

Abstract

Spiroindolines represent a privileged structure in medicinal chemistry, although stereocontrol around the spirocarbon can be a synthetic challenge. Here we present a palladium(0)-catalyzed intramolecular Mizoroki–Heck annulation reaction from (+)-Vince lactam-derived cyclopentenyl-tethered 2-bromo-N-methylanilines for the formation of N-methylspiroindolines. A series of 14 N-methylspiroindolines were synthesized in 59–81% yield with diastereoselectivity >98%, which was rationalized by density functional theory calculations and confirmed through X-ray crystallography. One spiroindoline was converted to an N- and C-terminal protected rigidified unnatural amino acid, which could be orthogonally deprotected.
Introduction
The spiroindoline scaffold is present in several natural products1 and has received interest for use in medicinal chemistry projects with targets related to cancer,2−4 antithrombosis,5 as well as in vivo imaging of neurodegenerative processes/disorders (Supporting Information).6 To further improve the utility of these unique scaffolds as tools in biotechnology and drug development projects new synthetic methods to access spiroindolines, preferably with high stereoselectivity, are needed. Several ways of producing spiroindolines exist in the literature, such as through interrupted Fischer indolization,7 isomerization/spirocyclization/transfer hydrogenation,8,9 photocatalytic [2,2]-addition,10 silver11,12 and gold13 catalyzed cyclization, as well as palladium-catalyzed dearomative ring-closing (Supporting Information).14 The Mizoroki–Heck reaction is one of the prominent methods for carbon–carbon bond formation, which is well-suited for utilization in the synthesis of sterically demanding spirocyclic structures, as demonstrated in the synthesis of spirooxindoles,15−17 spirobenzofurans,18 and spirolactones.19 This is due to the tendency of the intramolecular Mizoroki–Heck reaction to selectively yield the exo cyclization product upon the intramolecular reaction between an aryl halide and an alkene.20 This regioselectivity is observed in the construction of five- or six-membered rings even when the electronic properties of the alkene would suggest a different outcome, as the exo route is less sterically demanding. Even though the Mizoroki–Heck reaction has been applied for the synthesis of other spirocycles, palladium-catalyzed spiroindoline synthesis is less-explored. In this work, a 2,5-dimethylpyrrole-protected amine is used as a chiral auxiliary to construct the quaternary all-carbon spirocenter in the intramolecular Mizoroki–Heck synthesis of spiroindolines with high stereocontrol. This ability to achieve facial selectivity of the Mizoroki–Heck annulation through protection of the amine functionality with bulky protecting groups has earlier been investigated21 and used in our group for the preparation of spirooxindoles16,17 and spirobenzofurans.18 The essential chiral starting structure utilized in our new methodology, the stereopure (+)-Vince lactam (1), has previously garnered attention for its utilization in the synthesis of chiral, substituted cyclopentyl- and pentenyl rings in drug discovery projects. Some prominent examples are the synthesis of abacavir and peramivir, which are used for the treatment of viral diseases.22 Rigidified unnatural amino acids are also of importance to drug discovery programs, where they can function either as single molecular entities or be incorporated into proteins or peptides23,24 to impart improved pharmacokinetic characteristics, such as higher bioavailability and improved metabolic stability,25 of the medicinal compounds.
Herein, we report the synthesis of 14 novel spiroindolines in a diastereo- and regioselective manner through an intramolecular Mizoroki–Heck cyclization of a series of amino group functionalized cyclopentenyl-tethered N-methylbromoanilines. Diversification of substrates was achieved through varying the substitution of the anilines used for the spiroindoline synthesis. Through palladium(0)-catalyzed carbonylation chemistry, one compound was converted to an ester-protected unnatural amino acid which was orthogonally deprotected.
Results and Discussion
The study was initiated with the synthesis of N-allylaniline cyclization precursor 6 using a literature procedure for the first three synthetic steps (Scheme 1).21,18 This was achieved through acid-promoted ring opening of (1S)-(+)-2-azabicyclo[2.2.1]hept-5-en-3-one (1, (+)-Vince lactam) to get the optically pure ammonium chloride salt 2. The free amine was converted to the corresponding 2,5-dimethylpyrrole, a protecting group strategy chosen due to its proven ability to control the diastereoselectivity of a related intermolecular Mizoroki–Heck alkenylation/arylation of alkene 3.21 The air-sensitive intermediate 2 was without further purification subjected to a DBU-mediated double-bond isomerization to yield acrylic ester 3. The methyl ester was then reduced using DIBAL-H, followed by conversion of the allyl alcohol 4 to the corresponding allyl chloride 5 via an Appel reaction with triphenylphosphine and carbon tetrachloride. To yield the secondary amine cyclization precursor 6, allyl chloride 5 was substituted with 2-bromoaniline under microwave heating in a sealed reaction vial and with excess amounts of sodium hydride.
Scheme 1. Preparation of N-Allylaniline 6.

Reaction conditions for a: 1 (1 equiv), SOCl2 (1.1 equiv), MeOH, 100%. b: 1. 2 (1 equiv), hexane-2,5-dione (1 equiv), DIPEA (1 equiv), MeOH. 2. DBU (3 equiv), THF, 85%. c: 3 (1 equiv), DIBAL-H (3 equiv), THF, 84%. d: 4 (1 equiv), PPh3 (1.2 equiv), CCl4, DCM, 81%. e: 5 (1 equiv), NaH (3 equiv), 2-bromoaniline (2 equiv), THF, microwave heated at 80 °C, 63%.
With substrate 6 in hand, screening of reaction conditions for the Mizoroki–Heck cyclization was commenced. The results are presented in Table 1. A Pd(OAc)2/dppf catalytic system with Et3N as the base and DMF as the solvent was chosen as the starting point, as this has previously been successfully used for the synthesis of spiroxindoles using a related ring-closing protocol.16 Screening of the reaction temperature showed no improvement in conversion when raising the temperature from 80 to 100 °C or 120 °C (entries 1–4). Decreasing dppf loading from 10 to 5 mol % as well as using precatalyst Pd(dppf)Cl2 (5 mol %) resulted in a dramatic lowering of the conversion from 6 to 7 (entries 5–7). Our focus was then turned to other palladium ligands, where bis(tri-tert-butylphosphine)palladium (Pd(t-Bu3P)2) at higher loading (10 mol %) furnished an improved outcome with >99% conversion after 16 h at 80 °C and an isolated yield of 64% (entry 11). Old-fashioned PPh3 did not perform as well as a ligand in the reaction (entry 8), presumably because of the aryl bromide bond being relatively electron-rich due to the electron donating effect of the aniline nitrogen, making oxidative addition sluggish with this ligand.20 A couple of control experiments were also conducted, in which omission of palladium afforded <1% conversion (entry 12), and omission of base provided 3% conversion after 16 h (entry 13), thus verifying the necessity of these reaction components. During the optimization attempts, it was observed that both 6 and 7 were somewhat unstable under the reaction conditions, giving rise to unidentified byproducts and low yields, prompting us to investigate if functionalization of the secondary aniline could remedy these issues. Initially, benzylation and acetylation of the secondary aniline nitrogen were attempted, however unsuccessfully. N-Methylation of N-allylaniline 6 provided a way to synthesize N-methylallylaniline 8a (Scheme 2), which was evaluated with the best-performing conditions from the earlier reaction condition screening. For N-methylallylaniline 8a Pd(t-Bu3P)2 proved effective in catalyzing the reaction, providing >99% conversion after 16 h at 80 °C with an isolated yield of 83% (entry 14). The higher isolated yield with the N-methylated substrate (entry 14 compared to entry 11) indicates that our N-methylation strategy was successful in improving compound stability. Compared to entry 1, the Pd(OAc)2/dppf catalytic system performed relatively poorly with the N-methylallylaniline substrate (entry 15).
Table 1. Optimization of Reaction Conditions for Intramolecular Mizoroki–Heck Spirocyclization of 6/8aa.

| entry | starting material | Pd (mol %) | ligand (mol %) | temp (°C) | time (h) | NMR ratio (6:7 or 8a:9a)b | isolated yield of 7/9ac |
|---|---|---|---|---|---|---|---|
| 1 | 6 | Pd(OAc)2 (5) | dppf (10) | 80 | 48 | 11:89 | |
| 2 | 6 | Pd(OAc)2 (5) | dppf (10) | 100 | 16 | 25:75 | |
| 3 | 6 | Pd(OAc)2 (5) | dppf (10) | 120 | 5 | 19:81 | |
| 4 | 6 | Pd(OAc)2 (5) | dppf (10) | 120 | 16 | 22:78 | |
| 5 | 6 | Pd(OAc)2 (5) | dppf (5) | 80 | 5 | 83:17 | |
| 6 | 6 | Pd(OAc)2 (5) | dppf (5) | 80 | 16 | 82:18 | |
| 7 | 6 | Pd(dppf)Cl2 (5) | 80 | 16 | 90:10 | ||
| 8 | 6 | Pd(OAc)2 (5) | PPh3 (10) | 80 | 16 | 55:45 | |
| 9 | 6 | Pd(OAc)2 (5) | XPhos (10) | 80 | 16 | 96:4 | |
| 10 | 6 | Pd(t-Bu3P)2 (5) | 80 | 5 | 11:89 | ||
| 11 | 6 | Pd(t-Bu3P)2 (10) | 80 | 16 | 1:99 | 64% | |
| 12 | 6 | t-Bu3P (10) | 80 | 16 | 100:0 | ||
| 13d | 6 | Pd(t-Bu3P)2 (10) | 80 | 16 | 97:3 | ||
| 14 | 8a | Pd(t-Bu3P)2 (10) | 80 | 16 | 1:99 | 83% | |
| 15 | 8a | Pd(OAc)2 (5) | dppf (10) | 80 | 16 | 47:53 |
Reaction conditions: cyclization precursor 6 or 8a (0.1 mmol, 1 equiv), Et3N (2 equiv), DMF. Amounts of palladium precatalyst and ligands listed in the table.
Ratio between 6:7 or 8a:9a determined by 1H NMR of crude reaction mixture.
Isolated yields after column chromatography.
Experiment run without Et3N. In all screening reactions diastereoselectivity toward the anti product was >98% according to 1H NMR of the crude reaction mixture.
Scheme 2. Synthesis of Precursors 8a–8n for Intramolecular Cyclization through Allyl Chloride Substitution Followed by N-Methylation.

Reaction conditions: 1. 5 (1 equiv), aniline (2 equiv), NaH (3 equiv), dry THF, microwave heated at 80 °C. 2. MeI (1.2 equiv), 1.0 M KHMDS in dry THF (1.3 equiv), dry DMF. Isolated yields reported are for two steps (>95% purity as determined by 1H NMR).
Rewardingly, in all entries of Table 1, compound 7/9a was always observed as the diastereomer resulting from a stereocontrolled anti migratory insertion. As the secondary N-allylaniline starting materials were found to be unstable during preparation, the focus of our inquiry was shifted to N-methylallylaniline Mizoroki–Heck substrates. Cyclization precursors 8a–8n based on this concept were prepared in a two-step sequence, where allylic chloride substitution followed by N-methylation with methyl iodide and potassium bis(trimethylsilyl)amide (KHMDS) gave the desired 8a–8n in isolated yields ranging between 5–89% over two steps (Scheme 2). Unfortunately, some electron-deficient anilines furnished only low reactivity under the investigated conditions. Allylation of 4-amino-3-bromobenzoic acid and 2-bromo-5-nitroanline led to trace amounts of product formation as seen on LC-MS with allyl chloride 5 still remaining in the reaction mixture. 1-(3-Amino-4-bromophenyl)ethan-1-one afforded only low amounts of product with the partial conversion of allyl chloride 5 but also significant byproduct formation.
Having established the optimum reaction conditions, the scope of the intramolecular Heck–Mizoroki spirocyclization was investigated by varying the substitution of the aniline. The results are presented in Scheme 3. Analogous to what was observed during reaction optimization, only the diastereomer from an anti insertion was detected by 1H NMR for compounds 9a–9n. All substrates applied in the spirocyclization reaction performed satisfactorily with isolated yields ranging between 59 and 81%. No immediate pattern could be discerned concerning the outcome of electron-poor compared to electron-rich anilines. The chemoselectivity of the reaction was displayed in the synthesis of 9c, where the chloride was left intact with no signs of dehalogenation, and the compound could be isolated in 61% yield. Methyl-substituted aniline precursors 8e–8h were all well accommodated in the spirocyclization reaction giving moderate to good isolated yields. Interestingly, the 3-position of the aniline could be substituted with a methyl group without the additional steric hindrance around the reactive center negatively effecting the reactivity, with the isolated yield of 9e comparable to what was achieved for 4-, 5-, and 6-methylanilines 9f–9h. Dibromo substrates 8j and 8k afforded the corresponding spirocyclic products in 67% and 65% yield, respectively. However, debromination was detected in the spirocyclization reactions where in both cases debromination was seen only for the bromide in the 4-position (8j) and 5-position (8k). The debrominated product was in both cases observed as product 9a, and it is unknown whether this debromination occurs before or after ring closing. The hydride for this dehalogenation might originate from either triethylamine or DMF.26,271H NMR analysis of the crude reaction mixture of the cyclization reactions of substrates 8j and 8k displayed 9% and 12% debromination, respectively. Aminopyridine 8m was well-tolerated in the reaction, providing the spirocyclic 7-azaindoline 9m in a very good isolated yield (81%). Compound 9m was also subjected to X-ray crystallography studies (CCDC2144389), confirming the R-configuration of the spirocarbon in accordance with an anti insertion (Figure 1). The enantiomer 9n was prepared with excellent stereoselectivity and isolated in a yield similar to 9a. Thus, the results of Scheme 3 could be extrapolated to synthesize any of the enantiomers of compounds 9a–9m starting from (−)-Vince lactam.
Scheme 3. Spirocyclization of N-Methylallylanilines 8a–8n.

Reaction conditions for the synthesis of spiroindolines 9a–9n: cyclization precursor 8a–8n (1 equiv), Pd(t-Bu3P)2 (10 mol %), Et3N (2 equiv), DMF. Isolated yields determined after flash chromatography (>95% purity as determined by 1H NMR).
Figure 1.

PyMOL visual representation of the X-ray crystallography structure of compound 9m.
In order to access the benzyl-protected C-terminal end of our desired unnatural amino acid, aryl bromide 9k was subjected to a palladium-catalyzed benzyloxycarbonylation with a PdOAc2:XantPhos catalytic system (Scheme 4).28 The reaction was run in a two-chamber system, where one chamber is dedicated to the in situ production of carbon monoxide from Mo(CO)6, which can then diffuse to the other chamber where the carbon monoxide-consuming benzyloxycarbonylation can occur.29,30 Employing these conditions, the C- and N-protected unnatural amino acid 10 was synthesized and isolated in 64% yield.
Scheme 4. Benzyloxycarbonylation of 5-Bromospiro-indoline 9k.

Reaction conditions for 10: 9k (1 equiv), Pd(OAc)2 (2 mol %), XantPhos (4 mol %), Mo(CO)6 (2 equiv), DBU (3 equiv), Et3N (2 equiv), and benzyl alcohol used in both chambers, 64%.
The 2,5-dimethylpyrrole protected amine 10 was converted to primary amine 11 through a deprotection protocol employing excess amounts of hydroxylamine hydrochloride in a mixture of ethanol and water (Scheme 5).31
Scheme 5. Removal of 2,5-Dimethylpyrrole Protecting Group to Form Free Amine 11a.

Reaction conditions for 11: 10 (1 equiv), NH2OH·HCl (10 equiv), EtOH: H2O (2:1), 54%.
For the orthogonal deprotection of the carboxylic acid, hydrolysis of the benzyl ester with LiOH in a THF:water mixture was used to provide the free carboxylic acid in a yield of 83% (Scheme 6).
Scheme 6. Removal of Benzyl Protecting Group to Form Free Carboxylic Acid 12.

Reaction conditions for 12: 10 (1 equiv), LiOH (5 equiv), THF:H2O (3:1), 83%.
To further examine the mechanism leading to the diastereoselectivity of the reaction, computational studies were conducted.
Density functional theory calculations were performed for the Mizoroki–Heck annulation of 8a essentially as described previously for similar systems.16,21 The stereoconfiguration of the spirocenter is determined during the migratory insertion transition state (MI). Starting from the aryl-palladium complex, two diastereomeric π-complexes can form. It was found that the syn π-complex was slightly more stable than the anti π-complex. Assuming a fast equilibrium between the diastereomeric π-complexes, Curtin–Hammett conditions apply,32 and the diastereoselectivity will be governed by the energy difference between the two diastereomeric migratory insertions. The energy barrier for the migratory insertion leading to anti product 9a was found to be 2.9 kcal/mol lower than the insertion leading to the syn diastereomer (Figure 2, Supporting Information), in line with the Experimental Results. The transition states for the migratory insertion leading to the anti and syn products are shown in Figure 3.
Figure 2.

Relative free energies for Mizoroki–Heck reaction intermediates and transition states leading to syn and anti product calculated by DFT using B3LYP-D3. Additional ligands removed from palladium in the image for clarity. R = π complex intermediate, MI = migratory insertion transition state, I = σ complex intermediate, BHE = β-hydride elimination transition state, P = product.
Figure 3.
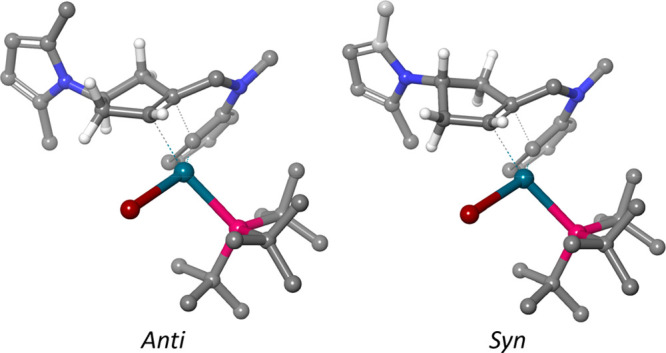
Diastereomeric transition states for migratory insertion illustrating how the 2,5-dimethylpyrrole substituent determines the ring conformation of the cyclopentene. This in turn makes the two trans protons in the top left structure antiperiplanar to the aryl palladium, resulting in a hyperconjugative stabilization of this diastereomeric migratory insertion. Note that the structure to the right is inverted to facilitate the structural comparison. Hydrogen atoms not belonging to the cyclopentenyl ring have been removed from the image for clarity.
Conclusion
A palladium-catalyzed Mizoroki–Heck method employing Pd(t-Bu3P)2 was developed for the diastereoselective spirocyclization of a series of N-methylallylanilines to form spiroindolines. This diastereoselectivity was confirmed by X-ray crystallography as well as 2D NOESY. Through a palladium-catalyzed carbonylation protocol, a bromo-substituted spiroindoline was converted to a C- and N-protected unnatural amino acid, where both the amine and carboxylic acid moieties were subsequently orthogonally deprotected.
Experimental Section
General Methods
1H-, 13C-, and 19F-NMR as well as 2D-NMR spectra were recorded on a Bruker 400 MHz instrument with chemical shifts reported in parts per million (ppm) and using the residual solvent peak as internal reference. High-resolution mass spectra (HRMS) was recorded using a mass spectrometer with electrospray ionization (ESI) with a 7-T hybrid linear ion trap. Thin-layer chromatography (TLC) with Supelco TLC plates with fluorescence indicator (254 nm) as well as LC/MS on a Thermo Fischer Scientific UltiMate 3000 HPLC system with an MSQ Plus mass spectrometer was used for monitoring of reactions. Manual flash chromatography was run using silica gel (230–400 mesh). Automated flash chromatography was conducted on a Biotage Isolera One flash purification instrument with Biotage Sfär cartridges. Automated reverse phase flash chromatography was performed on a Buchi Reveleris X2 flash chromatography purification system using a Claricep Flash Spherical C18 column. For conventional heating of reactions, DrySyn plates were used as heating mantle. For microwave reactions, the reactions were run in sealed microwave vials using an Anton Paar MonoWave 400 or Biotage Initiator microwave reactor. Optical rotation was recorded on a Rudolph Autopol II polarimeter. Palladium-catalyzed carbonylation reactions were run in two fused microwave vials (H-tubes).
(R)-1-(3-(Chloromethyl)cyclopent-3-en-1-yl)-2,5-dimethyl-1H-pyrrole (5)
PPh3 (11.9 g, 45.5 mmol) was added to a solution of allyl alcohol 4 (7.25 g, 37.9 mmol) in CCl4 (20 mL) and DCM (20 mL). The reaction was refluxed overnight. The reaction mixture was evaporated, and the crude product was purified by automated flash chromatography (5% EtOAc in i-hexane) to yield a light-yellow oil (6.44 g, 30.7 mmol, 81%). 1H NMR (400 MHz, acetonitrile-d3) δ 5.84–5.80 (m, 1H), 5.64 (s, 2H), 5.13–5.03 (m, 1H), 4.30–4.20 (m, 2H), 3.01–2.89 (m, 2H), 2.70–2.55 (m, 2H), 2.18 (s, 6H). 13C NMR (101 MHz, acetonitrile-d3) δ 139.5, 129.1, 128.4, 107.2, 53.4, 44.1, 40.5, 40.4, 13.6. HRMS: calcd. for C12H17ClN [M + H]+ 210.1050; found: 210.1051.
(R)-N-2-Bromo-((4-(2,5-dimethyl-1H-pyrrol-1-yl)cyclopent-1-en-1-yl)methyl)aniline (6)
To a suspension of sodium hydride (3 equiv, 2.10 mmol) in dry THF (1 mL) was added a dry THF solution (2 mL) of 2-bromoaniline (2 equiv, 1.4 mmol) slowly at 0 °C. After stirring for 30 min, allyl chloride 5 (1 equiv, 0.7 mmol) dissolved in dry THF (2 mL) was added, and the mixture was microwave heated at 80 °C for 2 h. Water (2 mL) was slowly added (pay attention to hydrogen gas evolution), and the product was extracted with EtOAc. The organic solvent was dried with MgSO4 and evaporated. The product was isolated by flash chromatography (5–10% EtOAc in iso-hexane) to yield the product 6 as an oil (152.3 mg, 0.44 mmol, 63%). [α]D25 = +4.7 (c = 1.06, THF). 1H NMR (400 MHz, DMSO-d6) δ 7.39 (dd, J = 7.8, 1.5 Hz, 1H), 7.13 (ddd, J = 8.2, 7.2, 1.5 Hz, 1H), 6.68 (dd, J = 8.2, 1.5 Hz, 1H), 6.51 (ddd, J = 7.8, 7.2, 1.5 Hz, 1H), 5.56 (s, 2H), 5.54–5.51 (m, 1H), 5.45 (t, J = 6.1 Hz, 1H), 5.02–4.91 (m, 1H), 3.95–3.82 (m, 2H), 2.91–2.75 (m, 2H), 2.53–2.38 (m, 2H), 2.07 (s, 6H). 13C NMR (101 MHz, acetonitrile-d3) δ 146.1, 141.1, 133.2, 129.5, 128.3, 124.6, 118.6, 113.0, 109.9, 107.1, 53.3, 44.4, 41.0, 40.3, 13.6. HRMS: calcd. for C18H22N2Br [M + H]+ 345.0966; found: 345.0974.
(1R,4S)-4-(2,5-Dimethyl-1H-pyrrol-1-yl)spiro[cyclopentane-1,3′-indolin]-2-ene (7)
In an oven-dried reaction vial, Pd(t-Bu3)2 (13.0 mg, 25.4 μmol) and Et3N (50.8 mg, 70 μL, 0.502 mmol) were taken together. The vial was evacuated and flashed with nitrogen three times. DMF (1 mL) was introduced in the reaction vial, and the solution was allowed to stir at room temperature for 5 min. Afterward, 6 (86.7 mg, 0.251 mmol) in 1 mL of DMF was introduced in the reaction mixture and stirred for 16 h at 80 °C. After completion of the reaction, the reaction mixture was quenched with water (5 mL), and the product was extracted with ethyl acetate (3 × 10 mL). The combined organic layer was washed with a saturated solution of brine and dried over anhydrous MgSO4, evaporated, and purified by column chromatography (0–12% EtOAc in pentane) to yield the product as an oil (42.5 mg, 0.160 mmol, 64%). [α]D= −16.76 (c= 0.99, THF). 1H NMR (400 MHz, DMSO-d6) δ 7.01–6.91 (m, 2H), 6.58 (td, J = 7.4, 1.0 Hz, 1H), 6.54–6.50 (m, 1H), 5.93 (dd, J = 5.5, 1.9 Hz, 1H), 5.88 (dd, J = 5.5, 2.5 Hz, 1H), 5.61–5.54 (m, 4H), 3.59 (dd, J = 9.3, 2.0 Hz, 1H), 3.41 (dd, J = 9.3, 2.1 Hz, 1H), 2.64 (dd, J = 13.4, 8.5 Hz, 1H), 2.19 (s, 6H), 1.99 (dd, J = 13.4, 8.1 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 151.1, 137.4, 134.7, 131.1, 127.6, 127.2, 122.4, 117.3, 108.9, 106.0, 60.1, 59.3, 57.4, 46.5, 13.8. HRMS: calcd. for C18H21N2 [M + H]+ 265. 1705; found: 265.1710.
General procedure 1 for the synthesis of N-methylallylanilines 8a–8n. To a suspension of sodium hydride (3 equiv) in dry THF (1 mL) was added a dry THF solution (2 mL) of aniline (2 equiv) slowly at 0 °C. After stirring for 30 min, allyl chloride 5 (1 equiv, 0.7 mmol) was added, and the mixture was microwave heated at 80 °C for 2 h. Water (2 mL) was slowly added (pay attention to hydrogen gas evolution), and the product was extracted with EtOAc. The organic solvent was dried with MgSO4 and evaporated. Automated flash chromatography was run to remove residual aniline from the mixture. The product was dissolved in 3 mL of dry DMF in a 20 mL closed vial with a septum. The vial was purged with nitrogen, and the mixture was cooled to 0 °C on ice. 1.0 M KHMDS in dry THF (1.3 equiv) was added dropwise to the cooled mixture, which was kept for stirring for 30 min. MeI (1.2 equiv) was added dropwise, and the mixture was stirred for 1 h. After completion of the reaction EtOAc (12 mL) was added, and the organic phase was washed with water (3 × 12 mL), followed by brine (12 mL). The organic phase was dried with MgSO4, and the solvent was evaporated. The product was isolated by automated flash chromatography.
(R)-2-Bromo-N-((4-(2,5-dimethyl-1H-pyrrol-1-yl)cyclopent-1-en-1-yl)methyl)-N-methylaniline (8a)
Compound 8a was synthesized according to general procedure 1. After automated flash chromatography (0–4% EtOAc in pentane), the product was isolated as a clear oil (104 mg, 0.29 mmol, 63% over two steps). [α]D25 = −11.3 (c = 0.1, THF). 1H NMR (400 MHz, acetonitrile-d3) δ 7.56 (dd, J = 7.9, 1.5 Hz, 1H), 7.29 (ddd, J = 8.0, 7.2, 1.5 Hz, 1H), 7.20 (dd, J = 8.0, 1.6 Hz, 1H), 6.93 (td, J = 7.9, 7.2, 1.6 Hz, 1H), 5.67–5.60 (m, 3H), 5.00 (m, 1H), 3.78–3.71 (m, 1H), 3.67–3.59 (m, 1H), 2.92–2.79 (m, 2H), 2.70 (s, 3H), 2.60–2.48 (m, 2H), 2.10 (s, 6H). 13C NMR (101 MHz, acetonitrile-d3) δ 152.0, 141.1, 134.6, 129.2, 128.2, 126.8, 125.5, 123.5, 120.9, 107.1, 57.0, 53.5, 42.2, 41.3, 40.1, 13.6. HRMS: calcd. for C19H24BrN2 [M + H]+ 359.1123; found: 359.1127.
(R)-3-Bromo-4-(((4-(2,5-dimethyl-1H-pyrrol-1-yl)cyclopent-1-en-1-yl)methyl)(methyl)amino)benzonitrile (8b)
Compound 8b was synthesized according to general procedure 1. After automated flash chromatography (0–20% EtOAc in pentane), the product was isolated as an oil (132 mg, 0.343 mmol, 37% over two steps). [α]D25 = −1.0 (c = 0.1, THF). 1H NMR (400 MHz, acetonitrile-d3) δ 7.89 (d, J = 1.7 Hz, 1H), 7.60 (dd, J = 8.5, 1.7 Hz, 1H), 7.20 (d, J = 8.5 Hz, 1H), 5.68–5.64 (m, 1H), 5.61 (s, 2H), 5.06–4.96 (m, 1H), 3.91 (d, J = 15.1 Hz, 1H), 3.80 (d, J = 15.1 Hz, 1H), 2.92–2.76 (m, 5H), 2.60–2.44 (m, 2H), 2.11 (s, 6H). 13C NMR (101 MHz, acetonitrile-d3) δ 156.0, 140.1, 138.5, 133.2, 128.3, 127.4, 122.8, 118.9, 118.1, 107.1, 106.6, 56.1, 53.5, 41.4, 41.1, 40.1, 13.6. HRMS: calcd. for C20H23BrN3 [M + H]+ 384.1075; found: 384.1061.
(R)-2-Bromo-4-chloro-N-((4-(2,5-dimethyl-1H-pyrrol-1-yl)cyclopent-1-en-1-yl)methyl)-N-methylaniline (8c)
Compound 8c was synthesized according to general procedure 1. After flash chromatography (0–10% EtOAc in pentane), the product was isolated as an oil (81.2 mg, 0.206 mmol, 29% over two steps). [α]D25 = −14.3 (c = 0.1, THF). 1H NMR (400 MHz, Acetonitrile-d3) δ 7.60 (d, J = 2.5 Hz, 1H), 7.30 (dd, J = 8.7, 2.5 Hz, 1H), 7.16 (d, J = 8.7 Hz, 1H), 5.66–5.59 (m, 3H), 5.05–4.94 (m, 1H), 3.73 (d, J = 14.5 Hz, 1H), 3.62 (d, J = 14.5 Hz, 1H), 2.90–2.78 (m, 2H), 2.68 (s, 3H), 2.58–2.46 (m, 2H), 2.09 (s, 6H). 13C NMR (101 MHz, acetonitrile-d3) δ 151.0, 140.7, 133.83, 129.1, 129.0, 128.3, 127.1, 124.4, 121.3, 107.1, 56.8, 53.4, 42.2, 41.2, 40.1, 13.6. HRMS: calcd. for C19H23N2ClBr [M + H]+ 393.0733; found: 393.0734.
(R)-2-Bromo-N-((4-(2,5-dimethyl-1H-pyrrol-1-yl)cyclopent-1-en-1-yl)methyl)-N-methyl-5-(trifluoromethyl)aniline (8d)
Compound 8d was synthesized according to general procedure 1. After flash chromatography (0–10% EtOAc in pentane), the product was isolated as an oil (40.0 mg, 0.094 mmol, 31% over two steps). [α]D25 = −26.0 (c = 0.1, THF). 1H NMR (400 MHz, acetonitrile-d3) δ 7.73 (dd, J = 8.3, 0.7 Hz, 1H), 7.44 (d, J = 1.9 Hz, 1H), 7.21 (dd, J = 1.9 Hz, 1H), 5.68–5.64 (m, 1H), 5.60 (s, 2H), 5.05–4.94 (m, 1H), 3.88–3.80 (m, 1H), 3.73–3.66 (m, 1H), 2.91–2.78 (m, 2H), 2.76 (s, 3H), 2.58–2.43 (m, 2H), 2.05 (s, 6H). 13C NMR (101 MHz, acetonitrile-d3) δ 152.6, 140.4, 135.6, 130.8 (q, J = 32.5 Hz), 128.3, 127.4, 125.1 (q, J = 271.0 Hz), 124.8 (q, J = 1.5 Hz), 121.6 (q, J = 3.7 Hz), 120.1 (q, J = 3.6 Hz), 107.1, 56.4, 53.4, 42.2, 41.2, 40.1, 13.5. HRMS: calcd. for C20H23N2F3Br [M + H]+ 427.0997; found: 427.1012.
(R)-2-Bromo-N-((4-(2,5-dimethyl-1H-pyrrol-1-yl)cyclopent-1-en-1-yl)methyl)-N,3-dimethylaniline (8e)
Compound 8e was synthesized according to general procedure 1. After flash chromatography (0–10% EtOAc in pentane), the product was isolated as an oil (114 mg, 0.305 mmol, 34% over two steps). [α]D25 = −23.01 (c = 0.1, THF). 1H NMR (400 MHz, acetonitrile-d3) δ 7.18 (dd, J = 7.8, 7.6 Hz, 1H), 7.05 (d, J = 7.8 Hz, 1H), 7.00 (d, J = 7.6 Hz, 1H), 5.65–5.59 (m, 3H), 5.04–4.94 (m, 1H), 3.71 (d, J = 14.2 Hz, 1H), 3.59 (d, J = 14.2 Hz, 1H), 2.91–2.79 (m, 2H), 2.66 (s, 3H), 2.60–2.47 (m, 2H), 2.39 (s, 3H), 2.08 (s, 6H). 13C NMR (101 MHz, acetonitrile-d3) δ 152.4, 141.3, 140.5, 128.4, 128.3, 126.8, 126.7, 124.1, 121.0, 107.1, 57.2, 53.5, 42.6, 41.3, 40.1, 24.3, 13.6. HRMS: calcd. for C20H26N2Br [M + H]+ 373.1279; found: 373.1285.
(R)-2-Bromo-N-((4-(2,5-dimethyl-1H-pyrrol-1-yl)cyclopent-1-en-1-yl)methyl)-N,4-dimethylaniline (8f)
Compound 8f was synthesized according to general procedure 1. After flash chromatography (0–10% EtOAc in pentane), the product was isolated as an oil (89.0 mg, 0.238 mmol, 29% over two steps). [α]D25 = −5.00 (c = 0.1, THF). 1H NMR (400 MHz, acetonitrile-d3) δ 7.35–7.31 (m, 1H), 7.04–7.01 (m, 2H), 5.56–5.52 (m, 3H), 4.97–4.86 (m, 1H), 3.62 (d, J = 14.3 Hz, 1H), 3.51 (d, J = 14.3 Hz, 1H), 2.83–2.71 (m, 2H), 2.58 (s, 3H), 2.51–2.39 (m, 2H), 2.18 (s, 3H), 2.01 (s, 6H). 13C NMR (101 MHz, acetonitrile-d3) δ 149.4, 141.3, 135.7, 134.8, 129.8, 128.3, 126.7, 123.3, 120.9, 107.1, 57.1, 53.5, 42.4, 41.3, 40.1, 20.3, 13.6. HRMS: calcd. for C20H26N2Br [M + H]+ 373.1279; found: 373.1279.
(R)-2-Bromo-N-((4-(2,5-dimethyl-1H-pyrrol-1-yl)cyclopent-1-en-1-yl)methyl)-N,5-dimethylaniline (8g)
Compound 8g was synthesized according to general procedure 1. After flash chromatography (0–10% EtOAc in pentane), the product was isolated as a white solid (139 mg, 0.372 mmol, 46% over two steps). [α]D25 = −13.33 (c = 0.1, THF). 1H NMR (400 MHz, acetonitrile-d3) δ 7.41 (d, J = 8.1 Hz, 1H), 7.04 (d, J = 1.8 Hz, 1H), 6.76 (dd, J = 8.1, 1.8 Hz, 1H), 5.65–5.59 (m, 3H), 5.05–4.95 (m, 1H), 3.77–3.70 (m, 1H), 3.65–3.58 (m, 1H), 2.91–2.79 (m, 2H), 2.68 (s, 3H), 2.59–2.46 (m, 2H), 2.27 (s, 3H), 2.09 (s, 6H). 13C NMR (101 MHz, acetonitrile-d3) δ 151.7, 141.1, 139.4, 134.2, 128.3, 126.7, 126.3, 124.2, 117.4, 107.0, 56.9, 53.5, 42.3, 41.3, 40.1, 21.1, 13.5. HRMS: calcd. for C20H26N2Br [M + H]+ 373.1279; found: 373.1283.
(R)-2-Bromo-N-((4-(2,5-dimethyl-1H-pyrrol-1-yl)cyclopent-1-en-1-yl)methyl)-N,6-dimethylaniline (8h)
Compound 8h was synthesized according to general procedure 1. After flash chromatography (0–10% EtOAc in pentane) the product was isolated as a white solid (110 mg, 0.295 mmol, 19% over two steps). [α]D25 = −12.31 (c = 0.1, THF). 1H NMR (400 MHz, acetonitrile-d3) δ 7.39 (d, J = 7.9 Hz, 1H), 7.16 (d, J = 7.7 Hz, 1H), 6.94 (dd, J = 7.9, 7.7 Hz, 1H), 5.73–5.68 (m, 1H), 5.63 (s, 2H), 5.08–4.97 (m, 1H), 3.99–3.59 (m, 2H), 2.95–2.81 (m, 2H), 2.73 (s, 3H), 2.70–2.52 (m, 2H), 2.35 (s, 3H), 2.15 (s, 6H). 13C NMR (101 MHz, acetonitrile-d3) δ 149.8, 142.3, 141.4, 132.4, 131.5, 128.3, 127.7, 126.2, 125.4, 107.1, 56.4, 53.8, 41.5, 40.1, 39.6, 19.9, 13.7. HRMS: calcd. for C20H26N2Br [M + H]+ 373.1279; found: 373.1288.
(R)-2-Bromo-N-((4-(2,5-dimethyl-1H-pyrrol-1-yl)cyclopent-1-en-1-yl)methyl)-4-fluoro-N-methylaniline (8i)
Compound 8i was synthesized according to general procedure 1. After flash chromatography (0–10% EtOAc in pentane), the product was isolated as an oil (19.0 mg, 0.050 mmol, 5% over two steps). [α]D25 = −16.0 (c = 0.1, THF). 1H NMR (400 MHz, acetonitrile-d3) δ 7.39 (dd, J = 8.4, 3.0 Hz, 1H), 7.23 (dd, J = 8.9, 5.6 Hz, 1H), 7.08 (ddd, J = 8.9, 8.1, 3.0 Hz, 1H), 5.65–5.62 (m, 1H), 5.61 (s, 2H), 5.04–4.94 (m, 1H), 3.74–3.67 (m, 1H), 3.61–3.55 (m, 1H), 2.91–2.79 (m, 2H), 2.66 (s, 3H), 2.57–2.47 (m, 2H), 2.08 (s, 6H). 13C NMR (101 MHz, acetonitrile-d3) δ 159.4 (d, J = 244.1 Hz), 148.5 (d, J = 2.8 Hz), 141.0, 128.3, 127.0, 124.5 (d, J = 8.8 Hz), 121.6 (d, J = 10.3 Hz), 121.2 (d, J = 25.3 Hz), 115.7 (d, J = 22.1 Hz), 107.1, 57.1, 53.5, 42.7, 41.3, 40.1, 13.5. HRMS: calcd. for C19H23N2BrF [M + H]+ 377.1029; found: 377.1031.
(R)-2,4-Dibromo-N-((4-(2,5-dimethyl-1H-pyrrol-1-yl)cyclopent-1-en-1-yl)methyl)-N-methylaniline (8j)
Compound 8j was synthesized according to general procedure 1. After flash chromatography (0–10% EtOAc in pentane), the product was isolated as an oil (105 mg, 0.238 mmol, 29% over two steps). [α]D25 = −7.20 (c = 0.1, THF). 1H NMR (400 MHz, acetonitrile-d3) δ 7.74 (d, J = 2.3 Hz, 1H), 7.43 (dd, J = 8.7, 2.3 Hz, 1H), 7.11 (d, J = 8.7 Hz, 1H), 5.66–5.59 (m, 3H), 5.04–4.94 (m, 1H), 3.74 (d, J = 14.5 Hz, 1H), 3.62 (d, J = 14.5 Hz, 1H), 2.90–2.78 (m, 2H), 2.69 (s, 3H), 2.58–2.44 (m, 2H), 2.08 (s, 6H). 13C NMR (101 MHz, acetonitrile-d3) δ 151.4, 140.7, 136.6, 132.0, 128.3, 127.2, 124.9, 121.6, 116.3, 107.1, 56.7, 53.4, 42.1, 41.2, 40.1, 13.6. HRMS: calcd. for C19H23N2Br2 [M + H]+ 437.0228; found: 437.0233.
(R)-2,5-Dibromo-N-((4-(2,5-dimethyl-1H-pyrrol-1-yl)cyclopent-1-en-1-yl)methyl)-N-methylaniline (8k)
Compound 8k was synthesized according to general procedure 1. After flash chromatography (0–10% EtOAc in pentane), the product was isolated as an oil (234 mg, 0.534 mmol, 66% over two steps). [α]D25 = −7.63 (c = 0.1, THF). 1H NMR (400 MHz, acetonitrile-d3) δ 7.45 (dd, J = 8.5, 1.0 Hz, 1H), 7.31 (d, J = 2.3 Hz, 1H), 7.07 (ddd, J = 8.5, 2.3, 1.0 Hz, 1H), 5.66–5.62 (m, 1H), 5.61 (s, 2H), 5.05–4.94 (m, 1H), 3.76 (d, J = 14.5 Hz, 1H), 3.64 (d, J = 14.5 Hz, 1H), 2.91–2.78 (m, 2H), 2.71 (s, 3H), 2.59–2.44 (m, 2H), 2.09 (s, 6H). 13C NMR (101 MHz, acetonitrile-d3) δ 153.3, 140.5, 136.0, 128.3, 128.1, 127.2, 126.5, 122.2, 119.4, 107.1, 56.5, 53.4, 42.1, 41.2, 40.1, 13.6. HRMS: calcd. for C19H23N2Br2 [M + H]+ 437.0228; found: 437.0209.
(R)-2-Bromo-N-((4-(2,5-dimethyl-1H-pyrrol-1-yl)cyclopent-1-en-1-yl)methyl)-5-methoxy-N-methylaniline (8l)
Compound 8l was synthesized according to general procedure 1. After flash chromatography (0–10% EtOAc in pentane), the product was isolated as an oil (30.0 mg, 0.077 mmol, 9%). [α]D25 = −11.90 (c = 0.1, THF). 1H NMR (400 MHz, acetonitrile-d3) δ 7.43 (d, J = 8.7 Hz, 1H), 6.75 (d, J = 2.9 Hz, 1H), 6.54 (dd, J = 8.7, 2.9 Hz, 1H), 5.67–5.63 (m, 1H), 5.61 (s, 2H), 5.05–4.95 (m, 1H), 3.79–3.72 (m, 4H), 3.68–3.60 (d, J = 14.3 Hz, 1H), 2.92–2.79 (m, 2H), 2.69 (s, 3H), 2.60–2.46 (m, 2H), 2.09 (s, 6H). 13C NMR (101 MHz, acetonitrile-d3) δ 160.9, 152.9, 141.1, 134.8, 128.3, 126.8, 111.0, 110.6, 109.9, 107.0, 56.8, 56.2, 53.5, 42.2, 41.3, 40.1, 13.6. HRMS: calcd. for C20H26ON2Br [M + H]+ 389.1229; found: 389.1218.
(R)-3-Bromo-N-((4-(2,5-dimethyl-1H-pyrrol-1-yl)cyclopent-1-en-1-yl)methyl)-N-methylpyridin-2-amine (8m)
Compound 8m was synthesized according to general procedure 1. After flash chromatography (0–10% EtOAc in pentane), the product was isolated as an oil (138 mg, 0.382 mmol, 47% over two steps). [α]D25 = −10.00 (c = 0.1, THF). 1H NMR (400 MHz, acetonitrile-d3) δ 8.17 (dd, J = 4.7, 1.7 Hz, 1H), 7.83 (dd, J = 7.7, 1.7 Hz, 1H), 6.78 (dd, J = 7.7, 4.7 Hz, 1H), 5.65–5.59 (m, 3H), 5.06–4.95 (m, 1H), 4.07–3.94 (m, 2H), 2.93–2.73 (m, 5H), 2.61–2.43 (m, 2H), 2.14 (s, 6H). 13C NMR (101 MHz, acetonitrile-d3) δ 160.7, 147.2, 143.5, 141.0, 128.3, 126.3, 119.0, 112.1, 107.1, 54.6, 53.6, 41.2, 40.1, 39.7, 13.7. HRMS: calcd. for C18H23N3Br [M + H]+ 360.1075; found: 360.1081.
(S)-2-Bromo-N-((4-(2,5-dimethyl-1H-pyrrol-1-yl)cyclopent-1-en-1-yl)methyl)-N-methylaniline (8n)
Compound 8n was synthesized according to general procedure 1 on a larger scale (allyl chloride 5, 680 mg, 3.24 mmol). After flash chromatography (0–4% EtOAc in pentane), the product was isolated as a white solid (1038 mg, 2.88 mmol, 89% over two steps). [α]D25 = 19.01 (c = 0.1, THF). 1H NMR (400 MHz, acetonitrile-d3) δ 7.56 (dd, J = 8.0, 1.5 Hz, 1H), 7.33–7.27 (m, 1H), 7.22 (dd, J = 8.1, 1.6 Hz, 1H), 6.97–6.91 (m, 1H), 5.66–5.62 (m, 1H), 5.61 (s, 2H), 5.05–4.95 (m, 1H), 3.75 (d, J = 14.0 Hz, 1H), 3.64 (d, J = 14.0 Hz, 1H), 2.92–2.80 (m, 2H), 2.70 (s, 3H), 2.58–2.48 (m, 2H), 2.09 (s, 6H). 13C NMR (101 MHz, Aacetonitrile-d3) δ 152.0, 141.1, 134.6, 129.3, 128.3, 126.8, 125.6, 123.5, 120.9, 107.1, 56.9, 53.5, 42.2, 41.3, 40.1, 13.6. HRMS: calcd. for C19H24BrN2 [M + H]+ 359.1123; found: 359.1130.
General procedure 2 for synthesis of N-methylspiroindolines 9a–9n. To an oven-dried reaction vial, Pd(t-Bu3P)2 (0.1 equiv) and Et3N (2 equiv) were added. The vial was sealed and purged three times with nitrogen. DMF (1 mL) was added to the vial, which was stirred at room temperature for 5 min. A solution of N-methylallylaniline (1 equiv) in DMF (1 mL) was added, and the mixture heated at 80 °C for 16 h. Ethyl acetate (10 mL) was added, and the organic phase washed with water (3 × 10 mL), followed by brine (10 mL). The organic phase was dried with MgSO4 and evaporated. The product was isolated by automated flash chromatography.
(R)-2-Bromo-N-((4-(2,5-dimethyl-1H-pyrrol-1-yl)cyclopent-1-en-1-yl)methyl)-N-methylaniline (9a)
Compound 9a was synthesized according to general procedure 2. After automated flash chromatography (0–12% EtOAc in pentane), the product was isolated as a white solid (67.0 mg, 0.241 mmol, 79%). [α]D25 = −324.1 (c = 0.1, THF). 1H NMR (400 MHz, Acetonitrile-d3) δ 7.09 (td, J = 7.7, 1.3 Hz, 1H), 7.00 (dd, J = 7.3, 1.0 Hz, 1H), 6.68 (td, J = 7.3, 1.0 Hz, 1H), 6.54 (d, J = 7.7 Hz, 1H), 5.96 (dd, J = 5.5, 2.0 Hz, 1H), 5.87 (dd, J = 5.5, 2.6 Hz, 1H), 5.65–5.57 (m, 3H), 3.45 (d, J = 9.1 Hz, 1H), 3.29 (d, J = 9.1 Hz, 1H), 2.73 (s, 3H), 2.66 (dd, J = 13.4, 8.5 Hz, 1H), 2.22 (s, 6H), 2.10 (dd, J = 13.4, 8.2 Hz, 1H). 13C NMR (101 MHz, acetonitrile-d3) δ 153.2, 138.0, 137.0, 132.9, 129.1, 128.7, 123.1, 119.2, 108.5, 107.3, 69.1, 61.5, 57.5, 46.7, 36.2, 14.2. HRMS: calcd. for C19H23N2 [M + H]+ 279.1861; found: 279.1860.
(1R,4S)-4-(2,5-Dimethyl-1H-pyrrol-1-yl)-1′-methylspiro[cyclopentane-1,3′-indolin]-2-ene-5′-carbonitrile (9b)
Compound 9b was synthesized according to general procedure 2. After automated flash chromatography (0–12% EtOAc in pentane), the product was isolated as a white solid (70.4 mg, 0.232 mmol, 77%). [α]D25 = −294.9 (c = 1.0, THF). 1H NMR (400 MHz, acetonitrile-d3) δ 7.42 (dd, J = 8.2, 1.7 Hz, 1H), 7.25 (d, J = 1.7 Hz, 1H), 6.50 (d, J = 8.2 Hz, 1H), 5.99 (dd, J = 5.5, 2.0 Hz, 1H), 5.84 (dd, J = 5.5, 2.6 Hz, 1H), 5.67–5.59 (m, 3H), 3.68 (d, J = 9.7 Hz, 1H), 3.50 (d, J = 9.7 Hz, 1H), 2.84 (s, 3H), 2.71 (dd, J = 13.6, 8.5 Hz, 1H), 2.21 (s, 6H), 2.08 (dd, J = 13.6, 8.1 Hz, 1H). 13C NMR (101 MHz, acetonitrile-d3) δ 155.8, 137.8, 137.2, 134.9, 133.8, 128.8, 126.8, 121.5, 107.3, 107.1, 99.1, 68.0, 61.3, 57.0, 47.3, 34.3, 14.2. HRMS: calcd. for C20H22N3 [M + H]+ 304.1814; found: 304.1819.
(1R,4S)-5′-Chloro-4-(2,5-dimethyl-1H-pyrrol-1-yl)-1′-methylspiro[cyclopentane-1,3′-indolin]-2-ene (9c)
Compound 9c was synthesized according to general procedure 2. After automated flash chromatography (0–12% EtOAc in pentane), the product was isolated as an oil (38.0 mg, 0.121 mmol, 61%). [α]D25 = −420.24 (c = 0.1, THF). 1H NMR (400 MHz, acetonitrile-d3) δ 7.06 (dd, J = 8.3, 2.2 Hz, 1H), 6.98 (d, J = 2.2 Hz, 1H), 6.48 (d, J = 8.3 Hz, 1H), 5.98 (dd, J = 5.5, 2.0 Hz, 1H), 5.85 (dd, J = 5.5, 2.6 Hz, 1H), 5.65–5.56 (m, 3H), 3.49 (d, J = 9.2 Hz, 1H), 3.33 (d, J = 9.2 Hz, 1H), 2.73 (s, 3H), 2.68 (dd, J = 13.6, 8.6 Hz, 1H), 2.21 (s, 6H), 2.09 (dd, J = 13.6, 8.1 Hz, 1H). 13C NMR (101 MHz, acetonitrile-d3) δ 152.0, 139.3, 137.3, 133.6, 128.8, 128.7, 123.4, 122.9, 109.3, 107.3, 69.1, 61.4, 57.5, 46.5, 36.0, 14.2. HRMS: calcd. for C19H22N2Cl [M + H]+ 313.1472; found: 313.1467.
(1R,4S)-4-(2,5-Dimethyl-1H-pyrrol-1-yl)-1′-methyl-4′-(trifluoromethyl)spiro[cyclopentane-1,3′-indolin]-2-ene (9d)
Compound 9d was synthesized according to general procedure 2. After flash chromatography (0–12% EtOAc in pentane), the product was isolated as an oil (19.0 mg, 0.055 mmol, 59%). [α]D25 = −462.16 (c = 0.1, THF). 1H NMR (400 MHz, acetonitrile-d3) δ 7.12 (d, J = 7.6 Hz, 1H), 6.96 (d, J = 7.6 Hz, 1H), 6.74 (s, 1H), 6.02 (dd, J = 5.5, 2.0 Hz, 1H), 5.88 (dd, J = 5.5, 2.6 Hz, 1H), 5.66–5.58 (m, 3H), 3.57 (d, J = 9.3 Hz, 1H), 3.41 (d, J = 9.3 Hz, 1H), 2.79 (s, 3H), 2.70 (dd, J = 13.6, 8.5 Hz, 1H), 2.21 (s, 6H), 2.13–2.08 (m, 1H). 13C NMR (101 MHz, acetonitrile-d3) δ 153.5, 141.5, 137.2, 133.8, 130.9 (q, J = 31.2 Hz), 128.8, 125.6 (q, J = 271.1 Hz, determined through HMBC), 123.5, 115.6 (q, J = 4.0 Hz), 107.3, 104.1 (q, J = 3.8 Hz), 68.7, 61.4, 57.5, 46.8, 35.5, 14.1. HRMS: calcd. for C20H22N2F3 [M + H]+ 347.1735; found: 347.1723.
(1R,4S)-4-(2,5-Dimethyl-1H-pyrrol-1-yl)-1′,4′-dimethylspiro[cyclopentane-1,3′-indolin]-2-ene (9e)
Compound 9e was synthesized according to general procedure 2. After flash chromatography (0–6% EtOAc in pentane), the product was isolated as a white solid (54.9 mg, 0.188 mmol, 66%). [α]D25 = −406.14 (c = 0.1, THF). 1H NMR (400 MHz, acetonitrile-d3) δ 6.99 (dd, J = 7.8, 7.6 Hz, 1H), 6.48 (dt, J = 7.6, 0.8 Hz, 1H), 6.40 (d, J = 7.8 Hz, 1H), 5.91 (dd, J = 5.5, 2.1 Hz, 1H), 5.81 (dd, J = 5.5, 2.6 Hz, 1H), 5.65 (s, 2H), 5.60–5.52 (m, 1H), 3.46 (d, J = 9.0 Hz, 1H), 3.22 (d, J = 9.0 Hz, 1H), 2.83 (dd, J = 14.4, 9.4 Hz, 1H), 2.70 (s, 3H), 2.23 (s, 6H), 2.22 (s, 3H), 1.94 (dd, J = 14.4, 7.0 Hz, 1H). 13C NMR (101 MHz, acetonitrile-d3) δ 154.0, 137.2, 134.9, 133.1, 132.1, 129.1, 128.8, 121.9, 107.2, 106.6, 70.3, 62.5, 57.8, 44.3, 36.2, 17.6, 14.2. HRMS: calcd. for C20H25N2 [M + H]+ 293.2018; found: 293.2013.
(1R,4S)-4-(2,5-Dimethyl-1H-pyrrol-1-yl)-1′,5′-dimethylspiro[cyclopentane-1,3′-indolin]-2-ene (9f)
Compound 9f was synthesized according to general procedure 2. After flash chromatography (0–12% EtOAc in pentane) the product was isolated as a white solid (40.9 mg, 0.140 mmol, 59%). [α]D25 = −381.23 (c = 0.1, THF). 1H NMR (400 MHz, acetonitrile-d3) δ 6.93–6.88 (m, 1H), 6.85 (dq, J = 1.8, 0.6 Hz, 1H), 6.45 (d, J = 7.9 Hz, 1H), 5.95 (dd, J = 5.5, 2.0 Hz, 1H), 5.85 (dd, J = 5.5, 2.6 Hz, 1H), 5.66–5.56 (m, 3H), 3.41 (d, J = 9.0 Hz, 1H), 3.24 (d, J = 9.0 Hz, 1H), 2.69 (s, 3H), 2.64 (dd, J = 13.4, 8.5 Hz, 1H), 2.24–2.21 (m, 9H), 2.09 (dd, J = 13.4, 8.1 Hz, 1H). 13C NMR (101 MHz, acetonitrile-d3) δ 151.2, 138.1, 137.3, 132.8, 129.3, 128.8, 128.5, 123.9, 108.6, 107.2, 69.4, 61.5, 57.6, 46.5, 36.7, 20.8, 14.2. HRMS: calcd. for C20H25N2 [M + H]+ 293.2018; found: 293.2010.
(1R,4S)-4-(2,5-Dimethyl-1H-pyrrol-1-yl)-1′,6′-dimethylspiro[cyclopentane-1,3′-indolin]-2-ene (9g)
Compound 9g was synthesized according to general procedure 2. After flash chromatography (0–12% EtOAc in pentane), the product was isolated as a white solid (69.0 mg, 0.236 mmol, 65%). [α]D25 = −449.31 (c = 0.1, THF). 1H NMR (400 MHz, acetonitrile-d3) δ 6.88 (d, J = 7.4 Hz, 1H), 6.51 (dq, J = 7.4, 0.7 Hz, 1H), 6.40–6.37 (m, 1H), 5.94 (dd, J = 5.5, 2.0 Hz, 1H), 5.85 (dd, J = 5.5, 2.6 Hz, 1H), 5.64 (s, 2H), 5.62–5.55 (m, 1H), 3.44 (d, J = 9.1 Hz, 1H), 3.27 (d, J = 9.1 Hz, 1H), 2.72 (s, 3H), 2.63 (dd, J = 13.4, 8.5 Hz, 1H), 2.27 (s, 3H), 2.22 (s, 6H), 2.09 (dd, J = 13.4, 8.2 Hz, 1H). 13C NMR (101 MHz, acetonitrile-d3) δ 153.4, 138.9, 138.2, 134.3, 132.7, 128.7, 122.9, 119.8, 109.4, 107.2, 69.3, 61.5, 57.2, 46.7, 36.2, 21.8, 14.2. HRMS: calcd. for C20H25N2 [M + H]+ 293.2018; found: 293.2023.
(1R,4S)-4-(2,5-Dimethyl-1H-pyrrol-1-yl)-1′,7′-dimethylspiro[cyclopentane-1,3′-indolin]-2-ene (9h)
Compound 9h was synthesized according to general procedure 2. After flash chromatography (0–6% EtOAc in pentane), the product was isolated as a white solid (69.0 mg, 0.236 mmol, 80%). [α]D25 = −433.33 (c = 0.1, THF). 1H NMR (400 MHz, acetonitrile-d3) δ 6.89–6.84 (m, 2H), 6.66 (t, J = 7.4 Hz, 1H), 5.94 (dd, J = 5.5, 2.0 Hz, 1H), 5.84 (dd, J = 5.5, 2.6 Hz, 1H), 5.63 (s, 2H), 5.60–5.54 (m, 1H), 3.45 (d, J = 9.8 Hz, 1H), 3.29 (d, J = 9.8 Hz, 1H), 2.92 (s, 3H), 2.61 (dd, J = 13.4, 8.5 Hz, 1H), 2.35 (s, 3H), 2.21 (s, 6H), 2.08 (dd, J = 13.4, 8.1 Hz, 1H). 13C NMR (101 MHz, Acetonitrile-d3) δ 151.1, 138.5, 138.1, 132.7, 132.2, 128.8, 121.5, 121.2, 120.6, 107.2, 70.5, 61.5, 57.6, 47.3, 40.2, 19.6, 14.2. HRMS: calcd. for C20H25N2 [M + H]+ 293.2018; found: 293.2016.
(1R,4S)-4-(2,5-Dimethyl-1H-pyrrol-1-yl)-5′-fluoro-1′-methylspiro[cyclopentane-1,3′-indolin]-2-ene (9i)
Compound 9i was synthesized according to general procedure 2. After flash chromatography (0–7% EtOAc in pentane), the product was isolated as an oil (8.00 mg, 0.027 mmol, 63%). [α]D25 = −334.79 (c = 0.1, THF). 1H NMR (400 MHz, acetonitrile-d3) δ 6.87–6.77 (m, 2H), 6.48 (dd, J = 8.4, 4.2 Hz, 1H), 5.99 (dd, J = 5.5, 2.0 Hz, 1H), 5.86 (dd, J = 5.5, 2.6 Hz, 1H), 5.66–5.56 (m, 3H), 3.45 (d, J = 9.1 Hz, 1H), 3.30 (d, J = 9.1 Hz, 1H), 2.70 (s, 3H), 2.67 (dd, J = 13.6, 8.6 Hz, 1H), 2.21 (s, 6H), 2.10 (dd, J = 13.6, 8.1 Hz, 1H). 13C NMR (101 MHz, acetonitrile-d3) δ 157.7 (d, J = 232.9 Hz), 149.7, 139.0 (d, J = 7.2 Hz), 137.3, 133.6, 128.8, 114.7 (d, J = 23.1 Hz), 110.7 (d, J = 23.9 Hz), 108.9 (d, J = 8.0 Hz), 107.3, 69.6, 61.4, 57.6, 46.3, 36.8, 14.2. HRMS: calcd. for C19H22N2F [M + H]+ 297.1767; found: 297.1767.
(1R,4S)-5′-Bromo-4-(2,5-dimethyl-1H-pyrrol-1-yl)-1′-methylspiro[cyclopentane-1,3′-indolin]-2-ene (9j)
Compound 9j was synthesized according to general procedure 2. After flash chromatography (0–12% EtOAc in pentane), the product was isolated as a yellowish oil (57.1 mg, 0.160 mmol, 67%). [α]D25 = −539.5 (c = 0.1, THF). 1H NMR (400 MHz, acetonitrile-d3) δ 7.20 (dd, J = 8.3, 2.1 Hz, 1H), 7.10 (d, J = 2.1 Hz, 1H), 6.44 (d, J = 8.3 Hz, 1H), 5.97 (dd, J = 5.5, 2.0 Hz, 1H), 5.85 (dd, J = 5.5, 2.6 Hz, 1H), 5.65–5.56 (m, 3H), 3.49 (d, J = 9.2 Hz, 1H), 3.33 (d, J = 9.2 Hz, 1H), 2.72 (s, 3H), 2.68 (dd, J = 13.7, 8.6 Hz, 1H), 2.21 (s, 6H), 2.08 (dd, J = 13.7, 8.0 Hz, 1H). 13C NMR (101 MHz, acetonitrile-d3) δ 152.4, 139.7, 137.3, 133.6, 131.6, 128.8, 126.1, 109.9, 109.9, 107.2, 69.0, 61.4, 57.5, 46.5, 35.9, 14.2. HRMS: calcd. for C19H22N2Br [M + H]+ 357.0966; found: 357.0966.
(1R,4S)-6′-Bromo-4-(2,5-dimethyl-1H-pyrrol-1-yl)-1′-methylspiro[cyclopentane-1,3′-indolin]-2-ene (9k)
Compound 9k was synthesized according to general procedure 2. After flash chromatography (0–12% EtOAc in pentane), the product was isolated as a yellowish oil (117 mg, 0.326 mmol, 65%). [α]D25 = −397.66 (c = 0.1, THF). 1H NMR (400 MHz, acetonitrile-d3) δ 6.88 (d, J = 7.8 Hz, 1H), 6.78 (dd, J = 7.8, 1.8 Hz, 1H), 6.65 (d, J = 1.8 Hz, 1H), 5.97 (dd, J = 5.5, 2.0 Hz, 1H), 5.84 (dd, J = 5.5, 2.6 Hz, 1H), 5.63 (s, 2H), 5.61–5.54 (m, 1H), 3.52 (d, J = 9.2 Hz, 1H), 3.35 (d, J = 9.2 Hz, 1H), 2.73 (s, 3H), 2.66 (dd, J = 13.5, 8.5 Hz, 1H), 2.20 (s, 6H), 2.08 (dd, J = 13.5, 8.2 Hz, 1H). 13C NMR (101 MHz, Acetonitrile-d3) δ 154.6, 137.5, 136.5, 133.4, 128.8, 124.6, 122.3, 121.2, 111.0, 107.3, 68.8, 61.4, 57.2, 46.7, 35.6, 14.2. HRMS: calcd. for C19H22N2Br [M + H]+ 357.0966; found: 357.0959.
(1R,4S)-4-(2,5-Dimethyl-1H-pyrrol-1-yl)-6′-methoxy-1′-methylspiro[cyclopentane-1,3′-indolin]-2-ene (9l)
Compound 9l was synthesized according to general procedure 2. After flash chromatography (0–6% EtOAc in pentane), the product was isolated as an oil (9.1 mg, 0.030 mmol, 61%). [α]D25 = −395.23 (c = 0.1, THF). 1H NMR (400 MHz, acetonitrile-d3) δ 6.87 (d, J = 8.0 Hz, 1H), 6.21 (dd, J = 8.0, 2.3 Hz, 1H), 6.12 (d, J = 2.3 Hz, 1H), 5.92 (dd, J = 5.5, 2.0 Hz, 1H), 5.84 (dd, J = 5.5, 2.6 Hz, 1H), 5.63 (s, 2H), 5.61–5.54 (m, 1H), 3.74 (s, 3H), 3.47 (d, J = 9.1 Hz, 1H), 3.30 (d, J = 9.1 Hz, 1H), 2.72 (s, 3H), 2.62 (dd, J = 13.4, 8.5 Hz, 1H), 2.21 (s, 6H), 2.07 (dd, J = 13.4, 8.1 Hz, 1H). 13C NMR (101 MHz, acetonitrile-d3) δ 161.9, 154.5, 138.3, 132.5, 129.4, 128.8, 123.5, 107.2, 103.5, 95.6, 69.5, 61.5, 56.9, 55.9, 46.8, 36.0, 14.2. HRMS: calcd. for C20H25N2O [M + H]+ 309.1967; found: 309.1959.
(1R,4S)-4-(2,5-Dimethyl-1H-pyrrol-1-yl)-1′-methyl-1′,2′-dihydrospiro[cyclopentane-1,3′-pyrrolo[2,3-b]pyridin]-2-ene (9m)
Compound 9m was synthesized according to general procedure 2. After flash chromatography (0–20% EtOAc in pentane), the product was isolated as a white solid (85.9 mg, 0.307 mmol, 81%). [α]D25 = −243.08 (c = 0.1, THF). 1H NMR (400 MHz, acetonitrile-d3) δ 7.86 (dd, J = 5.3, 1.6 Hz, 1H), 7.20 (dd, J = 7.1, 1.6 Hz, 1H), 6.49 (dd, J = 7.1, 5.3 Hz, 1H), 5.99 (dd, J = 5.5, 2.0 Hz, 1H), 5.86 (dd, J = 5.5, 2.6 Hz, 1H), 5.64 (s, 2H), 5.62–5.56 (m, 1H), 3.60 (d, J = 9.4 Hz, 1H), 3.42 (d, J = 9.4 Hz, 1H), 2.88 (s, 3H), 2.71 (dd, J = 13.5, 8.4 Hz, 1H), 2.21 (s, 6H), 2.08 (dd, J = 13.5, 8.1 Hz, 1H). 13C NMR (101 MHz, acetonitrile-d3) δ 163.3, 147.8, 137.5, 133.4, 130.1, 130.0, 128.8, 113.7, 107.3, 65.7, 61.4, 55.2, 47.4, 32.8, 14.2. HRMS: calcd. for C18H22N3 [M + H]+ 280.1814; found: 280.1815.
(1S,4R)-4-(2,5-Dimethyl-1H-pyrrol-1-yl)-1′-methylspiro[cyclopentane-1,3′-indolin]-2-ene (9n)
Compound 9n was synthesized according to general procedure 2. After flash chromatography (0–5% EtOAc in pentane), the product was isolated as a white solid (68.0 mg, 0.243 mmol, 73%). [α]D25 = 315.1 (c = 0.1, THF). 1H NMR (400 MHz, acetonitrile-d3) δ 7.09 (td, J = 7.8, 1.4 Hz, 1H), 7.01 (dd, J = 7.4, 1.4 Hz, 1H), 6.68 (ddd, J = 7.4, 0.9 Hz, 1H), 6.54 (d, J = 7.8 Hz, 1H), 5.96 (dd, J = 5.5, 2.0 Hz, 1H), 5.87 (dd, J = 5.5, 2.6 Hz, 1H), 5.64 (s, 2H), 5.63–5.56 (m, 1H), 3.46 (d, J = 9.1 Hz, 1H), 3.29 (d, J = 9.1 Hz, 1H), 2.74 (s, 3H), 2.66 (dd, J = 13.5, 8.5 Hz, 1H), 2.22 (s, 6H), 2.11 (dd, J = 13.5, 8.2 Hz, 1H). 13C NMR (101 MHz, acetonitrile-d3) δ 153.2, 138.0, 137.0, 132.9, 129.1, 128.8, 123.1, 119.1, 108.5, 107.2, 69.0, 61.5, 57.5, 46.7, 36.2, 14.2. HRMS: calcd. for C19H23N2 [M + H]+ 279.1861; found: 279.1861.
Benzyl-(1R,4S)-4-(2,5-dimethyl-1H-pyrrol-1-yl)-1′-methylspiro[cyclopentane-1,3′-indolin]-2-ene-6′-carboxylate (10)
To the first tube of a 5 mL H-tube equipped with Teflon-coated stirring bars were added 9k (100 mg, 0.280 mmol), palladium acetate (1.3 mg, 5.6 μmol), XantPhos (6.5 mg, 11 μmol), and Et3N (78 μL, 0.56 mmol). To the other was added Mo(CO)6 (148 mg, 0.560 mmol). The H-tube was sealed and purged three times with nitrogen gas. To each of the chambers was added benzyl alcohol (1 mL). DBU (125 μL, 0.840 mmol) was added to the Mo(CO)6-containing chamber, and the reaction was stirred at 120 °C for 2 h. Ethyl acetate (10 mL) was added, and the organic phase was washed with water (2 × 10 mL), followed by brine (10 mL). The organic phase was evaporated, and the crude product was purified by automatic flash chromatography (0–10% EtOAc in pentane) to provide the product as an oil (76 mg, 0.18 mmol, 64%). [α]D25 = −285.10 (c = 0.1, THF). 1H NMR (400 MHz, acetonitrile-d3) δ 7.48–7.43 (m, 1H), 7.43–7.32 (m, 5H), 7.11 (d, J = 1.5 Hz, 1H), 7.08 (dd, J = 7.6, 0.5 Hz, 1H), 6.01 (dd, J = 5.5, 2.0 Hz, 1H), 5.87 (dd, J = 5.5, 2.6 Hz, 1H), 5.64 (s, 2H), 5.62–5.56 (m, 1H), 5.32 (s, 2H), 3.54 (d, J = 9.2 Hz, 1H), 3.37 (d, J = 9.2 Hz, 1H), 2.78 (s, 3H), 2.70 (dd, J = 13.6, 7.5 Hz, 1H), 2.21 (s, 6H), 2.10 (dd, J = 13.6, 8.1 Hz, 1H). 13C NMR (101 MHz, acetonitrile-d3) δ 167.4, 153.4, 142.8, 137.8, 137.3, 133.7, 131.3, 129.6, 129.1, 128.9, 128.8, 123.1, 121.1, 108.3, 107.3, 68.8, 67.2, 61.4, 57.6, 46.6, 35.8, 14.2. HRMS: calcd. for C27H29N2O2 [M + H]+ 413.2229; found: 413.2233.
Benzyl (1R,4S)-4-Amino-1′-methylspiro[cyclopentane-1,3′-indolin]-2-ene-6′-carboxylate (11)
A reaction vial was loaded with hydroxylamine hydrochloride (84.2 mg, 1.21 mmol) and 10 (50.0 mg, 0.121 mmol). Next, EtOH:H2O (2:1, 1 mL) was added, and the mixture was heated to 100 °C and stirred at that temperature for 16 h. The crude product was isolated through automated reverse phase flash chromatography (5–100% MeCN in water (0.05% formic acid)) to provide the product as a white solid (21.7 mg, 0.0649 mmol, 54%). [α]D25 = −202.0 (c = 0.1, THF). 1H NMR (500 MHz, methanol-d4) δ 8.56 (bs, 2H), 7.47–7.41 (m, 3H), 7.41–7.36 (m, 2H), 7.36–7.30 (m, 1H), 7.16 (s, 1H), 6.98 (d, J = 7.6 Hz, 1H), 6.08 (dd, J = 5.6, 1.6 Hz, 1H), 5.92 (dd, J = 5.6, 1.9 Hz, 1H), 5.33 (s, 2H), 4.57–4.50 (m, 1H), 3.47 (d, J = 9.3 Hz, 1H), 3.36 (d, J = 9.3 Hz, 1H), 2.81 (s, 3H), 2.65 (dd, J = 14.2, 8.1 Hz, 1H), 2.07 (dd, J = 14.2, 5.6 Hz, 1H). 13C NMR (126 MHz, methanol-d4) δ 168.2, 153.8, 142.7, 142.0, 137.7, 131.8, 129.6, 129.3, 129.2, 123.2, 121.7, 109.0, 69.0, 67.7, 58.6, 57.5, 44.3, 35.8. HRMS: calcd. for C21H23N2O2 [M + H]+ 335.1760; found: 335.1766.
(1R,4S)-4-(2,5-Dimethyl-1H-pyrrol-1-yl)-1′-methylspiro[cyclopentane-1,3′-indolin]-2-ene-6′-carboxylic acid (12)
A reaction vial was loaded with 10 (68.3 mg, 0.166 mmol) and lithium hydroxide (20.0 mg, 0.835 mmol), and a THF:H2O mixture (3:1, 1 mL) was added. The mixture was heated to 50 °C and stirred at that temperature for 20 h. The reaction mixture was diluted with 10 mL water and washed with EtOAc (3 × 10 mL). The water phase was acidified with 2 M HCl and extracted with EtOAc (3 × 10 mL). The organic phase was dried with MgSO4, filtered, and evaporated to yield the product as a dark brown oil (44.4 mg, 0.138 mmol), 83%). The sample was too dark to obtain value for optical rotation. 1H NMR (400 MHz, chloroform-d) δ 7.57 (dd, J = 7.6, 1.4 Hz, 1H), 7.25 (d, J = 1.4 Hz, 1H), 7.04 (d, J = 7.6 Hz, 1H), 6.07 (dd, J = 5.5, 2.0 Hz, 1H), 5.93 (dd, J = 5.5, 2.5 Hz, 1H), 5.78 (s, 2H), 5.62–5.54 (m, 1H), 3.60 (d, J = 9.1 Hz, 1H), 3.39 (d, J = 9.1 Hz, 1H), 2.87 (s, 3H), 2.78 (dd, J = 13.8, 8.5 Hz, 1H), 2.28 (s, 6H), 2.25 (dd, J = 13.8, 8.1 Hz, 1H). 13C NMR (101 MHz, chloroform-d) δ 172.1, 151.6, 142.4, 136.5, 132.8, 129.6, 128.2, 122.0, 121.8, 108.9, 106.4, 68.3, 60.6, 56.9, 46.1, 35.9, 14.1. HRMS: calcd. for C20H23N2O2 [M + H]+ 323.1760; found: 323.1759.
Acknowledgments
The authors thank Uppsala University for financial support. The authors would also like to thank the Kjell and Märta Beijer Foundation and Luke Odell for valuable discussion regarding the project.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c04111.
Copies of all 1H NMR, 19F and 13C NMR spectra, NMR 2D-spectra, structural information, computational and X-ray crystallography data detailed in the experimental section (PDF)
Crystallographic information file for compound 9m(CIF)
PDB file for X-ray crystal structure of compound 9m (PDB)
ORTEP representation of X-ray crystal structure of compound 9m (PDF)
X-ray crystallography data for compound 9m (PDF)
Author Present Address
‡ Beactica AB, Virdings allé 2, SE-754 50 Uppsala, Sweden
The authors declare no competing financial interest.
Supplementary Material
References
- Trost B. M.; Brennan M. K. Asymmetric Syntheses of Oxindole and Indole Spirocyclic Alkaloid Natural Products. Synthesis 2009, 18, 3003–3025. 10.1055/s-0029-1216975. [DOI] [Google Scholar]
- Saraswati A. P.; Relitti N.; Brindisi M.; Osko J. D.; Chemi G.; Federico S.; Grillo A.; Brogi S.; McCabe N. H.; Turkington R. C.; et al. Spiroindoline-Capped Selective HDAC6 Inhibitors: Design, Synthesis, Structural Analysis, and Biological Evaluation. ACS Med. Chem. Lett. 2020, 11 (11), 2268–2276. 10.1021/acsmedchemlett.0c00395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brindisi M.; Senger J.; Cavella C.; Grillo A.; Chemi G.; Gemma S.; Cucinella D. M.; Lamponi S.; Sarno F.; Iside C.; et al. Novel Spiroindoline HDAC Inhibitors: Synthesis, Molecular Modelling and Biological Studies. Eur. J. Med. Chem. 2018, 157, 127–138. 10.1016/j.ejmech.2018.07.069. [DOI] [PubMed] [Google Scholar]
- Xu Z. G.; Li S. Q.; Meng J. P.; Tang D. Y.; He L. J.; Lei J.; Lin H. K.; Li H. Y.; Chen Z. Z. Functionalized Spiroindolines with Anticancer Activity through a Metal-Free Post-Ugi Diastereoselective One-Pot Cascade Reaction. Chem.—Eur. J. 2018, 24 (26), 6732–6736. 10.1002/chem.201801081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao J. X.; Wang T. C.; Ruel R.; Thibeault C.; L’Heureux A.; Schumacher W. A.; Spronk S. A.; Hiebert S.; Bouthillier G.; Lloyd J.; et al. Conformationally Constrained Ortho- Anilino Diaryl Ureas: Discovery of 1-(2-(1′-Neopentylspiro[Indoline-3,4′-Piperidine]-1-Yl)Phenyl) −3-(4-(Trifluoromethoxy)Phenyl)Urea, a Potent, Selective, and Bioavailable P2Y1 Antagonist. J. Med. Chem. 2013, 56 (22), 9275–9295. 10.1021/jm4013906. [DOI] [PubMed] [Google Scholar]
- Lindemann M.; Deuther-Conrad W.; Moldovan R.; Sekhar K. V. G. C.; Brust P.; Wenzel B. Do Spiroindolines Have the Potential to Replace Vesamicol as Lead Compound for the Development of Radioligands Targeting the Vesicular Acetylcholine Transporter?. Bioorg. Med. Chem. 2017, 25 (19), 5107–5113. 10.1016/j.bmc.2017.03.028. [DOI] [PubMed] [Google Scholar]
- Yang S. W.; Ho G.; Tulshian D.; Greenlee W. J.; Fernandez X.; McLeod R. L.; Eckel S.; Anthes J. Structure-Activity Relationships of 3-Substituted N-Benzhydryl-Nortropane Analogs as Nociceptin Receptor Ligands for the Treatment of Cough. Bioorg. Med. Chem. Lett. 2008, 18 (24), 6340–6343. 10.1016/j.bmcl.2008.10.088. [DOI] [PubMed] [Google Scholar]
- Pan Z.; Liu Y.; Hu F.; Liu Q.; Shang W.; Ji X.; Xia C. Enantioselective Synthesis of Spiroindolines via Cascade Isomerization/Spirocyclization/Dearomatization Reaction. Org. Lett. 2020, 22 (4), 1589–1593. 10.1021/acs.orglett.0c00181. [DOI] [PubMed] [Google Scholar]
- Xia Z. L.; Zheng C.; Wang S. G.; You S. L. Catalytic Asymmetric Dearomatization of Indolyl Dihydropyridines through an Enamine Isomerization/Spirocyclization/Transfer Hydrogenation Sequence. Angew. Chem., Int. Ed. 2018, 57 (10), 2653–2656. 10.1002/anie.201712435. [DOI] [PubMed] [Google Scholar]
- Zhu M.; Zheng C.; Zhang X.; You S. L. Synthesis of Cyclobutane-Fused Angular Tetracyclic Spiroindolines via Visible-Light-Promoted Intramolecular Dearomatization of Indole Derivatives. J. Am. Chem. Soc. 2019, 141 (6), 2636–2644. 10.1021/jacs.8b12965. [DOI] [PubMed] [Google Scholar]
- Zaman M.; Hasan M.; Peshkov A. A.; Van Hecke K.; Van der Eycken E. V.; Pereshivko O. P.; Peshkov V. A. Silver(I) Triflate-Catalyzed Protocol for the Post-Ugi Synthesis of Spiroindolines. Adv. Synth. Catal. 2020, 362 (1), 261–268. 10.1002/adsc.201901064. [DOI] [Google Scholar]
- Liang G.; Ji Y.; Liu H.; Pang Y.; Zhou B.; Cheng M.; Liu Y.; Lin B.; Liu Y. Silver Triflate/N-Fluorobenzenesulfonimide-Catalyzed Cycloisomerization of Tryptamine-Ynamide to Spiro[Indoline-3,4′-Piperidine] Induced by Cation-π-π Interactions between Substrate and Metal Ligand. Adv. Synth. Catal. 2020, 362 (1), 192–205. 10.1002/adsc.201901175. [DOI] [Google Scholar]
- Kumar A.; Vachhani D. D.; Modha S. G.; Sharma S. K.; Parmar V. S.; Van Der Eycken E. V. Post-Ugi Gold-Catalyzed Diastereoselective Domino Cyclization for the Synthesis of Diversely Substituted Spiroindolines. Beilstein J. Org. Chem. 2013, 9, 2097–2102. 10.3762/bjoc.9.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K. J.; Dai L. X.; You S. L. Palladium(0)-Catalyzed Dearomative Arylation of Indoles: Convenient Access to Spiroindolenine Derivatives. Org. Lett. 2012, 14 (14), 3772–3775. 10.1021/ol301663h. [DOI] [PubMed] [Google Scholar]
- Desrosiers J. N.; Hie L.; Biswas S.; Zatolochnaya O. V.; Rodriguez S.; Lee H.; Grinberg N.; Haddad N.; Yee N. K.; Garg N. K.; et al. Construction of Quaternary Stereocenters by Nickel-Catalyzed Heck Cyclization Reactions. Angew. Chem., Int. Ed. 2016, 55 (39), 11921–11924. 10.1002/anie.201606955. [DOI] [PubMed] [Google Scholar]
- Roy T.; Brandt P.; Wetzel A.; Bergman J.; Brånalt J.; Sävmarker J.; Larhed M. Selective Synthesis of Spirooxindoles by an Intramolecular Heck-Mizoroki Reaction. Org. Lett. 2017, 19 (10), 2738–2741. 10.1021/acs.orglett.7b01094. [DOI] [PubMed] [Google Scholar]
- Adeyemi A.; Wetzel A.; Bergman J.; Brånalt J.; Larhed M. Regio- and Stereoselective Synthesis of Spirooxindoles via Mizoroki-Heck Coupling of Aryl Iodides. Synlett 2019, 30 (1), 82–88. 10.1055/s-0037-1611360. [DOI] [Google Scholar]
- Adeyemi A.; Odell L. R.; Larhed M. Regio- A Nd Stereoselective Synthesis of Allylic Spiroethers (Spirobenzofuranes) via an Intramolecular Mizoroki-Heck Reaction. J. Org. Chem. 2020, 85 (12), 7648–7657. 10.1021/acs.joc.9b03329. [DOI] [PubMed] [Google Scholar]
- Fukuyama Y.; Yuasa H.; Tonoi Y.; Harada K.; Wada M.; Asakawa Y.; Hashimoto T. First Syntheses of 1,13- and 1,15-Dihydroxyherbertenes, and Herbertenolide by Applying Intramolecular Heck Reaction for the Construction of Adjacent Quaternary Centers. Tetrahedron 2001, 57 (45), 9299–9307. 10.1016/S0040-4020(01)00932-2. [DOI] [Google Scholar]
- Beletskaya I. P.; Cheprakov A. V. Heck Reaction as a Sharpening Stone of Palladium Catalysis. Chem. Rev. 2000, 100 (8), 3009–3066. 10.1021/cr9903048. [DOI] [PubMed] [Google Scholar]
- Wetzel A.; Bergman J.; Brandt P.; Larhed M.; Branalt J. Regio- and Stereoselective Synthesis of Functionalized Cyclopentene Derivatives via Mizoroki-Heck Reactions. Org. Lett. 2017, 19 (7), 1602–1605. 10.1021/acs.orglett.7b00325. [DOI] [PubMed] [Google Scholar]
- Singh R.; Vince R. 2-Azabicyclo[2.2.1]Hept-5-En-3-One: Chemical Profile of a Versatile Synthetic Building Block and Its Impact on the Development of Therapeutics. Chem. Rev. 2012, 112 (8), 4642–4686. 10.1021/cr2004822. [DOI] [PubMed] [Google Scholar]
- Narancic T.; Almahboub S. A.; O’Connor K. E. Unnatural Amino Acids: Production and Biotechnological Potential. World J. Microbiol. Biotechnol. 2019, 35 (4), 1–11. 10.1007/s11274-019-2642-9. [DOI] [PubMed] [Google Scholar]
- Blaskovich M. A. T. Unusual Amino Acids in Medicinal Chemistry. J. Med. Chem. 2016, 59 (24), 10807–10836. 10.1021/acs.jmedchem.6b00319. [DOI] [PubMed] [Google Scholar]
- Bhonsle J. B.; Clark T.; Bartolotti L.; Hicks R. P. A Brief Overview of Antimicrobial Peptides Containing Unnatural Amino Acids and Ligand-Based Approaches for Peptide Ligands. Curr. Top. Med. Chem. 2013, 13 (24), 3205–3224. 10.2174/15680266113136660226. [DOI] [PubMed] [Google Scholar]
- Brenda M.; Knebelkamp A.; Greiner A.; Heitz W. Novel Palladium-Catalyzed Biaryl Synthesis with Haloarenes. Synlett 1991, 11, 809–810. 10.1055/s-1991-20885. [DOI] [Google Scholar]
- Zawisza A. M.; Muzart J. Pd-Catalyzed Reduction of Aryl Halides Using Dimethylformamide as the Hydride Source. Tetrahedron Lett. 2007, 48 (38), 6738–6742. 10.1016/j.tetlet.2007.07.077. [DOI] [Google Scholar]
- Georgsson J.; Hallberg A.; Larhed M. Rapid Palladium-Catalyzed Synthesis of Esters from Aryl Halides Utilizing Mo(CO)6 as a Solid Carbon Monoxide Source. J. Comb. Chem. 2003, 5 (4), 350–352. 10.1021/cc0201086. [DOI] [PubMed] [Google Scholar]
- Nordeman P.; Odell L. R.; Larhed M. Aminocarbonylations Employing Mo(CO)6 and a Bridged Two-Vial System: Allowing the Use of Nitro Group Substituted Aryl Iodides and Aryl Bromides. J. Org. Chem. 2012, 77 (24), 11393–11398. 10.1021/jo302322w. [DOI] [PubMed] [Google Scholar]
- Ravn A. K.; Johansen M. B.; Skrydstrup T. Controlled Release of Reactive Gases: A Tale of Taming Carbon Monoxide. ChemPlusChem. 2020, 85 (7), 1529–1533. 10.1002/cplu.202000319. [DOI] [PubMed] [Google Scholar]
- Walia A.; Kang S.; Silverman R. B. Microwave-Assisted Protection of Primary Amines as 2,5-Dimethylpyrroles and Their Orthogonal Deprotection. J. Org. Chem. 2013, 78 (21), 10931–10937. 10.1021/jo401778e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeman J. I. The Curtin-Hammett Principle and the Winstein-Holness Equation: New Definition and Recent Extensions to Classical Concepts. J. Chem. Educ. 1986, 63 (1), 42–48. 10.1021/ed063p42. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.