

Abstract
Background: TASP1 encodes an endopeptidase activating histone methyltransferases of the KMT2 family. Homozygous loss-of-function variants in TASP1 have recently been associated with Suleiman-El-Hattab syndrome. We report six individuals with Suleiman-El-Hattab syndrome and provide functional characterization of this novel histone modification disorder in a multi-omics approach. Methods: Chromosomal microarray/exome sequencing in all individuals. Western blotting from fibroblasts in two individuals. RNA sequencing and proteomics from fibroblasts in one individual. Methylome analysis from blood in two individuals. Knock-out of tasp1 orthologue in zebrafish and phenotyping. Results: All individuals had biallelic TASP1 loss-of-function variants and a phenotype including developmental delay, multiple congenital anomalies (including cardiovascular and posterior fossa malformations), a distinct facial appearance and happy demeanor. Western blot revealed absence of TASP1. RNA sequencing/proteomics showed HOX gene downregulation (HOXA4, HOXA7, HOXA1 and HOXB2) and dysregulation of transcription factor TFIIA. A distinct methylation profile intermediate between control and Kabuki syndrome (KMT2D) profiles could be produced. Zebrafish tasp1 knock-out revealed smaller head size and abnormal cranial cartilage formation in tasp1 crispants. Conclusion: This work further delineates Suleiman-El-Hattab syndrome, a recognizable neurodevelopmental syndrome. Possible downstream mechanisms of TASP1 deficiency include perturbed HOX gene expression and dysregulated TFIIA complex. Methylation pattern suggests that Suleiman-El-Hattab syndrome can be categorized into the group of histone modification disorders including Wiedemann–Steiner and Kabuki syndrome.
Introduction
Homozygous loss-of-function variants in TASP1 (MIM *608270) have recently been associated with an autosomal recessive syndrome featuring developmental delay, happy demeanor, distinctive facial features and congenital anomalies (1). It has been termed ‘Suleiman-El-Hattab syndrome’ (MIM #618950).
TASP1 encodes taspase 1 (threonine aspartase 1, TASP1). This enzyme functions as a heterodimeric endopeptidase activating various nuclear factors, including histone methyltransferases of the evolutionarily conserved KMT2 family of proteins like KMT2A and KMT2B, which are part of the epigenetic machinery (2). Epigenetic marks are modifications of DNA (e.g. methylation of cytosine nucleotides) or histone tails influencing transcriptional activity of genes without altering the DNA sequence itself (3). Because of this central role in regulation of transcriptional activity, it is not surprising that variation in genes of the epigenetic machinery can result in Mendelian disorders. Haploinsufficiency of KMT2A has been associated with Wiedemann–Steiner syndrome [MIM #605130; (4)]. Haploinsufficiency of KMT2B has been associated with complex childhood-onset dystonia [MIM #617284; (5)]. Another member of the evolutionary conserved KMT2 family of histone methyltransferases is KMT2D. Haploinsufficiency of KMT2D has been associated with Kabuki syndrome 1 [MIM #147920; (6)].
To date, Suleiman-El-Hattab syndrome has been described in four individuals. We present here six individuals with Suleiman-El-Hattab syndrome including three unreported individuals and three previously reported individuals with updated phenotypic information. We further delineate the phenotypic and genotypic spectrum of this recently described syndrome, and additionally present both in vitro and in vivo functional work showing the consequences of biallelic TASP1 loss-of-function variants.
Results
Phenotypic characteristics
We present six individuals from five unrelated families with Suleiman-El-Hattab syndrome with biallelic putative loss-of-function variants in TASP1.
The six individuals were between 9 months and 10 years old. All had a neurodevelopmental phenotype featuring developmental delay with severe speech delay (older individuals remained non-verbal or only said few words) and motor delay (walking between ages two and four), hypotonia and microcephaly. Feeding difficulties were observed in all and most had failure to thrive. Happy demeanor was a common finding.
In terms of facial appearance, typical facial dysmorphies included thick and arched eyebrows, thick eyelids, periorbital fullness, synophrys, epicanthus, a broad nasal bridge, a wide mouth with a long smooth philtrum and a thin upper and thick lower lip, low-set and dysplastic ears and a preauricular skin tag (Fig. 1A, B, E, G, and I).
Figure 1.

Pedigrees and photographs of affected individuals, DeepGestalt composite image, western blot results, and schematic image of TASP1 gene and corresponding protein. Circles, female individual; crossed circle, deceased female individual; dot within circles/squares, heterozygous carrier of the respective TASP1 variant and double horizontal line, consanguineous couple; squares, male individual. (A) Individual 1 at 7 years of age showing prominent glabella, excessive forehead hair, thick and arched eyebrows with synophrys, epicanthus, hypertelorism, periorbital fullness with thick eyelids, broad nasal bridge, long and smooth philtrum, wide mouth, thin upper lip, thick lower lip, low-set dysplastic ears and webbed neck. (B) Individual 2 at 6 months of age showing prominent glabella, thick and arched eyebrows with synophrys, epicanthus, hyperterorism, periorbital fullness with thick eyelids, broad and flat nasal bridge, long and smooth philtrum, wide mouth, thin upper lip, thick lower lip, microretrognathia, low-set dysplastic ears and preauricular skin tag. (C) Pedigree of individuals 1 and 2. (D) Pedigree of individual 3. Parents elected not to publish the individual’s photographs. Facial features include prominent glabella, excessive forehead hair, arched and thick eyebrows with synophrys, epicanthus, downslanted palpebral fissures, hypertelorism, periorbital fullness with thick eyelids, broad nasal bridge, long and smooth philtrum, wide mouth, thin upper lip, thick lower lip, microretrognathia, high arched palate and low-set dysplastic ears. (E) Individual 4 at 2 years of age showing excessive forehead hair, arched and thick eyebrows with synophrys, epicanthus, downslanted palpebral fissures, hypertelorism, periorbital fullness with thick eyelids, broad nasal bridge, long and smooth philtrum, wide mouth, thin upper lip, thick lower lip, low-set dysplastic ears and webbed neck. (F) Pedigree of individual 4. (G) Individual 5 at 4 months of age showing excessive forehead hair, thick and arched eyebrows with synophrys, epicanthus, hypertelorism, periorbital fullness with thick eyelids, broad nasal bridge, thin upper lip, thick lower lip, microretrognathia, high arched palate, low-set dysplastic ears and preauricular skin tag. (H) Pedigree of individual 5. (I) Individual 6 at 4 months of age showing thick and arched eyebrows with synophrys, epicanthus, hypertelorism, periorbital fullness with thick eyelids, broad and flat nasal bridge, long and smooth philtrum, wide mouth, thin upper lip, thick lower lip, low-set and dysplastic ears and a short neck. (J) Pedigree of individual 6. (K) DeepGestalt composite image generated from facial images of individuals with genetically proven Suleiman-El-Hattab syndrome. The facial gestalt is distinct from all other descriptors of syndromes in the Face2Gene dataset (300+ syndromes). (L) TASP1 gene structure (NM_017714.3) and corresponding protein (NP_060184.2) with respective identified variants. Please note that the missense variant p.(Thr234Met) has been identified in a homozygous state in case 3 in Suleiman et al., 2019 [no updated data; (1)]. (M) Western blot. TASP1 protein (alpha-subunit p28) is not observed in primary fibroblasts carrying biallelic loss-of-function variants (TASP1Arg67*, individual 5; TASP1trunc, individual 6) compared with primary fibroblasts from a healthy donor (405VI wt). (N) TASP1 antibody specificity was verified by downregulating TASP1 gene expression using specific TASP1-targeting siRNAs. A clear reduction of TASP1 protein levels with siRNA transfection in HeLa cells was observed. NT, non-targeted siRNA; 405VI wt, primary fibroblasts of control and TASP1Arg67*, primary fibroblasts of individual 5.
Congenital anomalies were common including brain malformations (especially posterior fossa malformations; Fig. 2), cardiovascular malformations (ventricular and atrial septal defects and tetralogy of Fallot), limb and skeletal deformities and cryptorchidism in males. Hirsutism and recurrent respiratory infections were also commonly observed.
Figure 2.

Cranial magnetic resonance images of four individuals with variable degrees of posterior fossa malformations. (A) Images of individual 2 at age 8 months; sagittal T1 (A1), axial T2 (A2 and A3) and coronal T2 (A4); showing cystic dilatation of the fourth ventricle extending posteriorly and directly communicating with a massively enlarged posterior fossa, with associated hypoplasia/dysgenesis of cerebellar vermis and small cerebellar hemispheres and cephalad rotation of the vermian remnant (Dandy–Walker malformation). In addition, significant supratentorial hydrocephalus with dilated lateral and third ventricles is seen causing stretching of the overlying corpus callosum. (B) Images of individual 3 at age 5 months; sagittal T2 (B1 and B2), axial T2 (B3) and axial T1 (B4); showing a well-defined right cerebellopontine angle cyst 1.2 × 1.5 × 1.5 cm (arrow in B2, 3 and 4) and hypoplasia of the splenium of the corpus callosum (best seen in B1). (C) Images of individual 5 at age 3 years; sagittal T2 (C1), axial T2 (C2 and C3) and axial FLAIR (C4) show dilatation of the fourth ventricle that directly communicates with a mildly enlarged posterior fossa, with associated hypoplastic inferior cerebellar vermis. In addition, dilatation of lateral and third ventricles is seen, as well as periventricular white matter hyperintensity around the posterior horn of the lateral ventricle bilaterally (seen best in C4). (D) Images of individual 6 at age 6 years; sagittal T1 (D1), axial T2 (D2 and D3) and coronal T2 (D4) show dilatation of the fourth ventricle that directly communicates with a retrocerebellar cyst, with associated hypoplastic inferior vermis. In addition, dilatation of lateral and third ventricles is present.
Table 1 summarizes the phenotypic characteristics of the cohort and detailed clinical descriptions can be found in the Supplementary case reports.
Table 1.
Genotypic and phenotypic characteristics of individuals with Suleiman-El-Hattab syndrome
| Individual | 1 | 2 | 3 | 4 | 5 | 6 | Case 3 a | |
|---|---|---|---|---|---|---|---|---|
| Gender | Male | Male | Female | Female | Male | Female | Male | |
| Ethnicity | Arabian Peninsula | Arabian Peninsula | Arabian Peninsula | Arabian Peninsula | Turkish | North Africa | Arabian Peninsula | |
| Age | 7 years | 9 months | 19 months | 3 years | 6 years | 10 years | 3 years | |
| TASP1 allele 1 | Exon 5–11 deletion | Exon 5–11 deletion | Exon 5–11 deletion | Exon 5–11 deletion | c.199C > T p.(Arg67*) |
c.81del p.(Glu29Serfs*79) | c.701C > T p.(Thr234Met) |
|
| TASP1 allele 2 | Exon 5–11 deletion | Exon 5–11 deletion | Exon 5–11 deletion | Exon 5–11 deletion | c.199C > T p.(Arg67*) |
c.(598_599del) p.(Lys200Glufs*24) |
c.701C > T p.(Thr234Met) |
|
| Neurodevelopment | ||||||||
| Developmental delay | + | + | + | + | + | + | + | 7/7 |
| Hypotonia | + | + | + | + | + | − | + | 6/7 |
| Microcephaly | + | − | + | + | + | + | + | 6/7 |
| Seizures | − | − | − | − | + | − | + | 2/7 |
| Posterior fossa malformation | − | + | + | − | + | + | − | 4/7 |
| Dilated ventricles | − | + | − | − | + | + | + | 4/7 |
| Feeding and growth | ||||||||
| Feeding difficulties | + | + | + | + | + | + | + | 7/7 |
| Salivary drooling | + | − | + | + | − | − | + | 4/7 |
| Failure to thrive | + | + | + | + | − | − | + | 5/7 |
| Happy demeanor | + | NA (too young) | + | + | + | − | + | 5/6 |
| CVS malformation | ASD/VSD | TOF | PFO/VSD | PFO/VSD | VSD | VSD | − | 6/7 |
| Cryptorchidism | + | − | NA | NA | + | NA | + | 3/4 |
| Hirsutism | + | − | + | + | − | + | − | 4/7 |
| Recurrent respiratory infections | + | − | + | + | + | − | − | 4/7 |
| Preauricular skin tag | − | + | + | + | + | − | − | 4/7 |
| Single palmar crease | + | − | − | + | + | − | − | 3/7 |
| Facial features | ||||||||
| Prominent glabella | + | + | + | − | + | − | + | 4/7 |
| Excess forehead hair | + | − | + | + | + | − | − | 4/7 |
| Thick eyebrows | + | + | + | + | + | + | + | 7/7 |
| Arched eyebrows | + | + | + | + | + | + | + | 7/7 |
| Synophrys | + | + | + | + | + | + | + | 7/7 |
| Epicanthus | + | + | + | + | + | + | + | 7/7 |
| Hypertelorism | + | + | + | + | + | + | + | 7/7 |
| Downslanted eyes | − | − | + | + | − | − | − | 2/7 |
| Thick eyelids | + | + | + | + | + | + | + | 7/7 |
| Periorbital fullness | + | + | + | + | + | + | + | 7/7 |
| Broad nasal bridge | + | + | + | + | + | + | + | 7/7 |
| Long smooth philtrum | + | + | + | + | − | + | + | 6/7 |
| Wide mouth | + | + | + | + | − | + | + | 6/7 |
| Thin upper lip | + | + | + | + | + | + | + | 7/7 |
| Thick lower lip | + | + | + | + | + | + | + | 7/7 |
| Microretrognathia | − | + | + | − | + | − | − | 3/7 |
| High arched palate | − | − | + | − | + | − | − | 2/7 |
| Low-set ears | + | + | + | + | + | + | + | 7/7 |
| Dysplastic ears | + | + | + | + | + | + | + | 7/7 |
| Webbed neck | + | − | + | + | − | − | − | 3/7 |
| Ophthalmologic anomalies | − | − | − | Hyperopia | Strabismus, amblyopia, pale optic disc | Hyperopia | − | |
| Limb deformities | Brachydactyly, clinodactyly | Overlapping toes | − | Polydactyly | − | Short extremities, brachydactyly, broad thumb and first toe | − | |
| Skeletal deformities | − | − | − | − | − | Scoliosis, genu recurvatum | Short stature | |
| Skin | − | Lumbar skin dimple | Sacral hair tuft, | Congenital dermal melanocytosis | − | − | − | |
| Others | Hearing impairment | − | Umbilical hernia | Left hydronephrosis | Inguinal hernia | Choanal stenosis, anteriorly placed anus, spasticity of the lower limbs | − |
ASD, atrial septal defect; CNS, central nervous system; CVS, cardiovascular; NA, not applicable; PFO, patent foramen ovale; TOF, tetralogy of Fallot; VSD, ventricular septal defect. TASP1 transcript NM_017714.3, TASP1 protein NP_060184.2.
aDescribed in (1), no new phenotypic or functional data. Featured in the table and the discussion to present all reported individuals with Suleiman-El-Hattab syndrome.
The DeepGestalt algorithm could establish a distinct facial gestalt for Suleiman-El-Hattab syndrome [Fig. 1K; (7)]. The descriptor of photos of individuals genetically diagnosed with Suleiman-El-Hattab syndrome is distinct from all other descriptors of syndromes in the dataset (300+) achieving an area under the curve of 0.975 (95% confidence interval 88–93%).
Genotypic characteristics
Chromosomal microarray (CMA) in individual 1 revealed a homozygous deletion at 20p12.1 with a minimum deletion boundary of chr20:g.13 463 860–13 532 560 (hg19). This deletion encompasses part of TASP1. Breakpoint mapping revealed that this deletion is 149.4 kb in size spanning chr20:g.13 448 380–13 597 783 and includes exons 5–11 of TASP1 [NM_017714.3; (1)]. CMA in individual 2, brother of individual 1, revealed the same deletion in a homozygous state. CMA in individual 3 revealed a homozygous deletion at 20p21.2 with a minimum deletion boundary of ch20:g.13 447 411–13 606 048. Breakpoint mapping revealed the same boundaries as in individual 1 (chr20:g.13 448 380–13 597 783), including exons 5–11 of TASP1. CMA in individual 4 identified a homozygous deletion with a minimum deletion boundary of chr20:g.13 466 774–13 593 390. Breakpoint mapping revealed the same boundaries as in individual 1 (chr20:g.13 448 380–13 597 783), including exons 5–11 of TASP1 (1). For these individuals, parents were found to be heterozygous for the deletion, and none of the healthy siblings were found to have a homozygous deletion in this region (Fig. 1C, D and F). This intragenic deletion disrupts the active site of the enzyme (at amino acid position 234; NP_060184.2; see https://www.uniprot.org/uniprot/Q9H6P5/protvista) and is predicted to lead to a frameshift supporting the loss-of-function nature of this variant (1,8). It has previously been shown that individuals 1 and 4 share the same haplotype surrounding the deletion and that it is a founder deletion in the Arabian Peninsula (1).
Exome sequencing in individual 5 revealed a homozygous nonsense variant in TASP1 c.199C > T, p.(Arg67*) (chr20:g.13 605 846G > A). The variant p.(Arg67*) was reported once in a heterozygous state in the Genome Aggregation Database (gnomAD v.2.1.1, https://gnomad.broadinstitute.org/; allele frequency of 4 × 10−6) indicating that it is an extremely rare variant. This variant is predicted to either lead to nonsense-mediated decay or a truncated protein leading to a complete loss of function. Both parents are confirmed to be heterozygous carriers for this variant (Fig. 1H). Trio exome sequencing in individual 6 revealed compound-heterozygous frameshift variants c.81del, p.(Glu29Serfs*79) (chr20:g.13 610 645del) and c.598_599del, p.(Lys200Glufs*24) (chr20:g.13 539 731_13 539 732del). The father is heterozygous carrier of the p.(Glu29Serfs*79) variant and the mother is heterozygous carrier of the p.(Lys200Glufs*24) variant (Fig. 1J). Both variants have not been listed in over 140 000 individuals in gnomAD. Both variants are predicted to either lead to nonsense-mediated decay or a truncated protein. Figure 1L summarizes the localization of the different TASP1 pathogenic variants within the gene and protein.
Western blot
Western blot from primary fibroblasts from individuals 5 and 6 confirmed that TASP1 protein (alpha-subunit p28) is not observed in these cells compared with primary fibroblasts from a healthy donor (Fig. 1M). The specificity of the TASP1 antibody was verified by
downregulating TASP1 gene expression using specific siRNAs. A clear reduction of TASP1 protein levels with siRNA transfection in HeLa cells was observed (Fig. 1N). No difference in global levels of H3K4me3 histone mark could be detected in primary fibroblasts of individual 5 compared with primary fibroblasts from a healthy donor (Supplementary Fig. S1).
RNA sequencing and proteomics
RNA sequencing from fibroblasts of individual 5 revealed 20 significant differentially expressed genes (Supplementary Table S1). Notable differentially expressed genes were: GTF2A1 (adjusted P = 8.15 × 10−7), which was upregulated (log2fc = 0.55); HOXA4 (adjusted P = 5.02 × 10−4; log2fc = −5.42), HOXA7 (adjusted P = 7.86 × 10−4; log2fc = −5.4), HOXB2 (adjusted P = 9.5 × 10−3; log2fc = −1.8) and HOXA1 (adjusted P = 0.0312, log2fc = 5.15), which were all downregulated (Fig. 3A).
Figure 3.
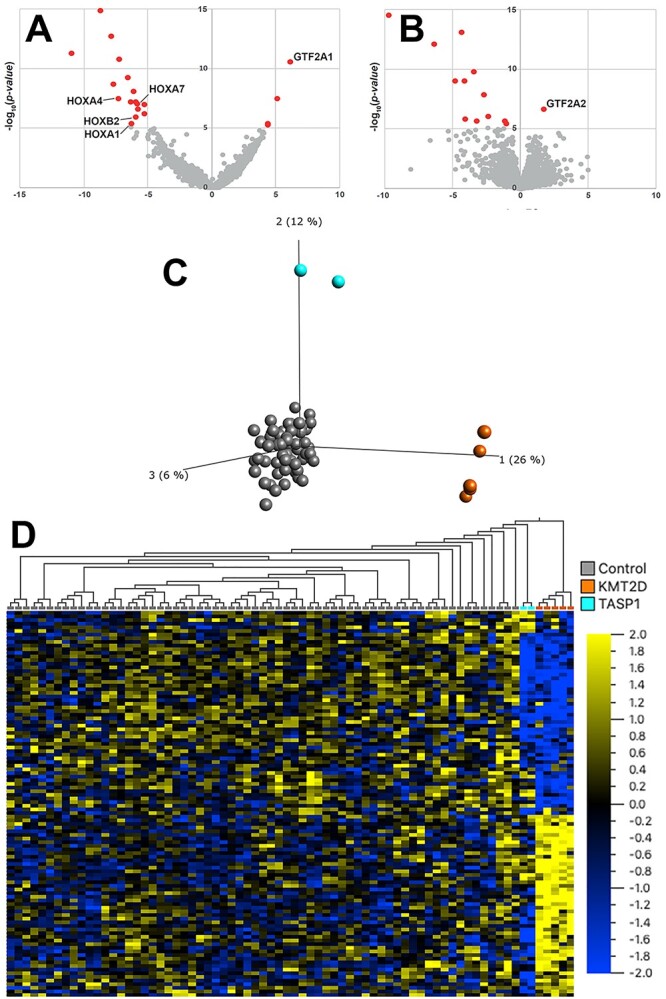
RNA sequencing, proteomics and DNAm (A) Volcano plot showing significant outliers of RNA sequencing with notable differentially expressed genes marked. (B) Volcano plot showing significant outliers of quantitative proteomics with notable differentially expressed protein marked. (C) Principal component analysis (PCA) based on DNAm values at 114 Kabuki syndrome 1 (KMT2D) signature sites clearly distinguishes the DNAm profile of healthy controls (n = 65 controls presented as grey dots), from DNAm profiles of the five individuals with Kabuki syndrome 1 (orange dots) on the first principal component (PC1) and the two individuals carrying TASP1 variants (blue; individuals 4 and 5) on PC2 (9). (D) Hierarchal clustering shows that, at Kabuki syndrome 1 signature sites, individuals carrying TASP1 variants have DNAm profiles that are intermediate when compared with the DNAm profiles of Kabuki syndrome 1 individuals and controls. Euclidean distance metric is used in the clustering dendrogram and the color gradient indicates the normalized β (DNAm) value ranging from −2.0 (blue) to +2.0 (yellow).
Quantitative proteomics from fibroblasts of individual 5 resulted in the identification of 13 significant aberrant protein levels (Supplementary Table S2). One notable significant outlier was GTF2A2 (adjusted P = 0.001770059; log2fc = 1.74), which was upregulated (Fig. 3B). TASP1 protein was not detected in the corresponding TMT-batch, hence proteomics was not able to provide any additional evidence on TASP1 protein stability.
DNA methylation pattern
Since the number of available TASP1 samples is not sufficient to establish a robust disorder-specific signature, we clustered the DNA methylation (DNAm) profiles of two individuals carrying homozygous loss-of-function TASP1 variants, individual 4 (deletion exons 5–11) and individual 5 (p.Arg67*), using the published Kabuki syndrome 1 signature (9). Principal component analysis (PCA) based on DNAm values at 114 Kabuki syndrome 1 signature sites clearly distinguishes the DNAm profile of healthy controls from DNAm profiles of the five individuals with Kabuki syndrome 1 on the first principal component (PC1) and the two individuals carrying TASP1 variants on PC2 (Fig. 3C). Hierarchal clustering shows that, at Kabuki syndrome 1 signature sites, individuals carrying TASP1 variants have distinct DNAm profiles that are intermediate when compared with the DNAm profiles of Kabuki syndrome 1 individuals and controls (Fig. 3D).
Zebrafish experiments
We were successfully able to target the single, tasp1 gene in Danio rerio and generate homozygous, F2 larvae with an indel variant predicting loss of function, c.313_314delTC, p.Ser105Glyfs*2 (ENSDART00000133661.3; Fig. 4A). Sectioning and staining of larval heads at 6 days post fertilization (dpf) showed wildtype EKW had an average brain area of 15,596 μm2 (Fig. 4B) compared with 14,747 μm2 in larvae homozygous for the tasp1 indel variant (Fig. 4C). A comparison of the cranial cartilages stained with Alcian blue in wildtype larvae (Fig. 4D) and homozygous, tasp1 indel larvae (Fig. 4G and J) showed that tasp1 crispant larvae had smaller trabeculae and shorter ethmoid cartilages compared with wildtype larvae. Wildtype larvae also had longer ceratobranchial and ceratohyal cartilages (Fig. 4E) compared with homozygous, tasp1 indel larvae (Fig. 4H and K). Wildtype larvae had longer lower jaw lengths and larger palatoquadrate and Meckel’s cartilages (Fig. 4F) compared with homozygous, tasp1 indel larvae (Fig. 4I and L).
Figure 4.

(A) Sequence of EKW control larva and a homozygous, tasp1 larva with c.313_314delTC, predicting p.Ser105Glyfs*2 (ENSDART00000133661.3). (B) and (C) Measurement of brain area in homozygous, tasp1 indel larvae shows a smaller size compared with wildtype control larvae. (B) Measurement of brain area from control larvae at 6 dpf. Larval brain area was measured from an average of five consecutive sections of brain containing diencephalon (red dotted line). (C) Measurement of brain area from homozygous, tasp1 indel larvae at 6 dpf. Larval brain area was measured from an average of five consecutive sections of brain containing diencephalon (red dotted line). This difference was not statistically significant. (D)–(L) Homozygous, tasp1 indel larvae show abnormal development and hypoplasia of the cranial cartilages at 6 dpf compared with wildtype controls. bb, basibranchial; bh, basihyal; cb, ceratobranchial; ch, ceratohyal; e, ethmoid plate; m, Meckel’s cartilage; pq, palatoquadrate and t, trabeculae cranii. (D) Dorsal view of cranial cartilages in 6 dpf EKW control larvae. (E) Lateral view of cranial cartilages in 6 dpf control larvae. (F) Ventral view of cranial cartilages in 6 dpf control larvae. (G) Dorsal view of 6 dpf tasp1 homozygous larvae with mild abnormality in cranial cartilages, showing ethmoid plate length is slighter shorter compared with EKW control. (H) Lateral view of 6 dpf tasp1 homozygous larvae with mild abnormality in cranial cartilages, showing shorter cartilage lengths compared with EKW control. (I) Ventral view of 6 dpf tasp1 homozygous larvae with mild abnormality in cranial cartilages, showing wider angle between ceratohyal cartilages compared with EKW control. (J) Dorsal view of 6 dpf tasp1 homozygous larvae with more severe abnormality in cranial cartilages, showing ethmoid plate is severely shorter and trabeculae cranii are abnormally positioned by growing further toward lateral compared with EKW control. (K) Lateral view of 6 dpf tasp1 homozygous larvae with severe abnormality in cranial cartilages, showing that trabeculae and other cartilages are abnormally positioned and appear to be more ventrally located and the ceratohyal cartilage appears to be shorter compared with EKW control. (L) Ventral view of 6 dpf tasp1 homozygous larvae with severe abnormality in cranial cartilages, showing shorter ceratobranchial and ceratohyal cartilages and atypical basibranchial and basihyal cartilages. Palatoquadrate and Meckel’s cartilage appear shorter in tasp1 homozygous larvae and thus the lower jaw length appears shorter than the upper jaw.
Discussion
Here, we report phenotypic, genotypic and functional data on the largest cohort of individuals with the recently characterized Suleiman-El-Hattab syndrome caused by biallelic loss-of-function variants in TASP1. The cohort presented here features six individuals with Suleiman-El-Hattab syndrome. Also included in the discussion and Table 1 is a seventh individual, previously described as case 3 in Suleiman et al. [no updated data; (1)].
These seven individuals have an overlapping phenotype of global developmental delay (7/7) with delayed motor development and severe speech delay (4/5 individuals at ages 3–10 years were non-verbal), microcephaly (6/7) and brain malformations (4/7), hypotonia (6/7), feeding difficulties (7/7), a happy demeanor (5/6; individual 2 was too young) and cardiovascular malformations (6/7), which were mostly mild [ventricular septal defect/atrial septal defects (ASD/VSD) with spontaneous resolution]. However, individual 2 had severe cardiac malformation (TOF) and required surgery (Table 1).
Notably, concerning brain malformations, 4/7 individuals had cystic malformations of the posterior fossa, with variable degrees ranging from small cerebellopontine angle arachnoid cyst, dilated fourth ventricle with enlarged posterior fossa along with cerebellar vermian hypoplasia, to the full extent of classic Dandy–Walker malformation associated with hydrocephalus [individuals 2, 3, 5 and 6; Fig. 2; (10,11)]. This extends the neurologic phenotype of Suleiman-El-Hattab syndrome and might further help in recognition of this syndrome. In addition, this supports the notion that imaging findings of cerebellar hypoplasia and Dandy–Walker malformation are often associated with underlying genetic causes and should prompt further evaluation (12).
Affected individuals feature a distinctive facial pattern, as also mathematically proven by the DeepGestalt algorithm, involving thick and arched eyebrows with synophrys, epicanthus, downslanted palpebral fissures, hypertelorism, broad nasal bridge, long and smooth philtrum, wide mouth with thin upper lip and thick lower lip, microretrognathia and ear anomalies [dysplastic, low-set; (7)]. This makes the syndrome recognizable by clinical geneticists and the syndrome is now featured in the Face2Gene online tool (https://www.face2gene.com).
TASP1 encodes the proenzyme taspase 1 (threonine aspartase 1, TASP1), which is cleaved into two subunits, alpha and beta, in an autocatalytic fashion, which in turn form the active heterodimeric enzyme, an endopetidase. The active enzyme uses the N-terminal threonine at amino acid position 234 (p.Thr234) of the mature beta subunit as the active-site nucleophile to proteolyze polypeptide substrates. This endopeptidase cleaves various nuclear factors after an aspartate, hence it is called threonine aspartase (taspase). For example, active TASP1 cleaves and hence activates histone methyltransferases of the KMT2 protein family like KMT2A and KMT2B. Cleavage of KMT2A (also known as MLL1) in turn is crucial for HOX and cyclin gene expression, which is important for cell cycle dynamics (2,13). In line with this, individual 5, who harbors a homozygous nonsense variant in TASP1 and has no detectable TASP1 protein on Western blot (Fig. 1M), showed significant RNA downregulation of HOX genes HOXA4, HOXA7, HOXA1 and HOXB2 (Fig. 3A). Coordinated HOX expression is central for a correct segmental body plan (14). Hence, dysregulation of HOX genes likely plays a role in the congenital and neurodevelopmental anomalies observed in TASP1-deficient individuals. However, it has to be pointed out that we tested the global level of the histone mark catalyzed by KMT2A histone methyltransferase (i.e. H3K4m3) and did not find any different levels between wildtype and TASP1 mutated cells (Supplementary Fig. S1) suggesting that the activity of KMT2A is not completely dependent of its TASP1-mediated proteolysis. This is consistent with the study of Zhao et al. (15–17), which showed that TASP1-dependent cleavage of KMT2A facilitates its degradation (and not its activity) to control the levels of KMT2A, reminiscent of that of TFIIA (see below). An alternative explanation for the unaffected levels of H3K4m3 in TASP1-mutated cell lines could be because of a compensatory phenomenon with other members of this histone methyltransferase family that do not required TASP1 cleavage. Hence, further studies need to be undertaken to unravel the intricate regulation of histone methyltransferases by TASP1.
Another substrate of TASP1 is transcription factor TFIIAα−β (GTF2A1), a general transcription factor with ubiquitous expression featuring an alpha and beta subunit. TFIIAα−β is bound to TFIIAγ (GTF2A2). When TASP1 cleaves the alpha and beta subunit of TFIIAα−β, the heterotrimeric complex TFIIAα/β/γ (encoded by GTF2A1 and GTF2A2, summarized as TFIIA) is susceptible to degradation by the proteasome impeding its transcriptional activity (2,17). Regulated turnover of TFIIA is key for correct embryonal cell proliferation and morphogenesis. Tasp1-deficient and Tfiia-noncleavable mice both show craniofacial anomalies such as jaw, eye and brain malformations (8). Intriguingly, on RNA sequencing of TASP1-deficient individual 5, GTF2A1 (TFIIAα−β) is upregulated; and on proteomics, GTF2A2 (TFIIAγ) is upregulated, probably illustrating the stable non-cleaved TFIIA complex (Fig. 3A and B). Hence, the neurodevelopmental phenotype including the central nervous system anomalies in Suleiman-El-Hattab syndrome could be because of TFIIA dysregulation including a positive feedback loop (GTF2A1 upregulation on RNA sequencing). Of course, it is a limitation, that RNA sequencing and proteomics data are only available from one individual. The findings are still biologically convincing. Nonetheless, further studies are needed for replication.
As TASP1 cleaves and hence activates histone methyltransferases of the KMT2 protein family, it is expected that defects in KMT2-genes result in syndromes with overlapping features of Suleiman-El-Hattab syndrome. Haploinsufficiency of KMT2A causes a phenotypically similar disorder, Wiedemann–Steiner syndrome (MIM #605130), also featuring developmental delay, facial anomalies, alongside hypertrichosis cubiti, growth failure, skeletal malformations, cardiac anomalies and microcephaly (4). Also individuals with haploinsufficiency of KMT2B (MIM #617284) can have, additionally to childhood-onset dystonia, facial anomalies such as elongated face, a bulbous nasal tip and microcephaly (18). Haploinsufficiency of KMT2D causes Kabuki syndrome 1 (MIM #147920), which is characterized by developmental delay, distinctive facial features, hirsutism, skeletal anomalies, persistence of fetal fingertip pads, recurrent infections, growth failure, seizures and congenital anomalies (6).
Intriguingly, Kabuki syndrome 1, Wiedemann–Steiner syndrome and Suleiman-El-Hattab syndrome share remarkable phenotypic overlap. All three are syndromic intellectual disability/developmental delay syndromes featuring a distinct facial gestalt with thick and arched eyebrows, hirsutism, growth failure and multiple congenital anomalies. KMT2A, KMT2B and KMT2D are all part of the highly conserved COMPASS family of histone methyltransferases. The members of the COMPASS protein family catalyze histone H3 lysine 4 (H3K4) methylation and are crucial for regulation of gene expression (2,19). As the KMT2-genes encode histone methyltransferases, we sought to investigate the effect of TASP1 deficiency on DNAm. Alterations of the epigenetic machinery can result in distinctive and reproducible DNAm patterns (‘episignatures’), as has recently been shown for 34 monogenic neurodevelopmental disorders, including Wiedemann–Steiner syndrome (KMT2A). A major advantage of this approach is that episignatures, which can be established by methylome analysis from peripheral blood, can serve as ‘biomarkers’ to classify variants of unknown significance in the respective genes (20). Because of the small number of investigated individuals, we could not establish a specific methylation pattern for Suleiman-El-Hattab syndrome but, interestingly, could show that the examined cases (blood DNA of individuals 4 and 5) have a distinct DNAm profile intermediate between Kabuki syndrome 1 and control profiles (Fig. 3C and D). In summary, our data suggest that Suleiman-El-Hattab syndrome can be grouped into the category of histone modification disorders.
We can also corroborate the effects of TASP1 loss-of-function by in vivo data. Already in 2006, knock-out mouse experiments revealed that Tasp1-deficient mice were smaller in size than their wild type counterparts and cell proliferation was reduced in mutant mice (14). Indeed, also in the seven individuals with Suleiman-El-Hattab syndrome, feeding difficulties and/or failure to thrive was a common finding (7/7 had feeding difficulties, 5/7 failure to thrive). Microcephaly was also reported in 6/7 individuals. Reduced function for the orthologous gene, tasp1, in zebrafish recapitulates this difference in growth, as tasp1 crispants have smaller head sizes in comparison with wildtype animals (Fig. 4B and C). Furthermore, tasp1 mutants showed abnormal formation of the cranial cartilages (Fig. 4D–L). In line with this finding, individuals with Suleiman-El-Hattab syndrome feature facial anomalies, including a broad nasal bridge, wide mouth and microretrognathia (Fig. 1K). TASP1 activates KMT2A, and it is interesting to note that zebrafish injected with antisense morpholinos targeting kmt2a also had significant craniofacial defects, with severe hypoplasia of the cartilaginous structures of the viscerocranium including complete loss of branchial arches 3–7, Meckel’s cartilage and the ceratohyal cartilages (21). Moreover, Tasp1 knock-out mice featured skeletal anomalies [vertebra, ribs and sternum; (14)]. In our cohort, major skeletal anomalies or dysplasia were not reported but, interestingly, there were three individuals with digital deformities (individuals 1, 4 and 6), and individual 6 also had shortened extremities. Altogether, our in vivo data complements and extends the in vivo data in the literature.
In conclusion, we further delineate the phenotypic and genotypic basis of Suleiman-El-Hattab syndrome by presenting the largest cohort of affected individuals of this recognizable syndrome. We provide functional insight in the pathophysiology of Suleiman-El-Hattab syndrome by suggesting perturbation of HOX gene expression and TFIIA complex dysregulation as possible downstream mechanisms of TASP1 deficiency. Furthermore, by methylation pattern analysis, we can show that Suleiman-El-Hattab syndrome can be categorized into the group of histone modification disorders including Wiedemann–Steiner and Kabuki syndrome.
Materials and methods
Recruitment of individuals
Individuals were seen in pediatric neurology and/or genetics departments of the respective institutions. Some of the individuals have been connected via GeneMatcher (22).
Molecular genetic testing
CMA for individual 1 was performed at Baylor Genetics Laboratory, Houston, TX, USA, as previously described (23). CMA for individual 2 was performed at Tawam Hospital Genetics Laboratory, Al Ain, United Arab Emirates, using CytoScan™ HD Array (Thermo Fisher Scientific, USA) as per the manufacturer’s instructions. CMA for individual 3 was performed at Centogene GmbH, Rostock, Germany, using CentoArrayCyto™. Breakpoint mapping in this individual was performed using gene specific primers (F: 5′ AAGGCACTCGCAAGTAACTG 3′; R: 5′ CACTGGAAAGACAGCTTGATGC 3′) to amplify the deleted allele (1). PCR product was later sequenced on a 3730xl capillary sequencer (Applied Biosystems, USA) to identify the breakpoints. CMA for individual 4 was performed at PreventionGenetics, Marshfield, Wisconsin, USA, as previously described (24). Exome sequencing for individual 5 was performed at Helmholtz Zentrum München, Neuherberg, Germany, as previously described (25,26). Sanger sequencing was used to test the individual’s parents. Trio exome sequencing for individual 6 and parents was done at Imagine Institute, Paris, France, as previously described (27).
Distinct facial gestalt
To establish a reproducible facial gestalt for Suleiman-El-Hattab syndrome, the facial recognition algorithm DeepGestalt was used [Face2Gene, FDNA Inc., USA; (7)]. The analysis of images was first pre-processed to achieve facial detection, landmark detection and alignment. After preprocessing, the input image was cropped into facial regions. Each region was fed into a Deep Convolutional Neural Network (DCNN) to obtain a softmax vector indicating its correspondence to each syndrome in the model. The analysis of visual facial data was used to form a mathematical representation of the face (facial descriptor), which can be readily compared with other such descriptors (i.e. facial gestalt of other syndromes). To enable visualization of these vectors, a composite image was produced.
Western blot
Primary fibroblasts 405VI wt (wildtype) from a healthy donor, primary fibroblasts TASP1Arg67* (individual 5), primary fibroblasts TASP1trunc (individual 6) and tumor cells HeLa (ATCC CCL-2) were cultivated in DMEM containing 100 U/ml penicillin, 100 μg/ml streptomycin and 10% fetal calf serum (FCS). All cell lines were incubated at 37°C in an atmosphere containing 5% CO2 and were regularly tested for mycoplasma. siRNAs were purchased from Horizon Discovery, UK: hTASP1 (L-004745-00-0005) and non-targeting siRNA (NT, D-001810-01-05) were used as control. Cells were transfected with 30 nm of siRNAs using Interferin (Polyplus, France) according to the manufacturer’s instructions and were processed 72 h later. Protein extracts were prepared and processed for SDS-PAGE as previously described (28). Membranes were then blotted with indicated antibodies: rabbit polyclonal Tasp1α (Proteintech, UK, 16739–1-AP, 1:500), mouse IgG1 β-catenin (BD Biosciences, USA, clone 14, 1:2000), rabbit polyclonal anti-Histone H3K4me3 (Abcam, UK, ab8580, 1:2000) and rabbit polyclonal anti-histone H4 (Active Motif, USA, 39 269, 1:2000).
RNA sequencing and proteomics
RNA sequencing of individual 5 was performed as strand-specific mRNA according to the TruSeq Stranded mRNA Sample Prep Guide on a NovaSeq 6000 platform (Illumina, USA) at Helmholtz Zentrum München, Neuherberg, Germany, as previously described (26). In order to detect differentially expressed genes, RNA sequencing data was analyzed using the method OUTRIDER in a cohort of 269 control samples (29).
Quantitative tandem mass tag (TMT) proteomics was performed at the BayBioMS core facility at the Technical University of Munich, Freising, Germany, and is described in detail on medRxiv (https://www.medrxiv.org/content/10.1101/2021.03.09.21253187v2.full, in revision).
DNAm pattern
Genomic DNA extracted from blood was treated with sodium bisulfite using the EpiTect PLUS Bisulfite Kit (Qiagen, Germany) and hybridized to the Illumina Infinium Human Methylation EPIC BeadChip (Illumina, USA) to interrogate more than 850 000 CpG sites at The Center for Applied Genomics (TCAG), Hospital for Sick Children Research Institute, Toronto, Ontario, Canada. The TASP1 samples with age-and sex-matched typically developing controls were run in two batches. The minfi Bioconductor package was used to preprocess data including quality control, Illumina normalization and background subtraction and extraction of β values. Probes with detection flaws, probes near single-nucleotide polymorphisms (SNPs) with minor allele frequencies above 1%, cross-reactive probes, probes with raw beta of 0 or 1 in >0.25% of samples, non-CpG probes and sex chromosome probes were removed according to the published pipeline (30). A total of n = 774 558 probes remained for interrogating DNAm levels. TASP1 samples (blood DNA of individuals 4 and 5) were compared with DNAm data from typically developing controls (n = 65) and individuals diagnosed with Kabuki syndrome 1 with a pathogenic KMT2D variant [n = 5; (9)].
Zebrafish
A Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) single guide (sg)RNA targeting exon 5 of tasp1 (ENSDART00000133661.3) was designed. sgRNA and Cas9 protein were co-injected into wildtype EKW zebrafish eggs and F0 founders with mosaicism for small indels in tasp1 at the target site were selected. Adult F1 heterozygotes with germline tasp1 indel variants were crossed to obtain F2, homozygous larvae. Larvae were cryosectioned and hematoxylin and eosin staining was performed. For measurements of brain size, 12-μm sections through the optic nerve of both eyes from wildtype and tasp1 indel homozygous larvae were selected and an average of five consecutive sections containing the diencephalon were measured using Image J software (https://imagej.nih.gov/ij/); measurements were averaged for 3 larvae from each group. Alcian blue staining was performed according to prior methods (31).
Supplementary Material
Contributor Information
Korbinian M Riedhammer, Institute of Human Genetics, Klinikum rechts der Isar, School of Medicine, Technical University of Munich, 81675 Munich, Germany; Department of Nephrology, Klinikum rechts der Isar, School of Medicine, Technical University of Munich, 81675 Munich, Germany.
Anna L Burgemeister, Genetikum, Genetic Counseling and Diagnostics, 70173 Stuttgart, Germany.
Vincent Cantagrel, Developmental Brain Disorders Laboratory, Université Paris Cité, Imagine Institute, INSERM UMR, 75015 Paris, France.
Jeanne Amiel, Department of Genetics, AP-HP, Necker Enfants Malades Hospital, Université Paris Cité, Imagine Institute, 75015 Paris, France.
Karine Siquier-Pernet, Developmental Brain Disorders Laboratory, Université Paris Cité, Imagine Institute, INSERM UMR, 75015 Paris, France.
Nathalie Boddaert, Département de radiologie pédiatrique, INSERM UMR 1163 and INSERM U1000, AP-HP, Necker Enfants Malades Hospital, 75015 Paris, France.
Jozef Hertecant, Division of Genetics and Metabolics, Department of Pediatrics, Tawam Hospital, Al Ain, United Arab Emirates.
Patricia L Kannouche, CNRS UMR 9019, Université Paris-Saclay, Equipe labellisée Ligue contre le Cancer, Gustave Roussy, 94805 Villejuif, France.
Caroline Pouvelle, CNRS UMR 9019, Université Paris-Saclay, Equipe labellisée Ligue contre le Cancer, Gustave Roussy, 94805 Villejuif, France.
Stephanie Htun, Department of Pediatrics, Division of Genetics, University of California, San Francisco, San Francisco, CA 94143, USA.
Anne M Slavotinek, Department of Pediatrics, Division of Genetics, University of California, San Francisco, San Francisco, CA 94143, USA.
Christian Beetz, Centogene GmbH, 18055 Rostock, Germany.
Dan Diego-Alvarez, Centogene GmbH, 18055 Rostock, Germany.
Kapil Kampe, Centogene GmbH, 18055 Rostock, Germany.
Nicole Fleischer, FDNA Inc., Boston, MA 02111, USA.
Zain Awamleh, Genetics and Genome Biology, Research Institute, The Hospital for Sick Children, Toronto, Ontario M5G 0A4, Canada.
Rosanna Weksberg, Genetics and Genome Biology, Research Institute, The Hospital for Sick Children, Toronto, Ontario M5G 0A4, Canada; Division of Clinical and Metabolic Genetics, The Hospital for Sick Children, Toronto, Ontario M5G 1X8, Canada; Department of Molecular Genetics, Institute of Medical Sciences, University of Toronto, Toronto, Ontario M5S 1A1, Canada.
Robert Kopajtich, Institute of Human Genetics, Klinikum rechts der Isar, School of Medicine, Technical University of Munich, 81675 Munich, Germany; Institute of Neurogenomics, Helmholtz Zentrum München, 85764 Neuherberg, Germany.
Thomas Meitinger, Institute of Human Genetics, Klinikum rechts der Isar, School of Medicine, Technical University of Munich, 81675 Munich, Germany.
Jehan Suleiman, Division of Neurology, Department of Pediatrics, Tawam Hospital, Al Ain, United Arab Emirates; Department of Pediatrics, College of Medicine and Health Sciences, United Arab Emirates University, Al Ain, United Arab Emirates.
Ayman W El-Hattab, Department of Clinical Sciences, College of Medicine, University of Sharjah, Sharjah, United Arab Emirates; Pediatrics Department, University Hospital Sharjah, Sharjah, United Arab Emirates; Genetics and Metabolic Department, KidsHeart Medical Center, Abu Dhabi, United Arab Emirates.
Acknowledgements
All authors thank the families for participating in the study.
Conflict of Interest statement. The authors declare that they have no competing interests.
Ethics approval, consent to participate and consent for publication
The study has been approved by the ethical committee of the Technical University of Munich (5369/12 S) and the further respective institutions and written informed consent under an IRB-approved protocol for sequencing and publication in a scientific journal (including photographs) was obtained by the legal guardians of each subject according to the Declaration of Helsinki. All animal experiments were performed under a protocol approved by the Institute for Animal Care and Use Committee at University of California, San Francisco (UCSF).
Availability of data and materials
Data and materials are available from the corresponding author upon reasonable request.
Funding
French National Research Agency (ANR-16-CE12-0005-01 to VC); National Eye Institute, National Institutes of Health (R01 EY032976-01 to AMS).
Authors’ contributions
KMR and AWEH performed data analysis, designed the study and wrote the manuscript. ALB, VC, JA, KSP, JH and JS provided phenotypic and genotypic data. NB, PLK, CP, SH, AMS, DDA, CB, KK, NF, ZA, RW, RK and TM did functional and data analyses. All authors critically reviewed and revised the manuscript.
References
- 1. Suleiman, J., Riedhammer, K.M., Jicinsky, T., Mundt, M., Werner, L., Gusic, M., Burgemeister, A.L., Alsaif, H.S., Abdulrahim, M., Moghrabi, N.N.et al. (2019) Homozygous loss-of-function variants of TASP1, a gene encoding an activator of the histone methyltransferases KMT2A and KMT2D, cause a syndrome of developmental delay, happy demeanor, distinctive facial features, and congenital anomalies. Hum. Mutat., 40, 1985–1992. [DOI] [PubMed] [Google Scholar]
- 2. Niizuma, H., Cheng, E.H. and Hsieh, J.J. (2015) Taspase 1: a protease with many biological surprises. Mol. Cell. Oncol., 2, e999513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bjornsson, H.T. (2015) The Mendelian disorders of the epigenetic machinery. Genome Res., 25, 1473–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jones, W.D., Dafou, D., McEntagart, M., Woollard, W.J., Elmslie, F.V., Holder-Espinasse, M., Irving, M., Saggar, A.K., Smithson, S., Trembath, R.C.et al. (2012) De novo mutations in MLL cause Wiedemann-Steiner syndrome. Am. J. Hum. Genet., 91, 358–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zech, M., Boesch, S., Maier, E.M., Borggraefe, I., Vill, K., Laccone, F., Pilshofer, V., Ceballos-Baumann, A., Alhaddad, B., Berutti, R.et al. (2016) Haploinsufficiency of KMT2B, encoding the lysine-specific histone methyltransferase 2B, results in early-onset generalized dystonia. Am. J. Hum. Genet., 99, 1377–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ng, S.B., Bigham, A.W., Buckingham, K.J., Hannibal, M.C., McMillin, M.J., Gildersleeve, H.I., Beck, A.E., Tabor, H.K., Cooper, G.M., Mefford, H.C.et al. (2010) Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet., 42, 790–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gurovich, Y., Hanani, Y., Bar, O., Nadav, G., Fleischer, N., Gelbman, D., Basel-Salmon, L., Krawitz, P.M., Kamphausen, S.B., Zenker, M.et al. (2019) Identifying facial phenotypes of genetic disorders using deep learning. Nat. Med., 25, 60–64. [DOI] [PubMed] [Google Scholar]
- 8. Takeda, S., Sasagawa, S., Oyama, T., Searleman, A.C., Westergard, T.D., Cheng, E.H. and Hsieh, J.J. (2015) Taspase1-dependent TFIIA cleavage coordinates head morphogenesis by limiting Cdkn2a locus transcription. J. Clin. Invest., 125, 1203–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Butcher, D.T., Cytrynbaum, C., Turinsky, A.L., Siu, M.T., Inbar-Feigenberg, M., Mendoza-Londono, R., Chitayat, D., Walker, S., Machado, J., Caluseriu, O.et al. (2017) CHARGE and Kabuki syndromes: gene-specific DNA methylation signatures identify epigenetic mechanisms linking these clinically overlapping conditions. Am. J. Hum. Genet., 100, 773–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kollias, S.S., Ball, W.S., Jr. and Prenger, E.C. (1993) Cystic malformations of the posterior fossa: differential diagnosis clarified through embryologic analysis. Radiographics, 13, 1211–1231. [DOI] [PubMed] [Google Scholar]
- 11. Barkovich, A.J., Kjos, B.O., Norman, D. and Edwards, M.S. (1989) Revised classification of posterior fossa cysts and cystlike malformations based on the results of multiplanar MR imaging. AJR Am. J. Roentgenol., 153, 1289–1300. [DOI] [PubMed] [Google Scholar]
- 12. Aldinger, K.A., Timms, A.E., Thomson, Z., Mirzaa, G.M., Bennett, J.T., Rosenberg, A.B., Roco, C.M., Hirano, M., Abidi, F., Haldipur, P.et al. (2019) Redefining the etiologic landscape of cerebellar malformations. Am. J. Hum. Genet., 105, 606–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hsieh, J.J., Cheng, E.H. and Korsmeyer, S.J. (2003) Taspase1: a threonine aspartase required for cleavage of MLL and proper HOX gene expression. Cell, 115, 293–303. [DOI] [PubMed] [Google Scholar]
- 14. Takeda, S., Chen, D.Y., Westergard, T.D., Fisher, J.K., Rubens, J.A., Sasagawa, S., Kan, J.T., Korsmeyer, S.J., Cheng, E.H. and Hsieh, J.J. (2006) Proteolysis of MLL family proteins is essential for taspase1-orchestrated cell cycle progression. Genes Dev., 20, 2397–2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhao, Z., Wang, L., Volk, A.G., Birch, N.W., Stoltz, K.L., Bartom, E.T., Marshall, S.A., Rendleman, E.J., Nestler, C.M., Shilati, J.et al. (2019) Regulation of MLL/COMPASS stability through its proteolytic cleavage by taspase1 as a possible approach for clinical therapy of leukemia. Genes Dev., 33, 61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hoiby, T., Mitsiou, D.J., Zhou, H., Erdjument-Bromage, H., Tempst, P. and Stunnenberg, H.G. (2004) Cleavage and proteasome-mediated degradation of the basal transcription factor TFIIA. EMBO J., 23, 3083–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhou, H., Spicuglia, S., Hsieh, J.J., Mitsiou, D.J., Hoiby, T., Veenstra, G.J., Korsmeyer, S.J. and Stunnenberg, H.G. (2006) Uncleaved TFIIA is a substrate for taspase 1 and active in transcription. Mol. Cell. Biol., 26, 2728–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Meyer, E., Carss, K.J., Rankin, J., Nichols, J.M., Grozeva, D., Joseph, A.P., Mencacci, N.E., Papandreou, A., Ng, J., Barral, S.et al. (2017) Mutations in the histone methyltransferase gene KMT2B cause complex early-onset dystonia. Nat. Genet., 49, 223–237. [DOI] [PubMed] [Google Scholar]
- 19. Shilatifard, A. (2012) The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu. Rev. Biochem., 81, 65–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Aref-Eshghi, E., Kerkhof, J., Pedro, V.P., Groupe, D.I.F., Barat-Houari, M., Ruiz-Pallares, N., Andrau, J.C., Lacombe, D., Van-Gils, J., Fergelot, P.et al. (2020) Evaluation of DNA methylation episignatures for diagnosis and phenotype correlations in 42 Mendelian neurodevelopmental disorders. Am. J. Hum. Genet., 106, 356–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Van Laarhoven, P.M., Neitzel, L.R., Quintana, A.M., Geiger, E.A., Zackai, E.H., Clouthier, D.E., Artinger, K.B., Ming, J.E. and Shaikh, T.H. (2015) Kabuki syndrome genes KMT2D and KDM6A: functional analyses demonstrate critical roles in craniofacial, heart and brain development. Hum. Mol. Genet., 24, 4443–4453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sobreira, N., Schiettecatte, F., Valle, D. and Hamosh, A. (2015) GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum. Mutat., 36, 928–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gambin, T., Yuan, B., Bi, W., Liu, P., Rosenfeld, J.A., Coban-Akdemir, Z., Pursley, A.N., Nagamani, S.C.S., Marom, R., Golla, S.et al. (2017) Identification of novel candidate disease genes from de novo exonic copy number variants. Genome. Med., 9, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Alabdullatif, M.A., Al Dhaibani, M.A., Khassawneh, M.Y. and El-Hattab, A.W. (2017) Chromosomal microarray in a highly consanguineous population: diagnostic yield, utility of regions of homozygosity, and novel mutations. Clin. Genet., 91, 616–622. [DOI] [PubMed] [Google Scholar]
- 25. Kremer, L.S., Bader, D.M., Mertes, C., Kopajtich, R., Pichler, G., Iuso, A., Haack, T.B., Graf, E., Schwarzmayr, T., Terrile, C.et al. (2017) Genetic diagnosis of Mendelian disorders via RNA sequencing. Nat. Commun., 8, 15824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Riedhammer, K.M., Stockler, S., Ploski, R., Wenzel, M., Adis-Dutschmann, B., Ahting, U., Alhaddad, B., Blaschek, A., Haack, T.B., Kopajtich, R.et al. (2021) De novo stop-loss variants in CLDN11 cause hypomyelinating leukodystrophy. Brain, 144, 411–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ucuncu, E., Rajamani, K., Wilson, M.S.C., Medina-Cano, D., Altin, N., David, P., Barcia, G., Lefort, N., Banal, C., Vasilache-Dangles, M.T.et al. (2020) MINPP1 prevents intracellular accumulation of the chelator inositol hexakisphosphate and is mutated in Pontocerebellar Hypoplasia. Nat. Commun., 11, 6087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Despras, E., Sittewelle, M., Pouvelle, C., Delrieu, N., Cordonnier, A.M. and Kannouche, P.L. (2016) Rad18-dependent SUMOylation of human specialized DNA polymerase eta is required to prevent under-replicated DNA. Nat. Commun., 7, 13326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brechtmann, F., Mertes, C., Matuseviciute, A., Yepez, V.A., Avsec, Z., Herzog, M., Bader, D.M., Prokisch, H. and Gagneur, J. (2018) OUTRIDER: a statistical method for detecting aberrantly expressed genes in RNA sequencing data. Am. J. Hum. Genet., 103, 907–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Choufani, S., Cytrynbaum, C., Chung, B.H., Turinsky, A.L., Grafodatskaya, D., Chen, Y.A., Cohen, A.S., Dupuis, L., Butcher, D.T., Siu, M.T.et al. (2015) NSD1 mutations generate a genome-wide DNA methylation signature. Nat. Commun., 6, 10207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wu, D., Mandal, S., Choi, A., Anderson, A., Prochazkova, M., Perry, H., Gil-Da-Silva-Lopes, V.L., Lao, R., Wan, E., Tang, P.L.et al. (2015) DLX4 is associated with orofacial clefting and abnormal jaw development. Hum. Mol. Genet., 24, 4340–4352. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data and materials are available from the corresponding author upon reasonable request.