

Abstract
Type 2 diabetes is a complex, systemic disease affected by both genetic and environmental factors. Previous research has identified genetic variants associated with type 2 diabetes risk; however, gene regulatory changes underlying progression to metabolic dysfunction are still largely unknown. We investigated RNA expression changes that occur during diabetes progression using a two-stage approach. In our discovery stage, we compared changes in gene expression using two longitudinally collected blood samples from subjects whose fasting blood glucose transitioned to a level consistent with type 2 diabetes diagnosis between the time points against those who did not with a novel analytical network approach. Our network methodology identified 17 networks, one of which was significantly associated with transition status. This 822-gene network harbors many genes novel to the type 2 diabetes literature but is also significantly enriched for genes previously associated with type 2 diabetes. In the validation stage, we queried associations of genetically determined expression with diabetes-related traits in a large biobank with linked electronic health records. We observed a significant enrichment of genes in our identified network whose genetically determined expression is associated with type 2 diabetes and other metabolic traits and validated 31 genes that are not near previously reported type 2 diabetes loci. Finally, we provide additional functional support, which suggests that the genes in this network are regulated by enhancers that operate in human pancreatic islet cells. We present an innovative and systematic approach that identified and validated key gene expression changes associated with type 2 diabetes transition status and demonstrated their translational relevance in a large clinical resource.
Introduction
The molecular changes that occur during disease pathogenesis are largely unknown in common complex systemic diseases like type 2 diabetes. Technologies such as RNA sequencing enable high throughput characterization of genome-wide regulatory profiles. This approach has been utilized in cross-sectional samples of cases and controls to identify regulatory variation and differentially expressed genes associated with disease phenotypes (1). At the same time, the development of large-scale DNA databanks linked to electronic health records (EHRs) provides an opportunity to evaluate the effects of markers of disease progression on the human phenome. Our two-stage integrative approach bridges these methodologies to uniquely empower discovery of novel disease mechanisms and support known clinically relevant genes.
The Cameron County Hispanic Cohort (CCHC, n = 4800) is a randomly sampled and longitudinally measured community cohort of Mexican Americans that provides the opportunity to assess changes in RNA abundance across time (2). Individuals living in Cameron County Texas suffer from a disproportionate burden of metabolic disease, motivating analyses of molecular changes associated with metabolic deterioration over time, measured here by changes in fasting blood glucose. The prevalence of type 2 diabetes in south Texas estimated from our randomly ascertained, low-income, community recruited cohort, CCHC, is 27.6%, considerably higher than that reported for Mexican Americans nationally, with a prediabetes prevalence of 32% (3). In a recent study of CCHC participants, we found that declining metabolic health had the greatest impact on type 2 diabetes risk, independent of obesity and family history of diabetes (4).
Furthermore, the availability of a large number of human metabolic phenotypes in Vanderbilt University Medical Center’s EHR-linked DNA databank, BioVU (n ~ 96 000 total genotyped samples), enables discovery of the effects of genes across the metabolic phenome in a clinical setting (5,6).
Based on twin and family studies, the estimated heritability of type 2 diabetes ranges from 20% to 80% (7,8). Previously, genetic linkage and genome wide-association studies have successfully identified many risk variants (9–13), but to date, reported loci explain less than 15% of heritability (9,14), with chip heritability estimates as high as 18% (15). Various studies have aimed to distinguish the missing heritable components from other components such as gene–environment interaction and epigenetics (16,17); however, no studies that we are aware of have focused on gene expression changes associated with the transition to type 2 diabetes.
The level of gene expression reflects both variation in DNA sequence and interaction with other factors, e.g. other genes, environment or epigenetic regulation. Therefore, the tissue-specific expression level of RNA in an individual at a given time represents both genetic and environmental conditions (18). Because gene expression levels reflect complex and interacting genetic and environmental effects, investigation of changes in the whole transcriptomic profile during disease progression provides a novel opportunity to increase our understanding of the molecular physiology of deteriorating metabolic health.
To better understand the role of RNA expression changes in the type 2 diabetes progression, we applied a novel two-stage approach to identify a set of genes related to disease transition status and to validate their function and broader clinical implication. This innovative approach is designed to facilitate discovery of robust associations even in a small sample size, due to two primary factors: (1) our data capture a unique point of disease progression where changes in gene expression are likely be large and robust and (2) we control for many potential confounders by comparing expression changes in the same individuals across time. Here, we leverage longitudinal genome-wide RNA sequence profiling and translate findings using a large-scale DNA biobank linked to comprehensive EHRs to improve understanding of the role of gene regulation in metabolic deterioration.
Results
Overview of study design
To elucidate biological processes involved in disease progression, we used a two-stage strategy to identify and validate genes associated with transition status (Fig. 1). In the discovery stage (Fig. 1C, left), we took advantage of longitudinal specimens and data from CCHC participants who were closely followed every 3–4 months to study metabolic measures over time. We used a nested case–control study design and used RNA-sequencing from two timepoints per individual to profile the transcriptome of transition cases, who developed fasting blood glucose levels diagnostic of type 2 diabetes between the two RNA measures, and controls, who maintained a non-diabetic fasting blood glucose level at both baseline and follow-up (step 1). To aggregate gene effects and improve power, we clustered genes into networks with correlated changes in expression between the two time points across individuals (step 2). After generating the networks, we examined the association between the network’s first principal component (eigengene, see Methods) and transition status to determine networks associated with metabolic deterioration (step 3). We evaluated the performance of our approach using an independent and external large-scale genome-wide association study (GWAS) repository of previously reported and mapped genes (step 4) and assessed the extent to which our significant networks map to known biological processes using Gene Ontology (GO) enrichment (step 5). To further elucidate the function and effect of identified genes on the metabolic phenome (Table 3 and Supplementary Material, Table S3), we conducted validation in a large-scale biobank (Fig. 1C, right). We applied PrediXcan, which estimates the genetically determined component of expression, and used these estimates to test a gene’s association with EHR endocrine/metabolic disease codes (step 6) (19). To establish the enrichment of associations with the metabolic phenome, we compared our observed results to a null distribution generated from 100 000 permutations (step 7). Finally, for novel genes in significant networks (i.e. never reported in prior GWAS), we assessed their role in type 2 diabetes risk by testing the association with genetically predicted expression in BioVU (step 8). Moreover, to follow-up identified genes, we used chromatin immunoprecipitation sequencing data from human islet and determined the enrichment of activated genes in significant networks (step 9).
Figure 1.

(A) Nested case–control design. CCHC is a randomly ascertained community-based cohort and comprises over 4700 individuals (approximately 60% female). All participants are followed longitudinally. (B) Elapsed time between measures for each participant, the shape indicates their health status and arrow color represent their final group. (C) Chart of study strategy.
Table 3.
Validation of 822 genes from network 5 in BioVU (P-value < 0.05)
| Phecode | Description | Counts | P-value* | Empirical null distribution | ||||
|---|---|---|---|---|---|---|---|---|
| 1% | 5% | 50% | 95% | 99% | ||||
| 275 | Disorders of mineral metabolism | 1632 | 1×10−5 | 1336 | 1364 | 1433 | 1502 | 1531 |
| 242 | Thyrotoxicosis with or without goiter | 820 | 9×10−5 | 663 | 681 | 723 | 766 | 784 |
| 279 | Disorders involving the immune mechanism | 806 | 2.7×10−4 | 650 | 668 | 713 | 758 | 777 |
| 260 | Protein-calorie malnutrition | 1376 | 4.8×10−4 | 1172 | 1196 | 1256 | 1316 | 1341 |
| 277 | Other disorders of metabolism | 967 | 2.1×10−3 | 817 | 837 | 884 | 932 | 952 |
| 255 | Disorders of adrenal glands | 1372 | 2.9×10−3 | 1167 | 1195 | 1261 | 1328 | 1356 |
| 251 | Other disorders of pancreatic internal secretion | 395 | 4.3×10−3 | 316 | 327 | 353 | 379 | 390 |
| 269 | Proteinuria | 207 | 6.1×10−3 | 154 | 162 | 180 | 198 | 205 |
| 252 | Disorders of parathyroid gland | 598 | 7.0×10−3 | 484 | 500 | 539 | 579 | 595 |
| 257 | Testicular dysfunction | 419 | 7.7×10−3 | 319 | 333 | 368 | 403 | 417 |
| 270 | Disorders of protein plasma/amino-acid transport and metabolism | 1715 | 9.3×10−3 | 1497 | 1529 | 1605 | 1682 | 1714 |
| 264 | Lack of normal physiological development | 958 | 1.2×10−2 | 817 | 839 | 889 | 939 | 961 |
| 250 | Diabetes mellitus | 3761 | 1.2×10−2 | 3359 | 3417 | 3561 | 3707 | 3768 |
| 245 | Thyroiditis | 771 | 1.5×10−2 | 636 | 655 | 705 | 755 | 776 |
| 259 | Other endocrine disorders | 762 | 1.9×10−2 | 650 | 668 | 709 | 752 | 769 |
| 241 | Nontoxic nodular goiter | 593 | 2.2×10−2 | 489 | 506 | 545 | 585 | 601 |
| 278 | Overweight, obesity and other hyperalimentation | 984 | 2.9×10−2 | 845 | 868 | 922 | 976 | 999 |
| 276 | Disorders of fluid, electrolyte and acid–base balance | 2111 | 3.5×10−2 | 1870 | 1908 | 2004 | 2101 | 2141 |
* P-values were generated empirically (see Methods) based on random sampling.
Demographic characteristics of discovery cohort
Demographic characteristics for the participants of this study are shown in Table 1. No significant difference was observed in sex distribution between the 24 cases (20% male) and 34 controls (15% male, P-value = 0.798). The age distribution was similar at baseline, with mean age 51.8 for cases and 54.1 for controls, respectively (P-value = 0.492). Cases had significantly higher body mass index (BMI) and fasting blood glucose at baseline than controls (P-value = 0.011 for BMI, and P-value = 0.039 for fasting blood glucose). There was a significant change in fasting blood glucose for both groups over time, but in different directions. Average fasting blood glucose for cases changed from 109.3 to 139.0 mg/dl (P-value <0.001) and for controls from 104.9 to 99.3 mg/dl (P-value = 0.002). Both groups had consistent BMI over the study period (BMI from 36.0 to 37.0 and P-value = 0.580 for cases and BMI from 32.1 to 31.7, P = 0.770 for controls). HbA1C measures are not available at every time for every subject, but all 53 subjects with available HbA1C had a normal value (<6.5%) at baseline. Ten cases (17 with an available measurement) had an abnormal (>6.5%) value at follow-up, while all controls (29 with an available measurement) had normal HbA1C at follow-up.
Table 1.
Demographic and metabolic characteristics of case and control groups, which were defined by changes in fasting blood glucose
| Cases (N = 24) | Controls (N = 34) | P-value | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Baseline | Follow-up | Baseline | Follow-up | Baselinea | Casesb | Controlsc | |||||
| Male (N, %) | 5 (21%) | 5 (15%) | 0.798 | ||||||||
| Age at baseline | 51.83 ± 11.97 | 54.12 ± 12.93 | 0.492 | ||||||||
| Follow-up time (months) | 19.65 ± 13.32 | 28.70 ± 10.96 | 0.067 | ||||||||
| Smokingd | 3 (13%) | 12 (35%) | 0.099 | ||||||||
| BMI (kg/m2) | 35.97 | ±5.79 | 37.01 | ±7.04 | 32.12 | ±5.11 | 31.74 | ±5.51 | 0.012 | 0.580 | 0.770 |
| Fasting glucose (mg/dl) | 109.29 | ±7.52 | 138.92 | ±33.20 | 104.94 | ±7.99 | 99.26 | ±6.32 | 0.039 | <0.001 | 0.002 |
aBaseline P-values were based on the comparison of the value at baseline between cases and controls, t-test for continuous variables and χ2 test for dichotomous variable.
bComparing the value at baseline and follow-up within case group, paired t-test was applied.
cComparing the value at baseline and follow-up within control group, paired t-test was applied.
dMore than 100 cigarettes in participant’s entire life before enrollment.
Network analysis and identification of disease-relevant networks (step 1–3)
A single-gene approach was found to be underpowered (see Supplementary Material, Tables S1 and S2), motivating the network analysis. We identified 17 networks (Table 2), with sizes varying from 116 to 6204 genes. The variance explained by the eigengene (i.e. the first principal component) for a network ranged from 23.0% to 65.0% (Fig. 1C).
Table 2.
Identified networks and their correlations with transition status
| Networks | Number of genes | Correlation with transition | Variation explained by eigengene |
|---|---|---|---|
| Network 1 | 717 | 0.22 | 55.7% |
| Network 2 | 3672 | 0.01 | 40.2% |
| Network 3 | 2490 | 0.02 | 51.1% |
| Network 4 | 133 | 0.19 | 52.7% |
| Network 5 | 822 | 0.25 | 51.9% |
| Network 6 | 263 | 0.01 | 36.7% |
| Network 7 | 3295 | 0.05 | 23.0% |
| Network 8 | 116 | 0.24 | 55.1% |
| Network 9 | 465 | -0.05 | 46.8% |
| Network 10 | 128 | -0.11 | 47.0% |
| Network 11 | 711 | -0.01 | 64.5% |
| Network 12 | 321 | 0.12 | 47.0% |
| Network 13 | 803 | -0.13 | 47.8% |
| Network 14 | 146 | -0.15 | 45.7% |
| Network 15 | 174 | -0.19 | 43.4% |
| Network 16 | 6204 | 0.08 | 60.6% |
| Network 17 | 838 | -0.15 | 45.7% |
The network membership score (NMS) (see Methods) provides an approach to evaluate a network’s association with transition. A single network, network 5, was found to be significantly associated with transition status (r = 0.26, P-value = 0.049), as assessed by the correlation between the network’s eigengene and transition status. Notably, for this network (see Supplementary Material, Fig. S1 for its topology), the NMS of each gene (see Table 2 and Supplementary Material, Fig. S2 for NMS distribution) was significantly correlated with the gene’s association (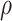 ) with transition status (
) with transition status (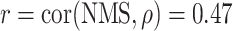 , P-value <1 × 10−15) across the 822 genes. This network is highly connected (Supplementary Material, Fig. S3) with mean connectivity = 50 and standard error of the mean (SEM) = 1.35, and within this network, the connectivity of a gene was significantly correlated with the gene’s association,
, P-value <1 × 10−15) across the 822 genes. This network is highly connected (Supplementary Material, Fig. S3) with mean connectivity = 50 and standard error of the mean (SEM) = 1.35, and within this network, the connectivity of a gene was significantly correlated with the gene’s association, 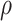 , with transition (r = 0.38, P = 3.8 × 10−29). Patterns of Δg for the top 38 genes (i.e. those genes with correlation coefficient to transition,
, with transition (r = 0.38, P = 3.8 × 10−29). Patterns of Δg for the top 38 genes (i.e. those genes with correlation coefficient to transition, 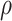 , greater than 0.30) were visualized using a heatmap (Fig. 2C). In the gene network significantly associated with transition status (network 5), the top four genes most significantly associated with transition were MIR3605, ASB9P1, NUDT16 and MKNK1-AS (
, greater than 0.30) were visualized using a heatmap (Fig. 2C). In the gene network significantly associated with transition status (network 5), the top four genes most significantly associated with transition were MIR3605, ASB9P1, NUDT16 and MKNK1-AS (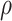 = 0.45, 0.31, 0.38 and 0.38, respectively; see Supplementary Material, Table S1). Finally, in addition to NMS (discussed above), weighted number of connections is a parameter of interest (see Supplementary Material, Fig. S3 for its intranetwork distribution). WDFY3 had the highest value for membership (0.98) and STX3 had the largest number of connections in the network (155.09). Connectivity and NMS were highly correlated (R2 = 0.85 and P = 7.88 × 10−232Fig. 2B), suggesting, as observed in standard co-expression analysis, that connectivity (a metric that determines the topology of the network) may be used as a measure of network membership (20). Furthermore, we conducted a sensitivity analysis using partial least squares discriminant analysis (PLS-DA) on the 822 genes of the implicated network with the log2 fold change (FC) residuals after regressing out the effects of age and BMI. We found that using the top two components from PLS-DA, there is a clear separation between the two groups (Supplementary Material, Fig. S4) and demonstrating that our observed association with T2D remains after adjustment (P-value = 0.02 for t-test on transition status of component 1).
= 0.45, 0.31, 0.38 and 0.38, respectively; see Supplementary Material, Table S1). Finally, in addition to NMS (discussed above), weighted number of connections is a parameter of interest (see Supplementary Material, Fig. S3 for its intranetwork distribution). WDFY3 had the highest value for membership (0.98) and STX3 had the largest number of connections in the network (155.09). Connectivity and NMS were highly correlated (R2 = 0.85 and P = 7.88 × 10−232Fig. 2B), suggesting, as observed in standard co-expression analysis, that connectivity (a metric that determines the topology of the network) may be used as a measure of network membership (20). Furthermore, we conducted a sensitivity analysis using partial least squares discriminant analysis (PLS-DA) on the 822 genes of the implicated network with the log2 fold change (FC) residuals after regressing out the effects of age and BMI. We found that using the top two components from PLS-DA, there is a clear separation between the two groups (Supplementary Material, Fig. S4) and demonstrating that our observed association with T2D remains after adjustment (P-value = 0.02 for t-test on transition status of component 1).
Figure 2.

(A) Illustration of our network methodology. In contrast to a conventional coexpression network, each node is a gene with its change in expression (Δg) over time. An edge between nodes is determined by the correlation in expression change across individuals between the corresponding nodes. Thus, the methodology aims to prioritize genes that may regulate the change in expression (Δg), rather than expression level, of partner genes. (B) Scatter plot of NMS and connectivity in network 5, showing significant correlation between the two metrics. The red line indicates the linear regression line. (C) Heatmap of key genes in network 5 (i.e. genes with correlation with transition >0.3). The color presents the log2 transformed fold change of follow-up over baseline for each gene, which we used to quantify Δg. Both subjects and genes are clustered by Ward’s minimum variance. The cluster analysis for genes is unsupervised and the subjects are forced to separate into two groups; cases and controls.
Performance of network methodology (step 4)
We found that the NMS for the significant network is significantly greater (Mann–Whitney U test P = 0.007) for known metabolic trait-associated genes identified by GWAS (Supplementary Material, Table S1) than for the remaining set of genes. As genes mapped in GWAS often play a functional role on the trait under study, this finding suggests that the network is capturing key aspects of metabolic biology (21–23). Notably, the network is also implicating novel genes, i.e. genes that have not been reported by previous GWAS, as perhaps expected given the methodology’s novel focus on identifying genes that influence changes in expression (Δg) (Fig. 1C).
GO enrichment for biological processes (step 5)
GO analysis implicated immune and inflammatory processes (Benjamini–Hochberg false discovery rate [FDR] < 0.05) (Fig. 3A), consistent with a number of recent studies that show the importance of chronic systemic inflammation or immunity preceding the onset of type 2 diabetes (Fig. 1C) (24–27).
Figure 3.

(A) Dot plot of gene ontology enrichment analysis. The diameter indicates the number of genes overlapping the gene ontology term and the color indicates the enrichment P-value. The figure was generated using the R package clusterProfiler. (B) Bar plot for the fold enrichment of 822 genes and their exonic regions in our identified network compared to all RefSeq genes and corresponding exonic regions in enhancer, poised enhancer, active enhancer and insulator regions. (C) Known GWAS T2D SNP (rs735949) overlaps islet transcription factor binding sites from ChIP-Seq data. The transcript structure of the gene ACSL1 from module 5 is also shown at bottom.
Enrichment of genetically determined expression associated with trait in a large-scale biobank (step 6–8)
The 822-gene network was found to be significantly enriched for associations with diabetes mellitus (enrichment P-value = 0.012), disorders of pancreatic internal secretion (P-value = 0.004), overweight, obesity and other hyperalimentation (P-value = 0.029), and most metabolism disorders (protein plasma/amino-acid transport and metabolism, P-value = 0.009; other metabolism disorder, P-value = 0.002). Lipid metabolism disorders were not observed to be significantly enriched in this gene set, nor was the phecode selected to be a negative control, secondary diabetes mellitus (Table 3 and Fig. 1C).
Notably, among the validated genes, we identified several that have never been reported by GWAS, including STX3, SIGKEC5, TMEM260 and MAPK3 (Supplementary Material, Table S1 for complete list).
Functional follow-up in human islets (step 9)
In human islet, ChromHMM analysis identified 16 different chromatin states. The specific chromatin states—as defined by (1) (high) H4K3me1 and (high or low) H3K27ac (which characterize strong enhancers and poised enhancers, respectively) and (2) high signal for the islet-specific transcription factors (28)—are enriched in the 822 genes (Fig. 3B and Supplementary Material, Fig. S5). Compared to all RefSeq genes, the 822 genes are more likely to overlap with these specific regulatory elements, i.e. 1.26-fold enrichment for gene regions and 1.56 for exon regions for poised enhancers, and 1.18-fold enrichment for gene regions and 0.99 for exon regions for strong enhancers (Fig. 3B and Supplementary Material, Fig. S5), indicating that islet regulatory regions near these genes are functionally active (Fig. 1C).
As a vignette to highlight the functional relevance of our discoveries, we mapped a single variant, rs735949 (a GWAS-identified T2D SNP intronic to Acyl-CoA Synthetase Long Chain Family Member 1, ACSL1, within our network (29)) to local genomic sequences or regulatory elements targeted by islet transcription factors (Supplementary Material, Table S1). We found that the variant also disrupts regulatory elements targeted by the five beta cell transcription factors PDX1, NKX6, NKX2, H2AZ and FOXA2 to influence ACSL1 islet transcription (Fig. 3C). Taken together, these findings in islets lend additional support for the functional relevance of these genes identified in blood for T2D biology.
Discussion
There are notable strengths to our study. First and foremost, this is a landmark, longitudinal study at the border of Mexico, uniquely capturing longitudinal molecular signatures of a major health disparity in a minority population with one of the highest burdens of type 2 diabetes worldwide. Our clinic in the heart of town and our commitment to community engagement allowed for extensive phenotypic characterization of this population. Most prospective studies of complex diseases focus on identifying baseline predictors for future incidence; for instance, several risk factors for the incidence of type 2 diabetes have been identified, e.g. adiponectin, C-reactive protein, and interleukin 6 (30–32). However, the development of a complex disease is often not adequately contextualized as a binary trait; indeed the development of the disease may take place over decades and in the context of distinct genetic and environmental contexts. Thus, longitudinal study designs that measure both genetic and environmental effects are warranted. Gene expression is dynamic over time and often reflects complex interactions between genetic factors and environmental conditions. RNA abundance is therefore well-suited to elucidate pathogenic mechanisms of complex disease like type 2 diabetes, as has been shown for studies of the inflammation process in humans (33,34). Furthermore, we took advantage of multiple independent data sets to validate our innovative discovery approach and findings, including exploring the translational effects of our identified genes in a large-scale biobank with linked EHRs.
The novelty of our longitudinal design presented opportunities and challenges for making robust and verifiable biological inferences. The discovery stage of our study included a relatively small sample size, which limited our power to identify genes from gene-level analysis of changes in expression genome-wide (see Supplemental Materials). This limitation prompted joint analysis of the effect of genes using a network methodology, in which we were well-powered to detect networks with modest to large effect size. Second, type 2 diabetes is a complex systemic disease with diverse clinical characteristics even among diagnosed patients. Here, we used fasting glucose to define the different phases of diabetic development, which, although a crucial element, may not capture genes related to other diagnostic criteria of diabetes such as HbA1C and 2-h post-load glucose levels. Third, our discovery stage analysis examined changes in expression in circulating blood samples, which may or may not be representative of the transcriptome in other tissues relevant to metabolic dysfunction. However, circulating whole blood is the diagnostic tissue for type 2 diabetes, is easily accessible and is practical for human population-based studies. Furthermore, it has been shown, using a broad collection of human tissues (35,36), that there is substantial sharing of regulatory variation across tissues. To directly address the issue of tissue specificity, in our validation stage, we demonstrated an enrichment of active genes of our identified genes in human beta islet cells. Lastly, our biobank validation stage is based on the associations between imputed gene expression and diagnosed phecodes. Therefore, the predictive power of the PrediXcan models may limit the accuracy of our imputed genetically regulated expression; however, these models have been tested on a broad spectrum of complex traits and shown notable power to discover trait-associated genes (37). Models trained in European ancestry samples are known to show decreased portability across ethnic groups. Cross-population PrediXcan analysis would decrease power but not create false positive results (38). Despite these challenges, we present an innovative approach that robustly identified and functionally validated a novel gene set in type 2 diabetes progression.
In the transcriptome discovery stage of this study, we followed 58 subjects with an initial fasting blood glucose measure <126 mg/dl for up to 5 years, during which 24 cases transitioned to fasting blood glucose levels diagnostic for diabetes over a mean follow-up of 2 years (Fig. 1C, step 1). In approximately the same time span, the 34 controls maintained fasting blood glucose levels <126 mg/dl. In this group of 58 individuals, we investigated patterns of gene expression via RNA sequencing data analysis at both baseline and follow-up and clustered genes with correlated changes in expression over time to identify 17 networks of genes (Fig. 1C, step 2). Our network methodology differs from conventional coexpression analysis. In particular, rather than focusing on cross-sectional expression profiles, our approach focuses on change in expression as the unit of analysis (i.e. the node) and identifies genes with correlated expression changes (i.e. edges). Only one network, containing 822 genes, was significantly associated with transition status (Fig. 1C, step 3). We found a significant association of NMS with gene member correlation coefficient to fasting blood glucose transition, suggesting that in this network, the greater a gene’s correlation with network variance, the greater the gene’s correlation with transition status. The heatmap of genes significantly correlated with case/control status from the network of interest, shown in Figure 1, demonstrates a visible separation and consistent pattern of RNA expression FC between cases and controls.
Due to the novelty of this approach, we evaluated the performance of the methodology using several external reference datasets including a repository of known GWAS findings and GO (Fig. 1C, steps 4–5). We found that our NMS and connectivity are significantly associated with an external gene prioritization scheme based on the GWAS catalog of metabolic traits (see ‘Assessing performance of network methodology…’ in Methods). This finding suggests that our methodology is able to capture key aspects of our current understanding of metabolic biology. Furthermore, subsequent GO enrichment analysis of our significant gene network highlights the role of inflammation and immunity in type 2 diabetes pathogenesis.
The identified network is large and likely contains both genes where changes in expression are causally associated with transition status or where transition has caused changes in expression, as well as genes where expression changes are merely correlated with causal changes due to, e.g. co-regulation. Thus, we sought to prioritize genes of highest interest that may warrant additional study. In this network, STX3 exhibited the highest connectivity, and WDFY3 had the highest membership score, suggesting that these genes are both highly representative of the aggregate effect of the network and may play a central role in biological processes that the network encompasses. WDFY3 encodes a phosphatidylinositol 3-phosphate-binding protein and has been reported to play a role in the WNT signaling pathway (39). The WNT signaling pathway is involved in many fundamental molecular functions, including lipid and glucose metabolism (40), and another gene in the WNT pathway, TCF7L2, is a known diabetic risk gene identified in many genome-wide association studies (11,13,41). In addition, STX3 is a member of the syntaxin family, which has a known function in the insulin synthesis and insulin signaling pathways (42–44), such as regulating insulin granular exocytosis and compound fusion in pancreatic beta cells (45). STX3 regulation was also found to be associated with type 2 diabetes pathogenesis in our independent biobank analyses.
Four genes within the network, MIR3605, ASB9P1, NUDT16 and MKNK1-AS, demonstrated the highest correlation with transition status. None of these genes have been previously implicated in genetic association analyses of type 2 diabetes. MIR3605 is a microRNA, a short non-coding RNA that participates in transcriptional regulation. Upregulation of MIR3605 has been observed in nasopharyngeal carcinoma tissue (46); notably, type 2 diabetes has been reported as a risk factor for the incidence of nasopharyngeal carcinoma (47), providing support for the existence of shared genetic risk for both diabetes and nasopharyngeal carcinoma. ASB9P1 is a pseudogene of ASB9, Ankyrin Repeat and SOCS Box Containing 9. ASB9 is involved in the regulation pathway of SOCS box, which inhibits the insulin signaling pathway and is connected to the development of insulin resistance (48,49). MKNK1-AS is the antisense RNA for MKNK1, MAP Kinase Interacting Serine/Threonine Kinase 1. The function of MKNK1 is related to cellular energy balance, and its expression is affected by high-fat diet (50,51). In animal studies, the Mknk1 knockout mice with high-fat diet express a better glucose tolerance and insulin sensitivity, suggesting that MKNK1 may be involved in insulin signaling and further insulin resistance (51). NUDT16 encodes Nudix hydrolase 16, a member of the Nudix hydrolase superfamily that plays a role in pyrimidine metabolism; however, its relationship with diabetes is still unclear.
To validate the biological relevance of our findings, we performed systematic validation of our findings using a large EHR-linked DNA biobank and beta islet chromatin immunoprecipitation data. In summary, we found significant enrichment of known metabolic trait genes, identified additional support for novel disease genes based on models of genetic regulation and observed enrichment of activated genes in beta islet cell ChiPseq data. Finally, we leveraged a large DNA biobank with linked EHR to evaluate the translational relevance of our genetic findings on the broader metabolic phenome.
In the biobank validation stage, we sought to (1) validate the role of our identified set of genes and (2) establish their broader clinical significance in type 2 diabetes risk and related disorders. It is important to note that the analysis in the transcriptome discovery stage cannot differentiate between expression changes resulting from deterioration of metabolic health and expression changes that are causally responsible for the observed metabolic changes. However, because genetic factors are intrinsic (i.e. not altered by disease), if genetic regulation of expression (as opposed to e.g. environmental or disease-state effects) is associated with metabolic disease in the biobank validation stage, this finding not only validates the role of the gene but also suggests that the observed expression changes may in fact be acting in a causal manner. To explore the effects of genetic regulation of the genes identified by the network analysis, we analyzed association between genetically regulated expression levels [based on PrediXcan models across all tissues derived from Genotype-Tissue Expression Project (GTEx) v6p] and clinical phenotypes in BioVU (Fig. 1C, step 6). We tested for enrichment for specific diabetes-related disorders, including pancreatic internal secretion disorder, obesity and other metabolic disorders (Fig. 1C, step 7). The 822 genes we identified were found to be significantly enriched for associations with diabetes mellitus and related metabolism disorders. Mineral metabolism disorders were the most significantly enriched; the association of mineral metabolism with diabetes and the possible mechanism of comorbidity have been discussed in previous studies (52–54). Bone fracture is a severe outcome of abnormal mineral metabolism, and risk increases 2-fold in type 2 diabetes patients (55). Additionally, we identified the disease category ‘other disorder of metabolism’, which includes metabolic syndrome and other lipoid metabolism disorders. Metabolic syndrome is a strong predictor for incidence of type 2 diabetes, with subjects with metabolic syndrome having three times greater type 2 diabetes incidence than those without metabolic syndrome (56). We used secondary diabetes mellitus as negative control and found that our identified genes are not significantly enriched in secondary diabetes mellitus (see Table 3). Collectively, the significant enrichment for associations between genes identified in our transcriptome discovery stage and phecodes in BioVU related to metabolic health such as diabetes mellitus, obesity and metabolic syndrome suggests that dysregulation (whether intrinsic or dynamic) of this set of genes may be pathogenic for metabolic health.
The tissue specificity of eQTLs is typically overestimated by simple overlap (due to incomplete power and reliance on a significance threshold), but more statistically sophisticated approaches have shown eQTL sharing across tissues to be substantial (57). This motivated inclusion of PrediXcan associations from all GTEx tissues in our study. Indeed, ChromHMM analysis of the 822 genes confirmed the functional relevance of our findings in human islet and showed an enrichment of enhancer regions in this cell type relative to all RefSeq genes.
Within the 822-gene network, genetically predicted expression levels of 31 novel genes that had not previously been identified by GWAS were significantly associated with type 2 diabetes in BioVU (Benjamini–Hochberg FDR <0.10, see Supplementary Material, Table S1). Of particular interest, SIGLEC5 encodes a protein of the sialic acid-binding immunoglobulin-like lectin (SIGLEC) family, with known function in inhibiting the activation of monocytes, macrophages and neutrophils (58). Recent evidence suggests the function of another protein in the SIGLEC family, Siglec-7, in improving β-cells’ function and reducing inflammation in pancreatic islets from diabetic patients (59). This novel gene finding is further supported by GO enrichment analysis, which found up to ~8% of genes in our significant network were associated with inflammatory process or immune response.
In summary, we present a novel approach for characterizing gene expression patterns associated with deteriorating glucose metabolism and assessing their translational relevance on the related metabolic phenome. Towards this end, we implemented an unsupervised approach using longitudinal transcriptome data to identify gene networks associated with transition of blood glucose levels to a diagnostic level for diabetes. This analysis identified a network of genes with effects on expression changes that are significantly associated with transition status. We evaluated the performance of the methodology by showing that the genes within the significant network overlap GWAS-implicated loci with glucose and insulin metabolism-related function. Furthermore, we showed that this network is significantly enriched for genes whose genetically regulated expression level is associated with type 2 diabetes and type 2 diabetes-related disorders in a large DNA databank, providing strong evidence that the gene network is enriched for genes that are a risk factor for, rather than a consequence of type 2 diabetes. Notably, this efficient approach enabled the discovery of a gene network associated with transition to diabetes in a limited sample size. Future, larger studies building on our study design and approach may further clarify the molecular mechanisms of diabetes development, establish the pattern of causality leading to the observed diabetes-associated expression changes and validate additional gene networks.
Materials and Methods
Transcriptome discovery stage
Study subjects
In this study, we employed a nested case–control study design. The CCHC was established on the Texas-Mexico border in 2004 (2). This randomly ascertained community cohort currently comprises over 4900 people and is approximately 60% female. All participants are followed longitudinally with 5-, 10- and 15-year follow-up visits. At each visit, extensive examinations included blood samples drawn following a confirmed 8-h fast. This study was approved by the Committee for the Protection of Human Subjects of the University of Texas Health Science Center at Houston.
Within this cohort, a nested sample of 286 people was selected based on having fasting glucose <126 mg/dl, when measured, HbA1C < 6.5%, and no prior diagnosis of type 2 diabetes. These individuals were then intensively followed up every 3–4 months over 5 years to track their metabolic status (60). From the 286, we selected 24 subjects whose fasting glucose transitioned to levels diagnostic for T2D over the study period (cases, with fasting blood glucose ≥126 mg/dl during follow-up) and 34 who did not transition (controls, with fasting blood glucose <126 mg/dl, and when measured, HbA1C < 6.5% during follow-up, see Fig. 1A and B). The fasting blood specimen taken at baseline and at the latest follow-up visits of these 58 subjects was subjected to RNA sequencing. Fasting blood glucose changes were defined using the time points at which RNA specimens were available. Specimens used for RNA sequencing for controls were taken after approximately the same or more elapsed time as cases (controls, average time 28.70 months; cases, average time 19.65 months, Table 1).
Experimental measurement
A fasting peripheral blood specimen was collected using PaxGene tubes for each participant at both baseline and at each follow-up visit every 3–4 months over a period of as many as 60 months (mean follow-up time 24.98 months, Table 1) (60). Specimens were stored at −80°C within 20 min of the draw. Fasting glucose was measured in a CLIA-approved laboratory. RNA was extracted from all specimens and stored at −80°C. RNA sequencing was performed with Illumina HiSeq 2000 at the Baylor Sequencing Center following standard protocols (61). The samples were sequenced in four batches with a mean RNA integrity (RIN) 8.11 (Supplementary Material, Fig. S8). The RNA sequencing library for each sample ranged in size from 6.9 to 49 million reads. All raw sequencing read libraries were checked with FastQC to ensure their sequencing quality (62), and all samples passed the quality requirements. All RNA sequencing reads were aligned to the human reference genome (hg19; Illumina iGenomes reference transcriptome, UCSC known genes) by Spliced Transcripts Alignment to a Reference (STAR, version 2.5.0a), an RNA sequence mapping tool, which considers both annotated and unannotated splice junctions as well as other mismatches or insertion/deletion during read alignment (63). Then, STAR was also used to count the number of mapped reads in each gene. Genes without mapped reads in over 10% of samples or with median absolute deviation equal to 0 were excluded from further analysis. In total, 21 298 genes were expressed in over 10% of subjects, had median absolute deviation greater than 0 and were included in further analysis, consistent with standard quality control approaches for RNA sequencing (64).
DEseq2 v1.30.1 was used to normalize the gene expression matrix (Fig. 1C step 1), considering the dispersion of gene expression and the sequencing depth of each sample (65). The dispersion was scaled to fit a smooth dispersion curve from empirical data, and sequencing depth was estimated using a median-of-ratios approach. We also considered the impact of hidden or unmeasured covariates on the gene expression difference between baseline and follow-up using Probabilistic Estimation of Expression Residual factors (see Supplementary Text) (66).
Network analysis of longitudinal expression profile
We used a modified weighted gene correlation network analysis (WGCNA) (version 1.6.8) to cluster genes into sparse networks (67) (Fig. 1C step 2). Although our methodology is novel, we leveraged well-established and commonly used network construction approaches and metrics (67). In contrast to conventional coexpression network analysis, each node is a gene g with its change in expression (Δg) between the two time points. An edge between two nodes g1 and g2 represents the correlation cor(Δg1, Δg2) across individuals (Fig. 2A). An edge, therefore, indicates an effect on expression change (Δg); the effect may be due to several factors, for example it may be a causal effect, due to coregulation, or it may be spurious. The input matrix was log2 transformed FC in normalized expression (from DEseq2) over time, which we used to quantify Δg.
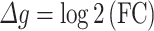 |
Explicitly, FC over time was defined as the ratio of the normalized gene expression at follow-up over the normalized gene expression at baseline (Fig. 1C). The topological overlap dissimilarity, a robust measure of interconnectedness which is based on shared network neighbors (68), was used to describe the connection between genes and as input for further steps. Hierarchical clustering and dynamic tree cut methods were performed in R to build the networks and to cut peripheral genes. Each network’s eigengene was defined as the first principal component of the network and calculated within WGCNA, and this eigengene was used to evaluate the correlation between network and transition status. To explore whether networks significantly associated with transition status remain significant after adjusting for covariates, we created residual measures of 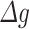 after regressing out the effects of the covariates age and BMI. We then performed a sensitivity analysis using PLS-DA with the residuals and evaluated the performance of top components on distinguishing transition status.
after regressing out the effects of the covariates age and BMI. We then performed a sensitivity analysis using PLS-DA with the residuals and evaluated the performance of top components on distinguishing transition status.
A hub gene, defined as a highly connected gene within a network, was identified by the weighted number of genes connected to it in the specific network. The degree of network membership for a gene g was calculated from the correlation between the gene’s expression change and the network’s eigengene (E), resulting in a NMS.
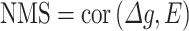 |
Both the weighted number of connections (‘connectivity’) and NMS were determined in an unsupervised fashion during the network construction, independently of transition status. For a gene g, we can determine its association with transition status using logistic regression.
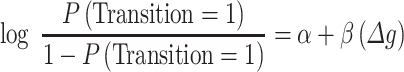 |
This allowed us to assess the association of 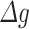 for a single gene within a network of interest on transition status (Fig. 1C step 3). The goal of this step is to prioritize potential genes of interest (e.g. those highlighted in Fig. 2C) and to determine the correlation of gene’s network membership and connectivity scores with association to transition status. All the genes within the identified networks, regardless of individual association with transition status, were carried forward to the validation stage.
for a single gene within a network of interest on transition status (Fig. 1C step 3). The goal of this step is to prioritize potential genes of interest (e.g. those highlighted in Fig. 2C) and to determine the correlation of gene’s network membership and connectivity scores with association to transition status. All the genes within the identified networks, regardless of individual association with transition status, were carried forward to the validation stage.
For network visualization, we used the R package igraph. We highlighted hub genes and their network partners with highest correlation (among the top 5% of all edges).
Assessing performance of network methodology using comprehensive GWAS repository
We evaluated whether networks significantly associated with transition status from our approach reflect known understanding of metabolic biology using an external dataset. We compiled a list of reported or mapped genes that have been identified by GWAS of metabolic traits, as curated in the EMBL-EBI GWAS repository (69) (downloaded on June 3, 2019). We included diseases and traits based on the American Diabetes Association’s definition of metabolic syndrome (70), including blood pressure, blood glucose, serum lipids, obesity, body mass index, waist-to-hip ratio, insulin resistance, and type 2 diabetes. We tested whether the NMS is significantly greater for metabolic trait associated genes (which defines a gene significance score, MetabolicScore = 1) than for the remaining set of genes (MetabolicScore = 0), using the non-parametric Mann–Whitney U test.
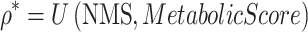 |
This test allowed us to assess the performance of the network methodology using an independent and external ‘gene significance’ scheme (MetabolicScore) for metabolic traits.
GO analysis
We performed enrichment analyses of networks significantly associated with transition status to identify enriched GO biological processes. We used the R package clusterProfiler to identify enriched annotations (with their gene count and P-value), using the set of genes in the networks significantly associated with transition status as input. All the three major gene ontologies from the GO project were used, including biological process, molecular function, and cellular component (71). We used Benjamini–Hochberg FDR < 0.05 as the significance threshold.
Biobank validation and functional follow-up stage
Study material
The biobank at Vanderbilt University Medical Center contains over 95 000 MEGAEX genotyped subjects with linked EHR (5). The genetically regulated expression of 23 000 genotyped subjects was imputed using PrediXcan (19) with models trained using GTEx V.6p data (72) (Fig. 1C step 4). The number of unique genes passing a threshold of R2 > 0.01 varied by tissue; the minimum number is 2041 from vagina and the maximum is 8023 from tibial nerve. In total, 17 481 unique genes passed this imputation threshold and were used in downstream associations between imputed genetically regulated expression and phecodes derived from ICD-9 codes. These effects were estimated genome-wide for the metabolic phenome (Table 3) using logistic regression with age and sex as covariates.
Enrichment analysis
To validate the networks of genes from the transcriptome discovery stage, we assessed their enrichment for gene-level associations with diabetes and related phenotypes in BioVU (Fig. 1C step 5). Phecodes tested for enrichment were chosen as the class of endocrine/metabolic diseases (as externally defined by the PheWAS Map 1.2), including diabetes mellitus (phecode 250), disorders of pancreatic internal secretion (251) overweight, obesity and other hyperalimentation (278) and other related metabolism disorders, including protein plasma/amino acid transport and metabolism (270), lipoid metabolism (272), mineral metabolism (275), goiter (240 and 241), thyroid disorder (244, 245, and 246), other metabolism disorders (277, including disorders of bilirubin excretion, metabolic syndrome, etc.) and other endocrine disorders. In addition, we also evaluated the enrichment for secondary diabetes mellitus (249), a condition characterized by the destruction of the beta-cells in pancreatic islets or the induction of insulin resistance resulting from an acquired disease, as a negative control. To obtain the empirical P-value, we counted the number of nominally significant associations, P-value < 0.05, between our identified genes and each selected phecode and measured the count against an empirical null distribution generated by random sampling. We simulated random draws of the same number of genes 100 000 times and established a null distribution from the observed number of times an association with a phecode of interest was seen by chance.
Functional follow-up in human islets
We analyzed ChIP-seq chromatin data for key islet transcription factors to show an approach to functional follow-up, in human islets, of the genes within the identified network that is correlated with transition status. These data (28) (downloaded from ArrayExpress: E-MTAB-1919) allowed us to test whether islet gene regulation might provide further functional insights into our discoveries. We applied a hidden Markov model approach to characterize chromatin states using ChromHMM (73). Binarized maps from the ChIP-seq bed files were generated using the BinaryBed function within ChromHMM. Following guidelines suggested by the ChromHMM authors, we tested numbers of states varying from 10 to 30 and found that the result with 16 states best describes the chromatin status. Sixteen chromatin states were identified using the LearnModel function with hg18 and default settings. Finally, the OverlapEnrichment function was used to evaluate the enrichment of different chromatin states in gene sets compared to RefSeq genes. For additional details and exact commands, please refer to GitHub repository (link provided under Code availability).
Supplementary Material
Acknowledgements
The authors would like to thank our cohort team, particularly Rocío Uribe, BSIE, and her team, who recruited and interviewed the participants, Marcela Morris, BS, for laboratory support, the data management team, and Norma Pérez-Olazarán, BBA, and Christina Villarreal, BA, for administrative support; Valley Baptist Medical Center, Brownsville, Texas, for providing us space for our Center for Clinical and Translational Science Clinical Research Unit; and the community of Brownsville and the participants who so willingly participated in this study in their city. E.R.G. is grateful to the President and Fellows of Clare Hall, University of Cambridge for fellowship support.
Conflict of Interest statement. The authors declare no competing interests. E.R.G. and J.E.B. receive an honorarium from Circulation Research, the official journal of the American Heart Association, as members of the Editorial Board.
Contributor Information
Hung-Hsin Chen, Vanderbilt Genetics Institute and Division of Genetic Medicine, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN 37232, USA.
Lauren E Petty, Vanderbilt Genetics Institute and Division of Genetic Medicine, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN 37232, USA.
Kari E North, Department of Epidemiology, University of North Carolina, Chapel Hill, NC 27599, USA.
Joseph B McCormick, The University of Texas Health Science Center at Houston (UTHealth) School of Public Health, Brownsville, TX 78520, USA.
Susan P Fisher-Hoch, The University of Texas Health Science Center at Houston (UTHealth) School of Public Health, Brownsville, TX 78520, USA.
Eric R Gamazon, Vanderbilt Genetics Institute and Division of Genetic Medicine, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN 37232, USA; Clare Hall, University of Cambridge, Cambridgeshire, UK.
Jennifer E Below, Vanderbilt Genetics Institute and Division of Genetic Medicine, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN 37232, USA.
Supplementary Material
Supplementary Material is available at HMGJ online.
Ethics approval and consent to participate
CCHC portion of this study was approved by the Committee for the Protection of Human Subjects of the University of Texas Health Science Center, Houston. BioVU data use was reviewed by the institutional review board at Vanderbilt University Medical Center and determined that the study does not qualify as ‘human subject’ research per §46.102(f)(2) under IRB# 151187.
Data Availability
The datasets used and/or analyzed for the current study are available on GEO (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE184050).
Code availability
Exact parameters and scripts used to run the analyses presented in this manuscript can be found at our GitHub repository (https://github.com/belowlab/CCHC-T2D-RNAseq).
Funding
Center for Clinical and Translational Sciences, National Institutes of Health Clinical and Translational Award (grant no. UL1 TR000371) from the National Center for Advancing Translational Sciences; predoctoral fellowship from American Heart Association (no. 18PRE34060101 to H-H.C.); NIH/NHGRI (R35HG010718, R01HG011138 to E.R.G.); NIH/NIGMS (R01GM140287 to E.R.G.); National Institutes of Health, National Heart, Lung, and Blood Institute (grant no. R01HL142302 to J.E.B., H-H.C., L.E.P., K.E.N., J.B.M, and S.P.F-H.). The dataset(s) used for the analyses described were obtained from Vanderbilt University Medical Center’s BioVU, which is supported by numerous sources: institutional funding, private agencies and federal grants. These include the NIH funded Shared Instrumentation Grant S10RR025141; and CTSA grants UL1TR002243, UL1TR000445 and UL1RR024975. Genomic data are also supported by investigator-led projects from NIH that include U01HG004798, R01NS032830, RC2GM092618, P50GM115305, U01HG006378, U19HL065962, R01HD074711; and additional funding sources listed at https://victr.vumc.org/biovu-funding/.
Authors’ contributions
H-H.C. conducted the computational analyses of expression data and wrote the manuscript. L.E.P. contributed to the experimental design and assisted in writing the manuscript. K.E.N. helped interpret results and write the manuscript. S.P.F-H. leads the Cameron County Hispanic Cohort, collected patient information and biological specimens, contributed to the experimental design and edited the manuscript. J.B.M. designed the study and oversaw RNA sequencing, sample selection and helped write the manuscript. E.R.G. contributed to study design, provided critical expertise on RNA sequencing and network analysis, helped with the computational analyses and interpretation of results and helped write the manuscript. J.E.B. oversaw all computational analyses, data interpretation and wrote the manuscript.
References
- 1. Jenkinson, C.P., Goring, H.H., Arya, R., Blangero, J., Duggirala, R. and DeFronzo, R.A. (2016) Transcriptomics in type 2 diabetes: bridging the gap between genotype and phenotype. Genom Data, 8, 25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fisher-Hoch, S.P., Rentfro, A.R., Salinas, J.J., Perez, A., Brown, H.S., Reininger, B.M., Restrepo, B.I., Wilson, J.G., Hossain, M.M., Rahbar, M.H.et al. (2010) Socioeconomic status and prevalence of obesity and diabetes in a Mexican American community, Cameron County, Texas, 2004–2007. Prev. Chronic Dis., 7, A53. [PMC free article] [PubMed] [Google Scholar]
- 3. Fisher-Hoch, S.P., Vatcheva, K.P., Rahbar, M.H. and McCormick, J.B. (2015) Undiagnosed diabetes and pre-diabetes in health disparities. PLoS One, 10, e0133135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wu, S., Fisher-Hoch, S.P., Reninger, B., Vatcheva, K. and McCormick, J.B. (2016) Metabolic health has greater impact on diabetes than simple overweight/obesity in Mexican Americans. J. Diabetes Res., 2016, 4094876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Roden, D.M., Pulley, J.M., Basford, M.A., Bernard, G.R., Clayton, E.W., Balser, J.R. and Masys, D.R. (2008) Development of a large-scale de-identified DNA biobank to enable personalized medicine. Clin. Pharmacol. Ther., 84, 362–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McGregor, T.L., Van Driest, S.L., Brothers, K.B., Bowton, E.A., Muglia, L.J. and Roden, D.M. (2013) Inclusion of pediatric samples in an opt-out biorepository linking DNA to de-identified medical records: pediatric BioVU. Clin. Pharmacol. Ther., 93, 204–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Meigs, J.B., Cupples, L.A. and Wilson, P.W. (2000) Parental transmission of type 2 diabetes: the Framingham Offspring Study. Diabetes, 49, 2201–2207. [DOI] [PubMed] [Google Scholar]
- 8. Poulsen, P., Kyvik, K.O., Vaag, A. and Beck-Nielsen, H. (1999) Heritability of type II (non-insulin-dependent) diabetes mellitus and abnormal glucose tolerance—a population-based twin study. Diabetologia, 42, 139–145. [DOI] [PubMed] [Google Scholar]
- 9. Ali, O. (2013) Genetics of type 2 diabetes. World J. Diabetes, 4, 114–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Barroso, I. (2005) Genetics of type 2 diabetes. Diabet. Med., 22, 517–535. [DOI] [PubMed] [Google Scholar]
- 11. Zeggini, E., Weedon, M.N., Lindgren, C.M., Frayling, T.M., Elliott, K.S., Lango, H., Timpson, N.J., Perry, J.R., Rayner, N.W., Freathy, R.M.et al. (2007) Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science, 316, 1336–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Diabetes Genetics Initiative of Broad Institute of, H, Mit, L.U., Novartis Institutes of BioMedical, R, Saxena, R., Voight, B.F., Lyssenko, V., Burtt, N.P., deBakker, P.I., Chen, H., Roix, J.J.et al. (2007) Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science, 316, 1331–1336. [DOI] [PubMed] [Google Scholar]
- 13. Sladek, R., Rocheleau, G., Rung, J., Dina, C., Shen, L., Serre, D., Boutin, P., Vincent, D., Belisle, A., Hadjadj, S.et al. (2007) A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature, 445, 881–885. [DOI] [PubMed] [Google Scholar]
- 14. Manolio, T.A., Collins, F.S., Cox, N.J., Goldstein, D.B., Hindorff, L.A., Hunter, D.J., McCarthy, M.I., Ramos, E.M., Cardon, L.R., Chakravarti, A.et al. (2009) Finding the missing heritability of complex diseases. Nature, 461, 747–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mahajan, A., Taliun, D., Thurner, M., Robertson, N.R., Torres, J.M., Rayner, N.W., Payne, A.J., Steinthorsdottir, V., Scott, R.A., Grarup, N.et al. (2018) Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet., 50, 1505–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yang, B.T., Dayeh, T.A., Volkov, P.A., Kirkpatrick, C.L., Malmgren, S., Jing, X., Renstrom, E., Wollheim, C.B., Nitert, M.D. and Ling, C. (2012) Increased DNA methylation and decreased expression of PDX-1 in pancreatic islets from patients with type 2 diabetes. Mol. Endocrinol., 26, 1203–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cornelis, M.C. and Hu, F.B. (2012) Gene-environment interactions in the development of type 2 diabetes: recent progress and continuing challenges. Annu. Rev. Nutr., 32, 245–259. [DOI] [PubMed] [Google Scholar]
- 18. Mohr, S. and Liew, C.C. (2007) The peripheral-blood transcriptome: new insights into disease and risk assessment. Trends Mol. Med., 13, 422–432. [DOI] [PubMed] [Google Scholar]
- 19. Gamazon, E.R., Wheeler, H.E., Shah, K.P., Mozaffari, S.V., Aquino-Michaels, K., Carroll, R.J., Eyler, A.E., Denny, J.C., Consortium, G.T., Nicolae, D.L.et al. (2015) A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet., 47, 1091–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Horvath, S. and Dong, J. (2008) Geometric interpretation of gene coexpression network analysis. PLoS Comput. Biol., 4, e1000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pietzner, M., Wheeler, E., Carrasco-Zanini, J., Cortes, A., Koprulu, M., Worheide, M.A., Oerton, E., Cook, J., Stewart, I.D., Kerrison, N.D.et al. (2021) Mapping the proteo-genomic convergence of human diseases. Science, 374, eabj1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Backman, J.D., Li, A.H., Marcketta, A., Sun, D., Mbatchou, J., Kessler, M.D., Benner, C., Liu, D., Locke, A.E., Balasubramanian, S.et al. (2021) Exome sequencing and analysis of 454,787 UK biobank participants. Nature, 599, 628–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nasser, J., Bergman, D.T., Fulco, C.P., Guckelberger, P., Doughty, B.R., Patwardhan, T.A., Jones, T.R., Nguyen, T.H., Ulirsch, J.C., Lekschas, F.et al. (2021) Genome-wide enhancer maps link risk variants to disease genes. Nature, 593, 238–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Donath, M.Y. and Shoelson, S.E. (2011) Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol., 11, 98–107. [DOI] [PubMed] [Google Scholar]
- 25. Romeo, G.R., Lee, J. and Shoelson, S.E. (2012) Metabolic syndrome, insulin resistance, and roles of inflammation—mechanisms and therapeutic targets. Arterioscler. Thromb. Vasc. Biol., 32, 1771–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Margaryan, S., Witkowicz, A., Arakelyan, A., Partyka, A., Karabon, L. and Manukyan, G. (2018) sFasL-mediated induction of neutrophil activation in patients with type 2 diabetes mellitus. PLoS One, 13, e0201087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Richard, C., Wadowski, M., Goruk, S., Cameron, L., Sharma, A.M. and Field, C.J. (2017) Individuals with obesity and type 2 diabetes have additional immune dysfunction compared with obese individuals who are metabolically healthy. BMJ Open Diabetes Res. Care, 5, e000379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pasquali, L., Gaulton, K.J., Rodriguez-Segui, S.A., Mularoni, L., Miguel-Escalada, I., Akerman, I., Tena, J.J., Moran, I., Gomez-Marin, C., van deBunt, M.et al. (2014) Pancreatic islet enhancer clusters enriched in type 2 diabetes risk-associated variants. Nat. Genet., 46, 136–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xue, A., Wu, Y., Zhu, Z., Zhang, F., Kemper, K.E., Zheng, Z., Yengo, L., Lloyd-Jones, L.R., Sidorenko, J., Wu, Y.et al. (2018) Genome-wide association analyses identify 143 risk variants and putative regulatory mechanisms for type 2 diabetes. Nat. Commun., 9, 2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Duncan, B.B., Schmidt, M.I., Pankow, J.S., Bang, H., Couper, D., Ballantyne, C.M., Hoogeveen, R.C. and Heiss, G. (2004) Adiponectin and the development of type 2 diabetes: the atherosclerosis risk in communities study. Diabetes, 53, 2473–2478. [DOI] [PubMed] [Google Scholar]
- 31. Li, S., Shin, H.J., Ding, E.L. and vanDam, R.M. (2009) Adiponectin levels and risk of type 2 diabetes: a systematic review and meta-analysis. JAMA, 302, 179–188. [DOI] [PubMed] [Google Scholar]
- 32. Pradhan, A.D., Manson, J.E., Rifai, N., Buring, J.E. and Ridker, P.M. (2001) C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA, 286, 327–334. [DOI] [PubMed] [Google Scholar]
- 33. Bar-Joseph, Z., Gitter, A. and Simon, I. (2012) Studying and modelling dynamic biological processes using time-series gene expression data. Nat. Rev. Genet., 13, 552–564. [DOI] [PubMed] [Google Scholar]
- 34. Calvano, S.E., Xiao, W., Richards, D.R., Felciano, R.M., Baker, H.V., Cho, R.J., Chen, R.O., Brownstein, B.H., Cobb, J.P., Tschoeke, S.K.et al. (2005) A network-based analysis of systemic inflammation in humans. Nature, 437, 1032–1037. [DOI] [PubMed] [Google Scholar]
- 35. GTEx Consortium, Laboratory Data Analysis, Coordinating Center-Analysis Working Group, Statistical Methods groups-Analysis Working Group, Enhancing, G.G., Fund, N.I.H.C., Nih/Nci, Nih/Nhgri, Nih/Nimh, Nih/Nida et al. (2017) Genetic effects on gene expression across human tissues. Nature, 550, 204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. GTEx Consortium (2015) Human genomics. The genotype-tissue expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science, 348, 648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gamazon, E.R., Segre, A.V., van deBunt, M., Wen, X., Xi, H.S., Hormozdiari, F., Ongen, H., Konkashbaev, A., Derks, E.M., Aguet, F.et al. (2018) Using an atlas of gene regulation across 44 human tissues to inform complex disease- and trait-associated variation. Nat. Genet., 50, 956–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Colbran, L.L., Gamazon, E.R., Zhou, D., Evans, P., Cox, N.J. and Capra, J.A. (2019) Inferred divergent gene regulation in archaic hominins reveals potential phenotypic differences. Nat Ecol Evol, 3, 1598–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kadir, R., Harel, T., Markus, B., Perez, Y., Bakhrat, A., Cohen, I., Volodarsky, M., Feintsein-Linial, M., Chervinski, E., Zlotogora, J.et al. (2016) ALFY-controlled DVL3 autophagy regulates Wnt Signaling, determining human brain size. PLoS Genet., 12, e1005919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jin, T. (2008) The WNT signalling pathway and diabetes mellitus. Diabetologia, 51, 1771–1780. [DOI] [PubMed] [Google Scholar]
- 41. Scott, L.J., Mohlke, K.L., Bonnycastle, L.L., Willer, C.J., Li, Y., Duren, W.L., Erdos, M.R., Stringham, H.M., Chines, P.S., Jackson, A.U.et al. (2007) A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science, 316, 1341–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kang, Y., Huang, X., Pasyk, E.A., Ji, J., Holz, G.G., Wheeler, M.B., Tsushima, R.G. and Gaisano, H.Y. (2002) Syntaxin-3 and syntaxin-1A inhibit L-type calcium channel activity, insulin biosynthesis and exocytosis in beta-cell lines. Diabetologia, 45, 231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Spurlin, B.A., Park, S.Y., Nevins, A.K., Kim, J.K. and Thurmond, D.C. (2004) Syntaxin 4 transgenic mice exhibit enhanced insulin-mediated glucose uptake in skeletal muscle. Diabetes, 53, 2223–2231. [DOI] [PubMed] [Google Scholar]
- 44. Lam, P.P., Leung, Y.M., Sheu, L., Ellis, J., Tsushima, R.G., Osborne, L.R. and Gaisano, H.Y. (2005) Transgenic mouse overexpressing syntaxin-1A as a diabetes model. Diabetes, 54, 2744–2754. [DOI] [PubMed] [Google Scholar]
- 45. Zhu, D., Koo, E., Kwan, E., Kang, Y., Park, S., Xie, H., Sugita, S. and Gaisano, H.Y. (2013) Syntaxin-3 regulates newcomer insulin granule exocytosis and compound fusion in pancreatic beta cells. Diabetologia, 56, 359–369. [DOI] [PubMed] [Google Scholar]
- 46. Li, S., Hang, L., Ma, Y. and Wu, C. (2016) Distinctive microRNA expression in early stage nasopharyngeal carcinoma patients. J. Cell. Mol. Med., 20, 2259–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Peng, X.S., Xie, G.F., Qiu, W.Z., Tian, Y.H., Zhang, W.J. and Cao, K.J. (2016) Type 2 diabetic mellitus is a risk factor for nasopharyngeal carcinoma: a 1:2 matched case-control study. PLoS One, 11, e0165131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rui, L., Yuan, M., Frantz, D., Shoelson, S. and White, M.F. (2002) SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J. Biol. Chem., 277, 42394–42398. [DOI] [PubMed] [Google Scholar]
- 49. Ueki, K., Kondo, T. and Kahn, C.R. (2004) Suppressor of cytokine signaling 1 (SOCS-1) and SOCS-3 cause insulin resistance through inhibition of tyrosine phosphorylation of insulin receptor substrate proteins by discrete mechanisms. Mol. Cell. Biol., 24, 5434–5446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhu, X., Dahlmans, V., Thali, R., Preisinger, C., Viollet, B., Voncken, J.W. and Neumann, D. (2016) AMP-activated protein kinase up-regulates mitogen-activated protein (MAP) kinase-interacting serine/threonine kinase 1a-dependent phosphorylation of eukaryotic translation initiation factor 4E. J. Biol. Chem., 291, 17020–17027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Moore, C.E., Pickford, J., Cagampang, F.R., Stead, R.L., Tian, S., Zhao, X., Tang, X., Byrne, C.D. and Proud, C.G. (2016) MNK1 and MNK2 mediate adverse effects of high-fat feeding in distinct ways. Sci. Rep., 6, 23476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liamis, G., Liberopoulos, E., Barkas, F. and Elisaf, M. (2014) Diabetes mellitus and electrolyte disorders. World J. Clin. Cases, 2, 488–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wahl, P., Xie, H., Scialla, J., Anderson, C.A., Bellovich, K., Brecklin, C., Chen, J., Feldman, H., Gutierrez, O.M., Lash, J.et al. (2012) Earlier onset and greater severity of disordered mineral metabolism in diabetic patients with chronic kidney disease. Diabetes Care, 35, 994–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ghodsi, M., Larijani, B., Keshtkar, A.A., Nasli-Esfahani, E., Alatab, S. and Mohajeri-Tehrani, M.R. (2016) Mechanisms involved in altered bone metabolism in diabetes: a narrative review. J. Diabetes Metab. Disord., 15, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hofbauer, L.C., Brueck, C.C., Singh, S.K. and Dobnig, H. (2007) Osteoporosis in patients with diabetes mellitus. J. Bone Miner. Res., 22, 1317–1328. [DOI] [PubMed] [Google Scholar]
- 56. Lorenzo, C., Okoloise, M., Williams, K., Stern, M.P., Haffner, S.M., Heart, S.A. and S. (2003) The metabolic syndrome as predictor of type 2 diabetes: the San Antonio heart study. Diabetes Care, 26, 3153–3159. [DOI] [PubMed] [Google Scholar]
- 57. Flutre, T., Wen, X., Pritchard, J. and Stephens, M. (2013) A statistical framework for joint eQTL analysis in multiple tissues. PLoS Genet., 9, e1003486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cornish, A.L., Freeman, S., Forbes, G., Ni, J., Zhang, M., Cepeda, M., Gentz, R., Augustus, M., Carter, K.C. and Crocker, P.R. (1998) Characterization of siglec-5, a novel glycoprotein expressed on myeloid cells related to CD33. Blood, 92, 2123–2132. [PubMed] [Google Scholar]
- 59. Dharmadhikari, G., Stolz, K., Hauke, M., Morgan, N.G., Varki, A., deKoning, E., Kelm, S. and Maedler, K. (2017) Siglec-7 restores beta-cell function and survival and reduces inflammation in pancreatic islets from patients with diabetes. Sci. Rep., 7, 45319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wu, S., McCormick, J.B., Curran, J.E. and Fisher-Hoch, S.P. (2017) Transition from pre-diabetes to diabetes and predictors of risk in Mexican-Americans. Diabetes Metab Syndr Obes, 10, 491–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kumar, R., Ichihashi, Y., Kimura, S., Chitwood, D.H., Headland, L.R., Peng, J., Maloof, J.N. and Sinha, N.R. (2012) A high-throughput method for Illumina RNA-Seq library preparation. Front. Plant Sci., 3, 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Andrews, S. (2010), FastQC: A Quality Control Tool for High Throughput Sequence Data [Online]. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/, in press.
- 63. Dobin, A., Davis, C.A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., Batut, P., Chaisson, M. and Gingeras, T.R. (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Conesa, A., Madrigal, P., Tarazona, S., Gomez-Cabrero, D., Cervera, A., McPherson, A., Szczesniak, M.W., Gaffney, D.J., Elo, L.L., Zhang, X.et al. (2016) A survey of best practices for RNA-seq data analysis. Genome Biol., 17, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Love, M.I., Huber, W. and Anders, S. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol., 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Stegle, O., Parts, L., Durbin, R. and Winn, J. (2010) A Bayesian framework to account for complex non-genetic factors in gene expression levels greatly increases power in eQTL studies. PLoS Comput. Biol., 6, e1000770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Langfelder, P. and Horvath, S. (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics, 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Li, A. and Horvath, S. (2009) Network module detection: affinity search technique with the multi-node topological overlap measure. BMC. Res. Notes, 2, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hindorff, L.A., Sethupathy, P., Junkins, H.A., Ramos, E.M., Mehta, J.P., Collins, F.S. and Manolio, T.A. (2009) Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. U. S. A., 106, 9362–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kahn, R., Buse, J., Ferrannini, E. and Stern, M. (2005) The metabolic syndrome: time for a critical appraisal. Joint statement from the American Diabetes Association and the European Association for the Study of diabetes. Diabetologia, 48, 1684–1699. [DOI] [PubMed] [Google Scholar]
- 71. Gene Ontology Consortium (2008) The gene ontology project in 2008. Nucleic Acids Res., 36, D440–D444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. GTEx Consortium (2013) The genotype-tissue expression (GTEx) project. Nat. Genet., 45, 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ernst, J. and Kellis, M. (2017) Chromatin-state discovery and genome annotation with ChromHMM. Nat. Protoc., 12, 2478–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed for the current study are available on GEO (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE184050).