

Abstract
Alternative splicing (AS) plays a key role in cancer: all its hallmarks have been associated with different mechanisms of abnormal AS. The improvement of the human transcriptome annotation and the availability of fast and accurate software to estimate isoform concentrations has boosted the analysis of transcriptome profiling from RNA-seq. The statistical analysis of AS is a challenging problem not yet fully solved. We have included in EventPointer (EP), a Bioconductor package, a novel statistical method that can use the bootstrap of the pseudoaligners. We compared it with other state-of-the-art algorithms to analyze AS. Its performance is outstanding for shallow sequencing conditions. The statistical framework is very flexible since it is based on design and contrast matrices. EP now includes a convenient tool to find the primers to validate the discoveries using PCR. We also added a statistical module to study alteration in protein domain related to AS. Applying it to 9514 patients from TCGA and TARGET in 19 different tumor types resulted in two conclusions: i) aberrant alternative splicing alters the relative presence of Protein domains and, ii) the number of enriched domains is strongly correlated with the age of the patients.
INTRODUCTION
Alternative splicing (AS) is a co- and pot-transcriptional process (1) by which a single pre-mRNA can lead to different mature mRNA (called isoforms or transcripts) by including, excluding, shortening, or lengthening exons and introns. Approximately 90% of the human genes present AS (2–4). In recent years, AS has been related to different disease processes in cancer and other pathologies (5,6). For example, a study indicates that the two isoforms provided by the gene ITSN1 (ITSN1-L and ITSN1-S) perform an opposite function in glioma progression (7). Aberrant AS leads to the expression of unusual mRNA that might produce either non-functional (8,9) or aberrant proteins (10,11).
RNA-Seq is nowadays the method of choice to study AS events. Algorithms to detect AS events –such as EventPointer (12), rMATS (13), SUPPA2 (14) or MAJIQ (15), among others– can be divided into two groups (16): those that find novel events and those that focus on known ones. The ability to discover novel events is, in some cases, a critical decision: novel events can be specific to the study and, therefore, can be considered for further analysis as possible biomarkers or even drivers of the disease. The main drawback of these algorithms is twofold: the computational burden is much larger, and it is difficult to jointly analyze data from disparate experiments. Algorithms that focus on known events—such as SUPPA2—use a reference transcriptome to find out splicing events and the isoform quantifications (provided by pseudo aligners such as Kallisto (17) or Salmon (18)) to quantify the found splicing events. Since the human transcriptome is being profusely annotated, novel events occur less often, and disease-specific events can also be included in the reference. On the other hand, alternative splicing can be also studied by analyzing the differential expression of the isoforms instead of alternative splicing events as it is done by 3D-RNA-Seq (19), Sleuth (20) or CuffDiff (21).
Accurate estimation of differential splicing is an active field of research. The presence of different sources of bias in RNA sequencing, the tiny differences of the corresponding isoforms (a few nucleotides in the alternative 3′ site for instance), or the lack of expression of the studied gene, make its study a challenging problem. For example, MAJIQ corrects the GC bias (22). Kallisto corrects its estimates using a fragment length correction (23) and showed that it also corrects the GC bias. Interestingly, rMATS, even though they tried different correction methods, none of them improved the computation of PSI. Besides, a complete analysis of the biological impact of alternative splicing is still an open question.
In this work, we present a new version of EventPointer (EP) that tries to solve these challenges. EP is an R Bioconductor package to identify AS events that includes either simple (case–control experiment) or complex experimental designs –such as time-course experiments, paired analysis, etc. The first version was developed to study microarrays. The second version (here referred to as EventPointer BAM) also analyzes RNAseq experiments and is able to find novel splicing events. The main improvements of this third version—the focus of this manuscript—are (i) the implementation of an algorithm to identify and state the statistical significance of known events using either Kallisto or Salmon pseudo-aligners, (ii) a bootstrap-based statistical method that provides better sensitivity and specificity than previous methods, (iii) the implementation of a protein domain enrichment analysis to predict functional consequences of the splicing and, finally, (iv) the option to design primers and probes for RT-PCR validation (either TaqMan, SYBR-green or semiquantitative). Also, we improved the speed of several algorithms and fix some minor bugs in the classification of the AS events.
Using a reference transcriptome opens the possibility to reanalyze previous data without the burden to map a large number of samples against the genome. We have studied the enrichment of domains based on splicing for all the samples in TCGA and TARGET (24). In addition to the identification of events differentially spliced, we analyzed the differential presence of protein domains between the tumor and its healthy counterparts. Interestingly, protein domains tend to be downregulated in most adult cancer types, and upregulated in tumors of youngsters and childhood. Besides, depleted protein domains are shared among the different cancer types. On the contrary, upregulated protein domains are usually cancer type-specific.
As a result, the new version of the EventPointer R package aims to provide a comprehensive solution to perform the analysis of AS in sequencing data. It affords estimates, statistics, and functional interpretation of the results of AS analysis. EP is available at Bioconductor. (https://bioconductor.org/packages/release/bioc/html/EventPointer.html)
MATERIALS AND METHODS
Data availability
The HVS dataset is available at the Sequence Read Archive (SRA) under the accession number SRS354082. The CX-4945 dataset is available at Gene Expression Omnibus with the accession number GSE104974.
All code to replicate the results of this work is available in GitHub (https://github.com/JFerrer-B/EventPointer_3.0_replicate).
Identification of known events
EP identifies and categorizes the splicing events based on the splicing graph of a gene. This splicing graph can be generated from the sequence reads (to find events ex novo by EP BAM), or from the provided transcriptome (by EP ST or EP ML) (25). Given the splicing graph, EP defines a splicing event as a triplet of subgraphs (Path 1, Path 2 and Reference Path) (Supplementary Figure S1). The events are classified into 7 main categories (cassette exons, alternative 3′splice site, alternative 5′splice site, intron retention, alternative last exon, alternative first exon, and mutually exclusive exons) (Figure 1A). EP classifies an event by checking the structure of its corresponding subgraph of triplets. Since many splicing events are non-canonical, events that do not match any of these categories are classified as ‘complex events’. For more details of how events are detected and classified see (25) and (Additional file 1). To ease the interpretation of ‘complex events’, EP subclassiflies these events by comparing the structure of its corresponding subgraph of triples with the features of the canonical events (Additional file 1).
Figure 1.

Overview of the new version of EventPointer (EP) complete pipeline. (A) EP identifies and classifies all possible alternative splicing events given a reference transcriptome. Here EP returns a .txt file with the information of all the events and the information of which isoform build up the path of each event (Events build info tables). (B) EP has two alternative pipelines to estimate the value of 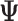 , namely: using only the maximum likelihood isoform expression (Isoform Expression ML Matrix) or harnessing the bootstraps returned by the pseudo-aligner (Isoform Expression* matrices). The former will return a unique matrix with the
, namely: using only the maximum likelihood isoform expression (Isoform Expression ML Matrix) or harnessing the bootstraps returned by the pseudo-aligner (Isoform Expression* matrices). The former will return a unique matrix with the 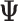 estimates (
estimates (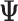 ML matrix) and the later a matrix with the
ML matrix) and the later a matrix with the 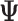 estimate for each bootstrap (
estimate for each bootstrap (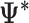 matrices). (C) The statistical significance of the ΔΨ between conditions is estimated based on a bootstrap test. (D) EP provides the option of primers design for PCR validation and the analysis of protein domain affected by splicing.
matrices). (C) The statistical significance of the ΔΨ between conditions is estimated based on a bootstrap test. (D) EP provides the option of primers design for PCR validation and the analysis of protein domain affected by splicing.
PSI (Ψ) computation
EventPointer uses the isoform expression obtained from pseudo-alignment (either Kallisto or Salmon) to compute Ψ. Let us assume that there are N isoforms included in Path 1 and that 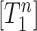 is the expression of the isoform ‘n’. Similarly,
is the expression of the isoform ‘n’. Similarly, 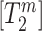 is the expression of the ‘m’ isoform out of the M isoforms that include the Path 2. By definition, the value of Ψ is the quotient of the concentrations of the isoforms that include the Path 1 over the concentrations of the isoforms that either include the Path 1 or the Path 2, i.e.
is the expression of the ‘m’ isoform out of the M isoforms that include the Path 2. By definition, the value of Ψ is the quotient of the concentrations of the isoforms that include the Path 1 over the concentrations of the isoforms that either include the Path 1 or the Path 2, i.e.
The expression to calculate the Ψ is defined by the following equations:
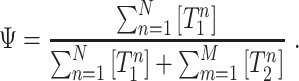 |
(1) |
The denominator of equation 1 is the sum of the concentrations of the isoforms that include Path 1 and Path 2, which in turn, are the isoforms that include the reference path.
The computation of Ψ is extremely fast since it only requires matrix multiplications: the expression matrix and a sparse indicial matrix whose elements state whether an isoform belongs to a path or not. This efficiency in the computation makes it possible to apply bootstrap statistics with a reasonable computational burden.
Statistical analysis based on bootstrap resampling
Bootstrapping is a statistical technique that estimates the distribution of a statistic by using random sampling with replacement. Bootstrapping Ψ values make it possible to estimate the distribution desired contrasts of Ψ (usually ΔΨ).
Thus, the first step consists of the calculation of the expression of the isoforms with Kallisto or Salmon. If the option of the bootstrap in these tools is selected, the expression of the isoforms (maximum likelihood) and their corresponding bootstraps are obtained. This bootstrap resampling (17) can be exploited to model the distribution of ΔΨ. In some studies, only the maximum likelihood estimate of the expression is available (for example, if the FASTQ files are not available). Our recommended pipeline is to use EventPointer ML in this case, and EventPointer ST if bootstrap data from pseudoalignment is available.
Once the isoform expression is provided, the value Ψ is computed as depicted in equation 1. For EventPointer ML the result is a matrix with the estimated Ψ values using maximum-likelihood (ΨML from now on, Figure 1B). For EventPointer ST, the result is an array with the Ψ values using each of the bootstrap estimates (Ψ* for now on, Figure 1B). Given these data, the last steps consist of the estimation of the distribution of the desired contrasts with their corresponding P-values:
Estimation of the distribution of ΔΨ
This process selects bootstrap samples from either ΨML or Ψ* for each condition separately, i.e. all the samples that share identical rows of the design matrix are sampled independently. For each bootstrap sample, the value of the desired contrasts is computed. This process is repeated nb times. For two given design and contrast matrices (D and C) the estimated value of the contrast is,
 |
(2) |
where  is the estimated value of
is the estimated value of  for each nb bootstraps and
for each nb bootstraps and  is the estimated value of the desired contrast.
is the estimated value of the desired contrast.
The distribution of the contrast is modeled by using a generalized lambda distribution (26). The estimation of the four parameters of this distribution (location 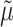 , dispersion
, dispersion 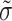 , and shape parameters
, and shape parameters 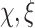 ) is done using the method of the moments to decrease the computational requirements. As a result, the algorithm provides an approximate density function for the contrast under study
) is done using the method of the moments to decrease the computational requirements. As a result, the algorithm provides an approximate density function for the contrast under study
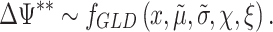 |
(3) |
This parametric approach makes it possible to estimate low P-values without the burden of using a large number of tens of or even hundreds of thousands of bootstrap samples (Figure 1C).
P-value calculation
As we are estimating the distribution of the desired contrast and not the null distribution, we compute the P-value harnessing the duality between the confidence intervals and the hypothesis tests (27). Thus, once the distribution of the contrast is estimated, the P-value can be obtained by focusing on the area of the tails of the distribution. Specifically, the P-value is two times one minus the maximum area of the tails of the density function, i.e.
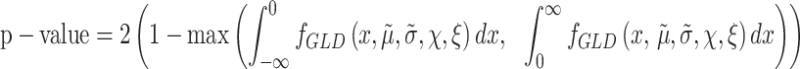 |
(4) |
In the case of using a composite hypothesis, i.e. the null hypothesis is defined by |ΔΨ| < θ, where θ is a threshold, the P-value is estimated focusing on the tails considering absolute values larger than the threshold. In this case,
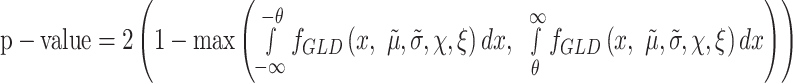 |
(5) |
If 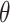 is large and the distribution of the contrast is close to the origin, the integrands are nulls, and the P-value could be larger than 1 (at most 2). The P-value should be clamped to one in these cases, or, as we have implemented, be modified by including a correction factor depending on the area of the null hypothesis. The formula for this correction is:
is large and the distribution of the contrast is close to the origin, the integrands are nulls, and the P-value could be larger than 1 (at most 2). The P-value should be clamped to one in these cases, or, as we have implemented, be modified by including a correction factor depending on the area of the null hypothesis. The formula for this correction is:
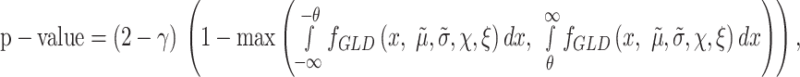 |
(6) |
where
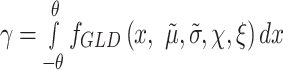 |
(7) |
With this correction, the P-value is bounded between zero and one.
For this computation, EP uses the estimate of the distribution of ΔΨ. This approach is different (but equivalent) to the estimation of the P-values using the null distribution. This approach is equivalent to computing the confidence interval ΔΨ with different α levels and selecting the minimum value α to reject the null hypothesis.
Protein domain enrichment
This analysis aims to evaluate if alternative splicing changes the proportion of splicing events encoding a specific protein domain and state its statistical significance. The approach is similar (but not identical) to a GO enrichment analysis. A positive ΔΨ increases the relative expression of a protein domain if it is included in the isoforms in Path 1. If ΔΨ is positive, and the domain is included in isoforms of path 2, the domain will be depleted and also the path where the protein domain is mapped. Using the isoforms that build each path of the events and the protein domains encoded by each transcript, EP builds an ExD matrix. The dimension of ExD is the number of events times the number of domains. Each entry E×Dij is 1 if domain j is encoded by path 1 of the event i and not by path 2 and is –1 if it is encoded by path 2 and not by path 1. E×Dij is 0 if domain j is encoded by both path 1 and path 2 or by neither of them. In the latter case, the relative presence of the domain will not be affected by splicing.
If the ΔΨ is positive, and the domain is included in Path 1 (the corresponding entry of the E×D matrix is a one), the relative usage of the domain increases. If the entry is –1 the relative usage of the domain decreases. Intuitively, if the relative presence of a domain increases the ΔΨ will be positive for the +1 entries of the ExD matrix and negative for the –1 entries (Figure 2). This will correspond to a positive correlation between the ΔΨ and the group of each event.
Figure 2.

Overview of the protein domain enrichment analysis. (A) Example of how the matrix E×D is built. Protein domain m is encoded only by path 1 of the event x corresponding to a 1 in the matrix E×D. In the second event, protein domain n is only encoded by path 2 resulting in a –1 in the matrix ExD and finally, protein domain p is encoded by both path 1 and path 2 of event z, and therefore, the E×D is 0. Thus, for each protein domain, events can be split into three groups, namely: events where the protein domain is only encoded by the path 2 (–1), only by the path 1 (1), and by both paths or none of them (0). (B) Then, a protein domain will be gained if: (i) events that encoded it by the path 1 have a positive ΔΨ and (ii) if the events that encoded it by the path 2 have a negative ΔΨ (blue boxplots). Therefore, a positive correlation between the ΔΨ and the categories of the events implies a gain of the protein domain. Likewise, a negative correlation implies a loss of it (green boxplots). Thus, a correlation between the ΔΨ and the group of each event is performed. SSF48371 in AML cancer (gain) and SSF50729 in LUAD cancer (loss) corresponding example boxplots are displayed.
The relative presence of the protein domain will decrease if the ΔΨ is positive for domains whose E×D is –1 and negative if E×D is + 1. This will correspond to a negative correlation between ΔΨ and the group of each event (Figure 2).
The statistical P-values of the enrichment are provided by computing a Spearman correlation test between ΔΨ of the events and the three categories of the columns of the E×D matrix.
Primers design
EP implements an algorithm that designs both primers and TaqMan probes to make a quantitative validation of alternative splicing events. The algorithm takes as input the splicing graph created by EP and the specific splicing event. It provides a set of possible groups of primers and TaqMan probes ranked according to a score.
EP creates a directed splicing graph (SG) using the gene structure. In this splicing graph, nodes are the genomic coordinates of the start and the end position of subexons (contiguous regions of the genome that belong to the same transcript). The beginning of the subexons forms the ‘a’ nodes and the end of the subexons forms the ‘b’ nodes. The edges of the graph represent exons if they join an ‘a’ node with a ‘b’ node or junctions if they connect a ‘b’ node with an ‘a’ node (Figure 3). Since ‘a’ nodes are connected by ‘b’ nodes and vice versa, this graph is bipartite. Internally, two additional nodes start and end are included in the graph but not shown in the figure for the sake of simplicity.
Figure 3.

Representation of a direct splicing graph from the structure of a gene. Nodes represent the start and end of subexons (contiguous regions of the genome that belong to the same set of transcripts). Edges that connect ‘a’ nodes with ‘b’ nodes represent subexons while edges that join ‘b’ nodes with ‘a’ nodes denote junctions (or contiguous regions in the exon as in alternative 3′ and 5′ sites). Black, red, and green specify the reference path, path 1 and path 2 respectively of the cassette exon located in exon 2. The numbers above each edge of the splicing graph are equivalent to optimal flux obtained from solving equation 11. The Flux of edge 1b-3a is fixed to 1 to obtain which combination of exons upstream and downstream fulfill the Full Flux Condition. Exons whose flux estimated from equation 11 is equal to the flux imposed in our path of interest are our candidate exons placing the primers. In this example, exon 1 for the upstream primer location, and exons 3 and 6 for the downstream primer location.
The splicing graph can be described with its corresponding incidence matrix. The incidence matrix B is a n × m matrix where n and m are the numbers of vertices and edges respectively, such that bi,j = −1 if the edge ej leaves vertex vi, 1 if it enters vertex vi and 0 otherwise.
The problem of detecting the primers can be split into two subproblems: (i) identify the genomic regions to place the primers (described below), and (ii) identify the actual primers that meet certain characteristics (for which we use primer3). The subproblem of finding suitable regions can be solved for each path of the event independently.
For each path, the (sub)exons where the primers are to be placed must have a minimal length (otherwise the primer cannot be placed). The primers must be placed upstream and downstream of the event. Besides, the candidate exons must fulfill the ‘Full Flux Condition’, i.e. the primer must amplify all the transcripts that traverse the path. It is also desirable that the primers are close enough to the event under study.
The Full Flux Condition problem is solved with a quadratic optimization problem (equation 8) that minimizes the sum of the squared fluxes subject to: (i) all the fluxes are positive, (ii) follow the graph structure and (iii) the studied path has a flux value of one.
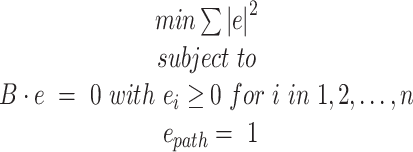 |
(8) |
Figure 3 exemplifies solving the full flux condition problem for a given splicing graph. In this case, the edge between nodes 1b and 3a is imposed to be one. This is equivalent to stating that the concentration of the transcripts that include this edge must be one. After running the optimization problem, some edges have flux equal to one and, other edges have flux smaller than one. Only the exons whose fluxes are 1 are potential candidates to interrogate the selected path. In this case, exons 1, 3 and 6.
Finally, it is also desirable to have the primers as close as possible to the studied event (it is better to place the primer in exon 3 than in exon 6) and the solutions are ranked accordingly.
After filtering out the valid exons to place the primers and solving for the alternate paths, EP sets a figure of merit to rank all the possible exon combinations. This score depends on the number of primers required (two or three) to measure the event, the length of the expected bands (if longer than a certain value will be penalized), the number of expected bands (in some cases, there can be more than two bands in semiquantitative RT-PCR), and the difference in the length of the bands. Taqman probes must be set on the exons of one of the paths and the exons of the reference.
Once the exons are known, primer3 (28) is called to calculate primer sequences and the TaqMan probes (one is placed on the reference and another probe in one of the paths of the event).
RESULTS
Exploiting pseudoaligner bootstraps improves the accuracy of AS analyses
Most algorithms quantify the splicing events in terms of the ‘proportion spliced-in’ (PSI or Ψ) and its statistical analysis relies on the increment of PSI (ΔΨ) between conditions. In a canonical event, Ψ measures the ratio between the expression of the isoforms that include the alternative region (a cassette exon, for example) with the expression of the isoforms that either include or exclude the alternative sequence –considering isoforms that share the reference region of the event. Ψ is zero if none of the isoforms that include the alternative region are expressed and Ψ is one if none of the isoforms that exclude the alternative region are expressed.
The estimate of the statistical distribution of Ψ (or its increment, ΔΨ) depends on many characteristics: the depth of sequencing, the expression of the spliced gene, the length of the regions specific to each of the splicing paths, the type of event, the influence of junction reads –more difficult to map– to state the value of Ψ, the ability to sequence that specific part of the transcriptome, etc. All of them (except the sequencing depth) are specific to each event and are very difficult to model properly. As a result, since each event has different characteristics, results are far from perfect if parametric statistics are used. To circumvent the difficulty of modeling the statistical behavior, EP now implements a bootstrap analysis to assert the statistical significance of the desired contrasts. Both Kallisto and Salmon have the option to provide bootstrap estimations of the isoform expression. EP exploits these bootstrap values to provide the statistical significance of the desired contrast (usually ΔΨ between normal and tumoral samples).
The new EP version implements two alternative pipelines depending on the availability of the bootstrap data provided by pseudo-aligners (Figure 1B). Both pipelines use bootstrap sampling to state the statistical significance of an event. The standard pipeline uses the bootstrap estimates of isoform expression provided by pseudo-aligners (either Salmon or Kallisto) to estimate the distribution of the desired contrasts (EventPointer ST –STandard method–). As the bootstrap data from pseudo-aligners is not always available, EP also implements another statistical analysis based on the Maximum-Likelihood isoform expression estimate (EventPointer ML). EventPointer ST method outperforms EventPointer ML in terms of accuracy, especially if the number of samples is small. The underlying reason for this improvement is that Kallisto or Salmon bootstraps indirectly provide an estimate of the reliability of the measured Ψ. The counterpart of EventPointer ST is that using pseudoalignment's bootstraps is more computing-intensive and is not always available.
Although using alignment's bootstraps is more computing-intensive, it is worth considering this pipeline as the accuracy of predictions improves, especially if the number of samples is small. The underlying reason for this improvement is that Kallisto or Salmon bootstraps indirectly provide an estimate of the reliability of the measured Ψ.
In some cases, despite ΔΨ being statistically significant, its value is very small and has little biological impact. To address this concern, EP mimics the ‘treat’ extension of the limma R package (29) to modify the simple null hypothesis (H0 : ΔΨ = 0) to a composite hypothesis (H0: |ΔΨ| < θ). A proper selection of the threshold provides events more interesting from a biological point of view and, especially if using semiquantitative PCR, easier to validate (see Methods).
EP describes the experiment by using design and contrast matrices. This modeling technique is very versatile as shows the widespread use of limma (30). We have adapted this modeling technique to use bootstrap statistics. As a result, EP achieves more reliable results than state-of-the-art methods. To substantiate these claims, we tested EventPointer against independent real and simulated data.
EP’s accuracy is high under different conditions of sequencing depth and read lengths and excels at shallow coverage
We analyzed the accuracy of EP (both ST and ML, with and without setting a threshold on ΔΨ) using the simulated data of SUPPA2 (14). Specifically, this dataset simulated 554 cassette exons (277 positives and 277 negatives) and 636 alternative splice-site events (318 positives and 318 negatives) between two conditions with three replicates for each condition. The simulations were carried out at different depths (120, 60, 25, and 10 Million (M) of reads) and for different read lengths (25, 50, 75 and 100 nt at a depth of 25 M of reads) using RSEM (31). We augmented the dataset with simulations with very shallow sequencing depth (5, 3, 2, and 1 M reads). Further, we applied SUPPA2, EventPointer BAM, rMATS, and MAJIQ pipelines to the same simulated data and compared its results with the ones obtained with EP. For all methods, we also applied a threshold option for a |ΔΨ| of 0.1. All methods except EventPointer BAM provide this option to compute the statistical significance. Thus, for EventPointer BAM, we set a P-value of 1 for those slicing events with a |ΔΨ| lower than 0.1.
The algorithms are considered to provide a positive detection if the P-value is smaller than 0.05 (as done in the SUPPA2 manuscript). MAJIQ does not return P-values but probabilities of change. We considered that MAJIQ returns a positive if and only if the probability of change is higher than 0.95 and the probability of no change is lower than 0.5.
For both cassettes and alternative 5′ or 3′ events, EventPointer ML (Supplementary Figure S3) is the most sensitive method (higher TPR) but is less specific (1-FPR) than SUPPA2 and EventPointer ST. Thus, harnessing the bootstrap data returned by Kallisto or Salmon (EventPointer ST) we obtain similar sensitivity with higher specificity (Supplementary Figure S3). Both SUPPA2 and EventPointer STP-values are prudently pessimistic: the expected FPR should be around 5% and the measured FPR is well below 5% for most simulations. The poor results in sensitivity of MAJIQ might be caused by the fact that MAJIQ only uses junction reads. On the contrary, the other methods use both exon and junction reads.
Further, when applying a threshold of 0.1 on |ΔΨ|, EventPointer ST improves its specificity keeping a high sensitivity (Supplementary Figure S4). In this case, using the threshold, MAJIQ improves its specificity—it is almost perfect—and the sensitivity does not degrade.
Receiver Operating Characteristic Curve (ROC) and the Precision-Recall Curve (PRC) are used to compare the algorithms without having to set a threshold on the P-value (for MAJIQ we considered the difference between the probability of change and no change to perform the ROC and the PRC). We used both methods to compare cassette and alternative 5′ and 3′ events (Supplementary Figures S5–S15). As expected, all methods perform better at higher depths (Figure 4A-B, Supplementary Figures S14 and S15) and with longer read lengths (Figure 4C-D, Supplementary Figures S18 and S19). SUPPA2 and EP’s outperforms the other methods and have also similar performance for deeply sequenced experiments and for any read length. EP’s approaches outperform SUPPA2 in the case of low depth, especially if a threshold is set on the |ΔΨ| (Supplementary Tables S1–S11). Summarizing, EP provides similar results to SUPPA2 under a wide range of conditions and outperforms SUPPA2 for shallow sequencing: the AUROC and AUPRC for EP methods is similar to SUPPA2 with half the number of reads (Supplementary Figures S16 and S17).
Figure 4.

Receiver operating characteristic curve (ROC curve) and Precision recall curve (PRC) from both simulated cassette exon and alternative splice site events at (A) depth of 1 M, (B) depth of 120 M with reads length of 100 nt and at (C) read length of 25 nt, and (D) read length of 100 nt at a sequencing depth of 25 M. Methods with the threshold variant are depicted in dotted lines. For all methods, the threshold was set to |ΔΨ| = 0.1. In panels B, C and D, a zoom of the left and right up corner of the ROC and PRC curves respectively is displayed.
EP provides versatile statistical modeling for simple and complex experiments
To test how EP works with real data, we analyzed two independent experiments. The first RNA-Seq data set, referred to as HVS, consists of an experiment of prostate cell lines split into two conditions (PC3E and GS689) with three replicates each (Supplementary Table S12). This data was used to show the accuracy of rMATS (13). The second RNA-Seq experiment, referred to as CX-4945, is depicted in (12). In this experiment, three different breast cancer cell lines (five replicates each) were exposed to CX-4945 (five replicates) and control (DMSO) (Supplementary Table S16). The experiment aims at deciphering how CX-4945 affects splicing. CX-4945 is a known casein kinase 2 (CK2) inhibitor (32), which has been proposed as a potential cancer treatment (33) and has been attested to regulate splicing in mammalian cells (34). Supplementary Tables S13 and S17 show the number of events found and reported as significant by each method in both experiments respectively.
We applied EventPointer (both new and previous versions), SUPPA2, rMATS and MAJIQ methods to the HVS data set. In this experiment, 32 out of 34 cassette exons were validated by PCR (13). The new versions of EP (both ST and ML) returned similar results as SUPPA2 (only one event was skipped by EP) in terms of the number of events reported correctly as significant. Moreover, the new version enhanced the results returned by EventPointer BAM, that missed four events and reported five incorrectly. rMATS and MAJIQ found all events. The former failed reporting only two events while the later reported only 26 as significant (with one false positive) and seven as negative (with six false negatives). Moreover, Ψ values corresponding to the PCR was available in (13) and compared with the Ψ estimates of each method by a Pearson correlation. The highest correlations correspond to the methods whose quantification step relies on events rather than in transcripts (rMATS, EventPointer BAM and MAJIQ). As expected, the correlations corresponding to EP and SUPPA2 are almost identical, since both methods use the transcript expression to estimate the value of Ψ (Table 1, Supplementary Figure S20). Supplementary Tables S14 and S15 show a further comparison between all the methods in terms of common events found and reported as significant.
Table 1.
Summary of the PCR-validated events found by each method for both HVS and CX-4945 data sets. TP, FP, TN and FN columns depict True Positives, False Positive, True Negatives and False Negative events respectively. For the HVS Data Set, it is also shown the correlation of the estimates of the Δ Ψ values with the Δ Ψ values using PCR. Since EventPointer and SUPPA2 compute the Ψ values using Kallisto, its estimates are identical and therefore the correlation
| HVS data set | CX-4945 data set | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Method | Events found | TP | FP | TN | FN | ρ(ΔΨPCR vs ΔΨRNAseq) | Events found | TP | FP | TN | FN |
| EventPointer ST | 33/34 | 31 | 2 | 0 | 0 | 0.82 | 20/29 | 18 | 2 | 0 | 0 |
| EventPointer ML | 33/34 | 31 | 2 | 0 | 0 | 0.82 | 20/29 | 18 | 2 | 0 | 0 |
| SUPPA2 | 34/34 | 32 | 2 | 0 | 0 | 0.82 | 17/29 | 8 | 0 | 2 | 7 |
| EventPointer BAM | 30/34 | 25 | 1 | 0 | 4 | 0.95 | 25/29 | 20 | 0 | 2 | 3 |
| rMATS | 34 /34 | 32 | 2 | 0 | 0 | 0.96 | 23/29 | 20 | 2 | 0 | 1 |
| MAJIQ | 34/34 | 26 | 1 | 1 | 6 | 0.92 | 28/29 | 21 | 2 | 0 | 5 |
We also tested the methods with the CX-4945 data set. One of the main advantages of EventPointer over other algorithms is the ability to describe the experiment using design and contrast matrices. Most algorithms (including SUPPA2, rMATS, etc.) only consider case-control experiments. In many cases, the experiment requires a more flexible description of it. In this work, 27 out of 29 selected events were validated by PCR. The new versions of EP (both ST and ML versions) were very sensitive as no positive events was missed. SUPPA2, EventPointer BAM, rMATS and MAJIQ reported as no significant 7, 3, 1 and 5 events, respectively (Table 1). Supplementary Tables S18 and S19 show a further comparison between all the methods in terms of common events found and reported as significant.
This experiment is not a simple case-control study and as SUPPA2, rMATS and MAJIQ cannot accommodate a complex design matrix to study it. To make the comparison, we considered the differences in AS regardless of the cell-type. Other approaches are also possible, such as an independent study of the three cell-lines followed by a side-by-side comparison of all of them, but in this case, there are 3 different case-control tests and therefore, interpretation is more difficult. This simple example illustrates the ability of EP to model a complex experiment using the design and contrast matrices framework.
It should be noted that in both experiments there are only two2 events as true negatives (Table 1) so the specificity of the algorithms is not completely assessed, i.e. an over-sensitive algorithm may appear to perform better.
Despite being based on bootstrap statistics, the whole pipeline for the new version of EP is reasonably fast and is not very memory demanding. For the HVS experiment, EP takes 4 h to pseudoalign the fastq files –using Kallisto–, 30 min to compute the splicing graph and detect the AS events (this step is not inherent to the experiment as it does not depend on the samples), and takes less than 2 minutes to quantify and perform the statistical analysis. The maximum memory requirement is below 4 Gb per core. On the other hand, EP BAM requires 8 h to map the samples –using STAR–, takes 30 h to build the splicing graph and 10 min to detect and quantify the AS events. It takes <1 min to perform the statistical analysis and its maximum memory need is 32GB for mapping reads (if using STAR). The memory requirements to build the splicing graph strongly depends on the samples and we found it difficult to estimate the memory requirements. In our experiments, required around 20 Gb per core but other experiments required even more (Supplementary material, Table S20).
Different analysis methods result in different classifications of events
All the compared methods use a splicing graph to detect and classify the events. The splicing graph is built using the BAM files in the case of rMATS, EP BAM and MAJIQ. In turn, SUPPA2, EP ML and EP ST build the splicing graph from a reference transcriptome. Based on the topology of the splicing graph, the methods classify the events according to the canonical classes. Unfortunately, the splicing graph of many genes is very complex and many splicing events do not match perfectly into the canonical classes. The definition of canonical events changes in the different methods to accommodate these complex events (for example, MAJIQ divides the canonical events (including the cassette events) into two different events—called Local Splice Variants—and provides the probabilities of change and no change of each of the junctions involved in each local splice variant).
In EventPointer, a splicing event is defined as a triplet of subgraphs {Reference Path, Path 1 and Path 2} of the splicing graph. These subgraphs are composed of sets of edges and nodes that share the following characteristics: (i) the flow traversing any edge of each subgraph is identical and (ii) the flow traversing any edge in Ref Path is the sum of the flows traversing Path 1 and Path 2. The detection of the events can be automated using graph theory. In the case of the splicing graph, the flow has a straightforward interpretation: the flow of an edge is the sum of the concentrations of the isoforms that share that edge. As a result, some isoforms share all the nodes and edges in Path 1, other different isoforms share all the nodes and edges in Path 2, and all of them share the nodes and edges in the reference path. An example of a cassette exon illustrates this definition. Path 1 consists of the nodes and edges that correspond to the inclusion of the alternative exon and its junctions. Path 2 is the edge that corresponds to the edge that skips the alternative exon. The Reference paths are, at least, the flanking exons of the skipping exon. (Supplementary Figure S2). This definition of splicing event is quite broad and eases the location of PCR primers in the reference path.
However, the classification of the events detected by EventPointer and SUPPA2 do not always match. The supplementary material illustrates examples of some of these disparities. If EventPointer ST or ML are compared to rMATS, or EventPointer BAM the disparities are even larger, since the splicing graph is different as it depends on the expression of isoforms in the samples, not only on the annotation.
We applied SUPPA2 and EP event detection approaches to the GRCH37.V74 reference transcriptome. EP detects a total of 130 957 events while SUPPA2 detects 179 108 events. EP and SUPPA2 share 90 769 common events. Table 2 shows how these events are classified by both methods. All the events classified as canonical by EP share this classification with SUPPA2 (Supplementary Figure S21). Many of the events classified as canonical in SUPPA2 are classified as complex in EP (see an example in supplementary Figure S22). To facilitate the interpretation of these events, EP has the option of subclassifying complex events according to their resemblance to the characteristics of canonical events. We compared this subclassification with SUPPA2 classification (supplementary Figure S21).
Table 2.
Number of events for each event type detected by EP and SUPPA2 in GRCH37.V74 reference transcriptome
| Type of event | EP | SUPPA2 | Type of event | EP | SUPPA2 |
|---|---|---|---|---|---|
| Alternative 3′ splice Site | 5326 | 16 910 | Alternative 5′ splice site | 3728 | 14 146 |
| Alternative First Exon | 3916 | 4651 | Alternative last exon | 1947 | 2182 |
| Cassette Exon | 8242 | 32 268 | Retained intron | 3939 | 7644 |
| Mutually Exlusive Exons | 79 | 509 | Complex event | 51 113 | 0 |
Supplementary Figures S22 and S25 show events whose classification is different or specific to one of the algorithms. For example, the event shown in Figure S25, is classified as a cassette exon by SUPPA2 and subclassified by EP as two events subtypes (a Cassette Exon plus plus a Retained Intron event). Using semiquantitative PCR for this event and placing the primers in the flanking exons, would produce a counterintuitive result of having 3 different bands in a cassette event.
MAJIQ uses a different approach to classify events: it evaluates the splicing by studying Local Splice Variants (LSV). MAJIQ considers a source or a target (somehow equivalent to the ‘common region’ in EP) that lead to different alternative 3′ splice sites or alternative 5′ splice sites respectively. The former is referred to as Single Source LSV (SS-LSV) and the later as Single Target LSV (ST-LSV). For example, a cassette exon is depicted by a SS-LSV and a ST-LSV. MAJIQ does not classify the events according to the standard categories (15).
EP analyses the domains disrupted by splicing, partially explaining its downstream effects
EP provides a function to identify which protein domains are affected by alternative splicing and perform an enrichment study on them (see Materials and Methods). In a previous work, we studied the effect of the CX-4945 treatment on triple-negative-breast-cancer cell lines. This compound inhibits the Casein Kinase (CK) domain. This domain is mapped to different Interpro (35) protein domains such as IPR011009 (Kinase-like_dom_sf), IPR000719 (Prot_kinase_dom), IPR017441 (Protein_kinase_ATP_BS), and IPR008271 (Ser/Thr_kinase_AS) (36). The result of the domain enrichment analysis reports that these domains have a lower relative presence in samples treated with CX-4945 than in control samples (negative value of the statistic) and that is statistically significant (Table 3, shaded rows).
Table 3.
Top-ranked enriched downregulated protein domains. The four specific protein domains related to the CK kinase family (IPR000719, IPR011009, IPR008271, and IPR017441) are ranked among the top ones. The Statistic column depicts the z-score of the enrichment analysis, the P-value column shows its corresponding P-value, Description column includes a brief description of each protein domain, and the Ranking column the position in the ranking of the most down-regulated protein domains
| PROTEIN DOMAIN | Statistic | P-value | Description | Ranking |
|---|---|---|---|---|
| IPR016024 | -14.18 | 1.51 e-45 | Armadillo-type fold | 1/5783 |
| IPR027417 | -12.95 | 2.80 e-38 | P-loop containing nucleoside triphosphate hydrolase | 2/5783 |
| IPR017986 | -11.23 | 3.35 e-29 | WD40-repeat-containing domain | 3/5783 |
| IPR011993 | -11.12 | 1.08 e-28 | PH domain-like | 4/5783 |
| IPR000719 | -10.75 | 2.07 e-14 | Protein kinase domain | 5/5783 |
| IPR011009 | -10.03 | 2.63 e-13 | Protein kinase-like domain | 6/5783 |
| ··· | ··· | ··· | ··· | ··· |
| IPR008271 | -8.13 | 4.46 e-09 | Serine/threonine-protein kinase, active site | 14/5783 |
| ··· | ··· | ··· | ··· | ··· |
| IPR017441 | -6.35 | 5.55 e-09 | Protein kinase, ATP binding site | 23/5783 |
In addition to these CK-related domains, Table 3 reports also other downregulated domains that were even more significant.Table Interestingly, IPR016024 (Armadillo-type fold) aims to bind large proteins and is known to be related with the regulation pathways of CK2A (37). IPR027417 (P-loop) which frequently appears in multiple nucleoside-binding protein folds, appears in MBNL1 protein (breast cancer metastasis suppressor) (38,39) and in pathways where CK2A is involved (40). IPR017986 (WD40-repeat) was found to act as protein-DNA interaction. Besides, WD40-repeat-containing domain appears in several oncoproteins from breast cancer cells (41) and it is altered in cells treated with CK2 inhibitors (42). Finally, IPR011993 is known to be related to lipid binding (35) and it is included in AKT—oncogene related to breast cancer and part of the CK2 signaling pathway (43,44). On the contrary, in this case the upregulated protein domains are less significant than the downregulated ones, suggesting that alternative splicing in this experiment is associated with the loss of protein domains—and likely, the corresponding functions (Supplementary Table S21).
Protein domains affected by alternative splicing in TCGA and TARGET are related to aging
The domain enrichment functionality was also used to analyze the impact of alternative splicing on protein domains for The Cancer Genome Atlas (TCGA) and Therapeutically Applicable Research to Generate Effective Treatments (TARGET) datasets. Specifically, for each type of cancer, we compared the pattern of splicing between normal and cancer samples and applied our protein domain enrichment analysis. We excluded cancer types with less than six normal samples (Supplementary Table S22). Only 3 TARGET cancer types (acute myeloid leukemia, AML; Wilms tumor, WT and rhabdoid timor, RT) remained after processing due to the absence of normal samples in the others.
In all, we analyzed 991 superfamilies (45) obtained from biomaRt (46) ensuring at least one AS event is related to each of them. We selected a local false discovery rate threshold of 0.1 to consider that a domain gained or lost presence in the tumor samples. This threshold is equivalent to a FDR of 4.5%.
517 superfamilies did not change their presence between normal and tumor samples across any type of cancer. All cancer types but THCA, WT, AML and RT present more downregulated than upregulated superfamilies (Figure 5, supplementary Table S22) which may indicate that one of the main effects of alternative splicing is the loss of functionality of the genes by transcribing isoforms that codify non-functional proteins or, directly, do not codify proteins (9,47). Conversely, the four remaining cancer types—THCA, WT, AML and RT—have more up-regulated superfamily domains. These differences may be related to aging: the TARGET dataset—WT, AML and RT—contains information on pediatric patients whilst TCGA dataset contains information on adult cancers. Interestingly, patients with THCA in the TCGA cohort are significantly younger than in any other cancer (the maximum P-value of pairwise comparisons using a one-tailed Wilcoxon.test smaller than 0.0048, Supplementary Table S23). These results suggest that cancer behavior is different in children and adults (48). Supplementary Figure S26 shows boxplots that relate the ratio between upregulated and downregulated domains number with the age of the patients for each cancer type and shows that there is a trend that relates aging and downregulation of protein domains.
Figure 5.

UpSet plot of the intersection of upregulated (panel A) and downregulated (panel B) superfamilies in 19 cancer types. The dark bar plot on the left shows the number of upregulated and downregulated superfamilies for each type of cancer respectively. The dot-matrix represents the different intersections. In A, sky blue: intersections of superfamilies upregulated in 1 or 2 cancer types, dark blue: intersections of superfamilies upregulated in at least three cancer types and up to 12. In B, green: intersection of superfamilies downregulated in one or two cancer types, orange: intersection of superfamilies downregulated in at least three cancer types and up to 12, pink: superfamilies downregulated in 12 or more cancer types. In both panels A and B, the colored bar chart at the top represents the number of superfamilies that each intersection contains. KICH: Kidney Chromophobe, BLCA: Bladder Urothelial Carcinoma, LUSC: Lung squamous cell carcinoma, READ: Rectum adenocarcinoma, KIRC: Kidney renal papillary cell carcinoma, COAD: Colon adenocarcinoma, HNSC: Head and Neck squamous cell carcinoma, KIRP: Kidney renal papillary cell carcinoma, LUAD: Lung adenocarcinoma, LIHC: Liver hepatocellular carcinoma, UCEC: Uterine Corpus Endometrial Carcinoma, PRAD: Prostate adenocarcinoma, STAD: Stomach adenocarcinoma, ESCA: Esophageal carcinoma, BRCA: Breast invasive carcinoma, THCA: Thyroid carcinoma, WT: Wilms Tumor, RT: Rhabdoid Tumor, AML: Acute Myeloid Leukemia.
We distinguished two cohorts: the adult cohort that includes all the TCGA samples but THCA, and the young cohort that includes the TARGET samples and THCA. The adult cohort, share several downregulated superfamilies. These findings are coherent with previous work done in (47) where they studied alternative splicing in several cancer sites and noticed the highly recurrent effect of protein domain losses. Their work compares these losses produced by alternative splicing to similar effects produced by somatic mutations.
In many superfamilies, the description is not sufficiently informative to hypothesize the functional implications of gaining or losing a domain. To guess the functional implications, we run a GO enrichment analysis (using a hypergeometric test (49), additional material) using the genes with any isoform annotated to each family. The predicted functions are the GO categories with a corresponding q-value lower than 0.1. GO redundancy was removed using the R package GOxploreR (50). From the final list, the top 10 GO terms with most significant P-valuess were selected. 8 different superfamilies were downregulated for all cancer types in the adult cohort (Additional File 2). Some relationship observed are already known and are related with cancer –even though these relationships might not be exclusive for cancer–, for example, SSF46966 (‘Spectrin repeat superfamily’), and SSF47031(‘Second domain of FERM superfamily’) are related to spectrin binding that is related to cytoskeleton proteins and growth factor signaling and its down-regulation is associated with most tumors and also common to other diseases such as hemolytic anemia (51,52). SSF48371(‘ARM repeat superfamily’) is related to the phosphatidylinositol 3-kinase pathway which is involved in proliferation, growth, and apoptosis (53). SSF49899 (‘Concanavalin A-like lectins/glucanases superfamily’), and SSF57196 (‘EGF/Laminin superfamily’) are related with the membrane which degradation is related with tumor invasion, tumor cell growth, and angiogenesis (54). SSF50729 (‘PH domain-like superfamily’) is related to the GTPase activity which is related to the RAS pathway (55).
On the contrary, the young cohort only shared 2 upregulated superfamilies (Additional File 3). It seems that upregulated superfamilies are cancer-specific (Additional file 4). From these families, some altered functions are associated with gene expression including epigenetic and transcriptomic alteration of chromatin accessibility and protein alterations due to phosphorylation. These alterations occur in the benefit of cell growth and internal organization for cell movement and can also be associated with cancer hallmarks (56–60), suggesting a different focus for cancer development if compared to the adult cohort.
Finally, we analyzed if there is any type of event leading to upregulated or downregulated domains in each cancer type. We observed that the most influential type of events are Retained intron –whose importance in cancer is already known (61)–, Alternative 3′ splice site and mutually exclusive exons. Interestingly, these types of events are related to downregulated domains in most types of cancers whilst they are only related to upregulated domains in the young cohort (Supplementary Figures S28 and S29).
We run the same enrichment analysis with Interpro domains and, as expected, the results were coherent with the previous findings (Supplementary Figure S27).
EventPointer provides primers and TaqMan probe sequences for the detection and validation of alternative splicing events
EP provides primers and TaqMan probes for PCR validation. The selection of the regions to place primers and TaqMan probes for validation is not straightforward. The primers on these regions must amplify only the transcripts involved in the event under study and this task may be quite involved in complex events. Similarly, one of the Taqman probes must be designed so that it interrogates only the transcripts in one of the paths (either Path 1 or Path 2) and the other probe is selected to interrogate all the isoforms in the event (usually in the reference path). With this selection of probes and primers, Ψ is the quotient of the expression between the signal of these two probes.
The definition of a splicing event in EP includes a ‘common region’ of the genome that shares the set of transcripts that form the event. This reference path makes it possible, at least theoretically, to validate all the events detected by EP using PCR as primers and probes can be placed in either path 1, path 2 or the reference.
EP ranks the regions to place the primers according to their suitability. Once the regions are selected, Primer3 (28) is used to compute the primers and probes. In some cases, the algorithm does not provide a result since no region meets the requirement of both EP and Primer3.
We have applied our primer design algorithm to the events found by EP in the GRCH37.V74 reference transcriptome. Primer3 provided design primers for 90,993 events (69%) out of the 130 957. In turn, the algorithm also found Taqman probes in 80283 events (88%) of them.
We applied EP’s primers design method to the 34 events validated in (13). EP detected 33 out of the 34 events (Table 1) and was able to design primers for 31 out of the 33 events. The primers proposed by EP and the primers used in (13) are placed on the same exons. Supplementary data file 5 includes the table with the primers used in (13) and the ones proposed by EP. It also includes an image from IGV showing where the primers are located of both rMATS and EP.
DISCUSSION
Previous versions of EventPointer were focused either on microarray data to find known events or RNA-seq to detect novel events (using as input the BAM files and the reconstructed splicing graph). The new version, described here, fills the gap to detect known events in RNA-seq using as input the concentrations from pseudo-aligners such as Kallisto or Salmon.
It could be argued that focusing on known events is a step back. We think that this is not the case: in many cases, the FASTQ or the BAM files from a study are not available (or have restricted access) because of privacy reasons and the previous EP version simply cannot be applied. Moreover, since the human transcriptome is being profusely annotated, novel events occur less often, and disease-specific events can also be included in the reference. Besides, the fact of sharing a common transcriptome makes it possible to compare results from different studies or to perform a meta-analysis. If this were not the case, matching the events from different samples is not trivial and many of the events would be lost in the translation. In an extreme case, the integration of different experiments could require reanalyzing all of them.
EP describes the experiments using design and contrast matrices. In some circumstances, case-control modeling can be adapted to study a more complex experiment. However, this adaptation implies a lack of statistical power since the number of samples for each sub-study is smaller than the initial one. The design-contrast matrix paradigm encompasses a larger number of experiments if compared to case-control modeling. We have developed a novel bootstrap statistical method to interrogate experiments described by design and contrast matrices.
We have compared the new EP version with the old EP version, SUPPA2, rMATS and MAJIQ algorithms using simulated data (generated by SUPPA2) and PCR validated events (for rMATS and the previous version of EP). The results show that, in the simulated data, both EP and SUPPA2 have high accuracy with different depth and read length conditions with an edge for EP if the sequencing depth is very shallow. Furthermore, although using bootstraps data from pseudo-alignment (EP ST) is more computing-intensive, it is worth considering this pipeline as the accuracy of predictions improves. EP includes the possibility of setting a threshold in the null hypothesis (mimicking the ‘treat’ function of the limma package) and results improve both in sensitivity and specificity. This threshold improves the behavior of other algorithms.
Since EP performance for shallow sequencing is remarkable, it would be interesting to study its application for single-cell sequencing in which the number of reads per cell is very small. Although, the small number of reads per cell makes it difficult to perform a transcriptome-wide study of splicing, using pseudo-bulk techniques (for example using DESeq2 (62)) this analysis could be achieved especially for well-expressed genes.
The correlation of PSI between the estimates of algorithms based on local analysis (rMATS, EP BAM and MAJIQ) and the estimate using PCR is almost perfect (above 0.9 for all the algorithms). Isoform-based algorithms (SUPPA2, EP ST, and EP ML) also have a very good correlation (above 0.8) but not as good as the previous ones. It seems, that the noise induced by other parts of the gene in the isoform concentration estimate negatively affects the estimation of PSI for local events. Nevertheless, the simulations show that isoform-based algorithms detect splicing events with higher sensitivity and specificity.
EP studies splicing functional impact using a protein domain enrichment analysis. It includes the PFAM, Interpro, or Superfamily categorizations, but could also be extended to other transcript-annotated information. We have applied this enrichment analysis to a previous experiment (12) and correctly identified altered functions known by literature, reporting 4 treatment-related Interpro domains as downregulated. We also analyzed the enrichment on TCGA and TARGET using the Superfamily and Interpro annotations. Both datasets showed striking differences: in TCGA—except THCA—protein domains tend to be depleted in cancer conditions and in TARGET, the domains tend to be enriched. We hypothesized that these differences are related to the age of the patients. The downregulated superfamilies common to the studied TCGA cancer types—except THCA—are related to cancer hallmarks such as extracellular organization, cell movement, membrane signaling, among others. Upregulated families in TARGET and THCA are also related with the hallmarks of cancer. Nevertheless, these down- and up-regulate superfamilies are also related to other diseases suggesting that these relationships are not exclusive for cancer.
Lastly, RT-PCR and TaqMan assays still stand as the gold-standard approach for validating alternative splicing events. EP eases this task by providing an algorithm that designs primers and probes for them. EP uses a specific definition of splicing events that makes it possible this automatic selection.
Summarizing, EventPointer analyzes AS both for case-control and complex experiments and includes the option of finding novel events. EP exploits the bootstrap estimates of isoform expression provided by pseudo-aligners. EP also includes the option of a domain analysis of proteins affected by AS and an algorithm for the design of primers for PCR validation. Thus, EP provides a one-step solution to perform the analysis of AS integrated into an R package and is available via Bioconductor.
DATA AVAILABILITY
EventPointer is publicly available as a Bioconductor R package (https://bioconductor.org/packages/release/bioc/html/EventPointer.html). The HVS dataset is available at the Sequence Read Archive (SRA) under theaccession number SRS354082. The CX-4945 dataset is available at Gene Expression Omnibus with the accession number GSE104974. Code to download the data and run the EventPonter pipeline are available at Code Ocean capulse (doi: 10.24433/CO.6711051.v1).
Supplementary Material
ACKNOWLEDGEMENTS
Author contributions: Development of the statistical methods based on bootstrap technique: D.O., J.A.F., F.C. and A.R.; validation with simulated and experimental data: J.A.F., and A.R.; development of the protein domain enrichment method: J.A.F. and A.R.; analysis and interpretation of the protein domain enrichment: M.G., J.A.F., C.C. and A.R.; development of the primers design method: P.S., J.A.F. and A.R.; all authors contribute to the writing and or revision of the manuscript. All authors read and approved the final manuscript.
Contributor Information
Juan A Ferrer-Bonsoms, Biomedical Engineering and Science Department, TECNUN, Universidad de Navarra, San Sebastián, Spain.
Marian Gimeno, Biomedical Engineering and Science Department, TECNUN, Universidad de Navarra, San Sebastián, Spain.
Danel Olaverri, Biomedical Engineering and Science Department, TECNUN, Universidad de Navarra, San Sebastián, Spain.
Pablo Sacristan, Biomedical Engineering and Science Department, TECNUN, Universidad de Navarra, San Sebastián, Spain.
César Lobato, Biomedical Engineering and Science Department, TECNUN, Universidad de Navarra, San Sebastián, Spain.
Carlos Castilla, Biomedical Engineering and Science Department, TECNUN, Universidad de Navarra, San Sebastián, Spain.
Fernando Carazo, Biomedical Engineering and Science Department, TECNUN, Universidad de Navarra, San Sebastián, Spain.
Angel Rubio, Biomedical Engineering and Science Department, TECNUN, Universidad de Navarra, San Sebastián, Spain.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
Editor project in the Accelerator Award Programme; Elkartek programme of the Basque Government [KK-2020/00008, F.J.P.]; Synlethal project [PIBA_2020_1_0055].
Conflict of interest statement. None declared.
REFERENCES
- 1. Drexler H.L., Choquet K., Churchman L.S.. Splicing kinetics and coordination revealed by direct nascent RNA sequencing through nanopores. Mol. Cell. 2020; 77:985–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pan Q., Shai O., Lee L.J., Frey B.J., Blencowe B.J.. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008; 40:1413–1415. [DOI] [PubMed] [Google Scholar]
- 3. Johnson J.M., Castle J., Garrett-Engele P., Kan Z., Loerch P.M., Armour C.D., Santos R., Schadt E.E., Stoughton R., Shoemaker D.D.. Genome-wide survey of human alternative Pre-mRNA splicing with exon junction microarrays. Science. 2003; 302:2141–2144. [DOI] [PubMed] [Google Scholar]
- 4. Black D.L. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 2003; 72:291–336. [DOI] [PubMed] [Google Scholar]
- 5. Oltean S., Bates D.O.. Hallmarks of alternative splicing in cancer. Oncogene. 2014; 33:5311–5318. [DOI] [PubMed] [Google Scholar]
- 6. Feng H., Li T., Zhang X.. Characterization of kinase gene expression and splicing profile in prostate cancer with RNA-Seq data. BMC Genomics. 2018; 19:564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shao Y., Chong W., Liu X., Xu Y., Zhang H., Xu Q., Guo Z., Zhao Y., Zhang M., Ma Y.et al.. Alternative splicing-derived intersectin1-L and intersectin1-S exert opposite function in glioma progression. Cell Death. Dis. 2019; 10:431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pickrell J.K., Pai A.A., Gilad Y., Pritchard J.K.. Noisy splicing drives mRNA isoform diversity in human cells. PLos Genet. 2010; 6:e1001236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tress M.L., Abascal F., Valencia A.. Alternative splicing may not be the key to proteome complexity. Trends Biochem. Sci. 2017; 42:98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Himeji D., Horiuchi T., Tsukamoto H., Hayashi K., Watanabe T., Harada M.. Characterization of caspase-8L: a novel isoform of caspase-8 that behaves as an inhibitor of the caspase cascade. Blood. 2002; 99:4070–4078. [DOI] [PubMed] [Google Scholar]
- 11. Ghadie M.A., Lambourne L., Vidal M., Xia Y.. Domain-based prediction of the human isoform interactome provides insights into the functional impact of alternative splicing. PLoS Comput. Biol. 2017; 13:e1005717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Romero J.P., Ortiz-Estévez M., Muniategui A., Carrancio S., De Miguel F.J., Carazo F., Montuenga L.M., Loos R., Pío R., Trotter M.W.B.et al.. Comparison of RNA-seq and microarray platforms for splice event detection using a cross-platform algorithm. BMC Genomics. 2018; 19:703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shen S., Park J.W., Lu Z.X., Lin L., Henry M.D., Wu Y.N., Zhou Q., Xing Y.. rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:E5593–E5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Trincado J.L., Entizne J.C., Hysenaj G., Singh B., Skalic M., Elliott D.J., Eyras E.. SUPPA2: fast, accurate, and uncertainty-aware differential splicing analysis across multiple conditions. Genome Biol. 2018; 19:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vaquero-Garcia J., Barrera A., Gazzara M.R., Gonzalez-Vallinas J., Lahens N.F., Hogenesch J.B., Lynch K.W., Barash Y.. A new view of transcriptome complexity and regulation through the lens of local splicing variations. Elife. 2016; 5:e11752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Carazo F., Romero J.P., Rubio A.. Upstream analysis of alternative splicing: a review of computational approaches to predict context-dependent splicing factors. Brief. Bioinform. 2019; 20:1358–1375. [DOI] [PubMed] [Google Scholar]
- 17. Bray N.L., Pimentel H., Melsted P., Pachter L.. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016; 34:525–527. [DOI] [PubMed] [Google Scholar]
- 18. Patro R., Duggal G., Love M.I., Irizarry R.A., Kingsford C.. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods. 2017; 14:417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Guo W., Tzioutziou N.A., Stephen G., Milne I., Calixto C.P.G., Waugh R., Brown J.W.S., Zhang R.. 3D RNA-seq: a powerful and flexible tool for rapid and accurate differential expression and alternative splicing analysis of RNA-seq data for biologists. RNA Biology. 2021; 18:1574–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pimentel H., Bray N.L., Puente S., Melsted P., Pachter L.. Differential analysis of RNA-seq incorporating quantification uncertainty. Nat. Methods. 2017; 14:687–690. [DOI] [PubMed] [Google Scholar]
- 21. Trapnell C., Hendrickson D.G., Sauvageau M., Goff L., Rinn J.L., Pachter L.. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013; 31:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Risso D., Schwartz K., Sherlock G., Dudoit S.. GC-content normalization for RNA-Seq data. BMC Bioinformatics. 2011; 12:480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Roberts A., Trapnell C., Donaghey J., Rinn J.L., Pachter L.. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 2011; 12:R22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Grossman R.L., Heath A.P., Ferretti V., Varmus H.E., Lowy D.R., Kibbe W.A., Staudt L.M.. Toward a Shared Vision for Cancer Genomic Data. N. Engl. J. Med. 2016; 375:1109–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Romero J.P., Muniategui A., De Miguel F.J., Aramburu A., Montuenga L., Pio R., Rubio A.. EventPointer: an effective identification of alternative splicing events using junction arrays. BMC Genomics. 2016; 17:467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chalabi Y., Diethelm W., Scott D.J.. Flexible distribution modeling with the generalized lambda distribution. Munich Pers. RePEc Arch. 2012; https://mpra.ub.uni-muenchen.de/43333/3/MPRA_paper_43333.pdf. [Google Scholar]
- 27. Panaretos V.M. Confidence intervals for model parameters. Statistics for Mathematicians. 2016; 131–150. [Google Scholar]
- 28. Untergasser A., Cutcutache I., Koressaar T., Ye J., Faircloth B.C., Remm M., Rozen S.G.. Primer3—new capabilities and interfaces. Nucleic Acids Res. 2012; 40:e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mccarthy D.J., Smyth G.K.. Testing significance relative to a fold-change threshold is a TREAT. Bioinformatics. 2009; 25:765–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K.. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015; 43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li B., Dewey C.N.. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011; 12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Siddiqui-Jain A., Drygin D., Streiner N., Chua P., Pierre F., O’Brien S.E., Bliesath J., Omori M., Huser N., Ho C.et al.. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010; 70:10288–10298. [DOI] [PubMed] [Google Scholar]
- 33. Chon H.J., Bae K.J., Lee Y., Kim J.. The casein kinase 2 inhibitor, CX-4945, as an anti-cancer drug in treatment of human hematological malignancies. Front. Pharmacol. 2015; 6:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim H., Choi K., Kang H., Lee S.Y., Chi S.W., Lee M.S., Song J., Im D., Choi Y., Cho S.. Identification of a novel function of CX-4945 as a splicing regulator. PLoS One. 2014; 9:94978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Blum M., Chang H.-Y., Chuguransky S., Grego T., Kandasaamy S., Mitchell A., Nuka G., Paysan-Lafosse T., Qureshi M., Raj S.et al.. The interpro protein families and domains database: 20 years on. Nucleic Acids Res. 2021; 49:D344–D354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bateman A. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019; 47:D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nitta R.T., Gholamin S., Feroze A.H., Agarwal M., Cheshier S.H., Mitra S.S., Li G.. Casein kinase 2α regulates glioblastoma brain tumor-initiating cell growth through the β-catenin pathway. Oncogene. 2015; 34:3688–3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen C.-J., Liu D.-Z., Yao W.-F., Gu Y., Huang F., Hei Z.-Q., Li X.. Identification of key genes and pathways associated with neuropathic pain in uninjured dorsal root ganglion by using bioinformatic analysis. J. Pain Res. 2017; 10:2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fish L., Pencheva N., Goodarzi H., Tran H., Yoshida M., Tavazoie S.F.. Muscleblind-like 1 suppresses breast cancer metastatic colonization and stabilizes metastasis suppressor transcripts. Genes Dev. 2016; 30:386–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Battistutta R., Cozza G., Pierre F., Papinutto E., Lolli G., Sarno S., Obrien S.E., Siddiqui-Jain A., Haddach M., Anderes K.et al.. Unprecedented selectivity and structural determinants of a new class of protein kinase CK2 inhibitors in clinical trials for the treatment of cancer. Biochemistry. 2011; 50:8478–8488. [DOI] [PubMed] [Google Scholar]
- 41. Chen Y., Wei H., Liu Y., Zheng S.. Promotional effect of microRNA-194 on breast cancer cells via targeting F-box/WD repeat-containing protein 7. Oncol. Lett. 2018; 15:4439–4444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li X., Zhao J., Xiong X., Li Y., Liu X., Wang T., Zhang H., Jiao Y., Jiang J., Zhang H.et al.. Hepatic F-box protein FBXW7 maintains glucose homeostasis through degradation of fetuin-A. Diabetes. 2018; 67:818–830. [DOI] [PubMed] [Google Scholar]
- 43. Barnett S.F., Defeo-Jones D., Fu S., Hancock P.J., Haskell K.M., Jones R.E., Kahana J.A., Kral A.M., Leander K., Lee L.L.et al.. Identification and characterization of pleckstrin-homology-domain-dependent and isoenzyme-specific akt inhibitors. Biochem. J. 2005; 385:399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Roskoski R. Signaling by kit protein-tyrosine kinase - The stem cell factor receptor. Biochem. Biophys. Res. Commun. 2005; 337:1–13. [DOI] [PubMed] [Google Scholar]
- 45. Gough J., Karplus K., Hughey R., Chothia C.. Assignment of homology to genome sequences using a library of hidden markov models that represent all proteins of known structure. J. Mol. Biol. 2001; 313:903–919. [DOI] [PubMed] [Google Scholar]
- 46. Durinck S., Spellman P.T., Birney E., Huber W.. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009; 4:1184–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Climente-González H., Porta-Pardo E., Godzik A., Eyras E.. The functional impact of alternative splicing in cancer. Cell Rep. 2017; 20:2215–2226. [DOI] [PubMed] [Google Scholar]
- 48. Vakkila J., Jaffe R., Michelow M., Lotze M.T.. Pediatric cancers are infiltrated predominantly by macrophages and contain a paucity of dendritic cells: a major nosologic difference with adult tumors. Clin. Cancer Res. 2006; 12:2049–2054. [DOI] [PubMed] [Google Scholar]
- 49. Rivals I., Personnaz L., Taing L., Potier M.C.. Enrichment or depletion of a GO category within a class of genes: which test. Bioinformatics. 2007; 23:401–407. [DOI] [PubMed] [Google Scholar]
- 50. Manjang K., Tripathi S., Yli-Harja O., Dehmer M., Emmert-Streib F.. Graph-based exploitation of gene ontology using GOxploreR for scrutinizing biological significance. Sci. Rep. 2020; 10:16672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yang P., Yang Y., Sun P., Tian Y., Gao F., Wang C., Zong T., Li M., Zhang Y., Yu T.et al.. βII spectrin (SPTBN1): biological function and clinical potential in cancer and other diseases. Int. J. Biol. Sci. 2020; 17:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Izdebska M., Zielińska W., Hałas-Wiśniewska M., Grzanka A.. Involvement of actin and actin-binding proteins in carcinogenesis. Cells. 2020; 9:2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sawyers C.L., Vivanco I.. The phosphatidylinositol 3-Kinase-AKT pathway in human cancer. Nat. Rev. Cancer. 2002; 2:489–501. [DOI] [PubMed] [Google Scholar]
- 54. Engbring J.A., Kleinman H.K.. The basement membrane matrix in malignancy. J. Pathol. 2003; 200:465–470. [DOI] [PubMed] [Google Scholar]
- 55. Maertens O., Cichowski K.. An expanding role for RAS GTPase activating proteins (RAS GAPs) in cancer. Adv. Biol. Regul. 2014; 55:1–14. [DOI] [PubMed] [Google Scholar]
- 56. Kurdistani S.K. Histone modifications as markers of cancer prognosis: a cellular view. Br. J. Cancer. 2007; 97:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bywater M.J., Pearson R.B., McArthur G.A., Hannan R.D.. Dysregulation of the basal RNA polymerase transcription apparatus in cancer. Nat. Rev. Cancer. 2013; 13:299–314. [DOI] [PubMed] [Google Scholar]
- 58. Resnik J.L., Reichart D.B., Huey K., Webster N.J.G., Seely B.L.. Elevated insulin-like growth factor i receptor autophosphorylation and kinase activity in human breast cancer. Cancer Res. 1998; 58:1159–1164. [PubMed] [Google Scholar]
- 59. Wolf K., Wu Y.I., Liu Y., Geiger J., Tam E., Overall C., Stack M.S., Friedl P.. Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat. Cell Biol. 2007; 9:893–904. [DOI] [PubMed] [Google Scholar]
- 60. Zoellner H., Chami B., Kelly E., Moore M.A.S.. Increased cell size, structural complexity and migration of cancer cells acquiring fibroblast organelles by cell-projection pumping. PLoS One. 2019; 14:e0224800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wong J.J.L., Schmitz U.. Intron retention: importance, challenges, and opportunities. Trends Genet. 2022; 38:789–792. [DOI] [PubMed] [Google Scholar]
- 62. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The HVS dataset is available at the Sequence Read Archive (SRA) under the accession number SRS354082. The CX-4945 dataset is available at Gene Expression Omnibus with the accession number GSE104974.
All code to replicate the results of this work is available in GitHub (https://github.com/JFerrer-B/EventPointer_3.0_replicate).
EventPointer is publicly available as a Bioconductor R package (https://bioconductor.org/packages/release/bioc/html/EventPointer.html). The HVS dataset is available at the Sequence Read Archive (SRA) under theaccession number SRS354082. The CX-4945 dataset is available at Gene Expression Omnibus with the accession number GSE104974. Code to download the data and run the EventPonter pipeline are available at Code Ocean capulse (doi: 10.24433/CO.6711051.v1).