

Abstract
Polygenic risk scores (PRSs) are useful for predicting breast cancer risk, but the prediction accuracy of existing PRSs in women of African ancestry (AA) remains relatively low. We aim to develop optimal PRSs for the prediction of overall and estrogen receptor (ER) subtype-specific breast cancer risk in AA women. The AA dataset comprised 9235 cases and 10 184 controls from four genome-wide association study (GWAS) consortia and a GWAS study in Ghana. We randomly divided samples into training and validation sets. We built PRSs using individual-level AA data by a forward stepwise logistic regression and then developed joint PRSs that combined (1) the PRSs built in the AA training dataset and (2) a 313-variant PRS previously developed in women of European ancestry. PRSs were evaluated in the AA validation set. For overall breast cancer, the odds ratio per standard deviation of the joint PRS in the validation set was 1.34 [95% confidence interval (CI): 1.27–1.42] with the area under receiver operating characteristic curve (AUC) of 0.581. Compared with women with average risk (40th–60th PRS percentile), women in the top decile of the PRS had a 1.98-fold increased risk (95% CI: 1.63–2.39). For PRSs of ER-positive and ER-negative breast cancer, the AUCs were 0.608 and 0.576, respectively. Compared with existing methods, the proposed joint PRSs can improve prediction of breast cancer risk in AA women.
Introduction
Breast cancer is the most common cancer in women in the United States and worldwide. It is a complex genetic disorder caused by high-penetrance genes, multiple common variants and non-genetic factors. In the last 10 years, genome-wide association studies (GWASs) have identified >180 breast cancer susceptibility loci (1–4). A polygenic risk score (PRS) is an additive linear combination of the effects of multiple single nucleotide polymorphisms (SNPs) from GWAS and can achieve a degree of risk stratification that is useful for risk-based programs of breast cancer screening and early detection. PRSs have been developed to predict breast cancer risk in non-Hispanic white, Asian and Latin American women (5–10). Recently, a large study has developed a 313-variant PRS for breast cancer risks in women of European ancestry (5). This PRS model distinguished breast cancer cases from controls [area under receiver operating characteristic (ROC) curve (AUC) = 0.630 overall], with a better discriminating capacity for estrogen receptor (ER)-positive breast cancer (AUC = 0.641) than for ER-negative breast cancer (AUC = 0.601).
African Americans have higher risk of developing early onset breast cancer and about 40% higher breast cancer mortality than other racial/ethnic groups in the United States (11), so it is very important to have risk-stratified screening in this population, especially for women aged 40–49 years. Currently, however, reliable PRS models do not exist for women of African ancestry (AA), including native Africans living in sub-Saharan Africa and the Africa diaspora. Most GWASs of breast cancer were conducted in women of European ancestry, and given the distinct allele frequencies and linkage disequilibrium (LD) structures across populations, PRSs developed in European ancestry populations have an attenuated, though statistically significant, predictive value when applied to AA populations (12,13). Recently, we showed that the 313-variant PRS exhibits reduced discriminating accuracy in AA, with AUC being 0.571, 0.588 and 0.562 for overall, ER-positive and ER-negative breast cancer, respectively (14).
To effectively use genetic information, such as allele frequencies and LD, in AA data, we adopted a forward stepwise logistic regression approach (5) to select genetic variants and then construct PRSs for AA women using individual genotypic and phenotypic data. The stepwise approach can retain SNPs significantly associated with the phenotype at a given threshold and effectively control the number of noise SNPs used for PRSs. As the sample sizes of existing AA datasets are much smaller than those from European-ancestry studies, using only AA data to develop a PRS may have limited accuracy. To further increase the prediction accuracy, we adopted the method of Márquez-Luna et al. (15) to develop joint PRSs by combining two components: (1) optimal PRS trained in women of AA by the stepwise logistic regression method, and (2) the 313-variant PRS that was previously developed in women of European ancestry. We used data in women of AA from four breast cancer GWAS consortia and the Ghana Breast Health Study (GBHS); the four consortia were: ROOT (The GWAS of Breast Cancer in the African Diaspora consortium), the African American Breast Cancer Epidemiology and Risk (AMBER) consortium, Breast Cancer Association Consortium (BCAC) and African American Breast Cancer consortium (AABC) (see Supplementary Material, Table S1).
Results
We have evaluated the three types of PRS methods described in Materials and Methods: (1) PRSs built by using genome-wide data in women of AA (PRSAFR), (2) the 313-variant PRS using effect sizes directly from previous European ancestry studies (PRSEUR) and (3) the joint and hybrid PRSs (PRSJoint). The evaluation was performed in an AA validation dataset.
PRSs built using AA data only (PRSAFR)
We built PRS models using preset P-value thresholds for filtering SNPs and selecting SNPs by a ‘hard-thresholding’ forward stepwise logistic regression in the AA training set (see Materials and Methods). Table 1 shows the comparison of the performance of these PRS models developed using AA data only and evaluated in an independent validation set. Using the forward stepwise regression approach, the prediction accuracy of PRSs increased as the P-value threshold increased from 10−5 to 0.1. The accuracy increased only slightly when the P-value cut-off changed from 0.05 to 0.1, whereas the number of SNPs selected for PRSs for three phenotypes increased by about 1.6-fold. Therefore, we used the PRS models with the P-value threshold of 0.05 for further analysis. The covariate-adjusted AUCs of PRSAFR, PRSAFR.ERp and PRSAFR.ERn were 0.535, 0.546 and 0.548 for overall, ER-positive and ER-negative breast cancer, respectively (16); PRSAFR, PRSAFR.ERp and PRSAFR.ERn denote the PRSs for overall, ER-positive and ER-negative using 29 569, 29 004 and 28 100 SNPs, respectively, selected by stepwise forward regression in the AA training dataset.
Table 1.
Comparison of the performance of PRS models developed using genome-wide approach in AA data: results in the validation set
| P-value cut-offa | SNPs entering model (n) | SNPs selected (n) | OR (95% CI)b | AUC (95% CI)b | |
|---|---|---|---|---|---|
| Overall breast cancer | |||||
| <10−5 | 288 | 62 | 1.04 (0.99–1.10) | 0.509 (0.495–0.524) | |
| <10−4 | 2053 | 428 | 1.03 (0.98–1.09) | 0.506 (0.489–0.522) | |
| <10−3 | 19 067 | 2351 | 1.07 (1.01–1.13) | 0.521 (0.507–0.535) | |
| <10−2 | 175 161 | 10 647 | 1.12 (1.06–1.18) | 0.535 (0.519–0.551) | |
| <0.05 | 829 335 | 29 569 | 1.13 (1.07–1.19) | 0.535 (0.519–0.551) | |
| <0.1 | 1 615 762 | 46 854 | 1.15 (1.09–1.22) | 0.541 (0.527–0.556) | |
| ER-positive | |||||
| <10−5 | 201 | 79 | 1.06 (0.99–1.13) | 0.517 (0.499–0.536) | |
| <10−4 | 2026 | 408 | 1.04 (0.97–1.12) | 0.512 (0.491–0.534) | |
| <10−3 | 20 186 | 2339 | 1.10 (1.03–1.18) | 0.529 (0.508–0.550) | |
| <10−2 | 178 697 | 10 493 | 1.19 (1.10–1.27) | 0.543 (0.523–0.562) | |
| <0.05 | 832 622 | 29 004 | 1.22 (1.13–1.31) | 0.546 (0.527–0.566) | |
| <0.1 | 1 624 378 | 45 997 | 1.22 (1.13–1.31) | 0.546 (0.527–0.565) | |
| ER-negative | |||||
| <10−5 | 209 | 50 | 1.13 (1.04–1.22) | 0.531 (0.508–0.554) | |
| <10−4 | 1872 | 419 | 1.08 (0.99–1.17) | 0.528 (0.506–0.550) | |
| <10−3 | 16 751 | 2230 | 1.03 (0.95–1.11) | 0.506 (0.482–0.531) | |
| <10−2 | 160 097 | 10 138 | 1.14 (1.05–1.23) | 0.535 (0.510–0.559) | |
| <0.05 | 784 928 | 28 100 | 1.20 (1.11–1.31) | 0.548 (0.525–0.572) | |
| <0.1 | 1 552 045 | 44 889 | 1.23 (1.13–1.33) | 0.551 (0.527–0.575) | |
aThe P-value cut-off used for selecting SNPs based on their marginal associations with cancer risk and then in stepwise regression in the training set.
bOR per 1 SD for the PRS. OR for association with breast cancer in the validation set was derived using logistic regression adjusting for age, consortium/study and 10 PCs. AUC of PRSs was calculated under the covariate-adjusted ROC model adjusting for age, consortium/study and 10 PCs of genotype data. PRS models highlighted were used for further analysis.
The PRS previously developed in women of European ancestry (PRSEUR)
Directly applying the PRS developed in data on women of European ancestry (Supplementary Material, Table S2) (PRSEUR) to our study sample of AA, we found that it was significantly associated with breast cancer risk, with varying prediction accuracy for the three breast cancer phenotypes (Table 2). We noticed that the PRSs trained in women of European ancestry (PRSEUR) had almost no correlation with the PRS developed with ‘hard-thresholding’ approach (PRSAFR) that used AA data only, suggesting that additional predictive power could be gained if combining these PRSs together (Supplementary Material, Table S3).
Table 2.
Performance of ancestry-specific and joint prediction PRS models in the validation set
Weight (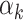 ) for each predictora ) for each predictora
|
OR (95% CI)a | P | AUC (95% CI)a | |
|---|---|---|---|---|
| Overall breast cancer | ||||
| PRSAFR (genome-wide threshold P < 0.05) | 1.13 (1.07–1.19) | 7.8 × 10−06 | 0.535 (0.519–0.551) | |
| PRS from European ancestry (PRSEUR)b | 1.30 (1.23–1.37) | 2.8 × 10−21 | 0.571 (0.557–0.585) | |
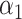 PRSAFR + PRSAFR + 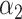 PRSEUR PRSEUR
|
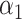 =0.31,
=0.31, 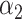 =0.69 =0.69 |
1.34 (1.27–1.41) | 3.4 × 10−25 | 0.577 (0.561–0.593) |
| PRShybridc | 1.34 (1.27–1.42) | 3.0 × 10−26 | 0.581 (0.566–0.597) | |
| ER-positive | ||||
| PRSAFR.ERp (genome-wide threshold P < 0.05) | 1.22 (1.13–1.31) | 2.7 × 10−7 | 0.546 (0.527–0.566) | |
| PRS from European ancestry (PRSEUR.ERp)b | 1.43 (1.33–1.53) | 6.1 × 10−24 | 0.597 (0.577–0.617) | |
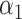 PRSAFR.ERp + PRSAFR.ERp + 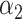 PRSEUR.ERp PRSEUR.ERp
|
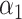 =0.35,
=0.35, 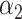 =0.65 =0.65 |
1.49 (1.39–1.60) | 1.1 × 10−28 | 0.608 (0.588–0.627) |
| ER-negative | ||||
| PRSAFR.ERn (genome-wide threshold P < 0.05) | 1.20 (1.11–1.31) | 1.1 × 10−5 | 0.548 (0.525–0.572) | |
| PRS from European ancestry (PRSEUR.ERn)b | 1.23 (1.13–1.34) | 8.7 × 10−7 | 0.557 (0.534–0.581) | |
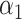 PRSAFR.ERn + PRSAFR.ERn + 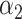 PRSEUR.ERn PRSEUR.ERn
|
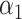 = 0.47,
= 0.47, 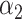 = 0.53 = 0.53 |
1.31 (1.21–1.43) | 1.1 × 10−10 | 0.576 (0.553–0.598) |
aWeight (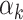 ) in the joint PRSs was estimated in validation set with a logistic regression model, including two-component PRSs (PRSAFR and PRSEUR) as predictors, and adjusting for age, consortium/study and 10 PCs; OR per 1 SD. AUC of PRSs was calculated under the covariate-adjusted ROC model adjusting for age, consortium/study and 10 PCs of genotype data.
) in the joint PRSs was estimated in validation set with a logistic regression model, including two-component PRSs (PRSAFR and PRSEUR) as predictors, and adjusting for age, consortium/study and 10 PCs; OR per 1 SD. AUC of PRSs was calculated under the covariate-adjusted ROC model adjusting for age, consortium/study and 10 PCs of genotype data.
bFor the 313 SNPs reported by Mavaddat et al. (5) for PRS in women of European ancestry, 307 SNPs appeared in our data of African ancestry.
cPRShybrid for overall cancer risk is a linear combination of the two joint PRSs for ER-positive and ER-negative breast cancer, with a weight of 0.62 for ER-positive and 0.38 for ER-negative cancer.
Table 3.
Associations between PRS percentiles and breast cancer risk in the validation set
| PRS category | No. control | Overall breast cancer | ER-positive | ER-negative | |||
|---|---|---|---|---|---|---|---|
| No. case | OR (95% CI)a | No. case | OR (95% CI)a | No. case | OR (95% CI)a | ||
| <5% | 156 | 100 | 0.79 (0.59–1.05) | 35 | 0.61 (0.41–0.92) | 26 | 0.74 (0.47–1.18) |
| 5–10% | 155 | 102 | 0.82 (0.62–1.09) | 28 | 0.52 (0.34–0.81) | 39 | 1.09 (0.73–1.63) |
| 0–10% | 311 | 202 | 0.81 (0.65–1.00) | 63 | 0.57 (0.42–0.78) | 65 | 0.92 (0.67–1.27) |
| 10–20% | 313 | 180 | 0.73 (0.58–0.91) | 77 | 0.72 (0.53–0.97) | 58 | 0.84 (0.60–1.18) |
| 20–40% | 624 | 422 | 0.85 (0.72–1.02) | 185 | 0.82 (0.66–1.04) | 111 | 0.80 (0.61–1.06) |
| 40–60% (ref.) | 624 | 486 | 1 (ref.) | 222 | 1 (ref.) | 141 | 1 (ref.) |
| 60–80% | 624 | 595 | 1.22 (1.03–1.44) | 266 | 1.18 (0.95–1.46) | 184 | 1.36 (1.06–1.74) |
| 80–90% | 312 | 350 | 1.45 (1.19–1.76) | 192 | 1.64 (1.29–2.09) | 94 | 1.39 (1.03–1.87) |
| 90–100% | 311 | 467 | 1.98 (1.63–2.39) | 256 | 2.20 (1.74–2.77) | 127 | 1.80 (1.37–2.38) |
| 90–95% | 155 | 216 | 1.83 (1.44–2.34) | 107 | 1.82 (1.35–2.45) | 55 | 1.61 (1.12–2.32) |
| >95% | 156 | 251 | 2.12 (1.67–2.69) | 149 | 2.58 (1.95–3.42) | 72 | 2.13 (1.52–3.00) |
aOR (95% CIs) were adjusted for age, consortium and 10 PCs.
The joint and hybrid PRS models
A joint PRS is a weighted linear combination of the two components PRSs, i.e. PRSJoint = α1 PRSAFR + α2 PRSEUR (see Materials and Methods). Table 3 shows the prediction performance of the joint and hybrid PRS models in the validation set. For each phenotype, the two-component joint PRS model performed better than individual PRSs. For overall breast cancer, adding the PRS developed in European ancestry population (PRSEUR) to the base model developed using the ‘hard-thresholding’ stepwise regression approach (PRSAFR), the AUC increased from 0.535 to 0.577. Similar results were observed for ER-positive and ER-negative breast cancer. Interestingly, the PRSs developed in European ancestry population contributed more to the two-component joint PRS model for overall (69%) and ER-positive breast cancer (65%). In contrast, the PRS developed using AA data (47%) has a similar contribution to the joint PRS of ER-negative disease as the PRS developed in European ancestry population (53%). The odds ratio (OR) per unit standard deviation (SD) was 1.49 [95% confidence interval (CI): 1.39–1.60] for the joint PRS of ER-positive breast cancer and 1.31 (95% CI: 1.21–1.43) for the joint PRS of ER-negative disease.
The joint PRS for overall breast had lower prediction accuracy (AUC = 0.577) than the joint PRSs for ER-positive (AUC = 0.608) and almost the same accuracy for ER-negative disease (AUC = 0.576). Therefore, we calculated the hybrid PRS for overall breast cancer that combines the PRSs of ER-positive and ER-negative diseases weighted by subtype proportions. The OR per SD of the hybrid PRS was 1.34 (95% CI: 1.27–1.42) with an AUC of 0.581. The SNPs and corresponding joint effect sizes used for the final joint and hybrid PRSs for the three phenotypes are listed in Supplementary Material, Tables S4–S6.
The contributing weights αk (k = 1, 2) of the two component PRSs (PRSAFR and PRSEUR) in the joint PRS models (Table 2) were estimated in the validation set with a logistic regression model, including the two-component PRSs, so there might be an overfitting problem. For the two-component joint PRS of overall breast cancer, the liability scale-adjusted R2 was 1.86%, which was very similar to the raw R2 of 1.91%. For ER-positive joint PRS, the adjusted and raw R2 were 3.60 and 3.66%, respectively. For ER-negative joint PRS, the adjusted and raw R2 were 1.13 and 1.21%, respectively. These analyses suggested that the bias owing to overfitting is minimal.
Table 3 showed associations between breast cancer risk and percentiles of the joint and hybrid PRSs. Women in the top 10 and 5% of the hybrid PRS had a 1.98-fold (95% CI: 1.63–2.39) and a 2.12-fold (95% CI: 1.67–2.69) elevated overall breast cancer risk compared with women at average risk (PRS in 40th–60th percentiles), respectively. For ER-positive breast cancer, compared with the population average, women in the top 10 and 5% of the joint PRS had a 2.20-fold (95% CI: 1.74–2.77) and a 2.58-fold (95% CI: 1.95–3.42) increased risk, respectively. For ER-negative breast cancer, those in the top 10% and 5% of the joint PRS had a 1.80-fold (95% CI: 1.37–2.38) and a 2.13-fold (95% CI: 1.52–3.00) increased risk, respectively, compared with women at average risk.
The joint and hybrid PRSs were significantly associated with breast cancer risk in women with and without family history of breast cancer (Table 4). We did not see any significant interaction between PRS and family history of breast cancer. In addition, family history was associated with about 1.76- to 2.05-fold increased risk of overall or subtype-specific breast cancer. We only observed slight attenuation of the association of family history with overall breast cancer and ER-negative cancer risk after adjusting for PRS (Table 4).
Table 4.
Associations between PRS and breast cancer risk by family history of breast cancer in the validation set
| Model | Overall breast cancer | ER-positive | ER-negative |
|---|---|---|---|
| OR (95% CI)a | OR (95% CI)a | OR (95% CI) a | |
| Association of PRS and cancer risk by family history | |||
| PRS unadjusted for family history | 1.31 (1.24–1.39) | 1.45 (1.35–1.56) | 1.31 (1.20–1.44) |
| PRS in women without family history | 1.30 (1.22–1.39) | 1.45 (1.33–1.57) | 1.31 (1.19–1.45) |
| PRS in women with family history | 1.34 (1.15–1.56) | 1.45 (1.21–1.74) | 1.29 (1.04–1.60) |
| P for testing interaction between PRS and family history | 0.829 | 0.965 | 0.779 |
| Association of family history and cancer risk | |||
| Family history unadjusted for PRS | 1.79 (1.52–2.11) | 2.05 (1.70–2.49) | 1.76 (1.39–2.23) |
| Family history adjusted for PRS | 1.76 (1.49–2.08) | 2.05 (1.68–2.49) | 1.72 (1.35–2.18) |
aFor PRS, ORs (95% CI) per 1 SD were presented. For family history, the OR comparing women with versus without family history of breast cancer. In all logistic regression models, age, consortium and 10 PCs were adjusted for.
We did not observe a statistically significant interaction between the joint/hybrid PRSs and age at diagnosis for overall or subtype-specific breast cancer risk (Supplementary Material, Fig. S1), although the association between PRS and overall or ER-positive breast cancer risk was weak for women aged 70 years or older.
We examined association of PRSs and breast cancer risk in two populations: Africans versus African Americans and African Barbadians. In both populations, PRSs were associated with breast cancer risk and there was no statistically significant interaction (Supplementary Material, Table S7). There was no significant interaction between ancestry groups (<80% AA vs. > 80% AA) and PRSs. There was a marginally significant heterogeneity effects of the PRSs for overall breast cancer and ER-negative breast cancer across the five consortia/studies but not for ER-positive PRS (Supplementary Material, Fig. S2). For overall breast cancer, the PRS had a moderate association in the ROOT and AABC consortia and had a stronger association in the AMBER consortium.
Absolute risk of developing breast cancer according to the PRS
Figure 1 shows the estimated lifetime and 10-year absolute risks of breast cancer for African Americans according to percentile of the PRSs. The absolute risk of overall breast cancer by age 80 years was 18.8% for women in the 99th percentile of the hybrid PRS and was 4.3% for women in the lowest first percentile. The absolute risk of ER-positive breast cancer by age 80 ranged from 2.3% in the lowest percentile of PRS to 17.6% in the highest percentile of PRS. For ER-negative breast cancer, the absolute risk by age 80 ranged from 1.3 to 4.8%. In contrast, the absolute risk of overall breast cancer by age 80 ranged from 3.2 to 31.3% for European American women in lowest and highest percentiles of the 313-variant PRS of European ancestry (5) (Supplementary Material, Fig. S3). The absolute risk by age 80 ranged from 2.4 to 31.6% for ER-positive and from 0.5 to 3.3% for ER-negative breast cancer among European Americans. The dotted line in Figure 1D illustrates the age at which women at different categories of the PRS reach a threshold of 10-year risk of 2%, which corresponds to the average risk for women aged 45 years in the United States. This threshold was reached at 35, 38 and 39 years for women whose PRSs were >99th, 95–99th and 90–95th percentiles, respectively.
Figure 1.

Cumulative lifetime and 10-year absolute risk of developing breast cancer among African Americans according to percentiles of the polygenic risk scores (PRSs). Cumulative lifetime absolute risk of developing (A) overall breast cancer, (B) estrogen receptor (ER)–positive breast cancer, and (C) ER-negative breast cancer. 10-year absolute risk of developing (D) overall breast cancer, (E) ER-positive breast cancer, and (F) ER-negative breast cancer. The pink dotted line in (d) demonstrates the 2% risk threshold that could be used to recommend screening age.
Discussion
In this study, we developed and validated joint PRSs of breast cancer among women of AA by pooling multiple studies and leveraging an existing PRS developed in European ancestry population. We adopted the method of Márquez-Luna et al. (15) to develop the joint PRSs that combined the PRS developed with only data from AA and the 313-variant PRS developed in women of European ancestry (5). With AUCs of 0.581, 0.608 and 0.576 for overall, ER-positive and ER-negative breast cancer, the joint PRSs provide a better predictive value than previous PRS models in AA women. Allman et al. evaluated a 77-variant PRS in African Americans and reported an AUC of 0.55 for overall breast cancer risk (12). Wang et al. reported an AUC of 0.531 for a 34-variant recalibrated PRS in women of AA (13). Recently, Du et al. evaluated the 313-variant PRS using the same dataset as the current study and reported AUCs of 0.571, 0.588 and 0.562 for overall, ER-positive, and ER-negative breast cancer, respectively (14). Although comparing with previous models, the improvements in AUCs are not large, the current PRSs can provide better risk stratification, making them suitable for clinical use.
The improved prediction value of the joint PRS models in women of AA may be because it has leveraged the strengths of two types of PRSs. The 313-variant PRS was developed with very large sample size of 94 075 breast cancer cases and 75 017 controls of European descent in BCAC (5), so it achieves high precision. The PRS model developed using ‘hard-thresholding’ genome-wide approach in AA datasets has the advantage that the training and validation dataset have the similar LD patterns. Of note, the contribution of the individual PRSs to the joint PRSs varied by breast cancer phenotypes. The 313-variant PRS has a better performance in predicting ER-positive than ER-negative breast cancer in both European and AA populations (5,14). Consistently, it also contributed more to the ER-positive joint PRS in this study. This may reflect that about 80% of breast cancer patients of European ancestry have ER-positive disease, so GWAS data in the BCAC contain more genetic information on ER-positive disease. In contrast, patients of African descent have a higher proportion of ER-negative disease than other populations. Probably because of this, the PRS trained in our combined AA dataset had about half contribution to the joint PRS for ER-negative risk.
We also observed that the subtype-specific PRSs performed better than the PRS for overall breast cancer risk. This is probably because of breast cancer etiology heterogeneity; many genetic variants have different effects on ER-positive and ER-negative breast cancers (4,17,18). Therefore, we generated a hybrid PRS for overall breast cancer risk, which is a weighted average of ER-positive and ER-negative joint PRSs. We found that the hybrid PRS had higher prediction accuracy than the corresponding joint PRS for overall breast cancer risk. If the finding that ‘the sum of the parts is greater than the whole’ can be confirmed in future studies, it could be a good strategy to estimate omnibus risk of breast cancer (19). Although an overall breast cancer risk model and an ER-negative model may be useful for clinical decision making regarding timing and frequency of breast cancer screening, an ER-positive model has the additional advantage of potentially identifying high-risk women who may benefit from chemoprevention with endocrine agents.
Although the joint PRS models have a better predictive performance than previous PRS models in AA women, the prediction accuracy is still lower than models reported for other racial/ethnic populations. Mavaddat et al. reported AUCs of 0.63 and 0.64 for their 313-variant and 3820-variant PRSs, respectively, for predicting overall breast cancer in women of European ancestry (5). Shieh et al. examined the performance of 71- and 180-variant PRS for overall breast cancer in a large Latino study and reported AUCs of 0.61 to 0.63 (10). Wen et al. examined a 67-variant PRS for overall breast cancer in East Asians and reported an AUC of 0.61 (9). In another PRS study of Asians, Ho et al. examined a 287-variant PRS and reported an AUC of 0.613 for overall breast cancer (20). The weaker performance of PRS in people of AA has been observed in other disease phenotypes (21). One study found that the prediction accuracy was 4.9-fold lower in Africans on average compared with that in European populations for 17 phenotypes, whereas the reduction in accuracy was 1.6-fold in Hispanic/Latino Americans, 1.7-fold in South Asians and 2.5-fold in East Asians (21). These observations are consistent with previous studies that showed that poorer PRS performance is related to genetic divergences between training and target populations (22,23). Therefore, several factors could account for this disparity, including relatively limited sample size, different LD patterns, allele frequencies and possible heterogeneity in effect sizes between populations.
To further improve the prediction accuracy of PRS in people of AA, it is important to include more racially/ethnically diverse individuals in medical genomic research. The ongoing Confluence project led by the US National Cancer Institute has prioritized large-scale genotyping for diverse populations (https://dceg.cancer.gov/research/cancer-types/breast-cancer/confluence-project), so it could improve the prediction accuracy of breast cancer PRS. Advances in methodologies in statistical genetics could also help to develop a better PRS utilizing information hidden in the existing GWAS datasets. For example, sophisticated methods that integrate additional biological information, genetic architecture and LD information can be promising to apply to diverse populations (24–26). For African Americans, an admixed population, global admixture proportion could help to predict cancer risk (15,27). We found the proportion of European ancestry was not associated with overall and ER-negative breast cancer (P > 0.3) but marginally significantly associated with ER-positive breast cancer (OR = 1.14 per a 25% increase in European ancestry, P = 0.011). Global admixture is essentially the same as the first principal component (PC) (r = 0.996), which was used to control for population stratification, so we did not use global admixture in our risk prediction model building. However, local ancestry, which is robust to population stratification, could also be tapped in future studies to gain statistical power to improve accuracy of genetic risk prediction (28–30).
The AUC, a discriminating accuracy metric, of the new PRS model is moderate, but the model could still provide meaningful risk stratification in the population. Women in the top fifth percentile of the new PRS have >2-fold elevated breast cancer risk compared with women at average risk. For women at average risk, the American Cancer Society strongly recommends initiating regular screening mammography at age 45 years, whose 10-year risk of developing breast cancer is about 2% (31). Based on the PRS, we estimated that about 10% of African American women have 10-year risk of 2% before they reach the age of 40. These women could start breast cancer screening earlier than age 40 and are possibly eligible for intensive screening programs or chemoprevention trials.
In summary, we proposed joint breast cancer PRSs in women of AA, which have moderate prediction value but are still not optimal. We found that the joint model can gain more information on ER-positive breast cancer prediction from the existing PRS developed in European ancestry population, while GWAS data from AA contribute more information to the prediction of ER-negative breast cancer.
Materials and Methods
Study participants and genotyping
This study included women of AA from four breast cancer GWAS consortia and a study in Ghana, with a combined sample size of 19 419 participants, including 9235 breast cancer cases and 10 184 controls. Data collections for individual studies of these consortia have been described previously (18,32–35). Sample size and selected characteristics for each consortium and study are summarized in Supplementary Material, Table S1. Women in the study sites in United States and Barbados were self-identified as African American or African Barbadian, whereas women in the African study sites were implied to be of AA. AA was confirmed using GWAS data. For each consortium/study in this project, individual protocols were approved by the relevant Institutional Review Boards (IRBs) at the participating centers. All participants provided written informed consent in accordance with the local IRBs.
Each consortium/study utilized a different GWAS array. Genotyping and quality control (QC) procedures have been described in detail in Supplementary Material, Table S1. The GWAS of Breast Cancer in the African Diaspora consortium (ROOT) consists of study participants from six studies (18), and samples were genotyped using the Illumina HumanOmni 2.5-8v1 array. After QC, 1657 cases (404 ER-positive, 374 ER-negative) and 2028 controls from the ROOT consortium remained in the analysis. AABC consists of nine epidemiological studies (32,36,37). Samples in AABC were genotyped using the Illumina Human 1M-Duo BeadChip. After QC, a total of 3005 cases (1517 ER-positive, 986 ER-negative) and 2713 controls remained in the analysis. AMBER consists of three studies (33). The AMBER samples were genotyped using the Illumina MEGA array, and after QC, 1406 cases (951 ER-positive, 385 ER-negative) and 2407 controls remained in the analysis. Nine studies with cases and controls of AA contributed samples to BCAC. Genotyping for BCAC was performed using Illumina OncoArray (with 260 K GWAS backbone) (38). After removing overlapped samples between BCAC (OncoArray) with AABC, AMBER and ROOT, a total of 2268 cases (1127 ER-positive, 613 ER-negative) and 1406 controls remained for the analysis. GBHS includes 899 cases (296 ER-positive, 277 ER-negative) and 1630 controls (34,35). Samples in GBHS were genotyped using Illumina Global Screening Array.
Training set and validation set
For pooling the samples from these studies, we conducted uniformed imputation using the cosmopolitan reference panel in the 1000 Genomes Project (1KGP) (Phase III release) within each consortium/study by the IMPUTE2 software (http://mathgen.stats.ox.ac.uk/impute/impute_v2.html) (39). After imputation, we filtered in variants (~15 million SNP or indel) with average minor allele frequency (MAF) > 0.01 and average imputation information score > 0.85. The distribution of imputation info across GWAS array is described in Supplementary Material, Table S1. We pooled datasets from the four AA consortia and the Ghana study into a combined dataset. PCs of genotype data were estimated using EIGENSTRAT in the pooled dataset (40,41). As shown in the scatter plots of the top five eigenvectors from the PC analysis (Supplementary Material, Fig. S4A and B), the first PC can distinguish participants from different continents (Africa vs. North America) and can indicate essentially the global proportion of AA. The third and fifth PCs can distinguish countries in Africa. We then randomly split the combined dataset into a training set (n = 13 598; 70%) and a validation set (n = 5821; 30%). Model development was conducted in the training set, while the performance of the PRS models were evaluated in the validation set.
Development of PRSs using genome-wide data in women of African ancestry
A PRS can be expressed as
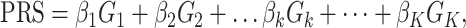 (1)
(1)
where βk is the per-allele log OR for breast cancer associated with SNP k and serves as the weight in PRS calculation, Gk is the allele dosage for SNP k and K is the total number of SNPs included in the PRS. This form of PRS assumes a log-additive genetic model for individual SNPs, which was considered as appropriate in previous PRS development (5–10). To find an optimal PRS, we need to determine which SNPs among all genome-wide variants should be included in the PRS according to association test results from the training dataset. We used a modified version of the model selection strategy outlined by Mavaddat and colleagues (5), which used a ‘hard-thresholding’ forward stepwise logistic regression. First, we performed single SNP-based association tests using multivariable logistic regression in the training set, adjusting for age, consortium/study and the top 10 PCs of genotype data. The per-allele log-ORs estimated in the single SNP-based analyses are called as ‘marginal’ effect sizes. We estimated the association for each of the three phenotypes (overall, ER-positive and ER-negative breast cancer) in parallel. The model development was also separately for each phenotype. In the ‘hard-thresholding’ approach, we selected SNPs in three steps. In step 1, we split each chromosome into 5 Mb bins and sorted SNPs by P-value within each bin. To avoid collinear problem in logistic regression, we filtered SNPs based on LD such that highly correlated SNPs (LD r2 > 0.9) with larger P-values were removed. In step 2, we selected SNPs by a series of stepwise forward logistic regression in 5 Mb bin. Only SNPs passing the prespecified P-value thresholds were included in the multivariable models. The SNP with the smallest (conditional) P-value was added sequentially to the model until no further SNPs could be added. We set P-value thresholds to be 10−5, 10−4, 10−3, 10−2, 0.05 and 0.1. In step 3, bins of the same chromosome were combined. SNPs on the boundary of two bins (2 Mb boundary) were filtered using LD and stepwise logistic regressions as described in steps 1 and 2. Finally, marginal beta coefficients for all selected SNPs across the genome were compiled together to calculate a PRS according to Eq. (1). We labeled this PRS as PRSAFR. For a high P-value threshold (e.g. 0.05), there are many (uncorrelated) SNPs on one chromosome and our sample size is limited, so the logistic model, including all SNPs, cannot be fit reliably.
The 313-variant PRS using effect sizes from European ancestry population (PRSEUR)
The 313-variant PRS was developed previously using data of European ancestry (5). Although its performance in AA populations is not optimal, it still offers moderate discriminatory ability (14). Therefore, we directly applied the weights (beta coefficients) from the 313-variant PRS in the validation set. Of the 313 variants, 6 variants were removed because of low MAF or imputation score, and the remaining 307 variants are shown in Supplementary Material, Table S2. Here, we use PRSEUR, PRSEUR.ERp and PRSEUR.ERn to denote the PRSs for overall, ER-positive and ER-negative phenotypes, respectively, where subscript ‘EUR’ indicates the weights are from European ancestry population.
Joint and hybrid PRS models
To improve risk prediction in diverse populations, Márquez-Luna et al. (15) proposed a multiethnic PRS method. The method combines PRS based on European training data with PRS based on training data from the target population (such as African Americans). Márquez-Luna and colleagues showed that the derived multiethnic PRS significantly improve prediction accuracy in the target population and is robust to overfitting (15). Here, we adapted this method to construct a joint PRS as a weighted linear combination of two PRSs:
PRSJoint = α1 PRSAFR + α2 PRSEUR, (2)
where PRSAFR and PRSEUR are PRSs described before, and the weights α1 and α2 are estimated in the validation set using a logistic regression model, including PRSAFR and PRSEUR as predictors, and adjusting for age, consortium/study and 10 PCs of genotypes. If we let α1 + α2 = 1, the weights represent the proportional contribution of the two PRSs on the joint PRS.
As the prediction accuracy of the joint PRS for overall breast cancer was relatively low compared with that of the joint PRS for ER-positive and very close to that of the joint PRS for ER-negative breast cancer, we also developed a hybrid PRS as a linear combination of the joint PRSs for ER-positive and for ER-negative breast cancer: PRShybrid = η PRSJoint.ERp + (1 − η) PRSJoint.ERn, where η = 0.62 was the proportion of ER-positive cases in our study samples.
Model evaluation in the validation set
For each PRS model described before, we evaluated its performance in the validation set. As the measure of the discriminating accuracy of a PRS, we calculated adjusted AUC using covariate-adjusted ROC regression (16) in which age, consortium and the top 10 PCs were adjusted for. The adjusted AUC quantifies the pure discriminating accuracy of a PRS without confounding from other covariates. In the evaluation of joint PRSs, we calculated liability scale-adjusted R2 (42), which roughly corrects for overfitting problem from estimating the contributing weights α1 and α2 in the validation set.
To estimate the strength of association, we fit multivariable logistic regression models and calculated OR and 95% CI per unit SD of PRS, adjusting for age, consortium and the top 10 PCs. We also categorized PRSs by percentile (<5, 5–10, 10–20, 20–40, 40–60, 60–80, 80–90, 90–95, > 95%) in controls and calculated adjusted OR for each category with 40–60% as the reference group. All analyses were done for overall, ER-positive and ER-negative breast cancer separately.
We examined whether age or first-degree family history of breast cancer modified the association between PRS and breast cancer risk by adding interaction terms in logistic regression models. We further examined whether the effect of PRS varied between Africans and African Americans/African Barbadians, between groups defined by African ancestry percentage (<80 vs. >80%) and between the five consortia/studies.
Calculation of absolute risks
We calculated the lifetime and 10-year absolute risks of developing breast cancer (overall and subtype-specific disease) based on population incidence rates and relative risk estimates for different PRS categories after taking into account the competing risk of dying from causes other than breast cancer, as described previously (6). The theoretical ORs for women in different PRS categories versus women in the 40th–60th percentiles were calculated using the method of Wen et al. (9) in which PRS was modeled as continuous predictor of breast cancer risk. Other inputs included age-specific breast cancer incidence rates in African Americans from Surveillance, Epidemiology and End Results (SEER, 2000–2017) (43) and the non-breast cancer mortality rates from Centers for Disease Control and Prevention (1999–2018) in the United States (44). Similarly, we calculated absolute risk of ER-positive and ER-negative breast cancer using subtype-specific incidence rates from SEER (43) and without accounting for the competing risk of other subtype. As a contrast, we also calculated the lifetime and 10-year absolute risks of developing breast cancer (overall and subtype-specific disease) for European Americans using existing PRS model in women of European ancestry (5) and breast cancer incidence rates in European Americans (43). Further details are provided in the Supplemental Material and Methods.
We conducted the analyses using R v.3.6.0 and Stata v.16. All tests of statistical significance were two-sided.
Supplementary Material
Acknowledgements
The ROOT investigators were supported by National Cancer Institute grants R01-CA228198, R01-CA142996, R01-CA89085 and P20-CA233307. D.H. and O.I.O. were also supported by Breast Cancer Research Foundation (BCRF-21-071). D.H. and G.G. were also partially supported by the National Cancer Institute (R03-CA227357 and R01-CA242929). K.L.N. and S.M.D. were supported by Basser Center for BRCA. F.Z. was supported by the Susan G. Komen Foundation (TREND21675016).
AABC was supported by a Department of Defense Breast Cancer Research Program Era of Hope Scholar Award to CAH (W81XWH-08-1-0383) and the Norris Foundation. Each of the participating AABC studies was supported by the following grants: MEC (National Institutes of Health grants R01-CA63464 and R37-CA54281); CARE (National Institute for Child Health and Development grant NO1-HD-3-3175, K05 CA136967); WCHS (US Army Medical Research and Material Command grant DAMD-17-01-0-0334, the National Institutes of Health grant R01-CA100598 and the Breast Cancer Research Foundation); SFBCS (National Institutes of Health grant R01-CA077305 and United States Army Medical Research Program grant DAMD17-96-6071); NC-BCFR (National Institutes of Health grant U01-CA069417); CBCS (National Institutes of Health Specialized Program of Research Excellence in Breast Cancer, grant number P50-CA58223, and Center for Environmental Health and Susceptibility National Institute of Environmental Health Sciences, National Institutes of Health, grant number P30-ES10126); PLCO (Intramural Research Program, National Cancer Institute, National Institutes of Health); NBHS (National Institutes of Health grant R01-CA100374). The Breast Cancer Family Registry (BCFR) was supported by the National Cancer Institute, National Institutes of Health under RFA-CA-06-503 and through cooperative agreements with members of the BCFR and Principal Investigators. The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the BCFR, nor does mention of trade names, commercial products or organizations imply endorsement by the U.S. Government or the BCFR. M.F.P was supported by Breast Cancer Research Foundation, Tower Cancer Research Foundation and a gift from Dr Richard Balch.
AMBER was supported by the National Cancer Institute grants P01-CA151135, R01-CA098663, R01-CA058420, UM1-CA164974, R01-CA100598, P50-CA58223, R01CA202981, U01-CA164974, R01-CA228357 and the University Cancer Research Fund of North Carolina. J.R.P. was supported by the Susan G. Komen Foundation and the Karin Grunebaum Foundation. Pathology data were obtained from numerous state cancer registries (AZ, CA, CO, CT, DE, DC, FL, GA, HI, IL, IN, KY, LA, MD, MA, MI, NJ, NY, NC, OK, PA, SC, TN, TX and VA). For the studies included in AMBER, individual protocols were approved by the relevant IRBs and by the IRBs of the participating cancer registries as required. The results reported do not necessarily represent the views of the National Institutes of Health or the state cancer registries.
BCAC is funded by Cancer Research UK (C1287/A16563, C1287/A10118), the European Union’s Horizon 2020 Research and Innovation Programme (grant numbers 634935 and 633784 for BRIDGES and B-CAST, respectively) and by the European Community’s Seventh Framework Programme under grant agreement number 223175 (grant number HEALTH-F2-2009-223175) (COGS). The EU Horizon 2020 Research and Innovation Programme funding source had no role in study design, data collection, data analysis, data interpretation or writing of the report. The Sister Study was funded by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences (Z01-ES044005).
GBHS authors acknowledge the research contributions of the Cancer Genomics Research Laboratory for their expertise, execution and support of this research in the areas of project planning, wet laboratory processing of specimens and bioinformatics analysis of generated data. This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under NCI Contract No. 75N910D00024. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services nor does mention of trade names, commercial products or organizations imply endorsement by the US Government. The success of this investigation would not have been possible without exceptional teamwork and the diligence of the field staff who oversaw the recruitment, interviews and collection of data from study participants. Special thanks are entitled to the following individuals: Korle Bu Teaching Hospital, Accra—Dr Adu-Aryee, Obed Ekpedzor, Angela Kenu, Victoria Okyne, Naomi Oyoe Ohene Oti and Evelyn Tay; Komfo Anoyke Teaching Hospital, Kumasi— Marion Alcpaloo, Bernard Arhin, Emmanuel Asiamah, Isaac Boakye and Samuel Ka-chungu; and Peace and Love Hospital, Kumasi—Samuel Amanama, Emma Abaidoo, Prince Agyapong, Thomas Agyei, Debora Boateng-Ansong, Margaret Frempong, Bridget Nortey Mensah, Richard Opoku and Kofi Owusu Gyimah. The study was further enhanced by surgical expertise provided by Dr Lisa Newman of the University of Michigan and by pathological expertise provided by Drs Stephen Hewitt and Petra Lenz of the National Cancer Institute and Dr Maire A. Duggan from the Cumming School of Medicine, University of Calgary, Canada. Study management assistance was received from Ricardo Diaz, Shelley Niwa and Usha Singh. Appreciation is also expressed to the many women who agreed to participate in the study and to provide information and biospecimens in hopes of preventing and improving outcomes of breast cancer in Ghana.
Conflict of Interest statement. None declared.
Contributor Information
Guimin Gao, Department of Public Health Sciences, The University of Chicago, Chicago, IL 60637, USA.
Fangyuan Zhao, Department of Public Health Sciences, The University of Chicago, Chicago, IL 60637, USA.
Thomas U Ahearn, Division of Cancer Epidemiology and Genetics, National Cancer Institute, National Institutes of Health, Bethesda, MD 20850, USA.
Kathryn L Lunetta, Department of Biostatistics, Boston University School of Public Health, Boston, MA 02118, USA.
Melissa A Troester, Department of Epidemiology, Gillings School of Global Public Health, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Zhaohui Du, Public Health Sciences Division, Fred Hutchinson Cancer Research Center, Seattle, WA 98109, USA.
Temidayo O Ogundiran, Department of Surgery, College of Medicine, University of Ibadan, Ibadan, Nigeria.
Oladosu Ojengbede, Centre for Population & Reproductive Health, College of Medicine, University of Ibadan, Ibadan, Nigeria.
William Blot, Division of Epidemiology, Department of Medicine, Vanderbilt Epidemiology Center, Vanderbilt-Ingram Cancer Center, Vanderbilt University School of Medicine, Nashville, TN 37232, USA.
Katherine L Nathanson, Department of Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA.
Susan M Domchek, Department of Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA.
Barbara Nemesure, Department of Family, Population and Preventive Medicine, Stony Brook University, Stony Brook, NY 11794, USA.
Anselm Hennis, Department of Family, Population and Preventive Medicine, Stony Brook University, Stony Brook, NY 11794, USA; University of the West Indies, Bridgetown, Bardados.
Stefan Ambs, Laboratory of Human Carcinogenesis, National Cancer Institute, Bethesda, MD 20892, USA.
Julian McClellan, Department of Public Health Sciences, The University of Chicago, Chicago, IL 60637, USA.
Mark Nie, Department of Public Health Sciences, The University of Chicago, Chicago, IL 60637, USA.
Kimberly Bertrand, Slone Epidemiology Center, Boston University, Boston, MA 02215, USA.
Gary Zirpoli, Slone Epidemiology Center, Boston University, Boston, MA 02215, USA.
Song Yao, Department of Cancer Prevention and Control, Roswell Park Comprehensive Cancer Center, Buffalo, NY 14203, USA.
Andrew F Olshan, Department of Epidemiology, Gillings School of Global Public Health, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Jeannette T Bensen, Department of Epidemiology, Gillings School of Global Public Health, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Elisa V Bandera, Cancer Prevention and Control Program, Rutgers Cancer Institute of New Jersey, New Brunswick, NJ 08903, USA.
Sarah Nyante, Department of Radiology, School of Medicine, University of North Carolina at Chapel Hill, Chapel Hill, NC 27514, USA.
David V Conti, Department of Preventive Medicine, Keck School of Medicine, University of Southern California, Los Angeles, CA 90033, USA.
Michael F Press, Department of Pathology, Keck School of Medicine, University of Southern California, Los Angeles, CA 90033, USA.
Sue A Ingles, Department of Preventive Medicine, Keck School of Medicine, University of Southern California, Los Angeles, CA 90033, USA.
Esther M John, Departments of Epidemiology & Population Health and of Medicine (Oncology) and Stanford Cancer Institute, Stanford University School of Medicine, Stanford, CA 94304, USA.
Leslie Bernstein, Biomarkers of Early Detection and Prevention, Department of Population Sciences, Beckman Research Institute, City of Hope Comprehensive Cancer Center, Duarte, CA 91010, USA.
Jennifer J Hu, Department of Public Health Sciences, University of Miami Miller School of Medicine, Miami, FL 33136, USA.
Sandra L Deming-Halverson, Division of Epidemiology, Department of Medicine, Vanderbilt Epidemiology Center, Vanderbilt-Ingram Cancer Center, Vanderbilt University School of Medicine, Nashville, TN 37232, USA.
Stephen J Chanock, Division of Cancer Epidemiology and Genetics, National Cancer Institute, National Institutes of Health, Bethesda, MD 20850, USA.
Regina G Ziegler, Division of Cancer Epidemiology and Genetics, National Cancer Institute, National Institutes of Health, Bethesda, MD 20850, USA.
Jorge L Rodriguez-Gil, Genomics, Development and Disease Section, Genetic Disease Research Branch, National Human Genome Research Institute, Bethesda, MD 20894, USA.
Lara E Sucheston-Campbell, Department of Veterinary Biosciences, College of Veterinary Medicine, The Ohio State University, Columbus, OH 43210, USA.
Dale P Sandler, Epidemiology Branch, National Institute of Environmental Health Sciences, National Institutes of Health, Research Triangle Park, NC 27709, USA.
Jack A Taylor, Epidemiology Branch, National Institute of Environmental Health Sciences, National Institutes of Health, Research Triangle Park, NC 27709, USA.
Cari M Kitahara, Radiation Epidemiology Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, National Institutes of Health, Bethesda, MD 20892, USA.
Katie M O’Brien, Epidemiology Branch, National Institute of Environmental Health Sciences, National Institutes of Health, Research Triangle Park, NC 27709, USA.
Manjeet K Bolla, Department of Public Health and Primary Care, Centre for Cancer Genetic Epidemiology, University of Cambridge, Cambridge CB1 8RN, UK.
Joe Dennis, Department of Public Health and Primary Care, Centre for Cancer Genetic Epidemiology, University of Cambridge, Cambridge CB1 8RN, UK.
Alison M Dunning, Department of Oncology, Centre for Cancer Genetic Epidemiology, University of Cambridge, Cambridge CB1 8RN, UK.
Douglas F Easton, Department of Public Health and Primary Care, Centre for Cancer Genetic Epidemiology, University of Cambridge, Cambridge CB1 8RN, UK; Department of Oncology, Centre for Cancer Genetic Epidemiology, University of Cambridge, Cambridge CB1 8RN, UK.
Kyriaki Michailidou, Biostatistics Unit, The Cyprus Institute of Neurology & Genetics, Nicosia 2371, Cyprus.
Paul D P Pharoah, Department of Public Health and Primary Care, Centre for Cancer Genetic Epidemiology, University of Cambridge, Cambridge CB1 8RN, UK; Department of Oncology, Centre for Cancer Genetic Epidemiology, University of Cambridge, Cambridge CB1 8RN, UK.
Qin Wang, Department of Public Health and Primary Care, Centre for Cancer Genetic Epidemiology, University of Cambridge, Cambridge CB1 8RN, UK.
Jonine Figueroa, Usher Institute of Population Health Sciences and Informatics, The University of Edinburgh Medical School, Edinburgh EH16 5TJ, UK; Cancer Research UK Edinburgh Centre, Edinburgh EH4 2XR, UK.
Richard Biritwum, University of Ghana, Accra, Ghana.
Ernest Adjei, Komfo Anokye Teaching Hospital, Kumasi, Ghana.
Seth Wiafe, School of Public Health, Loma Linda University, Loma Linda, CA 92350, USA.
Christine B Ambrosone, Department of Cancer Prevention and Control, Roswell Park Comprehensive Cancer Center, Buffalo, NY 14203, USA.
Wei Zheng, Division of Epidemiology, Department of Medicine, Vanderbilt Epidemiology Center, Vanderbilt-Ingram Cancer Center, Vanderbilt University School of Medicine, Nashville, TN 37232, USA.
Olufunmilayo I Olopade, Center for Clinical Cancer Genetics & Global Health, The University of Chicago, Chicago, IL 60637, USA.
Montserrat García-Closas, Division of Cancer Epidemiology and Genetics, National Cancer Institute, National Institutes of Health, Bethesda, MD 20850, USA.
Julie R Palmer, Slone Epidemiology Center, Boston University, Boston, MA 02215, USA.
Christopher A Haiman, Department of Preventive Medicine, Keck School of Medicine, University of Southern California, Los Angeles, CA 90033, USA.
Dezheng Huo, Department of Public Health Sciences, The University of Chicago, Chicago, IL 60637, USA; Center for Clinical Cancer Genetics & Global Health, The University of Chicago, Chicago, IL 60637, USA.
References
- 1. Lilyquist, J., Ruddy, K.J., Vachon, C.M. and Couch, F.J. (2018) Common genetic variation and breast cancer risk-past, present, and future. Cancer Epidemiol. Biomark. Prev., 27(4), 380–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shu, X., Long, J., Cai, Q., Kweon, S.S., Choi, J.Y., Kubo, M., Park, S. K. Bolla, M. K. Dennis, J. Wang, Q. et al. (2020) Identification of novel breast cancer susceptibility loci in meta-analyses conducted among Asian and European descendants. Nat. Commun., 11(1), 1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhang, H., Ahearn, T.U., Lecarpentier, J., Barnes, D., Beesley, J., Qi, G., Jiang, X. O’Mara, T.A. Zhao, N. Bolla, M.K. et al. (2020) Genome-wide association study identifies 32 novel breast cancer susceptibility loci from overall and subtype-specific analyses. Nat. Genet., 52(6), 572–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Amos, C.I., Dennis, J., Wang, Z., Byun, J., Schumacher, F.R., Gayther, S.A., Casey, G. Hunter, D.J. Sellers. T.A. Gruber, S.B. Dunning, A.M. et al. (2017) The OncoArray Consortium: a network for understanding the genetic architecture of common cancers. Cancer Epidemiol. Biomark. Prev., 26(1), 126–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mavaddat, N., Michailidou, K., Dennis, J., Lush, M., Fachal, L., Lee, A., Tyrer, J.P. Chen, T.H. Wang, Q. Bolla, M.K. et al. (2019) Polygenic risk scores for prediction of breast cancer and breast cancer subtypes. Am. J. Hum. Genet., 104(1), 21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mavaddat, N., Pharoah, P.D., Michailidou, K., Tyrer, J., Brook, M.N., Bolla, M.K., Wang, Q Dennis, J. Dunning, A.M. Shah, M. et al. (2015) Prediction of breast cancer risk based on profiling with common genetic variants. J. Natl. Cancer Inst., 107(5), djv036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pharoah, P.D., Antoniou, A.C., Easton, D.F. and Ponder, B.A. (2008) Polygenes, risk prediction, and targeted prevention of breast cancer. N. Engl. J. Med., 358(26), 2796–2803. [DOI] [PubMed] [Google Scholar]
- 8. Wacholder, S., Hartge, P., Prentice, R., Garcia-Closas, M., Feigelson, H.S., Diver, W.R., Thun, M.J., Cox, D.G., Hankinson, S.E., Kraft, P. et al. (2010) Performance of common genetic variants in breast-cancer risk models. N. Engl. J. Med., 362(11), 986–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wen, W., Shu, X.O., Guo, X., Cai, Q., Long, J., Bolla, M.K., Michailidou, K., Dennis, J., Wang, Q., Gao, Y.T. et al. (2016) Prediction of breast cancer risk based on common genetic variants in women of East Asian ancestry. Breast Cancer Res., 18(1), 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shieh, Y., Fejerman, L., Lott, P.C., Marker, K., Sawyer, S.D., Hu, D., Huntsman, S., Torres, J., Echeverry, M., Bohorquez, M.E. et al. (2020) A polygenic risk score for breast cancer in US Latinas and Latin American women. J. Natl. Cancer Inst., 112(6), 590–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. DeSantis, C.E., Ma, J., Gaudet, M.M., Newman, L.A., Miller, K.D., Goding Sauer, A., Jemal, A. and Siegel, R.L. (2019) Breast cancer statistics, 2019. CA Cancer J. Clin., 69(6), 438–451. [DOI] [PubMed] [Google Scholar]
- 12. Allman, R., Dite, G.S., Hopper, J.L., Gordon, O., Starlard-Davenport, A., Chlebowski, R. and Kooperberg, C. (2015) SNPs and breast cancer risk prediction for African American and Hispanic women. Breast Cancer Res. Treat., 154(3), 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang, S., Qian, F., Zheng, Y., Ogundiran, T., Ojengbede, O., Zheng, W., Blot, W., Nathanson, K.L., Hennis, A., Nemesure, B. et al. (2018) Genetic variants demonstrating flip-flop phenomenon and breast cancer risk prediction among women of African ancestry. Breast Cancer Res. Treat., 168(3), 703–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Du, Z., Gao, G., Adedokun, B., Ahearn, T., Lunetta, K.L., Zirpoli, G., Troester, M.A., Ruiz-Narvaez, E.A., Haddad, S.A., Pal Choudhury, P. et al. (2021) Evaluating polygenic risk scores for breast cancer in women of African ancestry. J. Natl. Cancer Inst., 113(9), 1168–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Márquez-Luna, C., Loh, P.R., South Asian Type 2 Diabetes C, Consortium STD and Price, A.L. (2017) Multiethnic polygenic risk scores improve risk prediction in diverse populations. Genet. Epidemiol., 41(8), 811–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Janes, H. and Pepe, M.S. (2009) Adjusting for covariate effects on classification accuracy using the covariate-adjusted receiver operating characteristic curve. Biometrika, 96(2), 371–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Milne, R.L., Kuchenbaecker, K.B., Michailidou, K., Beesley, J., Kar, S., Lindstrom, S., Hui, S., Lemacon, A., Soucy, P., Dennis, J. et al. (2017) Identification of ten variants associated with risk of estrogen-receptor-negative breast cancer. Nat. Genet., 49(12), 1767–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huo, D., Feng, Y., Haddad, S., Zheng, Y., Yao, S., Han, Y.J., Ogundiran, T.O., Adebamowo, C., Ojengbede, O., Falusi, A.G. et al. (2016) Genome-wide association studies in women of African ancestry identified 3q26.21 as a novel susceptibility locus for oestrogen receptor negative breast cancer. Hum. Mol. Genet., 25(21), 4835–4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gierach, G.L., Yang, X.R., Figueroa, J.D. and Sherman, M.E. (2013) Emerging concepts in breast cancer risk prediction. Curr Obstet Gynecol Rep., 2(1), 43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ho, W.K., Tan, M.M., Mavaddat, N., Tai, M.C., Mariapun, S., Li, J., Ho, P.J., Dennis, J., Tyrer, J.P., Bolla, M.K. et al. (2020) European polygenic risk score for prediction of breast cancer shows similar performance in Asian women. Nat. Commun., 11(1), 3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Martin, A.R., Kanai, M., Kamatani, Y., Okada, Y., Neale, B.M. and Daly, M.J. (2019) Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet., 51(4), 584–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Scutari, M., Mackay, I. and Balding, D. (2016) Using genetic distance to infer the accuracy of genomic prediction. PLoS Genet., 12(9), e1006288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Martin, A.R., Gignoux, C.R., Walters, R.K., Wojcik, G.L., Neale, B.M., Gravel, S.et al. (2017) Human demographic history impacts genetic risk prediction across diverse populations. Am. J. Hum. Genet., 100(4), 635–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vilhjalmsson, B.J., Yang, J., Finucane, H.K., Gusev, A., Lindstrom, S., Ripke, S., Genovese, G., Loh, P.R., Bhatia, G., Do, R. et al. (2015) Modeling linkage disequilibrium increases accuracy of polygenic risk scores. Am. J. Hum. Genet., 97(4), 576–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hu, Y., Lu, Q., Powles, R., Yao, X., Yang, C., Fang, F., Xu, X. and Zhao, H. (2017) Leveraging functional annotations in genetic risk prediction for human complex diseases. PLoS Comput. Biol., 13(6), e1005589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lloyd-Jones, L.R., Zeng, J., Sidorenko, J., Yengo, L., Moser, G., Kemper, K.E., Wang, H., Zheng, Z., Magi, R., Esko, T. et al. (2019) Improved polygenic prediction by Bayesian multiple regression on summary statistics. Nat. Commun., 10(1), 5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fejerman, L., John, E.M., Huntsman, S., Beckman, K., Choudhry, S., Perez-Stable, E., Burchard, E.G. and Ziv, E. (2008) Genetic ancestry and risk of breast cancer among U.S. Latinas. Cancer Res., 68(23), 9723–9728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Guan, Y. (2014) Detecting structure of haplotypes and local ancestry. Genetics, 196(3), 625–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen, W., Ren, C., Qin, H., Archer, K.J., Ouyang, W., Liu, N., Chen, X., Luo, X., Zhu, X., Sun, S. et al. (2015) A generalized sequential Bonferroni procedure for GWAS in admixed populations incorporating admixture mapping information into association tests. Hum. Hered., 79(2), 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Atkinson, E.G., Maihofer, A.X., Kanai, M., Martin, A.R., Karczewski, K.J., Santoro, M.L., Ulirsch, J.C., Kamatani, Y., Okada, Y., Finucane, H.K. et al. (2021) Tractor uses local ancestry to enable the inclusion of admixed individuals in GWAS and to boost power. Nat. Genet., 53(2), 195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Oeffinger, K.C., Fontham, E.T., Etzioni, R., Herzig, A., Michaelson, J.S., Shih, Y.C., Walter, L.C., Church, T.R., Flowers, C.R., LaMonte, S.J. et al. (2015) Breast cancer screening for women at average risk: 2015 guideline update from the American Cancer Society. JAMA, 314(15), 1599–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen, F., Chen, G.K., Stram, D.O., Millikan, R.C., Ambrosone, C.B., John, E.M., Bernstein, L., Zheng, W., Palmer, J.R., Hu, J.J. et al. (2013) A genome-wide association study of breast cancer in women of African ancestry. Hum. Genet., 132(1), 39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Palmer, J.R., Ambrosone, C.B. and Olshan, A.F. (2014) A collaborative study of the etiology of breast cancer subtypes in African American women: the AMBER consortium. Cancer Causes Control, 25(3), 309–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brinton, L.A., Awuah, B., Nat Clegg-Lamptey, J., Wiafe-Addai, B., Ansong, D., Nyarko, K.M., Wiafe, S., Yarney, J., Biritwum, R., Brotzman, M. et al. (2017) Design considerations for identifying breast cancer risk factors in a population-based study in Africa. Int. J. Cancer, 140(12), 2667–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nyante, S.J., Biritwum, R., Figueroa, J., Graubard, B., Awuah, B., Addai, B.W., Yarney, J., Clegg-Lamptey, J.N., Ansong, D., Nyarko, K. et al. (2019) Recruiting population controls for case-control studies in sub-Saharan Africa: the Ghana Breast Health Study. PLoS One, 14(4), e0215347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Feng, Y., Rhie, S.K., Huo, D., Ruiz-Narvaez, E.A., Haddad, S.A., Ambrosone, C.B., John, E.M., Bernstein, L., Zheng, W., Hu, J.J. et al. (2017) Characterizing genetic susceptibility to breast cancer in women of African ancestry. Cancer Epidemiol. Biomark. Prev., 26(7), 1016–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Feng, Y., Stram, D.O., Rhie, S.K., Millikan, R.C., Ambrosone, C.B., John, E.M., Bernstein, L., Zheng, W., Olshan, A.F., Hu, J.J. et al. (2014) A comprehensive examination of breast cancer risk loci in African American women. Hum. Mol. Genet., 23(20), 5518–5526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Michailidou, K., Lindstrom, S., Dennis, J., Beesley, J., Hui, S., Kar, S., Lemacon, A., Soucy, P., Glubb, D., Rostamianfar, A. et al. (2017) Association analysis identifies 65 new breast cancer risk loci. Nature, 551(7678), 92–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. The 1000 Genomes Project Consortium, Auton, A., Abecasis, G.R., Altshuler, D.M., Durbin, R.M., Bentley, D.R., Chakravarti, A., Clark, A.G., Donnelly, P., Chakravarti, A.et al. (2015) A global reference for human genetic variation. Nature, 526, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Patterson, N., Price, A.L. and Reich, D. (2006) Population structure and eigen analysis. PLoS Genet., 2(12), e190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Price, A.L., Patterson, N.J., Plenge, R.M., Weinblatt, M.E., Shadick, N.A. and Reich, D. (2006) Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet., 38(8), 904–909. [DOI] [PubMed] [Google Scholar]
- 42. Lee, S.H., Goddard, M.E., Wray, N.R. and Visscher, P.M. (2012) A better coefficient of determination for genetic profile analysis. Genet. Epidemiol., 36(3), 214–224. [DOI] [PubMed] [Google Scholar]
- 43. Surveillance, Epidemiology, and End Results (SEER) Program (www.seer.cancer.gov). SEER*Stat Database: Incidence - SEER Research Data (2000-2017), National Cancer Institute, DCCPS, Surveillance Research Program, released April 2020, based on the November 2019 submission.
- 44. Underlying Cause of Death 1999-2018 on CDC WONDER Online Database, released in 2020 [Internet]. [cited Nov 2, 2020]. Available from: http://wonder.cdc.gov/ucd-icd10.html.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.