

Abstract
BACKGROUND
Colorectal cancer (CRC) is the second leading cause of cancer-related mortality. Cancer stem cells (CSCs) in CRC, which are spared by many chemotherapeutics, have tumorigenic capacity and are believed to be the reason behind cancer relapse. So far, there have been no effective drugs to target colon CSCs. Diiminoquinone (DIQ) has shown promising effects on targeting colon cancer. However, there is limited research on the effects of DIQ on eradicating CSCs in CRC.
AIM
To investigate the anticancer potential of DIQ on colon CSCs in two-dimensional (2D) and three-dimensional (3D) models using colonospheres and patient-derived organoids.
METHODS
Various 2D methods have been used to assess the effect and the mechanism of DIQ on HCT116 and HT29 cell lines including cell proliferation and viability assays, migration and invasion assays, immunofluorescence staining, and flow cytometry. The potency of DIQ was also assessed in 3D culture using the sphere formation assay and colon cancer patient-derived organoid model.
RESULTS
Our results showed that DIQ significantly inhibited cell proliferation, migration, and invasion in HCT116 and HT29 cell lines. DIQ treatment induced apoptosis along with an accumulation of HCT116 and HT29 cancer cells in the sub-G1 region and an increase in reactive oxygen species in both CRC cell lines. DIQ reduced sphere-forming and self-renewal ability of colon cancer HCT116 and HT29 stem/progenitor cells at sub-toxic doses of 1 μmol/L. Mechanistically, DIQ targets CSCs by downregulating the main components of stem cell-related -catenin, AKT, and ERK oncogenic signaling pathways. Potently, DIQ displayed a highly significant decrease in both the count and the size of the organoids derived from colon cancer patients as compared to control and 5-fluorouracil conditions.
CONCLUSION
This study is the first documentation of the molecular mechanism of the novel anticancer therapeutic DIQ via targeting CSC, a promising compound that needs further investigation.
Keywords: Diiminoquinone, Anticancer activity, Colorectal cancer, Cancer stem cells, Patient-derived organoids, Colonospheres
Core Tip: Diiminoquinone has anticancer activity against colorectal cancer. Diiminoquinone targets chemoresistant cancer stem cells in three-dimensional in vitro models by downregulating the main components of stem cell-related -catenin, AKT, and ERK oncogenic signaling pathways. Our findings highlight diiminoquinone’s novel therapeutic potential against colorectal cancer.
INTRODUCTION
Colorectal cancer (CRC) ranks as the third most common cancer worldwide in 2020 in terms of incidence in men and women, and the second most common cause of cancer deaths in 2020 reaching 935000 deaths according to GLOBOCAN 2020 data[1].
Current medical treatment of CRC includes a wide array of systemic therapies, which include chemotherapeutics [such as 5-fluorouracil (5FU)], targeted therapy (such as epidermal growth factor receptor inhibitors), in addition to immunotherapy, depending on the stages of CRC. High mortality and recurrence rates of CRC are mainly correlated to metastasis, treatment resistance[2,3], and presence of chemoresistant cancer stem cells (CSCs)[4,5]. Although 5FU is the standard chemotherapy for CRC, either alone or in combination with other treatments, it has been ineffective and found to cause drug resistance[6,7]. Around 75% of patients with metastatic CRC receiving chemotherapy develop recurrence within 18 mo[8]. Although the field has witnessed several advances on the quest to control advanced and metastatic colon cancer with some newly developed drugs, there is still a pressing need to fully understand colon cancer biology, develop novel treatment approaches and pre-clinical models, and identify useful therapeutics targeting CSCs and chemoresistant cells and aiming at increasing patient survival.
Recently, we have witnessed the development of different types of in vitro three-dimensional (3D) culture systems to recapitulate the in vivo cancer growth[9,10]. 3D culture systems mainly include organoid models and multicellular spheroid models[11]. Cancer treatment, particularly using 3D culture for targeting CSC, is rapidly progressing toward personalized medicine taking into consideration the individual molecular biology and genetic variability of tumors. Introducing in vitro patient-derived organoid culture systems to 3D models have revolutionized CRC research[12,13].
CSCs are characterized by their self-renewal, pluripotency, and tumor expansion potential of differentiated cell populations with altered molecular and cellular phenotypes[14]. They are responsible for angiogenic induction and apoptotic resistance. This small subpopulation is associated with tumor invasion and metastasis, therapeutic resistance, cancer relapse, and poor prognosis in patients[15]. CSCs are present within solid tumors, and they are recognized to be resistant to chemotherapies such as 5FU or oxaliplatin[4]. Intriguingly, there are no effective drugs to target CSCs in CRC. Therefore, targeting this population holds hope for treatment response.
Studies have reported that some quinones, which are often secondary metabolites derived from plants, possess anticancer activity[16]. They are present and clinically used in a variety of cancer treatments, such as the anthracyclines daunorubicin, doxorubicin, and mitoxantrone, acting through the redox quinone-hydroquinone system[17]. Anthraquinones are a class of natural compounds that possess anticancer properties against various skin cancer cells and breast cancer cells[18]. Studies have shown that thymoquinone, which has a basic quinone structure, induces apoptosis and halts metastasis in CRC[19,20]. Also, iminoquinone exerts anticancer effects through inhibition of cell survival/proliferation and inhibition of oncogene expression[21].
The novel diiminoquinone (DIQ) compound has recently shown potent anticancer effects against the HCT116 CRC cell line as reported in our previous study[22]. The activity of DIQ is believed to be based on the structural similarities between quinones and diiminoquinones. Here, we investigated the anticancer activities and targeting mechanism(s) of DIQ against human colon CSCs using colonosphere cultures and patient-derived organoids. This study represents the first comprehensive documentation of the activity of DIQ against colon CSCs, findings that will provide the basis for proposing this stable and non-toxic compound for clinical testing and future discovery of new effective treatments for patients with colon cancer.
MATERIALS AND METHODS
Cell culture condition
Human CRC cell lines HCT116 and HT29 and non-tumorigenic fetal human intestinal FH74Int cell line were purchased from ATCC (ATCC, Manassas, VA, United States). HCT116 and HT29 cell lines were cultured and maintained in RPMI 1640 (Sigma-Aldrich, St. Louis, MO, United States) and L-glutamine (Sigma-Aldrich). FH74Int cells were grown in DMEM (Lonza, Verviers, Belgium) supplemented with 10 μg/mL insulin and 1% sodium pyruvate. Cell culture media was supplemented with antibiotics [1% penicillin-streptomycin (100 U/mL)], 10% heat-inactivated fetal bovine serum (FBS) (Sigma-Aldrich), and 5 μg/mL Plasmocin™ Prophylactic (InvivoGen, San Diego, CA, United States). Cells were maintained in an incubator at 37 ºC in a humidified atmosphere of 5% CO2 and 95% air and were routinely checked for mycoplasma contamination. All cells were mycoplasma free.
DIQ preparation and treatment
The purified compound DIQ (Figure 1) was synthesized by Professor Makhlouf Haddadin (Department of Chemistry, American University of Beirut)[22]. Stocks of the purified compound DIQ were prepared by dissolving 5 mg in 1 mL 100% dimethyl sulfoxide (Pan Biotech, Aidenbach, Germany). DIQ dilutions were stored at -20 °C. The stock solutions were then dissolved in cell culture medium such that the percentage of dimethyl sulfoxide on cells was less than 0.1%.
Figure 1.
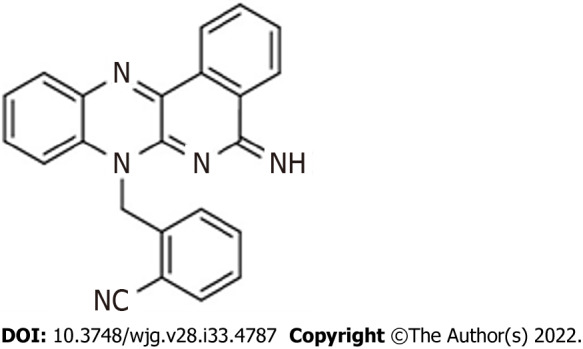
Chemical structure of the diiminoquinone compound.
MTT cell viability assay
The anti-proliferative effects of DIQ were measured in vitro by using MTT [3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide] (Sigma-Aldrich) assay according to the manufacturer’s instructions. HCT116 WT, HT29, and FHSInt74 cells were seeded in 96-well culture plates at a density of 10000 cells per well and incubated overnight. Then, the subconfluent cells were treated in triplicate with different concentrations of DIQ diluted in 100 μL complete media for 24, 48, and 72 h. For each time point, 10 μL of 5 mg/mL [in 1 × phosphate-buffered saline (PBS)] MTT reagent was added to each well and incubated at 37 °C for 4 h. The reduced MTT dye was solubilized with absolute isopropanol (Sigma-Aldrich) (100 μL/well) after which MTT optical density was measured at 595 nm by an ELISA reader (Multiskan Ex; ThermoFisher Scientific, Waltham, MA, United States). The percent cell proliferation with respect to control was determined for each drug dose.
Trypan blue exclusion assay
HCT116, HT29, and FHSInt74 cell lines were seeded in duplicates in 24-well culture plates at a density of 50000, 80000, and 100000 cells/300 μL complete media per well, respectively. Cells were incubated overnight then treated in duplicate with various concentrations of DIQ (1, 4, and 10 μmol/L) for 24, 48, and 72 h. Attached live cells were harvested by trypsin EDTA and added to the supernatant. The cell pellet was resuspended in 300 μL media. Live cells were counted using a hemocytometer. The percentage cell viability was expressed as percentage growth relative to the control condition of each time point and are derived from the mean of triplicates wells. Each experiment was repeated at least three times.
Wound healing assay
For wound healing, or scratch assay, CRC cells were seeded in 24-well plates and incubated until they reached 80%-90% confluence. Cells were then treated with 10 μg/mL mitomycin C (Sigma-Aldrich) to block cellular proliferation. A sterile 200 μL tip was used to scratch wounds of the same width on each monolayer. After washing the plates twice with PBS (Sigma-Aldrich) to remove the detached cells, the remaining cells were cultured in complete media with or without DIQ treatment at the IC50 concentration. Images using bright-field microscopy were subsequently taken at 0 h and 72 h to compare the wound width. The wound width was measured and expressed as a percentage of the relative wound width. The experiment was repeated three times with duplicate measurements in each experiment.
Transwell invasion assay
For the transwell invasion assay, 0.3 × 105 HCT116 and 0.5 × 105 HT29 cells were seeded in serum-free medium in the top chamber of 24-well inserts (pore size, 8 μm; Falcon, ThermoFisher Scientific) coated with 1:10 dilution in cold PBS of MatrigelTM (BD Bioscience, Franklin Lakes, NJ, United States). A medium supplemented with 10% FBS was used as a chemoattractant in the lower chamber. Cells with or without DIQ treatment were allowed to migrate through the membrane coated with MatrigelTM at 37 °C in a 5% CO2 incubator for 72 h. Non-migratory cells in the upper chamber were then gently scraped off with a cotton-tip applicator. Invading cells on the lower surface of the membrane were fixed and stained with hematoxylin and eosin. After staining, the total number of invading cells was counted using an inverted light microscope (10 × objective) from six consecutive fields for each well.
Reactive oxygen species
The level of intracellular reactive oxygen species (ROS) in HCT116 and HT29 was measured using the fluorescent probe dihydroethidium (DHE). For DHE staining, cells were seeded at a density of 50000 cells on coverslips in 24-well cell culture plates and allowed to become 40%-50% confluent. Following 48 h incubation with DIQ treatment at the IC50 dose, CRC cells were fixed in 4% formaldehyde for 20 min. After fixation, CRC cells were washed twice with 1 × PBS, then incubated with 20 μmol/L DHE dye (Invitrogen, Carlsbad, CA, United States). After 45 min staining, the DHE stain was removed, and the cells were washed with 1 × PBS. Mounting media with 4’,6-diamidino-2-phenylindole dye was added. Fluorescence images were taken immediately under a Zeiss LSM710 Laser confocal microscope (Carl Zeiss, Oberkochen, Germany) equipped with Zen software to process the images.
Cell cycle analysis
Cells were seeded at 5 × 105 cells in 6-well cell culture plates and incubated overnight prior to drug treatment for 24 and 72 h. Cells were then harvested and washed in PBS then fixed in 70% ice-cold ethanol added dropwise to the cell pellet while vortexing for 30 min on ice. To ensure that only DNA was stained, fixed cells were incubated for 30 min at 37 °C with 100 μL of propidium iodide (PI) (Sigma) solution [6 μL RNase, 30 μL PI (1 mg/mL)] in the dark in a flow tube (BD Falcon, ThermoFisher Scientific). A total of 10000 gated events were acquired in order to assess the proportions of cells in different stages of the cell cycle. Cell cycle analysis was performed by flow cytometry using Guava EasyCyte8 Flow Cytometer-Millipore. GuavaSoft™ 2.7 Software.
Annexin V-PI staining
HCT116 and HT29 cells were seeded at a density of 5 × 105 cells in 6-well cell culture plates and incubated overnight prior to drug treatment for 72 h. Cells were then harvested and washed in cold PBS. The pellet was resuspended in 100 μL binding buffer and stained with 5 μL annexin V-FITC and 5 μL PI in the dark for 30 min at room temperature. Then, 400 μL binding buffer was added, and apoptotic cells were analyzed with fluorescence-activated cell sorting flow cytometry.
Sphere formation assay
Self-renewal capacity is deemed to be one of the major defining hallmarks of stem/progenitor cells. Thus, to determine whether DIQ was able to target the self-renewing CSC pool, we investigated sphere formation capability over 5 generations. The sphere formation assay was used as previously reported by our laboratory[23,24]. Briefly, 1000 single cells/well in 96-well culture plate were suspended in cold MatrigelTM/serum-free medium (1:1) in a total volume of 10 μL. Cells were seeded uniformly in a circular manner around the bottom rim of the well and allowed to solidify in the incubator at 37 °C for 1 h. Subsequently, 100 μL of RPMI with 5% FBS treated with DIQ was added gently in the middle of each well. Each experimental condition was performed in duplicate. Spheres were replenished with warm media as in the original seeding every other day. Spheres were counted in the 96-well plate dishes after 8 to 12 d of sphere culture, and bright field images of the spheres were obtained using Axiovert microscope from Zeiss at × 10 magnification. Images were analyzed by Carl Zeiss Zen 2012 image software to determine sizes. Sphere-formation unit (SFU) was calculated for each generation as follows: SFU = number of spheres formed/number of cells originally plated. Results were represented as percentage of the SFU of each condition.
Sphere propagation assay
To enrich the stem-like population of cells, the media was aspirated from the well. Collected spheres using cold media were incubated in 300 μL of Trypsin/EDTA at 37 °C for 1-3 min and then passed through 27-gauge syringes three times. Single cells resulting from the dissociation of spheres were replated and treated at the same density of 1000 cells/well in 96-well culture plates as previously described. We believe that at least 5 generations of colonospheres were required to enrich the subpopulation of progenitor/stem-like colon cancer cells.
Immunofluorescence imaging of colonospheres
Spheres at generation 1 were collected with cold RPMI media and centrifuged to washout all MatrigelTM debris. After centrifugation, spheres were fixed in situ in 4% paraformaldehyde (PFA) at room temperature for 20 min. The PFA was aspirated gently, and spheres were permeabilized with 0.5% Triton X-100 for 30 min at room temperature. After carefully aspirating the permeabilization solution, spheres were blocked using the sphere blocking buffer [0.1% bovine serum albumin (BSA), 0.2% Triton X-100, 0.05% Tween-20, and 10% normal goat serum in PBS] for 2 h at room temperature. Spheres were washed in PBS then incubated overnight with different primary antibodies for assessment of treatment and characterization including Ki67, CD44, Gamma H2A histone family member X (γH2AX), cytokeratin (CK)19 and CK8 (refer to Table 1 for details on antibodies used). After gentle washing with PBS containing 0.1% Tween-20, spheres were incubated with Alexa-488 and/or 568-conjugated IgG (Invitrogen) for 2 h at room temperature. Spheres were mounted with the antifade Fluorogel II with 4’,6-diamidino-2-phenylindole (Abcam, Cambridge, United Kingdom). Confocal fluorescent images were acquired and analyzed using the Carl Zeiss LSM 710 Laser scanning microscope.
Table 1.
List of primary and secondary antibodies used in immunofluorescent staining
|
Antibody name
|
Species
|
Dilution
|
Catalog number
|
Company
|
| Primary antibodies | ||||
| CK8 | Mouse | 1:200 | 904801 | Biolegend |
| CK19 | Rabbit | 1:200 | ab15463 | Abcam |
| CD44 | Mouse | 1:100 | sc-7297 | Santa Cruz Biotechnology |
| Ki67 | Mouse | 1:50 | sc-23900 | Santa Cruz Biotechnology |
| p- HistoneH2AX | Rabbit | 1:200 | #9718S | Cell Signaling |
| Secondary antibodies | ||||
| Alexa fluoro 488 | Goat anti-mouse | 1:200 | A-28175 | Invitrogen |
| Alexa fluoro 568 | Goat anti-rabbit | 1:200 | A-11011 | Invitrogen |
| Phalloidin | 1:200 | R415 | Invitrogen | |
Western blot analysis
For two-dimensional (2D) western blot results, cells were plated in 12-well plates, treated with DIQ, and then collected. For 3D western blot results, HCT116 and HT29 cells were plated in 24-well plates (3 × 105 cells/well) with or without treatment to form spheres. At day 8-10, spheres were collected with cold RPMI media then washed with PBS to remove any residual media. Proteins were then extracted with RIPA lysis buffer (sc-24948; Santa Cruz Biotechnology, Dallas, TX, United States). Protein extracts were quantified using the DC Bio-Rad Protein Assay (Bio-Rad Laboratories, Hercules, CA, United States) according to the manufacturer’s protocol. Equal amounts of protein lysate were mixed with 5% β-mercaptoethanol and 2X Laemmli Sample Buffer (Bio-Rad Laboratories), electrophoresed in 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and then transferred to 0.45 μm nitrocellulose membrane (Bio-Rad Laboratories) for 2 h. Membranes were blocked for 1 h with 5% skim milk in tris-buffered saline with 0.1% tween 20, then blotted with primary antibodies (antibodies used are listed in Table 2) overnight at 4 ºC. The next day, membranes were washed three times with tris-buffered saline with 0.1% tween 20 and blotted with corresponding secondary antibodies for 1 h at room temperature. Hybridization with GAPDH-HRP (6C5) coupled antibody was performed for 1 h at room temperature as the housekeeping gene. Membranes were developed, and target proteins were detected using the enhanced chemiluminescence system (Bio-Rad Laboratories). Images were generated and quantified using ChemiDoc™ Imaging Systems (Bio-Rad Laboratories).
Table 2.
List of primary and secondary antibodies used in western blot experiments
|
Antibody name
|
Species
|
Dilution
|
Catalog number
|
Company
|
| Primary antibodies | ||||
| p53 | Rabbit | 1:50 | sc-6243 | Santa Cruz Biotechnology |
| p21 | Rabbit | 1:1000 | sc-2947 | Cell Signaling |
| CD133 | Rabbit | 1:500 | #64326S | Cell Signaling |
| β-catenin | Mouse | 1:200 | sc-7963 | Santa Cruz Biotechnology |
| PCNA | Mouse | 1:50 | sc-25280 | Santa Cruz Biotechnology |
| p-ERK | Mouse | 1:300 | sc-7383 | Santa Cruz Biotechnology |
| ERK | Rabbit | 1:300 | sc-93 | Santa Cruz Biotechnology |
| p-AKT | Rabbit | 1:1000 | #4060 | Cell Signaling |
| AKT | Rabbit | 1:1000 | #4691 | Cell Signaling |
| GAPDH-HRP (6C5) | Mouse | 1:20,000 | #MAB5476 | Abnova |
| Secondary antibodies | ||||
| Goat anti-mouse | Goat | 1:1000 | sc-516102 | Santa Cruz Biotechnology |
| Mouse anti-rabbit | Mouse | 1:1000 | sc-2357 | Santa Cruz Biotechnology |
Ethical consideration of patient derived-organoid culture
The study with all its experimental protocols was conducted under the Institutional Review Board approvals of the American University of Beirut and American University of Beirut Medical Center to obtain patient information and human colorectal tissue samples from consenting patients. All protocols were performed in accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinki) and in agreement with all ethical considerations of the Institutional Review Board for experiments involving human subjects. Oral consent was obtained from all patients, and confidentiality was maintained. For colectomy specimens, a core biopsy was taken from the area most likely to be involved with cancer according to the surgeon and pathologist recommendations.
Tissue processing and patient-derived organoid culture
Colon tumor tissue from patients was rinsed with PBS and manually minced using sterile scalpels. The majority of minced fragments was employed for organoid culturing; remaining fragments were transferred directly to 4% PFA for histological examination. According to the protocol described by Boehnke et al[25], minced fragments for organoid culturing were digested in advanced adDMEM/F12 (Gibco, ThermoFisher Scientific) supplemented with 1 × P/S, collagenase IV (Sigma-Aldrich), and amphotericin B (Sigma-Aldrich) at 37 °C for 1 h. During incubation, the tissue fragments were repeatedly suspended with a 100 μL micropipette. To exclude undigested tissue fragments, the suspension was filtered through a 100 μm cell strainer (Corning, Corning, NY, United States). The flowthrough was subjected to consecutive filtrations when needed. Isolated cells were seeded in 24-well plates with Matrigel at a cell density of 20000 single cells/well. Then, 20 μL drops were plated into the middle of the well. The plate was placed upside down in the 37 °C incubator for 15 min to allow the Matrigel to solidify. Finally, 300 μL of prewarmed human colon growth medium plus Y-27632 was added into each well. Cells were cultured with adDMEM/F12 additional with various factors added to maintain tumor biological traits and growth activity. Medium was changed every 2-3 d. Cultures were passaged when the aggregates reached 800 μm diameter. Organoids were counted at day 8-12 of each passage under Axiovert inverted microscope at × 10 magnification, and then images of organoids were taken at the same magnification. Images were then analyzed by Carl Zeiss Zen 2012 image software to determine size. The organoid formation count (OFC) was calculated at each generation by counting the number of organoids formed, starting with the same number of input cells in all conditions.
Passaging of the established patient-derived organoids
Organoids were collected when they reached the appropriate size and confluency for passaging (8-12 d after plating). To dissolve Matrigel, ice-cold medium was used, and organoids were collected. Organoids were then centrifuged at 200 g for 5 min at 4 °C. After that, the pellet was resuspended in 1 mL ice-cold adDMEM/F12 to dissolve residual Matrigel. After counting the cells in the pellet, the cells were resuspended in 90% cold Matrigel and seeded as a 5 μL drop in 96-well plate. Cells were cultured with adDMEM/F12 additional with various factors added to maintain the tumor’s biological traits and growth activity. Medium was changed every 2-3 d with or without DIQ treatment. Cultures were passaged when the aggregates reached 800 μm diameter. The previous steps were repeated for several generations.
Cell line-derived organoid protocol
Briefly, 5000 HCT116 and HT29 single cells/well in 96-well culture plate were suspended in cold MatrigelTM/serum-free medium (9:1) in a total volume of 5 μL as drops in the middle of individual wells of 96-well culture plates. Plated colon cells were allowed to solidify in the incubator at 37 °C for 30 min. Subsequently, 200 μL/well of advanced DMEM/F12 media with several factors, with or without DIQ treatment, was added. Each experimental condition was performed in duplicate. Organoids were replenished with warm media as in the original seeding every other day. Organoids were counted in the 96-well plate dishes after 8 to 12 d of organoid culture, and bright field images of the organoids were obtained using Axiovert microscope from Zeiss at × 10 magnification.
Immunofluorescence and morphological analysis of colonospheres and colorectal organoids
Spheres/organoids were collected with cold RPMI media and centrifuged to wash all MatrigelTM debris. After centrifugation, spheres/organoids were fixed in situ in 4% PFA at room temperature for 20 min. The PFA was aspirated gently, and spheres/organoids were permeabilized with 0.5% Triton X-100 for 30 min at room temperature. After carefully aspirating the permeabilization solution, spheres/organoids were blocked using the blocking buffer (0.1% BSA, 0.2% Triton X-100, 0.05% Tween-20, and 10% normal goat serum in PBS) for 2 h at room temperature. Spheres/organoids were washed in PBS then incubated overnight with different primary antibodies for assessment of treatment and characterization (refer to Table 1 for details on antibodies used). After gentle washing with PBS containing 0.1% Tween-20, spheres/organoids were incubated with Alexa-488 and/or 568-conjugated IgG (Invitrogen) for 2 h at room temperature. Spheres/organoids were mounted with the antifade Fluorogel II with 4’,6-diamidino-2-phenylindole (Abcam). Confocal fluorescent images were acquired and analyzed using the Carl Zeiss LSM 710 Laser scanning microscope. Paraffin embedding, microtome sectioning, and standard hematoxylin and eosin staining were all performed by the Histology Laboratory at the Diana Tamari Sabbagh building; all steps were performed at room temperature.
Animal experiments
Animal experiments were performed according to approved protocols by the Institutional Animal Care and Use Committee of the American University of Beirut. Mice were housed under optimum conditions of temperature and light set in specific pathogen-free animal housing. Mice were kept in plastic cages covered with sawdust and had unrestricted access to a commercial mouse diet (24% protein, 4.5% fat, 4% fiber) and water. Animals were sacrificed by cervical dislocation following deep anesthesia with isoflurane. For tumor induction in mice, a group of 6-8-wk old non-obese diabetic severe combined immunodeficiency male mice were inoculated subcutaneously into the flanks with 100 HCT116-derived spheres in a total volume of 50 μL growth media and Matrigel™ (1:1). Upon the detection of a palpable tumor post cell/sphere injection, group 1 injected with 3D spheres was treated with saline (control group), and group 2 injected with 3D spheres was treated with DIQ (20 mg/kg). Mice were treated three times/wk until tumor burden necessitated that we sacrificed the animals. Tumor size was measured every other day using Mitutoyo caliper throughout the study. Mice were monitored daily for signs of morbidity. Body weight recordings were carried out biweekly.
Statistical analysis
All statistical tests were performed using GraphPad Prism 7 (version 7.0; GraphPad Software Inc., La Jolla, CA, United States). Student’s t test, One-way or two-way analysis of variance tests were used in this study. In all statistical tests, the mean of treated groups was compared to the mean of control groups. Statistical significance was reported at P values of < 0.05. aP < 0.05; bP < 0.01; cP < 0.001. Experimental values were means ± standard error of the mean.
RESULTS
DIQ reduced the cell proliferation of human CRC cell lines in 2D in vitro models
To assess the effect of DIQ compound on the proliferation of human CRC cell lines cultured in 2D monolayers, we employed the MTT assay. Two human CRC cell lines, HCT116 and HT29, were treated with different concentrations of DIQ (1, 4, and 10 μmol/L) for 24, 48, and 72 h. The MTT results revealed that DIQ significantly inhibited the proliferation of HCT116 and HT29 human CRC cells at micromolar concentrations in a time- and dose-dependent manner (Figure 2A). Interestingly, a concentration of DIQ as low as 4 μmol/L was able to inhibit cell proliferation by approximately more than 30% at 24 h in HCT116 and HT29 cell lines and more than 50% cell reduction was observed at 48 h and 72 h in both cell lines. The mean IC50 values of DIQ was approximately 4 μmol/L in both cell lines (Figure 2A). The effect of DIQ on the viability of the human CRC cell lines was further confirmed by trypan blue exclusion method, and there was consistency between the MTT results and trypan blue exclusion assay (Figure 2B). Interestingly, DIQ treatment had relatively limited toxicity to the human non-tumorigenic intestinal FHS74Int cells when applied at doses up to 5 μmol/L and over a 72-h period (Supplementary Figure 1A).
Figure 2.

Diiminoquinone reduced the proliferation and the viability of HCT116 and HT29 colorectal cancer cell lines in a time- and dose-dependent manner. A: The anticancer effect of different concentrations of diiminoquinone (DIQ) on the proliferation of HCT116 and HT29 cells using the MTT assay was determined in triplicates at 24, 48, and 72 h. Results were expressed as the percentage of proliferation of the treated group compared to the control at every time point; B: The anticancer effect of different concentrations of DIQ on the viability of HCT116 and HT29 cells using the trypan blue exclusion assay was determined in triplicates at 24, 48, and 72 h. Results were expressed as percentage of viable cells of the treated group compared to the control at every time point. Data represent an average of three independent experiments and is reported as mean ± standard error of the mean (aP < 0.05, bP < 0.01, cP < 0.001).
DIQ inhibited migration and invasion of CRC cells
One of the most well-known properties of cancer cells is their ability to break away from their site and invade neighboring tissues[26]. Wound healing and transwell invasion assays were employed to evaluate the effects of DIQ on human CRC cell migration and invasion. DIQ at the corresponding IC50 concentration significantly suppressed and slowed down the cell migration ability of both cell lines at 72 h compared to the vehicle-treated control cells as determined by the wound healing assay (Figure 3A). The treatments failed to close the wound by more than 70% in both cell lines compared with control conditions, which were able to almost completely close the wound (Figure 3A). In addition, DIQ showed significant inhibitory potential on CRC cell invasion. The number of HCT116 and HT29 invasive cells were remarkably decreased in response to FBS in treated conditions reaching a value of less than two-fold compared to the control condition at 72 h (Figure 3B).
Figure 3.

Diiminoquinone reduced the migration and the invasion of HCT116 and HT29 colorectal cancer cells. A: HCT116 and HT29 cells were seeded in 24-well plates. A scratch was made on confluent cells using a 200 μL tip, and images were taken at 0 h and 72 h with or without the indicated treatment. Quantification of the distance of the wound closure was assessed over time. Representative images of wound healing assay at × 5 magnification (scale = 100 μm); B: Colorectal cancer cells were seeded onto the Matrigel-coated membrane in the top chamber of the transwell and were either treated or not with the indicated concentration in the presence of fetal bovine serum in the lower chamber. Cells that invaded to the lower chamber after 72 h were fixed, stained with hematoxylin and eosin, counted, and are represented as the number of invading cells compared to the control. Data represent an average of three independent experiments and is reported as mean ± standard error of the mean (aP < 0.05, bP < 0.01, cP < 0.001). DIQ: Diiminoquinone.
DIQ induced cell cycle arrest and apoptosis in CRC cells
To evaluate the underlying mechanism of growth inhibition by DIQ in CRC, the cell cycle distribution analysis of HCT116 and HT29 cells treated with the IC50 concentration of DIQ for 72 h was performed using flow cytometry. As shown in Figure 4A, DIQ treatment in HCT116 cells caused G1 arrest with concomitant decreases in the S and G2/M fractions mainly after 72 h. No changes in the cell cycle were noticed after treating both cell lines with DIQ for 24 h. DIQ effect on the HCT116 cell cycle was pronounced at 72 h. The proportion of HCT116 cells in G1 phase was increased from 45.6% in control cells to 60.2% in cells treated with DIQ for 72 h, while the proportion of cells in G2/M phase decreased from 35.2% to 21.5% (Figure 4A). However, in HT29 cells, DIQ treatment induced S phase (38.35%) cell cycle arrest after 72 h treatment and depleted cells at G1 and G2/M phases. Interestingly, upon treatment with 4 μmol/L DIQ, the percentage of HCT116 and HT29 cells in the sub-G1 phase significantly increased reaching 3.5- and 5.0-fold at 72 h, respectively, suggesting that the reduction in cell viability in response to DIQ could be due to cell death (Figure 4A). To further confirm whether growth inhibition was related to apoptosis, Annexin V and PI staining was performed. As shown in Figure 4A, after treating CRC cells with DIQ at the indicated concentrations for 72 h, the total apoptotic cell populations were significantly increased in both cell lines reaching 61% in HCT116 and 70% in HT29 cells.
Figure 4.

Diiminoquinone induced an accumulation of HCT116 and HT29 cancer cells in the sub-G1 region and apoptosis along with an increase in reactive oxygen species production. A: The distribution of phases of the cell cycle upon diiminoquinone (DIQ) treatment at 24 h and 72 h in HCT116 and HT29 cells using propidium iodide-based flow cytometric analysis of DNA content was shown. Data represent the average of three independent experiments and are reported as mean ± standard error of the mean (aP < 0.05, bP < 0.01, cP < 0.001); B: Reactive oxygen species (ROS) production in HCT116 and HT29 cells was detected by dihydroethidium (DHE) staining. Representative images of colorectal cancer cells exposed to DIQ stained with DHE for ROS content (red color). Cell nuclei were stained with 4’,6-diamidino-2-phenylindole (DAPI) (blue). A summary of the quantification of the red fluorescence intensity was represented; C: Lysates of colorectal cancer cells treated with 4 μmol/L DIQ were immunoblotted for p53, p21, β-catenin, p-ERK, ERK, p-AKT, AKT, and proliferating cell nuclear antigen (PCNA). Bands were detected by enhanced chemiluminescence and quantified using ChemiDoc MP Imaging System. Data represent an average of three independent experiments and is reported as mean ± standard error of the mean (aP < 0.05, bP < 0.01, cP < 0.001).
DIQ induced the production of ROS in CRC cells
Recently, targeting cancer via ROS-based mechanisms has been reported as a radical therapeutic approach[27]. To investigate the effect of DIQ on cellular stress and the involvement of oxidative stress in their anti-proliferative effect in CRC, ROS production was examined by DHE stain intensity. DHE is a fluorescent dye that can easily permeate cell membranes and has been widely used to quantify cellular O2·− and H2O2 by producing red fluorescent products. Our results showed that a significant increase of the DHE staining intensity was observed in treated cells at 48 h as compared to the control (Figure 4B). Thus, DIQ treatment induced ROS production in both CRC cell lines.
DIQ altered the expression of the cell cycle and proliferation markers in CRC cells
To determine the association between the observed cell cycle arrest and the increased ROS in HCT116 and HT29, western immunoblot analyses were performed on total cell extracts prepared from 2D-treated cells to detect possible changes in the expression of cell cycle and proliferation markers. As shown in Figure 4C, the expression levels of p53 and p21, which are cell cycle regulators of the G1 phase, were upregulated by 1.28-fold and 1.42-fold, respectively, in HCT116 upon DIQ treatment as compared to control conditions. Whereas, in HT29 treated cells, p53 was downregulated and p21 was significantly upregulated by 1.8-fold, suggesting that the inhibitory mechanism of DIQ is different in HCT116 and HT29 cells. The expression of the proliferation-associated proteins, such as AKT, p-AKT, ERK, p-ERK, and proliferating cell nuclear antigen (PCNA), were markedly decreased by DIQ treatment in both cell lines (Figure 4F). In addition, the expression levels of stem cell markers, CD133 and β-catenin, were also downregulated in both CRC cell lines.
DIQ targeted the enriched population of human CRC stem cells in 3D
We investigated colonosphere formation of HCT116 and HT29 cells, a salient feature of CSCs. To better visualize their sphere forming capabilities in 3D cultures, HCT116 and HT29 cells were cultured as single cells in Matrigel™ for 8-12 d in the presence of DIQ. The spheres were then visualized under an inverted light microscope, and bright-field images were taken (Figure 5). Cells that were able to form spheres in the first generation were collected and propagated by dissociating spheres into single cells and reseeding the same number of cells (1000 cells/well). The assay was performed until the fifth generation. Our data showed that both HCT116 and HT29 cells formed spheres, suggesting the presence of a unique population with stem cell-like properties. Notably, a clear dose-dependent attenuation of the SFU at generation 1 for both cell lines was observed when treated with different concentrations of DIQ (0.5 and 1 μmol/L). The SFU was always significantly and remarkably lower in drug-treated cells compared to that of the control condition by more than 50% (Figure 5). Consecutive propagations of formed spheres at each generation with successive treatment with DIQ were performed up to 5 generations. Interestingly, our results showed additional inhibition of the SFU upon DIQ treatment when the cells were propagated from generation 1 up to generation 5 spheres. We observed that 1 μmol/L of DIQ treatment decreased SFU of HCT116 cells by more than 10 times compared to the control (13.3%) at generation 5 reaching approximately 1%. Moreover, as shown in Figure 5, HT29 cells were more sensitive to DIQ, and there was an eradication of spheres at generation 4 (SFU = 0%) compared to the control (14.28%). In addition to assessing its effect on self-renewal capacity, DIQ significantly decreased the sizes of the spheres by more than 50% as compared to untreated control conditions. Further decrease in sphere sizes was recognized over the 5 generations in both cell lines depicting pronounced additive effect of the treatments on the formed spheres upon propagation (Figure 5). Thus, DIQ has led to fewer and smaller HCT116 and HT29 spheres. Interestingly, DIQ treatment did not show any significant effect on the size and SFU of FHS74Int-derived spheres over 5 generations (Supplementary Figure 2B). Taken together, these findings suggest that DIQ specifically targeted the colorectal CSC.
Figure 5.

Diiminoquinone reduced the sphere-forming and self-renewal ability of colon cancer stem/progenitor cells. A: HCT116 and B: HT29 cells were seeded at a density of 1000 single cells/well in Matrigel™ for 8 d with and without 1 μmol/L diiminoquinone (DIQ) at generation 1. Spheres were propagated for five generations in duplicates for each condition. Media or treatment was replenished every 2 d. Spheres were counted at day 8-12 of sphere culture. Results are expressed as sphere-formation unit, which was calculated according to the following formula: sphere-formation unit = (number of spheres counted/number of input cells) × 100. Quantification of the average size of generation 1 to generation 5 colon cancer spheres with or without treatment conditions. Spheres sizes were measured by Carl Zeiss Zen 2012 image software. Data represent an average diameter (μm) of 50 measured spheres. Representative bright field images of HCT116 and HT29 colon spheres in MatrigelTM taken by the Axiovert inverted microscope are shown. Data represent an average of three independent experiments and is reported as mean ± standard error of the mean (aP < 0.05, bP < 0.01, cP < 0.001).
DIQ induced apoptosis and inhibited proliferation of human CRC stem cells
Spheres collected at generation 1 were subjected to immunofluorescence analysis of the expression of the proliferation marker Ki67, cytokeratin epithelial markers, CK8 and CK19, and the stem cell marker CD44. Our data revealed that Ki67, CK8, and CK19 expression were significantly reduced in treated spheres derived from HCT116 and HT29 cell lines (Figure 6A-D). The downregulation of the CK19 marker in both HCT116 and HT29 spheres at generation 1 could be an indicator of an inhibition of the epithelial-mesenchymal transition process. Immunofluorescence staining showed high expression of CD44 in control spheres at generation 1 indicating enriched stemness in these cells. Treatment with DIQ showed a significant reduction of CD44 expression in HCT116 and HT29 colonospheres as compared to the control, which is in tune with the downregulation of the CRC stem marker CD133 data (Figure 6C and E). Finally, DIQ effect on DNA damage was studied by assessing the expression of γH2AX. Our results revealed that the expression of γH2AX was markedly increased in treated spheres in both cell types (Figure 6D).
Figure 6.

Diiminoquinone induced apoptosis and inhibited proliferation in colon cancer stem/progenitor cells. Representative immunofluorescence imaging of control and diiminoquinone (DIQ)-treated HCT116 and HT29 spheres collected at generation 1. A-D: Spheres stained for cytokeratin (CK)8 (green) and CK19 (red) (A), KI67 (B), CD44 (C), and gamma H2A histone family member X (γH2AX) (D) were obtained using confocal microscopy. The nuclei were stained with anti-fade reagent Fluorogel II with 4’,6-diamidino-2-phenylindole (DAPI). The quantification of the intensity of CK8, CK19, CD44, and Ki67 stain in control and DIQ-treated spheres was performed using Carl Zeiss Zen 2012 image software. Stain intensity was normalized to size. Scale bar 20 μm; E: Analysis of p53, p21, CD133, β-catenin, proliferating cell nuclear antigen (PCNA), p-ERK, ERK, p-AKT, and AKT protein expression in HCT116 and HT29 generation 1 spheres upon treatment is shown. GAPDH served as an internal control. Bands were detected by enhanced chemiluminescence using ChemiDoc MP Imaging System. Fold expression changes normalized to GAPDH and to total ERK and total AKT in the cases of p-ERK and p-AKT expression, respectively, are given. Data represent an average of three independent experiments and is reported as mean ± standard error of the mean (aP < 0.05, bP < 0.01, cP < 0.001).
To further assess the effect of DIQ on the enriched CSCs population, we were interested in determining the effect of DIQ on the expression of proliferation markers, stem cell markers, and Wnt signaling molecules of CSCs using western blot. Consistent with the western blot analyses of 2D CRC cells, the expression of the proliferation markers p-AKT and p-ERK were remarkably downregulated by DIQ treatment in both HCT116 and HT29-derived spheres confirming inhibitory effects of DIQ on the proliferation of 3D CSC colonospheres (Figure 6E). Western blot analysis revealed a decrease in the levels of the proliferation marker PCNA post treatment consistent with the data that DIQ decreased the size of HCT116 and HT29-derived spheres (Figure 6E). For the Wnt signaling studies, we investigated treatment effects on β-catenin, which plays an important role in colon cancer stemness properties. Western blot analysis showed a downregulation of β-catenin expression in treated compared to untreated spheres. Analysis of p53 and p21 protein expression in HCT116 spheres upon DIQ treatment showed upregulation of these proteins by 1.32-fold and 1.99-fold, respectively, further confirming apoptosis induction (Figure 6E). Only p21 expression was upregulated in HT29 cells by 1.26-fold as compared to non-treated spheres, whereas the expression of p53 was not affected by DIQ treatment in HT29 spheres.
DIQ had antitumor potential in non-obese diabetic severe combined immunodeficiency mice injected with HCT116 spheres
To investigate the antitumor effect of DIQ on targeting the CSC population of cells in vivo, we subcutaneously injected two groups of non-obese diabetic severe combined immunodeficiency mice with 100 spheres derived from HCT116 cells. Mice developed tumors in 2 wk and were then treated with 20 mg/kg DIQ three times per week for 21 d. DIQ treatment did not cause any change in the body weight or death of mice, indicating no toxicity.
DIQ significantly inhibited tumor growth in the treated group when compared to the control group particularly at day 21 (Figure 7). Interestingly, the average tumor volume was 403.2 mm3 in DIQ-treated mice at sacrifice, while it was 2158.5 mm3 in the control group (Figure 7).
Figure 7.

Diiminoquinone reduced tumor growth in non-obese diabetic severe combined immunodeficiency mice. Non-obese diabetic severe combined immunodeficiency (NOD-SCID) mice (5 mice/group) were injected with 100 HCT116 generation 1 spheres. Tumor growth was monitored by measuring the tumor volume during 21 d of treatment (3 times per week) with either 20 mg/kg diiminoquinone (DIQ) or physiologic saline. Representative images of control and DIQ-treated mice at day 21. Data represent an average of two independent experiments and is reported as mean ± standard error of the mean (aP < 0.05, bP < 0.01, cP < 0.001).
Assessment of the effect of DIQ treatment on the established colon cancer patient derived organoids: Organoids as models for DIQ assessment
We established a 3D organoid system from fresh tissue samples obtained from different stages of 5 random colon cancer consenting patients. As described in the methods section, a total of 20000 single cells derived from freshly digested tissues were plated per 20 μL droplets of 90% MatrigelTM in 24-well plates. Cells were plated depending on the total cell count that was successfully derived from the tissue specimens. Despite the expected challenges in modeling colon cancer, we succeeded in establishing colon organoids from patients undergoing colectomy. Organoids formed at generation 1 were dissociated, propagated to generation 2, and the effect of DIQ on the organoids formed was assessed. The growth of organoids was determined by the total number (OFC) and size (diameter) of the organoids formed. The response of colon cancer patient-derived organoids to DIQ was compared to that of 5FU, which is the standard first-line treatment option for CRC. This response was evaluated on 5 random treatment-naïve patients with different clinical data. We succeeded in establishing colon organoids and propagating them. The two different doses of DIQ (0.5 and 1 μmol/L) displayed a highly significant inhibition in the OFC and the size of tumor organoids derived from the 5 studied patients when they were compared to the control group in a dose-dependent manner (Figures 8-10). These results were consistent with the response of HCT116 and HT29 cell line-derived organoids to DIQ treatment. DIQ elicited a statistically significant decrease in both OFC and size of cell line-derived organoids (Supplementary Figure 2).
Figure 8.

Establishment and characterization of patient-derived organoids from colon cancer patient 1. A: Representative image of organoids derived from patient 1 stained with hematoxylin and eosin (HE); B: Representative bright-field images of organoids at generation (G)1, G2, and G6. Fresh tumor tissues were enzymatically digested, and single cells were plated in 90% growth factor-reduced Matrigel. G1 organoids were successfully propagated up to G6. Images were visualized by Axiovert inverted microscope at × 5, × 10, and × 20 magnification. Scale bar 100 μm; C: Immunofluorescent images of organoids stained with colon lineage epithelial markers cytokeratin (CK)19 and CK8 and stem cell marker CD44. The nuclei were stained with anti-fade Fluorogel II with 4’,6-diamidino-2-phenylindole (DAPI). Representative confocal microscopy images were acquired using a Zeiss LSM 710 laser scanning confocal microscope. Scale bar 100 μm; D: Representative bright-field images of G2 organoids treated with diiminoquinone (DIQ) (0.5 and 1 μmol/L) or 5-fluorouracil (5FU) (3 μmol/L). Organoid formation count (OFC) and size were calculated, and mean values were reported as mean ± standard error of the mean (aP < 0.05, bP < 0.01, cP < 0.001). Images were visualized by Axiovert inverted microscope at × 5 and × 20 magnification. Scale bar 100 μm.
Figure 10.

Diiminoquinone reduced the growth of the patient-derived organoids from different colon cancer patients. A: Representative bright field images of generation (G)1 organoids derived from patient 2 (grade 2; stage T3) grown with or without diiminoquinone (DIQ) or 5-fluorouracil (5FU). Organoid formation count (OFC) was calculated in duplicate wells per condition. The quantification of the average diameter was calculated. The data of OFC and size are presented in two separate graphs; B: Representative bright field images of G4 organoids derived from patient 4 (grade 2; stage T2) grown with or without DIQ or 5FU. OFC was calculated in duplicate wells per condition. The quantification of the average diameter size was calculated. The data of OFC and size are presented in two separate graphs; C: Representative bright field images of G2 organoids derived from patient 5 (grade 2; stage T3) grown with or without DIQ or 5FU. OFC was calculated in duplicate wells per condition. The quantification of the average diameter was calculated. The average mean of OFC and size are presented in two separate graphs. All mean values were reported as mean ± standard error of the mean (aP < 0.05, bP < 0.01, cP < 0.001). Scale bar, 100 μm.
In Patient 1, organoids were successfully propagated up to generation 6 as shown in Figure 8. Interestingly, an increase in the number of tumor organoids formed was observed with each propagation, thus indicating enrichment of stem cells and enhancement of the establishment of colon organoids. Characterization of the established patient 1-derived organoids was performed by studying the expression of the CRC epithelial lineage markers CK19 and CK8 and the stem cell marker CD44. Using immunofluorescence staining, the tumor organoids showed a positive expression of CK19, CK8, and CD44 confirming the presence of stem-like/progenitor CRC cells within the bulk of our patient-derived organoids.
As shown in Figure 9, a reduction in the OFC and size of the treated organoids was noticed in patient 3 with rectal mucinous adenocarcinoma (pT2 stage). Characterization of patient 3derived organoids and corresponding tissue was assessed by investigating the expression of CK19 and CD44 markers using immunofluorescence staining. These organoids mimicked the heterogeneity of corresponding tumor tissue. Strong expression of CK19 and CD44 was noted in organoids consistent with the corresponding tumor tissue (Figure 9). The co-expression of CK19 and CD44 was decreased upon treatment in a dose-dependent manner (Figure 9B). In Patients 2 and 5, both exhibited similar grade (grade 2) moderately differentiated sigmoid colon adenocarcinoma and were of the same stage (pT3). DIQ treatment at concentrations as low as 0.5 μmol/L displayed a decrease in the growth of the organoids in Patient 2 and an eradication of organoids in Patient 5 (Figure 10). Organoid formation was eradicated upon DIQ (0.5 and 1 μmol/L) treatment in Patient 4, who was diagnosed with moderately differentiated (grade 2) pT2 sigmoid colon adenocarcinoma. Interestingly, the effect of DIQ on the OFC and the size of the organoids was more potent than that of 5FU particularly in patients 2, 4, and 5 (Figure 10).
Figure 9.

Effect of diiminoquinone on established patient-derived organoids from colon cancer patient 3. A: Immunohistochemistry images of tissue derived from patient 3 stained with hematoxylin and eosin (HE). Immunofluorescent images of tissue stained with colon lineage epithelial markers cytokeratin (CK)19 and stem cell marker CD44. The nuclei were stained with anti-fade Fluorogel II with 4’, 6-diamidino-2-phenylindole (DAPI). Representative confocal microscopy images were acquired using a Zeiss LSM 710 laser scanning confocal microscope. Scale bar 20 μm; B: Immunofluorescent images of organoids derived from colon cancer patient 3 at generation (G)2 in the presence and absence of diiminoquinone (DIQ) treatment (0.5 and 1 μmol/L) stained with colon lineage epithelial markers CK19 and stem cell marker CD44. The nuclei were stained with anti-fade Fluorogel II with DAPI. Representative confocal microscopy images were acquired using a Zeiss LSM 710 laser scanning confocal microscope. Scale bar 20 μm. Representative bright-field images of organoids derived from colon cancer patient 3 at G2 in the presence and absence of DIQ treatment (0.5 and 1 μmol/L). Organoid formation count and size were calculated, and mean values were reported as mean ± standard error of the mean (aP < 0.05, bP < 0.01, cP < 0.001). Images were visualized by Axiovert inverted microscope at × 5 and × 20 magnification. Organoid formation count (OFC) and size of G were calculated, and mean values were reported as mean ± standard error of the mean (aP < 0.05, bP < 0.01, cP < 0.001). Scale bar 100 μm.
DISCUSSION
In this study, we investigated the anticancer activity of DIQ in 2D and 3D models of human colon cancer. DIQ reduced the sphere forming and self-renewal ability of CRC HCT116 and HT29 stem cells at sub-toxic doses. Mechanistically, DIQ targeted CSCs by reducing the proliferation marker Ki67 and CRC stem cell markers CD44 and CK19 as well as inducing DNA damage through increasing γH2AX expression and downregulating the main components of stem cell-related -catenin, AKT, and ERK oncogenic signaling pathways. DIQ displayed a highly significant decrease in both the count and the size of the organoids derived from colon cancer patients as compared to control and 5FU conditions. Furthermore, in 2D culture, DIQ significantly inhibited cell proliferation, migration, and invasion of HCT116 and HT29 cell lines. DIQ also induced apoptosis and an increase in ROS along with an accumulation of cells in the sub-G1 region. Consistent with the in vitro data, DIQ exhibited reduction in the tumor growth and proliferation in vivo.
Our major focus in this study was to evaluate the ability of DIQ to target CSCs in HCT116 and HT29 cells using a 3D sphere formation assay. CSCs are a rare subpopulation of stem-like tumor cells that are responsible for tumor relapse[28,29]. The increase of SFU in both CRC cell lines from generation 1 up to generation 5 suggests an enrichment in CSCs upon propagation, thus confirming the advantage of using the 3D sphere formation assay. Treatment of HCT116 and HT29 cells with DIQ at a concentration as low as 1 μmol/L targeted the subpopulation of stem/progenitor cells over five generations as reflected by the drastic decrease in the SFU and the sphere size in both cell lines. HT29 spheres were more sensitive to 1 μmol/L DIQ, and an eradication of HT29 spheres occurred at generation 3. The interesting finding of DIQ not affecting non-tumorigenic FHS74Int cells makes DIQ somewhat selective to cancer cells, which is the most essential aspect sought after in anticancer drugs.
To understand what molecular pathways could be targeted by DIQ, we focused mainly on the pathways implicated in CSCs. Multiple signaling systems are involved in resistance of CSCs to therapy. It is widely accepted that the Wnt/β-catenin pathway is the most relevant signaling pathway for colon cancer development. Wnt signaling contributes to stem cell development, tumorigenicity, and oncogenesis[30,31]. This pathway is mechanistically responsible for drug resistance of colon CSCs. Increasing evidence validates that this pathway can interact with other oncogenic signaling pathways, such as those involving MAPK, PI3K, AKT, and ERK, which are aberrantly activated in many human cancers[32,33]. Indeed, evidence has shown that AKT and ERK are overexpressed in human CRC[32]. Thus, identifying drugs that target these oncogenic pathways could make a solid rationale for the targeted therapy of cancers. The result of western blot analysis showed that the ratio of both phosphorylated AKT to total AKT (p-AKT/AKT) and phosphorylated ERK to total ERK (p-ERK/ERK), which are key players of AKT/ERK pathways, were decreased upon DIQ treatment in CRC spheres. These findings suggest that DIQ suppressed sphere growth and formation via dual inhibition of AKT/ERK dependent signaling pathways.
We additionally investigated the protein levels of the key stem cell markers in CRC, CD133, and β-catenin, which are involved in chemotherapy resistance. Interestingly, the expression of CD133 and β-catenin were dramatically downregulated after DIQ treatment. Moreover, upon DIQ treatment, there was a significant decrease in the expression of CD44 and CK19 in both CRC cell lines, which were highly expressed in control spheres, along with a decrease in the expression of Ki67 and PCNA. It is interesting to note that CK19, which is considered a tumor marker in CRC, is specifically and stably expressed in primary and metastatic CRC cells. Altogether, this suggests that DIQ could be considered a novel therapeutic compound for suppressing CSC self-renewal.
DIQ-mediated apoptosis and inhibition of cell cycle progression was dependent on the upregulation of p21, which is a known tumor suppressor[34], promotes ROS accumulation, binds to PCNA, and inhibits cell cycle progression[35]. ROS is one of the major inducer of DNA damage. Induction of ROS generation induces increased stress on cancer cells leading to cancer cell death. Interestingly, DIQ treatment induced ROS production in CRC cell lines suggesting that an increase in ROS might also be involved in the anticancer effects of DIQ.
Since DIQ-induced apoptosis in HCT116 colonospheres through an increase in TUNEL positivity as we previously reported[22], we assessed whether DNA damage was activated in the spheres derived from both cell lines. We evaluated the expression of γH2AX, which is a DNA double-strand damage biomarker and could be a classical cancer prognostic factor[36,37]. The loss of DNA damage in CRC is involved in the development of therapeutic resistance[37]. In addition, quinones and oxaliplatin have been shown to induce apoptosis of CRC cells by activating DBS and activating γH2AX expression[7,38]. Interestingly, DIQ increased the expression of γH2AX in both CRC cells; clearly emphasizing that DIQ is a potent inducer of DNA damage.
Interestingly, this study has also demonstrated effects of DIQ in patient-derived organoids. This model closely recapitulates tissue architecture and cellular composition and is used to assess the self-renewal and differentiation capacities of the organoid CSC, including growth kinetics and drug sensitivity[12,13]. Testing drug efficacy in colon patient-derived organoids holds great promise for personalized medicine and exhibits a significant potential to predict patient response and connect compound screening and clinical trials[39,40]. Since drug resistance to chemotherapy is a serious challenge in treating solid tumors, drug exposure studies on the patient-derived organoids help in choosing specific chemotherapy regimens for patients with malignant disease. Since chemotherapy response in CRC treatment varies between patients, the current study used patient-derived CRC organoids to evaluate the antineoplastic effect of DIQ in targeting stem cells. The established colon organoids expressed the CRC epithelial marker lineage CK19 and the CSC cell marker CD44. This observed co-expression recapitulates the architecture and the characteristics of colon tissues. Notably, DIQ caused a prominent inhibitory effect on the growth of CRC organoids from various patients at different CRC stages, emphasizing its antitumor potential in CRC patients. This effect was either more than or as potent as that of 5FU, emphasizing its inhibitory effect. The results of response of HCT116 and HT29 cell-derived organoids to DIQ treatments were consistent with that of patient-derived organoids. We, therefore, for the first time revealed that DIQ targeted the CSC in patient-derived colon organoids.
The present study has several limitations. The two major limitations in organoid establishment and subsequent applications were the small size of the patient tissue and the availability of tissues at the time of the study. As a clinical study, the patient sample size was relatively small. Additionally, the percentage success rate of deriving colon patient derived organoids was not more than 42%; only 5 out of 12 specimens were successfully established as colon organoids. This could possibly be due to limitations in tissue quality as well. A larger cohort is still required to further investigate and evaluate the effect of DIQ in translational medicine.
CONCLUSION
In conclusion, we demonstrated for the first time that DIQ reduces self-renewal capacity of colorectal tumors and prevents therapy resistance in patient-derived organoids through interfering with the stem cell Wnt/-catenin and AKT and ERK pathways that are involved in CRC tumorigenesis. Also, the effect of DIQ was involved in the major cell fate responses including apoptosis, cell cycle arrest, and stress response. DIQ inhibits the key processes of CRC tumorigenesis, including cell growth, proliferation, migration, and invasion. Our findings strongly suggest that DIQ could be a promising compound for treatment of CRC patients and could be clinically used as a non-toxic compound for targeting human colon cancer stem/progenitor cells.
ARTICLE HIGHLIGHTS
Research background
Colorectal cancer (CRC) is a multistep genetic disorder caused by sequential mutational events in signal transduction pathways occurring along with progression of the cancer. Quinone containing compounds have been reported as one of the promising novel anticancer therapeutics against CRC. However, the effects of diiminoquinone (DIQ) on CRC stem cells have not been extensively investigated yet.
Research motivation
To explore the promising anticancer effects of a novel quinone, DIQ, on CRC.
Research objectives
To investigate the anticancer potential of novel therapeutic DIQ on CRC using two-dimensional and three-dimensional models.
Research methods
MTT and trypan blue exclusion assays were employed to assess the anti-proliferative effect of DIQ on HCT116 and HT29 cell lines in vitro. Propidium iodide DNA and dihydroethidium staining were performed to determine cell cycle distribution and reactive oxygen species production in response to DIQ, respectively. Wound healing and transwell invasion assays were used to determine the invasion and migration ability of DIQ, respectively. Then, a sphere formation model was used to evaluate the potency of DIQ on targeting cancer stem cells in CRC cells for up to five generations. Immunofluorescent analysis and western blot were performed to elucidate the mechanism of action of DIQ in CRC. Organoid model was used to assess DIQ response on established organoids from fresh colorectal tissue samples from consenting patients.
Research results
DIQ reduced the self-renewal capacity of CRC cells and targeted the growth of colon cancer patient-derived organoids. DIQ downregulated the expression of key markers involved in the oncogenic stem cell Wnt/-catenin, AKT, and ERK signaling pathways that are involved in CRC tumorigenesis. Also, DIQ decreased proliferation, migration, and invasion and induced apoptosis, cell-cycle arrest, and reactive oxygen species.
Research conclusions
Our findings strongly suggest that DIQ could be a promising novel therapeutic for the treatment of CRC patients. This study represents the first documentation of the molecular mechanism of the novel anticancer therapeutics DIQ via targeting cancer stem cells, findings that have potential therapeutic implications for colon cancer patients.
Research perspectives
Further research on the DIQ mechanisms that are involved in CRC tumorigenesis is needed to be performed in the future. A larger cohort is still required to further investigate and evaluate the effects of DIQ in translational medicine.
ACKNOWLEDGEMENTS
We are thankful to all members of the Gali-Muhtasib and Abou-Kheir Laboratory and the staff of the core facilities in the DTS Building at the American University of Beirut for their technical help and support.
Footnotes
Institutional review board statement: All specimens from the patients were obtained after their informed consent. All experiments involving human subjects were performed in agreement with all ethical considerations of the Institutional Review Board.
Institutional animal care and use committee statement: Prior to any mouse experiment, all mice protocols were reviewed and approved by the Institutional Animal Care and Use Committee (American University of Beirut, Institutional Animal Care and Use Committee).
Conflict-of-interest statement: All the authors report no relevant conflicts of interest for this article.
ARRIVE guidelines statement: The ARRIVE Guidelines have been adopted. The authors have read the ARRIVE Guidelines, and the manuscript was prepared and revised according to the ARRIVE Guidelines.
Provenance and peer review: Invited article; Externally peer reviewed.
Peer-review model: Single blind
Peer-review started: March 18, 2022
First decision: June 11, 2022
Article in press: August 5, 2022
Specialty type: Medicine, research and experimental
Country/Territory of origin: Lebanon
Peer-review report’s scientific quality classification
Grade A (Excellent): 0
Grade B (Very good): B
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
P-Reviewer: Capello D, Italy; Fruhling P, Sweden S-Editor: Gong ZM L-Editor: Filipodia P-Editor: Li X
Contributor Information
Alissar Monzer, Department of Biology, American University of Beirut, Beirut 1107-2020, Lebanon.
Kevork Wakimian, Department of Anatomy, Cell Biology and Physiological Sciences, American University of Beirut, Beirut 1107-2020, Lebanon.
Farah Ballout, Department of Biology, American University of Beirut, Beirut 1107-2020, Lebanon.
Samar Al Bitar, Department of Biology, American University of Beirut, Beirut 1107-2020, Lebanon.
Amani Yehya, Department of Anatomy, Cell Biology and Physiological Sciences, American University of Beirut, Beirut 1107-2020, Lebanon.
Mariam Kanso, Department of Surgery, American University of Beirut Medical Center, Beirut 1107-2020, Lebanon.
Nour Saheb, Department of Pathology and Laboratory Medicine, American University of Beirut Medical Center, Beirut 1107-2020, Lebanon.
Ayman Tawil, Department of Pathology and Laboratory Medicine, American University of Beirut Medical Center, Beirut 1107-2020, Lebanon.
Samer Doughan, Department of Surgery, American University of Beirut Medical Center, Beirut 1107-2020, Lebanon.
Maher Hussein, Department of Surgery, American University of Beirut Medical Center, Beirut 1107-2020, Lebanon.
Deborah Mukherji, Department of Internal Medicine, American University of Beirut Medical Center, Beirut 1107-2020, Lebanon.
Walid Faraj, Department of Surgery, American University of Beirut Medical Center, Beirut 1107-2020, Lebanon.
Hala Gali-Muhtasib, Department of Biology and Center for Drug Discovery, American University of Beirut, Beirut 1107-2020, Lebanon.
Wassim Abou-Kheir, Department of Anatomy, Cell Biology and Physiological Sciences, American University of Beirut, Beirut 1107-2020, Lebanon. wa12@aub.edu.lb.
Data sharing statement
No additional data are available.
References
- 1.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Rawla P, Sunkara T, Barsouk A. Epidemiology of colorectal cancer: incidence, mortality, survival, and risk factors. Prz Gastroenterol. 2019;14:89–103. doi: 10.5114/pg.2018.81072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hsieh MH, Kung PT, Kuo WY, Ke TW, Tsai WC. Recurrence, death risk, and related factors in patients with stage 0 colorectal cancer: A nationwide population-based study. Medicine (Baltimore) 2020;99:e21688. doi: 10.1097/MD.0000000000021688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Phi LTH, Sari IN, Yang YG, Lee SH, Jun N, Kim KS, Lee YK, Kwon HY. Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018;2018:5416923. doi: 10.1155/2018/5416923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mitra A, Mishra L, Li S. EMT, CTCs and CSCs in tumor relapse and drug-resistance. Oncotarget. 2015;6:10697–10711. doi: 10.18632/oncotarget.4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Phua LC, Mal M, Koh PK, Cheah PY, Chan EC, Ho HK. Investigating the role of nucleoside transporters in the resistance of colorectal cancer to 5-fluorouracil therapy. Cancer Chemother Pharmacol. 2013;71:817–823. doi: 10.1007/s00280-012-2054-0. [DOI] [PubMed] [Google Scholar]
- 7.Ballout F, Monzer A, Fatfat M, Ouweini HE, Jaffa MA, Abdel-Samad R, Darwiche N, Abou-Kheir W, Gali-Muhtasib H. Thymoquinone induces apoptosis and DNA damage in 5-Fluorouracil-resistant colorectal cancer stem/progenitor cells. Oncotarget. 2020;11:2959–2972. doi: 10.18632/oncotarget.27426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brandi G, De Lorenzo S, Nannini M, Curti S, Ottone M, Dall'Olio FG, Barbera MA, Pantaleo MA, Biasco G. Adjuvant chemotherapy for resected colorectal cancer metastases: Literature review and meta-analysis. World J Gastroenterol. 2016;22:519–533. doi: 10.3748/wjg.v22.i2.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Edmondson R, Broglie JJ, Adcock AF, Yang L. Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay Drug Dev Technol. 2014;12:207–218. doi: 10.1089/adt.2014.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chaicharoenaudomrung N, Kunhorm P, Noisa P. Three-dimensional cell culture systems as an in vitro platform for cancer and stem cell modeling. World J Stem Cells. 2019;11:1065–1083. doi: 10.4252/wjsc.v11.i12.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fang Y, Eglen RM. Three-Dimensional Cell Cultures in Drug Discovery and Development. SLAS Discov. 2017;22:456–472. doi: 10.1177/1087057117696795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Young M, Reed KR. Organoids as a Model for Colorectal Cancer. Curr Colorectal Cancer Rep. 2016;12:281–287. doi: 10.1007/s11888-016-0335-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huch M, Knoblich JA, Lutolf MP, Martinez-Arias A. The hope and the hype of organoid research. Development. 2017;144:938–941. doi: 10.1242/dev.150201. [DOI] [PubMed] [Google Scholar]
- 14.Bao B, Ahmad A, Azmi AS, Ali S, Sarkar FH. Overview of cancer stem cells (CSCs) and mechanisms of their regulation: implications for cancer therapy. Curr Protoc Pharmacol. 2013;Chapter 14:Unit 14.25. doi: 10.1002/0471141755.ph1425s61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang JC. Cancer stem cells: Role in tumor growth, recurrence, metastasis, and treatment resistance. Medicine (Baltimore) 2016;95:S20–S25. doi: 10.1097/MD.0000000000004766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu JJ, Bao JL, Wu GS, Xu WS, Huang MQ, Chen XP, Wang YT. Quinones derived from plant secondary metabolites as anti-cancer agents. Anticancer Agents Med Chem. 2013;13:456–463. [PubMed] [Google Scholar]
- 17.Pereyra CE, Dantas RF, Ferreira SB, Gomes LP, Silva-Jr FP. The diverse mechanisms and anticancer potential of naphthoquinones. Cancer Cell Int. 2019;19:207. doi: 10.1186/s12935-019-0925-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang Q, Lu G, Shen HM, Chung MC, Ong CN. Anti-cancer properties of anthraquinones from rhubarb. Med Res Rev. 2007;27:609–630. doi: 10.1002/med.20094. [DOI] [PubMed] [Google Scholar]
- 19.Mehanna MM, Sarieddine R, Alwattar JK, Chouaib R, Gali-Muhtasib H. Anticancer Activity of Thymoquinone Cubic Phase Nanoparticles Against Human Breast Cancer: Formulation, Cytotoxicity and Subcellular Localization. Int J Nanomedicine. 2020;15:9557–9570. doi: 10.2147/IJN.S263797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guler EM, Sisman BH, Kocyigit A, Hatiboglu MA. Investigation of cellular effects of thymoquinone on glioma cell. Toxicol Rep. 2021;8:162–170. doi: 10.1016/j.toxrep.2020.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang W, Rayburn ER, Velu SE, Chen D, Nadkarni DH, Murugesan S, Zhang R. A novel synthetic iminoquinone, BA-TPQ, as an anti-breast cancer agent: in vitro and in vivo activity and mechanisms of action. Breast Cancer Res Treat. 2010;123:321–331. doi: 10.1007/s10549-009-0638-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Monzer A, Jabotian N, Ballout F, Zhu JS, Kurth MJ, Haddadin MJ, Gali-Muhtasib H. Novel 2-(5-Imino-5H-isoquinolones[3,4-b]quinoxalin-7-ylmethyl)-benzonitrile (DIQ3) and Other Related Derivatives Targeting Colon Cancer Cells: Syntheses and in Vitro Models. ACS Omega. 2019;4:3205–3212. doi: 10.1021/acsomega.8b02698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mouhieddine TH, Nokkari A, Itani MM, Chamaa F, Bahmad H, Monzer A, El-Merahbi R, Daoud G, Eid A, Kobeissy FH, Abou-Kheir W. Metformin and Ara-a Effectively Suppress Brain Cancer by Targeting Cancer Stem/Progenitor Cells. Front Neurosci. 2015;9:442. doi: 10.3389/fnins.2015.00442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abou-Kheir W, Hynes PG, Martin P, Yin JJ, Liu YN, Seng V, Lake R, Spurrier J, Kelly K. Self-renewing Pten-/- TP53-/- protospheres produce metastatic adenocarcinoma cell lines with multipotent progenitor activity. PLoS One. 2011;6:e26112. doi: 10.1371/journal.pone.0026112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boehnke K, Iversen PW, Schumacher D, Lallena MJ, Haro R, Amat J, Haybaeck J, Liebs S, Lange M, Schäfer R, Regenbrecht CR, Reinhard C, Velasco JA. Assay Establishment and Validation of a High-Throughput Screening Platform for Three-Dimensional Patient-Derived Colon Cancer Organoid Cultures. J Biomol Screen. 2016;21:931–941. doi: 10.1177/1087057116650965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang WG, Sanders AJ, Katoh M, Ungefroren H, Gieseler F, Prince M, Thompson SK, Zollo M, Spano D, Dhawan P, Sliva D, Subbarayan PR, Sarkar M, Honoki K, Fujii H, Georgakilas AG, Amedei A, Niccolai E, Amin A, Ashraf SS, Ye L, Helferich WG, Yang X, Boosani CS, Guha G, Ciriolo MR, Aquilano K, Chen S, Azmi AS, Keith WN, Bilsland A, Bhakta D, Halicka D, Nowsheen S, Pantano F, Santini D. Tissue invasion and metastasis: Molecular, biological and clinical perspectives. Semin Cancer Biol. 2015;35 Suppl:S244–S275. doi: 10.1016/j.semcancer.2015.03.008. [DOI] [PubMed] [Google Scholar]
- 27.Shi X, Zhang Y, Zheng J, Pan J. Reactive oxygen species in cancer stem cells. Antioxid Redox Signal. 2012;16:1215–1228. doi: 10.1089/ars.2012.4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu Z, Pestell TG, Lisanti MP, Pestell RG. Cancer stem cells. Int J Biochem Cell Biol. 2012;44:2144–2151. doi: 10.1016/j.biocel.2012.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rich JN. Cancer stem cells: understanding tumor hierarchy and heterogeneity. Medicine (Baltimore) 2016;95:S2–S7. doi: 10.1097/MD.0000000000004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nwabo Kamdje AH, Takam Kamga P, Tagne Simo R, Vecchio L, Seke Etet PF, Muller JM, Bassi G, Lukong E, Kumar Goel R, Mbo Amvene J, Krampera M. Developmental pathways associated with cancer metastasis: Notch, Wnt, and Hedgehog. Cancer Biol Med. 2017;14:109–120. doi: 10.20892/j.issn.2095-3941.2016.0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma J, Cheng J, Gong Y, Tian L, Huang Q. Downregulation of Wnt signaling by sonic hedgehog activation promotes repopulation of human tumor cell lines. Dis Model Mech. 2015;8:385–391. doi: 10.1242/dmm.018887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ye Q, Cai W, Zheng Y, Evers BM, She QB. ERK and AKT signaling cooperate to translationally regulate survivin expression for metastatic progression of colorectal cancer. Oncogene. 2014;33:1828–1839. doi: 10.1038/onc.2013.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Braicu C, Buse M, Busuioc C, Drula R, Gulei D, Raduly L, Rusu A, Irimie A, Atanasov AG, Slaby O, Ionescu C, Berindan-Neagoe I. A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer. Cancers (Basel) 2019;11 doi: 10.3390/cancers11101618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Al Bitar S, Gali-Muhtasib H. The Role of the Cyclin Dependent Kinase Inhibitor p21cip1/waf1 in Targeting Cancer: Molecular Mechanisms and Novel Therapeutics. Cancers (Basel) 2019;11 doi: 10.3390/cancers11101475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Waga S, Hannon GJ, Beach D, Stillman B. The p21 inhibitor of cyclin-dependent kinases controls DNA replication by interaction with PCNA. Nature. 1994;369:574–578. doi: 10.1038/369574a0. [DOI] [PubMed] [Google Scholar]
- 36.Mah LJ, El-Osta A, Karagiannis TC. gammaH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia. 2010;24:679–686. doi: 10.1038/leu.2010.6. [DOI] [PubMed] [Google Scholar]
- 37.Palla VV, Karaolanis G, Katafigiotis I, Anastasiou I, Patapis P, Dimitroulis D, Perrea D. gamma-H2AX: Can it be established as a classical cancer prognostic factor? Tumour Biol. 2017;39:1010428317695931. doi: 10.1177/1010428317695931. [DOI] [PubMed] [Google Scholar]
- 38.Chiu SJ, Lee YJ, Hsu TS, Chen WS. Oxaliplatin-induced gamma-H2AX activation via both p53-dependent and -independent pathways but is not associated with cell cycle arrest in human colorectal cancer cells. Chem Biol Interact. 2009;182:173–182. doi: 10.1016/j.cbi.2009.08.019. [DOI] [PubMed] [Google Scholar]
- 39.Fatehullah A, Tan SH, Barker N. Organoids as an in vitro model of human development and disease. Nat Cell Biol. 2016;18:246–254. doi: 10.1038/ncb3312. [DOI] [PubMed] [Google Scholar]
- 40.Azar J, Bahmad HF, Daher D, Moubarak MM, Hadadeh O, Monzer A, Al Bitar S, Jamal M, Al-Sayegh M, Abou-Kheir W. The Use of Stem Cell-Derived Organoids in Disease Modeling: An Update. Int J Mol Sci. 2021;22 doi: 10.3390/ijms22147667. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No additional data are available.