

ABSTRACT
Here, we report the assembled and annotated genome of the freshwater diatom Fragilaria crotonensis SAG 28.96. The 61.85-Mb nuclear genome was assembled into 879 contigs, has a GC content of 47.40%, contains 26,015 predicted genes, and shows completeness of 81%.
ANNOUNCEMENT
Fragilaria crotonensis is broadly distributed in freshwater systems, including both oligotrophic and hypereutrophic lakes, and serves as a biological indicator of eutrophication (1–5). F. crotonensis is an important member of Lake Erie’s phytoplankton because it has historically bloomed in summer (6) and remains a dominant member seasonally (7–11). To facilitate diatom-focused omics studies of Lake Erie and other lakes, we report the assembled and annotated F. crotonensis SAG 28.96 genome. The 61.85-Mb genome was assembled into 879 contigs, with 26,015 predicted genes and a GC content of 47.40%. The genome is predicted to be 81% complete (Table 1).
TABLE 1.
General features of the F. crotonensis SAG 28.96 nuclear genome
| Parametera | Finding for Fragilaria crotonensis |
|---|---|
| Genome size (Mb) | 61.85 |
| GC content (%) | 47.40 |
| No. of contigs | 879 |
| N50 (bp) | 89,148 |
| L50 (contigs) | 206 |
| Total no. of predicted genes | 26,015 |
| No. of annotated genes | 11,422 |
| No. of unannotated genes | 14,593 |
| Avg gene length (bp) | 1,283.73 |
| Coding density | 0.54 |
| Completeness (%) | 81 |
| Sequencing depth (×) | 58 |
Genome size, GC content, number of contigs, and N50 and L50 values were determined via tQUAST-LG (v5.0.2). Genome completeness was assessed via BUSCO (v5.2.2) using the Stramenopile markers data set. Coding density is defined as follows: ([average gene length [bp] × total number of genes]/genome size [bp]). Sequencing depth is defined as follows: (total number of pooled reads [bp]/genome size [bp]).
Nonaxenic unialgal cultures of F. crotonensis SAG 28.96 (Culture Collection of Algae at the University of Göttingen, Göttingen, Germany) were cultured and collected as reported previously (8). DNA was extracted using standard phenol-chloroform methods with ethanol precipitation (12) and was quantified using the Qubit double-stranded DNA (dsDNA) HS assay kit (Invitrogen). Short-read sequencing was performed using an Illumina NovaSeq 6000 system (65 million paired-end 250-bp reads) at the Clinical Genomics Center (Oklahoma Medical Research Foundation, Oklahoma City, OK) with libraries prepared using the Illumina TruSeq PCR-free LT kit (350-bp insert). Long-read sequencing was performed in-house using a MinION MK1B R9.4.1 flow cell (N50, 17.815 kb; total number of reads, 642,517; total read length, 5.38 Gb) with high-molecular-weight DNA prepared with the ligation sequencing kit SQK-LSK109 (Oxford Nanopore Technologies) (13).
Assembly and gene prediction were performed using a previously established pipeline (14). Briefly, bases were called for Nanopore reads with Guppy (v4.0.15) (15). Adapters were trimmed using Porechop (v0.2.4) (16) with reads trimmed for quality (Q scores of 9) and length (500 bp) using NanoFilt (v2.7.1) (17). Illumina reads were trimmed using CLC Genomics Workbench (v20.0, with default settings). The assembly was performed using Canu (v2.1) (18). Contigs were polished using Pilon (v1.23) (19) with read mappings generated using Bowtie2 (v2.2.3) (20). Redundant contigs due to heterogeneity in diploid genomes were removed using Redundans (v0.14a) (21). Removal of bacterial contamination was performed using the Kaiju web server (22). Genome completeness was assessed by BUSCO (v5.2.2) using the Stramenopile database (23). Genes were called using BRAKER (24) with F. crotonensis transcriptomic data (25) that were assembled in CLC Genomics Workbench and mapped to the assembly using Hisat2 (26). Translated amino acid sequences were uploaded to the eggNOG-mapper web server to predict function (27). Contigs lacking coding sequences or those containing only bacterial genes were removed, along with the organellular genomes. tRNAs were predicted using tRNA-scan-SE (v2.0.6) (28). Genome statistics were determined using QUAST-LG (v5.0.2) (29).
Until recently, diatom research primarily relied on two model marine diatom genomes (30, 31). There are now 22 fully characterized Bacillariophyta genomes available, but only 6 are freshwater (Fig. 1). A lack of representative freshwater diatom genomes is a gap in the field because differences in physiology exist. There are further morphological distinctions stemming from evolutionary divergence. As a result, there is a need to sequence not only freshwater diatom taxa but also a greater variety of morphologically and evolutionarily distinct diatoms to facilitate future diatom omics studies.
FIG 1.
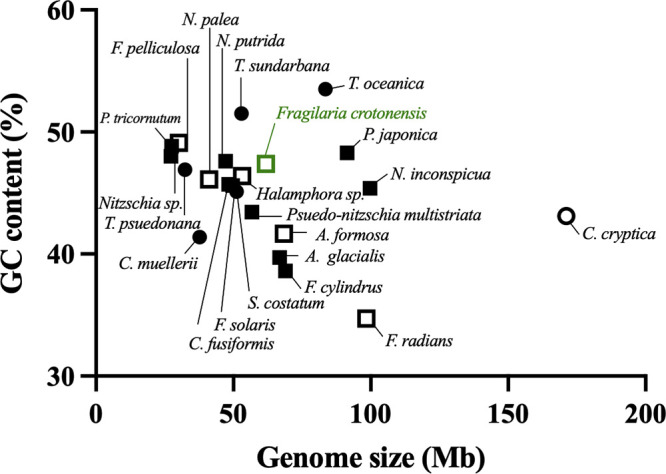
Variability of genome size and GC content of 21 Bacillariophyta genomes sequenced, annotated, and available to date in the NCBI taxonomy database, in addition to the newly sequenced F. crotonensis genome. Diatoms classified as estuarine/marine are indicated by filled symbols (n = 15), while freshwater diatoms are indicated by open symbols (n = 7). Centric diatoms are indicated by circles (n = 6), while pennate diatoms are indicated by squares (n = 16). The genome of F. crotonensis SAG 28.96 is indicated in green. An unclassified Bacillariophyta genome and a Licmophora abbreviata (environmentally assembled sample) genome are not included in this graph.
Data availability.
The annotated nuclear genome was deposited in GenBank under the accession number JAKSYS000000000. Data are available under BioProject accession number PRJNA807324 and BioSample accession number SAMN25978007.
ACKNOWLEDGMENTS
We thank Veronica Brown for assistance.
This work was funded through an Illumina-University of Tennessee Knoxville Genomics Core minigrant, the Bowling Green State University Great Lakes Center for Fresh Waters and Human Health supported by the NSF (grant OCE-1840715) and NIH (grant 1P01ES028939-01), and an NSF Graduate Research Fellowship Program grant to B.N.Z. (grant DGE-19389092).
We declare no conflicts of interest.
Contributor Information
Steven W. Wilhelm, Email: wilhelm@utk.edu.
Jason E. Stajich, University of California, Riverside
REFERENCES
- 1.Saros JE, Michel TJ, Interlandi SJ, Wolfe AP. 2005. Resource requirements of Asterionella formosa and Fragilaria crotonensis in oligotrophic alpine lakes: implications for recent phytoplankton community reorganizations. Can J Fish Aquat Sci 62:1681–1689. doi: 10.1139/f05-077. [DOI] [Google Scholar]
- 2.Morales E, Rosen B, Spaulding S. 2013. Fragilaria crotonensis. https://diatoms.org/species/fragilaria_crotonensis. Accessed 31 January 2022.
- 3.Spaulding SA, Otu MK, Wolfe AP, Baron JS. 2015. Paleolimnological records of nitrogen deposition in shallow, high-elevation lakes of Grand Teton National Park, Wyoming, USA. Arctic Antarctic Alpine Res 47:703–717. doi: 10.1657/AAAR0015-008. [DOI] [Google Scholar]
- 4.Wolfe AP, Cooke CA, Hobbs WO. 2006. Are current rates of atmospheric nitrogen deposition influencing lakes in the eastern Canadian Arctic? Arctic Antarctic Alpine Res 38:465–476. doi: 10.1657/1523-0430(2006)38[465:ACROAN]2.0.CO;2. [DOI] [Google Scholar]
- 5.Davis CC. 1964. Evidence for the eutrophication of Lake Erie from phytoplankton records. Limnol Oceanogr 9:275–283. doi: 10.4319/lo.1964.9.3.0275. [DOI] [Google Scholar]
- 6.Hartig JH. 1987. Factors contributing to development of Fragilaria crontonensis Kitton pulses in Pigeon Bay waters of western Lake Erie. J Great Lakes Res 13:65–77. doi: 10.1016/S0380-1330(87)71628-1. [DOI] [Google Scholar]
- 7.Saxton MA, D'Souza NA, Bourbonniere RA, McKay RML, Wilhelm SW. 2012. Seasonal Si:C ratios in Lake Erie diatoms: evidence of an active winter diatom community. J Great Lakes Res 38:206–211. doi: 10.1016/j.jglr.2012.02.009. [DOI] [Google Scholar]
- 8.Zepernick BN, Gann ER, Martin RM, Pound HL, Krausfeldt LE, Chaffin JD, Wilhelm SW. 2021. Elevated pH conditions associated with Microcystis spp. blooms decrease viability of the cultured diatom Fragilaria crotonensis and natural diatoms in Lake Erie. Front Microbiol 12:598736. doi: 10.3389/fmicb.2021.598736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wallen DG. 1996. Adaptation of the growth of the diatom Fragilaria crotonensis (Kitton) and the phytoplankton assemblage of Lake Erie to chromium toxicity. J Great Lakes Res 22:55–62. doi: 10.1016/S0380-1330(96)70934-6. [DOI] [Google Scholar]
- 10.Reavie ED, Barbiero RP, Allinger LE, Warren GJ. 2014. Phytoplankton trends in the Great Lakes, 2001–2011. J Great Lakes Res 40:618–639. doi: 10.1016/j.jglr.2014.04.013. [DOI] [Google Scholar]
- 11.Allinger LE, Reavie ED. 2013. The ecological history of Lake Erie as recorded by the phytoplankton community. J Great Lakes Res 39:365–382. doi: 10.1016/j.jglr.2013.06.014. [DOI] [Google Scholar]
- 12.Martin RM, Wilhelm SW. 2020. Phenol-based RNA extraction from polycarbonate filters. Protocols.io. doi: 10.17504/protocols.io.bivuke6w. [DOI] [Google Scholar]
- 13.Jain M, Olsen HE, Paten B, Akeson M. 2016. The Oxford Nanopore MinION: delivery of Nanopore sequencing to the genomics community. Genome Biol 17:239. doi: 10.1186/s13059-016-1103-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gann ER, Truchon AR, Papoulis SE, Dyhrman ST, Gobler CJ, Wilhelm SW. 2022. Aureococcus anophagefferens (Pelagophyceae) genomes improve evaluation of nutrient acquisition strategies involved in brown tide dynamics. J Phycol 58:146–160. doi: 10.1111/jpy.13221. [DOI] [PubMed] [Google Scholar]
- 15.Wick RR, Judd LM, Holt KE. 2019. Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol 20:129. doi: 10.1186/s13059-019-1727-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wick RR, Judd LM, Gorrie CL, Holt KE. 2017. Completing bacterial genome assemblies with multiplex MinION sequencing. Microb Genom 3:e000132. doi: 10.1099/mgen.0.000132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Coster W, D'Hert S, Schultz DT, Cruts M, Van Broeckhoven C. 2018. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 34:2666–2669. doi: 10.1093/bioinformatics/bty149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. 2017. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 27:722–736. doi: 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, Earl AM. 2014. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pryszcz LP, Gabaldón T. 2016. Redundans: an assembly pipeline for highly heterozygous genomes. Nucleic Acids Res 44:e113. doi: 10.1093/nar/gkw294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Menzel P, Ng KL, Krogh A. 2016. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat Commun 7:11257. doi: 10.1038/ncomms11257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31:3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 24.Hoff K, Lomsadze A, Borodovsky M, Stanke M. 2019. Whole-genome annotation with BRAKER. Methods Mol Biol 1962:65–95. doi: 10.1007/978-1-4939-9173-0_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hackl T, Martin R, Barenhoff K, Duponchel S, Heider D, Fischer MG. 2020. Four high-quality draft genome assemblies of the marine heterotrophic nanoflagellate Cafeteria roenbergensis. Sci Data 7:29. doi: 10.1038/s41597-020-0363-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. 2019. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37:907–915. doi: 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huerta-Cepas J, Forslund K, Coelho LP, Szklarczyk D, Jensen LJ, von Mering C, Bork P. 2017. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol Biol Evol 34:2115–2122. doi: 10.1093/molbev/msx148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chan PP, Lowe TM. 2019. tRNAscan-SE: searching for tRNA genes in genomic sequences. Methods Mol Biol 1962:1–14. doi: 10.1007/978-1-4939-9173-0_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mikheenko A, Prjibelski A, Saveliev V, Antipov D, Gurevich A. 2018. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 34:i142–i150. doi: 10.1093/bioinformatics/bty266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Armbrust EV, Berges JA, Bowler C, Green BR, Martinez D, Putnam NH, Zhou S, Allen AE, Apt KE, Bechner M, Brzezinski MA, Chaal BK, Chiovitti A, Davis AK, Demarest MS, Detter JC, Glavina T, Goodstein D, Hadi MZ, Hellsten U, Hildebrand M, Jenkins BD, Jurka J, Kapitonov VV, Kröger N, Lau WWY, Lane TW, Larimer FW, Lippmeier JC, Lucas S, Medina M, Montsant A, Obornik M, Parker MS, Palenik B, Pazour GJ, Richardson PM, Rynearson TA, Saito MA, Schwartz DC, Thamatrakoln K, Valentin K, Vardi A, Wilkerson FP, Rokhsar DS. 2004. The genome of the diatom Thalassiosira pseudonana: ecology, evolution, and metabolism. Science 306:79–86. doi: 10.1126/science.1101156. [DOI] [PubMed] [Google Scholar]
- 31.Bowler C, Allen AE, Badger JH, Grimwood J, Jabbari K, Kuo A, Maheswari U, Martens C, Maumus F, Otillar RP, Rayko E, Salamov A, Vandepoele K, Beszteri B, Gruber A, Heijde M, Katinka M, Mock T, Valentin K, Verret F, Berges JA, Brownlee C, Cadoret J-P, Chiovitti A, Choi CJ, Coesel S, De Martino A, Detter JC, Durkin C, Falciatore A, Fournet J, Haruta M, Huysman MJJ, Jenkins BD, Jiroutova K, Jorgensen RE, Joubert Y, Kaplan A, Kröger N, Kroth PG, La Roche J, Lindquist E, Lommer M, Martin-Jézéquel V, Lopez PJ, Lucas S, Mangogna M, McGinnis K, Medlin LK, Montsant A, et al. 2008. The Phaeodactylum genome reveals the evolutionary history of diatom genomes. Nature 456:239–244. doi: 10.1038/nature07410. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The annotated nuclear genome was deposited in GenBank under the accession number JAKSYS000000000. Data are available under BioProject accession number PRJNA807324 and BioSample accession number SAMN25978007.