

Abstract
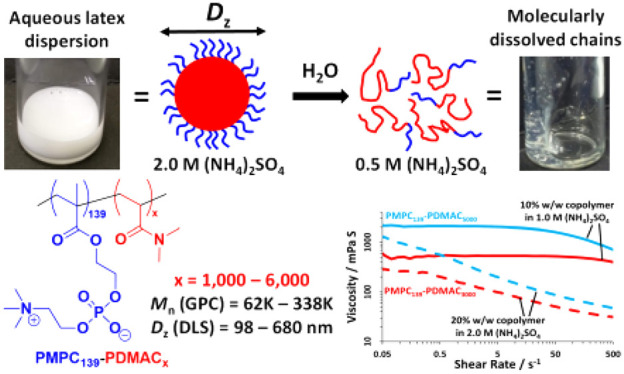
We report the synthesis of sterically-stabilized diblock copolymer particles at 20% w/w solids via reversible addition–fragmentation chain transfer (RAFT) aqueous dispersion polymerization of N,N′-dimethylacrylamide (DMAC) in highly salty media (2.0 M (NH4)2SO4). This is achieved by selecting a well-known zwitterionic water-soluble polymer, poly(2-(methacryloyloxy)ethyl phosphorylcholine) (PMPC), to act as the salt-tolerant soluble precursor block. A relatively high degree of polymerization (DP) can be targeted for the salt-insoluble PDMAC block, which leads to the formation of a turbid free-flowing dispersion of PDMAC-core particles by a steric stabilization mechanism. 1H NMR spectroscopy studies indicate that relatively high DMAC conversions (>99%) can be achieved within a few hours at 30 °C. Aqueous GPC analysis indicates high blocking efficiencies and unimodal molecular weight distributions, although dispersities increase monotonically as higher degrees of polymerization (DPs) are targeted for the PDMAC block. Particle characterization techniques include dynamic light scattering (DLS) and electrophoretic light scattering (ELS) using a state-of-the-art instrument that enables accurate ζ potential measurements in a concentrated salt solution. 1H NMR spectroscopy studies confirm that dilution of the as-synthesized dispersions using deionized water lowers the background salt concentration and hence causes in situ molecular dissolution of the salt-intolerant PDMAC chains, which leads to a substantial thickening effect and the formation of transparent gels. Thus, this new polymerization-induced self-assembly (PISA) formulation enables high molecular weight water-soluble polymers to be prepared in a highly convenient, low-viscosity form. In principle, such aqueous PISA formulations are highly attractive: there are various commercial applications for high molecular weight water-soluble polymers, while the well-known negative aspects of using a RAFT agent (i.e., its cost, color, and malodor) are minimized when targeting such high DPs.
Introduction
It is well-known that reversible addition-fragmentation chain transfer (RAFT) polymerization enables the synthesis of a wide range of functional vinyl polymers with good control over the molecular weight distribution.1−4 There are many literature examples of RAFT solution homopolymerization and, in some cases, mean degrees of polymerization (DP) up to (and even beyond) 10,000 have been targeted.5−7 This latter aspect is interesting for two reasons. First, in the case of water-soluble polymers (e.g., polyacrylamide), such high molecular weights are useful for commercial applications such as flocculants, binders, or thickeners.8−10 Second, the main disadvantage of RAFT chemistry is that the chain transfer agent is an organosulfur compound, which is relatively expensive and confers both malodor and color.11 Since the mean DP is inversely proportional to the concentration of this RAFT agent,12 targeting very high DPs minimizes the problems associated with its use, which could make a decisive difference to the feasibility of industrial scale-up.13
However, the synthesis of high molecular weight water-soluble polymers via RAFT aqueous solution polymerization leads to extremely viscous reaction mixtures. For example, gel formation was reported by Destarac and co-workers when preparing polyacrylamide with a mean DP of around 10,000.6 Such gels can be difficult to remove from the reaction vessel after the polymerization, and heat dissipation during polymerization can become inefficient. In principle, this problem could be addressed by conducting such polymerizations in highly salty media. Under such conditions, the water-soluble polymer chains become insoluble, which leads to the formation of low-viscosity, free-flowing particulate dispersions rather than highly viscous or gel-like aqueous solutions.14 Indeed, this approach is used to prepare high molecular weight polyacrylamide in the form of particles via conventional free radical polymerization conducted in aqueous solution in the presence of 2.0 M ammonium sulfate.15−17
Polymerization-induced self-assembly (PISA) involves the growth of an insoluble block from a soluble precursor block in a suitable solvent. In the case of an aqueous PISA formulation, the growing second block becomes water-insoluble while the first block remains water-soluble: the resulting amphiphilic diblock copolymer chains undergo micellar nucleation and ultimately sterically-stabilized diblock copolymer nanoparticles are produced. Depending on the aqueous solubility of the monomer used to generate the hydrophobic block, aqueous PISA formulations can involve either RAFT aqueous emulsion polymerization18−20 or RAFT aqueous dispersion polymerization.21−26 In both cases, the hydrophobic block normally remains insoluble at the end of the polymerization. However, there are several literature examples in which a temperature switch leads to in situ nanoparticle dissolution to yield molecularly dissolved diblock copolymer chains.27−29 We hypothesized that a similar approach might involve the synthesis of a salt-intolerant water-soluble polymer in highly salty media to produce low-viscosity particles. Subsequent dilution using pure water would then lower the salt concentration in the aqueous continuous phase, which should lead to the molecular dissolution of the high molecular weight copolymer chains within the particle cores and hence a strong thickening effect (see Scheme 1).
Scheme 1. Schematic Cartoon and Corresponding Digital Images to Illustrate the Sterically Stabilized Diblock Copolymer Particles in the Presence of 2.0 M Ammonium Sulfate Obtained after RAFT Aqueous Dispersion Polymerization of a Suitable Water-Soluble Monomer to Form the “Salted Out” Red Chains.
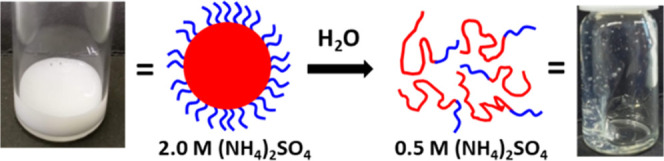
A four-fold dilution with deionized water lowers the salt concentration of the initial aqueous dispersion and results in molecular dissolution of these particles, with the concomitant formation of a highly viscous transparent aqueous solution.
According to well-established principles in colloid science, steric stabilization is much more likely to be effective than charge stabilization for such aqueous PISA syntheses.30−32 Clearly, such formulations would require a steric stabilizer that remains soluble in the presence of substantial amounts of salt to confer effective colloidal stabilization. According to the literature, suitable salt-tolerant water-soluble polymeric stabilizers are likely to be either certain types of polyelectrolytes16,17,33,34 or polybetaines.35−37
Herein, we report the RAFT aqueous dispersion polymerization of N,N′-dimethylacrylamide (DMAC) in highly salty media using poly(2-(methacryloyloxy)ethyl phosphorylcholine) (PMPC) as a salt-tolerant steric stabilizer. According to the literature, PMPC remains soluble even in the presence of 5.0 M NaCl.35 This approach is then extended to include polyelectrolytic steric stabilizers.
Experimental Section
Materials
2-(Methacryloyloxy)ethyl phosphorylcholine (MPC) was kindly donated by Biocompatibles (U.K.), and ammonium sulfate was purchased from Alfa Aesar (U.K.) and Eisen-Golden Laboratories (CA). 2-Ethylhexanoyl tert-butyl peroxide (T21S) was obtained from AkzoNobel (Netherlands), potassium hydroxide was obtained from LabChem (PA), and 2,2′-azobis(2-imidazolinylpropane) dihydrochloride (VA-044) was obtained from Fluorochem (U.K.). N,N′-Dimethylacrylamide (DMAC), ascorbic acid (AsAc), potassium persulfate (KPS), 4,4′-azobis(4-cyanopentanoic acid) (ACVA), azobis(isobutyl)amidine dihydrochloride (AIBA), CH3COOH, NaH2PO4, NaNO3, KOH, a 50% solution of 2-acrylamido-2-methyl-1-propanesulfonic acid sodium salt (AMPS), phosphate buffer solution (PBS) tablets, and D2O were purchased from Sigma-Aldrich (U.K.). Each of these chemicals was used as received.
2-(Acryloyloxy)ethyl trimethylammonium chloride (ATAC) was donated by BASF (Germany) in the form of an 80% w/w aqueous solution. 4-Cyano-4-(2-phenylethanesulfanylthiocarbonyl)-sulfanylpentanoic acid (PETTC) was prepared and purified as reported elsewhere,38 as was 2-(((butylthio)carbonothioyl)thio)-2-methylpropanoic acid (BDMAT).39,40 All solvents were purchased from Fisher Scientific (U.K.) and were used as received.
Synthesis Protocols
Synthesis of the PMPC139 Precursor via RAFT Solution Polymerization of 2-(Methacryloyloxy)ethyl phosphorylcholine (MPC) in Methanol at 64 °C
PETTC (250 mg, 0.74 mmol), MPC (26.10 g, 88.4 mmol), ACVA (41 mg, 150 μmol), and methanol (49.0 g, corresponding to a 35% w/w solution) were weighed into a 250 mL round-bottom flask charged with a magnetic flea and this reaction solution was degassed using nitrogen gas for 45 min at 20 °C. The sealed flask was immersed into an oil bath set at 64 °C for 210 min, and the polymerization was subsequently quenched by exposing the reaction mixture to air while cooling to 20 °C. The final MPC conversion was 75%, as judged by 1H NMR spectroscopy (calculated by comparing the integrated vinyl signals assigned to the MPC monomer at 5.6–6.2 ppm to the integrated polymethacrylic backbone signals at 0.6–2.4 ppm). The reaction solution was precipitated into a ten-fold excess of acetone. The crude PMPC precursor was redissolved in methanol, and the precipitation was repeated. After dissolution using deionized water, the resulting aqueous polymer solution was freeze-dried overnight. The degree of polymerization was 135 ± 10, as judged by 1H NMR spectroscopy (calculated by comparing the integrated aromatic signals assigned to the RAFT end-group at 7.1–7.4 ppm to the integrated polymethacrylic backbone signals at 0.6–2.4 ppm). RAFT end-group analysis using UV spectroscopy indicated a mean degree of polymerization of 139 ± 1 (the Beer–Lambert plot for PETTC is provided in Figure S1). Aqueous GPC analysis indicated an Mn of 17 kg mol–1 and an Mw/Mn of 1.18 (see below for eluent and calibration details).
Preparation of 2.0 M Ammonium Sulfate Solution and Redox Initiator Solutions
Ammonium sulfate (26.43 g) was added to a 100 mL volumetric flask, which was subsequently filled with water to obtain a 2.0 M solution. The required molarity, refractive index, and dynamic viscosity for an aqueous solution of 2.0 M ammonium sulfate were calculated by interpolation of tabulated solution properties reported at 20 °C as recorded in Table S1.41 The interpolated dynamic viscosity was estimated to 25 °C using the ratio of the dynamic viscosity for water at 20 and 25 °C. The following numerical values for a 2.0 M aqueous solution of ammonium sulfate were used in this study: molality = 2.32 mol kg–1; refractive index = 1.370, and dynamic viscosity = 1.367 × 10–3 kg m–1 s–1. The relative permittivity of a 2.0 M aqueous solution of ammonium sulfate was assumed to be that of pure water. According to the literature, the addition of salt leads to a lower relative permittivity compared to that of water.42 However, this systematic error is not considered to be important relative to the likely error incurred when calculating the ζ potential for electrosterically-stabilized nanoparticles43 (see Results and Discussion for further details).
KPS (30.0 mg) was dissolved in an aqueous solution of 2.0 M ammonium sulfate (30 g) to make up a 0.1% w/w KPS stock solution. Similarly, AsAc (30.0 mg) was dissolved in an aqueous solution of 2.0 M ammonium sulfate (30 g) to make up a 0.1% w/w AsAc stock solution. Each stock solution was stored in a refrigerator at 4 °C prior to use.
Synthesis of PMPC139–PDMACx Diblock Copolymer Particles via RAFT Aqueous Dispersion Polymerization of N,N′-Dimethylacrylamide (DMAC) in 2 M Ammonium Sulfate at 30 °C
A typical protocol for the synthesis of PMPC139–PDMAC5000 spheres at 20% w/w solids was conducted as follows. The PMPC139 precursor (140 mg, 4.0 μmol), DMAC (1.972 g, 19.9 mmol), the 0.1% aqueous solution of KPS (270 mg, 1.0 μmol), and an aqueous solution of 2.0 M ammonium sulfate (8.00 g) were weighed into a 25 mL round-bottom flask charged with a magnetic flea and this reaction solution was degassed using nitrogen gas for 30 min at 20 °C. The sealed flask was immersed into an oil bath set at 30 °C, and a 0.1% aqueous solution of AsAc (170 mg, 1.0 μmol) was added to initiate the DMAC polymerization. After 18 h, the polymerization was subsequently quenched by exposing the reaction mixture to air while cooling to 20 °C. The final DMAC conversion was more than 99%, as judged by 1H NMR spectroscopy (as calculated by comparing the integrated vinyl signals assigned to the DMAC monomer at 5.6–6.7 ppm to the integrated polyacrylamide backbone signals at 1.1–2.7 ppm). Aqueous GPC analysis indicated an Mn of 262 kg mol–1 and an Mw/Mn of 1.97 (see below for eluent and calibration details).
Synthesis of PATAC195–PDMAC1000 Diblock Copolymer Particles via RAFT Aqueous Dispersion Polymerization
PETTC (290 mg, 0.85 mmol), ATAC (80% w/w in water) (41.35 g, 170 mmol), AIBA (46 mg, 170 μmol), and methanol (50.1 g, corresponding to a 40% w/w solution) were weighed into a 250 mL round-bottom flask charged with a magnetic flea, and this reaction solution was degassed using nitrogen gas for 45 min at 20 °C. The sealed flask was immersed into an oil bath set at 56 °C for 120 min, and the polymerization was then quenched by exposing the reaction mixture to air while cooling to 20 °C. The final ATAC conversion was 97%, as judged by 1H NMR spectroscopy (calculated by comparing the integrated vinyl signals assigned to the ATAC monomer at 5.8–6.4 ppm to the integrated polyacrylic backbone signals at 1.3–2.7 ppm). Excess water was added, and the methanol was removed under reduced pressure. Afterward, the reaction solution was purified by dialysis over 3 days with regular water changes. The resulting aqueous polymer solution was freeze-dried overnight. The degree of polymerization was 195, as judged by 1H NMR spectroscopy (calculated by comparing the integrated aromatic signals assigned to the RAFT end-group at 7.1–7.4 ppm to the integrated polyacrylic backbone signals at 1.3–2.7 ppm). Aqueous GPC analysis indicated an Mn of 34 kg mol–1 and an Mw/Mn of 1.20 (see below for eluent and calibration details).
Subsequently, the PATAC195 precursor (120 mg, 3.2 μmol), DMAC (312 mg, 3.15 mmol), a 0.1% aqueous solution of VA-044 (339 mg, 1.0 μmol), and an aqueous solution of 2.0 M sulfate (3.55 g) were weighed into a 10 mL round-bottom flask charged with a magnetic stirrer, and this reaction solution was degassed using nitrogen gas for 30 min at 20 °C. The sealed flask was immersed into an oil bath set at 48 °C to initiate the DMAC polymerization. After 18 h, the polymerization was quenched by exposing the reaction mixture to air while cooling to 20 °C. The final DMAC conversion was more than 99%, as judged by 1H NMR spectroscopy (as calculated by comparing the integrated vinyl signals assigned to the DMAC monomer at 5.6–6.7 ppm to the integrated polyacrylamide backbone signals at 1.1–2.7 ppm). Aqueous GPC analysis indicated an Mn of 68 kg mol–1 and an Mw/Mn of 1.95 (see below for eluent and calibration details).
Synthesis of PAMPS250–PDMAC1000 Diblock Copolymer Particles via RAFT Aqueous Dispersion Polymerization
BDMAT (150 mg, 0.59 mmol), AMPS (55% w/w) (61.92 g, 149 mmol), T21S (25.7 mg, 119 μmol), and 1.0 M PBS (23.5 g, corresponding to a 40% w/w solution) were weighed into a 250 mL round-bottom flask charged with a magnetic flea, and this reaction solution was degassed using nitrogen gas for 45 min at 20 °C. The sealed flask was immersed into an oil bath set at 90 °C for 150 min and the polymerization was subsequently quenched by exposing the reaction mixture to air while cooling to 20 °C. The final AMPS conversion was 99% as judged by 1H NMR spectroscopy (calculated by comparing the integrated vinyl signals assigned to the AMPS monomer at 5.6–6.2 ppm to the integrated polyacrylic backbone signals at 1.2–2.3 ppm). The reaction solution was purified by dialysis against water for three days. The resulting aqueous polymer solution was freeze-dried overnight. The degree of polymerization was 250 as judged by 1H NMR spectroscopy (calculated by comparing the integrated methyl signals assigned to the RAFT end-group at 0.8–0.9 ppm to the integrated acrylic backbone signals at 1.2–2.3 ppm). Aqueous GPC analysis indicated an Mn of 28 kg mol–1 and an Mw/Mn of 1.35 (see below for eluent and calibration details).
Subsequently, the PAMPS250 precursor (220 mg, 3.8 μmol), DMAC (379 mg, 3.82 mmol), a 0.1% aqueous solution of VA-044 (344 mg, 1.3 μmol), and an aqueous solution of 2.0 M (NH4)2SO4 (2.05 g) were weighed into a 10 mL round-bottom flask charged with a magnetic stirrer, and this reaction solution was degassed using nitrogen gas for 30 min at 20 °C. The sealed flask was immersed into an oil bath set at 48 °C to initiate the DMAC polymerization. After 18 h, the polymerization was quenched by exposing the reaction mixture to air while cooling to 20 °C. The final DMAC conversion was more than 99% as judged by 1H NMR spectroscopy (calculated by comparing the integrated vinyl signals assigned to the DMAC monomer at 5.6–6.7 ppm to the integrated polyacrylamide backbone signals at 1.1–2.7 ppm). Aqueous GPC analysis indicated an Mn of 74 kg mol–1 and an Mw/Mn of 1.54 (see below for eluent and calibration details).
Preparation of Dilute Aqueous Dispersions for DLS and ζ Potential Studies
Aqueous dispersions of PMPC139–PDMAC1000, PATAC195–PDMAC1000, and PAMPS250–PDMAC1000 particles in 2.0 M ammonium sulfate were diluted to 0.1% w/w using 2.0 M ammonium sulfate, which had been adjusted to an apparent pH of 3.
Preparation of Titrant Solutions
A stock titrant solution was prepared gravimetrically comprising an aqueous solution of 1.0 M KOH (22.97 g), deionized water (99.98 g), and ammonium sulfate (30.40 g). Deionized water was filtered through a 0.2 μm polyethersulfone syringe filter prior to use.
Characterization Methods
1H NMR Spectroscopy
Spectra were recorded at 25 °C in D2O using a 400 MHz Bruker Avance-400 spectrometer with 64 scans being averaged per spectrum.
Gel Permeation Chromatography (GPC)
Molecular weights and dispersities were determined for the various homopolymers and diblock copolymers using an Agilent 1260 Infinity GPC instrument. The setup comprised a pump, a degasser, three columns in series (PL-Aquagel Mixed-H, OH-30, and OH-40), and a refractive index detector. The column and detector temperature was set at 30 °C, and the flow rate was 1.0 mL min–1. Calibration was achieved using nine near-monodisperse poly(ethylene oxide) standards (2.1–969 kDa), and data were analyzed using Agilent Technologies GPC/SEC software. For PMPCx and its copolymers, the eluent was an aqueous solution containing 0.20 M NaNO3 and 0.05 M Trizma buffer at pH 7.0. For PATACx and its copolymers, the eluent was an aqueous solution containing 0.50 M CH3COOH and 0.30 M NaH2PO4 at pH 2.0. For PAMPSx and its copolymers, the eluent was a 70% v/v aqueous solution containing 0.20 M NaNO3 and 0.01 M NaH2PO4 at pH 7.0, with 30% v/v methanol.
Potentiometric Titration
An acidified aqueous dispersion (25.0 mL) was placed in a 250 mL glass beaker and stirred with a magnetic flea. Titrant solution was passed through a 0.2 μm polyethersulfone syringe filter into a volumetric 50 mL burette and a standard glass pH electrode was immersed within the aqueous dispersion. A total titrant of 7.0 mL was added in aliquots of no more than 0.5 mL, with smaller aliquots being used where necessary to accurately determine the equivalence point. The apparent pH of the aqueous dispersion was recorded after adding each aliquot, with pH equilibration being achieved within 30 s of each addition. All measurements were performed at 22 ± 1 °C. Approximately 0.25 mL of the aqueous dispersion was removed at suitable intervals for electrophoretic light scattering (ELS) and dynamic light scattering (DLS) analyses. No attempt was made to remove dissolved CO2 or to prevent its dissolution into the samples. It was assumed that the samples were near to or at the saturation level of dissolved CO2.
Dynamic Light Scattering (DLS)
Hydrodynamic diameters were determined using a Zetasizer Nano ZS (Malvern Panalytical, Malvern, U.K.). Samples were analyzed without further dilution, and three measurements were made in each case at a scattering angle of 173°. The instrument was configured to automatically determine the experimental duration and optical attenuation. The experimental correlation functions were analyzed using the cumulants method to yield the z-average hydrodynamic diameter (Dz) and polydispersity index (PDI). The Stokes–Einstein equation was employed, which is valid for dilute, noninteracting, and monodisperse spheres. Method 1 involved using a quartz cuvette (10 mm path length; volume = 1.00 mL) and 0.05% w/w aqueous copolymer dispersions were analyzed at 20 °C. Method 2 involved using disposable polystyrene semi-micro cuvettes (4 mm path length; volume = 0.25 mL) and 0.1% w/w aqueous copolymer dispersions were analyzed at 25 °C.
Electrophoretic Light Scattering (ELS)
Electrophoretic mobilities were determined by electrophoretic light scattering (NG-ELS) using an instrument provided by Enlighten Scientific LLC (Hillsborough, NC). The functional design and operation of this instrument are similar to the original phase analysis light scattering (PALS) apparatus44 that employed a crossed-beam optical configuration in contrast to the more common reference beam configuration used for other ELS instruments. The electrode assembly used for the NG-ELS equipment is similar to that described by Uzgiris.45 Disposable polystyrene semi-micro cuvettes (4 mm path length; volume = 0.25 mL) were used as the sample holders. Two identical parallel plate platinized platinum46 electrodes, placed 4 mm apart, were used to provide the driving voltage. The sample temperature was determined using a miniature NTC-type thermistor placed in direct contact with an ≈0.1% w/w aqueous copolymer dispersion. This temperature sensor was positioned at the mid-point between the electrodes and approximately 1 mm above the intersection point of the two laser beams. Temperature control was achieved by placing the sample cuvette in an aluminum block that ensured efficient heat transfer with the (cooler) water circulating through channels within the block. The water temperature depended on the amount of Joule heating of the sample and hence on both the sample conductivity and the voltage applied across the electrodes. Complex impedance analysis of the electrode waveform was used to quantify electrode polarization and Joule heating. Mobility measurements were made using sinusoidal electrode signal waveforms with a nominal amplitude of 4.5 V at frequencies of 64 and 128 Hz. Small adjustments (up to ± 0.3 V) to the amplitude were made prior to data collection to ensure that the cell temperature remained at 25 ± 1 °C during each measurement. The scattered light was analyzed using both the PALS and the laser Doppler electrophoresis (LDE) methods simultaneously. Data were collected for 1 min, and the same data set was used to calculate the electrophoretic mobility by each method. For each sample, five independent measurements were made at each electrode signal frequency, yielding a total of ten measurements per sample from which a mean value was calculated.
Small-Angle X-Ray Scattering (SAXS)
SAXS patterns were recorded for 1.0% w/w aqueous copolymer dispersions at Diamond Light Source (station I22, Didcot, U.K.) using a monochromatic X-ray beam (λ = 0.124 nm), a two-dimensional (2D) Pilatus 2M pixel detector (Dectris, Switzerland), and a q range of 0.02–2.00 nm–1, where q = (4π sin θ)/λ corresponds to the modulus of the scattering vector, and θ is half of the scattering angle. SAXS data were reduced (integrated, normalized, and background-subtracted) using Dawn software supplied by Diamond Light Source. The X-ray scattering intensity for water was used for absolute scale calibration of the scattering patterns with data manipulation via SAXS Utilities software. Irena SAS macros for Igor Pro were utilized for modeling.
Optical Microscopy (OM)
Images were recorded at ×400 magnification using a Cole-Parmer bifocal compound microscope equipped with a Moticam-BTW digital camera.
UV Absorption Spectroscopy
Spectra were recorded between 200 and 400 nm using a PC-controlled UV-1800 spectrophotometer at 25 °C using a 1 cm path length quartz cell. A Beer–Lambert curve was constructed using a series of five PETTC solutions in methanol. The absorption maximum at 306 nm assigned to the trithiocarbonate group was used for this calibration plot, and PETTC concentrations were selected such that the absorbance remained below 1.1. The molar extinction coefficient for PETTC was determined to be 10,900 ± 100 mol–1 dm3 cm–1; hence the mean DP for the PMPC homopolymer was calculated to be 139 ± 1.
Rheology
An MCR 502 rheometer (Anton Paar, Gratz, Austria) equipped with a Couette geometry was used for rotational rheology experiments. Measurements were performed at 20 °C and shear sweeps were conducted from 0.05 to 500 s–1 using approximately 10 mL of either 20% w/w aqueous copolymer dispersions or 10% w/w aqueous copolymer solutions.
Results and Discussion
The RAFT solution polymerization of MPC was conducted in methanol at 64 °C using a trithiocarbonate-based RAFT agent (PETTC). The mean degree of polymerization (DP) of the PMPC homopolymer was determined via end-group analysis using UV spectroscopy to be 139 ± 1. Aqueous GPC analysis indicated a relatively narrow molecular weight distribution (Mw/Mn = 1.18) for the precursor. However, the poly(ethylene oxide) calibration standards used for GPC analysis meant that only relative Mn values could be obtained. The RAFT aqueous dispersion polymerization of DMAC was conducted in the presence of 2.0 M ammonium sulfate at 30 °C using the PMPC139 precursor as a salt-tolerant steric stabilizer block, as outlined in Scheme 2.
Scheme 2. Reaction Scheme for the Synthesis of PMPC139–PDMACx (x = 500 to 7000) Diblock Copolymer Particles via RAFT Aqueous Dispersion Polymerization of DMAC at 30 °C in the Presence of 2.0 M Ammonium Sulfate.

Conditions: targeting 20% w/w solids using a PMPC139–TTC/KPS molar ratio of 4.0 and a [KPS]/[AsAc] molar ratio of 1.0.
PMPC was chosen as the steric stabilizer block because its zwitterionic structure is known to confer aqueous solubility even at 5.0 M NaCl.35 PDMAC was chosen as the core-forming block because it is a non-ionic water-soluble polymer that becomes water-insoluble in the presence of added salt.47 Moreover, the resulting PMPC–PDMAC diblock copolymer chains were anticipated to be amenable to aqueous GPC analysis.48 Inspecting the data presented in Table 1, using 2.0 M ammonium sulfate should be sufficient to produce an aqueous dispersion polymerization formulation since the PMPC precursor and the DMAC monomer are soluble in the aqueous continuous phase and the growing PDMAC chains should become insoluble. Accordingly, the series of aqueous PISA syntheses shown in Table 2 were conducted at 30 °C using a low-temperature persulfate/ascorbic acid (KPS/AsAc) redox initiator while targeting 20% w/w solids at pH 3.
Table 1. Aqueous Solubility of MPC Monomer, DMAC Monomer, PMPC139 Homopolymer, and PDMAC500 Homopolymer at 5.0% w/w Solids in the Presence of 0–4.0 M Ammonium Sulfate as Judged by Visual Inspection at pH 7 and 25 °C.
| aqueous
(NH4)2SO4 concentration/mol dm–3 |
|||||
|---|---|---|---|---|---|
| 0 | 1.0 | 2.0 | 3.0 | 4.0 | |
| MPC monomer | soluble | soluble | soluble | soluble | soluble |
| PMPC139 | soluble | soluble | soluble | soluble | soluble |
| DMAC monomer | soluble | soluble | soluble | soluble | insoluble |
| PDMAC500 | soluble | soluble | insoluble | insoluble | insoluble |
Table 2. Summary of Conversion, GPC, and DLS Data Obtained for the RAFT Aqueous Dispersion Polymerization of DMAC at 30 °C Using a PMPC139 Precursor at 20–30% w/w Solids.
| calculated
for PDMAC block: |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| solids/w/w% | PDMAC DP (x) | conversiona/% | DPa | Mna/kg mol–1 | GPC Mnb/kg mol–1 | Mw/Mnb | Dzc/nm | PDIc | physical appearance |
| 20 | 500 | >99 | 520 | 52 | 31 | 1.96 | 70 | 0.09 | translucent gel |
| 1000 | >99 | 1000 | 99 | 62 | 1.76 | 98 | 0.12 | translucent gel | |
| 2000 | >99 | 1980 | 196 | 131 | 1.65 | 240 | 0.10 | free-flowing & turbid | |
| 3000 | >99 | 3060 | 303 | 150 | 1.82 | 253 | 0.17 | free-flowing & turbid | |
| 4000 | >99 | 4020 | 398 | 168 | 1.97 | 350 | 0.27 | free-flowing & turbid | |
| 5000 | >99 | 4910 | 487 | 262 | 1.97 | 560 | 0.31 | free-flowing & turbid | |
| 6000 | >99 | 6090 | 604 | 338 | 2.10 | 680 | 0.40 | free-flowing & turbid | |
| 7000 | >98 | 6630 | 657 | unstable dispersion | |||||
| 25 | 5000 | >99 | 5110 | 506 | unstable dispersion | ||||
| 30 | 5000 | macroscopic precipitation | |||||||
Determined by 1H NMR spectroscopy (comparison between the integrated vinyl signals assigned to DMAC monomer at 5.6–6.7 ppm, the integrated PDMAC methine proton signal at 2.2–2.7 ppm, and the PMPC139 azamethylene signal at 3.6 ppm).
Determined by aqueous GPC using a series of near-monodisperse poly(ethylene oxide) calibration standards.
Dz denotes z-average diameter and PDI denotes polydispersity index as determined by DLS according to method 1 (see main text).
The PMPC139 precursor afforded colloidally stable dispersions of increasing turbidity when targeting PDMAC DPs ranging from 500 to 6000. However, precipitation was observed when targeting PDMAC DPs above 6000 or when the target copolymer concentration was increased to 25% w/w solids. The PDMAC core block DPs were determined relative to that of the stabilizer block by end-group analysis using 1H NMR spectroscopy. Reasonably good agreement (within experimental error) with the target PDMAC DPs was confirmed by comparing the integrated methine proton signal on the PDMAC backbone at 2.2–2.7 ppm and the PMPC139 azamethylene signal at 3.6 ppm. Comparing these NMR-derived Mn values to those determined by GPC analysis suggests a significant systematic error for the latter technique. This is understandable because poly(ethylene oxide) calibration standards are unlikely to be accurate for the analysis of PDMAC-rich diblock copolymers. Macroscopic precipitation was observed for the attempted synthesis conducted at 30% w/w solids. Essentially full DMAC conversion (>98%) was obtained for each of these syntheses as judged by 1H NMR spectroscopy studies. Targeting PDMAC DPs ≤1000 produced translucent gels, but lowering the solid concentration led to free-flowing dispersions. These gels are most likely caused by these relatively short PDMAC chains not being fully desolvated in the presence of 2.0 M ammonium sulfate.
There is a systematic increase in particle diameter when targeting higher PDMAC DPs. Similar observations have been reported for various other PISA formulations that produce kinetically-trapped spheres.33,49,50 Moreover, there is also good evidence that targeting larger particles (i.e., higher PDMAC DPs) leads to a progressive broadening of the particle size distribution.
Aqueous GPC data obtained for the series of PMPC139–PDMACx diblock copolymers shown in Table 2 are summarized in Figure 1. Unimodal molecular weight distributions are obtained in each case, with a systematic shift to higher Mn observed when targeting higher PDMAC DPs, which can also be observed in the normalized GPC traces (see Figure S2). However, dispersities are above 1.50, which indicates imperfect RAFT control. We have reported similar dispersities when targeting relatively high DPs for the core-forming block in other PISA formulations.51−54 Because Mn values are calculated relative to a series of near-monodisperse poly(ethylene oxide) calibration standards, a significant systematic error is expected in this case. Indeed, the theoretical Mn for PMPC139–PDMAC6000 is 636 kg mol–1, whereas the corresponding experimental GPC value is 338 kg mol–1.
Figure 1.

Aqueous GPC curves recorded for the PMPC139 precursor and a series of PMPC139–PDMACx diblock copolymers prepared by chain extension via RAFT aqueous dispersion polymerization of DMAC at 30 °C in the presence of 2.0 M ammonium sulfate. Mn values are calculated relative to a series of near-monodisperse poly(ethylene oxide) calibration standards (see Figure S2 for the corresponding normalized GPC curves).
Small-angle X-ray scattering (SAXS) was used to characterize selected PMPC139–PDMACx particles (where x = 1000 or 3000), see Figure 2. A well-known spherical micelle model55−57 was used to provide a satisfactory data fit to an I(q) vs q plot. This approach indicated a volume-average diameter (Dv) of 67 ± 8 nm for the PMPC139–PDMAC1000 particles and 213 ± 38 nm for the PMPC139–PDMAC3000 particles. As expected, these diameters are somewhat lower than the corresponding z-average diameters reported by DLS (see Table 2).58
Figure 2.
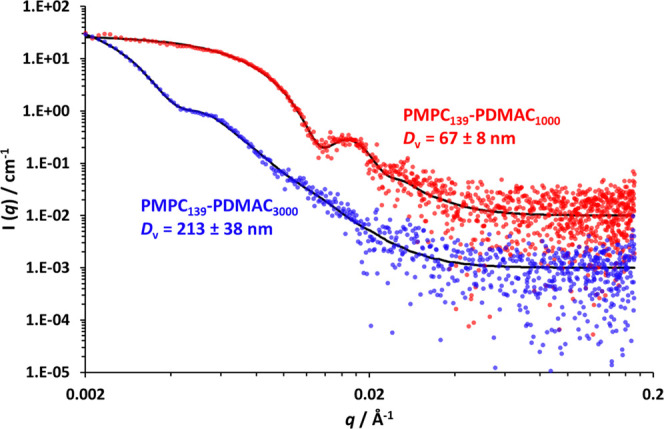
SAXS patterns recorded for 1.0% w/w aqueous dispersions of PMPC139–PDMACx particles (where x = 1000 or 3000) at 25 °C. The black solid lines denote the data fit obtained using a well-known spherical micelle model.55−57Dv denotes the volume-average diameter. The red pattern has been scaled by a factor of ten relative to the blue pattern for the sake of clarity.
1H NMR spectroscopy was used to study the kinetics of the DMAC polymerization at 30 °C when targeting PMPC139–PDMAC5000 particles at 20% w/w solids (see Figure 3a). Periodic sampling of the reaction mixture confirmed that a DMAC conversion of 98% was achieved within 2 h under such conditions, while the corresponding linear semilogarithmic plot indicated first-order kinetics with respect to monomer. Each aliquot taken from the reaction mixture was also subjected to analysis by aqueous GPC (see Figure 3b). Each GPC curve was unimodal, and there was a clear shift in the entire molecular weight distribution toward higher molecular weight, indicating a reasonably high blocking efficiency for the PMPC139 precursor and hence well-defined diblock copolymer chains. This is perhaps more apparent for the normalized GPC curves (see Figure S3). However, dispersities increased monotonically with monomer conversion and always remained above 1.50.
Figure 3.

(a) Conversion vs time curve and corresponding semilogarithmic plot determined by 1H NMR spectroscopy for the RAFT aqueous dispersion polymerization of DMAC at 30 °C in 2.0 M ammonium sulfate when targeting a PDMAC DP of 5000 at 20% w/w solids. (b) Aqueous GPC curves obtained by periodic sampling of the reaction mixture to monitor the evolution in the molecular weight distribution (see Figure S3 for the corresponding normalized GPC curves).
In principle, transmission electron microscopy (TEM) can be used to assign the morphology of diblock copolymer particles prepared via PISA. In practice, the particles prepared herein are unstable with respect to dilution with deionized water (see below). On the other hand, dilution using an aqueous solution of 2.0 M ammonium sulfate is also problematic because this leads to extensive salt crystal formation during TEM grid preparation. In view of these technical problems, we examined the PMPC139–PDMAC5000 particles by optical microscopy, see Figure 4. This technique indicates the presence of a population of micron-sized particles, but it is insensitive to the submicron-sized particle populations indicated by DLS and SAXS studies.
Figure 4.

Optical microscopy image recorded for PMPC139–PDMAC5000 particles prepared at 20% w/w solids by RAFT aqueous dispersion polymerization of DMAC at 30 °C in the presence of 2.0 M ammonium sulfate.
1H NMR spectroscopy was employed to investigate the extent of solvation of the core-forming PDMAC block before and after particle dissolution on dilution of a 20% w/w aqueous dispersion with deionized water. Accordingly, PMPC139–PDMAC5000 particles were prepared in D2O in the presence of 2.0 M ammonium sulfate using the same reaction conditions outlined in Scheme 2. 1H NMR spectra were recorded for the initial aqueous dispersion and the resulting aqueous solutions after up to a four-fold dilution using D2O (see Figure 5). The lower five spectra were normalized to the signal assigned to the two azamethylene protons (−CH2N(CH3)3) adjacent to the quaternary amine within the PMPC block, which remains fully solvated at all salt concentrations. The uppermost spectrum was recorded for a PDMAC500 homopolymer in D2O; the signals marked a and b correspond to the methylene and methine backbone protons, and c corresponds to the two equivalent pendent methyl groups.
Figure 5.

1H NMR spectra recorded for a PDMAC500 (red spectrum) and a PMPC139 (blue spectrum) homopolymers in the absence of salt, as well as a PMPC139–PDMAC5000 diblock copolymer prepared at 20% w/w in D2O in the presence of 2.0 M ammonium sulfate, see the lowest black spectrum. As the 20% w/w PMPC139–PDMAC5000 dispersion is diluted with further D2O, both the background salt concentration and the copolymer concentration are systematically reduced (see four other black spectra). Just a two-fold dilution of the turbid dispersion is sufficient to cause molecular dissolution of the particles as the PDMAC block becomes solvated in 1.0 M ammonium sulfate. A further two-fold dilution of this transparent solution with D2O results in PMPC139–PDMAC5000 chains dissolved in 0.5 M ammonium sulfate, for which the PDMAC signals are now indistinguishable from those of PDMAC500 homopolymer in water (compare the uppermost black spectrum with the red spectrum).
Clearly, signal a is almost completely attenuated in the presence of 2.0 M ammonium sulfate. However, this signal becomes much more prominent as the ammonium sulfate concentration is lowered, indicating a much higher degree of hydration for the PDMAC block on dilution. Similar observations were made for signals b and c. However, the former signal overlaps with other signals, while the latter is only partially suppressed in the presence of 2.0 M ammonium sulfate. The spectra recorded using 0.5 M ammonium sulfate and the pure PDMAC homopolymer were almost identical, which suggests that this polymer is essentially fully solvated at this salt concentration. This indicates that lowering the ammonium sulfate concentration from 2.0 to 0.5 M via four-fold dilution of the as-synthesized 20% w/w aqueous dispersions of PMPC139–PDMAC5000 particles using deionized water should be sufficient to cause complete particle dissolution.47,54
Rotational rheology experiments were conducted on samples using shear sweeps from 0.05 to 500 s–1 at 20 °C. The viscosities of a range of 10% w/w aqueous solutions comprising molecularly-dissolved PMPC139–PDMACx chains in the presence of 1.0 M ammonium sulfate obtained after two-fold dilution of the as-synthesized dispersions using deionized water are shown in Figure 6a. A monotonic increase in solution viscosity is observed at all shear rates when increasing the PDMAC DP for the molecularly-dissolved chains. The viscosity of the aqueous solution remains relatively constant for shear rates ranging from 0.05 to 5.0 s–1, with shear-thinning behavior being observed at higher shear rates. Figure 6b compares the viscosities of as-synthesized 20% w/w aqueous dispersions of PMPC139–PDMACx particles (where x = 3000 or 5000) in 2.0 M ammonium sulfate with the corresponding two 10% w/w aqueous copolymer solutions in 1.0 M ammonium sulfate. Clearly, the viscosity of each dispersion is significantly lower than that of the more dilute solution at all shear rates. Moreover, the two dispersions are much more strongly shear-thinning at higher shear, leading to an order of magnitude reduction in viscosity at 5 s–1. Similar behavior has been reported in the literature when comparing colloidal particles with the corresponding solvated copolymer chains.60,61
Figure 6.

(a) Viscosity vs shear rate data obtained by rotational rheology studies of 10% w/w aqueous solutions of molecularly-dissolved PMPC139–PDMACx chains in the presence of 1.0 M ammonium sulfate. (b) Viscosity vs shear rate data obtained by rotational rheology studies of 20% w/w aqueous dispersions of either PMPC139–PDMAC3000 or PMPC139–PDMAC5000 particles in 2.0 M ammonium sulfate compared to that for 10% w/w aqueous solutions of the same two copolymers in the presence of 1.0 M ammonium sulfate.
Two alternative steric stabilizers were also evaluated for the RAFT aqueous dispersion polymerization of DMAC conducted in the presence of 2.0 M ammonium sulfate. To complement the zwitterionic nature of the salt-tolerant PMPC139 steric stabilizer, we evaluated a cationic polyelectrolyte (PATAC195) and an anionic polyelectrolyte (PAMPS250), see chemical structures shown in Scheme 3. Both these polyelectrolytes have been reported to exhibit salt-tolerant behavior.34,62−64 A PDMAC DP of 1000 was targeted, and 1H NMR spectroscopy studies of the final reaction mixtures confirmed that more than 99% DMAC conversion was obtained in each case.
Scheme 3. Chemical Structures of the Cationic PATAC195 and Anionic PAMPS250 Precursors Used to Stabilize PDMAC-Rich Diblock Copolymer Particles Prepared via RAFT Aqueous Dispersion Polymerization of DMAC in 2.0 M Ammonium Sulfate.

It is common practice to estimate the ζ potential of colloidal particles in aqueous solution as a function of pH. However, the correct interpretation of the experimental data for sterically-stabilized particles dispersed in highly salty aqueous media can be problematic for two reasons. First, the relatively high ionic strength reduces the hydrogen ion activity, which affects the accuracy of the glass Ag/AgCl reference electrode typically used to measure the pH. Moreover, additional errors may be incurred owing to a change in the junction potential of such electrodes when in contact with such salty media. Second, the steric stabilizer chains at the particle–liquid interface provide a permeable medium through which the solution phase can flow. If electrical charge arises from the ionic groups within such steric stabilizer chains, the electrokinetic models commonly used to calculate ζ potential from electrophoretic mobility become invalid.65 To overcome these technical problems, we used a state-of-the-art instrument to determine apparent ζ potentials for the three types of PDMAC-rich particles prepared in high salt using the PMPC139, PATAC195, or PAMPS250 precursor in turn (see Supporting Information for further information).
In the present study, the electrophoretic mobility of the particles was measured as a function of the addition of varying amounts of KOH. The apparent pH was determined using a glass Ag/AgCl reference electrode without any compensation to offset the effect of the high ionic strength on the electrode response (although a temperature sensor within the electrode assembly did enable temperature compensation). Accordingly, ζ potentials calculated using the Smoluchowski model66 are regarded as apparent zeta potentials. The hydrodynamic z-average diameter of the particles was also determined in the presence of 2.0 M ammonium sulfate as a function of pH during these measurements.
The apparent ζ potentials determined by electrophoretic light scattering for each of these three dispersions as a function of added KOH is shown in Figure 7. As expected, the electrophoretic footprint for each type of particle is dictated by the chemical nature of the steric stabilizer chains. Thus the cationic PATAC195–PDMAC1000 particles exhibit positive apparent ζ potentials of +15.8 ± 1.1 mV, whereas the anionic PAMPS250–PDMAC1000 particles exhibit negative apparent ζ potentials of −25.9 ± 1.5 mV. Finally, the zwitterionic PMPC250–PDMAC1000 particles exhibit apparent ζ potentials close to zero (+1.1 ± 1.2 mV). Similar observations have been reported for other PMPC-stabilized nano-objects at low salt.67,68
Figure 7.

Apparent ζ potentials observed on addition of varying volumes of aqueous 0.2 M KOH solution in the presence of 2.0 M ammonium sulfate for 0.1% w/w aqueous dispersions of PATAC195–PDMAC1000 (red triangles), PMPC139–PDMAC1000 (green squares), or PAMPS250–PDMAC1000 (blue circles) particles.
If these particles were hard spheres, the conventional interpretation for such electrophoretic observations is that the PAMPS250–PDMAC1000 particles possess a sufficiently high anionic ζ potential to prevent aggregation, the PATAC195–PDMAC1000 particles possess a moderate cationic ζ potential that is likely to retard but not prevent aggregation, while the PMPC250–PDMAC1000 particles are essentially uncharged and hence likely to be colloidally unstable. However, this is a naïve and incorrect interpretation, not least because the salt-tolerant PAMPS250, PATAC195, and PMPC139 chains confer additional steric stabilization.69
Conclusions
We report the synthesis of a series of sterically-stabilized diblock copolymer particles via RAFT aqueous dispersion polymerization of DMAC in highly salty media. This is achieved by selecting a suitable salt-tolerant water-soluble polymer to act as an effective steric stabilizer. Such stabilizers can possess zwitterionic (e.g., PMPC), cationic (e.g., PATAC), or anionic (e.g., PAMPS) character, which leads to the corresponding diblock copolymer particles exhibiting essentially zero, negative, or positive apparent ζ potentials, respectively. It is non-trivial to make such aqueous electrophoresis measurements in highly salty media. Indeed, such experiments require state-of-the-art instrumentation. Relatively high DPs can be targeted for the salt-insoluble block to ensure that this component dominates the formulation. This approach enables high molecular weight water-soluble polymers to be prepared in a highly convenient low-viscosity form. Subsequent dilution using deionized water lowers the background salt concentration and causes in situ molecular dissolution of the particles, which leads to a substantial thickening effect and the formation of highly viscous transparent aqueous solutions. In principle, such aqueous PISA formulations are highly attractive: there are various potential commercial applications for high molecular weight water-soluble polymers while the well-known negative aspects of using RAFT agents (i.e., their cost, color, and malodor) are minimized. For example, the organosulfur content of the dry PMPC139–PDMAC6000 diblock copolymer targeted herein is only ≈0.03%, which corresponds to just ≈ 63 ppm for a 20% w/w aqueous copolymer dispersion.
Acknowledgments
The authors thank EPSRC for a CASE PhD studentship for the first author. BASF is thanked for additional financial support for this project and for permission to publish these results. S.P.A. acknowledges an EPSRC Particle Technology Fellowship Grant (EP/R003009) and Dr. S. Ebbens for his contribution to this project. Dr. O. J. Deane and Dr. R. R. Gibson are thanked for their assistance in developing the aqueous GPC protocol, and Dr. A. Czajka is thanked for his help in modeling the SAXS data. Dr. A. L. Lewis (formerly of Biocompatibles U.K.) is acknowledged for the kind donation of the MPC monomer. Finally, we thank Dr. S. J. Byard for performing the preliminary experiments that led to the current study.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.macromol.2c01071.
UV absorption spectra and calibration plot; dynamic viscosities of various ammonium sulfate solutions; normalized GPC curves obtained for both final copolymers and during kinetic experiments; and further technical details regarding the determination of ζ potentials in highly salty media (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Moad G.; Rizzardo E.; Thang S. H. Living Radical Polymerization by the RAFT Process. Aust. J. Chem. 2005, 58, 379–410. 10.1071/CH05072. [DOI] [Google Scholar]
- Moad G.; Rizzardo E.; Thang S. H. Living Radical Polymerization by the RAFT Process - A First Update. Aust. J. Chem. 2006, 59, 669–692. 10.1071/CH06250. [DOI] [Google Scholar]
- Moad G.; Rizzardo E.; Thang S. H. Living Radical Polymerization by the RAFT Process - A Second Update. Aust. J. Chem. 2009, 62, 1402. 10.1071/CH09311. [DOI] [Google Scholar]
- Moad G.; Rizzardo E.; Thang S. H. Living Radical Polymerization by the RAFT Process – A Third Update. Aust. J. Chem. 2012, 65, 985. 10.1071/CH12295. [DOI] [Google Scholar]
- Carmean R. N.; Becker T. E.; Sims M. B.; Sumerlin B. S. Ultra-High Molecular Weights via Aqueous Reversible-Deactivation Radical Polymerization. Chem 2017, 2, 93–101. 10.1016/j.chempr.2016.12.007. [DOI] [Google Scholar]
- Read E.; Guinaudeau A.; Wilson D. J.; Cadix A.; Violleau F.; Destarac M. Low Temperature RAFT/MADIX Gel Polymerisation: Access to Controlled Ultra-High Molar Mass Polyacrylamides. Polym. Chem. 2014, 5, 2202–2207. 10.1039/c3py01750h. [DOI] [Google Scholar]
- Carmean R. N.; Sims M. B.; Figg C. A.; Hurst P. J.; Patterson J. P.; Sumerlin B. S. Ultrahigh Molecular Weight Hydrophobic Acrylic and Styrenic Polymers through Organic-Phase Photoiniferter-Mediated Polymerization. ACS Macro Lett. 2020, 9, 613–618. 10.1021/acsmacrolett.0c00203. [DOI] [PubMed] [Google Scholar]
- Pelton R. H.; Allen L. H. The Effects of Some Electrolytes on Flocculation with a Cationic Polyacrylamide. Colloid Polym. Sci. 1983, 261, 485–492. 10.1007/BF01419832. [DOI] [Google Scholar]
- Klimchuk K. A.; Hocking M. B.; Lowen S. Water-Soluble Acrylamide Copolymers. IX. Preparation and Characterization of the Cationic Derivatives of Poly(Acrylamide-Co-N, N-Dimethylacrylamide), Poly(Acrylamide-Co-Methacrylamide), and Poly(Acrylamide-Co-N-t-Butylacrylamide). J. Polym. Sci., Part A: Polym. Chem. 2001, 39, 2525–2535. 10.1002/pola.1229. [DOI] [Google Scholar]
- Guyot A.; Chu F.; Schneider M.; Graillat C.; McKenna T. F. High Solid Content Latexes. Prog. Polym. Sci. 2002, 27, 1573–1615. 10.1016/S0079-6700(02)00014-X. [DOI] [Google Scholar]
- Perrier S. 50th Anniversary Perspective: RAFT Polymerization - A User Guide. Macromolecules 2017, 50, 7433–7447. 10.1021/acs.macromol.7b00767. [DOI] [Google Scholar]
- Chiefari J.; Chong Y. K.; Ercole F.; Krstina J.; Jeffery J.; Le T. P. T.; Mayadunne R. T. A.; Meijs G. F.; Moad C. L.; Moad G.; Rizzardo E.; Thang S. H. Living Free-Radical Polymerization by Reversible Addition - Fragmentation Chain Transfer: The RAFT Process. Macromolecules 1998, 31, 5559–5562. 10.1021/ma9804951. [DOI] [Google Scholar]
- Destarac M. Industrial Development of Reversible-Deactivation Radical Polymerization: Is the Induction Period Over?. Polym. Chem. 2018, 9, 4947–4967. 10.1039/C8PY00970H. [DOI] [Google Scholar]
- Destarac M.; Wilson D. J.; Silvia S.. Controlled Radical Polymerization in Water-in-Water Dispersion. US10160821B2, 2018.
- Wu Y. M.; Wang Y. P.; Yu Y. Q.; Xu J.; Chen Q. F. Dispersion Polymerization of Acrylamide with 2-Acrylamido-2-Methyl-1- Propane Sulfonate in Aqueous Solution. J. Appl. Polym. Sci. 2006, 102, 2379–2385. 10.1002/app.24494. [DOI] [Google Scholar]
- Guo A.; Geng Y.; Zhao L.; Li J.; Liu D.; Li P. Preparation of Cationic Polyacrylamide Microsphere Emulsion and Its Performance for Permeability Reduction. Pet. Sci. 2014, 11, 408–416. 10.1007/s12182-014-0355-0. [DOI] [Google Scholar]
- Cho M. S.; Yoon K. J.; Song B. K. Dispersion Polymerization of Acrylamide in Aqueous Solution of Ammonium Sulfate: Synthesis and Characterization. J. Appl. Polym. Sci. 2002, 83, 1397–1405. 10.1002/app.2300. [DOI] [Google Scholar]
- Ferguson C. J.; Hughes R. J.; Pham B. T. T.; Hawkett B. S.; Gilbert R. G.; Serelis A. K.; Such C. H. Effective Ab Initio Emulsion Polymerization under RAFT Control. Macromolecules 2002, 35, 9243–9245. 10.1021/ma025626j. [DOI] [Google Scholar]
- Ferguson C. J.; Hughes R. J.; Nguyen D.; Pham B. T. T.; Gilbert R. G.; Serelis A. K.; Such C. H.; Hawkett B. S. Ab Initio Emulsion Polymerization by RAFT-Controlled Self-Assembly. Macromolecules 2005, 38, 2191–2204. 10.1021/ma048787r. [DOI] [Google Scholar]
- Zhang X.; Boissé S.; Zhang W.; Beaunier P.; D’Agosto F.; Rieger J.; Charleux B. Well-Defined Amphiphilic Block Copolymers and Nano-Objects Formed in Situ via RAFT-Mediated Aqueous Emulsion Polymerization. Macromolecules 2011, 44, 4149–4158. 10.1021/ma2005926. [DOI] [Google Scholar]
- Warren N. J.; Armes S. P. Polymerization-Induced Self-Assembly of Block Copolymer Nano-Objects via RAFT Aqueous Dispersion Polymerization. J. Am. Chem. Soc. 2014, 136, 10174–10185. 10.1021/ja502843f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charleux B.; Delaittre G.; Rieger J.; D’Agosto F. Polymerization-Induced Self-Assembly: From Soluble Macromolecules to Block Copolymer Nano-Objects in One Step. Macromolecules 2012, 45, 6753–6765. 10.1021/ma300713f. [DOI] [Google Scholar]
- Boissé S.; Rieger J.; Belal K.; Di-Cicco A.; Beaunier P.; Li M. H.; Charleux B. Amphiphilic Block Copolymer Nano-Fibers via RAFT-Mediated Polymerization in Aqueous Dispersed System. Chem. Commun. 2010, 46, 1950–1952. 10.1039/B923667H. [DOI] [PubMed] [Google Scholar]
- D’Agosto F.; Rieger J.; Lansalot M. RAFT-Mediated Polymerization-Induced Self-Assembly. Angew. Chem., Int. Ed. 2019, 59, 8368–8392. 10.1002/anie.201911758. [DOI] [PubMed] [Google Scholar]
- Liu G.; Qiu Q.; Shen W.; An Z. Aqueous Dispersion Polymerization of 2-Methoxyethyl Acrylate for the Synthesis of Biocompatible Nanoparticles Using a Hydrophilic RAFT Polymer and a Redox Initiator. Macromolecules 2011, 44, 5237–5245. 10.1021/ma200984h. [DOI] [Google Scholar]
- Sugihara S.; Ma’Radzi A. H.; Ida S.; Irie S.; Kikukawa T.; Maeda Y. In Situ Nano-Objects via RAFT Aqueous Dispersion Polymerization of 2-Methoxyethyl Acrylate Using Poly(Ethylene Oxide) Macromolecular Chain Transfer Agent as Steric Stabilizer. Polymer 2015, 76, 17–24. 10.1016/j.polymer.2015.08.051. [DOI] [Google Scholar]
- An Z.; Shi Q.; Tang W.; Tsung C. K.; Hawker C. J.; Stucky G. D. Facile RAFT Precipitation Polymerization for the Microwave-Assisted Synthesis of Well-Defined, Double Hydrophilic Block Copolymers and Nanostructured Hydrogels. J. Am. Chem. Soc. 2007, 129, 14493–14499. 10.1021/ja0756974. [DOI] [PubMed] [Google Scholar]
- Figg C. A.; Simula A.; Gebre K. A.; Tucker B. S.; Haddleton D. M.; Sumerlin B. S. Polymerization-Induced Thermal Self-Assembly (PITSA). Chem. Sci. 2015, 6, 1230–1236. 10.1039/C4SC03334E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham V. J.; Derry M. J.; Fielding L. A.; Musa O. M.; Armes S. P. RAFT Aqueous Dispersion Polymerization of N-(2-(Methacryloyloxy)Ethyl)Pyrrolidone: A Convenient Low Viscosity Route to High Molecular Weight Water-Soluble Copolymers. Macromolecules 2016, 49, 4520–4533. 10.1021/acs.macromol.6b00820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einarson M. B.; Berg J. C. Electrosteric Stabilization of Colloidal Latex Dispersions. J. Colloid Interface Sci. 1993, 155, 165–172. 10.1006/jcis.1993.1022. [DOI] [Google Scholar]
- Romero-Cano M. S.; Martín-Rodríguez A.; De las Nieves F. J. Electrosteric Stabilization of Polymer Colloids with Different Functionality. Langmuir 2001, 17, 3505–3511. 10.1021/la001659l. [DOI] [Google Scholar]
- Bremmell K. E.; Jameson G. J.; Biggs S. Polyelectrolyte Adsorption at the Solid/Liquid Interface Interaction Forces and Stability. Colloids Surf., A 1998, 139, 199–211. 10.1016/S0927-7757(98)00281-7. [DOI] [Google Scholar]
- Byard S. J.; Blanazs A.; Miller J. F.; Armes S. P. Cationic Sterically Stabilized Diblock Copolymer Nanoparticles Exhibit Exceptional Tolerance toward Added Salt. Langmuir 2019, 35, 14348–14357. 10.1021/acs.langmuir.9b02789. [DOI] [PubMed] [Google Scholar]
- Huang B.; Jiang J.; Kang M.; Liu P.; Sun H.; Li B. G.; Wang W. J. Synthesis of Block Cationic Polyacrylamide Precursors Using an Aqueous RAFT Dispersion Polymerization. RSC Adv. 2019, 9, 12370–12383. 10.1039/C9RA02716E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi M.; Terayama Y.; Ishikawa T.; Hoshino T.; Kobayashi M.; Ogawa H.; Masunaga H.; Koike J. I.; Horigome M.; Ishihara K.; Takahara A. Chain Dimension of Polyampholytes in Solution and Immobilized Brush States. Polym. J. 2012, 44, 121–130. 10.1038/pj.2011.116. [DOI] [Google Scholar]
- Jhan Y. Y.; Tsay R. Y. Salt Effects on the Hydration Behavior of Zwitterionic Poly(Sulfobetaine Methacrylate) Aqueous Solutions. J. Taiwan Inst. Chem. Eng. 2014, 45, 3139–3145. 10.1016/j.jtice.2014.08.022. [DOI] [Google Scholar]
- Doncom K. E. B.; Warren N. J.; Armes S. P. Polysulfobetaine-Based Diblock Copolymer Nano-Objects via Polymerization-Induced Self-Assembly. Polym. Chem. 2015, 6, 7264–7273. 10.1039/C5PY00396B. [DOI] [Google Scholar]
- Jones E. R.; Semsarilar M.; Blanazs A.; Armes S. P. Efficient Synthesis of Amine-Functional Diblock Copolymer Nanoparticles via RAFT Dispersion Polymerization of Benzyl Methacrylate in Alcoholic Media. Macromolecules 2012, 45, 5091–5098. 10.1021/ma300898e. [DOI] [Google Scholar]
- Bray C.; Peltier R.; Kim H.; Mastrangelo A.; Perrier S. Anionic Multiblock Core Cross-Linked Star Copolymers: Via RAFT Polymerization. Polym. Chem. 2017, 8, 5513–5524. 10.1039/C7PY01062A. [DOI] [Google Scholar]
- Lai J. T.; Filla D.; Shea R. Functional Polymers from Novel Carboxyl-Terminated Trithiocarbonates as Highly Efficient RAFT Agents. Macromolecules 2002, 35, 6754–6756. 10.1021/ma020362m. [DOI] [Google Scholar]
- Weast R.CRC Handbook of Chemistry and Physics, 66th ed.; CRC Press: Florida, 1992; Vol. 268. [Google Scholar]
- Peyman A.; Gabriel C.; Grant E. H. Complex Permittivity of Sodium Chloride Solutions at Microwave Frequencies. Bioelectromagnetics 2007, 28, 264–274. 10.1002/bem.20271. [DOI] [PubMed] [Google Scholar]
- Sin J. S. Ion Partitioning Effect on the Electrostatic Interaction between Two Charged Soft Surfaces. Colloids Surf., A 2021, 628, 127296 10.1016/j.colsurfa.2021.127296. [DOI] [Google Scholar]
- Miller J. F.; Schätzel K.; Vincent B. The Determination of Very Small Electrophoretic Mobilities in Polar and Nonpolar Colloidal Dispersions Using Phase Analysis Light Scattering. J. Colloid Interface Sci. 1991, 143, 532–554. 10.1016/0021-9797(91)90286-H. [DOI] [Google Scholar]
- Uzgiris E. E. Laser Doppler Methods in Electrophoresis. Prog. Surf. Sci. 1981, 10, 53–164. 10.1016/0079-6816(81)90006-X. [DOI] [Google Scholar]
- Feltham A. M.; Spiro M. Platinized Platinum Electrodes. Chem. Rev. 1971, 71, 177–193. 10.1021/cr60270a002. [DOI] [Google Scholar]
- Byard S. J.Synthesis and Characterisation of Stimulus-Responsive Diblock Copolymer Nano-Objects Prepared by RAFT Aqueous Dispersion Polymerisation, Synthesis and Characterisation of Stimulus-responsive Diblock Copolymer Nano-objects Prepared by RAFT Aqueous Dispersion Polymerisation, PhD Thesis, University of Sheffield, 2019. [Google Scholar]
- Warren N. J.; Muise C.; Stephens A.; Armes S. P.; Lewis A. L. Near-Monodisperse Poly(2-(Methacryloyloxy)Ethyl Phosphorylcholine)-Based Macromonomers Prepared by Atom Transfer Radical Polymerization and Thiol-Ene Click Chemistry: Novel Reactive Steric Stabilizers for Aqueous Emulsion Polymerization. Langmuir 2012, 28, 2928–2936. 10.1021/la204083z. [DOI] [PubMed] [Google Scholar]
- Williams M.; Penfold N. J. W.; Armes S. P. Cationic and Reactive Primary Amine-Stabilised Nanoparticles via RAFT Aqueous Dispersion Polymerisation. Polym. Chem. 2016, 7, 384–393. 10.1039/C5PY01577D. [DOI] [Google Scholar]
- Cockram A. A.; Bradley R. D.; Lynch S. A.; Fleming P. C. D.; Williams N. S. J.; Murray M. W.; Emmett S. N.; Armes S. P. Optimization of the High-Throughput Synthesis of Multiblock Copolymer Nanoparticles in Aqueous Media: Via Polymerization-Induced Self-Assembly. React. Chem. Eng. 2018, 3, 645–657. 10.1039/C8RE00066B. [DOI] [Google Scholar]
- Derry M. J.; Fielding L. A.; Warren N. J.; Mable C. J.; Smith A. J.; Mykhaylyk O. O.; Armes S. P. In Situ Small-Angle X-Ray Scattering Studies of Sterically-Stabilized Diblock Copolymer Nanoparticles Formed during Polymerization-Induced Self-Assembly in Non-Polar Media. Chem. Sci. 2016, 7, 5078–5090. 10.1039/C6SC01243D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byard S. J.; Williams M.; McKenzie B. E.; Blanazs A.; Armes S. P. Preparation and Cross-Linking of All-Acrylamide Diblock Copolymer Nano-Objects via Polymerization-Induced Self-Assembly in Aqueous Solution. Macromolecules 2017, 50, 1482–1493. 10.1021/acs.macromol.6b02643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker B. R.; Derry M. J.; Ning Y.; Armes S. P. Exploring the Upper Size Limit for Sterically Stabilized Diblock Copolymer Nanoparticles Prepared by Polymerization-Induced Self-Assembly in Non-Polar Media. Langmuir 2020, 36, 3730–3736. 10.1021/acs.langmuir.0c00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham V. J.; Armes S. P.; Musa O. M. Synthesis, Characterisation and Pickering Emulsifier Performance of Poly(Stearyl Methacrylate)-Poly(N-2-(Methacryloyloxy)Ethyl Pyrrolidone) Diblock Copolymer Nano-Objects via RAFT Dispersion Polymerisation in n-Dodecane. Polym. Chem. 2016, 7, 1882–1891. 10.1039/C6PY00138F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilavsky J.; Jemian P. R. Irena: Tool Suite for Modeling and Analysis of Small-Angle Scattering. J. Appl. Crystallogr. 2009, 42, 347–353. 10.1107/S0021889809002222. [DOI] [Google Scholar]
- Pedersen J. S. Form Factors of Block Copolymer Micelles with Spherical, Ellipsoidal and Cylindrical Cores. J. Appl. Crystallogr. 2000, 33, 637–640. 10.1107/S0021889899012248. [DOI] [Google Scholar]
- Muratov A.; Moussäd A.; Narayanan T.; Kats E. I. A Percus-Yevick Description of the Microstructure of Short-Range Interacting Metastable Colloidal Suspensions. J. Chem. Phys. 2009, 131, 054902 10.1063/1.3179667. [DOI] [PubMed] [Google Scholar]
- Chu B.; Liu T. Characterization of Nanoparticles by Scattering Techniques. J. Nanoparticle Res. 2000, 2, 29–41. 10.1023/A:1010001822699. [DOI] [Google Scholar]
- Tan B. H.; Tam K. C.; Dupin D.; Armes S. P. Rheological Behavior of Acid-Swellable Cationic Copolymer Latexes. Langmuir 2010, 26, 2736–2744. 10.1021/la9027699. [DOI] [PubMed] [Google Scholar]
- Jesson C. P.; Pearce C. M.; Simon H.; Werner A.; Cunningham V. J.; Lovett J. R.; Smallridge M. J.; Warren N. J.; Armes S. P. H2O2 Enables Convenient Removal of RAFT End-Groups from Block Copolymer Nano-Objects Prepared via Polymerization-Induced Self- Assembly in Water. Macromolecules 2017, 50, 182–191. 10.1021/acs.macromol.6b01963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Chen D.; Yue Y.; Zhang W.; Wang P. Dispersion Copolymerization of Acrylamide with Acrylic Acid in an Aqueous Solution of Ammonium Sulfate: Synthesis and Characterization. J. Appl. Polym. Sci. 2006, 102, 3685–3690. 10.1002/app.24259. [DOI] [Google Scholar]
- Lu J.; Peng B.; Li M.; Lin M.; Dong Z. Dispersion Polymerization of Anionic Polyacrylamide in an Aqueous Salt Medium. Pet. Sci. 2010, 7, 410–415. 10.1007/s12182-010-0086-9. [DOI] [Google Scholar]
- Bai S.; Wang Y.; Liu B.; Zhu Y.; Guo R. Dispersion Copolymerization of Acrylamide and Sodium 2-Acrylamido-2-Methylpropanesulfonate in Aqueous Salt Solution Stabilized with a Macro-RAFT Agent. Colloids Surf., A 2018, 553, 446–455. 10.1016/j.colsurfa.2018.05.082. [DOI] [Google Scholar]
- Miller J. F. Determination of Protein Charge in Aqueous Solution Using Electrophoretic Light Scattering: A Critical Investigation of the Theoretical Fundamentals and Experimental Methodologies. Langmuir 2020, 36, 8641–8654. 10.1021/acs.langmuir.0c01694. [DOI] [PubMed] [Google Scholar]
- Hunter R. J.Zeta Potential in Colloid Science; Academic Press: New York, 1981. [Google Scholar]
- Konno T.; Kurita K.; Iwasaki Y.; Nakabayashi N.; Ishihara K. Preparation of Nanoparticles Composed with Bioinspired 2-Methacryloyloxyethyl Phosphorylcholine Polymer. Biomaterials 2001, 22, 1883–1889. 10.1016/S0142-9612(00)00373-2. [DOI] [PubMed] [Google Scholar]
- Ahmed M.; Ishihara K.; Narain R. Calcium Mediated Formation of Phosphorylcholine-Based Polyplexes for Efficient Knockdown of Epidermal Growth Factor Receptors (EGFR) in HeLa Cells. Chem. Commun. 2014, 50, 2943–2946. 10.1039/C4CC00181H. [DOI] [PubMed] [Google Scholar]
- Napper D. H.Polymeric Stabilization of Colloidal Dispersions; Academic Press: London, 1983. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.