

Abstract

Recently, we reported a library of 82 compounds, selected from different databanks through virtual screening and docking studies, and pointed to 6 among them as potential repurposed dual binders to both the catalytic site and the secondary binding pockets of subunit A of ricin (RTA). Here, we report additional molecular modeling studies of an extended list of compounds from the original library. Rounds of flexible docking followed by molecular dynamics simulations and further rounds of MM-PBSA calculations using a more robust protocol, enabled a better investigation of the interactions of these compounds inside RTA, the elucidation of their dynamical behaviors, and updating the list of the most important residues for the ligand binding. Four compounds were pointed as potential repurposed ricin inhibitors that are worth being experimentally investigated.
Introduction
Ricin is a highly toxic plant toxin present in the seeds of Ricinus communis, a bush native from the Mediterranean area, Eastern Africa and India, which can also be found in all tropical regions today. Also known as castor oil plant, R. communis is the source of the ricin oil, a raw material extensively used in the production of lubricants, green fuel, drugs, and cosmetics.1 However, the high toxicity of ricin occasionally causes accidental intoxication of workers involved in the oil extraction. Also, the castor oil cake, a byproduct of the ricin oil extraction, is rich in minerals and other nutrients, being, for this reason, used as a fertilizer and to feed livestock. As this byproduct is not always totally free of ricin, intoxication or even death of livestock also happens as well.
Due to its easiness of obtention, water solubility, and toxicity higher than that of the nerve agents, ricin has also been used as a chemical-biological warfare agent for assassination and terrorism purposes. Many reports of this misuse can be found in the literature,2−4 with the most notorious being the assassination, in London, of the Bulgarian journalist Georgi Markov in September 1978. The murderer used an umbrella adapted with a hypodermic needle at its extremity to inject a tiny sphere stuffed with around 0.4 mg of ricin, and covered with wax, in Markov’s leg, causing his death 4 days later. Literature also reports the tentative of aerolization or mass production of ricin by terrorist groups in 19892 and 2010,4 besides several cases of letters contaminated with ricin being delivered to public authorities in the USA in the decades 2000 and 2010.2,5
Ricin is a N-glycosidase classified as a type II ribosome-inactivating protein (RIP). This family of proteins found in bacteria and plants is capable of inhibiting the protein synthesis in eukaryotic cells, playing an important defense role against pathogens and insects.6 Despite sharing similar mechanisms of action and being structurally related, the RIP types I and III are not as cytotoxic as ricin and the other RIP type II, such as abrin. This happens because those RIPs lack the B domain responsible to enable their entrance into the cells.6
The mechanism of action of ricin comprises a permanent damage to the rRNA due to the abstraction of the adenine 4324 (A-4324) from the loop GAGA of the 28S rRNA, which is highly conserved in eukaryotic cells. This causes an interruption in the protein synthesis and lead to cell death.7,8 The ricin unit responsible for this is known as RTA (or Ricin Toxin A) which unites to RTB (Ricin Unit B) through a disulfide bond to compose the whole ricin structure. Once inside the endoplasmic reticulum of the cell, RTA is separated from RTB, due to the action of the enzyme disulfide isomerase, and moves to the cytosol where it will promote the rRNA damage. To date, there is no antidote against such action yet and the vaccines developed so far to prevent ricin intoxication are not effective.8,9
The search for antidotes against ricin has already afforded many compounds capable of binding to the catalytic site of RTA (comprising residues Val81, Gly121, Glu177, and Arg180) and work as competitive inhibitors.10,11 However, no activity below the micromolar range was achieved yet, with the most promising compounds reported so far (Figure 1) presenting IC50 between 6 and 20 μM.11 The publication of the crystallographic structure of NNCP (Figure 1) complexed at the catalytic site of RTA in the Protein Data Bank (PDB) (https://www.rcsb.org/) under the ID: 4HUO, elucidated the binding mode (see Figure S1a) of those compounds.11 Besides it also revealed that they are not capable of also binding to the secondary binding pocket of RTA (comprising residues Asp75, Asn78, Asp96, and Asp100). This pocket had been previously revealed by Ho et al. (2009)12 through the crystallographic structure of RTA complexed with the model cyclic tetranucleotide inhibitor C2X [Figure S1b], which mimics the recognition loop of the 28S rRNA [PDB (https://www.rcsb.org/) ID: 3HIO].12 This cyclic tetranucleotide was designed by Ho et al. (2009)12 with the goal of establishing the catalytic site features contributing to the RTA catalytic activity. It is also a transition state structure meant to guide the design and synthesis of potent RIP inhibitors. Analysis of 4HUO [Figure S1b] shows clearly that there is still enough room for the drug design/discovery of new and more potent inhibitors, targeting at the same time the catalytic site and the secondary binding pocket of RTA.
Figure 1.

Structures of the best competitive inhibitors of RTA currently reported in the literature.
Recently, we proposed the repurposing approach as a promising strategy to follow in the search for more effective antidotes against ricin intoxication capable of binding into both the catalytic site and the secondary pocket of RTA.13 On this line, we performed virtual screening (VS) searches in the FDA-approved drugs data set, available at Cheminfo (https://chemoinfo.ipmc.cnrs.fr/), the approved drugs library available at DrugBank (https://www.drugbank.ca/), and the PubChem database (https://pubchem.ncbi.nlm.nih.gov/). This search enabled the selection of 6795 potential binders to RTA which, after additional refinements, including drug-likeness, afforded a library of 82 compounds. Further molecular dynamics (MD) simulations on the first 15 compounds selected from this library enabled pointing 6 (Figure S2) as potential antidotes against RTA.13 Here, we moved forward on the investigation of this library through additional theoretical studies on the 6 molecules shown in Figure S2 plus 9 more selected compounds (Figure 2). Flexible docking of these compounds enabled drawing their fingerprints inside RTA and plotting the most important residues for the ligand binding. Also, longer MD simulations of 500 ns of the best poses of these compounds inside RTA enabled reevaluating the former results and selecting the ones that should keep the interactions inside the catalytic site and the secondary pocket, besides ranking them according to the potential of inhibiting RTA.
Figure 2.

2D structures of the 9 additional compounds selected for the current study.
Methodology
Protein Preparation
The receptor used to perform our studies was the three-dimensional structure of RTA in complex with the cyclic tetranucleotide inhibitor C2X available in the PDB (https://www.rcsb.org/) under the code: 3HIO.12 This structure was downloaded and optimized using the default configuration of the quickprep tool of the MOE package (https://www.chemcomp.com/Products.htm) in order to remove gaps, optimize bond lengths and angles, calculate charges, and properly protonate residues and ligand according to the physiologic pH. Crystallographic water molecules were also removed, and only ligand and RTA were kept in the structure for the theoretical studies.
Ligand Preparation
The 3D structures of the ligands studied here (Figures 2 and S2) were constructed using the builder tool of the MOE package (https://www.chemcomp.com/Products.htm) and added to a MOE databank named as “ligands.mdb”. Afterward, each entry of the databank was “washed”, using the compute/molecule/wash tool of the MOE package (https://www.chemcomp.com/Products.htm), in order to optimize bond lengths, angles, and charges and also to afford the dominant species of each ligand under physiologic pH.
Docking Studies
The entries of the ligands.mdb file described above were submitted to rounds of docking calculations on the active site of the crystallographic structure of RTA (PDB ID: 3HIO) using the dock module of the MOE package (https://www.chemcomp.com/Products.htm). The docking grid was defined to include all residues present in the whole binding pocket of C2X and the docking placement method used was Triangle Matcher with induced fit refinement and the generate fingerprints options active. First, the 100 best ranked poses (lowest energies) of each ligand were collected using the London dG scoring function, and afterward, those poses were re-scored using the GBVI/WSA dG function with the 20 best results being collected for further analysis of the fingerprints of each ligand inside RTA. This docking protocol was validated by re-docking of the crystallographic structure of C2X inside 3HIO, where the five best ranked poses were collected. The docking energy of NNCP inside RTA was obtained after energy minimization of the crystallographic structure 4HUO. Regarding NNCT, once it is reported in the literature that it binds the same way as NNCP,11 the docking energy was calculated through energy minimization of 4HUO after changing the structure of NNCP to NNCT by adding an −OH group in the para position of the NNCP phenyl ring.
MD Simulations
The dynamics tool of the MOE package (https://www.chemcomp.com/Products.htm) was used to run the MD simulations. The Compute/Simulations/Dynamics path was used to prepare each system according to the parameters of the NAMD14 software and using the forcefield AMBER10:EHT,15 with a cutoff of 10 for electrostatic and 8, 10 for VdW interactions. Each complex was centered in a cubic box containing around 9000 water molecules and neutralized with NaCl ions. The rounds of simulations involved first 10 ps of an energy minimization step followed by 100 ps of NPT and 200 ps of NVT. Afterward, production steps of 500 ns of free MD simulation were performed. The MD simulation results were analyzed using the md_analysis tool and the database viewer (DBV) menu of MOE (https://www.chemcomp.com/Products.htm). The trajectory files of each system, used for analysis of the MD simulations, were generated after superposition of the initial and simulated frames of protein and ligand.
MM-PBSA Calculations
Hybrid sequential QM/MM–MD methods are among the most accurate ways to estimate the free energy associated to the protein–ligand binding.16 However, as many configurations are generated in large MD simulations, the number of QM calculations required is too high because in every step, an energy evaluation of the system is needed. Therefore, a great computational effort is necessary to carry out this kind of simulation. Aiming, then, to reduce the number of QM calculations without the loss of the relevant information from the simulation, new methods based on the statistical inefficiency and wavelet analysis for selecting MD conformations had been reported in the literature.17 However, the computational cost involved is still high. One alternative to reduce this computational cost with an acceptable impact on the accuracy of the calculations is the use of the MM-PBSA approach,18 which has become one of the most used methods for the estimation of free energy in biological systems.19
Despite not explicitly calculating the entropy effect, the MM-PBSA method considers bonded and nonbonded interactions (vacuum potential energy), as well as polar and nonpolar terms (free energy of solvation).18 The Poisson–Boltzmann equation is used to estimate the solvation energy term (usually with dielectric constant set to 1), while the surface area (SASA) method18 is used to calculate the nonpolar solvation energy term.
Here, the MM-PBSA calculations were performed as before13 using the g_mmpbsa tool, compatible with GROMACS 2019.4 software.18 For this, it was necessary first to run 100 ns of MD simulation of the best poses of the ligands inside RTA obtained in the docking studies in order to afford the trajectory frames needed to feed the g_mmpbsa tool. Those MD simulations were performed following the same protocol used before13 and described in detail in the Supporting Information. The g_mmpbsa tool was used to predict the binding free energy of the ligands that showed capable of keeping interactions in both pockets of RTA during the 500 ns of MD simulations with NAMD.14
Results and Discussion
As mentioned in the Introduction, we studied in this work the 6 molecules pointed before as potential antidotes against ricin (Figure S2)13 plus 9 additional compounds (Figure 2) originally selected by VS and pointed by docking studies as potential dual binders to RTA, but not investigated through MD simulations. By extending the study to these 9 compounds, we advanced one more step toward the ultimate goal of investigating through MD simulations the whole library of 82 compounds formerly selected by VS.13 These 9 compounds were selected based on a visual inspection to ensure a structural diversity that represents the most of the whole library.
Docking Study
The total energy values of the five poses returned from the re-docking of C2X over its crystallographic structure (3HIO) ranged from −15.23 to −15.62 kcal·mol–1, while the root-mean-square deviation (RMSD) values ranged from 1.66 to 2.01 Å. Because a RMSD < 2.0 Å is considered valid according to the literature recommendation,20 the poses obtained during our re-docking studies were good enough to validate the docking protocol used. The superposition with the lowest RMSD observed (1.66 Å) is shown in Figure S3.
It is well known that an appropriate theoretical strategy for selecting docking poses is crucial for the subsequent steps of MD simulations in order to avoid conformational changes in the time scale of few nanoseconds, which could compromise the MD simulation viability. For this reason, distinct selecting poses approaches have been reported in literature.21 In the current work, as we are looking for potential dual binders to RTA, we selected the best ranked pose (lowest energy) of each ligand showing interactions with at least one residue of both the catalytic site and the secondary pocket of RTA. The only exception was C2X which the pose selected was the one with lowest RMSD from the re-docking, shown in Figure S3. The docking results obtained for the selected poses are summarized in Table 1.
Table 1. Interactions of the Best Poses of Each Ligand after Induced Fit Docking inside RTAa.

Residues of the catalytic site are shown in red, while the ones of the secondary pocket are shown in blue. D = donor to side chain; A = Acceptor from sidechain; I = Ionic attraction; d = donor to backbone; a = acceptor from backbone; and R = Arene attraction.
As shown in Table 1, no ligand presented more negative energy value compared to C2X. This was already expected once this compound mimics the sarcin–ricin recognition loop of the 28S rRNA. However, the best poses of most ligands presented total energy more negative than NNCP and NNCT. This suggests a higher affinity for the RTA active site than the reference ligands and, therefore, a higher potential to act as more effective RTA inhibitors. Regarding the 9 additional compounds included in this work, all ranked in the same range of values (between −7.44 and −9.74 kcal·mol–1) as the 6 compounds repurposed before,13 with the most promising among all these molecules being CID 135977982 and CID 136132835 ranking, respectively, −9.74 and −9.72 kcal·mol–1. This means that they might also be good options of repurposed ligands against RTA.
Results in Table 1 also show that among the residues not belonging to the catalytic or the secondary pockets, Arg213 and Arg258 showed up in interactions with most of the ligands. These two residues are located in the border of the active site of RTA which includes the catalytic site and the secondary pocket, but in opposition to them, as illustrated in Figure S4, which shows the best pose of CID 135977982 inside RTA. As far as we know Arg213 and Arg258 have not been reported yet in the literature as important residues for the binding of inhibitors to RTA.
MD Simulations
The plots of total energy for the 18 complexes studied by MD simulation (Figures S5–S10) show that all systems stabilized since the beginning of the simulation time, with an average energy ranging between −0.9 × 10–6 and −1.3 × 10–6 kcal·mol–1. Also, the RMSD plots calculated after superposition of initial and simulated structures for RTA and ligands (Figure 3), show that the fluctuations never passed 6.0 and 2.5 Å for the ligands and protein, respectively, except for plazomicin that was not capable of stabilizing inside RTA and left after 50 ns of MD simulation (see also movie: Plazomicin_RTA.mpg in the Supporting Information). For the other 17 systems, the total energy and RMSD results point to stability over the simulated time and accommodation of the ligands inside RTA.
Figure 3.

RMSD plots for the 18 systems during the MD simulations. Black lines = RTA, red lines = ligands.
Table 2 lists all H-bonds formed during the MD simulations with their respective percentages of occupancy, while the plots for the ligands capable of dual biding to RTA during more than 10% of the simulated time are shown in Figures S11–S14. In those Figures, the graphs in red lines represent the number of H-bonds formed with residues of the catalytic site of RTA, while the graphs in blue lines represent H-bonds formed with residues of the secondary pocket. As can be seen, most of the interactions pointed in the docking studies (Table 1) were confirmed by the MD simulations, with some additional interactions showing up during the MD simulations. It can also be seen in Table 2 that both Arg213 and Arg258 show up h-bonding to C2X with occupancies > 10%. Arg258 also shows up h-bonding to many other ligands with high percentages of occupancy. This corroborates the docking findings and suggests that these two residues might be key for the binding into RTA.
Table 2. H-Bonds Formed during the MD Simulationsa.


Residues of the catalytic site are shown in red, while residues of the secondary pocket are shown in blue.
Analysis of Table 2 and Figure S11 show that C2X was capable of maintaining H-bonds with the residues of both the catalytic site and the secondary pocket during the whole simulated time, showing occupancies over 10% with at least three residues from each site. Also, the RMSD plot of C2X (see its red line in Figure 3) shows stabilization around 3.5 Å, what means a good accommodation inside RTA. This is well illustrated in the movie: C2X_RTA.mpg in Supporting Information and by the frames shown in Figure 4 (where the external loop of C2X was omitted for better clarity). As can be seen, the cytosine and guanosine portions of C2X, found, respectively, in the catalytic site and the secondary pocket of RTA in the crystallographic structure (PDB code 3HIO), stay inside those cavities during the whole simulation time. Therefore, our theoretical results confirmed that C2X is capable of binding and stabilizing inside RTA, mimicking the recognition loop of 28S rRNA. This corroborates the results reported by Ho et al. (2009)12 and the mechanism of action reported in the literature for RTA.12
Figure 4.
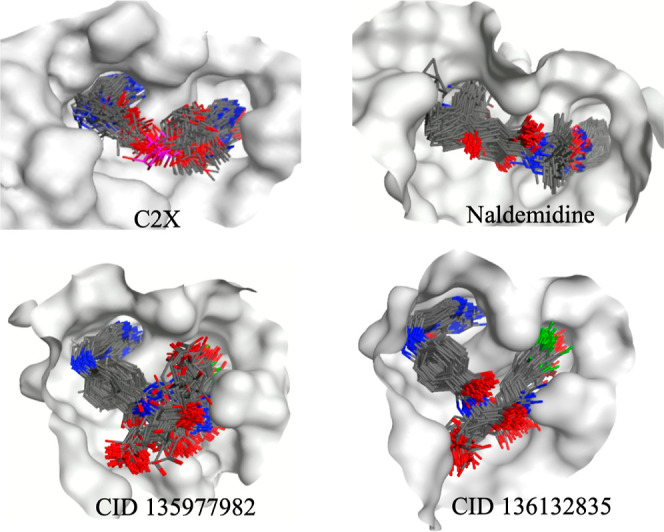
Superposition of frames of C2X, naldemedine, CID 135977982, and CID 136132835 collected during the MD simulations. Hydrogen atoms and the external loop of C2X were omitted for better clarity. The receptor surface is represented in gray.
Only CID 20044260, CID 22659428, naldemedine, CID 135977982, and CID 136132835 kept H-bonds with residues of both the catalytic site and the secondary pocket during the whole simulated time (see Figures S11–S14). Analysis of the dynamical behavior of those compounds (see the respective movies supplied as the Supporting Information) revealed that only naldemedine, CID 135977982, and CID 136132835 showed a good accommodation inside RTA as can be also observed in the superposition of frames collected during the MD simulations shown in Figure 4. This is also reflected in Figure 3 which show very little RMSD fluctuations for these compounds. CID 20044260 and CID 22659428, besides showing larger RMSD fluctuations (Figure 3), also did not accommodate well inside RTA as can be seen in the superposition of frames collected during the MD simulations shown in Figure S15 and the respective movies in the Supporting Information.
The large RMSD fluctuations observed in some systems are due to unstable interactions between protein and ligand which make both to move more. Larger fluctuation means worse behavior and, therefore, not good accommodation of the ligand inside RTA. They reflect changes in positions of the ligands during the MD simulations and the corresponding effect in the RTA behavior. These movements can be observed in the movies of the MD simulations supplied as Supporting Information.
Analysis of Figure S15 and the movies supplied as Supporting Information show that most of the no dual binders did not stabilize well inside RTA during the MD simulations. Regarding NNCP and NNCT, while the pterinic rings stabilized inside the catalytic site, the other moiety of those molecules did not find where to anchor and remained unstable the whole simulation. This is a possible reason for the IC50 in the range of μM reported for these compounds.11 A similar behavior was also observed for CID 136023163, CID 18498053, CID 20044260, deferoxamine, CID 18309602, ertapenem, leucovorin, and CID 22659482. Ceftaroline, on the other hand, moved from the active site since the beginning of the simulation and despite staying close to RTA, did not accommodate well inside it. The only ligands in Figure S15 that showed stable dynamic behavior inside RTA were nilotinib and CID 18493267 which, despite not behaving as dual binders, stabilized during the MD simulation, keeping interactions inside the secondary pocket and at the entrance of the catalytic site (see movies: Nilotinib_RTA.mpg and CID18493267_RTA.mpg supplied as the Supporting Information). This means that these two ligands might also bind strongly to RTA, avoiding the interactions with the loop GAGA of the rRNA 28S.
MM-PBSA Calculations
The bars plot in Figure 5 show the values of binding energies predicted through MM-PBSA calculations for the reference compounds and the potential dual binders pointed in the MD simulations. As expected, all of them presented negative binding energies, with a good correlation with the docking and MD results. This fact confirms their affinities for RTA. It is interesting to notice, however, that CID 135977982 and CID 136132835 showed better (more negative) binding energies than C2X. This is probably due to the cyclic part of C2X which did not establish stable interactions inside RTA and fluctuated during the MD simulation (see movie C2X_RTA.mpg in the Supporting Information). Those fluctuations might be adding unfavorable interactions which contribute to increase the binding energy value.
Figure 5.

MM-PBSA results for RTA complexed with the reference compounds and the potential dual binders.
Comparison of Results for the 6 Potential Dual Binders Pointed Before
The flexible docking studies performed with MOE corroborated the rigid docking results obtained before using Molegro Virtual Docker (MVD)13 with all repurposed ligands showing energetically favorable poses (negative values) that can H-bond to at least one residue of each binding site of RTA. Also, the values of docking energy with both methods followed the same tendency, with the only outlier being deferoxamine (see Table 3). This might be due to the higher flexibility of this compound that would favor finding a more energetically favorable pose during the flexible docking in comparison to the rigid docking. Despite this, in general, the results suggest that both docking protocols might be suitable for finding appropriate poses for the further MD simulations steps.
Table 3. Rigid and Flexible Docking Results for the 6 Potential Dual Binders Pointed Before.

Differently from the docking studies, not much corroboration was found when comparing the MD protocols applied to the 6 first potential dual binders. The results obtained for the extended MD simulations suggest that the 50 ns of free MD simulation ran before might not be enough to fully characterize the dynamical behavior of these compounds inside RTA. As shown in the H-bond plots in Figures 4 and S11, none of the 6 compounds was capable of keeping H-bonds with residues of the catalytic site and the secondary pocket during the whole simulated time. Deferoxamine was the one with best performance but lost consistency after 200 ns. CID 18498053, CID 18309602, and leucovorin established H-bonds mainly with the secondary pocket, while CID 136023163 and plazomicin lost the H-bonds with the RTA during the first third of the simulated time. Plazomicin was not even capable of stabilizing in another position inside RTA and left before 100 ns of simulation, as can be seem in the RMSD plots shown in Figure 3 and in the movie Plazomicin_RTA.mpg in the Supporting Information.
Pterin Derivatives
It is important to notice that the compounds CID 135977982, CID 136023163, and CID 136132835 (Figures 2 and S2) are all pterin derivatives, differing from each other only by the substitution pattern in the phenol ring. CID 136023163 brings two iodine atoms in the ortho positions to the OH group, while CID 136132835 brings one fluorine in one ortho position and an iodine in the other, and CID 135977982 has only one fluorine atom in ortho to the OH group. Despite this single difference, the best pose of CID 136023163 was not capable of stabilizing inside RTA forming H-bonds with the catalytic site and secondary pocket (see Figure S15 and the movie CID136023163-RTA.mpg supplied as Supporting Information), while the other two analogues displayed the best binding results of our study, showing a binding mode very similar to the reference compound C2X (see Figure S14 and the movies CID135977982-RTA.mpg and CID136132835_RTA.mpg supplied as Supporting Information). This probably happened because the two large iodine atoms did not allow the stabilization of the phenol ring of CID 136023163 in the secondary pocket. Such result adds some valuable structure activity relationship (SAR) information to this project that certainly will help in the selection/design of the ideal ring size on this part of the molecule to achieve an effective and stable binding.
Conclusions
Our theoretical study allowed a refinement of the protocol used before13 for the theoretical investigation of repurposed drugs selected by VS, as potential antidotes against ricin and also moving forward on the theoretical investigation of the library selected before.13 Despite no significant changes were observed using the new docking protocol, the extension of the time of MD simulation for 500 ns showed a fundamental step to corroborate the docking results and filter the ligands which are really capable of performing stable interactions inside RTA. This refinement enabled pointing with more confidence four compounds to further in vitro assays: CID 135977982, CID 136132835, and naldemidine, as potential dual binders to RTA, and nilotinib and CID 18493267, as single binders to the secondary pocket of RTA. Despite not interacting directly with residues of the catalytic site, these compounds seem to be capable of interacting with residues at its entrance, blocking the catalytic activity of RTA. This might trigger a stronger inhibition. We believe that these five compounds will present IC50 values in the nM range after experimentally evaluated. Our study also corroborated the complex RTA/C2X as a consistent model of the RTA binding to the loop GAGA of the 28S rRNA. This validates the PDB structure 3HIO reported by Ho and con-workers12 for the drug design against RTA. On this sense also the discovery of consistent H-bonds with residues Arg213 and Arg258 observed for most of the ligands in both the docking and MD simulations, opens new opportunities for the drug design of new antidotes against ricin. As far as we know, this was not described in the literature before. Still on the context of drug design, the limitation imposed by the two iodine atoms in the phenol ring of the pterin derivatives adds useful information for the design of more effective pterin derivatives as antidotes against ricin.
Acknowledgments
The authors wish to thank the Chemical Computing Group for technical support with MOE; Calcul Quebec for the access to high performance hardware facilities; and INRS—Centre Armand-Frappier Santé biotechnologie for the infrastructure.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c04819.
Binding modes of NNCP and C2X inside RTA; 2D structures of the 6 compounds pointed before by VS for repurposing; protocol used for MD simulations using GROMACS; redocking of C2X; best pose of CID 135977982 inside RTA; energy plots after MD simulations; H-bonds formed by the dual binders during the MD simulations; and superposition of frames collected during the MD simulations (PDF)
MD simulation movies (ZIP)
The authors declare the following competing financial interest(s): M.L.D. is an employee of Chemical Computing Group ULC, creators of MOE software used to perform most of the modeling work described herein.
The authors declare the following competing financial interest(s): Michael L. Drummond is employee of Chemical Computing Group ULC, creators of MOE software used to perform most of the modeling work described herein.
Supplementary Material
References
- a Doan L. G. Ricin: Mechanism of toxicity, clinical manifestations, and vaccine development. A review. J. Toxicol., Clin. Toxicol. 2004, 42, 201–208. 10.1081/CLT-120030945. [DOI] [PubMed] [Google Scholar]; b Sousa R. B.; Lima K. S. C.; Santos C. G. M.; França T. C. C.; Nepovimova E.; Kuca K.; Dornelas M. R.; Lima A. L. S. A New Method for Extraction and Analysis of Ricin Samples through MALDI-TOF-MS/MS. Toxins 2019, 11, 201. 10.3390/toxins11040201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audi J.; Belson M.; Patel M.; Schier J.; Osterloh J. Ricin Poisoning - A comprehensive review. JAMA 2005, 294, 2342. 10.1001/jama.294.18.2342. [DOI] [PubMed] [Google Scholar]
- a Janik E.; Ceremuga M.; Saluk-Bijak J.; Bijak M. Biological toxins as the potential tools for bioterrorism. Int. J. Mol. Sci. 2019, 20, 1181. 10.3390/ijms20051181. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Knight B. Ricin - a potent homicidal poison. Br. Med. J. 1979, 1, 350–351. [PMC free article] [PubMed] [Google Scholar]
- Pita R.; Romero A. Toxins as Weapons: A Historical Review. Forensic Sci. Rev. 2014, 26, 85–96. [PubMed] [Google Scholar]
- Musshoff F.; Madea B. Ricin poisoning and forensic toxicology. Drug Test. Anal. 2009, 1, 184–191. 10.1002/dta.27. [DOI] [PubMed] [Google Scholar]
- a Zhou K.; Fu Z.; Chen M.; Lin Y.; Pan K. Structure of trichosanthin at 1.88 Å resolution. Proteins: Struct., Funct., Bioinf. 1994, 19, 4–13. 10.1002/prot.340190103. [DOI] [PubMed] [Google Scholar]; b Funatsu G.; Islam M. R.; Minami Y.; Sung-Sil K.; Kimura M. Conserved amino acid residues in ribosome-inactivating proteins from plants. Biochimie 1991, 73, 1157–1161. 10.1016/0300-9084(91)90160-3. [DOI] [PubMed] [Google Scholar]; c Endo Y.; Tsurugi K.; Yutsudo T.; Takeda Y.; Ogasawara T.; Igarashi K. Site of action of a Vero toxin (VT2) from Escherichia coli O157:H7 and of Shiga toxin on eukaryotic ribosomes. Eur. J. Biochem. 1988, 171, 45–50. 10.1111/j.1432-1033.1988.tb13756.x. [DOI] [PubMed] [Google Scholar]; d May M. J.; Hartley M. R.; Roberts L. M.; Krieg P. A.; Osborn R. W.; Lord J. M. Ribosome inactivation by ricin A chain: a sensitive method to assess the activity of wild-type and mutant polypeptides. EMBO J. 1989, 8, 301–308. 10.1002/j.1460-2075.1989.tb03377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord J. M.; Roberts L. M.; Robertus J. D. Ricin: structure, mode of action, and some current applications. FASEB J. 1994, 8, 201–208. 10.1096/fasebj.8.2.8119491. [DOI] [PubMed] [Google Scholar]
- Olson M. A.; Carra J. H.; Roxas-Duncan V.; Wannemacher R. W.; Smith L. A.; Millard C. B. Finding a new vaccine in the ricin protein fold. Protein Eng., Des. Sel. 2004, 17, 391–397. 10.1093/protein/gzh043. [DOI] [PubMed] [Google Scholar]
- a Gal Y.; Alcalay R.; Sabo T.; Noy-Porat T.; Epstein E.; Kronman C.; Mazor O. Rapid assessment of antibody-induced ricin neutralization by employing a novel functional cell-based assay. J. Immunol. Methods 2015, 424, 136–139. 10.1016/j.jim.2015.05.005. [DOI] [PubMed] [Google Scholar]; b Hu W.-g.; Yin J.; Chau D.; Hu C. C.; Lillico D.; Yu J.; Negrych L. M.; Cherwonogrodzky J. W. Conformation-Dependent High-Affinity Potent Ricin-Neutralizing Monoclonal Antibodies. BioMed Res. Int. 2013, 2013, 471346. 10.1155/2013/471346. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Legler P. M.; Brey R. N.; Smallshaw J. E.; Vitetta E. S.; Millard C. B. Structure of RiVax: a recombinant ricin vaccine. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 826–830. 10.1107/s0907444911026771. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Roy C. J.; Brey R. N.; Mantis N. J.; Mapes K.; Pop I. V.; Pop L. M.; Ruback S.; Killeen S. Z.; Doyle-Meyers L.; Vinet-Oliphant H. S.; et al. Thermostable ricin vaccine protects rhesus macaques against aerosolized ricin: Epitope-specific neutralizing antibodies correlate with protection. Proc. Natl. Acad. Sci. U.S.A. 2015, 112, 3782. 10.1073/pnas.1502585112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Pruet J. M.; Saito R.; Manzano L. A.; Jasheway K. R.; Wiget P. A.; Kamat I.; Anslyn E. V.; Robertus J. D. Optimized 5-membered heterocycle-linked pterins for the inhibition of Ricin Toxin A. ACS Med. Chem. Lett. 2012, 3, 588–591. 10.1021/ml300099t. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wiget P. A.; Manzano L. A.; Pruet J. M.; Gao G.; Saito R.; Monzingo A. F.; Jasheway K. R.; Robertus J. D.; Anslyn E. V. Sulfur incorporation generally improves Ricin inhibition in pterin-appended glycine-phenylalanine dipeptide mimics. Bioorg. Med. Chem. Lett. 2013, 23, 6799–6804. 10.1016/j.bmcl.2013.10.017. [DOI] [PubMed] [Google Scholar]
- Saito R.; Pruet J. M.; Manzano L. A.; Jasheway K.; Monzingo A. F.; Wiget P. A.; Kamat I.; Anslyn E. V.; Robertus J. D. Peptide-Conjugated Pterins as Inhibitors of Ricin Toxin A. J. Med. Chem. 2013, 56, 320–329. 10.1021/jm3016393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho M.-c.; Sturm M. B.; Almo S. C.; Schramm V. L. Transition state analogues in structures of ricin and saporin ribosome-inactivating proteins. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 20276–20281. 10.1073/pnas.0911606106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Botelho F. D.; dos Santos M. C.; Gonçalves A. D.; Kuca K.; Valis M.; LaPlante S. R.; França T. C. C.; de Almeida J. S. F. D. Ligand-Based Virtual Screening, Molecular Docking, Molecular Dynamics, and MM-PBSA Calculations towards the Identification of Potential Novel Ricin Inhibitors. Toxins 2020, 12, 746. 10.3390/toxins12120746. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Botelho F. D.; Santos M. C.; Gonçalves A. S.; França T. C.; LaPlante S. R.; de Almeida J. S. Identification of novel potential ricin inhibitors by virtual screening, molecular docking, molecular dynamics and MM-PBSA calculations: a drug repurposing approach. J. Biomol. Struct. Dyn. 2020, 40, 5309. 10.1080/07391102.2020.1870154. [DOI] [PubMed] [Google Scholar]
- Nelson M. T.; Humphrey W.; Gursoy A.; Dalke A.; Kalé L. V.; Skeel R. D.; Schulten K. NAMD: a parallel, object-oriented molecular dynamics program. Int. J. Supercomput. Appl. High Perform. Comput. 1996, 10, 251–268. 10.1177/109434209601000401. [DOI] [Google Scholar]
- Case D. A.; Darden T.; Cheatham T.; Simmerling C. L.; Wang J.; Duke R. E.; Luo R.; Crowley M.; Walker R. C.; Zhang W.. Amber 10; University of California, 2008.
- Lipparini F.; Mennucci B. Hybrid QM/classical models: Methodological advances and new applications. Chem. Phys. Rev. 2021, 2, 041303. 10.1063/5.0064075. [DOI] [Google Scholar]
- Gonçalves M. A.; Santos L. S.; Prata D. M.; Peixoto F. C.; da Cunha E. F.; Ramalho T. C. Optimal wavelet signal compression as an efficient alternative to investigate molecular dynamics simulations: application to thermal and solvent effects of MRI probes. Theor. Chem. Acc. 2017, 136, 15. 10.1007/s00214-016-2037-z. [DOI] [Google Scholar]
- Kumari R.; Kumar R.; Lynn O. S. D. D.; Lynn A. g _ mmpbsa - A GROMACS tool for MM-PBSA and its optimization for high-throughput binding energy calculations. J. Chem. Inf. Model. 2014, 54, 1951. 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- Homeyer N.; Gohlke H. Free energy calculations by the Molecular Mechanics Poisson-Boltzmann Surface Area method. Mol. Inf. 2012, 31, 114. 10.1002/minf.201100135. [DOI] [PubMed] [Google Scholar]
- Kontoyianni M.; McClellan L. M.; Sokol G. S. Evaluation of Docking Performance: Comparative Data on Docking Algorithms. J. Med. Chem. 2004, 47, 558. 10.1021/jm0302997. [DOI] [PubMed] [Google Scholar]
- a da Cunha E. F. F.; Ramalho T. C.; Reynolds R. C. Binding Mode Analysis of 2,4-diamino-5-methyl-5-deaza-6-substituted Pteridines with Mycobacterium tuberculosis and Human Dihydrofolate Reductases. J. Biomol. Struct. Dyn. 2008, 25, 377–385. 10.1080/07391102.2008.10507186. [DOI] [PubMed] [Google Scholar]; b Farahani M. D.; França T. C. C.; Alapour S.; Shahout F.; Boulon R.; Iddir M.; Maddalena M.; Ayotte Y.; LaPlante S. R. Jumping from Fragment to Drug via Smart Scaffolds. ChemMedChem 2022, 17, e202200092 10.1002/cmdc.202200092. [DOI] [PubMed] [Google Scholar]; c França T. C. C.; Pascutti P. G.; Ramalho T. C.; Figueroa-Villar J. D. A three-dimensional structure of Plasmodium falciparum serine hydroxymethyltransferase in complex with glycine and 5-formyl-tetrahydrofolate. Homology modeling and molecular dynamics. Biophys. Chem. 2005, 115, 1–10. 10.1016/j.bpc.2004.12.002. [DOI] [PubMed] [Google Scholar]; d Santos M. C.; Botelho F. D.; Gonçalves A. S.; Kuca K.; Nepovimova E.; Cavalcante S. F. A.; Lima A. L. S.; França T. C. C. Theoretical assessment of the performances of commercial oximes on the reactivation of acetylcholinesterase inhibited by the nerve agent A-242 (novichok). Food Chem. Toxicol. 2022, 165, 113084. 10.1016/j.fct.2022.113084. [DOI] [PubMed] [Google Scholar]; e Guimarães A. P.; Oliveira A. A.; da Cunha E. F. F.; Ramalho T. C.; França T. C. C. Design of New Chemotherapeutics Against the Deadly Anthrax Disease. Docking and Molecular Dynamics Studies of Inhibitors Containing Pyrrolidine and Riboamidrazone Rings on Nucleoside Hydrolase from Bacillus anthracis. J. Biomol. Struct. Dyn. 2011, 28, 455–469. 10.1080/07391102.2011.10508588. [DOI] [PubMed] [Google Scholar]; f França T. C. C.; Wilter A.; Ramalho T. C.; Pascutti P. G.; Figueroa-Villar J. D. Molecular dynamics of the interaction of Plasmodium falciparum and human Serine Hydroxymethyltransferase with 5-formyl-6-hydrofolic acid analogues: Design of new potential antimalarials. J. Braz. Chem. Soc. 2006, 17, 1383–1392. 10.1590/S0103-50532006000700028. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.