

Abstract
Mixed human cytomegalovirus (HCMV) strain infections are frequent in lung transplant recipients (LTRs). To date, the influence of the donor (D) and recipient (R) HCMV serostatus on intra-host HCMV strain composition and viral population dynamics after transplantation is only poorly understood. Here, we investigated ten pre-transplant lungs from HCMV-seropositive donors and 163 sequential HCMV-DNA-positive plasma and bronchoalveolar lavage samples from fifty LTRs with multiviremic episodes post-transplantation. The study cohort included D+R+ (38 per cent), D+R− (36 per cent), and D−R+ (26 per cent) patients. All samples were subjected to quantitative genotyping by short amplicon deep sequencing, and twenty-four of them were additionally PacBio long-read sequenced for genotype linkages. We find that D+R+ patients show a significantly elevated intra-host strain diversity compared to D+R− and D−R+ patients (P = 0.0089). Both D+ patient groups display significantly higher viral population dynamics than D− patients (P = 0.0061). Five out of ten pre-transplant donor lungs were HCMV DNA positive, whereof three multiple HCMV strains were detected, indicating that multi-strain transmission via lung transplantation is likely. Using long reads, we show that intra-host haplotypes can share distinctly linked genotypes, which limits overall intra-host diversity in mixed infections. Together, our findings demonstrate donor-derived strains as the main source of increased HCMV strain diversity and dynamics post-transplantation. These results foster strategies to mitigate the potential transmission of the donor strain reservoir to the allograft, such as ex vivo delivery of HCMV-selective immunotoxins prior to transplantation to reduce latent HCMV.
Keywords: human cytomegalovirus, intra-host strain diversity, mixed infections, lung transplant, viral strain dynamics, PacBio sequencing
Introduction
Human cytomegalovirus (HCMV), a double-stranded DNA virus of the β-herpesvirus family, establishes a lifelong latent infection with reactivation episodes. Multiple HCMV strains (i.e. mixed infections) can be acquired during a person’s lifetime (Meyer-König et al. 1998; Puchhammer-Stöckl et al. 2006) and are specifically frequent in transplant patients, where HCMV strains can be donor- or recipient-derived (D/R) or both (Puchhammer-Stöckl and Görzer 2011). While infections in healthy adults are typically asymptomatic, they can lead to severe outcomes in those with immature or compromised immune systems (Kabani and Ross 2020; Limaye, Babu, and Boeckh 2020; Griffiths and Reeves 2021). Simultaneous presence of multiple strains in an individual provide an opportunity for viral recombination, selection for antiviral drug-resistant mutants and might consequently impact viral pathogenicity (Renzette et al. 2014). In fact, HCMV-infection-related increased morbidity and mortality remain a high risk for lung transplant recipients (LTRs) (Almaghrabi, Omrani, and Memish 2017).
With about 236 kb in size, HCMV consists of predominantly conserved regions across strains and some hypervariable genes spread across the genome (Dolan et al. 2004; Sijmons et al. 2015). These variable loci result in a limited number of distinct genotypes that have been extensively used to study population-level HCMV diversity found within and between hosts, primarily focusing on glycoproteins such as gB, gN, gO, and gH (Wang et al. 2021). Various techniques, including restriction fragment length polymorphism analysis (Huang et al. 1980), targeted amplicon sequencing (Coaquette et al. 2004; Puchhammer-Stöckl et al. 2006; Sowmya and Madhavan 2009; Hasing et al. 2021), and whole-genome sequencing (Cunningham et al. 2010; Renzette et al. 2011; Sijmons et al. 2015; Lassalle et al. 2016; Hage et al. 2017), have been used. With the increasing sequencing depth of next-generation sequencing platforms, the detection of low-frequency variants, i.e. minors, became possible (Görzer et al. 2010). Currently, there is mounting evidence that HCMV exists as a heterogeneous collection of genomes with variations in composition and distribution between anatomical compartments (Renzette et al. 2013; Hage et al. 2017) and over time (Görzer et al. 2010; Hage et al. 2017; Suárez et al. 2020; Dhingra et al. 2021). However, in samples with mixed strains, the determination of individual consensus sequences using short reads presents a challenge. Genotype read-matching, short motif read-matching, and genotype-specific single nucleotide polymorphism matching of hypervariable genes have been applied as strategies to identify multi-strain infections (Renzette et al. 2011; Suárez et al. 2019b; Camiolo et al. 2021). In 2019, Cudini et al. introduced a computational method to reconstruct individual sequences within a sample from short-read data. They showed that the high nucleotide diversity of HCMV samples is due to mixed infections (Cudini et al. 2019). Another possibility to determine individual sequences in a sample is by single-read sequencing of long reads with an average read length of >10 kb. Given that HCMV DNA in clinical samples needs to be enriched by polymerase chain reaction (PCR) prior to long-read sequencing, read lengths are limited to the respective amplicon size. We recently demonstrated that the true diversity in mixed populations of patient samples can be underestimated by short-read sequencing, since haplotype sequences sharing long stretches of sequence identity even in amplicon target regions can be missed, highlighting the utility of long reads (Brait, Külekçi, and Goerzer 2022).
Despite extensive research on HCMV strain diversity, much less is known about strain dynamics in LTRs, which can harbour heterogeneous viral populations (Görzer et al. 2008). In principle, dynamics can change by introducing new strains to the population, by reactivation of latent strains, by de novo mutation, by recombination, or through the change in relative frequencies of present strains. It has been shown that reinfection with donor strains and reactivations of recipient strains can occur similarly often, although this is difficult to distinguish in mixed-infected patients (Manuel et al. 2009b). Also, multiple strain transmissions from the donor organ to the recipient and a shift in strain predominance over time were found to be common (Hage et al. 2017; Hasing et al. 2021), suggesting a complex dynamic post-transplantation.
Here, we combine Illumina deep-sequence and PacBio long-read sequence data from 163 specimens of fifty LTRs of different D/R risk groups with recurrent HCMV infection to examine within-patient HCMV strain diversity and dynamics. This retrospective study points out major contributors to viral diversity in lung transplant patients, leading to increased dynamics post-transplantation.
Results
PCR genotyping success rates of five genomic regions and overall genotype distribution
For this study, fifty LTRs with at least two HCMV DNAemia episodes with >102 copies/ml either in the bronchoalveolar lavage (BAL) or ethylenediaminetetraacetic acid-plasma (EP) or both and one sample with >103 copies/ml during the follow-up period of at least 185 days post-transplantation were included (details are provided in Materials and methods). Genotypes of up to five polymorphic loci, namely UL6, UL73 (gN), UL74 (gO), UL139, and UL146 (Fig. 1), were assessed in 163 specimens consisting of eighty-nine BAL and seventy-four EP samples by Illumina deep sequencing as described previously (Brait, Külekçi, and Goerzer 2022). This resulted in genotyping success rates for the five loci ranging between 67 and 85 per cent (Fig. 2A). Despite a comparable viral load distribution, BAL samples had a higher genotyping success rate for all loci than EP samples (Fig. 2). A detailed summary of PCR performances and the number of genotypes per locus are provided in Supplementary Table S1. We observed a trend towards higher viral loads with increasing numbers of maximally detected genotypes (Supplementary Fig. S1A). Considering the slightly different PCR genotyping success rates of the different loci, we also analysed each region separately (Supplementary Fig. S1B). Here, the association between higher viral loads and increasing genotype numbers was significant for gN (P = 0.0093), UL6 (P = 0.0490), and UL146 (P = 0.0031) but not for gO and UL139. Given the well-known gN/gO linkage disequilibrium, this observed divergence may be explained by the higher sensitivity of the gN-specific PCR compared to the gO-specific PCR, as reflected in the PCR success rates of 85 and 72 per cent, respectively (Fig. 2A) (Mattick et al. 2004). The samples with the highest genotype numbers (≥3) did not have the highest viral loads. Of all samples with ≥2 genotypes at any locus, 79 per cent (49 out of 62) showed a single genotype in one of the five regions. These data indicate that the applied genotyping PCRs are highly sensitive also for samples with low viral loads starting from 102 copies/ml and thus are well suitable for identifying mixed genotypes in clinical samples.
Figure 1.

Schematic illustration of the locations of short (UL) and long (F) amplicon target regions along the HCMV genome of strain Merlin (NCBI: AY446894). The first track shows the overall HCMV genome structure that consists of a unique long (UL) and a unique short (US) region. Above, forward and reverse primers shown as triangles, and the size of the long amplicons (F5, F1, F3, and F4) are depicted. Next, a zoom-in into these four long amplicons (green horizontal lines with amplicon sizes) is shown separately. The mean pairwise nucleotide identity for over 200 published sequences for these regions is shown in three colours reflecting the height of the bars: green (100 per cent), brown (≥30 per cent to <100 per cent), and red (≤30 per cent). All annotated CDSs are shown as filled arrows, and those used for short- and long-read genotyping are in orange and green, respectively. Lastly, blue lines below each long amplicon indicate short amplicon regions (UL6, UL73, UL74, UL146, and UL139) and the respective amplicon sizes. Graphs were generated with Geneious Prime 2019.0.3.
Figure 2.
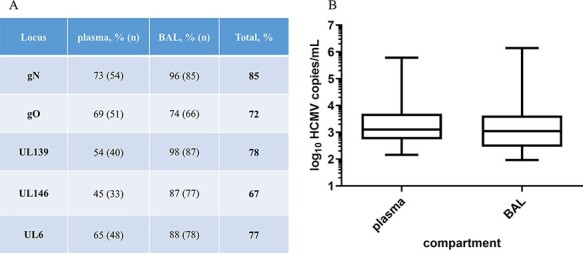
Genotyping performances for the five regions gN (UL73), gO (UL74), UL139, UL146, and UL6 and the viral load distribution for plasma and BAL samples. (A) Genotyping success in per cent genotyped sample/total number of samples of this compartment. In total, gN has the highest PCR success rate and UL146 the lowest. (B) Box whisker plots of the viral load of plasma and BAL samples of the total cohort. The viral load distribution is not different between sample types (P = 0.301, n = 163, Mann–Whitney test).
We detected each genotype of the five loci in one and up to twenty LTRs, except for UL146-3, UL146-5, and UL146-6, which we did not find in any sample (Supplementary Fig. S2). Each genotype was also found as a major genotype (defined as >70 per cent of all reads) in at least one sample. No significant differences between genotypes and viral loads were found for any of the five loci, and this is illustrated for gO in Supplementary Fig. S3.
Donor and recipient HCMV-seropositive patients display higher genotype diversity
We aimed to analyse the genotype diversity among the three HCMV serostatus combination groups, whereof nineteen were D + R+ (38 per cent), followed by eighteen D−R+ (36 per cent) and thirteen D+R− LTR patients (26 per cent). Similar HCMV DNA loads were observed between all three D/R serostatus combinations (Fig. 3A). For each patient, the maximum number of genotypes detected at any of the five loci and in any BAL and EP sample was counted (Fig. 3B). More than one genotype was found in 84 per cent of D+R+ patients compared to 62 and 50 per cent in D+R− and D−R+ patients, respectively. D+R+ patients were significantly more frequently infected with >2 genotypes (6/19) than D+R− and D−R+ patients (1/31) (P = 0.0089; Fisher’s exact test). Moreover, the D+R+ patient group shows the highest numbers of genotypes, with up to four genotypes in two LTRs (Fig. 3B).
Figure 3.
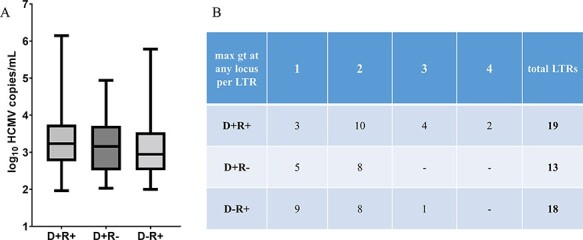
Viral load and maximum number of genotypes (max gt) in LTRs with For Review Only different HCMV serostatus combinations of D and R. (A) No significant difference in viral load between the three HCMV serostatus combination groups is observed (P = 0.2626, n = 163, Kruskal–Wallis test plus post hoc Dunn’s multiple comparisons test). (B) The maximum number of genotypes (gt) at any of the five loci detected for each LTR is provided (n = 50).
Donor HCMV-seropositive patients exhibit higher genotype dynamics over time
Next, we analysed how the genotype composition within a patient changes over time in the three serostatus patient groups. In 36/50 LTRs, genotyping data of at least two episodes of HCMV DNAemia in the same compartment were available and allowed the analysis of intra-host genotype dynamics over time (Fig. 4A). In total, twenty-six BAL and fourteen EP sample pairs were analysed (Supplementary Table S2). The median time difference between the paired samples was 179 days (range = 14–531 days). LTRs with at least one sample with ≥2 genotypes at any loci or differing genotypes at different time points were defined as mixed infected (n = 27). On the contrary, in single infected patients, one genotype was identified longitudinally (n = 9). We defined the total genotype dynamics over time as a change in two variables: (1) increase in genotype number and (2) change in relative genotype frequency. The former describes an increase in genotype number between Time points 1 and 2, while the latter accounts for a predominance change in the major genotype (defined as >70 per cent of all reads) between the two episodes.
Figure 4.
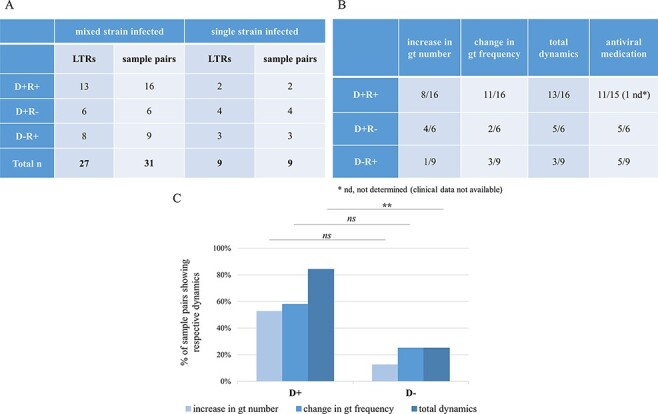
Intra-host genotype dynamics over time in LTRs of the three HCMV serostatus combination groups. (A) The number of LTRs and the number of sample pairs comprising the dynamics cohort are categorised into the three serostatus groups. LTRs from whom samples from two pairs were available were analysed. (B) The table shows the number of mixed sample pairs that presented the respective dynamics, increase in gt number, or change in gt frequency, or both (total dynamics). Antiviral medication was considered when given between or at the time of the analysed time points as a fraction of all pairs in this group. (C) For statistical tests, one sample pair per patient (the earliest one tested) was included (n = 27, Fisher’s exact test; ** for P < 0.01). Total dynamics was significantly higher in D+ patients compared to D− patients (P = 0.0061), while the increase in gt number and change in gt frequency did not reach significance (P = 0.0899 and P = 0.2087; respectively). gt, genotype; ns, non-significant.
D+ patients showed significantly higher total dynamics than D− patients (P = 0.0061) (Fig. 4B, C) and a trend in the increase in genotype numbers (P = 0.0899). In 69 per cent of D+R+ patients, a change in predominance was observed compared to 33 per cent each in the other two patient groups. Antiviral medication, which may influence strain dynamics, was similarly frequent between single- and mixed-infected LTRs (P > 0.9999, n = 34), between mixed-infected D+ and D− LTRs (P = 0.1972, n = 26), and between LTRs with and without total dynamics (P > 0.9999, n = 26).
Two patterns of genotype predominance dynamics over time
The predominance change in relative genotype frequency displayed two distinct patterns. First, an exchange of the pre-existing genotype (thereby an initially minor genotype becomes the major), and second, the introduction of a new genotype that overtakes the previously predominant genotype (whereby the initially major genotype becomes undetectable or a minor). While paired BAL samples (n = 10) presented both predominance patterns equally frequent, all plasma sample pairs (n = 6) displayed the second pattern, indicating the introduction of a new predominant genotype (Supplementary Table S2). Overall, a change in predominance was detected in the earliest 62 days after the first episode.
The pre-transplant donor lung may harbour multiple genotypes
To learn more about the donor lung as a potential HCMV source, we analysed the middle lobe parts of ten lungs of HCMV-seropositive donors for HCMV DNA positivity and genotype diversity. Of note, these donor lungs are from a separate group of individuals and do not correspond to any of the fifty LTRs analysed in this study. For each middle lobe, one to eight different locations were collected (Supplementary Table S3A). Each individual tissue piece was stored in buffer before being processed to a single cell suspension (for details see Materials and methods). In total, sixty lung pieces and the corresponding storage buffer samples were tested for HCMV positivity. We detected HCMV DNA in 8/60 cell suspensions and in 15/60 storage buffer samples with viral loads ranging between 2 × 102 – 6 × 103 and 1 × 102 – 1 × 103 copies/ml, respectively. Although more viral DNA in the cells than in the storage buffer might be expected, the viral loads of both sample types were surprisingly similar. Frequent detection of HCMV DNA in storage buffer is most likely due to the release of HCMV DNA from damaged cells and/or blood vessels during collection. In summary, five out of the ten lung donors were HCMV DNA positive. Two out of ten donors were HCMV DNA positive in the cells only, two donors were positive in the storage buffer only, and one donor was positive in both cells and storage buffer. In the cellular fractions, we detected HCMV DNA in up to four out of eight different locations (Supplementary Table S3A). These findings indicate focal HCMV DNA distribution in the analysed lung tissues. All HCMV-DNA-positive samples were subjected to short amplicon genotyping. We detected up to three genotypes in a single donor sample, and three out of four donors displayed mixed genotypes (Supplementary Table S3B).
Long-read PacBio sequencing for haplotype analysis
Thus far, we have assessed HCMV strain diversity and dynamics based on sequencing data of short polymorphic regions to define the respective HCMV genotypes. To expand our understanding on within-host HCMV strain diversity, we quantitatively determined the individual haplotypes in a subset of twenty-three BALs and one EP sample from twenty LTRs by long-read PacBio sequencing. Sample selection was restricted by the viral load sensitivity limit of the long-read PCR (for details refer to Materials and methods). Herein, the term haplotype refers to a single HCMV genome of >6 kb covering multiple non-adjacent genotype-defining regions.
Long amplicons were generated from four regions, F5, F1, F3, and F4 (Fig. 1), and subjected to long-read PacBio sequencing similarly as described previously (Brait, Külekçi, and Goerzer 2022). The amplicon fragments range between 6.2 and 7.9 kb in length and cover all polymorphic genes (dN, non-synonymous substitutions per non-synonymous site ≥0.086) we used for genotyping and fifteen more. These include seven of the most divergent HCMV genes (dN ≥ 0.034) and two less variable genes, UL55 (gB) and UL75 (gH), extensively sequenced in previous studies due to their known functional importance (Sijmons et al. 2015). Detailed information on PacBio-determined haplotypes is presented in Supplementary Table S4A. In Fig. 5A, the assignments of the identified haplotype sequences to the most similar reference sequences are displayed. Ten LTRs were single haplotype infected and nine LTRs were infected with up to two haplotypes, and in one patient (LTR-45), up to three haplotypes were detected. In total, we identified 116 individual haplotypes in 20 LTRs, and for each, sequences with identities >98 per cent were found in the National Center for Biotechnology Information (NCBI) Nucleotide (nr/nt) database (Supplementary Table 4B), except for four haplotypes sharing only 93–96 per cent identity. Notably, for 38 per cent of the 165 reference sequences, corresponding haplotype sequences were found in our cohort, illustrating a substantial inter-host haplotype diversity. Despite a high inter-host variability, we found haplotypes in different patients sharing sequence identities of up to 99.9 and 99.8 per cent in F3 and F5, respectively (6A_hap1, 12B/F_hap1) and 99.7 per cent in F1 (16B_hap2, 45B/D_hap2). Haplotypes of F4 were more diverse with a maximum of 98.5 per cent sequence identity between haplotypes 11O_hap1 and 37B_hap1.
Figure 5.
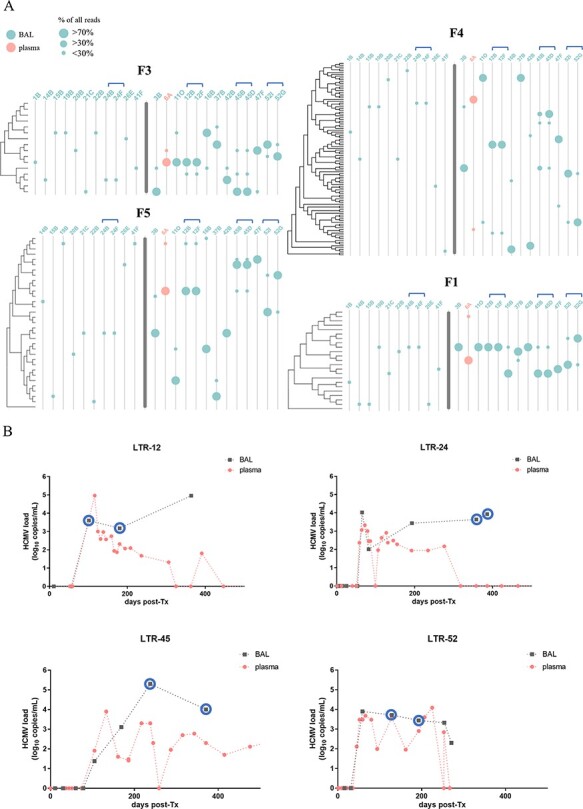
A: Graphical representation of the 144 detected haplotypes of the four long amplicon regions. The trees cluster representative haplotypes of the respective regions that differ in >150 nucleotides based on all publicly available HCMV strains (accession numbers are provided in Supplementary Table S8A). Distances are in units of the number of base differences per sequence. Trees were generated in MEGA X using the unweighted pair group method with arithmetic mean (UPGMA) method and displayed with Evolview. Single and mixed-infected samples are separated by a bold grey line. Samples 24B/F, 12B/F, 45B/D, and 52I/G are follow-up samples of the same patients and are indicated with blue brackets. (B) HCMV viral loads of longitudinal samples from BAL and plasma from LTRs 12, 24, 45, and 52. Blue circles indicate samples that have been subjected to PacBio long-read sequencing. post-Tx, post-transplantation.
Rapid intra-host dynamics, but stable haplotype sequences over time
For four LTRs, two follow-up BAL samples each were included (Fig. 5A; indicated with blue brackets) to assess the haplotype dynamics over time. The course of HCMV DNAemia for these four patients is provided in Fig. 5B. First, one patient (LTR-24) was single haplotype infected, showing the same haplotypes in both samples (24B, 24F; time between samples (∆): 28 days). Second, the longitudinal samples 52I and 52G (∆: 64 days) of LTR-52 illustrate the change in the predominance of the two haplotypes from Time points 1 to 2 throughout all four regions. Third, Samples 12F and 12B (∆: 79 days) of LTR-12 show the same two haplotypes with consistent relative frequencies for both time points. Fourth, for patient LTR-45, a change in the predominance of the major haplotype is only observed in region F4 (∆: 133 days). Alignments of the within-patient haplotype sequences show 100 per cent sequence identity except for the minor haplotypes of Samples 45B and 45D for the region F1 (45B/D_F1_hap2) and a suspected fourth haplotype for region F4 (45B_F4_hap4). Here, a closer look reveals that 45B/D_F1_hap2 consists of a mixture of two sub-haplotypes with twelve nucleotide differences (Supplementary Table S4C). Separation of the two sub-haplotype sequences shows that both sub-haplotypes are present in Samples 45B and 45D and the respective sequences are 100 per cent identical in both samples. The sequence of haplotype 45B_F4_hap4, in contrast, is different. Visual inspection of the individual PacBio circular consensus sequence (ccs) reads revealed two different groups of ccs, both sharing sequence similarity with haplotype sequences 45B_F4_hap1 and 45B_F4_hap2 (Supplementary Fig. S4). The two groups with twelve and fifteen ccs reads each display recombined sequences with breakpoints upstream and downstream of the coding sequence (CDS) of UL144, respectively. PCR-mediated recombination cannot be ruled out; thus, these sequences were excluded from further analysis.
Genotypes shared between intra-host haplotypes limit the overall diversity in mixed samples
To investigate how linkage combinations between non-adjacent polymorphic regions contribute to intra-host diversity in mixed strain samples, we compared the number of genotypes of eight highly polymorphic genes (dN ≥ 0.082) with the number of haplotypes in these regions. In total, ten mixed-infected LTRs with seventy-five distinct haplotypes were analysed. Genes used for genotype determination were gN and gO in F3; UL139, UL144, and UL146 in F4; and UL6, UL9, and UL11 in F5 (Fig. 1). For region F3, in all samples, the number of genotypes matched the number of haplotypes, possibly because of the well-described linkages between gN and gO genotypes restricting recombination and consequently different linkage combinations (Mattick et al. 2004). All haplotypes in our cohort showed previously described gN/gO linkages. In three LTRs, fewer genotypes than haplotypes were observed in target regions F4 and F5, suggesting different linkage patterns (Supplementary Table S5). First, we identified two F5 haplotypes in sample 3B, both sharing the UL11-2 genotype with only one nucleotide difference in the 429-bp-long region used for UL11 genotyping. Pairwise alignments of the two F5 haplotypes clearly show substantial differences across the full 6.4 kb sequence with stretches of high identity in UL11 CDS (Supplementary Fig. S5A). Second, we found two F4 haplotypes in sample 11O but only one UL139-2 genotype with fourteen nucleotide differences (98 per cent identity) across the genotyping region (Supplementary Fig. S5B). Lastly, in both samples of LTR-45 (45B/45D), three F4 haplotypes, but two UL139, UL144, and UL146 genotypes, and three F5 haplotypes, but two UL6, UL-9, and UL11 genotypes were found. Again, haplotypes differed in one and up to eleven nucleotides in the respective genotyping regions, but considerable differences up- and downstream confirmed distinct haplotypes (Supplementary Fig. S5C, D). Interestingly, while the same UL139 genotype was shared between hap1 and hap2, the same UL144 and UL146 genotypes were found between hap2 and hap3, which is indicative of past recombination. Of note, intra-host haplotypes in our cohort shared up to 40 per cent identical sequence sections, which is ideal for homologous recombination (Supplementary Fig. S6).
While linkage patterns of non-adjacent genotypes within a haplotype region can unambiguously be defined by long-read sequencing, linkages of non-adjacent haplotype sequences might be assessed according to their relative frequencies (Brait, Külekçi, and Goerzer 2022). We refer to this as a statistical linkage of haplotypes (relative frequencies of all haplotypes are provided in Supplementary Table S4A). As illustrated in Fig. 6B for patient LTR-52, it can be assumed that the major (hap1) and minor (hap2) haplotypes, respectively, are present in the same HCMV genome. Moreover, the statistical linkage remained stable even after the predominance change from Time point 1 to 2. For samples with varying relative frequencies of the distinct haplotypes of each region, direct statistical linking is not reasonable. The longitudinal samples 45B/45D, shown in Fig. 6A, exemplify this problem clearly. First, in both samples, the relative haplotype frequencies were similar for the regions F5, F1, and F3, but not for F4. Second, the major haplotypes of F5, F1, and F3 did not change over time, but the major haplotype of F4 did. Third, relative frequencies of the haplotypes hap2 and hap3 were only similar for F5, F1, and F3, but not for F4. Thus, the three individual haplotypes of F5, F1, and F3 can be statistically linked, but no further linkage with the F4 haplotypes is feasible.
Figure 6.
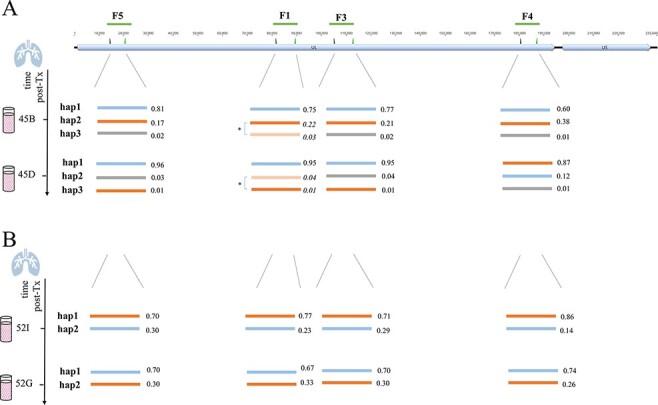
Schematic presentation of haplotypes of longitudinally PacBio-sequenced BAL samples of LTR-45 (A) and LTR-52 (B). The top track shows the overall HCMV genome structure that consists of a UL and a US region with the long amplicon target regions (F5, F1, F3, and F4) depicted as green lines. Haplotypes determined for each of the four long amplicon regions are presented as lines of different colours. The values next to each haplotype are the relative frequencies of the respective haplotypes. (A) The two sub-haplotypes of LTR-45 of region F1 (marked with an asterisk) were initially detected as a single haplotype (hap2), since they only differed in twelve nucleotides across the whole fragment (further details in ‘Results’ section and Supplementary Table S4C). Here, they are shown as separate haplotypes and the calculated relative frequencies are shown in italic. (B) For LTR-52, similar relative frequencies of both haplotypes of each region suggest their statistical linkage. A change in predominance is observed between both time points and supports that orange and blue haplotypes are linked, respectively. nt, nucleotide.
Discussion
In this study, we have comprehensively assessed the diversity and viral population dynamics of mixed HCMV strains in patients with multiviremic HCMV episodes after lung transplantation. We identify the donor lung as a critical source for a complex and dynamic HCMV strain population independent of the recipients’ HCMV serostatus, and we demonstrate that distinct genotype linkage combinations may not necessarily increase the overall intra-host diversity.
Sensitive and quantitative assessment of HCMV genome diversity directly from clinical material is important to study the composition and dynamics of mixed infections robustly. Due to the low amount of starting HCMV DNA in clinical specimens, PCR-amplicon enrichment followed by short-read Illumina deep sequencing was performed. Previous studies using PCR-based approaches confirmed that the detection of mixed strains could depend on the viral load, the genetic loci, and clinical specimen type analysed (Görzer et al. 2008; Sowmya and Madhavan 2009; Manuel et al. 2009a). We were able to analyse samples with viral loads starting from 102 copies/ml and detect minor variants down to 1 per cent, making this approach explicitly more sensitive for detection of mixed infections compared to a whole-genome sequencing approach (Suárez et al. 2019b). The five genes (UL6, gN, gO, UL139, and UL146) we chose are among the twelve highest polymorphic ones with a dN ≥ 0.086, are predicted to encode for products of immunomodulation, and are known to be necessary for cell entry (Bradley et al. 2008; Sijmons et al. 2015; Wang et al. 2021). While the high overall genotyping success rates (67–85 per cent) confirm that these target regions are very well suitable to determine genotypic diversity, the data also show that HCMV DNA isolated from BAL samples is better amplifiable than from plasma samples (Fig. 2A). This is most likely due to the high fragmentation of plasma HCMV DNA as previously suggested (Tong et al. 2017; Brait, Külekçi, and Goerzer 2022). This also explains why amplicons of >6 kb, which were used for long-read sequencing, could hardly be generated from plasma-derived HCMV DNA, despite similar viral load concentrations as in BAL samples.
In our cohort of fifty lung transplant patients, we found representatives of almost all genotype sequences (41 out of 44), yet with varying prevalences. No specific genotype was associated with significantly higher or lower viral loads, suggesting that genotypic variations of the five genes analysed do not affect the HCMV replication efficiency (Fig. 2—supplement 2). Three UL146 genotypes, gt3, gt5, and gt6, were not present in our study cohort (Fig. 2—supplement 3). This is well in accordance with prior studies also reporting a low frequency for these genotypes (Bradley et al. 2008; Berg et al. 2021). Thus, the high number of distinct UL146 genotypes in the context of a limited sample size and potential differences in geographical frequencies of genotypes as previously shown (Bradley et al. 2008; Suárez et al. 2019b) may account for the missing of these three UL146 genotypes in our study cohort. On the other hand, differences in the pathophysiological properties among distinct UL146 genotypes may also play a role in the observed genotype distribution pattern. UL146 open reading frame (ORF) encodes a viral α (CXC)-chemokine, which is suggested to recruit neutrophils for viral dissemination (Lüttichau 2010). A recent study investigated the functional variability of distinct recombinant UL146 proteins, showing that UL146 polymorphisms differentially affect chemokine receptor binding affinity, which could influence HCMV dissemination and pathogenesis (Heo et al. 2015). Hence, it might be speculated that the absence of UL146 gt3, gt5, and gt6 in this cohort is due to a lower virulence compared to the other genotypes. In general, cellular CXC chemokines are divided into two groups depending on the presence or absence of an ELR (Glu-Leu-Arg) motif prior to the CXC motif with an angiogenesis promoting and inhibiting function, respectively. Interestingly, all UL146 genotypes that have been detected in our cohort share this ELR motif but not the missing genotypes gt5 and gt6 (Heo et al. 2008). High levels of ELR chemokines have been suggested to be major mediators of lung disease processes such as bronchiolitis obliterans syndrome (BOS), adult respiratory distress syndrome, and pulmonary fibrosis (Keane et al. 1997; Keane et al. 2002; Belperio et al. 2005). It can be speculated that increased activity of viral ELR chemokine homologues in the lung could change the balance between angiogenic or angiostatic chemokines in favour of aberrant angiogenesis. This might play a role in why HCMV replication is a risk factor for BOS development (Paraskeva et al. 2011) and might be worth to be addressed in future studies.
A main objective of this study was to identify the relationship between pre-transplant D/R HCMV serostatus and strain diversity and dynamics post-transplantation. The finding that D+R+ patients harbour a higher number of genotypes compared to the other two D/R groups (Fig. 3B) indicates that both donor- and recipient-derived strains contribute to the observed diversity. Although several previous studies found that reinfection with donor-derived strains are more common than reactivation of recipient virus (Chou 1986; Grundy et al. 1988; Sunwen and Norman 1988), others have argued that infections post-transplantation are approximately equally donor- and recipient-derived (Manuel et al. 2009b). Moreover, community-acquired infections post-Tx cannot be ruled out completely but is considered very unlikely in LTRs as the CMV infection incidence in D−R− LTRs is low. Reports on the detection of HCMV in tissues of non-immunocompromised individuals (Kraat et al. 1992; Schonian, Crombach, and Maisch 1993; Hendrix et al. 1997; Meyer-König et al. 1998; Kytö et al. 2005) further underline the potential of HCMV transmission with the allograft, yet the extent of transmission remained controversial. In this study, we could demonstrate HCMV DNA positivity in 5/10 pre-transplant lungs of HCMV-seropositive donors, with up to three HCMV genotypes present in small HCMV-DNA-positive tissue sections of a single donor lung. Assuming that the detected HCMV DNA reflects replicating rather than latent HCMV DNA, these findings indicate focal points of HCMV reactivation of either a single or multiple HCMV strains. Of note, in the HCMV-positive cases, reactivation must have started already before surgical organ removal, which is likely given that all donors were in the intensive care unit and under ventilation for >24 h. Interestingly, the focal distribution of HCMV DNA in the human lung resembles the murine CMV infection model in which the authors also observed a ‘patchwork pattern’ of focal reactivation and recurrence (Kurz et al. 1999; Kurz and Reddehase 1999). Taken together, our data strongly support the findings that HCMV transmission by the allograft of an HCMV-seropositive donor is very likely, yet either the extent thereof or whether it occurs or not may vary among donors. In this study, 2/15 D+R+ patients analysed longitudinally were solely infected with a single HCMV strain despite an overall higher diversity in this patient group. Nevertheless, our findings point towards the donor’s lung as an important contributor to HCMV strain transmission in both HCMV-seropositive and -seronegative recipients. Strategies to target this reservoir in pre-transplant lungs of HCMV-seropositive donors are promising. Recently, the ‘shock and kill’ approach in which latent cells are targeted and cleared has been applied to donor lungs. Treatment with the immunotoxin F49A-FTP during ex vivo lung perfusion has been shown to reduce reactivation to 76 per cent compared to 15 per cent in non-treated but perfused controls (Ribeiro et al. 2022). While off-target effects have been evaluated by comparing cell viability and inflammation markers in treated and non-treated lungs, potential effects of a fully functioning immune system after implantation are unknown. More studies are needed to address concerns about safety and efficacy in real use and to generate optimised protocols that allow EVLP beyond the limited preservation time window of up to 24 h (Takahashi et al. 2021). In future, this therapy can be integrated into already established EVLP platforms that are currently used in some lung transplant centres to assess and screen lungs pre-transplantation.
We could further show that D+ patients display higher viral population dynamics of their mixed HCMV strain populations post-transplantation compared to D− patients, also arguing for the donor-derived strains as the likely cause. It can be speculated that sequential reactivation and dissemination of donor-derived strains post-transplantation can ultimately increase the host’s latency reservoir and that gradual reseeding of the lung as well as dissemination into other tissues can contribute to the distribution of HCMV strains (Collins-McMillen et al. 2018). Over time, these processes might result in locally and temporally different replication levels of the distinct strains. This will either be observed as an increase in the genotype numbers and/or as a change in predominance. The latter dynamic pattern occurs more frequently in D+R+ patients, which may be explained by its generally richer pool of strains. A previous study demonstrating the sequential occurrence of different predominant strains in D+R− patients receiving organs from the same donor supports our view of sequential and/or stochastic reactivation of donor strains from the graft (Hasing et al. 2021), as has also been proposed by the murine CMV model (Reddehase et al. 1994). Additionally, it is presumable that initial replication of the major strain triggers a strain-specific immune response that might be well controlled thereafter while the initially less prevalent strain becomes predominant in further episodes due to limited cross-protection (Klein et al. 1999; Wang et al. 2021). It is also thinkable that antiviral medication affects viral population dynamics, for example by reducing minor populations. However, since all study patients received antiviral prophylaxis and pre-emptive therapy was similarly often administered to all patient groups, a medication effect on the observed differences in dynamics can hardly be evaluated. Notably, no antiviral resistance mutations were found in the four out of fifty patients that were tested for resistance mutations during routine diagnosis.
In-depth short amplicon sequencing over polymorphic regions is a highly sensitive strategy to decipher mixed genotype infections but may underestimate the true strain diversity due to the small genomic regions analysed and to the lack of information on linkage patterns. To partially overcome these limitations, we additionally performed long amplicon haplotype sequencing on a subset of twenty-four samples of twenty patients, of whom ten patients were mixed infected. The four haplotype target regions (F1, F3, F4, and F5) cover about 10-fold larger genomic regions than our short amplicon approach while including all five genes used for genotyping. In thirteen samples, where both methods have been applied, the number of genotypes and haplotypes matched. The advantage of long reads to resolve distinct haplotypes in mixed populations became particularly obvious in patients in whom we found haplotypes with partially shared genotypes (Supplementary Fig. S5). Hence, the lack of information on the arrangement of genotypes on individual genomes by short amplicon genotyping approaches might help to understand why correlating specific genetic variants to clinical observations may lead to inconsistent results (Wang et al. 2021). In one patient, the same genotype assignments were shared between hap1 and hap2 in one locus (UL139) and hap2 and hap3 in other loci (UL144 and UL146), suggesting that hap2 had arisen by intra-host recombination between hap1 and hap3. The low number of single nucleotide polymorphisms between the same genotypes, which could have been accumulated over time in the patient, would have supported this conclusion. However, the substantial differences across the full-length haplotype sequences make a recent intra-patient generation of a recombinant haplotype unlikely since within-host haplotype sequences are highly stable on short timescales, as shown in our study and by others (Hage et al. 2017; Cudini et al. 2019; Dhingra et al. 2021; Götting et al. 2021). Additionally, almost identical sequences of the respective haplotypes can be found in the database, further pointing towards transmission from another host. Also, identical haplotypes were observed across patients of our cohort, and in one patient (LTR-41), we identified a previously described non-recombinant strain, supporting the view that HCMV diversification has occurred early in human history (Mattick et al. 2004; Bradley et al. 2008; Suárez et al. 2019b). Both approaches used in our study, short- and long-read sequencing, suggest an upper limit to intra-host HCMV genetic diversity with a maximum of four genotypes and three haplotypes, respectively, in our cohort of lung transplant patients. Although long-read sequencing of longitudinal samples is suitable for directly proving within-patient recombination, no direct evidence was seen in the analysed genomic regions. Hence, from our data, it appears that the overall within-host HCMV diversity predominantly results from a mixture of genomically distinct strains rather than from newly emerging variants, which confirms previous studies (Cudini et al. 2019; Suárez et al. 2019b; Suárez et al. 2020).
Although we observe an upper limit of HCMV strain diversity in our patient samples, we cannot completely exclude that we have underestimated the level of multi-strain infections. First, despite the application of a highly sensitive sequencing approach, only minors with viral loads above the sensitivity limit of the PCR can be successfully amplified. This could potentially introduce a bias for minor populations and might explain that we observe higher viral loads with an increased number of strains (Supplementary Fig. S1). Second, strains distinguishable at genomic locations outside our target regions would not have been detected, a limitation of our approach compared to whole-genome sequencing. Third, HCMV prevalences differ among distinct geographic locations (Cannon 2010; Zuhair 2019), and this may influence the extent of intra-host multi-strain populations as it has recently been shown in breast milk specimens from African women (Suárez et al. 2019a). Fourth, it could not be excluded that antiviral prophylaxis, which has been given to all patients in our study cohort, may have led to a reduction in the number of detectable strains.
Determination of mixed strain infections can be of clinical importance as their presence is suggested to result in poorer outcomes for transplant patients (Puchhammer-Stöckl and Görzer 2011). Our short-range PCR approach that allows using samples with low viral loads and clinical material with fragmented DNA is suitable for this purpose and could be extended to include regions relevant for antiviral resistance (Limaye, Babu, and Boeckh 2020) or immune modulation (Vietzen et al. 2021). Early detection of mutant strains that might initially be in low quantities could be of clinical relevance. Long-read sequencing for haplotype determination, on the contrary, might be applied selectively to samples with high viral loads for the determination of recombined strains (Brait, Külekçi, and Goerzer 2022).
In conclusion, by HCMV genotype and haplotype determination of clinical samples, we demonstrate the extent to which the donor lung can contribute to HCMV strain diversity and dynamics after transplantation. We suggest that rapid intra-host dynamics of a limited number of HCMV strains might allow quick adaptation to changing environments and less so by enhancing diversity through recombination. Understanding the forces affecting HCMV population diversity and dynamics is an essential step for treatment and vaccine development.
Materials and methods
Study design and sample collection
For this retrospective study, we analysed routine HCMV DNAemia monitoring data for EP and BAL specimens of LTRs post-transplantation. Between 2016 and the end of 2018, 286 patients received a lung transplant at the Medical University of Vienna. As a standard regimen, all LTRs receive immunosuppressive treatment and antiviral prophylaxis for at least 3 months post-transplantation. To investigate strain dynamics, we selected LTRs with (1) at least two active HCMV infection episodes (>102 copies/ml) in either EP or BAL and (2) with at least one sample with >103 copies/ml (to maximise the chance for long-range amplicon PCR). We defined positive HCMV DNAemia sample points as distinct episodes if, between two sample points, (1) the viral load declined below 102 copies/ml or (2) if there was a time interval of 3 months. All EP and BAL specimens were stored at −20°C. Of the total 339 specimens from fifty-three LTRs that were initially selected, eighty-one specimens were not available. DNA extraction of sixty-seven was unsuccessful, and PCR amplification of twenty-eight samples resulted in no amplicons. Consequently, three LTRs were excluded. Two to fifteen samples were collected from each LTR, and sampling time points ranged between 4 and 1,476 days post-transplantation. Clinical information about the patients of the final cohort was retrieved from medical records.
DNA extraction and viral load quantification
DNA was isolated from 250 µl of BAL or EP samples using the QIAamp Viral RNA Mini Kit (Qiagen) as described in the manufacturer’s protocol and eluted in 35 µl elution buffer. HCMV DNA was quantified by in-house HCMV-specific qPCR as previously described (Kalser et al. 2017) and stored at 4°C or −20°C for further steps.
Short- and long-range amplicon PCR
Short-range amplicon PCRs for the five polymorphic regions gN, gO, UL6, UL139, and UL146 of HCMV-DNA-positive samples were performed as described previously (Brait, Külekçi, and Goerzer 2022), but with minor modifications. To save limited DNA material, UL6 and the first step of the nested UL146 PCR were multiplexed. Also, gO PCRs, consisting of three primers, were multiplexed. For samples with low viral loads (<103 copies/ml) and sufficient material availability, the usual DNA template amount of 5 µl was doubled. The viral load sensitivity of our long-range PCR was tested for BAL and EP samples (Supplementary Table S6). Based on that, BAL samples with >5 × 103 copies/ml and EP samples with >5 × 104 copies/ml were initially amplified with long-range PCR. In addition to previously described amplicons F3 and F4 (Brait, Külekçi, and Goerzer 2022), F1 and F5 amplicons were designed and tested to cover more polymorphic regions with long reads. F1, F3, and F4 PCR primers were multiplexed. PCRs of samples with lower viral loads and EP samples, in general, were performed with the doubled template amount of 20 µl, if available. Samples with unsuccessful long-range PCR were genotyped using short-range PCR. All primers are depicted in Fig. 1, and Supplementary Table S7 lists detailed descriptions of PCR primers. PCR product lengths were confirmed on analytical agarose gels and concentrations quantified by Qubit prior to Illumina or PacBio library preparation.
Illumina and PacBio sequencing
Library preparation and sequencing for Illumina sequencing were performed as described previously with a few adaptations (Brait, Külekçi, and Goerzer 2022). Briefly, amplicons were pooled equimolarly and 2 ng input DNA was used to generate a 4-nM library using the Nextera XT library preparation and index kit, followed by paired-end sequencing (150 cycles, v2 and v2 Micro kits) on an Illumina MiSeq with automatic adapter trimming. Fastq raw reads that passed default Illumina filters (pass-filter reads) were imported into CLC Genomics Workbench 21.0 (Qiagen) for further analysis.
According to the manufacturer’s protocol, pooled long-range amplicons were purified using the Qiaquick Spin Kit and eluted in 50 µl elution buffer. After quantification and purity determination with the NanoDrop 1,000 tool (Peqlab) and Qubit 2.0 fluorometer (Thermo Fisher), samples were submitted to the Next Generation Sequencing Facility at Vienna BioCenter Core Facilities. No size selection for the amplicons was performed before SMRT bell amplicon library preparation. Sequencing was performed on a PacBio-Sequel system for 20 h. Twenty-four samples were multiplex sequenced on two lanes.
Bioinformatical workflows for PacBio and Illumina reads
PacBio CCS generation
CCS reads were generated from PacBio raw reads using the ccs tool (https://github.com/PacificBiosciences/ccs) with a minimum predicted read quality of 0.99 (>99 per cent accuracy) and a minimum of three full passes per strand. To avoid CCS of potential PCR-mediated heteroduplexes of different strains, the by-strand option was used, whereby CCS for forward and reverse strands is generated and analysed separately. Next, CCS was demultiplexed using lima (https://github.com/pacificbiosciences/barcoding/) and the resulting bam files were imported into CLC Genomics Workbench 21.0.3 (Qiagen) for further analysis.
HCMV haplotype determination
For each of the four long-range amplicon regions, we compiled reference databases to be used for read mapping. First, we extracted the corresponding amplicon region from 236 publicly available HCMV whole-genome sequences and aligned them using MUSCLE 3.8.31. Pairwise distances were calculated in MEGA X 10.2.4, and only sequences with >150 nucleotide differences in the respective region were filtered for the final reference databases. This final cut-off was chosen as it resulted in sufficiently distinct reference sequences to avoid mapping of reads to multiple reference sequences. These resulted in 33, 23, 16, and 93 reference sequences for F5, F1, F3, and F4, respectively (Supplementary Table S8A). Trees were generated in MEGA X 10.2.4 using the UPGMA method and displayed with Evolview v3 (Subramanian et al. 2019) (Fig. 5A).
Firstly, human reads were excluded by random mapping against the latest reference genome GRCh38 (accession GCA_000001405.28) as described previously (Brait, Külekçi, and Goerzer 2022), and reads were length trimmed according to the amplicon length (F1 7.5–7.8 kb; F4 6.45–6.75 kb; F1 7.8–8.1 kb, and F5 6.2–6.5 kb). Using the Long Read Support plugin for CLC Genomics (Qiagen), reads were mapped against the filtered reference sequences with default settings and extractions of the consensus haplotype sequences were generated (low coverage threshold: 3, ambiguity code N insertion with the noise threshold set to 0.3 and the minimum nucleotide count to 3). As a control step, all initial, length-trimmed reads were re-mapped to the new consensus haplotype(s), and the new consensus sequence was extracted. All the final haplotypes of each sample were aligned and checked for their uniqueness. Potential chimaeras were inspected visually by checking if final haplotypes could be reconstructed by combining segments of two more abundant haplotypes. Lastly, all unmapped reads of the first mapping of each amplicon were again mapped to all final haplotypes to confirm that all unmapped reads for a certain amplicon belong to any of the other three amplicons.
HCMV genotype determination
Q30 quality-trimmed Illumina fastq reads were filtered for reads >80 bp in length and human genomic reads were excluded. Default mapping parameters were used for match/mismatch scores, insertion/deletion costs, and a length fraction of 0.3 and a similarity fraction of 0.95. Only mappings with >10 reads and a consensus length of at least 75 per cent of the reference sequence were chosen to extract consensus sequences. A noise threshold of 0.3 and a minimum nucleotide count of 3 were applied to insert ambiguity codes (N). All the resulting genotype consensus sequences were aligned and screened visually, and unique sequences were counted as genotypes. PacBio-derived haplotypes were genotyped using blastn (match/mismatch= 2/-3, gap costs = existence 5, extension 2) and a self-assembled BLAST database (Supplementary Table S8B).
For genotyping, five highly polymorphic regions of UL6 (CDS: 127-388), gO (CDS: 691-987), gN (CDS: 1-379), UL139 (CDS: 1-414, genome position: 187077-186395), and UL146 (CDS: 1-360, genome position: 181341-180571) were used based on sequences provided previously (Sijmons et al. 2015; Suárez et al. 2019b). In total, forty-seven reference sequences were used. For long amplicon reads, genotyping with thirty-four additional references was performed for regions gH (CDS: 1-177), UL144 (CDS: 1-500), UL9 (CDS: 57-430), UL10 (CDS: 0-762), UL11 (CDS: 194-623), and gB cleavage site (CDS: 1138-1619). All listed positions are in reference to strain Merlin (GenBank: AY446894.2). Reference sequences for genotyping are provided in Supplementary Table S8B.
Donor lung tissue processing
Donor lungs were collected between 2020 and 2021. Lung tissues were removed for pre-transplantation size adjustment and would have been discarded otherwise. Middle lobe sections of 1–2 cm3 of ten HCMV-seropositive donors were collected in phosphate-buffered saline (PBS) and stored at 4°C for processing on the same day (solution termed supernatant, short SN). Bigger sections were divided, and a part was submerged in RNAlater™ solution (Invitrogen) for storage at −20°C. Next, sections were minced with sterile single-use scalpels, weighted in a 50-ml falcon tube, and digested with Hanks’ balanced salt solution supplemented with 0.15 per cent Collagenase D (Roche) for 60 min at 37°C on an orbital shaker at 300 rpm. The tissue solution was diluted with PBS, vortexed vigorously, and strained through a 70-µm cell strainer to obtain a single cell suspension. After centrifugation (500 rpm, 10 min, 4°C), red blood cells (RBC) were lysed with RBC Lysis buffer solution (Invitrogen™ eBioscience™) and the remaining cells were counted using an automated cell counter (Nexcelom). Dead cells were excluded using trypan blue. Both 1 ml of SN and the single cell suspension (at most 106 cells) were separately transferred into 2 ml of lysis buffer and eluted in 50 μl elution buffer using the bead-based NucliSens EasyMagextractor (BioMérieux). HCMV-specific quantitative polymerase chain reaction (qPCR)-positive samples were genotyped using short-range amplicon PCRs and the next generation sequencing (NGS) workflow described earlier.
Statistical analysis
Statistical calculations were performed in GraphPad Prism version 9.0.0. P < 0.05 was considered statistically significant in all tests.
Supplementary Material
Acknowledgements
We acknowledge the support and technical assistance of Barbara Jilka, Andreas Rohorzka, Sylvia Malik, Michaela Binder, Barbara Dalmatiner, and Gabriele Sigmund. Lastly, we want to thank Sylvia Knapp for her valuable advice.
Contributor Information
Büsra Külekci, Center for Virology, Medical University of Vienna, Kinderspitalgasse 15, Vienna 1090, Austria.
Stefan Schwarz, Department of Thoracic Surgery, Medical University of Vienna, Währinger Gürtel 18-20, Vienna 1090, Austria.
Nadja Brait, Cluster of Microbial Ecology, Groningen Institute for Evolutionary Life Sciences, University of Groningen, Nijenborgh 7, Groningen 9747 AG, The Netherlands.
Nicole Perkmann-Nagele, Division of Clinical Virology, Medical University of Vienna, Währinger Gürtel 18-20, Vienna 1090, Austria.
Peter Jaksch, Department of Thoracic Surgery, Medical University of Vienna, Währinger Gürtel 18-20, Vienna 1090, Austria.
Konrad Hoetzenecker, Department of Thoracic Surgery, Medical University of Vienna, Währinger Gürtel 18-20, Vienna 1090, Austria.
Elisabeth Puchhammer-Stöckl, Center for Virology, Medical University of Vienna, Kinderspitalgasse 15, Vienna 1090, Austria.
Irene Goerzer, Center for Virology, Medical University of Vienna, Kinderspitalgasse 15, Vienna 1090, Austria.
Data availability
Raw sequence data have been deposited in the NCBI Sequence Read Archive under BioProject ID PRJNA803978. Haplotype sequences generated in this study and with identities <98 per cent to publicly available sequences were submitted to GenBank with the Accession No. OM835733–OM835736 (Supplementary Table S4B).
Supplementary data
Supplementary data are available at Virus Evolution online.
Funding
Austrian Science Fund (P31503-B26 to I.G.).
Conflict of interest:
None declared.
Declarations
This study was approved by the Ethics Committee of the Medical University of Vienna under EK-number 1321/2017. All data were pseudonymised before analyses.
References
- Almaghrabi R. S., Omrani A. S., and Memish Z. A. (2017) ‘Cytomegalovirus Infection in Lung Transplant Recipients’, Expert Review of Respiratory Medicine, 11: 377–83.doi: 10.1080/17476348.2017.1317596 [DOI] [PubMed] [Google Scholar]
- Belperio J. A. et al. (2005) ‘Role of CXCR2/CXCR2 Ligands in Vascular Remodeling during Bronchiolitis Obliterans Syndrome’, Journal of Clinical Investigation, 115: 1150.doi: 10.1172/JCI200524233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg C. et al. (2021) ‘The Frequency of Cytomegalovirus non-ELR UL146 Genotypes in Neonates with Congenital CMV Disease Is Comparable to Strains in the Background Population’, BMC Infectious Diseases, 21: 1–12.doi: 10.1186/s12879-021-06076-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley A. J. et al. (2008) ‘Genotypic Analysis of Two Hypervariable Human Cytomegalovirus Genes’, Journal of Medical Virology, 80: 1615.doi: 10.1002/JMV.21241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brait N., Külekçi B., and Goerzer I. (2022) ‘Long Range PCR-based Deep Sequencing for Haplotype Determination in Mixed HCMV Infections’, BMC Genomics, 23: 31.doi: 10.1186/s12864-021-08272-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camiolo S. et al. (2021) ‘GRACy: A Tool for Analysing Human Cytomegalovirus Sequence Data’, Virus Evolution, 7: 1.doi: 10.1093/VE/VEAA099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon M. J., Schmid D. S., and Hyde T. B. (2010) ‘Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection’, Reviews in medical virology, 20: 202–213.doi: 10.1002/rmv.655 [DOI] [PubMed] [Google Scholar]
- Chou S. (1986) ‘Acquisition of Donor Strains of Cytomegalovirus by Renal-transplant Recipients’, The New England Journal of Medicine, 314: 1418–23.doi: 10.1056/NEJM198605293142205 [DOI] [PubMed] [Google Scholar]
- Coaquette A. et al. (2004) ‘Mixed Cytomegalovirus Glycoprotein B Genotypes in Immunocompromised Patients’, Clinical Infectious Diseases, 39: 155–61.doi: 10.1086/421496 [DOI] [PubMed] [Google Scholar]
- Collins-McMillen D. et al. (2018) ‘Molecular Determinants and the Regulation of Human Cytomegalovirus Latency and Reactivation’, Viruses, 10: 444.doi: 10.3390/v10080444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cudini J. et al. (2019) ‘Human Cytomegalovirus Haplotype Reconstruction Reveals High Diversity Due to Superinfection and Evidence of Within-host Recombination’, Proceedings of the National Academy of Sciences of the United States of America, 116: 5693–8.doi: 10.1073/pnas.1818130116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham C. et al. (2010) ‘Sequences of Complete Human Cytomegalovirus Genomes from Infected Cell Cultures and Clinical Specimens’, Journal of General Virology, 91: 605–15.doi: 10.1099/vir.0.015891-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhingra A. et al. (2021) ‘Human Cytomegalovirus Multiple-strain Infections and Viral Population Diversity in Haematopoietic Stem Cell Transplant Recipients Analysed by High-throughput Sequencing’, Medical Microbiology and Immunology, 210: 291–304.doi: 10.1007/s00430-021-00722-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolan A. et al. (2004) ‘Genetic Content of Wild-type Human Cytomegalovirus’, Journal of General Virology, 85: 1301–12.doi: 10.1099/vir.0.79888-0 [DOI] [PubMed] [Google Scholar]
- Görzer I. et al. (2010) ‘Deep Sequencing Reveals Highly Complex Dynamics of Human Cytomegalovirus Genotypes in Transplant Patients over Time’, Journal of Virology, 84: 7195–203.doi: 10.1128/jvi.00475-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2008) ‘Virus Load Dynamics of Individual CMV-genotypes in Lung Transplant Recipients with Mixed-genotype Infections’, Journal of Medical Virology, 80: 1405–14.doi: 10.1002/jmv.21225 [DOI] [PubMed] [Google Scholar]
- Götting J. et al. (2021) ‘Human Cytomegalovirus Genome Diversity in Longitudinally Collected Breast Milk Samples’, Frontiers in cellular and infection microbiology, 11: 66424.doi: 10.3389/fcimb.2021.664247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths P., and Reeves M. (2021) ‘Pathogenesis of Human Cytomegalovirus in the Immunocompromised Host’, Nature Reviews. Microbiology, 19: 759–73.doi: 10.1038/s41579-021-00582-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy J. E. et al. (1988) ‘Symptomatic Cytomegalovirus Infection in Seropositive Kidney Recipients: Reinfection with Donor Virus Rather than Reactivation of Recipient Virus’, The Lancet, 332: 132–5.doi: 10.1016/S0140-6736(88)90685-X [DOI] [PubMed] [Google Scholar]
- Hage E. et al. (2017) ‘Characterization of Human Cytomegalovirus Genome Diversity in Immunocompromised Hosts by Whole-genome Sequencing Directly from Clinical Specimens’, Journal of Infectious Diseases, 215: 1673–83.doi: 10.1093/infdis/jix157 [DOI] [PubMed] [Google Scholar]
- Hasing M. E. et al. (2021) ‘Donor Cytomegalovirus Transmission Patterns in Solid Organ Transplant Recipients with Primary Infection’, Journal of Infectious Diseases, 223: 827–37.doi: 10.1093/infdis/jiaa450 [DOI] [PubMed] [Google Scholar]
- Hendrix R. M. G. et al. (1997) ‘Widespread Presence of Cytomegalovirus DNA in Tissues of Healthy Trauma Victims’, Journal of Clinical Pathology, 50: 59–63.doi: 10.1136/JCP.50.1.59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo J. et al. (2015) ‘Novel Human Cytomegalovirus Viral Chemokines, vCXCL-1s, Display Functional Selectivity for Neutrophil Signaling and Function’, Journal of Immunology (Baltimore, Md. : 1950), 195: 227–36.doi: 10.4049/JIMMUNOL.1400291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2008) ‘Polymorphisms within Human Cytomegalovirus Chemokine (UL146/UL147) and Cytokine Receptor Genes (UL144) are Not Predictive of Sequelae in Congenitally Infected Children’, Virology, 378: 86–96.doi: 10.1016/J.VIROL.2008.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang E. ‐S. et al. (1980) ‘Cytomegalovirus: Genetic Variation of Viral Genomes’, Annals of the New York Academy of Sciences, 354: 332–46.doi: 10.1111/j.1749-6632.1980.tb27976.x [DOI] [PubMed] [Google Scholar]
- Kabani N., and Ross S. A. (2020) ‘Congenital Cytomegalovirus Infection’, The Journal of Infectious Diseases, 221: S9–S14.doi: 10.1093/INFDIS/JIZ446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalser J. et al. (2017) ‘Differences in Growth Properties among Two Human Cytomegalovirus Glycoprotein O Genotypes’, Frontiers in Microbiology, 8: 1609.doi: 10.3389/fmicb.2017.01609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane M. P. et al. (1997) ‘The CXC Chemokines, IL-8 and IP-10, Regulate Angiogenic Activity in Idiopathic Pulmonary Fibrosis’, Journal of Immunology (Baltimore, Md. : 1950), 159: 1437–43. [PubMed] [Google Scholar]
- ——— et al. (2002) ‘Imbalance in the Expression of CXC Chemokines Correlates with Bronchoalveolar Lavage Fluid Angiogenic Activity and Procollagen Levels in Acute Respiratory Distress Syndrome’, Journal of Immunology (Baltimore, Md. : 1950), 169: 6515–21.doi: 10.4049/JIMMUNOL.169.11.6515 [DOI] [PubMed] [Google Scholar]
- Klein M. et al. (1999) ‘Strain-Specific Neutralization of Human Cytomegalovirus Isolates by Human Sera’, Journal of Virology, 73: 878.doi: 10.1128/jvi.73.2.878-886.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraat Y. J. et al. (1992) ‘Detection of Latent Human Cytomegalovirus in Organ Tissue and the Correlation with Serological Status’, Transplant International : Official Journal of the European Society for Organ Transplantation, 5: 613–6.doi: 10.1007/978-3-642-77423-2_180 [DOI] [PubMed] [Google Scholar]
- Kurz S. K. et al. (1999) ‘Focal Transcriptional Activity of Murine Cytomegalovirus during Latency in the Lungs’, Journal of Virology, 73: 482–94.doi: 10.1128/JVI.73.1.482-494.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz S. K., and Reddehase M. J. (1999) ‘Patchwork Pattern of Transcriptional Reactivation in the Lungs Indicates Sequential Checkpoints in the Transition from Murine Cytomegalovirus Latency to Recurrence’, Journal of Virology, 73: 8612–22.doi: 10.1128/JVI.73.10.8612-8622.1999/ASSET/E499D82E-79D0-401F-8736-B1786D388FBF/ASSETS/GRAPHIC/JV1090703006.JPEG [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kytö V. et al. (2005) ‘Cytomegalovirus Infection of the Heart Is Common in Patients with Fatal Myocarditis’, Clinical Infectious Diseases, 40: 683–8.doi: 10.1086/427804 [DOI] [PubMed] [Google Scholar]
- Lassalle F. et al. (2016) ‘Islands of Linkage in an Ocean of Pervasive Recombination Reveals Two-speed Evolution of Human Cytomegalovirus Genomes’, Virus Evolution, 2: 1.doi: 10.1093/ve/vew017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limaye A. P., Babu T. M., and Boeckh M. (2020) ‘Progress and Challenges in the Prevention, Diagnosis, and Management of Cytomegalovirus Infection in Transplantation’, Clinical Microbiology Reviews, 34: 1–37.doi: 10.1128/CMR.00043-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüttichau H. R. (2010) ‘The Cytomegalovirus UL146 Gene Product vCXCL1 Targets Both CXCR1 and CXCR2 as an Agonist’, The Journal of Biological Chemistry, 285: 9137.doi: 10.1074/JBC.M109.002774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuel O. et al. (2009a) ‘Impact of Genetic Polymorphisms in Cytomegalovirus Glycoprotein B on Outcomes in Solid-organ Transplant Recipients with Cytomegalovirus Disease’, Clinical Infectious Diseases, 49: 1160–6.doi: 10.1086/605633 [DOI] [PubMed] [Google Scholar]
- ——— et al. (2009b) ‘An Assessment of Donor-to-recipient Transmission Patterns of Human Cytomegalovirus by Analysis of Viral Genomic Variants’, Journal of Infectious Diseases, 199: 1621–8.doi: 10.1086/598952 [DOI] [PubMed] [Google Scholar]
- Mattick C. et al. (2004) ‘Linkage of Human Cytomegalovirus Glycoprotein gO Variant Groups Identified from Worldwide Clinical Isolates with gN Genotypes, Implications for Disease Associations and Evidence for N-terminal Sites of Positive Selection’, Virology, 318: 582–97.doi: 10.1016/j.virol.2003.09.036 [DOI] [PubMed] [Google Scholar]
- Meyer-König U. et al. (1998) ‘Simultaneous Infection of Healthy People with Multiple Human Cytomegalovirus Strains’, Lancet, 352: 1280–1.doi: 10.1016/S0140-6736(05)70487-6 [DOI] [PubMed] [Google Scholar]
- Paraskeva M. et al. (2011) ‘Cytomegalovirus Replication within the Lung Allograft Is Associated with Bronchiolitis Obliterans Syndrome’, American Journal of Transplantation : Official Journal of the American Society of Transplantation and the American Society of Transplant Surgeons, 11: 2190–6.doi: 10.1111/J.1600-6143.2011.03663.X [DOI] [PubMed] [Google Scholar]
- Puchhammer-Stöckl E., and Görzer I. (2011) ‘Human Cytomegalovirus: An Enormous Variety of Strains and Their Possible Clinical Significance in the Human Host’, Future Virology, 6: 259–71.doi: 10.2217/fvl.10.87 [DOI] [Google Scholar]
- Puchhammer-Stöckl E. et al. (2006) ‘Emergence of Multiple Cytomegalovirus Strains in Blood and Lung of Lung Transplant Recipients’, Transplantation, 81: 187–94.doi: 10.1097/01.tp.0000194858.50812.cb [DOI] [PubMed] [Google Scholar]
- Reddehase M. J. et al. (1994) ‘The Conditions of Primary Infection Define the Load of Latent Viral Genome in Organs and the Risk of Recurrent Cytomegalovirus Disease’, The Journal of Experimental Medicine, 179: 185–93.doi: 10.1084/JEM.179.1.185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renzette N. et al. (2011) ‘Extensive genome-wide variability of human cytomegalovirus in congenitally infected infants’, PLoS pathogens, 7: e1001344.doi: 10.1371/journal.ppat.1001344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2013) ‘Rapid intrahost evolution of human cytomegalovirus is shaped by demography and positive selection’, PLoS genetics, 9: e1003735.doi: 10.1371/journal.pgen.1003735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2014) ‘Human cytomegalovirus intrahost evolution-a new avenue for understanding and controlling herpesvirus infections’, Current opinion in virology, 8: 109–115.doi: 10.1016/j.coviro.2014.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro R. V. P. et al. (2022) ‘Ex Vivo Treatment of Cytomegalovirus in Human Donor Lungs Using a Novel Chemokine-based Immunotoxin’, Journal of Heart and Lung Transplantation, 41: 287–97.doi: 10.1016/j.healun.2021.10.010 [DOI] [PubMed] [Google Scholar]
- Schonian U., Crombach M., and Maisch B. (1993) ‘Assessment of Cytomegalovirus DNA and Protein Expression in Patients with Myocarditis’, Clinical Immunology and Immunopathology, 68: 229–33.doi: 10.1006/CLIN.1993.1123 [DOI] [PubMed] [Google Scholar]
- Sijmons S. et al. (2015) ‘High-throughput Analysis of Human Cytomegalovirus Genome Diversity Highlights the Widespread Occurrence of Gene-disrupting Mutations and Pervasive Recombination’, Journal of Virology, 89: 7673–95.doi: 10.1128/JVI.00578-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowmya P., and Madhavan H. N. (2009) ‘Analysis of Mixed Infections by Multiple Genotypes of Human Cytomegalovirus in Immunocompromised Patients’, Journal of Medical Virology, 81: 861–9.doi: 10.1002/jmv.21459 [DOI] [PubMed] [Google Scholar]
- Suárez N. M. et al. (2020) ‘Whole-Genome Approach to Assessing Human Cytomegalovirus Dynamics in Transplant Patients Undergoing Antiviral Therapy’, Frontiers in cellular and infection microbiology, 10: 267.doi: 10.3389/fcimb.2020.00267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2019a) ‘Multiple-Strain Infections of Human Cytomegalovirus with High Genomic Diversity are Common in Breast Milk from Human Immunodeficiency Virus-Infected Women in Zambia’, The Journal of Infectious Diseases, 220: 792–801.doi: 10.1093/infdis/jiz209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2019b) ‘Human Cytomegalovirus Genomes Sequenced Directly from Clinical Material: Variation, Multiple-strain Infection, Recombination, and Gene Loss’, Journal of Infectious Diseases, 220: 781–91.doi: 10.1093/infdis/jiz208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian B. et al. (2019) ‘Evolview V3: A Webserver for Visualization, Annotation, and Management of Phylogenetic Trees’, Nucleic Acids Research, 47: W270–W275.doi: 10.1093/nar/gkz357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunwen C., and Norman D. J. (1988) ‘The Influence of Donor Factors Other than Serologic Status on Transmission of Cytomegalovirus to Transplant Recipients’, Transplantation, 46: 89–93.doi: 10.1097/00007890-198807000-00016 [DOI] [PubMed] [Google Scholar]
- Takahashi M. et al. (2021) ‘Strategies to Prolong Homeostasis of Ex Vivo Perfused Lungs’, The Journal of Thoracic and Cardiovascular Surgery, 161: 1963–73.doi: 10.1016/J.JTCVS.2020.07.104 [DOI] [PubMed] [Google Scholar]
- Tong Y. et al. (2017) ‘Determination of the Biological Form of Human Cytomegalovirus DNA in the Plasma of Solid-organ Transplant Recipients’, Journal of Infectious Diseases, 215: 1094–101.doi: 10.1093/infdis/jix069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vietzen H. et al. (2021) ‘Extent of Cytomegalovirus Replication in the Human Host Depends on Variations of the HLA-E/UL40 Axis’, MBio, 12: 1–12.doi: 10.1128/MBIO.02996-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. Y. et al. (2021) ‘Common Polymorphisms in the Glycoproteins of Human Cytomegalovirus and Associated Strain-Specific Immunity’, Viruses, 13: 1106.doi: 10.3390/v13061106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuhair M. et al. (2019) ‘Estimation of the worldwide seroprevalence of cytomegalovirus: A systematic review and meta-analysis’, Reviews in medical virology, 29: e2034.doi: 10.1002/rmv.2034 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequence data have been deposited in the NCBI Sequence Read Archive under BioProject ID PRJNA803978. Haplotype sequences generated in this study and with identities <98 per cent to publicly available sequences were submitted to GenBank with the Accession No. OM835733–OM835736 (Supplementary Table S4B).