

Abstract
According to data from 2020, Slovakia has long been among the top five countries with the highest incidence rate of colorectal cancer (CRC) worldwide, and the rate is continuing to rise every year. In approximately 80% of CRC cases, allelic loss (loss of heterozygosity, LOH) occurs in the long arm of chromosome 18q. The most important genes that can be silenced by 18q LOH or mutations are small mothers against decapentaplegic homolog (SMAD) 2 and SMAD4, which are intracellular mediators of transforming growth factor (TGF)-β superfamily signals. TGF-β plays an important role in the pro-oncogenic processes, including such properties as invasion, epithelial-mesenchymal transition (commonly known as EMT), promotion of angiogenesis, and immunomodulatory effects. Several recent studies have reported that activation of TGF-β signaling is related to drug resistance in CRC. Because the mechanisms of drug resistance are different between patients in different stages of CRC, personalized treatment is more effective. Therefore, knowledge of the activation and inhibition of factors that affect the TGF-β signaling pathway is very important.
Keywords: Small mothers against decapentaplegic homologs, Transforming growth factor-beta, Colorectal cancer, Marker, Signaling pathway
Core Tip: A thorough understanding of the complete transforming growth factor (TGF)-β/small mothers against decapentaplegic homolog signaling pathway is important for defining its functions during pathological processes of colorectal cancer. Inhibitors specifically targeting TGF-β pathway mediators that reduce the expression of a particular protein may lead to fewer/milder adverse effects. However, the dual role of the TGF-β pathway in the onset and progression of cancer complicates the physiological/pathological and, thus, clinical situation. In recent years, research has shown that modification of members of this pathway is a promising approach for clinical procedures. Long-term treatment should emphasize personalized and targeted therapy.
INTRODUCTION
According to data from 2020, Slovakia has long been among the top five countries with the highest incidence rate of colorectal cancer (CRC) worldwide, and this rate continues to rise every year[1]. Although significant progress has been made in the diagnosis, screening, and treatment of patients with advanced CRC, therapeutic options are still limited, requiring the discovery of additional markers to act as prognostic predictors[2].
Up to 60%-65% of colorectal tumors have no family history (sporadic) and are the result of somatic mutations and epigenetic changes due to factors such as a lifestyle with limited physical activity, alcoholism and smoking[3]. CRC can arise as a result of these genetic and epigenetic aberrations (Figure 1): Chromosomal instability (CIN; 65%-85%), methylation of the CpG island (CIMP; 10%-20%), and DNA microsatellite instability (MSI; 12%-15%)[4]. Some authors have noted that patients with a tumor-bearing the CpG island methylator phenotype will have a worse prognosis compared to patients with a CIMP-negative tumor[5-7]. The instability of DNA microsatellite regions is characterized by mutations in the genome that arise due to defects in mismatch repair genes and can affect and inactivate tumor suppressor genes, leading to malignant transformation[8,9]. CIN is caused by the gain or loss of whole or large parts of chromosomes, leading to karyotype variability between cells. CIN results in chromosome imbalance (aneuploidy), subchromosomal genomic amplification, and loss of heterozygosity (LOH)[10].
Figure 1.
Representation of individual colorectal cancer subtypes.
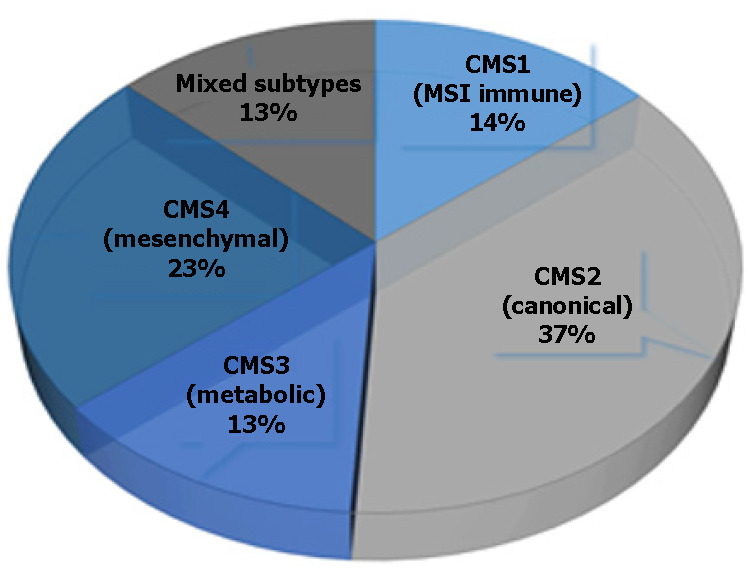
This type of classification, based on a single molecular marker, is not very informative in the early diagnosis of CRC; thus, a combination of several molecular markers has been proposed as a better classification approach for patients with CRC. Moreover, the joint efforts of the CRC Subtyping Consortium have led to a formal proposal for the stratification of CRC cases into the following four molecular subtypes (referred to as CMS1-4)[11,12] (Figure 2; Table 1).
Figure 2.

Three genetic and epigenetic aberrations of colorectal cancer formation. LOH: Loss of heterozygosity; TGF: Transforming growth factor.
Table 1.
Characteristics of individual colorectal cancer subtypes
|
CMS1
|
CMS2
|
CMS3
|
CMS4
|
|
(MSI immune)
|
(Canonical)
|
(Metabolic)
|
(Mesenchymal)
|
| CIMP high | CIN | CIN, CIMP low | CIN |
| Hypermethylation | |||
| SCNA-low | SCNA-high | SCNA-intermediate | SCNA-high |
| BRAF mutant | KRAS mutant | ||
| Activation of immune cells | WNT, MYC activation | Metabolic deregulation | TGF- activation |
| Worse survival after relapse | Superior survival after relapse | Worse relapse-free and overall survival |
SCNA: Somatic copy number alteration; CIN: Chromosomal instability; CIMP: Methylation of the CpG island; TGF: Transforming growth factor.
CMS1 is usually a right-sided (proximal) tumor, commonly diagnosed in older age females, and is associated with worse survival after relapse. This subtype is characterized by hypermethylation of CpG islands, which causes loss of tumor suppressor function and has a low prevalence of somatic copy number alterations (referred to as SCNAs). The hypermethylation of promoter regions of the MMR genes causes MSI[11].
CMS2 is mainly located on the left side (distal part of the colon) and is often diagnosed in men, with a better prognosis and a higher survival rate, even after relapse. This gene expression profile is characterized by low mutation rate. CMS2 also represents over-activation of epidermal growth factor (EGF)-related signaling pathways, with higher expression of the epidermal growth factor receptor (EGFR)[13]. Finally, Guinney et al[11] reported that CMS2 has more copy number gains in oncogenes and losses in tumor suppressor genes than the other CMSs.
CMS3 is another right-sided subtype and is the most frequently diagnosed in patients with evident metabolomics disease[13]. Although KRAS mutation is present in every CMS, it occurs more frequently in CMS3[11].
CMS4 tumors exhibit extremely low levels of hypermutation and are defined by an activated transforming growth factor (TGF)-β pathway and by epithelial-mesenchymal transition (EMT), making them generally more chemoresistant[13]. CMS4 tumors tend to be diagnosed at more advanced stages (III and IV); indeed, the poor prognosis of CMS4 (compared to the relatively favorable prognoses of CMS1 and CMS2) in non-metastatic disease have been demonstrated[11].
The basic characteristics of each CRC subtype, CMS1-4, are summarized in Table 1.
Approximately 80% of colorectal tumors have loss of an allele in the long arm of chromosome 18q, followed by LOH on chromosome 17p (75%-80%), 8p (40%), 5q (30%), and finally 22q (20%-30%). Allelic loss in chromosome 18q has been reported in 70% of cases of primary CRC with late-stage adenomas and shows a strong correlation with poor prognosis [14]. Patients with 18q LOH have a particularly poor prognosis in stage Ⅱ disease, leading to the conclusion that stage II adjuvant therapy is important for these patients[15].
There are many candidate tumor suppressor genes in 18q, including small mothers against decapentaplegic homolog (SMAD) 2, SMAD4, netrin receptor DCC (DCC), and Cdk5 and Abl enzyme substrate 1 (CABLES1 )[16]. The most important genes that can be silenced by 18q LOH or mutations are SMAD2 and SMAD4, which are intracellular mediators of TGF-β superfamily signaling[17].
TGF-β SUPERFAMILY SIGNALING
TGF-β superfamily signaling is mainly divided into the following two subfamilies: TGF-β-activin-nodal and bone morphogenetic protein (BMP). The TGF-β ligand (comprised of the TGF-β1, -β2, and -β3 isoforms) is a multifunctional member of the cytokine family, playing an important role in such cellular responses as cell proliferation, differentiation, and pathological processes. TGF-β itself plays a key role in the processes of EMT and fibrosis[18].
The canonical (SMAD-dependent) TGF-β signaling pathway (Figure 3) utilizes serine/threonine kinase receptors (TGF-βRI/TGF-βRII) in the plasma membrane and phosphorylates their cytoplasmic effectors SMAD2 and SMAD3. TGF-βRI receptors differ from TGF-βRII by the presence of an N-terminal glycine/serine-rich (GS) domain, which regulates kinase activity and SMAD binding. TGF-βRII receptor phosphorylates serine and threonine residues within the GS domain of TGF-βRI, and activated TGF-βRI receptor phosphorylates the distal C-termini of SMAD2 and SMAD3. An anchor of SMAD receptor activation, a SMAD cofactor that directly interacts with SMAD2/3, is required to anchor SMAD2/3 proteins to the TGF-β receptor. After phosphorylation, SMAD2 and SMAD3 dimers form heteromeric complexes with SMAD4 and then translocate to the nucleus. They act as transcription factors, mediate the expression of various genes, and promote various biological functions in the tumor microenvironment, resulting in tumor suppression[19].
Figure 3.

Transforming growth factor-beta superfamily signal transduction. TGF: Transforming growth factor; EMT: Epithelial-mesenchymal transition; ERK: Extracellular signal-regulated kinase; BMP: Bone morphogenetic protein; SMAD: Small mothers against decapentaplegic homolog.
A conserved branch of the TGF-β superfamily involves BMP signaling. BMP canonical signaling is triggered upon the binding of soluble ligands to serine-threonine kinase receptors, BMPRI and BMPRII, in the plasma membrane. Activated BMP receptors stimulate various intracellular signaling pathways. This canonical pathway is characterized by phosphorylation of SMAD1/5/8, which subsequently forms a gene-regulatory complex with SMAD4. Alternative BMP signaling can occur via the non-canonical pathway and is due to the presence of multiple intracellular kinases (Figure 3)[20].
While TGF-β-induced extracellular matrix production promotes tumor development, the inhibitory response to TGF suppresses tumor formation. Thus, the level of TGF-β receptor activation can alter the outcome of TGF-β signaling from suppression to oncogenesis. The TGF-β/SMAD signaling pathway has a dual effect; during tumor initiation and early stages, it stops the cell cycle and triggers apoptosis and in later stages, it promotes tumorigenesis and increases tumor progression and invasiveness[21]. TGF-β signaling causes cell cycle arrest and death during tumor initiation, acting as a tumor suppressor. However, it has also been demonstrated to increase tumor cell proliferation, EMT, and stem-like activity during tumor progression, as well as fibrosis, inflammation, and angiogenesis[22-24].
TGF-β AND ITS ROLE IN TUMOR SUPPRESSION
TGF-β signaling regulates cell proliferation mainly by inhibiting cell cycle progression through a mechanism that arrests the cell in the G1 phase. In most epithelial, endothelial, and hematopoietic cells, this arrest occurs through the activation of cyclin-dependent kinase (CDK) inhibitors, such as p21CIP1 and p15INK4b. TGF-β signaling also inhibits c-Myc oncogene transcription as well as DNA-binding protein inhibitors (ID1-3) and nuclear factors, which play key roles in cell differentiation and progression from the G1 to S phase of the cell cycle[25].
The canonical TGF-β signaling pathway can induce apoptosis by modulating the expression of various members of the B-cell lymphoma 2 (Bcl-2) family such as death receptor fibroblast death-associated antigen (FAS), DNA damage-inducible (GADD) 45-β, and kinase associated with by death (DAPK), which depends on the type of cells where the signaling takes place. It can also induce growth arrest and modulate caspases to induce intrinsic and extrinsic apoptosis[18].
TGF-β AND ITS ROLE IN TUMOR PROMOTION
In later stages of cancer, TGF-β may adversely promote tumor progression and metastasis[18]. The TGF-β signaling pathway activates the promoter activity of the translation inhibitory protein 4E-BP1 (regulator of eukaryotic translation initiation factor-4F (eIF4E) through SMAD4, thereby suppressing translation, cell growth and proliferation [26]. TGF-β also promotes the secretion of matrix metalloproteases (MMPs), mainly MMP-2 and MMP-9, and inhibits the activity of their tissue inhibitors (TIMPs)[27].
Fibrotic processes are well known to play a key role in promoting malignancy, and TGF-β is one of the most prominent inducers of fibrotic processes. During fibrosis, abundant ECM components accumulate due to activated myofibroblasts. In tumor tissue, solidified stroma stimulates tumor cell proliferation, migration, and survival. Fibrosis plays a vital role in EMT regulation, promotes angiogenesis and hypoxia, and inhibits anti-tumor immunity. Ultimately, the degree of tissue fibrosis is related to tumor aggression and poor patient prognosis[28].
TGF-β collaborates closely with BMP during fibrosis, due to their structural similarity and shared signal transmission modality. Their role is to regulate fibrosis-causing processes, like EMT. The interaction of TGF-β and BMP to form a complex with SMAD4, together with SMAD7 which elicits an inhibitory effect, affects the balance between the activation of SMADs that are members of the TGF-β signaling pathway (SMAD2/3) and SMADs that are part of the BMP signaling pathway (SMAD1/5/8). Therefore, many studies report antagonistic roles of TGF-β and BMP[29,30]; according to them, BMP activity is antifibrotic. Fewer studies support the opposite trend. Specifically, Katsuno et al[31] determined that BMP signaling can promote TGF-β signaling through the activation of protein arginine N-methyltransferase (PRMT1), which methylates SMAD6/7. SMAD6/7, in turn, activates SMAD1/3/5, resulting in the promotion of EMT during fibrosis and the maintenance of the tumor cell phenotype in malignancies[29,31].
TGF-β/SMAD RECEPTORS
Each of the isoforms of TGF-β (-1, -2, -3) binds to serine/threonine kinases, which belong to the group of transmembrane receptors and can bind to TGF-βI and TGF-βII. The name of TGF-βRI is also an activin-like receptor kinase (ALK). Seven types of TGF-βRIs have been identified to date (ALK1-7), five types of TGF-βRIIs (TGF-βRII, BMPRII, ACVRII, ACVRIIB, and AMHRII), and two types of TGF-βRIIIs (betaglycan and endoglin). All TGF-βRs consist of a C-terminal cytoplasmatic domain of a serine/threonine kinase, an internal transmembrane region, and an N-terminal domain, which binds ligands[18].
TGF-β receptors, SMAD proteins, and their mutation or inactivation have been described in many publications, along with their role in the progression of malignancies[32,33]. The loss of TGF-β tumor suppressor functions, which play a key role in inhibition in normal epithelial cells as well as in tumor cells, leads to oncogenic processes. Many human cancers, including CRC, are resistant to TGF-β-mediated growth inhibition, however. This resistance may be due to mutation or functional inactivation of TGF-βRI, decreased expression of TGF-βRI or TGF-βRII, and inactivation mutations of individual members of the TGF-β signaling pathway, such as SMAD2 and SMAD4[25]. Reportedly, approximately 20%-30% of CRCs contain mutations of TGF-βRII, and mostly involve colon cancer cells with MSI. One of the most frequent MSI mutations detected occurs in a coding polyadenine tract in exon 3 of the TGF-βRII gene. Some studies have even suggested that one of the important factors contributing to CRC transformation is the inactivation of TGFβR2, which increases cell proliferation due to prolonged activation of cdk4 expression[34-36].
Not only TGF-βRII but also TGF-βRI may contain a similar hypermutable polyadenine sequence resulting from mismatch repair defects, and the mutant allele (known as TGF-βRI6A) has been described to predispose to colon cancer, with a reported frequency of 100%[37].
SMADs
The mammalian TGF-β receptor family contains five SMAD substrates (SMAD1, SMAD2, SMAD3, SMAD5 and SMAD8); these are commonly referred to as receptor-regulated SMADs or R-SMADs[19]. Bone morphogenetic protein (BMP) and anti-Müeller receptors have high affinity for SMAD 1, 5, and 8 and TGF-β, activin and nodal receptors bind SMAD 2 and 3 proteins. SMAD4 belongs to the co-SMAD group, the second class of the SMAD family, which serves as a common partner for all R-SMADs such as SMAD2, SMAD3, SMAD1, SMAD5, and SMAD8 to form heterotrimeric complexes. These heterotrimeric SMAD complexes are subsequently translocated to nuclei, where they bind to specific promoters to act as DNA-specific transcriptional regulators of target genes[38]. SMAD6 and SMAD7 have inhibitory roles in the TGF-β/SMAD signaling pathway[39,40].
SMAD proteins are composed of approximately 500 amino acids and consist of two globular domains [MAD homology (MH) 1 and MH2] joined by a linker region. The N-terminal domain of MH1 is highly conserved in all R-SMADs and SMAD4, but not in SMADs 6 and 7, and contains a hairpin structure with DNA-binding capability. The MH2 domain contains hydrophobic elements that bind to TGF-βR and BMPR transmembrane receptors. The linker region is quite different between the different subgroups, whereas the C-terminal domain (MH2) is identical in all SMAD proteins[19]. The MH2 domain is involved in SMAD protein homooligomerization and heterooligomerization, cytoplasmic anchoring, and transcription. In normal (healthy) and premalignant cells, the TGF-β tumor signaling pathway has a suppressive role, but this pathway can be inhibited, damaged, or even used by cancer cells to promote oncogenic functions[38]. The known roles of individual SMAD proteins during the onset and progression of CRC are summarized in Table 2.
Table 2.
Roles of individual small mothers against decapentaplegic homolog proteins in the onset and progression of colorectal cancer
|
Type of SMAD
|
Role in colorectal cancer
|
References
|
| SMAD1 | Participates in the modification of cell growth, differentiation, apoptosis and other processes that are essential in the regulation of the body’s immune system | [39-42] |
| Promotes epithelial-mesenchymal transition process | ||
| By increasing the expression of ATG5 induces autophagy | ||
| SMAD2 | Inhibits the expression of related functional genes, cell proliferation and regulates the transcriptional response that promotes cell apoptosis | [43,44] |
| Expression of SMAD2 is correlated with patient survival | ||
| SMAD3 | In the formation of a tumor, depending on the stage of the cancer, it plays the double role of an oncogene or a tumor suppressor gene | [45-48] |
| Reduces its expression through mir-4429, and inhibits the appearance, development and metastasis of cancer cells | ||
| SMAD4 | Plays a very important role in the transduction of the TGF-β signaling pathway | [32,49] |
| Maintains the cell cycle in the G1 phase, which leads to abnormal tumor proliferation | ||
| Is a tumor suppressor gene | ||
| High mutation rate of SMAD4 in CRC patients was associated with poor prognosis, but not with clinical stage | ||
| SMAD5 | Mediates TGF-β superfamily ligand signaling pathway and thus influences cancer progression | [50] |
| No relevant studies on the role of SMAD5 in CRC patients have been found in the last 5 years | ||
| SMAD6 | Regulates TGF-β signaling pathway, promotes angiogenesis, stimulates extracellular matrix, and inhibits immunity, thus contributing to tumor growth, diffusion, and metastasis | [51] |
| No relevant studies on the role of SMAD6 in CRC patients have been found in the last 5 years | ||
| SMAD7 | Plays a dual role in different tumor stages, acting as a tumor suppressor gene by inhibiting proliferation and promoting apoptosis in the early stage, and increasing invasion in the late stage, promoting epithelial-mesenchymal transition, which correlates with the degree of malignancy | [52,53] |
SMAD: Small mothers against decapentaplegic homolog; ATG5: Autophagy-related gene 5; CRC: Colorectal cancer; TGF: Transforming growth factor.
In 65% of colon adenocarcinomas and 50% of rectal adenocarcinomas, mutations in any of the 43 genes that encode proteins of the TGF-β pathway superfamily have been described[19]. Many proteins interact with the SMADs to modulate their activity. Therefore, by regulating these proteins, we can influence the process of carcinogenesis[41].
Role of SMAD2/3
Many studies describe the significant role of SMAD2/3 in the EMT process, which is activated by the TGF-β signaling pathway. The most important difference between SMAD2 and SMAD3 is that the MH1 region of SMAD2 has two more amino acid fragments than SMAD3. Due to this amino acid difference, SMAD3 can directly bind to DNA and has transcriptional activity, whereas SMAD2 lacks transcriptional activity[42,43].
Although SMAD3 is highly homologous to SMAD2, the roles of SMAD2 and SMAD3 are different in the TGF-β signaling process. SMAD3 plays a very important role as a mediator of EMT, as demonstrated by inhibition or knockdown of SMAD3, which blocked cell migration induced by the TGF-β signaling pathway. Therefore, regulation of SMAD3 protein expression is a very important regulatory step in EMT prevention[44].
The results of Liu et al[45] point to other important differences between SMAD2 and SMAD3. SMAD2 is mostly located in the cytoplasm, whereas a large amount of SMAD3 is distributed in the nucleus. Western blot analysis was performed in that study, which provided evidence to support the conclusion that in the absence of TGF-β activation, endogenous SMAD2 is found mainly in the cytoplasm, while large amounts of SMAD3 are found in the nucleus of human embryonic stem cells, kidney cells, and skin fibroblast cells. This otherness in different cell compartments of SMAD2 and SMAD3 proteins may reflect their activity in TGF-beta-induced signal transduction.
Analyses of tissue and experiments with explanted tissue have revealed strongly reduced phosphorylated SMAD3 and increased levels of its inhibitor SMAD7 in Crohn’s disease tissue and a moderate reduction in ulcerative colitis (UC) tissue[46]. UC poses a high risk of developing CRC; however, the molecular mechanisms underlying the transition from UC to CRC are unclear[47].
Wang et al[48] showed that it was possible to increase the transcriptional activity of SMAD3, phosphorylation of SMAD2, and reduction of SMAD7 expression by knocking out signal transducer and activator of transcription 3 (STAT3), which ultimately led to the suppression of tumor progression in CRC. STAT3 is a member of the STAT protein family and can promote oncogenesis of CRC through various pathways.
Liu et al[49] reported that treatment with exogenous interleukin 6 (IL-6) stimulated STAT3 activation, increased TGF-β-induced SMAD3 and Snail expression, and inhibited the EMT process, suggesting that the Janus kinase/signal transducer and activator of transcription 3 (JAK/STAT3) pathway is required for TGF-β-induced EMT and cancer cell migration and invasion by upregulating SMAD3 and Snail expression. Moreover, Xu et al[50] showed that the expression of SMAD2 is correlated with patient survival. Their results demonstrated that the MIR22 host gene (MIR22HG) has been shown to play a role in suppressing colorectal tumors by binding competitively to SMAD2, thereby preventing the interaction between SMAD2 and SMAD4. These data suggest that the MIR22HG silencing promotes the EMT process and thus tumorigenicity in CRC.
Many papers have been published in recent years that link the action of the TGF-β signaling pathway to other pathways. The mitogen-activated protein kinase (MAPK) pathway may phosphorylate a group of proteins that are responsible for altering cell behavior, or conversely, proteins of this pathway may be activated by extracellular molecules such as cytokines produced by the TGF-β signaling pathway. The extracellular signal-regulated kinase (ERK) pathway inhibits the TGF-β pathway by phosphorylating SMAD2 and SMAD3 without translocating them to the nucleus[51,52].
Despite the important roles of SMAD2 and SMAD3 in the TGF-β signaling process, the prevalence of mutations was estimated up to 6%. Fleming et al[53] showed that the percentage of mutations increased with the combined prevalence of SMAD4, SMAD2, and SMAD3 mutations to 14.8% in primary sporadic CRCs.
Lin et al[54] described that nitrilase 1 (NIT1) suppresses the proliferation of CRC cells through a positive feedback loop between NIT1 and the TGFβ/SMAD signaling pathway because SMAD3 transcriptionally upregulates at the transcriptional level. NIT1 belongs to the carbon-nitrogen hydrolase superfamily and plays an important role in the suppression of CRC.
Role of SMAD4
A key component of TGF-β signaling is SMAD4, which plays an important role as a so-called switch in deciding whether to stop the cell cycle or progress to the spread of cancer[32]. Impaired TGF-β signaling due to the deletion of SMAD4 is detected in 16%-25% of CRCs[55]. Sadeghi et al[56] found SMAD4 mutations in 33.3% of analyzed tissues collected from patients with CRC.
Most SMAD4 mutations occur in the MH2 domain, although this domain represents only 41.5% of the coding sequence of the entire SMAD4 protein[56-58]. The MH2 domain is essential for homodimerization and heterooligomerization with SMAD2 or SMAD3 proteins. Therefore, mutations in this region can cause blocks to the growth, inhibition, and apoptosis that is otherwise generally induced by TGF-β. Moreover, SMAD4 mutations promote inflammation by TGF-β and thus may expand genetically damaged cells during tumorigenesis[56]. The most frequent mutation of the SMAD4 gene has been described in CRC which leads to the formation of a salt bridge between Arg361 and Asp351 and which affects homodimerization and heterooligomerization with SMAD2 and SMAD3[59,60].
Sadeghi et al[56] further described in their publication that the other significant mutations in CRC are at codons 264 and 271 of SMAD4 protein, which are located in the linker domain, a region required for subcellular localization and transcriptional activation.
Analyzes of tissue sections by immunohistochemical methods of carcinomas from various organs, including the gastrointestinal tract have shown a loss of SMAD4 expression in > 50% of colorectal carcinomas, which is associated with lymph node metastases. SMAD4 loss has been seen in 58% of pancreatic adenocarcinomas, 27% of appendiceal adenocarcinomas, 16% of cholangiocarcinomas, 10% of lung adenocarcinomas, and < 5% of esophageal, breast, gastric, and mucinous ovarian adenocarcinomas[61]. Although the LOH on chromosome 18q can be the main cause of SMAD4 loss in CRC, there are other posttranscriptional and posttranslational mechanisms that may contribute to SMAD4 protein loss or dysfunction, such as ubiquitylation, sumoylation, or interference with regulatory microRNA (miRNA)[62].
Regarding the correlation between SMAD proteins and clinicopathological characteristics, Yang et al[63] showed that SMAD4 concentrations in CRC patients were significantly higher in the N0 stage compared to patients with NI stage. Regarding patients in advanced stages (TNM III-IV), reduced concentrations of SMAD4 were recorded in them compared to patients in early stages (TNM I-II). In addition, SMAD4 was significantly decreased in patients who were older than 65 years.
Szeglin et al[64] determined probes and corresponding genes from analysis of SMAD-binding elements (SBEs) that were correlated with SMAD4 expression. They subsequently confirmed that a SMAD4-modulated gene profile predicted disease-free survival in stage II and III CRC. According to them, this gene profile has prognostic potential in selected CRC patients.
Role of SMAD7
SMAD7 acts as an inhibitor of SMAD in the TGF-β/SMAD pathway and may prevent TGF-β-dependent SMAD2/SMAD4 complex formation and inhibit SMAD2 accumulation in the nucleus (Figure 4). SMAD7 may also promote the dephosphorylation and inactivation of TGF-βRI with cooperation of the E3 ubiquitin ligase SMURF1/2. SMAD7 may also localize to the nucleus and limit the binding of the SMAD2-3/SMAD4 complex to specific SMAD-responsive DNA sequences[65]. So, SMAD7 plays an important role in both the cytoplasm and the nucleus, thereby maintaining the balance in the TGF-β induced signaling pathway. Inactivation of any component in this pathway will result in accelerated cell growth and dysregulation of apoptosis signals, leading to uncontrolled cell growth and differentiation, and the induction of cancer cells[66]. Therefore, overproduction of SMAD7 leads to significantly decreased EMT in response to TGF-β[67].
Figure 4.

Inhibitory effect of small mothers against decapentaplegic homolog 7 on the process of colorectal cancer development. TGF: Transforming growth factor; SMAD: Small mothers against decapentaplegic homolog.
Several studies have reported the significant role of SMAD7 in sporadic CRC. According to results published by Li et al[66], SMAD7 is a target of miR-424, which is implicated in the regulation of SMAD7 expression via the circTBL1XR1/miR-424 axis.
Boulay and colleagues, in 264 biopsy samples from CRC patients, showed that the deletion of SMAD7 is less common than deletion of SMAD4 and SMAD2, and patients with such a SMAD7 deletion have a significantly better prognosis than patients without a deletion. Their findings demonstrated that patients with SMAD7 deletions had a low ratio of death risk and relapse, which clearly defined SMAD7 as a negative prognostic marker in CRC patients[68,69].
SMAD7 and SMAD4 genes are deregulated in CRC, whereas there is a markedly higher increase in SMAD7 expression (~ 11.3-fold) than SMAD4 expression (approximately 2-fold) in tumor cells[70]. SMAD7 protein expression is closely related to Dukes’ stage, CRC invasion depth, and lymph node metastases, and positively correlates with CRC expression[66].
Less frequently, it has been reported that SMAD7 also has an anti-cancer effect. Gastrointestinal carcinomas, such as CRC, are characterized by frequent genetic alterations in SMAD components. Furthermore, depending on the stage of the tumor, SMAD7 activity can transition from tumor-suppressive to tumor-promoting (i.e., early vs advanced). Given the opposing roles of TGF signaling, these seemingly contradicting functions are not surprising[71,72].
REGULATION OF TGF-β SIGNALING PATHWAY BY NON-CODING RNAs
Genes that encode proteins represent less than 2% of the total human genome, while approximately 90% of the human genome consists of non-coding RNAs (ncRNAs) that do not encode proteins. ncRNAs are divided into two larger groups[73]; in one are the housekeeping ncRNAs, including the very abundant rRNAs and tRNAs, and in the other are the regulatory ncRNAs, including long ncRNAs (lncRNAs), microRNAs (miRNAs), circular RNAs (circRNAs), PIWI-interacting RNAs, small tRNA-derived RNAs (tRFs), small nuclear RNAs (snoRNAs), siRNAs and others. The most studied classes of ncRNAs are lncRNAs, miRNAs, and circRNAs. These types of ncRNAs very significantly regulate or are regulated by the TGF-β signaling pathway[74].
LNCRNAs AS REGULATORS IN CRC
lncRNAs influence gene expression through several mechanisms, such as silencing of the X chromosome, modification of chromatin, imprinting of the genome, activation of transcription, and nuclear transport. Imbalance in regulation of lncRNA transcription has been associated with apoptosis, angiogenesis, proliferation, invasion, metastasis and drug resistance of CRC[74].
The lncRNAs cancer susceptibility candidate 9 (CASC9) and small nucleolar RNA host gene 6 (SNHG6) can positively regulate the TGF-β pathway in CRC. CASC9, in particular, increases the stabilization of TGF-β2 mRNA[75], and a study by Zhang et al[76] showed that it targets miRNA-542-3p and could also increase chemoresistance. The lncRNA SNHG6, on the other hand, targets miR-26a-5p and increases the resistance of CRC cells to 5-fluorouracil (5-FU).
The lncRNA nuclear paraspeckle assembly transcript 1 (NEAT1) has been verified to participate in the development and progression of colon cancer[77].
CTBP1-AS2 has an important role in CRC proliferation and metastasis. While CTBP1-AS2 has been shown to significantly promote activation of the TGF-β/SMAD2/3 signaling pathway, miR93-5p (a downstream molecule of CTBP1-AS2) has been shown to target the 3′-untranslated region (UTR) of TGF-β. Furthermore, investigations of the functionally of miR-93-5p showed that its overexpression exerts an anti-cancer effect by inhibiting the TGF-β/SMAD2/3 pathway[78].
miRNAs AS REGULATORS IN CRC
miRNA regulates the transcription of genes encoding proteins at the post-transcriptional level. They perform this task by binding to complementary sequences located in the 3′-UTRs of their target mRNAs[79]. miRNAs are also competitively inhibited by lncRNAs[24].
In TGF-β signaling, miRNAs can play a stimulatory role, as shown in cells treated with anti-metabolites and anti-microtubule medicines; this is similar to what has been reported in cases of chemo-resistance against DNA damaging agents. In particular, miR-423-5p, miR-552, miR-34a, and the miR-17-92 cluster (miR-17, miR-18a, miR-19a, miR-19b, miR-20a, and miR-92a) are examples of miRNAs that regulate TGF-β signaling in CRC. Furthermore, SMAD2, SMAD4, and TGF-βRII genes are markedly associated with miRNA-155 and miR-22, both of which strongly correlate with tumor properties, suggesting clinical utility in immunotherapy[24]. miR-4666-3p and miR-329 act as tumor suppressor genes, affecting TGF-βR1 and thus preventing the activation of the TGF-β1/Smad pathway[80]. Finally, miR-147 overexpression has been shown to inhibit EMT and the TGF-β/SMAD pathway in colon cancer cells[81].
circRNAs AS REGULATORS IN CRC
circRNAs are formed by back-splicing of linear RNA and connections via covalent linkage. circRNAs can prevent miRNAs from binding to the 3’-UTR sequence of a particular gene, by attachment to miRNAs, ultimately regulating gene expression by activating mRNA cleavage or subsequent translation[82].
circPTEN1 is significantly downregulated in CRC and its expression is positively correlated with patient prognosis. circPTEN1 binds to the MH2 domain of SMAD4 and prevents the interaction between SMAD4 and SMAD2/3, which leads to suppression of translocation of the SMAD complex into the nucleus, followed by the activation of the transcription of downstream genes that regulate the EMT by the TGF-β signaling pathway[83].
circPACRGL acts as a miR-142-3p/miR-506-3p sponge to promote TGF-β1 expression and, thus, promote the differentiation of N1 to N2 neutrophils[84].
Gaining a more comprehensive understanding of the role of ncRNAs in CRC may lead to new approaches in the treatment of this disease; however, currently, only a limited number of identified and characterized lncRNAs and circRNAs with a confirmed regulatory role in CRC are known. There remains an urgent need to investigate the role of other lncRNAs and circRNAs that may facilitate the prognosis, diagnosis and treatment of CRC.
TREATMENT OF CRC
Over the last 10 years, researchers have developed a new anticancer therapy for patients with advanced or metastatic cancer. Several recent studies have shown that drug resistance in the treatment of various cancers, including CRC, is associated with the activation of TGF-β signaling[24]. 5-FU, an anticancer agent that belongs to the category of antimetabolites, is widely used to regulate metabolic pathways that are essential for cancer cell proliferation and survival. 5-FU is a standard chemotherapeutic used for the treatment of CRC patients, and a large proportion of these patients relapse or metastasize during the course of treatment. In patients with CRC, drug resistance is a key cause of chemotherapy failure and disease progression[85,86]. Recent research suggests that SMAD4 expression levels correlate with the prognosis and response to 5-FU and can help guide therapeutic decisions regarding its administration[87,88]. Reduced concentrations of SMAD3 or loss of SMAD4 suppress the expression of tumor suppressor genes that are induced by the TGF-β signaling pathway, which in turn leads to the expression of anti-apoptotic proteins Bcl-2 and Bcl-Wand increased survival of cancer cells resistance to 5-fluorouracil in CRC[89].
The role of TGF-β/SMAD signaling in tumor radiotherapy is controversial. It has been described in some studies that fibrosis is induced by upregulation of SMAD2/3 after radiation exposure. Reactive oxygen species (ROS) are involved in irradiation (IR)-induced fibrosis through TGF-β signaling. SMAD molecules that are activated by the TGF-β signaling pathway regulate ROS production by upregulating NADP oxidase 4[89,90]. Mutations in some genes, such as tumor protein p53, Ras, SMAD4, and EMT, are important in radioresistance or radiosensitization and can be controlled by SMAD-dependent or SMAD-independent TGF-β pathways[91]. Publications in recent years suggest that TGF-β signaling through various mechanisms, especially through miRNA-mediated regulation, plays an important role in the resistance of tumor cells to DNA-damaging agents. In CRC, miR-34a interacts directly with the 3’-untranslated region of SMAD4 and suppresses TGF-β/SMAD4 signaling. In patients with oxaliplatin-resistant CRC, miR-34a is downregulated to increase macroautophagy by activating the TGF-β/SMAD pathway[92,93].
ANTI-TGF-β THERAPIES
The objective of targeting TGF-β signaling as a therapeutic approach to treat cancer is supported by a plethora of findings from genetic and preclinical studies. Several strategies have been tested thus far that aim to block the TGF-β signaling pathway (Figure 5). These include: (1) Preventing TGF-β production or expression of its receptor by antisense oligonucleotides (ASOs; short synthetic single-stranded nucleic acids that bind to RNA to regulate gene expression); (2) preventing TGF-β activation via integrin-blocking antibodies, in which the antibodies compete with the TGF-β ligand to bind to its receptor, as well as the ability to block the activation of latent TGF-β (both steps are crucial for TGF-β to elicit its protumorogenic and immunosuppressive responses); (3) inhibiting the interaction between TGF-β and its receptor with neutralizing antibodies to TGF-β, blocking antibodies to TGF-βRII or ligand traps (engineered soluble forms of the receptor that compete with the cell-bound receptor); (4) preventing intracellular TGF-β receptor signal transduction via small-molecule kinase inhibitors, which bind to the ATP-binding domain of TGF-β kinase and inhibit ATP kinase activity, thereby blocking the downstream signaling cascade[94]; (5) immune checkpoint inhibitors (ICIs), which have essential roles in modulating the immune system. This group includes monoclonal antibodies that send inhibitory signals to T cells, enhancing T cells’ antitumor immune response and improving antitumor defense. In addition to immunoregulatory cells such as regulatory T cells (Tregs), M2 macrophages, and myeloid-derived suppressor cells (MDSCs), the cytokine TGF-β also has the ability to control and modulate T cell functions. This is facilitated by the release of molecules that are able to activate specific ICIs. In this way, activation of inhibitory immune checkpoints, such as cytotoxic T-lymphocyte-associated protein-4 (CTLA-4), programmed cell death-1/Ligand (PD-1/PD-L1), lymphocyte-activation gene 3 (LAG3), or T-cell immunoglobulin-and mucin domain-3-containing molecule 3 (TIM-3) can disrupt cytotoxic T-lymphocyte (CTL) proliferation in CRC and reduce the immune response against cancer[95]; (6) vaccine-based approaches to modulate TGF-β signaling, which have been applied with the aim of facilitating the immune destruction of cancer cells in many different tumor types. It is important to realize that tumors are able to prevent the activation of the immune system by hiding tumor cell antigens and also suppress the immune system. Thus, cancer vaccines will help to activate and maintain an anti-tumor immune response; and (7) adoptive cell therapy, which is a form of passive immunotherapy that involves transferring immune cells or molecules to the host[96].
Figure 5.

Inhibition strategies of transforming growth factor-β signaling pathway and miRNAs targets for colorectal cancer treatment. TGF: Transforming growth factor; SMAD: Small mothers against decapentaplegic homolog.
Many of these agents have been or are being evaluated in clinical trials to treat CRC (Table 3).
Table 3.
Clinical trials of drugs for the treatment of colorectal cancer (United States National Library of Medicine; ClinicalTrials.gov)
|
Clinical trials (phase)
|
Drug
|
Target
|
Mechanism of action
|
| Antisense oligonucleotides | |||
| NCT00844064 (I) | AP12009 (trabedersen) | TGF-β2 | By binding to TGF-βII mrna, its expression is reduced |
| Antibodies | |||
| NCT04952753 (II) | NIS793 | TGF-β | Reduction of active cytokine, reduction of SMAD2/3 phosphorylation, and reduction of TGF-β target gene expression |
| NCT02947165 (I) | NIS793 | TGF-β | Reduction of active cytokine, reduction of SMAD2/3 phosphorylation, and reduction of TGF-β target gene expression |
| NCT01646203(I) | IMC-TR1 | TGF-βRII | Reduction of active cytokine, reduction of SMAD2/3 phosphorylation, and reduction of TGF-β target gene expression |
| Ligand traps | |||
| NCT03436563 (I/II) | M7824 | TGF-βRII | Bifunctional anti-PD-L1/TGF-βRII trap fusion protein |
| NCT02517398(I) | Bintrafusp alfa | TGF-βRII and PD-L1 | First-in-class bifunctional fusion protein composed of a mab against PD-L1 fused to the extracellular domain of the TGF-β receptor II |
| NCT04856787 (II/III) | SHR-1701 | TGF-βRII | Bifunctional anti-PD-L1/TGF-βRII agent |
| Small molecule receptor kinase inhibitors | |||
| NCT04031872 (I/II) | LY3200882; capecitabine | TGF-βRI | By blocking ATP binding to TGF-βR, receptor kinase activity and signal transduction are reduced |
| NCT05400122 (I) | Vactosertib | TGF-βRI | Inhibits the activity of TGF-βR1 |
| NCT03724851 (I/II) | Vactosertib + Pembroli-zumab | TGF-βRI | Inhibits the activity of TGF-βR1 |
| NCT03470350 (I/II) | Galunisertib | TGF-βRI | Inhibits the activity of TGF-βR1 |
| Immune checkpoints | |||
| NCT04540159 | TGF-β1 | Measuring the level Active TGF-β1 by flow-cytometric analysis in the intraabdominal ascites | |
| Adoptive cell therapy | |||
| NCT03431311 (I/II) | ACT | TGF-βII | ACT with Radium-1 TCR + T cells transiently redirected against the TGF-βRII frameshift antigen which is expressed in MSI+ colon cancer. |
| NCT05040568 (I) | CB-NK-TGF-βR2-/NR3C1 | Immunotherapy with ex vivo preactivated and expanded CB-NK cells in combination with cetuximab | |
ACT: Adoptive cell therapy; TGF: Transforming growth factor.
SMALL MOLECULE INHIBITORS OF SMAD EXPRESSION AND PHOSPHORYLATION
Since SMAD molecules have an important role in the TGF-β signaling pathway, great efforts have been made for the search of SMAD activation inhibitors. Indeed, it has been shown that SMAD3 silencing can suppress cancer cell growth and metastasis by increasing the cancer-killing activity of natural killer (NK) cells. Thus, the selective inhibition of the SMAD3 protein with a potent, low toxicity drug could provide a promising anticancer treatment. Some compounds have shown good inhibitory activity against SMAD 2 or SMAD3 through direct or indirect downregulation of their respective expressions and phosphorylations[97].
Peptide aptamers or DNA aptamers are artificial short peptides, respectively single-stranded DNA or RNA nucleotides, which are antibody-like in function. Aptamers can bind specific molecules with high specificity and affinity. SMAD2-and SMAD3-binding aptamers have also been established. Upon binding to SMAD2 or SMAD3, the aptamer prevents their binding and complex formation, thereby arresting TGF-β signaling[98,99]. Aptamers also have the potential to be used more frequently in clinical practice, from disease diagnosis to targeted delivery of therapeutic agents. Their simplicity in manufacturing and lengthy shelf life significantly improve this potential[100].
The specific inhibitor of SMAD3 (SIS3) is a synthetic substance that specifically inhibits the phosphorylation of SMAD3 and thus its binding to SMAD4[101]. Furthermore, targeting the inhibition of SMAD3 is currently considered a promising therapeutic strategy in the treatment of cancer[102].
MEDICATION THERAPEUTIC STRATEGIES THROUGH THE TGF-Β /SMAD SIGNALING PATHWAY
The effects of several potential molecules that induce tumor growth or inhibit the proliferation and metastasis of carcinoma cells through regulation of the TGF-β/SMAD signaling pathway have been described[103]. Baicalein is a major flavonoid, originally extracted from the edible medicinal plants of Scutellaria baicalensis and S. lateriflora. Baicalein reduces the concentrations of phosphorylated SMAD2 and SMAD3, without affecting the total levels of SMAD2 and SMAD3 and thus inhibits the TGF-β/SMAD2/3 signaling pathway in fibroblasts in vitro and in vivo without affecting SMAD 1, 5, and 8 in the BMP signaling pathway[104].
Ginseng is valued as the most important medicinal plant in traditional Chinese medicine. The major constituents of ginseng are ginsenosides. Ginsenoside Rg3 has an inhibitory effect on the TGF-β/SMAD and ERK signaling pathways in keloid fibroblasts and increases mRNA expression levels in SMAD7[105]. Dai et al[106] showed that ginsenoside Rb2 can inhibit the expression of SMAD4 and phosphorylated SMAD2/3 in CRC cells.
Kaempferol is a natural flavanol, a type of flavonoid, found in a variety of plants and plant-derived foods, including kale, beans, tea, spinach, and broccoli. It binds to the TβRI, leading to its inactivation. This results in inhibition of the TGF-β/SMAD signaling pathway due to reduced phosphorylation of SMAD2/3[107].
Loureirin B, a flavonoid extracted from Dracaena cochinchinensis, is used in traditional Chinese medicine (TCM). Loureirin B upregulates the expression of MMP-1, MMP-3, MMP-9, and MMP-13 and thus causes degradation of extracellular matrix, inhibits the phosphorylation of SMAD2 and SMAD3 and thus effectively suppresses the TGF-β/SMAD pathway[108].
Galangin is a polyphenolic compound derived primarily from different medicinal herbs, the effect of which is the downregulation of SMAD2 and SMAD3 phosphorylation without altering the expression of total SMAD2, SMAD3, SMAD4, SMAD6, and SMAD7[109].
Celastrol is a pharmacologically active substance extracted from Tripterygium wilfordii Hook F, which is used in TCM to treat cancer and other inflammatory diseases[110]. Zhang et al[111] showed that celastrol reduces the levels of SMAD4 and phosphorylated SMAD2/3. Together, celastrol may inhibit CRC through TGF-β, which is a promising treatment for CRC.
Qingjie Fuzheng granules are TCM comprising a 4-herb mixture, composed of Hedyotis diffusa Willd, malt, Astragalus, and S. barbata D. Don significantly inhibits the expression of several key proteins in the canonical TGF-β/SMAD pathway, including TGF-β, phosphorylated SMAD2/3, and SMAD4. This inhibition leads to a decrease in the ratio of N-cadherin to E-cadherin, indicating that EMT is inhibited [111].
CONCLUSION
Antitumor immunity is mediated by macrophages, NK cells, granulocytes (polymorphonuclear leukocytes, PMNs), T cells, and antibodies. In recent years, the particular role of PMNs in regulation of adaptive immunity, especially in cancer, has emerged. PMNs in cancer are functionally diverse, with some authors describing their antitumor activity, but the number of publications in which the authors confirm their negative regulation of immune responses and their presence in cancer patients associated with poor prognosis and therapeutic outcomes is increasing. These cells suppress the physiological role of T and B lymphocytes and NK cells, and also promote tumor progression and metastasis through non-immune mechanisms. Cytokines produced by tumor cells [vascular endothelial growth factor (VEGF), TGF-β] also play a similar role when they inhibit T cell development and function. TGF-β, as an immunosuppressive factor, significantly affects the proliferation, activation, and differentiation of immune effector cells. Epigenetic changes that may be affected by the TGF-β pathway in CRC should be carefully studied because the mechanisms of drug resistance are different between patients in different stages of cancer and personalized treatment is more effective. Therefore, knowledge of the activation and inhibition of factors that affect the TGF-β signaling pathway is very important.
ACKNOWLEDGEMENTS
This review is a summary work consisting of the results of many authors. We would like to thank all the authors whose work we have included in this review article and apologize to the authors whose relevant work has not been included in this review article.
Footnotes
Conflict-of-interest statement: The authors declare having no conflict of interests for this article.
Provenance and peer review: Unsolicited article; Externally peer reviewed.
Peer-review model: Single blind
Corresponding Author’s Membership in Professional Societies: Slovak Surgical Society, No. 14119.
Peer-review started: May 12, 2022
First decision: June 19, 2022
Article in press: August 16, 2022
Specialty type: Biochemistry and molecular biology
Country/Territory of origin: Slovakia
Peer-review report’s scientific quality classification
Grade A (Excellent): A
Grade B (Very good): B, B
Grade C (Good): C, C, C
Grade D (Fair): D
Grade E (Poor): 0
P-Reviewer: Gao W, China; Ghazy A, Egypt; Kiuchi J, Japan; Liang H, China; Luo ZW, China S-Editor: Chen YL L-Editor: A P-Editor: Zhang XD
Contributor Information
Jana Maslankova, Department of Medical and Clinical Biochemistry, Faculty of Medicine, Pavol Jozef Safarik University in Kosice, Kosice 04011, Slovakia.
Ivana Vecurkovska, Department of Medical and Clinical Biochemistry, Faculty of Medicine, Pavol Jozef Safarik University in Kosice, Kosice 04011, Slovakia.
Miroslava Rabajdova, Department of Medical and Clinical Biochemistry, Faculty of Medicine, Pavol Jozef Safarik University in Kosice, Kosice 04011, Slovakia.
Jana Katuchova, First Department of Surgery, Medical Faculty of Safarik University, Kosice 04011, Kosicky kraj, Slovakia. jana.katuchova@upjs.sk.
Milos Kicka, First Department of Surgery, Medical Faculty of Safarik University, Kosice 04011, Kosicky kraj, Slovakia.
Michala Gayova, Department of Burns and Reconstructive Surgery, Medical Faculty at Safarik University and University Hospital, Kosice 04011, Slovakia.
Vladimir Katuch, Department of Neurosurgery, Medical Faculty at Safarik University and University Hospital, Kosice 04011, Slovakia.
References
- 1.Xi Y, Xu P. Global colorectal cancer burden in 2020 and projections to 2040. Transl Oncol. 2021;14:101174. doi: 10.1016/j.tranon.2021.101174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lech G, Słotwiński R, Słodkowski M, Krasnodębski IW. Colorectal cancer tumour markers and biomarkers: Recent therapeutic advances. World J Gastroenterol. 2016;22:1745–1755. doi: 10.3748/wjg.v22.i5.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahmad R, Singh JK, Wunnava A, Al-Obeed O, Abdulla M, Srivastava SK. Emerging trends in colorectal cancer: Dysregulated signaling pathways (Review) Int J Mol Med. 2021;47 doi: 10.3892/ijmm.2021.4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang W, Kandimalla R, Huang H, Zhu L, Li Y, Gao F, Goel A, Wang X. Molecular subtyping of colorectal cancer: Recent progress, new challenges and emerging opportunities. Semin Cancer Biol. 2019;55:37–52. doi: 10.1016/j.semcancer.2018.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang F, Xia X, Yang C, Shen J, Mai J, Kim HC, Kirui D, Kang Y, Fleming JB, Koay EJ, Mitra S, Ferrari M, Shen H. SMAD4 Gene Mutation Renders Pancreatic Cancer Resistance to Radiotherapy through Promotion of Autophagy. Clin Cancer Res. 2018;24:3176–3185. doi: 10.1158/1078-0432.CCR-17-3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reynolds IS, Furney SJ, Kay EW, McNamara DA, Prehn JHM, Burke JP. Meta-analysis of the molecular associations of mucinous colorectal cancer. Br J Surg. 2019;106:682–691. doi: 10.1002/bjs.11142. [DOI] [PubMed] [Google Scholar]
- 7.Gallois C, Laurent-Puig P, Taieb J. Methylator phenotype in colorectal cancer: A prognostic factor or not? Crit Rev Oncol Hematol. 2016;99:74–80. doi: 10.1016/j.critrevonc.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 8.Andre T, Amonkar M, Norquist JM, Shiu KK, Kim TW, Jensen BV, Jensen LH, Punt CJA, Smith D, Garcia-Carbonero R, Sevilla I, De La Fouchardiere C, Rivera F, Elez E, Diaz LA Jr, Yoshino T, Van Cutsem E, Yang P, Farooqui M, Le DT. Health-related quality of life in patients with microsatellite instability-high or mismatch repair deficient metastatic colorectal cancer treated with first-line pembrolizumab versus chemotherapy (KEYNOTE-177): an open-label, randomised, phase 3 trial. Lancet Oncol. 2021;22:665–677. doi: 10.1016/S1470-2045(21)00064-4. [DOI] [PubMed] [Google Scholar]
- 9.De' Angelis GL, Bottarelli L, Azzoni C, De' Angelis N, Leandro G, Di Mario F, Gaiani F, Negri F. Microsatellite instability in colorectal cancer. Acta Biomed. 2018;89:97–101. doi: 10.23750/abm.v89i9-S.7960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pino MS, Chung DC. The chromosomal instability pathway in colon cancer. Gastroenterology. 2010;138:2059–2072. doi: 10.1053/j.gastro.2009.12.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, Soneson C, Marisa L, Roepman P, Nyamundanda G, Angelino P, Bot BM, Morris JS, Simon IM, Gerster S, Fessler E, De Sousa E Melo F, Missiaglia E, Ramay H, Barras D, Homicsko K, Maru D, Manyam GC, Broom B, Boige V, Perez-Villamil B, Laderas T, Salazar R, Gray JW, Hanahan D, Tabernero J, Bernards R, Friend SH, Laurent-Puig P, Medema JP, Sadanandam A, Wessels L, Delorenzi M, Kopetz S, Vermeulen L, Tejpar S. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21:1350–1356. doi: 10.1038/nm.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fontana E, Eason K, Cervantes A, Salazar R, Sadanandam A. Context matters-consensus molecular subtypes of colorectal cancer as biomarkers for clinical trials. Ann Oncol. 2019;30:520–527. doi: 10.1093/annonc/mdz052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stintzing S, Wirapati P, Lenz HJ, Neureiter D, Fischer von Weikersthal L, Decker T, Kiani A, Kaiser F, Al-Batran S, Heintges T, Lerchenmüller C, Kahl C, Seipelt G, Kullmann F, Moehler M, Scheithauer W, Held S, Modest DP, Jung A, Kirchner T, Aderka D, Tejpar S, Heinemann V. Consensus molecular subgroups (CMS) of colorectal cancer (CRC) and first-line efficacy of FOLFIRI plus cetuximab or bevacizumab in the FIRE3 (AIO KRK-0306) trial. Ann Oncol. 2019;30:1796–1803. doi: 10.1093/annonc/mdz387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nguyen HT, Duong HQ. The molecular characteristics of colorectal cancer: Implications for diagnosis and therapy. Oncol Lett. 2018;16:9–18. doi: 10.3892/ol.2018.8679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang W, Wang GQ, Sun XW, Chen G, Li YF, Zhang LY, Qiu HB, Huang CY, Zhan YQ, Zhou ZW. Prognostic values of chromosome 18q microsatellite alterations in stage II colonic carcinoma. World J Gastroenterol. 2010;16:6026–6034. doi: 10.3748/wjg.v16.i47.6026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vacante M, Borzì AM, Basile F, Biondi A. Biomarkers in colorectal cancer: Current clinical utility and future perspectives. World J Clin Cases. 2018;6:869–881. doi: 10.12998/wjcc.v6.i15.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Samanta D, Datta PK. Alterations in the Smad pathway in human cancers. Front Biosci (Landmark Ed) 2012;17:1281–1293. doi: 10.2741/3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baba AB, Rah B, Bhat GR, Mushtaq I, Parveen S, Hassan R, Hameed Zargar M, Afroze D. Transforming Growth Factor-Beta (TGF-β) Signaling in Cancer-A Betrayal Within. Front Pharmacol. 2022;13:791272. doi: 10.3389/fphar.2022.791272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gough NR, Xiang X, Mishra L. TGF-β Signaling in Liver, Pancreas, and Gastrointestinal Diseases and Cancer. Gastroenterology. 2021;161:434–452.e15. doi: 10.1053/j.gastro.2021.04.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frey P, Devisme A, Schrempp M, Andrieux G, Boerries M, Hecht A. Canonical BMP Signaling Executes Epithelial-Mesenchymal Transition Downstream of SNAIL1. Cancers (Basel) 2020;12 doi: 10.3390/cancers12041019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karathanasi V, Tosios KI, Nikitakis NG, Piperi E, Koutlas I, Trimis G, Sklavounou A. TGF-β1, Smad-2/-3, Smad-1/-5/-8, and Smad-4 signaling factors are expressed in ameloblastomas, adenomatoid odontogenic tumors, and calcifying cystic odontogenic tumors: an immunohistochemical study. J Oral Pathol Med. 2013;42:415–423. doi: 10.1111/jop.12016. [DOI] [PubMed] [Google Scholar]
- 22.Gu S, Feng XH. TGF-β signaling in cancer. Acta Biochim Biophys Sin (Shanghai) 2018;50:941–949. doi: 10.1093/abbs/gmy092. [DOI] [PubMed] [Google Scholar]
- 23.Luo J, Chen XQ, Li P. The Role of TGF-β and Its Receptors in Gastrointestinal Cancers. Transl Oncol. 2019;12:475–484. doi: 10.1016/j.tranon.2018.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang M, Zhang YY, Chen Y, Wang J, Wang Q, Lu H. TGF-β Signaling and Resistance to Cancer Therapy. Front Cell Dev Biol. 2021;9:786728. doi: 10.3389/fcell.2021.786728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seoane J, Gomis RR. TGF-β Family Signaling in Tumor Suppression and Cancer Progression. Cold Spring Harb Perspect Biol. 2017;9 doi: 10.1101/cshperspect.a022277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hao Y, Baker D, Ten Dijke P. TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int J Mol Sci. 2019;20 doi: 10.3390/ijms20112767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muscella A, Vetrugno C, Cossa LG, Marsigliante S. TGF-β1 activates RSC96 Schwann cells migration and invasion through MMP-2 and MMP-9 activities. J Neurochem. 2020;153:525–538. doi: 10.1111/jnc.14913. [DOI] [PubMed] [Google Scholar]
- 28.Piersma B, Hayward MK, Weaver VM. Fibrosis and cancer: A strained relationship. Biochim Biophys Acta Rev Cancer. 2020;1873:188356. doi: 10.1016/j.bbcan.2020.188356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dituri F, Cossu C, Mancarella S, Giannelli G. The Interactivity between TGFβ and BMP Signaling in Organogenesis, Fibrosis, and Cancer. Cells. 2019;8 doi: 10.3390/cells8101130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ning J, Zhao Y, Ye Y, Yu J. Opposing roles and potential antagonistic mechanism between TGF-β and BMP pathways: Implications for cancer progression. EBioMedicine. 2019;41:702–710. doi: 10.1016/j.ebiom.2019.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Katsuno Y, Qin J, Oses-Prieto J, Wang H, Jackson-Weaver O, Zhang T, Lamouille S, Wu J, Burlingame A, Xu J, Derynck R. Arginine methylation of SMAD7 by PRMT1 in TGF-β-induced epithelial-mesenchymal transition and epithelial stem-cell generation. J Biol Chem. 2018;293:13059–13072. doi: 10.1074/jbc.RA118.002027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao M, Mishra L, Deng CX. The role of TGF-β/SMAD4 signaling in cancer. Int J Biol Sci. 2018;14:111–123. doi: 10.7150/ijbs.23230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang T, Liu W, Meng W, Zhao H, Yang Q, Gu SJ, Xiao CC, Jia CC, Fu BS. Downregulation of miR-542-3p promotes cancer metastasis through activating TGF-β/Smad signaling in hepatocellular carcinoma. Onco Targets Ther. 2018;11:1929–1939. doi: 10.2147/OTT.S154416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fricke F, Mussack V, Buschmann D, Hausser I, Pfaffl MW, Kopitz J, Gebert J. TGFBR2dependent alterations of microRNA profiles in extracellular vesicles and parental colorectal cancer cells. Int J Oncol. 2019;55:925–937. doi: 10.3892/ijo.2019.4859. [DOI] [PubMed] [Google Scholar]
- 35.Grady WM. Polymerase Slippage Restoration of Frameshifted TGFBR2 in Colorectal Cancer: A Novel Paradigm. Gastroenterology. 2015;148:1276–1279. doi: 10.1053/j.gastro.2015.04.023. [DOI] [PubMed] [Google Scholar]
- 36.Jung B, Staudacher JJ, Beauchamp D. Transforming Growth Factor β Superfamily Signaling in Development of Colorectal Cancer. Gastroenterology. 2017;152:36–52. doi: 10.1053/j.gastro.2016.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou R, Huang Y, Cheng B, Wang Y, Xiong B. TGFBR1*6A is a potential modifier of migration and invasion in colorectal cancer cells. Oncol Lett. 2018;15:3971–3976. doi: 10.3892/ol.2018.7725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sinha A, Iyengar PV, Ten Dijke P. E3 Ubiquitin Ligases: Key Regulators of TGFβ Signaling in Cancer Progression. Int J Mol Sci. 2021;22 doi: 10.3390/ijms22020476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.de Ceuninck van Capelle C, Spit M, Ten Dijke P. Current perspectives on inhibitory SMAD7 in health and disease. Crit Rev Biochem Mol Biol. 2020;55:691–715. doi: 10.1080/10409238.2020.1828260. [DOI] [PubMed] [Google Scholar]
- 40.Miyazawa K, Miyazono K. Regulation of TGF-β Family Signaling by Inhibitory Smads. Cold Spring Harb Perspect Biol. 2017;9 doi: 10.1101/cshperspect.a022095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tecalco-Cruz AC, Ríos-López DG, Vázquez-Victorio G, Rosales-Alvarez RE, Macías-Silva M. Transcriptional cofactors Ski and SnoN are major regulators of the TGF-β/Smad signaling pathway in health and disease. Signal Transduct Target Ther. 2018;3:15. doi: 10.1038/s41392-018-0015-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen HY, Chiang YF, Huang JS, Huang TC, Shih YH, Wang KL, Ali M, Hong YH, Shieh TM, Hsia SM. Isoliquiritigenin Reverses Epithelial-Mesenchymal Transition Through Modulation of the TGF-β/Smad Signaling Pathway in Endometrial Cancer. Cancers (Basel) 2021;13 doi: 10.3390/cancers13061236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ma X, Gao Y, Chen Y, Liu J, Yang C, Bao C, Wang Y, Feng Y, Song X, Qiao S. M2-Type Macrophages Induce Tregs Generation by Activating the TGF-β/Smad Signalling Pathway to Promote Colorectal Cancer Development. Onco Targets Ther. 2021;14:5391–5402. doi: 10.2147/OTT.S336548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen N, Balasenthil S, Reuther J, Frayna A, Wang Y, Chandler DS, Abruzzo LV, Rashid A, Rodriguez J, Lozano G, Cao Y, Lokken E, Chen J, Frazier ML, Sahin AA, Wistuba II, Sen S, Lott ST, Killary AM. DEAR1 is a chromosome 1p35 tumor suppressor and master regulator of TGF-β-driven epithelial-mesenchymal transition. Cancer Discov. 2013;3:1172–1189. doi: 10.1158/2159-8290.CD-12-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu L, Liu X, Ren X, Tian Y, Chen Z, Xu X, Du Y, Jiang C, Fang Y, Liu Z, Fan B, Zhang Q, Jin G, Yang X, Zhang X. Smad2 and Smad3 have differential sensitivity in relaying TGFβ signaling and inversely regulate early lineage specification. Sci Rep. 2016;6:21602. doi: 10.1038/srep21602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Monteleone G, Kumberova A, Croft NM, McKenzie C, Steer HW, MacDonald TT. Blocking Smad7 restores TGF-beta1 signaling in chronic inflammatory bowel disease. J Clin Invest. 2001;108:601–609. doi: 10.1172/JCI12821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Romano M, DE Francesco F, Zarantonello L, Ruffolo C, Ferraro GA, Zanus G, Giordano A, Bassi N, Cillo U. From Inflammation to Cancer in Inflammatory Bowel Disease: Molecular Perspectives. Anticancer Res. 2016;36:1447–1460. [PubMed] [Google Scholar]
- 48.Wang G, Yu Y, Sun C, Liu T, Liang T, Zhan L, Lin X, Feng XH. STAT3 selectively interacts with Smad3 to antagonize TGF-β signalling. Oncogene. 2016;35:4388–4398. doi: 10.1038/onc.2015.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu RY, Zeng Y, Lei Z, Wang L, Yang H, Liu Z, Zhao J, Zhang HT. JAK/STAT3 signaling is required for TGF-β-induced epithelial-mesenchymal transition in lung cancer cells. Int J Oncol. 2014;44:1643–1651. doi: 10.3892/ijo.2014.2310. [DOI] [PubMed] [Google Scholar]
- 50.Xu J, Shao T, Song M, Xie Y, Zhou J, Yin J, Ding N, Zou H, Li Y, Zhang J. MIR22HG acts as a tumor suppressor via TGFβ/SMAD signaling and facilitates immunotherapy in colorectal cancer. Mol Cancer. 2020;19:51. doi: 10.1186/s12943-020-01174-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Haque S, Morris JC. Transforming growth factor-β: A therapeutic target for cancer. Hum Vaccin Immunother. 2017;13:1741–1750. doi: 10.1080/21645515.2017.1327107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Boye A. A cytokine in turmoil: Transforming growth factor beta in cancer. Biomed Pharmacother. 2021;139:111657. doi: 10.1016/j.biopha.2021.111657. [DOI] [PubMed] [Google Scholar]
- 53.Fleming NI, Jorissen RN, Mouradov D, Christie M, Sakthianandeswaren A, Palmieri M, Day F, Li S, Tsui C, Lipton L, Desai J, Jones IT, McLaughlin S, Ward RL, Hawkins NJ, Ruszkiewicz AR, Moore J, Zhu HJ, Mariadason JM, Burgess AW, Busam D, Zhao Q, Strausberg RL, Gibbs P, Sieber OM. SMAD2, SMAD3 and SMAD4 mutations in colorectal cancer. Cancer Res. 2013;73:725–735. doi: 10.1158/0008-5472.CAN-12-2706. [DOI] [PubMed] [Google Scholar]
- 54.Lin C, Zhang J, Lu Y, Li X, Zhang W, Lin W, Zheng L. NIT1 suppresses tumour proliferation by activating the TGFβ1-Smad2/3 signalling pathway in colorectal cancer. Cell Death Dis. 2018;9:263. doi: 10.1038/s41419-018-0333-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Razzaque MS, Atfi A. TGIF1-Twist1 axis in pancreatic ductal adenocarcinoma. Comput Struct Biotechnol J. 2020;18:2568–2572. doi: 10.1016/j.csbj.2020.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Najjar Sadeghi R, Saeedi N, Sahba N, Sadeghi A. SMAD4 mutations identified in Iranian patients with colorectal cancer and polyp. Gastroenterol Hepatol Bed Bench. 2021;14:S32–S40. [PMC free article] [PubMed] [Google Scholar]
- 57.McCarthy AJ, Chetty R. Smad4/DPC4. J Clin Pathol. 2018;71:661–664. doi: 10.1136/jclinpath-2018-205095. [DOI] [PubMed] [Google Scholar]
- 58.Zi Z. Molecular Engineering of the TGF-β Signaling Pathway. J Mol Biol. 2019;431:2644–2654. doi: 10.1016/j.jmb.2019.05.022. [DOI] [PubMed] [Google Scholar]
- 59.Liao X, Hao Y, Zhang X, Ward S, Houldsworth J, Polydorides AD, Harpaz N. Clinicopathological characterization of SMAD4-mutated intestinal adenocarcinomas: A case-control study. PLoS One. 2019;14:e0212142. doi: 10.1371/journal.pone.0212142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mehrvarz Sarshekeh A, Advani S, Overman MJ, Manyam G, Kee BK, Fogelman DR, Dasari A, Raghav K, Vilar E, Manuel S, Shureiqi I, Wolff RA, Patel KP, Luthra R, Shaw K, Eng C, Maru DM, Routbort MJ, Meric-Bernstam F, Kopetz S. Association of SMAD4 mutation with patient demographics, tumor characteristics, and clinical outcomes in colorectal cancer. PLoS One. 2017;12:e0173345. doi: 10.1371/journal.pone.0173345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ritterhouse LL, Wu EY, Kim WG, Dillon DA, Hirsch MS, Sholl LM, Agoston AT, Setia N, Lauwers GY, Park DY, Srivastava A, Doyle LA. Loss of SMAD4 protein expression in gastrointestinal and extra-gastrointestinal carcinomas. Histopathology. 2019;75:546–551. doi: 10.1111/his.13894. [DOI] [PubMed] [Google Scholar]
- 62.Silva VR, Santos LS, Dias RB, Quadros CA, Bezerra DP. Emerging agents that target signaling pathways to eradicate colorectal cancer stem cells. Cancer Commun (Lond) 2021;41:1275–1313. doi: 10.1002/cac2.12235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang L, Liu Z, Tan J, Dong H, Zhang X. Multispectral imaging reveals hyper active TGF-β signaling in colorectal cancer. Cancer Biol Ther. 2018;19:105–112. doi: 10.1080/15384047.2017.1395116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Szeglin BC, Wu C, Marco MR, Park HS, Zhang Z, Zhang B, Garcia-Aguilar J, Beauchamp RD, Chen XS, Smith JJ. A SMAD4-modulated gene profile predicts disease-free survival in stage II and III colorectal cancer. Cancer Rep (Hoboken) 2022;5:e1423. doi: 10.1002/cnr2.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Troncone E, Marafini I, Stolfi C, Monteleone G. Involvement of Smad7 in Inflammatory Diseases of the Gut and Colon Cancer. Int J Mol Sci. 2021;22 doi: 10.3390/ijms22083922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li N. CircTBL1XR1/miR-424 axis regulates Smad7 to promote the proliferation and metastasis of colorectal cancer. J Gastrointest Oncol. 2020;11:918–931. doi: 10.21037/jgo-20-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Alidoust M, Hamzehzadeh L, Khorshid Shamshiri A, Afzaljavan F, Kerachian MA, Fanipakdel A, Aledavood SA, Allahyari A, Bari A, Moosanen Mozaffari H, Goshayeshi L, Pasdar A. Association of SMAD7 genetic markers and haplotypes with colorectal cancer risk. BMC Med Genomics. 2022;15:8. doi: 10.1186/s12920-021-01150-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Boulay JL, Mild G, Lowy A, Reuter J, Lagrange M, Terracciano L, Laffer U, Herrmann R, Rochlitz C. SMAD7 is a prognostic marker in patients with colorectal cancer. Int J Cancer. 2003;104:446–449. doi: 10.1002/ijc.10908. [DOI] [PubMed] [Google Scholar]
- 69.Stolfi C, De Simone V, Colantoni A, Franzè E, Ribichini E, Fantini MC, Caruso R, Monteleone I, Sica GS, Sileri P, MacDonald TT, Pallone F, Monteleone G. A functional role for Smad7 in sustaining colon cancer cell growth and survival. Cell Death Dis. 2014;5:e1073. doi: 10.1038/cddis.2014.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rosic J, Dragicevic S, Miladinov M, Despotovic J, Bogdanovic A, Krivokapic Z, Nikolic A. SMAD7 and SMAD4 expression in colorectal cancer progression and therapy response. Exp Mol Pathol. 2021;123:104714. doi: 10.1016/j.yexmp.2021.104714. [DOI] [PubMed] [Google Scholar]
- 71.Stolfi C, Marafini I, De Simone V, Pallone F, Monteleone G. The dual role of Smad7 in the control of cancer growth and metastasis. Int J Mol Sci. 2013;14:23774–23790. doi: 10.3390/ijms141223774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Troncone E, Monteleone G. Smad7 and Colorectal Carcinogenesis: A Double-Edged Sword. Cancers (Basel) 2019;11 doi: 10.3390/cancers11050612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Diamantopoulos MA, Tsiakanikas P, Scorilas A. Non-coding RNAs: the riddle of the transcriptome and their perspectives in cancer. Ann Transl Med. 2018;6:241. doi: 10.21037/atm.2018.06.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jia Z, An J, Liu Z, Zhang F. Non-Coding RNAs in Colorectal Cancer: Their Functions and Mechanisms. Front Oncol. 2022;12:783079. doi: 10.3389/fonc.2022.783079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Luo K, Geng J, Zhang Q, Xu Y, Zhou X, Huang Z, Shi KQ, Pan C, Wu J. LncRNA CASC9 interacts with CPSF3 to regulate TGF-β signaling in colorectal cancer. J Exp Clin Cancer Res. 2019;38:249. doi: 10.1186/s13046-019-1263-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang H, Wang J, Yu T, Lu J, Yu Z. Silencing LncRNA CASC9 inhibits proliferation and invasion of colorectal cancer cells by MiR-542-3p/ILK. PLoS One. 2022;17:e0265901. doi: 10.1371/journal.pone.0265901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Azizidoost S, Ghaedrahmati F, Anbiyaee O, Ahmad Ali R, Cheraghzadeh M, Farzaneh M. Emerging roles for lncRNA-NEAT1 in colorectal cancer. Cancer Cell Int. 2022;22:209. doi: 10.1186/s12935-022-02627-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li Q, Yue W, Li M, Jiang Z, Hou Z, Liu W, Ma N, Gan W, Li Y, Zhou T, Chen S. Downregulating Long Non-coding RNAs CTBP1-AS2 Inhibits Colorectal Cancer Development by Modulating the miR-93-5p/TGF-β/SMAD2/3 Pathway. Front Oncol. 2021;11:626620. doi: 10.3389/fonc.2021.626620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jung G, Hernández-Illán E, Moreira L, Balaguer F, Goel A. Epigenetics of colorectal cancer: biomarker and therapeutic potential. Nat Rev Gastroenterol Hepatol. 2020;17:111–130. doi: 10.1038/s41575-019-0230-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chao HM, Wang TW, Chern E, Hsu SH. Regulatory RNAs, microRNA, long-non coding RNA and circular RNA roles in colorectal cancer stem cells. World J Gastrointest Oncol. 2022;14:748–764. doi: 10.4251/wjgo.v14.i4.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ning X, Wang C, Zhang M, Wang K. Ectopic Expression of miR-147 Inhibits Stem Cell Marker and Epithelial-Mesenchymal Transition (EMT)-Related Protein Expression in Colon Cancer Cells. Oncol Res. 2019;27:399–406. doi: 10.3727/096504018X15179675206495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vakhshiteh F, Hassani S, Momenifar N, Pakdaman F. Exosomal circRNAs: new players in colorectal cancer. Cancer Cell Int. 2021;21:483. doi: 10.1186/s12935-021-02112-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zheng L, Liang H, Zhang Q, Shen Z, Sun Y, Zhao X, Gong J, Hou Z, Jiang K, Wang Q, Jin Y, Yin Y. circPTEN1, a circular RNA generated from PTEN, suppresses cancer progression through inhibition of TGF-β/Smad signaling. Mol Cancer. 2022;21:41. doi: 10.1186/s12943-022-01495-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shang A, Gu C, Wang W, Wang X, Sun J, Zeng B, Chen C, Chang W, Ping Y, Ji P, Wu J, Quan W, Yao Y, Zhou Y, Sun Z, Li D. Exosomal circPACRGL promotes progression of colorectal cancer via the miR-142-3p/miR-506-3p- TGF-β1 axis. Mol Cancer. 2020;19:117. doi: 10.1186/s12943-020-01235-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Che J, Pan L, Yang X, Liu Z, Huang L, Wen C, Lin A, Liu H. Thymidine phosphorylase expression and prognosis in colorectal cancer treated with 5-fluorouracil-based chemotherapy: A meta-analysis. Mol Clin Oncol. 2017;7:943–952. doi: 10.3892/mco.2017.1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kugimiya N, Nishimoto A, Hosoyama T, Ueno K, Takemoto Y, Harada E, Enoki T, Hamano K. JAB1-STAT3 activation loop is associated with recurrence following 5-fluorouracil-based adjuvant chemotherapy in human colorectal cancer. Oncol Lett. 2017;14:6203–6209. doi: 10.3892/ol.2017.6994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lin Z, Zhang L, Zhou J, Zheng J. Silencing Smad4 attenuates sensitivity of colorectal cancer cells to cetuximab by promoting epithelialmesenchymal transition. Mol Med Rep. 2019;20:3735–3745. doi: 10.3892/mmr.2019.10597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Oyanagi H, Shimada Y, Nagahashi M, Ichikawa H, Tajima Y, Abe K, Nakano M, Kameyama H, Takii Y, Kawasaki T, Homma KI, Ling Y, Okuda S, Takabe K, Wakai T. SMAD4 alteration associates with invasive-front pathological markers and poor prognosis in colorectal cancer. Histopathology. 2019;74:873–882. doi: 10.1111/his.13805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Vu T, Yang S, Datta PK. MiR-216b/Smad3/BCL-2 Axis Is Involved in Smoking-Mediated Drug Resistance in Non-Small Cell Lung Cancer. Cancers (Basel) 2020;12 doi: 10.3390/cancers12071879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dai YH, Li XQ, Dong DP, Gu HB, Kong CY, Xu Z. P27 Promotes TGF-β-Mediated Pulmonary Fibrosis via Interacting with MTORC2. Can Respir J. 2019;2019:7157861. doi: 10.1155/2019/7157861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cheng Q, Li C, Yang CF, Zhong YJ, Wu D, Shi L, Chen L, Li YW, Li L. Methyl ferulic acid attenuates liver fibrosis and hepatic stellate cell activation through the TGF-β1/Smad and NOX4/ROS pathways. Chem Biol Interact. 2019;299:131–139. doi: 10.1016/j.cbi.2018.12.006. [DOI] [PubMed] [Google Scholar]
- 92.Sun C, Wang FJ, Zhang HG, Xu XZ, Jia RC, Yao L, Qiao PF. miR-34a mediates oxaliplatin resistance of colorectal cancer cells by inhibiting macroautophagy via transforming growth factor-β/Smad4 pathway. World J Gastroenterol. 2017;23:1816–1827. doi: 10.3748/wjg.v23.i10.1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chen L, Zhu Q, Lu L, Liu Y. MiR-132 inhibits migration and invasion and increases chemosensitivity of cisplatin-resistant oral squamous cell carcinoma cells via targeting TGF-β1. Bioengineered. 2020;11:91–102. doi: 10.1080/21655979.2019.1710925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Teixeira AF, Ten Dijke P, Zhu HJ. On-Target Anti-TGF-β Therapies Are Not Succeeding in Clinical Cancer Treatments: What Are Remaining Challenges? Front Cell Dev Biol. 2020;8:605. doi: 10.3389/fcell.2020.00605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Makaremi S, Asadzadeh Z, Hemmat N, Baghbanzadeh A, Sgambato A, Ghorbaninezhad F, Safarpour H, Argentiero A, Brunetti O, Bernardini R, Silvestris N, Baradaran B. Immune Checkpoint Inhibitors in Colorectal Cancer: Challenges and Future Prospects. Biomedicines. 2021;9 doi: 10.3390/biomedicines9091075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lynch D, Murphy A. The emerging role of immunotherapy in colorectal cancer. Ann Transl Med. 2016;4:305. doi: 10.21037/atm.2016.08.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang QM, Tang PM, Lian GY, Li C, Li J, Huang XR, To KF, Lan HY. Enhanced Cancer Immunotherapy with Smad3-Silenced NK-92 Cells. Cancer Immunol Res. 2018;6:965–977. doi: 10.1158/2326-6066.CIR-17-0491. [DOI] [PubMed] [Google Scholar]
- 98.Huang J, Chen X, Fu X, Li Z, Huang Y, Liang C. Advances in Aptamer-Based Biomarker Discovery. Front Cell Dev Biol. 2021;9:659760. doi: 10.3389/fcell.2021.659760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lee J, Ryu M, Bae D, Kim HM, Eyun SI, Bae J, Lee K. Development of DNA aptamers specific for small therapeutic peptides using a modified SELEX method. J Microbiol. 2022 doi: 10.1007/s12275-022-2235-4. [DOI] [PubMed] [Google Scholar]
- 100.Alhamhoom Y, As Sobeai HM, Alsanea S, Alhoshani A. Aptamerbased therapy for targeting key mediators of cancer metastasis (Review) Int J Oncol. 2022;60 doi: 10.3892/ijo.2022.5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ji X, Wang H, Wu Z, Zhong X, Zhu M, Zhang Y, Tan R, Liu Y, Li J, Wang L. Specific Inhibitor of Smad3 (SIS3) Attenuates Fibrosis, Apoptosis, and Inflammation in Unilateral Ureteral Obstruction Kidneys by Inhibition of Transforming Growth Factor β (TGF-β)/Smad3 Signaling. Med Sci Monit. 2018;24:1633–1641. doi: 10.12659/MSM.909236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wu N, Lian G, Sheng J, Wu D, Yu X, Lan H, Hu W, Yang Z. Discovery of a novel selective water-soluble SMAD3 inhibitor as an antitumor agent. Bioorg Med Chem Lett. 2020;30:127396. doi: 10.1016/j.bmcl.2020.127396. [DOI] [PubMed] [Google Scholar]
- 103.Zhang T, Wang XF, Wang ZC, Lou D, Fang QQ, Hu YY, Zhao WY, Zhang LY, Wu LH, Tan WQ. Current potential therapeutic strategies targeting the TGF-β/Smad signaling pathway to attenuate keloid and hypertrophic scar formation. Biomed Pharmacother. 2020;129:110287. doi: 10.1016/j.biopha.2020.110287. [DOI] [PubMed] [Google Scholar]
- 104.Zhang YF, Zhou SZ, Cheng XY, Yi B, Shan SZ, Wang J, Li QF. Baicalein attenuates hypertrophic scar formation via inhibition of the transforming growth factor-β/Smad2/3 signalling pathway. Br J Dermatol. 2016;174:120–130. doi: 10.1111/bjd.14108. [DOI] [PubMed] [Google Scholar]
- 105.Tang M, Bian W, Cheng L, Zhang L, Jin R, Wang W, Zhang Y. Ginsenoside Rg3 inhibits keloid fibroblast proliferation, angiogenesis and collagen synthesis in vitro via the TGFβ/Smad and ERK signaling pathways. Int J Mol Med. 2018;41:1487–1499. doi: 10.3892/ijmm.2018.3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dai G, Sun B, Gong T, Pan Z, Meng Q, Ju W. Ginsenoside Rb2 inhibits epithelial-mesenchymal transition of colorectal cancer cells by suppressing TGF-β/Smad signaling. Phytomedicine. 2019;56:126–135. doi: 10.1016/j.phymed.2018.10.025. [DOI] [PubMed] [Google Scholar]
- 107.Li H, Yang L, Zhang Y, Gao Z. Kaempferol inhibits fibroblast collagen synthesis, proliferation and activation in hypertrophic scar via targeting TGF-β receptor type I. Biomed Pharmacother. 2016;83:967–974. doi: 10.1016/j.biopha.2016.08.011. [DOI] [PubMed] [Google Scholar]
- 108.Bai X, He T, Liu J, Wang Y, Fan L, Tao K, Shi J, Tang C, Su L, Hu D. Loureirin B inhibits fibroblast proliferation and extracellular matrix deposition in hypertrophic scar via TGF-β/Smad pathway. Exp Dermatol. 2015;24:355–360. doi: 10.1111/exd.12665. [DOI] [PubMed] [Google Scholar]
- 109.Zhang Y, Shan S, Wang J, Cheng X, Yi B, Zhou J, Li Q. Galangin inhibits hypertrophic scar formation via ALK5/Smad2/3 signaling pathway. Mol Cell Biochem. 2016;413:109–118. doi: 10.1007/s11010-015-2644-3. [DOI] [PubMed] [Google Scholar]
- 110.Shi J, Li J, Xu Z, Chen L, Luo R, Zhang C, Gao F, Zhang J, Fu C. Celastrol: A Review of Useful Strategies Overcoming its Limitation in Anticancer Application. Front Pharmacol. 2020;11:558741. doi: 10.3389/fphar.2020.558741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhang L, Liu J, Lin S, Tan J, Huang B, Lin J. Qingjie Fuzheng Granule Inhibited the Migration and Invasion of Colorectal Cancer Cells by Regulating the lncRNA ANRIL/let-7a/TGF-β1/Smad Axis. Evid Based Complement Alternat Med. 2020;2020:5264651. doi: 10.1155/2020/5264651. [DOI] [PMC free article] [PubMed] [Google Scholar]