

Abstract
Many neurodevelopmental disorders (NDDs) are the result of mutations on the X chromosome. One severe NDD resulting from mutations on the X chromosome is CDKL5 deficiency disorder (CDD). CDD is an epigenetic, X-linked NDD characterized by intellectual disability (ID), pervasive seizures and severe sleep disruption, including recurring hospitalizations. CDD occurs at a 4:1 ratio, with a female bias. CDD is driven by the loss of cyclin-dependent kinase-like 5 (CDKL5), a serine/threonine kinase that is essential for typical brain development, synapse formation and signal transmission. Previous studies focused on male subjects from animal models, likely to avoid the complexity of X mosaicism. For the first time, we report translationally relevant behavioral phenotypes in young adult (8–20 weeks) females and males with robust signal size, including impairments in learning and memory, substantial hyperactivity and increased susceptibility to seizures/reduced seizure thresholds, in both sexes, and in two models of CDD preclinical mice, one with a general loss-of-function mutation and one that is a patient-derived mutation.
Introduction
Numerous genetic factors have been shown to confer risk for neurodevelopmental disorders (NDDs), including autism spectrum disorder (ASD) and intellectual disabilities (IDs). Many NDDs are the result of mutations on the X chromosome, as it accounts for ~1000 more gene products than its ancestral homologue, the Y chromosome (1–4). One NDD resulting from a mutation on the X chromosome is CDKL5 deficiency disorder (CDD) (5–7), a monogenic, severe NDD characterized by developmental delays, IDs, seizures and treatment-resistant epilepsy and sleep disruption (8–12). CDD is driven by the loss of cyclin-dependent kinase-like 5 (CDKL5) which encodes a serine/threonine kinase that is essential for normal brain development (5,13), synapse formation and neuronal signal transmission. CDD is 4:1 in females, as they are missing one CDKL5 with the other X allele remaining intact (2X alleles in XX) whereas males are ultra-rare and more severe missing the one and only CDKL5 (1X allele in XY). CDD now has a uniform consensus as a unique NDD, distinct from Rett or Angelman syndromes and/or ASD, as it was previously classified as an atypical form of Rett syndrome; however, CDD’s seizure profile is comorbid with ID, ASD and other problems with development. CDD was previously classified as the early-onset-seizure variant of Rett syndrome as a catalogue of CDKL5 sequence variations, including pathogenic mutations, nonpathogenic polymorphisms and sequence variations of uncertain significance can be found at the RettBASE website (http://mecp2.chw.edu.au). Its discrete diagnosis is warranted, as one study of CDD (13) demonstrated that less than a quarter of those affected met clinical criteria for early-onset Rett syndrome. Finally, we now have an understanding of CDD’s distinct genetic cause as from those of Rett syndrome.
Unfortunately, with respect to CDD, there are no approved therapies or treatments, and standard of care is not effective at managing seizures or preventing neurodevelopmental delays nor failure to make gains in motor and cognitive milestones. To examine the specific and functional loss of CDKL5, mouse models were generated to investigate and discover the functional effects of Cdkl5 deficiency and to evaluate potential therapeutics. The first model from Zhou’s laboratory deleted exon 6 and observed substantial behavioral alterations that caused hyperactivity, abnormal social approach and reduced contextual fear conditioning (14) in male subjects. In 2019, a novel mouse line was created that used a common patient variant R59X to create a patient-specific knock-in model line. This novel model, also only tested in males, reported hyperactivity, reduced contextual fear conditioning, unusual motor coordination and less directed social behavior (15). Laboratories are continuing to pursue the cell-type specificity of these phenotypes and myoclonic events in aged mice (16–18). Neither of these mouse models exhibited spontaneous behavioral seizures when evaluated as young adults, which is not unreasonable, given there has been a failure to detect this key phenotype on the C57BL/6J background strain of mice for other NDDs and seizure disorder models, such as Angelman Syndrome (19). C57BL/6J mice have been repeatedly illustrated to be seizure resistant, published by our group (20) and others (19,21–37).
Given the latest advances in gene editing technology, CDKL5, MECP2, CNKSR2 and CASK deletion-related disorders have been of emerging interest to translational researchers. In X-linked disorders, mutations are usually de novo (not inherited) and female patients are the affected population because as heterozygotes they have both a healthy allele and a mutant allele within their XX genotype (38). Embryonic lethality is likely high in males. Females are mosaics, having a mixture of cells expressing either their mother’s or father’s X-linked genes (39–41). Often, cell mosaicism is advantageous, ameliorating the deleterious effects of X-linked mutations and contributing to physiological diversity. As a consequence, most X-linked mutations produce male-only diseases. Yet, in some cases, the dynamic interaction between cells in mosaic females lead to female-specific disease manifestations (41). As a result of random X chromosome inactivation (XCI), these patients end up with ~50% of their cells expressing the mutant allele, whereas the present healthy allele is epigenetically silenced. Multiple studies of XCI have revealed that genes expressed from the active X chromosome have a unique epigenetic signature when compared with the inactive X chromosome (42). More interestingly, genes that ‘escape’ XCI and express genes from the inactive chromosome resemble the epigenetics of the active allele with a signature that is defined by the presence of active histones and the absence of promoter methylation. Recently, our group developed a strategy to recruit epigenetic modifiers to the promoter of genes on the inactive X chromosome, resulting in increased transcription and reactivation of Cdkl5 (43). Corroborative findings using a conditional mouse model that restored Cdkl5 after the early stages of brain development highlighted the potential for disease reversal in CDD and suggested a broad therapeutic time window (44).
In addition to epigenetic editing (45,46), other novel precision-based therapeutics that target the underlying genetic loss of CDKL5/Cdkl5 via cutting-edge technologies are being investigated including conditional genetic rescue using tamoxifen inducible technology (44), deep brain stimulation (47), Cdkl5 protein substitution therapy (48), AAV delivered gene replacement therapy of CDKL5/Cdkl5 (49) and approaches that combine gene editing strategies, such as treatment with small-molecule inhibitors of DNA methylation and/or an antisense oligonucleotide against X–inactive-specific transcript RNA (50). Traditional therapeutics are also being assessed, including anti-seizure medications, cannabis derivatives, neurosteroids, anti-neuroinflammatries GSK-3beta/HDAC6 inhibitors and sodium channel blockers (51–55).
To assess the efficacy of these novel therapeutics, our laboratory has been focused on identifying rigorous, reproducible, objective, translationally relevant phenotypes or outcome measures. We have tailored focus on outcome indices in mice and rats that are translatable to the clinic and include a range of behavioral domains, including motor, learning and memory, seizures, neuroanatomy by magnetic resonance imaging, electroencephalographic (EEG) signatures during seizures, sensorimotor tasks, and sleep. We also evaluate the correlation of these metrics with various timepoints across the lifespan, from development to age-related declines (20,56–60). Our strategy has been reported and validated using models of Angelman and Phelan McDermid syndromes and an Scn1a-regulator model of Dravet syndrome/epilepsy (30,57,60,61).
This report is the first description of translationally relevant phenotypes in young adult (8–16 week) mice of both sexes, using two etiologically distinct preclinical CDD models. Observations included robust impairments in touchscreen pairwise discrimination and spatial learning and memory, hyperactivity and increased susceptibility to seizures. Findings are critical as they can be utilized as outcome measure indices for a wide variety of novel gene editing and small molecule therapeutics that are currently in the pipeline as potential interventions for CDD.
Results
Given that 90% of those affected by CDD are females (53,62) and that the majority of CDD mouse model research has focused on male mice, we performed an in-depth translationally relevant behavioral and physiological comparison of two different lines that lack functional CDKL5 in both sexes. Western blots confirmed lack of expression in Cdkl5exon6−/y males and about 42% reduction in expression in Cdkl5exon6−/+ females versus Cdkl5exon6+/y males and Cdkl5exon6+/+ females, respectively. Lack of expression in Cdkl5R59X−/y males and a 49% reduction of expression in Cdkl5R59X+/− females were verified in whole-brain protein lysate Western blotting (Supplementary Material, Fig. S1), corroborating what had been previously described (14,63). Volumetric analysis of brain regions associated with neurological impairments in CDD and quantification of the expression of two markers of neuroinflammation (TMEM119 and GFAP) via fluorescent immunohistochemistry on sagittal brain sections showed no phenotypes in either model (Supplementary Material, Figs. S2, S3, S4, and S5).
Motor function is highly translational and consists of many nuanced components, including gross exploration and motor abilities, balance, coordination, gait and fine motor skills. We utilized a tailored motor battery to perform a comprehensive outcome assessment that may be useful for clinical trials. Cdkl5 mutants exhibited a large main effect of genotype using total activity by a two-way ANOVA (Fig. 1A; F(3, 40) = 22.36, P < 0.0001; Fig. 1E; F(3, 40) = 18.63, P < 0.0001), exhibiting hyperactivity when compared with wild-type, sex-matched littermate controls. Holm-Sidak-corrected post hoc analysis for multiple comparisons highlighted the significant effect in Cdkl5exon6−/y males (P < 0.0001) differing from Cdkl5exon6+/y, at every 5 min time bin across the 30 min activity assay (0–5 min, P < 0.0005; 6–10 min, P < 0.0001; 11–15 min, P < 0.0001; 16–20 min, P < 0.0001; 21–25 min, P < 0.0001; and 26–30 min, P < 0.0001). In females, Holm-Sidak-corrected post hoc analysis for multiple comparisons revealed the significance, Cdkl5exon6−/+ (P = 0.0287) versus Cdkl5exon6+/+ mice, limited to the first two 5 min time bins across the 30 min activity assay (0–5 min, P = 0.0468; 6–10 min, P < 0.002). As hypothesized, males were more affected due to the total loss of the CDKL5 protein. Cdkl5R59X+/y and −/y males exhibited significance using total activity following post hoc analysis (P < 0.0001) whereas females did not pass post hoc correction analysis (P = 0.955). Cdkl5R59X−/y differed from Cdkl5R59X+/y during four of six 5 min time bins across the 30 min activity assay (0–5 min, P = 0.0764; 6–10 min, P < 0.0001; 11–15 min, P < 0.0001; 16–20 min, P < 0.0001; 21–25 min, P < 0.0001; and 26–30 min, P < 0.0001). Activity summed over the 30 min session illustrated robust Cdkl5exon6 and Cdkl5R59X main effects of genotype by one-way ANOVA (Fig. 1B; F(3, 40) = 13.33, P < 0.0001; Fig. 1F; F(3, 40) = 20.97, P < 0.0001). Thus, mice of both models exhibiting hyperactivity when compared with their wild-type, sex-matched littermate controls. Holm-Sidak-corrected post hoc analysis for multiple comparisons, highlighted significant effects in Cdkl5exon6−/y (P < 0.0001) versus Cdkl5exon6+/y males and Cdkl5exon6−/+ (P = 0.0338) versus Cdkl5exon6+/+ females. Males and females with the R59X mutation were hyperactive when analyzed by Holm-Sidak-corrected post hoc analysis for multiple comparisons in Cdkl5R59X−/y (P < 0.0001) versus Cdkl5R59X+/y males and Cdkl5R59−/+ (P = 0.0268) versus Cdkl5R59X+/+ females. Cdkl5exon6 and Cdkl5R59X exhibited a large main effect of genotype by a two-way ANOVA using horizontal activity in a novel open field (Fig. 1C; F(3, 40) = F(3, 40) = 22.15, P < 0.0001; Fig. 1G; F(3, 40) = 10.75, P < 0.0001), with deletion and knock-in mice of both models exhibiting hyperactivity compared with wild-type, sex-matched littermate controls. Holm-Sidak-corrected post hoc analysis for multiple comparisons highlighted the significant effect of Cdkl5exon6−/y males (P < 0.0001) differed from Cdkl5exon6+/y during five of the six 5 min time bins across the 30 min activity assay (0–5 min, P = 0.0512; 6–10 min, P < 0.0006; 11–15 min, P < 0.0006; 16–20 min, P < 0.0002; 21–25 min, P < 0.0172; and 26–30 min, P < 0.0001). In females, horizontal activity showed hyperactivity in the heterozygous females Cdkl5exon6−/+ as compared with wild-type (P = 0.0183). Further, when analyzed by Holm-Sidak-corrected post hoc analysis for multiple comparisons, highlights were clear in two middle bins of the six 5 min time bins across the 30 min activity assay (10–15 min, P = 0.0512; 16–20 min, P < 0.0006). The R59X model showed hyperactivity in both males (P = 0.0006) and females (P = 0.0183). Holm-Sidak-corrected post hoc analysis for multiple comparisons found mutant males were hyperactive during five of six 5 min time bins across the 30 min assay (0–5 min, P = 0.0512; 6–10 min, P < 0.0006; 11–15 min, P < 0.0006; 16–20 min, P < 0.0002; 21–25 min, P < 0.0172; and 26–30 min, P < 0.0001). In females, horizontal activity also showed hyperactivity when Cdkl5R59X−/+ mice were compared with wild-type (P = 0.0183). Furthermore, when analyzed by Holm–Sidak-corrected post hoc analysis for multiple comparisons, highlights were clear in two middle bins of the six 5 min time bins across the 30 min activity assay (10–15 min, P = 0.0069; 16–20 min, P = 0.0039).
Figure 1.

Hyperactivity and motor learning deficits in Cdkl5 deletion and knock-in mice. (A–C) Cdkl5exon6−/+ females and Cdkl5exon6−/y males exhibited increased activity by total activity over 5 min time bins, summed total activity and horizontal activity metrics. (D) Cdkl5exon6−/y males, but not Cdkl5exon6−/+ females, had significantly lower latencies to fall off the rotarod compared with wild-type, sex-matched littermate controls. (E–G) Cdkl5R59X−/+ females showed significantly increased activity by summed total activity, and horizontal activity metrics but not total activity over 5 min time bins. Cdkl5R59X−/y males showed increased activity by all three activity metrics. (H) Cdkl5R59X−/y males had lower latencies to fall off the rotarod compared with wild-type, sex-matched littermate controls. Data are expressed as mean ± S.E.M. *P < 0.05, **P < 0.001 and ***P < 0.0001 indicate when the Cdkl5 mutant mice differ from wild-type, sex-matched littermate controls.
A corroborating assay of motor abilities, coordination and motor learning is the rotarod. As observed previously by others (14,64,65), Cdkl5exon6 and Cdkl5R59X mutants exhibited large main effects of genotype using a two-way ANOVA (Cdkl5exon6Fig. 1D; F(3, 40) = 5.890, P < 0.0002; Cdkl5R59XFig. 1H; F(3, 40) = 3.835, P < 0.0167), with affected mice of both models exhibiting shorter latencies to fall off the accelerating rod, illustrating the deficits in motor coordination and balance when compared with wild-type, sex-matched littermate controls. Cdkl5exon6 but not Cdkl5R59X mutants exhibited a main effect of time using a two-way ANOVA (Cdkl5exon6Fig. 1D; F(3, 40) = 3.228, P < 0.05; Cdkl5R59XFig. 1H; F(3, 40) = 1.045, P = 0.353), suggesting a motor learning deficit in one of the two models.
Cognitive deficits were observed in the Y-maze, an assay of learning and memory that has been validated by experiments in which scopolamine (0.56 and 1 mg/kg; 30 min pre, i.p.) produced significant reductions in spontaneous alternation behavior relative to vehicle-treated C57Bl/6J mice (communication with Dr Sukoff-Rizzo and open access https://www.jax.org/MNBF). These data alongside our y-axis validation data highlight inter-laboratory reliability and reproducibility. An overall genotype effect was observed in both Cdkl5exon6 and Cdkl5R59X, in which mutant mice made fewer alternation triads than wild-type, sex-matched littermates (Fig. 2A; F(3, 40) = 11.34, P < 0.001 and Fig. 2E; F(3, 39) = 12.45, P < 0.001). Cdkl5exon6−/y males (P < 0.001) and Cdkl5exon6−/+ females (P = 0.0154) differed from Cdkl5exon6+/y males and Cdkl5exon6+/+ females, respectively, by making fewer alternation triads as described in Materials and Methods section. As would be predicted from the hyperactivity described above, both Cdkl5exon6 and Cdkl5R59X (Fig. 2B; F(3, 40) = 3.475, P = 0.0184 and Fig. 2F; F(3, 39) = 3.049, P = 0.0399) mutant mice made a greater number of total transitions. Holm–Sidak-corrected post hoc analysis for multiple comparisons did not find an effect of a single between-group comparison. Each genotype had an effect size greater than ‘1’, considered large Cohen’s d for effect size in Y-maze spontaneous alternation (Supplementary Material, Table S2).
Figure 2.
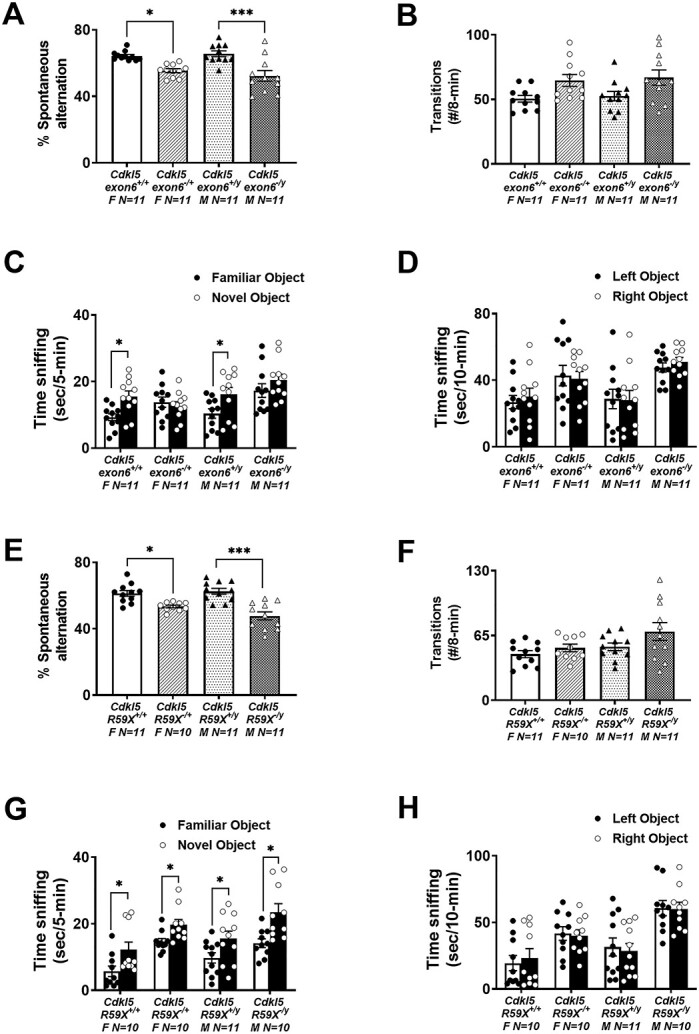
Cognitive deficits in Cdkl5 deletion and knock-in mice. (A) Cdkl5exon6−/+ females and Cdkl5exon6−/y males exhibited working memory impairments by lower percentage of spontaneous alternation in Y-maze. (B) The total transitions made by Cdkl5exon6−/+ females and Cdkl5exon6−/y males were similar to their wild-type, sex-matched littermate controls. (C) Cdkl5exon6−/+ females and Cdkl5exon6−/y males exhibited a lack of recognition memory following a 1 h delay compared with wild-type, sex-matched littermate controls that spent more time with the novel object. (D) All groups explored the two objects similarly during the familiarization phase. (E) Cdkl5R59X−/+ females and Cdkl5R59X−/y males exhibited working memory impairments by lower percentage of spontaneous alternation in Y-maze. (F) The total transitions made by Cdkl5R59X−/+ females and Cdkl5R59X−/y males made were similar to wild-type, sex-matched littermate controls. (G) Cdkl5R59X−/+ females and Cdkl5R59X−/y males exhibited intact recognition memory following a 1 h delay similar to wild-type, sex-matched littermate controls. (H) All groups explored the two objects similarly during the familiarization phase. Data are expressed as mean +/− S.E.M. *P < 0.05, **P < 0.001 and ***P < 0.0001 indicate when the Cdkl5 mutant mice differ from wild-type, sex-matched littermate controls.
A corroborating assay of learning and memory, novel object recognition (NOR), was manually scored by a highly trained observer blinded to genotype. Scores indicated both Cdkl5exon6 and Cdkl5R59X wild-type mice of both sexes spent more time investigating the novel object versus the familiar object, as expected (Cdkl5exon6+/yFig. 2C; t(20) = 2.317, P < 0.003; Cdkl5exon6−/+ t(20) = 3.194, P < 0.0046 and Fig. 2G; Cdkl5R59X−/y t(20) = 2.164, P < 0.0139; Cdkl5R59X+/+ t(20) = 2.323, P < 0.028). Scores from the Cdkl5exon6 line indicated that mutant mice of both sexes did not spend more time investigating the novel object versus the familiar object (Cdkl5exon6−/yFig. 2C; t(20) = 1.141, P = 0.2673; Cdkl5exon6−/+ t(20) = 0.5904, P = 0.5618), highlighting a corroborating deficit in learning and memory. However, scores from the Cdkl5R59X line indicate that mutant mice spent more time investigating the novel object versus the familiar object (Cdkl5R59X−/yFig. 2G; t(20) = 3.201, P < 0.005; Cdkl5R59X−/+ t(20) = 2.723, P = 0.0134), indistinguishable from control mice. This finding is a unique observation of differing phenotypes across the two mouse models in which the etiology of Cdkl5 loss differs. Our methods utilized were consistent with other NOR literature (56,66). Data illustrating no preference for the left or right objects and sufficient time spent investigating the objects is also shown, to confirm a lack of inherent preference bias (Fig. 2D; Cdkl5exon6+/y; t(20) = 0.0818, P > 0.05; Cdkl5exon6+/+ t(20) = 0.5358, P < 0.05; Cdkl5exon6−/y t(20) = 0.9218, P > 0.05; Cdkl5exon6−/+ t(20) = 0.2359, P > 0.05; and Fig. 2H; Cdkl5R59X+/y t(20) = 0.7402, P > 0.05; Cdkl5R59X+/+ t(20) = 0.4422, P > 0.05; Cdkl5R59X−/y t(20) = 0.9306, P > 0.05; Cdkl5R59X−/+ t(20) = 0.2443, P > 0.05).
We utilized innovative computerized touchscreen assays with high translational relevance to evaluate visual discrimination attributable to cortical circuitry and glutamatergic neurotransmission (67–70). During pre-training stages, we analyzed trials/sessions across genotypes for both Cdkl5 mutant lines (Table 1). All mice successfully completed the pre-training criteria. Cdkl5exon6−/+ female mice completed significantly more trials on both habituation days (habituation day 1; Cdkl5exon6−/+; t(19) = 3.776, P = 0.0013; habituation day 2: Cdkl5exon6−/+; t(19) = 3.776, P = 0.0013) and Cdkl5exon6−/y males completed more trials on habituation day 1 but not on day 2 (habituation day 1; Cdkl5exon6−/y; t(19) = 3.720, P = 0.0015; habituation day 2: Cdkl5exon6−/y; t(19) = 3.720, P = 0.0015). Cdkl5 R59X−/+ female mice completed significantly more trials on habituation day 1 but not on day 2 (habituation day 1; Cdkl5R59X−/+; t(19) = 4.235, P = 0.0004; habituation day 2: Cdkl5R59X−/+; t(19) = 1.035, P = 0.3136) and Cdkl5 R59X−/y males completed more trials on both habituation days [habituation day 1; Cdkl5R59X−/y; t(19) = 3.037, P = 0.0068; habituation day 2: Cdkl559X−/y; t(19) = 3.082, P = 0.0061]. As shown in Table 1, in Stage 2, all Cdkl5-mutant mice completed significantly more trials compared with wild-type, sex-matched littermate controls (Cdkl5exon6−/+; t(19) = 4.323, P = 0.0004; Cdkl5exon6−/y; t(19) = 3.256, P = 0.0042; Cdkl5R59X−/+; t(19) = 4.370, P = 0.0003; Cdkl5R59X−/y; t(19) = 5.641, P < 0.0001). In Stage 3, the Cdkl5exon6 deletion mice completed significantly more trials (Cdkl5exon6−/+; t(19) = 4.502, P = 0.0002; Cdkl5exon6−/y; t(19) = 3.727, P = 0.0014; Cdkl5R59X−/+; t(19) = 1.771, P = 0.0935; Cdkl5R59X−/y; t(19) = 1.891, P = 0.0748). In Stage 4, Cdkl5exon6−/y required significantly more days to reach criterion when compared with their wild-type, sex-matched littermate controls (Cdkl5exon6−/+; t(19) = 0.1111, P = 0.9127; Cdkl5exon6−/y; t(19) = 02.970, P = 0.0079; Cdkl5R59X−/+; t(19) = 0.7902, P = 0.4392; Cdkl5R59X−/y; t(19) = 0.09384, P = 0.9262).
Table 1.
Pre-training performance of Cdkl5 deletion and knock-in mice. All Cdkl5 mutant mice successfully initiated trials during 2 days of habituation, met the criteria of completing 30+ trials for 2 consecutive days in stage 3 and required similar number of days to reach criterion in stage 4 when compared with wild-type, sex-matched littermate controls
| Pre-training stages | Cdkl5 exon6 +/+ F N = 10 | Cdkl5 exon6 −/+ F N = 11 | Cdkl5 exon6 +/y M N = 10 | Cdkl5 exon6 −/y M N = 11 | Cdkl5 R59X +/+ F N = 10 | Cdkl5 R59X −/+ F N = 11 | Cdkl5 R59X +/y M N = 10 | Cdkl5 R59X −/y M N = 11 |
|---|---|---|---|---|---|---|---|---|
| Stage 1: Habituation day 1 number of trials | 11.10 ± 1.51 | 23.73 ± 2.87 | 15.10 ± 1.16 | 34.91 ± 4.96 | 9.80 ± 0.95 | 19.00 ± 1.88 | 19.60 ± 5.64 | 42.82 ± 5.18 |
| Stage 1: Habituation day 2 number of trials | 34.10 ± 10.13 | 86.55 ± 17.79 | 67.30 ± 14.03 | 99.09 ± 18.13 | 47.70 ± 8.12 | 63.27 ± 12.28 | 82.90 ± 8.14 | 138 ± 15.33 |
| Stage 2: Number of trials | 27.60 ± 3.49 | 59.45 ± 6.26 | 26.50 ± 5.99 | 58.55 ± 7.64 | 27.80 ± 2.928 | 48.18 ± 3.56 | 32.10 ± 3.37 | 72.18 ± 6.03 |
| Stage 3: Number of trials | 53.13 ± 7.31 | 94.18 ± 5.61 | 57.97 ± 6.68 | 90.00 ± 5.51 | 56.59 ± 7.50 | 76.45 ± 8.34 | 51.87 ± 5.66 | 69.20 ± 6.86 |
| Stage 4: Days to reach criterion | 3.70 ± 0.47 | 3.64 ± 0.34 | 2.90 ± 0.10 | 4.18 ± 0.40 | 2.20 ± 0.20 | 2.46 ± 0.25 | 2.60 ± 0.16 | 2.64 ± 0.34 |
During visual discrimination acquisition, mutant male and female mice from the exon 6 deletion exhibited robust learning and memory impairments (Fig. 3B; Cdkl5exon6−/y; t(20) = 3.936, P < 0.0009; Fig. 3A; Cdkl5exon6−/+ [t(20) = 3.950, P < 0.0009), requiring significantly more sessions to a stringent criterion of completing at least 30 trials, at an accuracy of 80% or higher, on two consecutive days, when compared with wild-type, sex-matched littermate controls. Analysis of survival curves, that as, percentage of mice that reached the 80% accuracy criterion on each training day, indicated that the percentage of mice that reached this criterion was significantly higher in wild-type, sex matched controls (Fig. 3D; Cdkl5exon6−/y; log-rank Mantel–Cox test X2(54) = 11.78, P < 0.0006; Fig. 3C; Cdkl5exon6−/+ log-rank Mantel–Cox test X2(52) = 10.78, P < 0.001). Analysis of additional parameters indicated that Cdkl5exon6−/y male mice required more trials to reach the criterion, compared with the Cdkl5exn6+/y controls (Fig. 3F; Cdkl5exon6−/y; t(20) = 4.249, P < 0.0001; Fig. 3E; Cdkl5exon6−/+t(20) = 1.167, P < 0.248) and required greater correction trials, suggesting a slower rate of learning (Fig. 3H; Cdkl5exon6−/y; t(20) = 6.34, P < 0.0001; Fig. 3G; Cdkl5exon6−/+t(20) = 3.806, P < 0.0004).
Figure 3.

Delayed touchscreen learning in Cdkl5 deletion mice. (A, B) Cdkl5exon6−/+ females and Cdkl5exon6−/y males took significantly longer to reach a criterion of completing at least 30 trials with 80% or higher accuracy on two consecutive days compared with wild-type, sex-matched littermates. (C, D) The percentage of mice that reached criterion was significantly higher in wild-type, sex-matched controls than Cdkl5exon6−/+ females and Cdkl5exon6−/y males. (E, F) Cdkl5exon6−/+ females and Cdkl5exon6−/y males completed fewer trials per session. (G, H) Cdkl5exon6−/+ females and Cdkl5exon6−/y males required higher correction trials to reach criterion. Data are expressed as mean ± S.E.M. *P < 0.05, **P < 0.001 and ***P < 0.0001 indicate when the Cdkl5 mutant mice differ from wild-type, sex-matched littermate controls.
Mutant male and female R59X mice also exhibited robust learning and memory impairments (Fig. 4BCdkl5R59X−/y; t(19) = 7.195, P < 0.0001; Fig. 4A; Cdkl5R59X−/+ t(19) = 3.742, P = 0.0014), requiring significantly more sessions to a stringent criterion of completing at least 30 trials, at an accuracy of 80% or higher, on two consecutive days, when compared with wild-type, sex-matched littermate controls. Analysis of survival curves indicated that the percentage of mice that reached this criterion was significantly higher in wildtype, sex-matched controls (Fig. 4D; Cdkl5R59X−/y; log-rank Mantel–Cox test X2(54) = 17.61, P < 0.0001; Fig. 4C; Cdkl5R59X−/+ log-rank Mantel–Cox test X2(53) = 10.73, P = 0.0011). Analysis of additional parameters indicated that Cdkl5R59X−/y male mice required more trials to reach criterion, compared with Cdkl5R59X+/y controls (Fig. 4F; Cdkl5R59X−/y; t(19) = 4.866, P < 0.0001; Fig. 4E; Cdkl5R59X−/+ t(19) = 2.635, P = 0.011) and required greater correction trials, suggesting a slower rate of learning (Fig. 4H; Cdkl5R59X−/y; t(19) = 9.920, P < 0.0001; Fig. 4G; Cdkl5R59X−/+ t(19) = 6.949, P < 0.0001).
Figure 4.

Delayed touchscreen learning in Cdkl5 knock-in mice. (A, B) Cdkl5R59X−/+ females and Cdkl5R59X−/y males took significantly longer to reach a criterion of completing at least 30 trials with 80% or higher accuracy on two consecutive days compared with wild-type, sex-matched littermates. (C, D) The percentage of mice that reached criterion was significantly higher in wild-type, sex-matched littermate controls than Cdkl5R59X−/+ females and Cdkl5R59X−/y males. (E, F) Cdkl5R59X−/+ females and Cdkl5R59X−/y males completed fewer trials per session. (G, H) Cdkl5R59X−/+ females and Cdkl5R59X−/y males required higher correction trials to reach criterion. Data are expressed as mean ± S.E.M. *P < 0.05, **P < 0.001 and ***P < 0.0001 indicate when the Cdkl5 mutant mice differ from wild-type, sex-matched littermate controls.
Behavioral assessment of seizure threshold in female mice of both lines was performed with injections of 80 mg/kg of pentylenetetrazol (PTZ) as described previously (20,30,59,71–73). PTZ-induced convulsions were used to gauge susceptibility to primary generalized seizures and as a gross approximation of excitation–inhibitory balance. Latencies to myoclonic jerk and generalized clonic seizure (loss of righting reflex) (Fig. 5A; Cdkl5exon6−/+; t(15) = 2.660, P = 0.0178; Fig. 5B; Cdkl5exon6−/+; t(15) = 2.410, P = 0.0292), were faster and shorter. They illustrate preliminary characterization of seizure, subthreshold epileptiform activity and imbalances in the excitatory/inhibitory homeostasis. Administration of PTZ, a non-competitive, GABAA antagonist, leads to brain hyperexcitability. Female Cdkl5R59X−/+ mice also exhibited faster latencies to myoclonic jerk and loss of righting reflex/time to clonic seizure (Fig. 5C; Cdkl5R59X−/+; t(17) = 2.309, P = 0.0338; Fig. 5D; Cdkl5R59X−/+; t(17) = 3.095, P = 0.0066). Multiple previous studies in male hemizygous Cdkl5 mutant mice have failed to show spontaneous seizures leading to epilepsy in mice younger than 4 months of age (15,17,18,74,75).
Figure 5.

Increased seizure susceptibility in Cdkl5 deletion and knock-in female mice. (A, B) Reduced latencies to first jerk and generalized clonic seizures were observed in Cdkl5exon6−/+ females after an i.p. injection of 80 mg/kg PTZ over the course of a 30 min trial. (C, D) Reduced latencies to first jerk and generalized clonic seizures were observed in Cdkl5R59X−/+ females after an i.p. injection of 80 mg/kg PTZ over the course of a 30 min trial. Data are expressed as mean ± S.E.M. *P < 0.05 indicates when the Cdkl5 mutant mice differ from wild-type, sex-matched littermate controls.
Discussion
The X chromosome is highly enriched for genes that are critical in brain function and linked to neurological diseases. This is highlighted by the fact that there are ~140 known genes that are causative for X-linked IDs (3,4). CDD is an X-linked NDD with debilitating ID and pervasive seizures leading to epileptic encephalopathy and severe sleep disruption. The goal of the proposed studies was to provide quantifiable translational phenotypes in females and males using two in vivo model systems, one with an exon deletion and one with a patient-derived knock-in mutation. We discovered hyperactivity in both sexes, in both lines, and substantial learning and memory deficits in multiple assays that capture cognitive dysfunction. We also report, for the first time, seizure susceptibility, confirming the biology underlying the behavioral phenomenon in this unique rare genetic NDD. Although no regional, cellular level neuroanatomical changes were observed via immunohistochemistry to correlate behavioral abnormalities with distinct structural changes, more sensitive methodologies, aimed at global brain connectivity, white matter tracts and subtler volumetric differences are being pursued in an ongoing work. Numerous other reports illustrate volumetric reductions or enlargements in brain subregions correlated with behavioral deficits (59,73,76–86), suggesting that our global investigations will yield detectable brain abnormalities.
Additionally, it is now well known that the role of microglia in typical and atypical neurodevelopment is critically important (87–89) and inhibition of microglia activation alleviates phenotypes resulting from Cdkl5 mutations (50). However, as no differences in microglia were observed, in this study that utilized a single time point and it is quite possible that the effect of Cdkl5 mutations on microglia activation and reactivity may be nuanced or occur at critical windows of development, which were not observed in our initial characterization (88,90,91). Previous research has behaviorally and biochemically characterized male mice from numerous CDD models, including the two lines used here, which do not spontaneously cease as young adults nor during the average duration of their lifespan. Less work has been performed on female mice, resulting in a relative lack of informative and quantifiable translational phenotypes with sizeable effects. The first observation of disturbance-associated seizure-like events in heterozygous female mice across two independent mouse models of CDD was recently reported as onsetting in >4-month-old female mice and increased with aging (18). Another report of spontaneous seizure-like events in mouse models of CDD, highlighted epileptic spasms, the most frequent and persistent seizure type in CDD patients, in two mouse models of CDD carrying heterozygous mutations, Cdkl5R59X−/+ and Cdkl5exon6−/+. Spasm-like events were reported in a significant proportion of aged heterozygous female mice carrying either of the two Cdkl5 mutations, most frequently associated with generalized slow-wave activity and tended to occur in clusters during sleep (17).
Here, uniquely, our team focused on sensitive behavioral assays, with a large signal-to-noise ratio across three major functional domains (motor, cognition, behavioral seizures) which will allow for the assessment of future novel therapeutic interventions and information for IND approval(s). The studies were the first to identify translationally relevant cross-species phenotypes with ultrasensitive, touchscreen technology for cognitive deficits used similarly to the NIH Toolbox in clinical trials of ID. We also identified hyperactivity, and heightened seizure susceptibility in females in two models of CDD preclinical mice (Tables 2 and S1). It is our opinion that the work performed herein can provide information on assay sensitivity. In other disorders, we discovered that gait analysis is a more reliable and sensitive motor outcome measure than rotarod for motor and balance in Angelman syndrome (92). We reached this conclusion after using ~70–100 cohorts of Angelman mouse models. Additional cohorts with interventional assessments would need to be analyzed to determine the most reliable and sensitive assays for mouse models of CDD.
Table 2.
Summary of major outcomes tested in Cdkl5 deletion and knock-in mice
| Outcomes | Cdkl5 exon6 −/+ | Cdkl5 exon6 −/y | Cdkl5 R59X −/+ | Cdkl5 R59X −/y | Consistent across sexes | Consistent across strains |
|---|---|---|---|---|---|---|
| Hyperactivity | Significant increase | Significant increase | Significant increase | Significant increase | Yes | Yes |
| Cognition | Significant deficit | Significant deficit | Significant deficit | Significant deficit | Yes | Yes |
| Hyperexcitability | Significant increase | Not tested | Significant increase | Not tested | Only tested in females | Yes |
Clinically relevant outcome measures are required to demonstrate the utility of innovative therapeutic designs, as well as to validate other traditional medicinal therapies that may be in the discovery pipeline of biotechnology and pharmaceutical companies. The first model from Zhou’s laboratory, a deletion of Cdkl5 exon 6, demonstrated substantial behavioral alterations in hyperactivity, abnormal social approach and reduced contextual fear conditioning in deletion males (14). These data were collected on male mice, presumably, to avoid the confounding effects of X-linked mosaicism in the females. However, this perpetuates an inherent lack of translational relevance to the affected females of the CDD community. Our data presented herein indicate robust hyperactivity deficits in both sexes of both the exon 6 deletion and the R59X knock-in mice. The robust signal of hyperactivity was corroborated in both lines and both sexes. We cannot compare fear conditioning data to earlier reports, as many behavioral neuroscience laboratories, including ours, consider hyperactivity a confounding variable when evaluating/interpreting the reduction in freezing percentages in the fear conditioning data in addition to the underlying neural circuitry which is fear memory associated, such as in PTSD (93–95), over general intellect. The novel mouse model that recreated the patient variant R59X, also reported hyperactivity, reduced contextual fear conditioning, abnormal motor coordination and less directed social behavior (15). Similar comments as above regarding females and hyperactivity confounding during fear learning apply to this characterization. Both of these laboratories are continuing to pursue the critical work of examining cell-type specificity of these phenotypes (16,63,96) and screening various therapeutics highlighting ex vivo slice electrophysiology and behavioral assays. Our data corroborate the earlier findings, extend the results to females, define a novel, robust cortical-based cognitive phenotype in the touchscreen, a spatial-based working memory deficit in the Y-maze and report abnormal hyperexcitability providing more functional phenotypes as well as translational relevance.
Over the past 5 years, our group has optimized sensitive assays across functional domains that allow for the assessment of novel therapeutic interventions (26). In addition, we also employ gold-standard behavioral assays such as open field, rotarod, NOR and the Y-maze. In this study, we used our highly translational, touchscreen assay to assess cortical-dependent learning via pairwise visual discrimination, such as other laboratories (58,67–70,97–103), as touchscreen assays are ideal for simple and complex cognition assessments with limited motor confounds. We have utilized these assays in rat and mouse models of Angelman syndrome (79,100) and of Phelan–McDermid syndrome, (Shank3+/− mutations) both of which have motor hypo-dysfunction (79,104). To date, in our group, every time hyperactivity has been observed, animals perform better, likely because they have more attempts to ‘try’ the task. One neural circuit primarily involved in motor activity begins in the cerebral cortex, connects to basal ganglia, which connect to the thalamus, and finally loops to the motor cortex. Within this circuit, there are mini circuits with substantia nigra and the subthalamic nucleus. Further, there are both direct and indirect pathways that project through this circuitry. Irrespective of subcircuitry and pathways, the basal ganglia (i.e. caudate, putamen and globus pallidus) exert a modulating effect on this neural circuit and affecting the behavioral manifestation of movement. The basal ganglia are primarily innervated with dopaminergic neurons. Concordantly, ADHD, tic disorders and Huntington’s disease have all been associated with either morphometric abnormalities (i.e. usually reductions) or dopaminergic abnormalities in basal ganglia regions (105). Gross morphometric abnormalities in these CDD models is ongoing.
Circuits important for simple to complex cognition measured by the touchscreen require the prefrontal cortex and include the dorsal frontal striatal, orbitofrontal-striatal and frontal-cerebellar circuits. Dorsal frontal striatal circuitry has been linked to cognitive control whereas orbitofrontal-striatal loops have been related to set shifting or reversal of choice processing (69,106–111). Frontal-cerebellar circuits have been implicated in timing and multiple models of autism (86,112–117). Neurobiological dysfunction in any of these circuits could lead to symptoms of NDDs such as CDD, as behavioral control could be disturbed by (1) deficits in the prefrontal cortex itself or (2) problems in the circuits relaying information to the prefrontal cortex, leading to reduced signaling for control. Future studies will differentiate between these interlinked reciprocal circuits with the prefrontal cortex in females with CDD (118–120). If such a differentiation can be achieved, it might permit a neurobiological subtyping of hyperactivity attributable to heightened perseverance versus hyperactivity attributable to a lack of impulse control versus hyperactivity attributable to motor spasticity and/or myoclonic seizure such as events.
We have also utilized touchscreen learning and memory testing in a model of another NDD, Coffin–Siris syndrome, mice with mutations in Arid1b+/− which have gross physical delays, and are substantially smaller than their age-, sex-matched and wild-type littermates. Interestingly, despite their smaller stature (analogous to the patient community), Arid1b+/− have normal motor activity levels and performed similar to typical WT during visual discrimination (35). A fascinating, unique observation of these Cdkl5 model lines is that although they exhibit substantial hyperactivity, indicated by ~30% increased transitions (Fig. 2D and H) in the Y-maze, ~50% increased activity appeared in the open field (Fig. 1B and F) data and increased trial number in the touchscreen (Figs 3E and F and 4E and F), they exhibited clear and robust learning impairments and learning delays, contrasting other NDD models with variations in activity, including Chd8 and Arid1b (72,73) and fragile X syndrome (101). This means that despite more attempts at the learning and memory tasks, their ability is deficient. Not only does this study, for the first time, report cognitive dysfunction in females, but it reports that these learning and memory impairments are not confounded by their inherent hyperactivity. This is the first observation, to our knowledge, of such a unique detectable, sizeable impairment uninfluenced by the physical ability, activity and/or sensory processing of the subject mice.
Bahi-Buisson and colleagues (5–7,121) have several reports on the early onset and natural history of epilepsy in patients with CDKL5 mutations describing early epilepsy in the first stage (median age of 4 weeks), epileptic encephalopathy in the second stage and tonic seizures and late myoclonic epilepsy in the third. Data from the intricate, nuanced indices of EEG, during light/dark cycles, and active versus sleep stages will be highly informative when powered, analyzed and ready for public reporting. Although we included data on seizure susceptibility and general hyperexcitability, we felt it premature to report traces from EEG. We discovered a low survival rate following survival surgery for wireless, telemetry implants for EEG recordings with 4 of 14 mutation animals surviving and 4 of 8 WT. Therefore, we have chosen to dedicate a second report describing the profound loss of subjects and eventually when we have adequate power in sample size, we will report the neuro, sleep and respiratory physiology alongside global neuroanatomy findings in a follow-up report.
Recently, anecdotal and published reports of myoclonic seizure events, provoked by handling, had been observed in aged female Cdkl5 mice by 42 weeks of age (17,18). These new studies monitored video and four-channel EEG recordings, within a cohort of >42-week-old female Cdkl5exon6−/+ and Cdkl5R59X−/+ mutant mice compared with wild-type littermates, such as the design herein, except our mice are never aged beyond 20 weeks. Mulcahey et al. (17) observed a high frequency of epileptic spasms. These events appeared to myoclonic, in nature, as they were characterized by sudden-onset, brief myoclonic jerks involving the limbs or trunk and correlated with the amplitude of the epileptiform activity on EEG (17). The data presented herein corroborate and extend that Cdkl5 knockout mice display decreased latency to the first stages of seizure progression upon low-dose PTZ administration (15), suggesting that seizure susceptibility is altered in the absence of CDKL5. Yet, our PTZ data contrast with other reports that used high-dose kainic acid seizure induction of overt seizures since Cdkl5 hemizygous knockout males exhibited similar latencies to wild-type littermates, following KA (74). These methodologies while similar in theory (decreasing GABA, PTZ and increasing glutamate, KA) are different as the behavioral seizure event following PTZ onsets rapidly and occurs over a short duration (19,20,122). This is the first study of seizures in females observing myoclonic jerks involving sudden and repetitive movement of the head and neck with or without tail stiffening at ~20 weeks of age and conclude similar results as other reports that show seizures increase with aging (18). However, as seizures increase in frequency and seizure thresholds are reduced, concomitant with natural aging in C57BL/6J mice, we are enthusiastic for our ongoing bioinformatic analysis using extended EEG recording data to quantify and analyze comprehensive epileptiform, power spectrum and seizure characterization in both models in both sexes across many developmental and aging timepoints, prior to 20 weeks of age, as the PTZ susceptibility suggest these substantial aging paradigms may not be essential for phenotypic detection and are less translational by lifespan timepoints.
Summary
Our report demonstrated objective, quantifiable translational behavioral phenotypes in females and males using two in vivo model systems, one with an exon deletion and one with a patient-derived knock-in mutation of CDD. We discovered hyperactivity, substantial learning and memory deficits using touchscreen technology which captures translational cognitive dysfunction, and hyperexcitability by reduced seizure thresholds.
Materials and Methods
Animals
Cdkl5 exon6 mice congenic on C57BL/6J were generated by crossing Cdkl5exon6−/+ females (JAX stock #021967) with inbred C57BL/6J males. Cdkl5R59X mice congenic on C57BL/6J were generated by crossing Cdkl5R59X−/+ females (JAX stock #028856) with inbred C57BL/6J males. To identify mice, neonates were labelled by paw tattoo on postnatal day (PND) 2–3 using non-toxic animal tattoo ink (Ketchum Manufacturing Inc., Brockville, ON, Canada). At PND 5–7, tails of pups were clipped (0.5 cm) for genotyping, following the UC Davis Institutional Animal Care and use Committees (IACUC) policy regarding tissue collection. Genotyping was performed with RED Extract-N-Amp (Sigma Aldrich, St. Louis, MO) using primers 34 149 5′-GGAAGAAATGCCAAATGGAG-3′,34 150 5′-GGAGACCTGAAGAGCAAAGG-3, 34 151 5′-CCCTCTCAGTAAGGCAGCAG-3, and 34 152 5′-TGGTTTTGAGGTGGTTCACA-3 for Cdkl5exon6, and 5′-GCTGCTTACATTAGGAGAGACTGC-3′ and 5′-GTCACATGACCAGCCAGCGT-3′ for Cdkl5R59X. After weaning on PND21, mice were socially housed in groups of 2–4 by sex.
Tissue extraction
Adult wild-type and mutant Cdkl5 mice, of both sexes, were cervically dislocated and brains were rapidly extracted. The cerebral cortex, hippocampus, and cerebellum were dissected and flash frozen over dry ice. Tissue was later homogenized and separated for genomic DNA, RNA or protein.
Western blots
Protein was extracted using Pierce RIPA buffer (ThermoFisher, Waltham, MA) with protease and phosphatase inhibitor cocktail (ThermoFisher, Waltham, MA). Protein concentrations were measured by the Pierce bicinchoninic acid assay kit (ThermoFisher, Waltham, MA). Around 25 μg of whole brain protein lysate per sample was separated on 4–20% stacking TGX gels (BioRad, Hercules, CA) and transferred overnight at 30 V onto Immobilon-FL PVDF membranes (Millipore Sigma, Burlington, MA). PVDF membranes were blocked for 1 h with Intercept (PBS) blocking buffer (LI-COR, Lincoln, NE). Following blocking, PVDF membranes were incubated with anti-CDKL5 (1:1000, MRC-PPU Reagents, S957D) and anti-beta actin (1:10°000, Sigma-Aldrich, A5441) antibodies in a 1:1 dilution of Intercept and 0.2% PBS Tween (PBST) at 4°C overnight. Following incubation, membranes were washed in 0.1% PBST 5× for 5 min. Membranes were then incubated with IRDye 800 donkey anti-goat IgG secondary antibody (1:15°000, LI-COR Biosciences, 926-32214) and IRDye 680 goat anti-mouse IgG secondary antibody (1:20°000, LI-COR Biosciences, 926-68 070) in the same intercept dilution as described previously. Following incubation, membranes were washed 5× in 0.1% PBST and 1× in PBS before storing in fresh PBS. Membranes were imaged on an Odyssey CLx imager (LI-COR, Lincoln, NE). Quantitative analysis was performed using the Image Studio Lite software (LI-COR, Lincoln, NE).
Subjects for behavior and neurophysiology
All mice were housed in Techniplast cages (Techniplast, West Chester, PA, USA). Cages were housed in ventilated racks in a temperature- (68–72°F) and humidity (~25%)-controlled colony room on a 12:12 light/dark cycle. Standard rodent chow and tap water were available ad libitum. In addition to standard bedding, a Nestlet square, shredded brown paper and a cardboard tube (Jonesville Corporation, Jonesville, MI) were provided in each cage. All experimental procedures were performed in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals and were approved by the institutional IACUC #21494 (PI, Silverman) of the University of California, Davis.
Order of testing
Two cohorts of mice were tested per strain as follows: Cohort 1 of Cdkl5exon6 deletion mice was sampled from 11 litters. The order and age of testing were as follows: (1) open field at 6 weeks of age, (2) DigiGait at 6 weeks of age, (3) rotarod at 7 weeks of age, (4) spontaneous alternation at 8 weeks of age, (5) novel object recognition at 8 weeks of age and (6) touchscreen pairwise discrimination from 12 to 20 weeks of age. Cohort 2 was sampled from 4 litters and tested in PTZ-induced seizures at 8 weeks of age. Cohort 1 of Cdkl5R59X knock-in mice was sampled from 13 litters. The order and age of testing were as follows: (1) open field at 6 weeks of age, (2) DigiGait at 6 weeks of age, (3) rotarod at 7 weeks of age, (4) spontaneous alternation at 8 weeks of age, (5) novel object recognition at 8 weeks of age and (6) touchscreen pairwise discrimination from 12 to 20 weeks of age. Cohort 2 was sampled from 3 litters and tested in PTZ-induced seizures at 8 weeks of age.
Behavioral assays
Open field
General exploratory locomotion in a novel open field arena was evaluated as previously described (72,73,104). Briefly, each subject was tested in a VersaMax animal activity monitoring system (Accuscan, Columbus, OH, USA) for 30 min in a ~30 lux testing room. Total distance traversed, horizontal activity, vertical activity and time spent in the center were automatically measured to assess gross motor abilities in mice. Open field parameters (total distance traveled, horizontal activity, vertical activity and center time) were analyzed within sex using a two-way repeated measures ANOVA, with genotype as the between-group factor and time as the within-group factor. Multiple comparisons for time bins were conducted.
Rotarod
Motor coordination, balance and motor learning were tested with an accelerating rotarod (Ugo Basile, Gemonio, Italy) as previously described (123). Mice were placed on a rotating cylinder that slowly accelerated from 5 to 40 revolutions per min over 5 min. Mice were given three trials per day with a 60 min inter-trial rest interval and tested for 3 consecutive days for a total of nine trials. Performance was scored as latency to fall off the cylinder. Latency to fall was analyzed with two-way repeated measures ANOVA, with genotype as the between-group factor and day as the within-group factor. Multiple comparisons for days were conducted.
Spontaneous alternation in a Y-maze
Spontaneous alternation was assayed using methods as previously described (124) in mice. The Y-shaped apparatus was made of non-reflective matte white finished acrylic (P95 White, Tap Plastics, Sacramento, CA, USA). Subjects were placed in the middle of the apparatus and transitions between the three arms were scored by an investigator blind to genotype. Mice are placed midway of the start arm, facing the center of the Y for an 8 min test period and the sequence of entries into each arm are recorded via a ceiling mounted camera integrated with behavioral tracking software (Noldus Ethovision). Percentage spontaneous alternation is calculated as the number of triads (entries into each of the three different arms of the maze in a sequence of three without returning to a previously visited arm) relative to the number of alternation opportunities. One-way ANOVA was used to detect differences in alternation. Multiple comparisons were corrected for using Holm–Sidak post hoc methods for sex.
Novel object recognition
The NOR test was conducted as previously (26,60,73,125) described in opaque matte white (P95 White, Tap Plastics, Sacramento, CA, USA) arenas (41 cm l × 41 cm w × 30 cm h). The assay consisted of four sessions: a 30 min habituation session, a second 10 min habituation phase, a 10 min familiarization session and a 5 min recognition test. On day 1, each subject was habituated to a clean empty arena for 30 min. Then, 24 h later each subject was returned to the empty arena for an additional 10 min habituation session. The mouse was then removed from the testing arena and was placed in a clean temporary holding cage, while two identical objects were placed in the arena. Subjects were returned to the testing arena and given a 10 min familiarization period in which they had time to investigate the two identical objects. After the familiarization phase, subjects were returned to their holding cages for a 1 h interval period. One familiar object and one novel object were placed in the arena, where the two identical objects had been located during the familiarization phase. After the 1 h interval, each subject was returned to the arena for a 5 min recognition test. The familiarization session and the recognition test were recorded using the Ethovision XT video tracking software (version 9.0, Noldus Information Technologies, Leesburg, VA, USA). Sniffing was defined as head facing the object with the nose point within 2 cm or less from the object. Time spent sniffing each object was scored by an investigator blind to both genotype and treatment. Recognition memory was defined as spending significantly more time sniffing the novel object compared with the familiar object via a Student’s paired t-test. Total time spent sniffing both objects was used as a measure of general exploration. Time spent sniffing two identical objects during the familiarization phase confirmed the lack of an innate side bias. Objects used were plastic toys: a small soft plastic orange safety cone and a hard plastic magnetic cone with ribbed sides, as previously described (26,60,73,125).
Touchscreen pairwise discrimination
An efficient pre-training regimen was validated based on previously published work (58,72). The reinforcer was 20 μl of a palatable liquid nutritional supplement (Strawberry Ensure Plus, Abbott, IL, USA) diluted to 50% with water. A standard tone cue was used to signal the delivery of the reinforcer during pre-training and acquisition. Prior to pre-training, subject mice were weighed and placed on a restricted diet of 2–4 g of rodent chow per mouse per day, to induce a 15% weight loss. Body weight was carefully monitored throughout the experiment, to ensure that a minimum of 85% of free feeding body weight was maintained for each mouse. The pre-training consisted of four stages. Stage 1 consisted of two days of habituation to the chamber under overhead lighting (60 lux). During stage 2, subjects were trained to collect the liquid food reward. It was a single 45 min session in which entering and exiting the magazine triggers reinforcement under overhead lighting. During stage 3, mice were trained to touch the stimulus on the screen to dispense the reward. Subjects were trained in daily 45 min sessions during which an image (a random picture from a selection of 40 images) was presented in one of the two windows and remained on the screen until it was touched. Mice must complete 30 trials/day for two consecutive days to advance to the next stage. During stage 4, mice were trained not to touch an incorrect side of the screen. Subjects were trained in 45 min daily sessions in which touching the blank side was discouraged with a 5 s time-out during which the overhead lighting turned off. Completion of at least 30 trials, at an average accuracy of 80%, on two consecutive days, is required for advancement. Images used in stages 3 and 4 were not used in the subsequent discrimination task. Only mice that completed all stages of pre-training were advanced to the pairwise visual discrimination task. Bussey–Saksida touchscreen apparatus for mice (Campden Instruments Ltd/Lafayette Instruments, Lafayette, IL, USA) was used according to a procedure modified from original methods described previously (98,99,102,104). Subjects were trained to discriminate between two novel images, a spider and an airplane presented in a spatially pseudo-randomized manner in the two windows of the touchscreen. Completion of at least 30 trials, at an average accuracy of 80%, on two consecutive days, is required for completion. Each 45 min session consisted of unlimited number of trials separated by 15 s intertrial intervals. Designation of the correct and incorrect images were counterbalanced across mice within each genotype. Correct responses were reinforced. Each incorrect response was followed by a correction trial in which the images were presented in an identical manner to the previous trial until a correct response was made. Days to reach criterion, percentage of mice reaching criterion on each day, number of errors, correction errors and total trials were compared between genotypes, as described previously (104). Genotypes, within sex, were analyzed with paired t-test. Log-rank Mantel–Cox test was used to analyze the percentage of animals that reached criteria in the survival/completion analysis for the touchscreen test.
Pentylenetetrazol-induced seizures
In a separate cohort, behavioral assessment of seizure threshold in mice was performed with intraperitoneal injections of 80 mg/kg of pentylenetetrazole (Sigma Aldrich, St. Louis, MO, USA) as described previously (20,30,59,124). Dosing was conducted in a dim light setting (~30 lux). Directly after administration of PTZ, subjects were placed in a clean, empty cage and subsequent seizure stages were live-scored for 30 min. Seizure stages were scored using a modified Racine scale where 0 = normal exploratory behavior, 1 = immobility, 2 = generalized spasm, 3 = Straub’s tail, 4 = forelimb clonus, 5 = generalized clonus, 6 = clonic-tonic seizure, and 7 = full tonic extension/death. Time to each stage was taken in seconds and analyzed between genotypes using a Student’s t-test. This seizure assay was only performed in females.
Perfusions
At the conclusion of behavior testing, mice were euthanized by CO2 asphyxiation followed by bilateral thoracotomy. Mice were perfused with 10 ml of PBS (HyClone) followed by 10 ml of 4% paraformaldehyde (Fisher Healthcare, Pittsburg, PA). Brains were harvested, fixed in formalin for 24 h, then transferred to 30% sucrose (Fisher Chemical, Fair Lawn, NJ) for 24–48 h at 4°C. Brains were then frozen for 3 min in 100% isopropanol bath on dry ice at −77°C and then stored at −80°C until further processing.
Immunohistochemistry
Brains were serially sectioned at 30 μm in the sagittal plane using a Cryotome FSE (Precisionary Instruments) and placed into a 12-well plate filled with PBS with 0.1% sodium azide (NaN3) with 8–12 sections per well. The wells were incubated in PBS with 10% SEA BLOCK blocking buffer (Thermo Scientific) for 1 h on an orbital shaker at 60 rpm. Sections were transferred into the primary antibody solution (5% SEA BLOCK in PBS with anti-TMEM119: 1:250, Cell Signaling, 90 840; anti-GFAP: 1:2000, Abcam, ab10062; and anti-NeuN: 1:1000, EMD Millipore, ABN90) and placed on an orbital shaker at 4°C overnight. Sections were then washed with 0.1% PBST three times and placed into a secondary antibody solution (5% SEA BLOCK in PBS with Alexa Fluor 488 Goat Anti-Rabbit IgG (H+L): 1:2000, Invitrogen; Alexa Fluor 594 Goat Anti-Mouse IgG (H+L): 1:2000, Invitrogen; and Alexa Fluor 647 Goat Anti-Guinea Pig IgG (H+L)) on an orbital shaker at room temperature for 1 h. After incubation, the wells were incubated with Hoechst (1:2000) for 5 min on an orbital shaker, then transferred to 0.1% PBST and washed three times. To utilize the autodetection software on the AxioScan, sections were immersed in 0.01% Sudan Black in 70% EtOH, gently agitated for 1 min, and then transferred into PBS prior to mounting. The sections were then mounted onto microscope slides (Thermo Scientific) sequentially from lateral to midline and coverslipped using Fluoromount (Sigma). Whole brain sections were then scanned at 20× and stitched together using a Zeiss AxioScan. Image analysis was performed in Zen 2 (blue edition) (Zeiss) and ImageJ.
Statistical analysis
All statistical analysis were carried out using the Prism 9 software (GraphPad Software, San Diego, CA). All significance levels were set at P < 0.05 and all t-tests were two-tailed. Outliers were identified and excluded using Grubb’s test and D’Agostino and Pearson tests were used to check assumptions of normality. Cohen’s d was used for effect size calculations. Post hoc comparisons were performed following a significant main effect or interaction and were conducted using Holm-Sidak’s multiple comparisons test. Data are presented as mean ± standard error of the mean (S.E.M) unless otherwise noted.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank the Stem Cell and MIND Institute vivarium teams for maintaining the mouse colonies.
Conflict of Interest statement. The authors declare that they have no competing interests.
Contributor Information
Anna Adhikari, MIND Institute, University of California Davis School of Medicine, Sacramento, CA, USA; Department of Psychiatry and Behavioral Sciences, University of California Davis School of Medicine, Sacramento, CA, USA.
Fiona K B Buchanan, Department of Neurology, University of California Davis School of Medicine, Sacramento, CA, USA; Stem Cell Program and Gene Therapy Center, University of California Davis School of Medicine, Sacramento, CA, USA.
Timothy A Fenton, MIND Institute, University of California Davis School of Medicine, Sacramento, CA, USA; Department of Psychiatry and Behavioral Sciences, University of California Davis School of Medicine, Sacramento, CA, USA.
David L Cameron, Department of Neurology, University of California Davis School of Medicine, Sacramento, CA, USA; Stem Cell Program and Gene Therapy Center, University of California Davis School of Medicine, Sacramento, CA, USA.
Julian A N M Halmai, Department of Neurology, University of California Davis School of Medicine, Sacramento, CA, USA; Stem Cell Program and Gene Therapy Center, University of California Davis School of Medicine, Sacramento, CA, USA.
Nycole A Copping, MIND Institute, University of California Davis School of Medicine, Sacramento, CA, USA; Department of Psychiatry and Behavioral Sciences, University of California Davis School of Medicine, Sacramento, CA, USA.
Kyle D Fink, MIND Institute, University of California Davis School of Medicine, Sacramento, CA, USA; Department of Neurology, University of California Davis School of Medicine, Sacramento, CA, USA; Stem Cell Program and Gene Therapy Center, University of California Davis School of Medicine, Sacramento, CA, USA.
Jill L Silverman, MIND Institute, University of California Davis School of Medicine, Sacramento, CA, USA; Department of Psychiatry and Behavioral Sciences, University of California Davis School of Medicine, Sacramento, CA, USA.
Funding
MIND Institute’s Intellectual and Developmental Disabilities Research Center Pilot Program Award (P50HD103526 to K.D.F.); UPenn Orphan Disease Center: LouLou Foundation (CDKL5-19-D-104-01 to K.D.F.); Caley J. Brown Foundation (to K.D.F.); MIND Institute’s Intellectual and Developmental Disabilities Research Center Grant (P50HD103526 to J.L.S. and A.A.).
References
- 1. Des Portes, V. (2013) X-linked mental deficiency. Handb. Clin. Neurol., 111, 297–306. [DOI] [PubMed] [Google Scholar]
- 2. Laumonnier, F., Bonnet-Brilhault, F., Gomot, M., Blanc, R., David, A., Moizard, M.P., Raynaud, M., Ronce, N., Lemonnier, E., Calvas, P.et al. (2004) X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am. J. Hum. Genet., 74, 552–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Skuse, D. (2003) X-linked genes and the neural basis of social cognition. Novartis Found. Symp., 251, 84–98discussion 98-108; 109-111, 281-197. [PubMed] [Google Scholar]
- 4. Turkmen, A. and Lin, B. (2021) Detecting X-linked common and rare variant effects in family-based sequencing studies. Genet. Epidemiol., 45, 36–45. [DOI] [PubMed] [Google Scholar]
- 5. Bahi-Buisson, N. and Bienvenu, T. (2012) CDKL5-related disorders: from clinical description to molecular genetics. Mol. Syndromol., 2, 137–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bahi-Buisson, N., Kaminska, A., Boddaert, N., Rio, M., Afenjar, A., Gerard, M., Giuliano, F., Motte, J., Heron, D., Morel, M.A.et al. (2008) The three stages of epilepsy in patients with CDKL5 mutations. Epilepsia, 49, 1027–1037. [DOI] [PubMed] [Google Scholar]
- 7. Bahi-Buisson, N., Villeneuve, N., Caietta, E., Jacquette, A., Maurey, H., Matthijs, G., Van Esch, H., Delahaye, A., Moncla, A., Milh, M.et al. (2012) Recurrent mutations in the CDKL5 gene: genotype-phenotype relationships. Am. J. Med. Genet. A, 158A, 1612–1619. [DOI] [PubMed] [Google Scholar]
- 8. Ho, N.T., Kroner, B., Grinspan, Z., Fureman, B., Farrell, K., Zhang, J., Buelow, J., Hesdorffer, D.C. and Rare Epilepsy Network Steering, C (2018) Comorbidities of rare epilepsies: results from the rare epilepsy network. J. Pediatr., 203, 249, e245–258. [DOI] [PubMed] [Google Scholar]
- 9. Arican, P., Gencpinar, P. and Olgac Dundar, N. (2019) A new cause of developmental and epileptic encephalopathy with continuous spike-and-wave during sleep: CDKL5 disorder. Neurocase, 25, 59–61. [DOI] [PubMed] [Google Scholar]
- 10. Lo Martire, V., Alvente, S., Bastianini, S., Berteotti, C., Silvani, A., Valli, A., Viggiano, R., Ciani, E. and Zoccoli, G. (2017) CDKL5 deficiency entails sleep apneas in mice. J. Sleep Res., 26, 495–497. [DOI] [PubMed] [Google Scholar]
- 11. Hagebeuk, E.E., van denBossche, R.A. and deWeerd, A.W. (2013) Respiratory and sleep disorders in female children with atypical Rett syndrome caused by mutations in the CDKL5 gene. Dev. Med. Child Neurol., 55, 480–484. [DOI] [PubMed] [Google Scholar]
- 12. Lee, K.Z. and Liao, W. (2018) Loss of CDKL5 disrupts respiratory function in mice. Respir. Physiol. Neurobiol., 248, 48–54. [DOI] [PubMed] [Google Scholar]
- 13. Fehr, S., Wilson, M., Downs, J., Williams, S., Murgia, A., Sartori, S., Vecchi, M., Ho, G., Polli, R., Psoni, S.et al. (2013) The CDKL5 disorder is an independent clinical entity associated with early-onset encephalopathy. Eur. J. Hum. Genet., 21, 266–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang, I.T., Allen, M., Goffin, D., Zhu, X., Fairless, A.H., Brodkin, E.S., Siegel, S.J., Marsh, E.D., Blendy, J.A. and Zhou, Z. (2012) Loss of CDKL5 disrupts kinome profile and event-related potentials leading to autistic-like phenotypes in mice. Proc. Natl. Acad. Sci. U. S. A., 109, 21516–21521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yennawar, M., White, R.S. and Jensen, F.E. (2019) AMPA receptor dysregulation and therapeutic interventions in a mouse model of CDKL5 deficiency disorder. J. Neurosci., 39, 4814–4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tang, S., Terzic, B., Wang, I.J., Sarmiento, N., Sizov, K., Cui, Y., Takano, H., Marsh, E.D., Zhou, Z. and Coulter, D.A. (2019) Altered NMDAR signaling underlies autistic-like features in mouse models of CDKL5 deficiency disorder. Nat. Commun., 10, 2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mulcahey, P.J., Tang, S., Takano, H., White, A., Davila Portillo, D.R., Kane, O.M., Marsh, E.D., Zhou, Z. and Coulter, D.A. (2020) Aged heterozygous Cdkl5 mutant mice exhibit spontaneous epileptic spasms. Exp. Neurol., 332, 113388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Terzic, B., Cui, Y., Edmondson, A.C., Tang, S., Sarmiento, N., Zaitseva, D., Marsh, E.D., Coulter, D.A. and Zhou, Z. (2021) X-linked cellular mosaicism underlies age-dependent occurrence of seizure-like events in mouse models of CDKL5 deficiency disorder. Neurobiol. Dis., 148, 105176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Born, H.A., Dao, A.T., Levine, A.T., Lee, W.L., Mehta, N.M., Mehra, S., Weeber, E.J. and Anderson, A.E. (2017) Strain-dependence of the Angelman Syndrome phenotypes in Ube3a maternal deficiency mice. Sci. Rep., 7, 8451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Copping, N.A., Adhikari, A., Petkova, S.P. and Silverman, J.L. (2019) Genetic backgrounds have unique seizure response profiles and behavioral outcomes following convulsant administration. Epilepsy Behav., 101, 106547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ferraro, T.N., Golden, G.T., Smith, G.G. and Berrettini, W.H. (1995) Differential susceptibility to seizures induced by systemic kainic acid treatment in mature DBA/2J and C57BL/6J mice. Epilepsia, 36, 301–307. [DOI] [PubMed] [Google Scholar]
- 22. Ferraro, T.N., Smith, G.G., Schwebel, C.L., Doyle, G.A., Ruiz, S.E., Oleynick, J.U., Lohoff, F.W., Berrettini, W.H. and Buono, R.J. (2010) Confirmation of multiple seizure susceptibility QTLs on chromosome 15 in C57BL/6J and DBA/2J inbred mice. Physiol. Genomics, 42A, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jazrawi, S.P. and Horton, R.W. (1986) Brain adrenoceptor binding sites in mice susceptible (DBA/2J) and resistant (C57 Bl/6) to audiogenic seizures. J. Neurochem., 47, 173–177. [DOI] [PubMed] [Google Scholar]
- 24. Spyrou, N.A., Prestwich, S.A. and Horton, R.W. (1984) Synaptosomal [3H]GABA uptake and [3H]nipecotic acid binding in audiogenic seizure susceptible (DBA/2) and resistant (C57 B1/6) mice. Eur. J. Pharmacol., 100, 207–210. [DOI] [PubMed] [Google Scholar]
- 25. Hertz, L., Schousboe, A., Formby, B. and Lennox-Buchthal, M. (1974) Some age-dependent biochemical changes in mice susceptible to seizures. Epilepsia, 15, 619–631. [DOI] [PubMed] [Google Scholar]
- 26. Adhikari, A., Copping, N.A., Beegle, J., Cameron, D.L., Deng, P., O'Geen, H., Segal, D.J., Fink, K.D., Silverman, J.L. and Anderson, J.S. (2021) Functional rescue in an Angelman syndrome model following treatment with lentivector transduced hematopoietic stem cells. Hum. Mol. Genet., 30, 1067–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chung, L., Bey, A.L., Towers, A.J., Cao, X., Kim, I.H. and Jiang, Y.H. (2018) Lovastatin suppresses hyperexcitability and seizure in Angelman syndrome model. Neurobiol. Dis., 110, 12–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ciarlone, S.L., Grieco, J.C., D'Agostino, D.P. and Weeber, E.J. (2016) Ketone ester supplementation attenuates seizure activity, and improves behavior and hippocampal synaptic plasticity in an Angelman syndrome mouse model. Neurobiol. Dis., 96, 38–46. [DOI] [PubMed] [Google Scholar]
- 29. Copping, N.A., McTighe, S.M., Fink, K.D. and Silverman, J.L. (2021) Emerging gene and small molecule therapies for the neurodevelopmental disorder angelman syndrome. Neurotherapeutics, 18, 1535–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Copping, N.A. and Silverman, J.L. (2021) Abnormal electrophysiological phenotypes and sleep deficits in a mouse model of Angelman syndrome. Mol. Autism, 12, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dodge, A., Peters, M.M., Greene, H.E., Dietrick, C., Botelho, R., Chung, D., Willman, J., Nenninger, A.W., Ciarlone, S., Kamath, S.G.et al. (2020) Generation of a novel rat model of Angelman syndrome with a complete Ube3a gene deletion. Autism Res., 13, 397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Goto, M., Saito, Y., Honda, R., Saito, T., Sugai, K., Matsuda, Y., Miyatake, C., Takeshita, E., Ishiyama, A., Komaki, H.et al. (2015) Episodic tremors representing cortical myoclonus are characteristic in Angelman syndrome due to UBE3A mutations. Brain Dev., 37, 216–222. [DOI] [PubMed] [Google Scholar]
- 33. Mandel-Brehm, C., Salogiannis, J., Dhamne, S.C., Rotenberg, A. and Greenberg, M.E. (2015) Seizure-like activity in a juvenile Angelman syndrome mouse model is attenuated by reducing Arc expression. Proc. Natl. Acad. Sci. U. S. A., 112, 5129–5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Miura, K., Kishino, T., Li, E., Webber, H., Dikkes, P., Holmes, G.L. and Wagstaff, J. (2002) Neurobehavioral and electroencephalographic abnormalities in Ube3a maternal-deficient mice. Neurobiol. Dis., 9, 149–159. [DOI] [PubMed] [Google Scholar]
- 35. Philpot, B.D., Thompson, C.E., Franco, L. and Williams, C.A. (2011) Angelman syndrome: advancing the research frontier of neurodevelopmental disorders. J. Neurodev. Disord., 3, 50–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tan, W.H., Bacino, C.A., Skinner, S.A., Anselm, I., Barbieri-Welge, R., Bauer-Carlin, A., Beaudet, A.L., Bichell, T.J., Gentile, J.K., Glaze, D.G.et al. (2011) Angelman syndrome: mutations influence features in early childhood. Am. J. Med. Genet. A, 155A, 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tan, W.H. and Bird, L.M. (2016) Angelman syndrome: current and emerging therapies in 2016. Am. J. Med. Genet. C. Semin. Med. Genet., 172, 384–401. [DOI] [PubMed] [Google Scholar]
- 38. Ahn, J. and Lee, J. (2008) X chromosome: X inactivation. Nature Education, 1(1):24. [Google Scholar]
- 39. Hagerman, R.J. and Hagerman, P.J. (2006) X inactivation and cellular mosaicism. JAMA, 296, 930–931author reply 931. [DOI] [PubMed] [Google Scholar]
- 40. Migeon, B.R. (2006) The role of X inactivation and cellular mosaicism in women's health and sex-specific diseases. JAMA, 295, 1428–1433. [DOI] [PubMed] [Google Scholar]
- 41. Migeon, B.R. (2020) X-linked diseases: susceptible females. Genet Med, 22, 1156–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Renthal, W., Boxer, L.D., Hrvatin, S., Li, E., Silberfeld, A., Nagy, M.A., Griffith, E.C., Vierbuchen, T. and Greenberg, M.E. (2018) Characterization of human mosaic Rett syndrome brain tissue by single-nucleus RNA sequencing. Nat. Neurosci., 21, 1670–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Halmai, J., Deng, P., Gonzalez, C.E., Coggins, N.B., Cameron, D., Carter, J.L., Buchanan, F.K.B., Waldo, J.J., Lock, S.R., Anderson, J.D.et al. (2020) Artificial escape from XCI by DNA methylation editing of the CDKL5 gene. Nucleic Acids Res., 48, 2372–2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Terzic, B., Davatolhagh, M.F., Ho, Y., Tang, S., Liu, Y.T., Xia, Z., Cui, Y., Fuccillo, M.V. and Zhou, Z. (2021) Temporal manipulation of Cdkl5 reveals essential postdevelopmental functions and reversible CDKL5 deficiency disorder-related deficits. J. Clin. Invest., 131, e143655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. O'Geen, H., Bates, S.L., Carter, S.S., Nisson, K.A., Halmai, J., Fink, K.D., Rhie, S.K., Farnham, P.J. and Segal, D.J. (2019) Ezh2-dCas9 and KRAB-dCas9 enable engineering of epigenetic memory in a context-dependent manner. Epigenetics Chromatin, 12, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Segal, D.J. (2019) Grand challenges in gene and epigenetic editing for neurologic disease. Front. Genome Ed., 1, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hao, S., Wang, Q., Tang, B., Wu, Z., Yang, T. and Tang, J. (2021) CDKL5 deficiency augments inhibitory input into the dentate gyrus that can be reversed by deep brain stimulation. J. Neurosci., 41, 9031–9046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Trazzi, S., De Franceschi, M., Fuchs, C., Bastianini, S., Viggiano, R., Lupori, L., Mazziotti, R., Medici, G., Lo Martire, V., Ren, E.et al. (2018) CDKL5 protein substitution therapy rescues neurological phenotypes of a mouse model of CDKL5 disorder. Hum. Mol. Genet., 27, 1572–1592. [DOI] [PubMed] [Google Scholar]
- 49. Gao, Y., Irvine, E.E., Eleftheriadou, I., Naranjo, C.J., Hearn-Yeates, F., Bosch, L., Glegola, J.A., Murdoch, L., Czerniak, A., Meloni, I.et al. (2020) Gene replacement ameliorates deficits in mouse and human models of cyclin-dependent kinase-like 5 disorder. Brain, 143, 811–832. [DOI] [PubMed] [Google Scholar]
- 50. Carrette, L.L.G., Wang, C.Y., Wei, C., Press, W., Ma, W., Kelleher, R.J., 3rd and Lee, J.T. (2018) A mixed modality approach towards Xi reactivation for Rett syndrome and other X-linked disorders. Proc. Natl. Acad. Sci. U. S. A., 115, E668–E675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Loi, M., Gennaccaro, L., Fuchs, C., Trazzi, S., Medici, G., Galvani, G., Mottolese, N., Tassinari, M., Giorgini, R.R., Milelli, A.et al. (2021) Treatment with a GSK-3beta/HDAC dual inhibitor restores neuronal survival and maturation in an in vitro and in vivo model of CDKL5 deficiency disorder. Int. J. Mol. Sci., 22, 5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Galvani, G., Mottolese, N., Gennaccaro, L., Loi, M., Medici, G., Tassinari, M., Fuchs, C., Ciani, E. and Trazzi, S. (2021) Inhibition of microglia overactivation restores neuronal survival in a mouse model of CDKL5 deficiency disorder. J. Neuroinflammation, 18, 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Olson, H.E., Daniels, C.I., Haviland, I., Swanson, L.C., Greene, C.A., Denny, A.M.M., Demarest, S.T., Pestana-Knight, E., Zhang, X., Moosa, A.N.et al. (2021) Current neurologic treatment and emerging therapies in CDKL5 deficiency disorder. J. Neurodev. Disord., 13, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Barbiero, I., Bianchi, M. and Kilstrup-Nielsen, C. (2021) Therapeutic potential of pregnenolone and pregnenolone methyl ether on depressive and CDKL5 deficiency disorders: Focus on microtubule targeting. J. Neuroendocrinol., 34, e13033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Aledo-Serrano, A., Gomez-Iglesias, P., Toledano, R., Garcia-Penas, J.J., Garcia-Morales, I., Anciones, C., Soto-Insuga, V., Benke, T.A., Del Pino, I. and Gil-Nagel, A. (2021) Sodium channel blockers for the treatment of epilepsy in CDKL5 deficiency disorder: findings from a multicenter cohort. Epilepsy Behav., 118, 107946. [DOI] [PubMed] [Google Scholar]
- 56. Adhikari, A., Copping, N.A., Onaga, B., Pride, M.C., Coulson, R.L., Yang, M., Yasui, D.H., LaSalle, J.M. and Silverman, J.L. (2018) Cognitive deficits in the Snord116 deletion mouse model for Prader-Willi syndrome. Neurobiol. Learn. Mem., 165, 106874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Copping, N. and Silverman, J. (2020) Abnormal electrophysiological and sleep deficits in a mouse model of Angelman Syndrome Mol. Autism, 12, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Copping, N.A., Berg, E.L., Foley, G.M., Schaffler, M.D., Onaga, B.L., Buscher, N., Silverman, J.L. and Yang, M. (2016) Touchscreen learning deficits and normal social approach behavior in the Shank3B model of Phelan-McDermid Syndrome and autism. Neuroscience, 345, 155–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Copping, N.A., Christian, S.G.B., Ritter, D.J., Islam, M.S., Buscher, N., Zolkowska, D., Pride, M.C., Berg, E.L., LaSalle, J.M., Ellegood, J.et al. (2017) Neuronal overexpression of Ube3a isoform 2 causes behavioral impairments and neuroanatomical pathology relevant to 15q11.2-q13.3 duplication syndrome. Hum. Mol. Genet., 26, 3995–4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dhamne, S.C., Silverman, J.L., Super, C.E., Lammers, S.H.T., Hameed, M.Q., Modi, M.E., Copping, N.A., Pride, M.C., Smith, D.G., Rotenberg, A.et al. (2017) Replicable in vivo physiological and behavioral phenotypes of the Shank3B null mutant mouse model of autism. Mol Autism, 8, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Haigh, J.L., Adhikari, A., Copping, N.A., Stradleigh, T., Wade, A.A., Catta-Preta, R., Su-Feher, L., Zdilar, I., Morse, S., Fenton, T.A.et al. (2021) Deletion of a non-canonical regulatory sequence causes loss of Scn1a expression and epileptic phenotypes in mice. Genome Med., 13, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mangatt, M., Wong, K., Anderson, B., Epstein, A., Hodgetts, S., Leonard, H. and Downs, J. (2016) Prevalence and onset of comorbidities in the CDKL5 disorder differ from Rett syndrome. Orphanet J. Rare Dis., 11, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tang, S., Wang, I.J., Yue, C., Takano, H., Terzic, B., Pance, K., Lee, J.Y., Cui, Y., Coulter, D.A. and Zhou, Z. (2017) Loss of CDKL5 in glutamatergic neurons disrupts hippocampal microcircuitry and leads to memory impairment in mice. J. Neurosci., 37, 7420–7437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Okuda, K., Takao, K., Watanabe, A., Miyakawa, T., Mizuguchi, M. and Tanaka, T. (2018) Comprehensive behavioral analysis of the Cdkl5 knockout mice revealed significant enhancement in anxiety- and fear-related behaviors and impairment in both acquisition and long-term retention of spatial reference memory. PLoS One, 13, e0196587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sivilia, S., Mangano, C., Beggiato, S., Giuliani, A., Torricella, R., Baldassarro, V.A., Fernandez, M., Lorenzini, L., Giardino, L., Borelli, A.C.et al. (2016) CDKL5 knockout leads to altered inhibitory transmission in the cerebellum of adult mice. Genes Brain Behav., 15, 491–502. [DOI] [PubMed] [Google Scholar]
- 66. Gulinello, M., Mitchell, H.A., Chang, Q., Timothy O'Brien, W., Zhou, Z., Abel, T., Wang, L., Corbin, J.G., Veeraragavan, S., Samaco, R.C.et al. (2018) Rigor and reproducibility in rodent behavioral research. Neurobiol. Learn. Mem., 165, 106780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Brigman, J.L., Daut, R.A., Saksida, L., Bussey, T.J., Nakazawa, K. and Holmes, A. (2015) Impaired discrimination learning in interneuronal NMDAR-GluN2B mutant mice. Neuroreport, 26, 489–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Brigman, J.L., Feyder, M., Saksida, L.M., Bussey, T.J., Mishina, M. and Holmes, A. (2008) Impaired discrimination learning in mice lacking the NMDA receptor NR2A subunit. Learn. Mem., 15, 50–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Marquardt, K., Josey, M., Kenton, J.A., Cavanagh, J.F., Holmes, A. and Brigman, J.L. (2019) Impaired cognitive flexibility following NMDAR-GluN2B deletion is associated with altered orbitofrontal-striatal function. Neuroscience, 404, 338–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kenton, J.A., Castillo, R., Holmes, A. and Brigman, J.L. (2018) Cortico-hippocampal GluN2B is essential for efficient visual-spatial discrimination learning in a touchscreen paradigm. Neurobiol. Learn. Mem., 156, 60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Born, H.A., Martinez, L.A., Levine, A.T., Harris, S.E., Mehra, S., Lee, W.L., Dindot, S.V., Nash, K.R., Silverman, J.L., Segal, D.J.et al. (2021) Early developmental EEG and seizure phenotypes in a full gene deletion of ubiquitin protein Ligase E3A rat model of angelman syndrome. eNeuro, 8, 0345–0361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ellegood, J., Petkova, S.P., Kinman, A., Qiu, L.R., Adhikari, A., Wade, A.A., Fernandes, D., Lindenmaier, Z., Creighton, A., Nutter, L.M.J.et al. (2021) Neuroanatomy and behavior in mice with a haploinsufficiency of AT-rich interactive domain 1B (ARID1B) throughout development. Mol. Autism, 12, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Gompers, A.L., Su-Feher, L., Ellegood, J., Copping, N.A., Riyadh, M.A., Stradleigh, T.W., Pride, M.C., Schaffler, M.D., Wade, A.A., Catta-Preta, R.et al. (2017) Germline Chd8 haploinsufficiency alters brain development in mouse. Nat. Neurosci., 20, 1062–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Amendola, E., Zhan, Y., Mattucci, C., Castroflorio, E., Calcagno, E., Fuchs, C., Lonetti, G., Silingardi, D., Vyssotski, A.L., Farley, D.et al. (2014) Mapping pathological phenotypes in a mouse model of CDKL5 disorder. PLoS One, 9, e91613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Okuda, K., Kobayashi, S., Fukaya, M., Watanabe, A., Murakami, T., Hagiwara, M., Sato, T., Ueno, H., Ogonuki, N., Komano-Inoue, S.et al. (2017) CDKL5 controls postsynaptic localization of GluN2B-containing NMDA receptors in the hippocampus and regulates seizure susceptibility. Neurobiol. Dis., 106, 158–170. [DOI] [PubMed] [Google Scholar]
- 76. Allemang-Grand, R., Ellegood, J., Spencer Noakes, L., Ruston, J., Justice, M., Nieman, B.J. and Lerch, J.P. (2017) Neuroanatomy in mouse models of Rett syndrome is related to the severity of Mecp2 mutation and behavioral phenotypes. Mol. Autism, 8, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Berg, E.L., Copping, N.A., Rivera, J.K., Pride, M.C., Careaga, M., Bauman, M.D., Berman, R.F., Lein, P.J., Harony-Nicolas, H., Buxbaum, J.D.et al. (2018) Developmental social communication deficits in the Shank3 rat model of phelan-mcdermid syndrome and autism spectrum disorder. Autism Res., 11, 587–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Berg, E.L., Jami, S.A., Petkova, S.P., Berz, A., Fenton, T.A., Lerch, J.P., Segal, D.J., Gray, J.A., Ellegood, J., Wohr, M.et al. (2021) Excessive laughter-like vocalizations, microcephaly, and translational outcomes in the Ube3a deletion rat model of Angelman syndrome. J. Neurosci., 41, 8801–8814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Berg, E.L., Pride, M.C., Petkova, S.P., Lee, R.D., Copping, N.A., Shen, Y., Adhikari, A., Fenton, T.A., Pedersen, L.R., Noakes, L.S.et al. (2020) Translational outcomes in a full gene deletion of ubiquitin protein ligase E3A rat model of Angelman syndrome. Transl. Psychiatry, 10, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ellegood, J., Anagnostou, E., Babineau, B.A., Crawley, J.N., Lin, L., Genestine, M., DiCicco-Bloom, E., Lai, J.K., Foster, J.A., Penagarikano, O.et al. (2015) Clustering autism: using neuroanatomical differences in 26 mouse models to gain insight into the heterogeneity. Mol. Psychiatry, 20, 118–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ellegood, J., Babineau, B.A., Henkelman, R.M., Lerch, J.P. and Crawley, J.N. (2013) Neuroanatomical analysis of the BTBR mouse model of autism using magnetic resonance imaging and diffusion tensor imaging. NeuroImage, 70, 288–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ellegood, J., Nakai, N., Nakatani, J., Henkelman, M., Takumi, T. and Lerch, J. (2015) Neuroanatomical phenotypes are consistent with autism-like behavioral phenotypes in the 15q11-13 duplication mouse model. Autism Res., 8, 545–555. [DOI] [PubMed] [Google Scholar]
- 83. Petkova, S.P., Pride, M., Klocke, C., Fenton, T.A., White, J., Lein, P.J., Ellegood, J., Lerch, J.P., Silverman, J.L. and Waldau, B. (2020) Cyclin D2-knock-out mice with attenuated dentate gyrus neurogenesis have robust deficits in long-term memory formation. Sci. Rep., 10, 8204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Portmann, T., Yang, M., Mao, R., Panagiotakos, G., Ellegood, J., Dolen, G., Bader, P.L., Grueter, B.A., Goold, C., Fisher, E.et al. (2014) Behavioral abnormalities and circuit defects in the basal ganglia of a mouse model of 16p11.2 deletion syndrome. Cell Rep., 7, 1077–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Silverman, J.L. and Ellegood, J. (2018) Behavioral and neuroanatomical approaches in models of neurodevelopmental disorders: opportunities for translation. Curr. Opin. Neurol., 31, 126–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Steadman, P.E., Ellegood, J., Szulc, K.U., Turnbull, D.H., Joyner, A.L., Henkelman, R.M. and Lerch, J.P. (2014) Genetic effects on cerebellar structure across mouse models of autism using a magnetic resonance imaging atlas. Autism Res., 7, 124–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Bilbo, S.D. (2018) The diverse culinary habits of microglia. Nat. Neurosci., 21, 1023–1025. [DOI] [PubMed] [Google Scholar]
- 88. Dziabis, J.E. and Bilbo, S.D. (2021) Microglia and sensitive periods in brain development. Curr. Top. Behav. Neurosci., 10.1007/7854_2021_242. [DOI] [PubMed] [Google Scholar]
- 89. Smith, C.J. and Bilbo, S.D. (2019) Microglia sculpt sex differences in social behavior. Neuron, 102, 275–277. [DOI] [PubMed] [Google Scholar]
- 90. Mao, Y., Evans, E.E., Mishra, V., Balch, L., Eberhardt, A., Zauderer, M. and Gold, W.A. (2021) Anti-Semaphorin 4D rescues motor, cognitive, and respiratory phenotypes in a Rett syndrome mouse model. Int. J. Mol. Sci., 22, 9465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Veselinovic, A., Petrovic, S., Zikic, V., Subotic, M., Jakovljevic, V., Jeremic, N. and Vucic, V. (2021) Neuroinflammation in autism and supplementation based on omega-3 polyunsaturated fatty acids: a narrative review. Medicina (Kaunas), 57, 893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Petkova, S., Adhikari, A., Berg, E., Fenton, T., Duis, J. and Silverman, J. (2022) Gait as a quantitative transaltional outcome measure. Autism Res.. 10.1002/aur.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Silverman, J.L., Nithianantharajah, J., Der-Avakian, A., Young, J.W. and Sukoff Rizzo, S.J. (2020) Lost in translation: at the crossroads of face validity and translational utility of behavioral assays in animal models for the development of therapeutics. Neurosci. Biobehav. Rev., 116, 452–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Sukoff Rizzo, S.J., Anderson, L.C., Green, T.L., McGarr, T., Wells, G. and Winter, S.S. (2018) Assessing healthspan and lifespan measures in aging mice: optimization of testing protocols, replicability, and rater reliability. Curr. Protoc. Mouse Biol., 8, e45. [DOI] [PubMed] [Google Scholar]
- 95. Sukoff Rizzo, S.J. and Silverman, J.L. (2016) Methodological considerations for optimizing and validating behavioral assays. Curr. Protoc. Mouse Biol., 6, 364–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Lupori, L., Sagona, G., Fuchs, C., Mazziotti, R., Stefanov, A., Putignano, E., Napoli, D., Strettoi, E., Ciani, E. and Pizzorusso, T. (2019) Site-specific abnormalities in the visual system of a mouse model of CDKL5 deficiency disorder. Hum. Mol. Genet., 28, 2851–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Bussey, T.J., Padain, T.L., Skillings, E.A., Winters, B.D., Morton, A.J. and Saksida, L.M. (2008) The touchscreen cognitive testing method for rodents: how to get the best out of your rat. Learn. Mem., 15, 516–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Graybeal, C., Bachu, M., Mozhui, K., Saksida, L.M., Bussey, T.J., Sagalyn, E., Williams, R.W. and Holmes, A. (2014) Strains and stressors: an analysis of touchscreen learning in genetically diverse mouse strains. PLoS One, 9, e87745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Horner, A.E., Heath, C.J., Hvoslef-Eide, M., Kent, B.A., Kim, C.H., Nilsson, S.R., Alsio, J., Oomen, C.A., Holmes, A., Saksida, L.M.et al. (2013) The touchscreen operant platform for testing learning and memory in rats and mice. Nat. Protoc., 8, 1961–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Leach, P.T. and Crawley, J.N. (2018) Touchscreen learning deficits in Ube3a, Ts65Dn and Mecp2 mouse models of neurodevelopmental disorders with intellectual disabilities. Genes Brain Behav., 17, e12452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Leach, P.T., Hayes, J., Pride, M., Silverman, J.L. and Crawley, J.N. (2016) Normal performance of Fmr1 mice on a touchscreen delayed nonmatching to position working memory Task. eNeuro, 3, 0143–0162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Morton, A.J., Skillings, E., Bussey, T.J. and Saksida, L.M. (2006) Measuring cognitive deficits in disabled mice using an automated interactive touchscreen system. Nat. Methods, 3, 767. [DOI] [PubMed] [Google Scholar]
- 103. Yang, M., Lewis, F.C., Sarvi, M.S., Foley, G.M. and Crawley, J.N. (2015) 16p11.2 Deletion mice display cognitive deficits in touchscreen learning and novelty recognition tasks. Learn. Mem., 22, 622–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Durston, S., vanBelle, J. and deZeeuw, P. (2011) Differentiating frontostriatal and fronto-cerebellar circuits in attention-deficit/hyperactivity disorder. Biol. Psychiatry, 69, 1178–1184. [DOI] [PubMed] [Google Scholar]
- 105. Brigman, J.L., Daut, R.A., Wright, T., Gunduz-Cinar, O., Graybeal, C., Davis, M.I., Jiang, Z., Saksida, L.M., Jinde, S., Pease, M.et al. (2013) GluN2B in corticostriatal circuits governs choice learning and choice shifting. Nat. Neurosci., 16, 1101–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Brigman, J.L., Powell, E.M., Mittleman, G. and Young, J.W. (2012) Examining the genetic and neural components of cognitive flexibility using mice. Physiol. Behav., 107, 666–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Graybeal, C., Feyder, M., Schulman, E., Saksida, L.M., Bussey, T.J., Brigman, J.L. and Holmes, A. (2011) Paradoxical reversal learning enhancement by stress or prefrontal cortical damage: rescue with BDNF. Nat. Neurosci., 14, 1507–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Marquardt, K., Saha, M., Mishina, M., Young, J.W. and Brigman, J.L. (2014) Loss of GluN2A-containing NMDA receptors impairs extra-dimensional set-shifting. Genes Brain Behav., 13, 611–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Marquardt, K., Sigdel, R. and Brigman, J.L. (2017) Touch-screen visual reversal learning is mediated by value encoding and signal propagation in the orbitofrontal cortex. Neurobiol. Learn. Mem., 139, 179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Thompson, S.M., Josey, M., Holmes, A. and Brigman, J.L. (2015) Conditional loss of GluN2B in cortex and hippocampus impairs attentional set formation. Behav. Neurosci., 129, 105–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Kelly, E., Escamilla, C.O. and Tsai, P.T. (2021) Cerebellar dysfunction in autism spectrum disorders: deriving mechanistic insights from an internal model framework. Neuroscience, 462, 274–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Amaral, D.G., Schumann, C.M. and Nordahl, C.W. (2008) Neuroanatomy of autism. Trends Neurosci., 31, 137–145. [DOI] [PubMed] [Google Scholar]
- 114. Li, J., Chen, X., Zheng, R., Chen, A., Zhou, Y. and Ruan, J. (2021) Altered cerebellum spontaneous activity in juvenile autism spectrum disorders associated with clinical traits. J. Autism Dev. Disord., 10.1007/s10803-021-05167-6. [DOI] [PubMed] [Google Scholar]
- 114. Palmen, S.J., vanEngeland, H., Hof, P.R. and Schmitz, C. (2004) Neuropathological findings in autism. Brain, 127, 2572–2583. [DOI] [PubMed] [Google Scholar]
- 115. Scott, J.A., Schumann, C.M., Goodlin-Jones, B.L. and Amaral, D.G. (2009) A comprehensive volumetric analysis of the cerebellum in children and adolescents with autism spectrum disorder. Autism Res., 2, 246–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Simmons, D.H., Titley, H.K., Hansel, C. and Mason, P. (2021) Behavioral tests for mouse models of autism: an argument for the inclusion of cerebellum-controlled motor behaviors. Neuroscience, 462, 303–319. [DOI] [PubMed] [Google Scholar]
- 117. Babinski, D.E. and Kujawa, A. (2021) Identifying neural markers of peer dysfunction in girls with ADHD. J. Psychiatr. Brain Sci, 6, e210022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Seymour, K.E., Reinblatt, S.P., Benson, L. and Carnell, S. (2015) Overlapping neurobehavioral circuits in ADHD, obesity, and binge eating: evidence from neuroimaging research. CNS Spectr., 20, 401–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Wang, S., Yang, Y., Xing, W., Chen, J., Liu, C. and Luo, X. (2013) Altered neural circuits related to sustained attention and executive control in children with ADHD: an event-related fMRI study. Clin. Neurophysiol., 124, 2181–2190. [DOI] [PubMed] [Google Scholar]
- 120. Bahi-Buisson, N., Nectoux, J., Rosas-Vargas, H., Milh, M., Boddaert, N., Girard, B., Cances, C., Ville, D., Afenjar, A., Rio, M.et al. (2008) Key clinical features to identify girls with CDKL5 mutations. Brain, 131, 2647–2661. [DOI] [PubMed] [Google Scholar]
- 121. Huang, H.S., Burns, A.J., Nonneman, R.J., Baker, L.K., Riddick, N.V., Nikolova, V.D., Riday, T.T., Yashiro, K., Philpot, B.D. and Moy, S.S. (2013) Behavioral deficits in an Angelman syndrome model: effects of genetic background and age. Behav. Brain Res., 243, 79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Flannery, B.M., Silverman, J.L., Bruun, D.A., Puhger, K.R., McCoy, M.R., Hammock, B.D., Crawley, J.N. and Lein, P.J. (2015) Behavioral assessment of NIH Swiss mice acutely intoxicated with tetramethylenedisulfotetramine. Neurotoxicol. Teratol., 47, 36–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Vogel Ciernia, A., Pride, M.C., Durbin-Johnson, B., Noronha, A., Chang, A., Yasui, D.H., Crawley, J.N. and LaSalle, J.M. (2017) Early motor phenotype detection in a female mouse model of Rett syndrome is improved by cross-fostering. Hum. Mol. Genet., 26, 1839–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. McTighe, S.M., Mar, A.C., Romberg, C., Bussey, T.J. and Saksida, L.M. (2009) A new touchscreen test of pattern separation: effect of hippocampal lesions. Neuroreport, 20, 881–885. [DOI] [PubMed] [Google Scholar]
- 125. Bussey, T.J., Muir, J.L., Everitt, B.J. and Robbins, T.W. (1997) Triple dissociation of anterior cingulate, posterior cingulate, and medial frontal cortices on visual discrimination tasks using a touchscreen testing procedure for the rat. Behav. Neurosci., 111, 920–936. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.