysis. Supernatant media was also removed from each microwell following culture and polarization of the MDMs and evaluated by multi‐analyte bead analysis. To achieve adequate sample size, media from three replicate microwells were combined for supernatant analysis. Samples were then centrifuged to remove any residual cells and the media was interrogated for secretion of classical M1‐ and M2‐associated cytokines using commercial bead array. Figure 2A,B depict the mRNA and secreted protein data from these experiments. As demonstrated in these figures, we were able to detect and analyze mRNA as well as secreted protein expression in all experimental conditions in Stacks. At the mRNA level, expression of the M1 markers (CCL5, CXCL‐9, 10, 11, and 12) was increased in the M1‐polarized MDMs while the M2 markers (CCL‐2, 18, 22, IL‐10, and MRC1) were concordantly increased in the M2‐polarized MDMs (Figure 2B). Secreted protein analysis of M1‐associated (CXCL‐9, 10, 11) and M2‐associated (CCL‐17, 22, and 24) cytokines in M1 and M2‐polarized MDMs was also consistent with MDM polarization status (Figure 2C). 17
The M1/M2 markers that we utilized in these experiments are well‐established biomarkers of macrophage polarization. 18 , 19 , 20 , 21 While there is considerable variability in the degree of up‐ and down‐regulation reported in the literature for each biomarker, there is a tight consistency on the direction (up or down) that each marker changes with polarization. Our findings are in accordance with these characteristic biomarker profiles and we were able to demonstrate expected changes in M1/M2 genes expression at both the mRNA and secreted protein levels. Furthermore, our replicate values demonstrated statistically significant differences between the different populations of MDMs. This data therefore confirms both effective polarization of primary MDMs into functional phenotypes in Stacks and accurate analysis of mRNA and secreted protein expression of polarized immune cell subsets within the Stacks system.
Next, we designed a multi‐parameter flow cytometry assay as an orthogonal approach to further establish MDM polarization patterns at a cellular level, and adapted flow cytometry protocols to Stacks to allow investigation of microscale specimens. To achieve this end, we performed immune fluorescent labeling and washing of the cells in‐chip followed by digestion of the hydrogel to release cells for acquisition as a single‐cell suspension on a BD LSR II instrument. Performing immune labeling of cells contained by hydrogel in‐chip allowed for the reduction of material transfers, and minimized direct manipulation of cells by avoiding pipetting and washing steps with centrifugation and resuspension that could all lead to cell loss in traditional macroscale manipulation. The median of total CD11b+/live/single/cell events acquired was 234 cells (68 to 877, Min to Max). This acquisition range is in accordance with conventional flow cytometry guidelines recommended for rare‐event analysis. 22 , 23 Histograms in Figure 2D show that M1‐polarized CD11b+ macrophages expressed increased levels of surface CD80 expression compared to M0 unpolarized or M2‐polarized cultures. Conversely, MDMs under M2‐polarizing conditions increased CD200R expression compared to M0 and M1 cultures. CD14 expression was retained in M1 polarization while diminished on M2‐polarized cells. These findings, which are also in‐line with the phenotypic profiles of the polarized and unpolarized MDMs in conventional culture platforms, provide further confirmation of successful culture and polarization of primary MDMs in Stacks. In addition, these results confirm that effective single‐cell multi‐parameter immunophenotypic analysis can be performed on cells cultured in Stacks.
Taken together, our qPCR, multi‐analyte bead assay, and flow cytometry analysis collectively confirm that primary cells can be cultured within Stacks and that their phenotype can be successfully manipulated using conventional protocols. Orthogonal multi‐analyte analysis of gene expression can then be performed using standard assays to evaluate expression profiles at both the mRNA and protein levels.
3.2. Cell differentiation in Stacks
In order to accurately model an environment that is as complex and diverse as the TME, an in vitro platform must be able to support the culture and differentiation of a wide range of cell types. We therefore also evaluated whether we could utilize the Stacks platform to differentiate and analyze a multipotent stem cell population known as mesenchymal stem cells (MSCs). MSCs can be derived from multiple sources including bone marrow and adipose tissue and can be differentiated into cell types such as adipocytes, osteoblasts, and fibroblasts. 24 MSCs and MSC‐derived adipocytes are crucial components of TMEs and have been shown to support the growth, survival, and invasiveness of tumor cells. 25 , 26 , 27 Adipocytes also secrete multiple cytokines and factors (collectively called adipokines) that are immunologically active, highlighting their important role in the TME. 28 In the current study, we demonstrated the ability to perform a long‐term culture and differentiation of primary human‐derived MSCs in Stacks. Primary human MSCs were derived from bone marrow aspirates from two donors, seeded onto a collagen‐based extracellular matrix (ECM) in Stacks, and treated with control or adipogenic media for 14 days. Adipogenic differentiation was then evaluated by in‐chip staining with LipidSpot (a fluorescent dye that stains lipid droplets) and Hoechst and visualized with confocal microscopy (Figure 3A,B). The percentage of cells that had differentiated into adipocytes was quantified by enumeration of LipidSpot‐positive cells and total number of nuclei using the Spot Detection function on NIS‐Elements. As demonstrated in Figures 3B,C, our analysis demonstrated a clear differentiation of the MSCs in Stacks that could be detected visually and quantified using automated image analysis. The role of 3D culture in adipocyte biology is increasingly supported, however, the source of pre‐adipocytes and 3D culture methods vary greatly in current literature. 28 , 29 , 30 , 31 Our findings, which are in accordance with these reports showing robust differentiation into adipocytes under 3D conditions, 27 , 28 , 29 , 30 further demonstrate that these methods can be applied toward human bone marrow‐derived adipogenic MSCs under microscale conditions within a collagen ECM. Furthermore, these findings also highlight the ability to perform long‐term culture of primary human adipocytes in Stacks. Ultimately, these findings establish a foundation for future investigations to generate a multicellular TME that more accurately recapitulates in vivo conditions.
FIGURE 3.
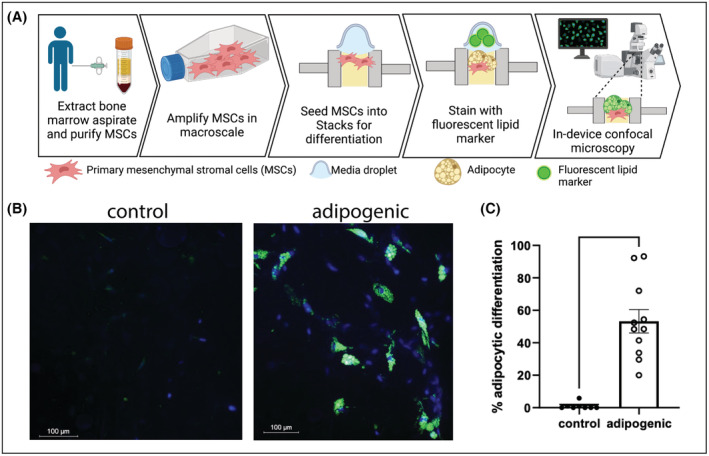
Differentiation of primary human bone marrow mesenchymal stem cells (MSCs) in Stacks. (A) MSCs from two separate donors were seeded into Stacks and cultured for 14 days under control or adipogenic conditions. (B) Adipocytic differentiation was shown by the development of lipid droplets, identified with LipidSpot™ staining (green) and Hoechst nuclear staining (blue) by confocal microscopy. (C) LipidSpot™ ‐positive cells and total nucleated cells were quantified using automated image analysis and expressed as a percentage of LipidSpot™‐positive cells. Each data point represents percentage of LipidSpot‐positive cells in one microwell; *p < .05.
3.3. Cytotoxicity assays
Given the important roles for the TME in therapeutic response and resistance, analysis of cytotoxicity is crucial for TME‐focused research. We therefore investigated whether Stacks could be utilized to perform cytotoxicity assays using different categories of tumor‐directed agents. (Figure 4A). For this assay, we seeded an androgen‐independent 22Rv1 prostate tumor cell line on a collagen‐fibronectin matrix in Stacks and allowed cells to adhere and grow for 3 days. The tumor cells were then treated with one of four escalating doses of docetaxel chemotherapy for 48 h. Following treatment, the tumor cells were stained with Calcein AM in‐chip and relative viability was established by measuring the fluorescence intensity of the Calcein AM over control conditions using a microplate reader. As Figure 4B demonstrates, we were able to detect a clear dose–response curve for docetaxel treatment in Stacks with low variability between replicates. Although the concentrations of docetaxel required to achieve a corresponding treatment effect were higher in Stacks than in traditional culture platforms, these differences are attributable to the considerably higher number of cells per volume of media (approximately twofold higher) in the microscale platform vs conventional platforms as well differences in cellular metabolism and proliferation in microscale cultures. 32 Additionally, multiple studies have demonstrated protective effects of 3D ECM on cytotoxicity and viability. 33
FIGURE 4.
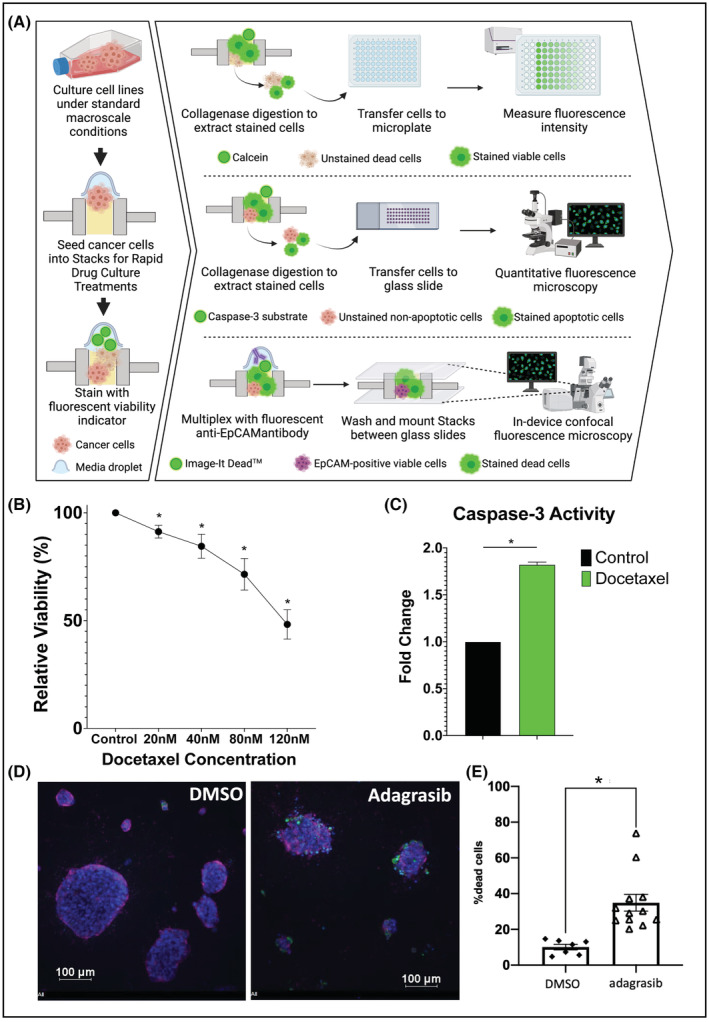
Cytotoxicity assays on cell lines in Stacks. (A) Workflow for cytotoxicity assays on cell lines. (B) Dose titration curve of 22rv1 prostate cancer cells treated with docetaxel for 48 h followed by analysis of viable cell number using quantification of Calcein AM staining with a microplate reader. Data expressed as percent of viable cells as compared to control condition (n = 3). (C) Quantification of caspase‐3 activity in DU145 prostate cancer cells treated with 20 nM docetaxel for 48 h. Data expressed as fold change of caspase‐3 activity in docetaxel treated cells as compared to control condition (n = 3). (D) The H358 lung cancer cell line expressing the KRAS G12C oncogene was cultured in Stacks and treated with vehicle or a KRAS G12C inhibitor (n = 2). Cells were stained with Hoechst (blue), anti‐EpCAM‐Alexa647 antibody (purple) and Image‐IT Dead™ reagent (green), and imaged in‐device using confocal fluorescence microscopy. (E) Dead cells and total number of cells was quantified with automated image analysis and the percentage of dead cells was significantly induced by inhibition of KRAS G12C in H358 lung cancer cells. Each data point represents percentage of Image‐IT Dead™‐positive cells in one microwell; *p < .05.
We next evaluated the detection of apoptotic markers using a caspase‐3 activity assay. Tumor cells were seeded on matrix in Stacks and allowed to adhere and grow prior to docetaxel treatment. Following treatment, cells were stained in‐chip with substrate that fluoresces upon cleavage by caspase‐3. Matrix was then digested using collagenase and cells were transferred to individual wells on silicon isolator affixed to a glass slide. Spot detection function was used to identify individual cells and generate statistics on signal intensity values in each cell and in sample as a whole. Figure 4C represents caspase‐3 activity in DU145 cells both with and without docetaxel treatment. As illustrated in this figure, there was an increase in caspase‐3 activity following docetaxel treatment, confirming that apoptotic activity could also be effectively detected and measured in Stacks.
In addition to analysis of the cytotoxic effects of chemotherapy, we also evaluated whether we could analyze the cytotoxic effects of targeted cancer‐directed therapies since this approach is becoming increasingly utilized for cancer treatment. To accomplish this, we seeded the H358 lung cancer cell line expressing the KRAS G12C oncogene on the collagen‐fibronectin matrix in Stacks, and treated with vehicle or adagrasib, a novel KRAS G12C small molecule inhibitor for 48 h. Adagrasib has been shown to be effective against several KRAS G12C models including the H358 cell line and a similar drug sotorasib. 34 , 35 Cells were then stained in‐chip with Image‐IT Dead (IID) Green live/dead dye along with anti‐EpCAM antibody and Hoechst33342, prior to fixation and imaging with confocal microscopy (Figure 4D). The spot detection function on NIS Elements was used to establish the frequency of IID‐positive dead cells per well and demonstrated adagrasib‐induced cytotoxicity in H358 cells under 3D microfluidic conditions (Figure 4E), thereby supporting a role for Stacks in evaluation of targeted treatments within the TME. The previous study of adagrasib did evaluate cytotoxicity in the H358 lung cancer cell line under 3D conditions by culturing H358 cells in spheroids using ultra‐low attachment plates without the addition of ECM. 35 Our findings are consistent with the prior study, and additionally demonstrate that adagrasib retains cytotoxicity against H358 cells cultured under 3D conditions on a collagen‐based ECM, an important finding given the protective effects of ECM on cell viability. 33 , 35 Furthermore, the cytotoxicity assay used in this study is compatible with immunofluorescent staining for cell surface markers (in this case, the epithelial marker EpCAM).
We also sought to determine whether we could use Stacks for multiparameter assessment of cellular features and the effects of drug treatments on primary patient‐derived cancer organoids (PDCOs) embedded and cultured in 3D matrix. Figure 5A shows the process of PDCO culture, treatment, and drug response assessment in‐device. PDCOs were cultured from primary tumor tissue from prostatectomy specimen as previously described 11 and were treated with vehicle, docetaxel, or bicalutamide, followed by fluorescent staining in Stacks. PDCOs were then mounted and imaged directly in‐chip by confocal microscopy (Figure 5B). Representative images and enumeration by manual counts in representative z‐stacks (Figure 5C) show that the EpCAM‐positive epithelial cells in PDCOs accumulated IID live/dead dye intracellularly following docetaxel and bicalutamide treatment compared to PDCO treated with DMSO vehicle, in accordance with previous reports of drug treatment of primary prostate PDCOs in a conventional cell culture platform. 36
FIGURE 5.
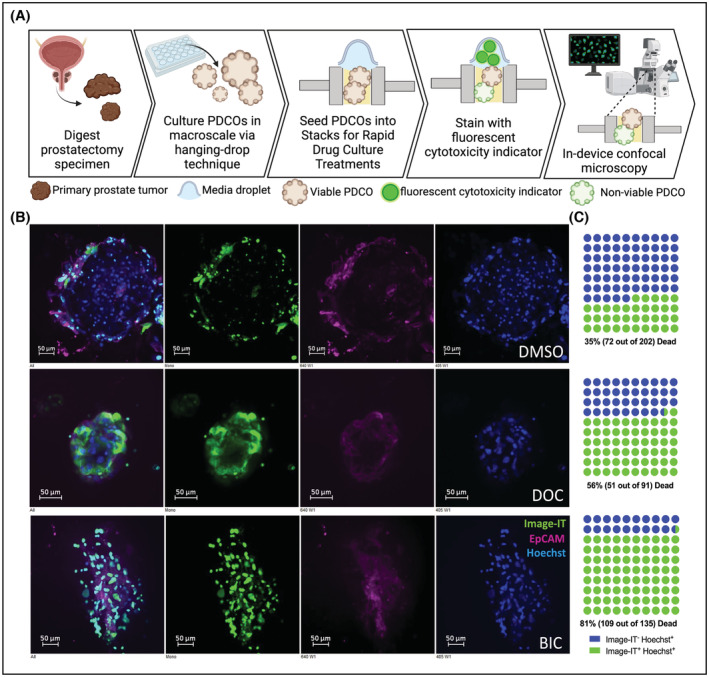
Drug treatment of primary prostate PDCOs. (A) PDCOs were derived from prostatectomy specimen from patients with primary prostate cancer. PDCOs were cultured in hanging droplets and transferred into STACKs for drug treatment. Fluorescent images (B) represent patient biopsy‐derived PDCOs after 72 h of treatment with DMSO, or 10 nM Docetaxel (DOC) or 100uM Bicalutamide (BIC), stained with Hoechst (blue), Image‐IT Dead™ Green (green), and EpCAM‐Alexa647 (purple). (C) Dot plots show manual counts of Hoechst+/Image‐IT− (blue) and Hoechst+/Image‐IT+(green) cellular content in representative z‐stack layers.
These cytotoxicity studies collectively demonstrate the versatility of the Stacks platform for evaluation of treatment effects including targeted, hormonal, and chemotherapeutic agents. The analysis can be performed using confocal microscopy in‐chip to minimize sample loss and disruption of 3D architecture or it can be effectively performed by transfer of samples to a microplate to increase the efficiency of the analysis. Analysis of multiplex phenotypic markers can be useful to evaluate cell‐type specific differential responses in the context of heterogenous microenvironments. Endpoints are also versatile and can include relative cell viability as well as evaluation of molecular pathways such as caspase‐3 activity.
3.4. Cell migration
Cell migration is a process that is critical to the roles of the TME in cancer progression. Immune‐tumor interactions, blood vessel recruitment, and tumor invasion all depend on effective migration. We therefore performed direct analysis of cell migration in Stacks. To evaluate these protocols, we used primary T cells as our model since this is a heterogeneous population that is highly motile. Figure 6A depicts the process for analysis of T cell migration toward tumor cells in 3D matrix in Stacks platform. The primary T cells were obtained from peripheral blood samples and stimulated with anti‐CD3 and anti‐CD28 beads. 22Rv1 tumor cells were seeded on 3D matrix in Stacks culture wells and allowed to adhere and proliferate. A different Stacks plate containing wells with collagen‐fibronectin matrix was attached on top of the 22Rv1 plate. T cells were then seeded on 3D matrix of top plate and allowed to migrate toward the 22Rv1 cells for 24 h. The T cell plate was then removed and cells were fixed in each well. The T cells were stained for CD4 and CD8 expression and the entire 3D well was imaged using confocal microscopy; representative images of a single Z‐layer and a 3D reconstruction of an entire well are shown in Figure 6B. Within NIS‐Elements software, images were analyzed with a spot detection macro on the nuclear channel to first identify individual cells, and then binary layer parameter restrictions were applied to classify cells as either CD4+ or CD8+. Average migration distance of each population was quantified by determining the distance of each cell from the top of the well. This distance was then summed for all CD4+ and CD8+ cells in each well and then divided by the total number of each cell population on the well (Figure 6C,D).
FIGURE 6.
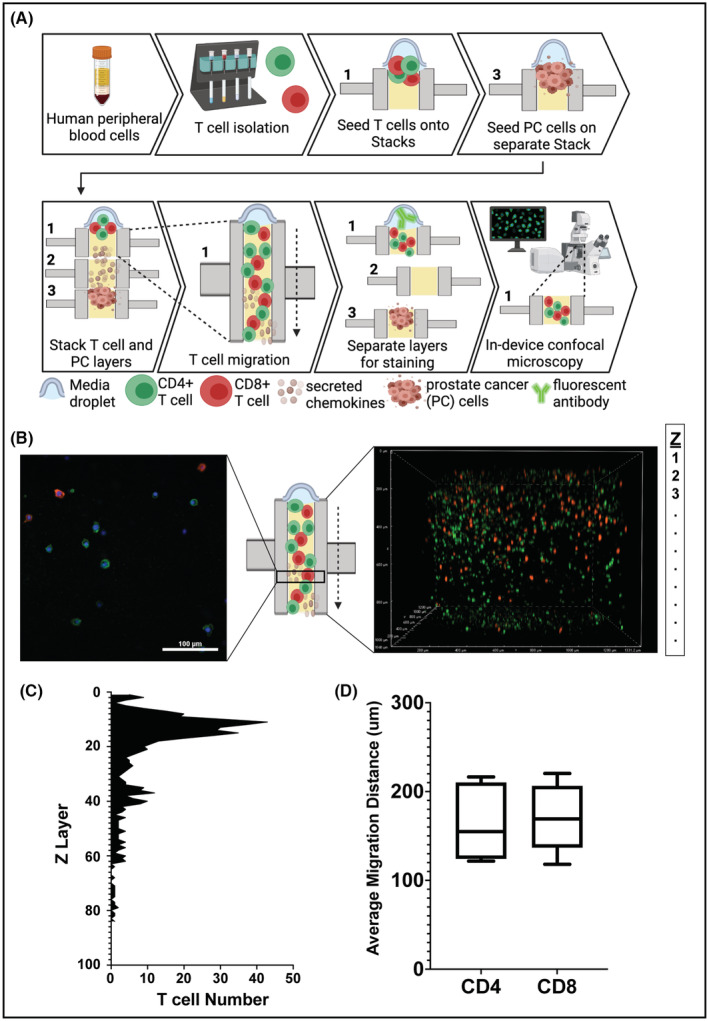
Analysis of cell invasion/migration in Stacks. (A) Workflow for primary T cell isolation, culture, and analysis of tumor‐directed migration. (B) Confocal fluorescence microscopy images of primary T cells in Stacks following 24 h migration. (Left) Axial slice of microwell; Hoechst (blue), CD4 (green), CD8 (red). (Right) 3D image of the entire well. Green cells are CD4+ T cells and red cells are CD8+ T cells. (C) Histogram representation of the total number of T cells (CD4+ and CD8+) identified in each z‐layer. (D) Box and whiskers plot with line at mean of migration distance of CD4+ and CD8+ T cells in Stacks (n = 6).
As illustrated in Figure 6B, we were able to clearly visualize the T cells throughout the 3D matrix in the microwells. The CD4 (green) and CD8 (red) staining was distinct among the two populations, enabling separate analysis of each subtype. Figure 6C shows the distribution of the T cells throughout the gel based on the quantification performed by spot detection. The migration pattern illustrated in this figure, which shows a “non‐migratory” population in the top layers and a collection of “migratory” T cells throughout the lower layers, is representative of all evaluated patient samples and was consistent across experimental repeats. For each well, we were able to derive the mean migration distance of each T cell subset as one method to compare T cell migration among replicate wells and among different patients. (Figure 6B). Our analysis of mean migration demonstrated directional migration of the CD4 and CD8 T cell populations toward the tumor cells with similar average migration distances for each donor. When compared to a mono‐culture T cell population, we observed a trend of increased directional migration of both T cell populations toward the tumor cells, indicating that the T cells were actively responding to tumor‐derived factors in the media (Supporting Information 1, Figure S3). These findings illustrate how the Stacks platform can be utilized to visualize and quantify cellular migration within multicellular models. While we analyzed T cells in co‐culture model in these experiments, these protocols could be applied to other cell types with the ability to modify the configurations of TME models as needed. In addition, endpoints other than mean migration, such as threshold migration, migration pattern, etc. could also be utilized to quantify/analyze cell migration depending on the particular needs of each experiment.
3.5. Leveraging the multiplexed and high throughput capabilities of Stacks
As detailed above, the notable features of the Stacks platform include multi‐culture of multiple cell types within a single system, efficient utilization of limited, and multiplexed analysis of complex culture models. These features are highly advantageous for the investigation of the TME, which is a complex multi‐cellular environment with diverse biologic functions. Therefore, having established that mRNA and protein expression, cellular phenotype, treatment cytotoxicity, 3D spatial architecture, and cell migration of both cell line and primary cells could be effectively analyzed using the Stacks platform, we next aimed to leverage the unique capabilities of Stacks to perform multiplexed and high‐throughput investigation of multi‐culture TME models using scarce patient biospecimens (Figure 7A). For our multiplexed experiment, we isolated autologous monocytes and T cells from a single patient blood sample and performed integrated analysis of mono‐, co‐, and tri‐culture models involving 22Rv1 tumor cells. The monocytes were differentiated to MDMs in‐chip and left unpolarized (M0) or polarized to M1‐ or M2‐like phenotypes using IFN‐γ and IL‐4, respectively. T cells were activated using CD3/CD28 activator beads. The M0 MDMs, activated T cells, and 22Rv1 tumor cells were cultured in mono‐(MDM mono, T cell mono, 22Rv1 mono), co‐ (MDM co‐T cell, MDM co‐22Rv1, T cell co‐22Rv1), and tri‐culture (MDM‐T cell‐22Rv1) configurations. Each of the models that included tumor cells were left untreated or treated with 80 nM of docetaxel (10 total conditions) for 48 h. Following culture, each cell population was isolated and analyzed for mRNA expression in all conditions using qPCR analysis (Figure 7B), and media was interrogated for corresponding protein expression of select genes using a multi‐analyte bead assay (Figure 7C). MDMs, T cells, and tumor cells were also stained with cell type‐specific surface markers and imaged in‐chip using fluorescent confocal microscopy (Figure 7D). Lastly, the docetaxel treated and untreated tumor cells were analyzed for relative viability in mono‐, co‐, and tri‐culture conditions using microplate reader analysis (Figure 7E). This entire experiment, which involved three cell types (two primary and one cell line), cultured in a total of 17 unique conditions (including tri‐culture) in triplicate, with multiplexed analysis of cell‐specific mRNA and protein expression, secreted factor concentration, and treatment cytotoxicity using four distinct conventional analysis modalities required less than 15 ml of blood from a single donor and could be performed by a single operator.
FIGURE 7.
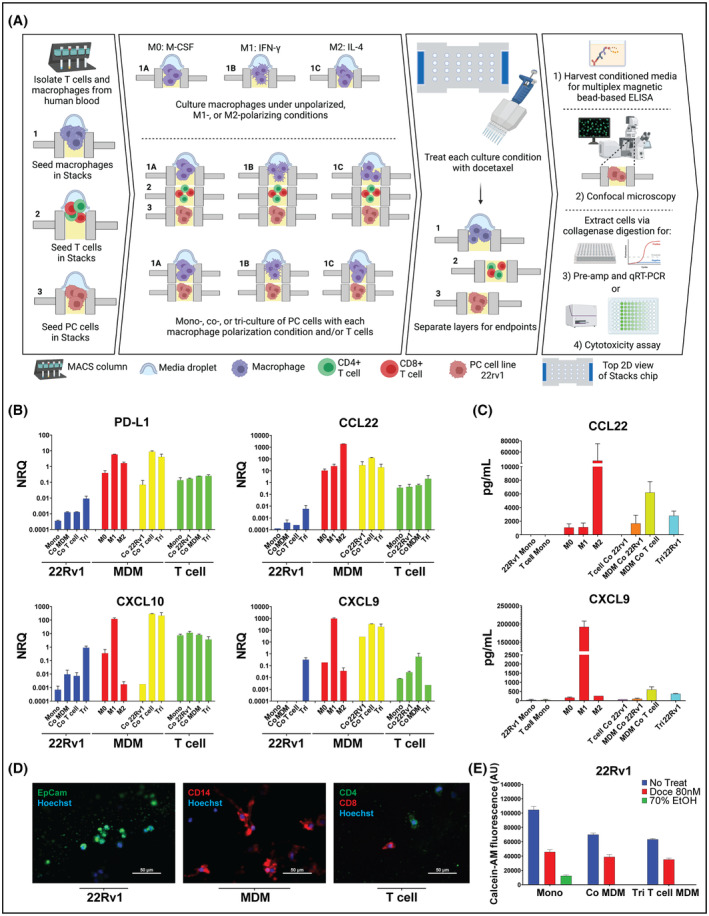
Multiplexed assay in Stacks. (A) Workflow of the isolation, culture, treatment, and analysis of cells for the multiplexed assay (B) mRNA expression of select genes measured by qRT‐PCR in each cell type (22Rv1, MDM, and T cell) each cultured in six different experimental conditions. Colors reflect the cell type from which the mRNA was obtained: 22Rv1 (in blue), Polarized MDM (in red) (M0, M1, M2), MDM (in yellow), and T cells. (in green). Data expressed as the Normal Relative Quantity (NRQ) (C) Concentration of select cytokines in media from each experimental condition using multi‐analyte bead assay. The bar colors between these two graphs are uniform in the cell type and culture condition that they represent. (D) Representative confocal microscopy images of the three above‐described cell types: 22rv1 (image on left) stained for EpCAM (in red) and Hoechst (in blue); MDM (image in middle) stained for CD14 (in red), and Hoechst (in blue); and T cells (image on right) stained for CD4 (in green), CD8 (in red), and Hoechst (in blue). (E) Quantification of viable 22Rv1 tumor cells in treated (80 nM docetaxel for 48 h) or DMSO conditions in monoculture (Mono), co‐culture with MDMs (Co MDM), and in tri‐culture with MDMs and T cells (Tri) using Calcein‐AM staining. Green bar represents 70% EtOH‐treated positive control for cell death. Data collected on a plate‐reader and expressed as total Calcein‐AM fluorescence arbitrary units (AU). Data for A–E are from a single experiment with at least two biologic replicates analyzed per condition.
As demonstrated in Figure 7B, we saw distinct mRNA expression profiles in each cell type and we were able to evaluate the impact of each culture condition on mRNA expression. For the MDMs, the mRNA and protein secretion profiles in the unpolarized and polarized mono‐culture conditions (which served as the internal controls) (Figures 7B,C) appropriately mirrored the findings of our single‐layer experiments (Figures 2B,C), supporting the accuracy of the expression analysis in the tri‐culture Stack. In co‐culture, the expression profiles of MDMs cultured with the tumor cells were similar to the profiles of the M2‐polarized macrophages. This was an expected finding since prostate tumor cells are known to polarize MDMs to an M2‐like phenotype. 36 , 37 In addition, MDMs co‐cultured with activated T cells resembled M1‐polarized macrophages, which was also expected since activated T cells secrete large quantities of the M1‐polarizing cytokine, IFN‐γ. 38 For the T cells and tumor cells in mono‐culture, we saw overall low expression of CCL22 and CXCL9‐10 at both the mRNA and secreted protein levels. This was again expected since macrophages are the major source of these cytokines within the TME. 37 Confocal imaging of each layer demonstrated detectable expression of the distinct cell‐specific markers for tumor cells (EpCAM+), MDMs (CD14+), and T cells (CD4+ and CD8+) in the separate Stacks layers (Figure 7D). These findings confirmed that in addition to evaluating each layer for mRNA expression levels, we could also visualize protein expression and identify specific cell types following multi‐culture experiments in Stacks. This capability enables layer‐specific immunophenotypic analysis as well as orthogonal evaluation of gene expression, analysis of cellular migration, and visualization of 3D architecture in Stacks cultures.
When we analyzed the tri‐culture data, we found that culturing the cells in tri‐culture resulted in differential gene expression profiles in the tri‐culture conditions compared to the mono‐ and co‐culture conditions. We also found that there was a trend toward a decrease in the number of viable tumor cells moving from mono‐culture to co‐culture to tri‐culture. This same trend was much less pronounced following docetaxel treatment, resulting in the appearance of a relatively smaller effect of docetaxel treatment in tri‐culture compared to the other treatment conditions in this experiment. (Figure 7E). This data suggests that expanding culture models beyond standard in vitro mono‐culture models has meaningful impacts on gene expression, culture dynamics, and chemosensitivity. Given the cellular complexity of the TME, such findings may prove to be more representative of cell behavior in multicellular niches, in vivo. However, further studies will be needed to confirm these findings.
Overall, this multiplexed experiment provides a demonstration of numerous key advantages of the Stacks platform over traditional platforms, including the capacity for investigation of more than two cell types (not possible with traditional Transwell systems) in a co‐culture setting, efficient utilization of limited resources (microwells require <1/10th the volume of biomaterials of a 96‐well plate), in‐chip processing and confocal imaging of 3D structures in all layers (not possible with traditional Transwell systems), and diverse multiplexed, multi‐analyte analysis using conventional tools generating orthogonal data sets from limited clinical biospecimen.
Our next aim was to demonstrate the high‐throughput assay capacity of the Stacks system. To accomplish this aim, we performed a co‐culture drug screening assay using prostate tumor cells that were cultured with primary MDMs. The primary MDMs were derived from monocytes of three individual donors and were differentiated and cultured within Stacks. The MDMs from each patient donor were co‐cultured with three independent prostate tumor cell lines (22Rv1, LNCaP, and C42B) to generate a total of nine unique co‐culture configurations (Figure 8A). Replicates from each model were then treated with one of three escalating doses of docetaxel or left untreated (a total of four conditions) for a total of 36 experimental conditions. In addition, we also set up 12 monoculture control conditions (untreated and treated conditions at each dose level for all three cell types alone) and three total positive biological controls for plate reader calibration (cells treated with 70% EtOH controls). All conditions were performed in triplicate for a total of 151 wells. Following 48 h of docetaxel treatment, tumor cells were isolated and viable cell number was established by measuring Calcein uptake with microplate reader analysis. The data presented in Figure 8B demonstrates a clear dose‐dependent effect of docetaxel treatment in all conditions. In addition, we were able to identify a potential patient‐specific effect (co‐culture of MDMs from patient 693 appeared to increase LNCaP sensitivity to docetaxel while other donor MDMs did not) as well as a potential cell‐type effect (MDMs from all patients appeared to protect C42B at the 120 nM dose). Since TME elements, including infiltrating macrophages, are well known to contribute to therapeutic resistance, experiments such as these could provide key sentinel data for further biologic investigation of the pathways that regulate TME‐mediated therapeutic resistance in cancer and evaluation of patient‐specific effects. In addition, it is worth noting that this experiment was performed by a single operator, demonstrating the feasibility of high throughput multi‐culture assays in Stacks.
FIGURE 8.
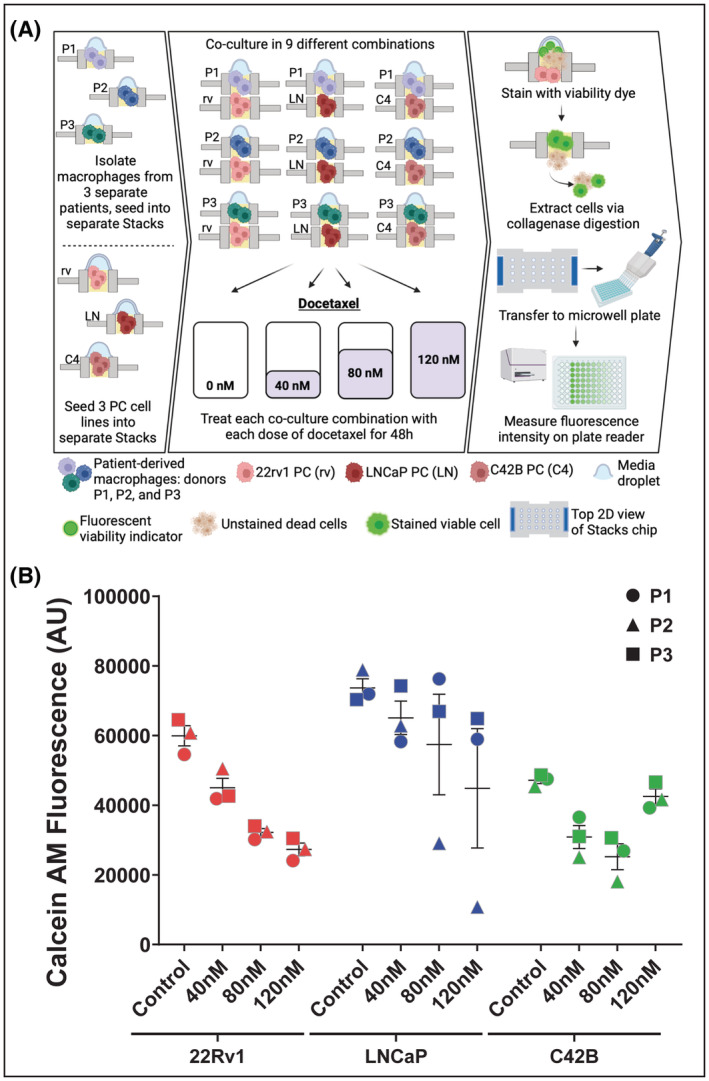
High throughput Stacks assay. (A) Workflow of isolation, culture, treatment, and analysis of high throughput TME docetaxel screening assay across 36 separate experimental conditions (51 total conditions with controls). (B) Quantification of viable cell number across all co‐culture treatment conditions using Calcein‐AM staining. Data organized by tumor cell line and docetaxel treatment concentration (48 h treatment). Data points are the average of at least two replicates of each experimental condition. The shapes indicate the patient from whom the MDMs were derived (Circle‐ Pt 1, Triangle‐ Pt 2, Square‐ Pt 3). Each color represents the type of tumor cell line across conditions (Red‐ 22Rv1, Blue‐ LNCaP, Green‐ C42B). Data expressed as total Calcein‐AM fluorescence arbitrary units (AU).
4. DISCUSSION
The TME is a highly complex, multi‐cellular environment with an array of critical roles in cancer progression and therapeutic resistance. 4 , 38 , 39 Although there has been considerable advancement in our understanding of the dynamic crosstalk within the tumor niche, there is still a clear need to refine models to more faithfully reflect the native TME network in vitro. 40 In this manuscript, we present TME‐relevant data for a reconfigurable microfluidic cell culture platform, known as Stacks, which provides numerous advantages for TME‐focused research over traditional in vitro systems. In prior publications, we detailed the design of the Stacks system and presented initial data supporting cell culture and analysis within Stacks. 10 , 12 However, these studies did not address a number of key features of the Stacks system, including primary cell culture, multi‐culture of >2 cell populations, flow cytometry analysis of cells recovered from Stacks, or analysis of secreted factors, cell migration, and treatment cytotoxicity. These studies also did not address the multiplexed and high‐throughput capabilities of Stacks, which are key features of this platform. We therefore aimed to expand the application of this technology to enable comprehensive TME‐directed research using primary cells in Stacks.
Using MDMs, MSCs, and PDCOs in mono‐, co‐, and tri‐culture systems, we were able to demonstrate that integrated analysis of mRNA expression, secreted factor production, and immunophenotype could be performed on a diverse array of primary TME populations in Stacks. We also demonstrated multiple methods of assessing cell viability, cytotoxicity, and apoptosis using fluorescent markers that can be detected in‐chip using confocal microscopy or by transferring cells to a microplate for high‐throughput analyses. With confocal imaging, we showed that these functional markers can be multiplexed with cell surface protein analysis to identify cell‐specific effects, evaluate spatial distribution of cytotoxicity within a 3D structure, and to visualize and quantify spatial processes such as cell migration. Furthermore, we demonstrated how these assays could be integrated into multi‐cellular TME models with multiplexed, high‐throughput capabilities.
While the first five figures of this manuscript individually demonstrate data that could be potentially generated in conventional platforms, they collectively illustrate how this single platform can support a broad range of investigative needs. Specifically, Stacks can be utilized to culture cell lines and primary cells in both 2D and 3D with minimal sample input. An array of multi‐analyte and multi‐functional assays including immunophenotyping, gene expression analysis, cytotoxicity assays, cell migration evaluation, etc. can be integrated with subsequent in‐chip or out‐of‐chip analysis most optimal for experimental priorities and feasibility. This broad functionality is one of the key advantage of Stacks over other available platforms, which typically do not offer this degree of assay flexibility. Additionally, the Stacks platform supports efficient utilization of scarce primary material as well as the culture of up to 6 independent cell populations within a single system. These are important advantages for TME research compared to traditional cell culture platforms, which typically require much larger inputs, necessitating a reliance on cell lines, and are limited in modeling complex, multi‐cellular environments. Although other microfluidic platforms may enable efficient utilization of biomaterials and support multi‐culture, the open and reconfigurable design of Stacks enables considerable flexibility to add, remove, and reorganize layers to investigate spatio‐temporal dynamics of cell culture. Furthermore, the design of the Stacks platform is conducive for injection molding supporting large‐scale fabrication and the layout of the wells is designed to accommodate standard multi‐channel pipettes to allow high efficiency and high throughput experimentation. These collective capabilities make Stacks truly unique among available macroscale and microscale platforms as well as a potentially valuable asset for cancer research.
The significance of expanding the applications of the Stacks platform is that each of the expanded applications has high relevance for TME research. Our tri‐culture data demonstrated that expanding beyond two cell types in in vitro tumor models can alter the biology of the various cell types within these models, therefore multi‐cultures may more faithfully reflect the in vivo biology of complex, multi‐cellular networks compared to mono‐ or co‐culture systems. Since multiple TME subpopulations play critical roles in therapeutic efficacy and resistance, the multi‐culture capabilities of Stacks may also provide a platform to perform rapid drug screening in the context of multi‐cellular TME networks, which could support the identification of therapies with higher translational relevance over traditional mono‐culture drug screening. Furthermore, with the high throughput and multiplexed capabilities of the system, numerous pathways can be evaluated simultaneously, potentially leading to more rapid biologic advances. Lastly, our data on T cell migration demonstrated how Stacks can be utilized to analyze cell migration and invasion in the context of multi‐cellular models. Cell migration is a critical function for tumor cell metastasis and immune cell infiltration, and the ability to analyze this process in multi‐cellular models could potentially advance immunotherapy and tumor metastasis research. 41 , 42
Overall, we believe that this manuscript validates the Stacks platform as a versatile tool for TME research and highlights some of the more unique features of the platform. The protocols demonstrated here can provide a foundation for further method development by other investigators utilizing this system, with considerable room to customize assay conditions to meet specific experimental needs. We look forward to the increased utilization of Stacks for TME research and anticipate that this platform will support meaningful gains in the field of cancer research.
AUTHOR CONTRIBUTIONS
David Kosoff, Nan Sethakorn, Erika Heninger, David J. Beebe, Peiman Hematti, and David F. Jarrardcontributed to the conceptualization and methodology of this study. David Kosoff, Nan Sethakorn, Erika Heninger, Matthew T. Breneman, Emma Recchia, and Adeline B. Ding contributed to data curation and data analysis. David Kosoff, David J. Beebe, Peiman Hematti, and David F. Jarrard acquired funding and supervised the research. David Kosoff, Nan Sethakorn, and Erika Heninger prepared the original draft of the manuscript. All Authors have reviewed & edited the manuscript.
DISCLOSURES
David Beebe holds equity in Stacks to the Future LLC, which licensed some of the technology used in this study.
Supporting information
Figure S1
Table S1
ACKNOWLEDGMENTS
We thank the UWCCC Flow Cytometry Laboratory, the Optical Imaging Core Facility, and the Small Molecule Screening Facility for use of shared equipment and excellent technical support. This work was supported by the Assistant Secretary of Defense for Health Affairs through the Prostate Cancer Research Program, Award No. W81XWH‐18‐1‐0273 to DK. The U.S. Army Medical Research Acquisition Activity, 820 Chandler Street, Fort Detrick MD 21702‐5014 is the awarding and administering acquisition office. Opinions, interpretations, conclusions, and recommendations are those of the author and are not necessarily endorsed by the Department of Defense. NS was supported by the United States Department of Veterans Affairs Advanced Fellowship in Women's Health and the University of Wisconsin Department of Hematology T32 HL07899 Fellowship funded by the National Institute of Health. This work was supported by the shared resources grant ‘University of Wisconsin Carbone Cancer Center Support Grant P30 CA014520’ by the National Institutes of Health including support for facilities and core services at the UWCCC Flow Cytometry Lab, Small Molecule Screening Facility, and the Translational Science Biocore. Figures were created with BioRender.com.
Sethakorn N, Heninger E, Breneman MT, et al. Integrated analysis of the tumor microenvironment using a reconfigurable microfluidic cell culture platform. The FASEB Journal. 2022;36:e22540. doi: 10.1096/fj.202200684RR
Nan Sethakorn and Erika Heninger should be considered the joint first author.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- 1. Buchheit CL, Weigel KJ, Schafer ZT. Cancer cell survival during detachment from the ECM: multiple barriers to tumour progression. Nat Rev Cancer. 2014;14:632‐641. [DOI] [PubMed] [Google Scholar]
- 2. Swann JB, Smyth MJ. Immune surveillance of tumors. J Clin Invest. 2007;117:1137‐1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646‐674. [DOI] [PubMed] [Google Scholar]
- 4. De Palma M, Lewis CE. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell. 2013;23:277‐286. [DOI] [PubMed] [Google Scholar]
- 5. DeNardo DG, Barreto JB, Andreu P, et al. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell. 2009;16:91‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Escamilla J, Schokrpur S, Liu C, et al. CSF1 receptor targeting in prostate cancer reverses macrophage‐mediated resistance to androgen blockade therapy. Cancer Res. 2015;75:950‐962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Orimo A, Weinberg RA. Stromal fibroblasts in cancer: a novel tumor‐promoting cell type. Cell Cycle. 2006;5:1597‐1601. [DOI] [PubMed] [Google Scholar]
- 8. Weber CE, Kuo PC. The tumor microenvironment. Surg Oncol. 2012;21:172‐177. [DOI] [PubMed] [Google Scholar]
- 9. Bejarano L, Jordāo MJC, Joyce JA. Therapeutic targeting of the tumor microenvironment. Cancer Discov. 2021;11:933‐959. [DOI] [PubMed] [Google Scholar]
- 10. Yu K, Chen B, Aran D, et al. Comprehensive transcriptomic analysis of cell lines as models of primary tumors across 22 tumor types. Nat Commun. 2019;10:3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Heninger E, Kosoff D, Rodems TS, et al. Live cell molecular analysis of primary prostate cancer organoids identifies persistent androgen receptor signaling. Med Oncol. 2021;38:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kosoff D, Yu J, Suresh V, Beebe DJ, Lang JM. Surface topography and hydrophilicity regulate macrophage phenotype in milled microfluidic systems. Lab Chip. 2018;18:3011‐3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yu J, Berthier E, Craig A, et al. Reconfigurable open microfluidics for studying the spatiotemporal dynamics of paracrine signalling. Nat Biomed Eng. 2019;3:830‐841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dominici M, Le Blanc K, Mueller I, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8:315‐317. [DOI] [PubMed] [Google Scholar]
- 15. Kim J, Hematti P. Mesenchymal stem cell‐educated macrophages: a novel type of alternatively activated macrophages. Exp Hematol. 2009;37:1445‐1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chinnadurai R, Rajakumar A, Schneider AJ, Bushman WA, Hematti P, Galipeau J. Potency analysis of mesenchymal stromal cells using a Phospho‐STAT matrix loop analytical approach. Stem Cells. 2019;37:1119‐1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor‐associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549‐555. [DOI] [PubMed] [Google Scholar]
- 18. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958‐969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Murray PJ, Allen JE, Biswas SK, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11:889‐896. [DOI] [PubMed] [Google Scholar]
- 22. Donnenberg AD, Donnenberg VS. Rare‐event analysis in flow cytometry. Clin Lab Med. 2007;27:627‐652, viii. [DOI] [PubMed] [Google Scholar]
- 23. Roederer M. How many events is enough? Are you positive? Cytometry A. 2008;73:384‐385. [DOI] [PubMed] [Google Scholar]
- 24. Pittenger MF, Discher DE, Péault BM, Phinney DG, Hare JM, Caplan AI. Mesenchymal stem cell perspective: cell biology to clinical progress. NPJ Regen Med. 2019;4:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Diedrich JD, Rajagurubandara E, Herroon MK, Mahapatra G, Huttemann M, Podgorski I. Bone marrow adipocytes promote the Warburg phenotype in metastatic prostate tumors via HIF‐1alpha activation. Oncotarget. 2016;7:64854‐64877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hardaway AL, Herroon MK, Rajagurubandara E, Podgorski I. Marrow adipocyte‐derived CXCL1 and CXCL2 contribute to osteolysis in metastatic prostate cancer. Clin Exp Metastasis. 2015;32:353‐368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wessely A, Waltera A, Reichert TE, Stockl S, Grassel S, Bauer RJ. Induction of ALP and MMP9 activity facilitates invasive behavior in heterogeneous human BMSC and HNSCC 3D spheroids. FASEB J. 2019;33:11884‐11893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wolff A, Frank M, Staehlke S, Peters K. A comparative study on the Adipogenic differentiation of mesenchymal stem/stromal cells in 2D and 3D culture. Cell. 2022;11:1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Emont MP, Yu H, Jun H, et al. Using a 3D culture system to differentiate visceral adipocytes in vitro. Endocrinology. 2015;156:4761‐4768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shen JX, Couchet M, Dufau J, et al. 3D adipose tissue culture links the organotypic microenvironment to improved adipogenesis. Adv Sci (Weinh). 2021;8:e2100106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Daya S, Loughlin AJ, Macqueen HA. Culture and differentiation of preadipocytes in two‐dimensional and three‐dimensional in vitro systems. Differentiation. 2007;75:360‐370. [DOI] [PubMed] [Google Scholar]
- 32. Paguirigan AL, Beebe DJ. From the cellular perspective: exploring differences in the cellular baseline in macroscale and microfluidic cultures. Integr Biol (Camb). 2009;1:182‐195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Edmondson R, Broglie JJ, Adcock AF, Yang L. Three‐dimensional cell culture systems and their applications in drug discovery and cell‐based biosensors. Assay Drug Dev Technol. 2014;12:207‐218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Skoulidis F, Li BT, Dy GK, et al. Sotorasib for lung cancers with KRAS p.G12C mutation. N Engl J Med. 2021;384:2371‐2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hallin J, Engstrom LD, Hargis L, et al. The KRAS(G12C) inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS‐mutant cancers in mouse models and patients. Cancer Discov. 2020;10:54‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Linxweiler J, Hammer M, Muhs S, et al. Patient‐derived, three‐dimensional spheroid cultures provide a versatile translational model for the study of organ‐confined prostate cancer. J Cancer Res Clin Oncol. 2019;145:551‐559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. House IG, Savas P, Lai J, et al. Macrophage‐derived CXCL9 and CXCL10 are required for antitumor immune responses following immune checkpoint blockade. Clin Cancer Res. 2020;26:487‐504. [DOI] [PubMed] [Google Scholar]
- 38. Giraldo NA, Sanchez‐Salas R, Peske JD, et al. The clinical role of the TME in solid cancer. Br J Cancer. 2019;120:45‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Correia AL, Bissell MJ. The tumor microenvironment is a dominant force in multidrug resistance. Drug Resist Updat. 2012;15:39‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wu M, Swartz MA. Modeling tumor microenvironments in vitro. J Biomech Eng. 2014;136:021011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bravo‐Cordero JJ, Hodgson L, Condeelis J. Directed cell invasion and migration during metastasis. Curr Opin Cell Biol. 2012;24:277‐283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chaplin DD. Overview of the immune response. J Allergy Clin Immunol. 2010;125:S3‐S23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Table S1
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.