

Abstract
Increased neoantigens in hypermutated cancers with DNA mismatch repair deficiency (dMMR) are proposed as the major contributor to the high objective response rate in anti-PD-1 therapy. However, the mechanism of drug resistance is not fully understood. Using tumor models defective in the MMR gene Mlh1 (dMLH1), we show that dMLH1 tumor cells accumulate cytosolic DNA and produce IFN-β in a cGAS-STING-dependent manner, which renders dMLH1 tumors slowly progressive and highly sensitive to checkpoint blockade. In neoantigen-fixed models, dMLH1 tumors potently induce T-cell priming and lose resistance to checkpoint therapy independent of tumor mutational burden. Accordingly, loss of STING or cGAS in tumor cells decreases tumor-infiltration of T cells and endows resistance to checkpoint blockade. Clinically, down-regulation of cGAS/STING in human dMMR cancers correlates with poor prognosis. We conclude that DNA sensing within tumor cells is essential for dMMR-triggered anti-tumor immunity. This study provides new mechanisms and biomarkers for anti-dMMR-cancer immunotherapy.
Keywords: Cytosolic DNA, mismatch repair, MLH1, DNA sensing, cGAS, STING, checkpoint blockade, MSI, T cell-infiltration, Cancer
Graphical Abstract
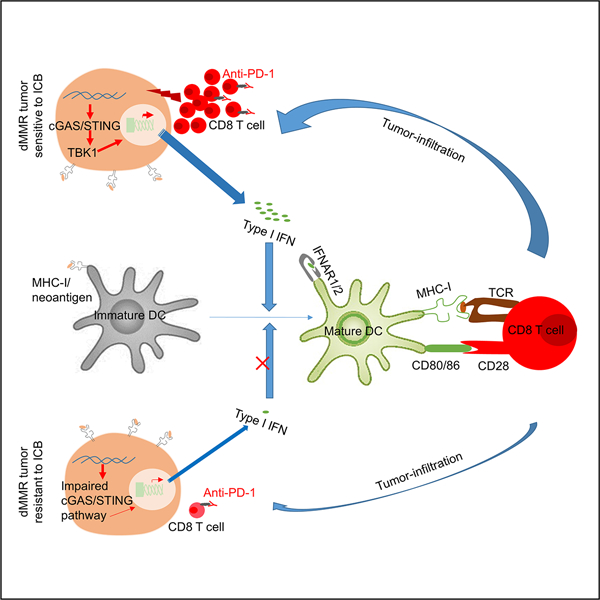
eTOC Blurb:
About 50% of patients with dMMR cancers are objectively responsive to immunotherapy. In addition to neoantigens, Lu et al find that dMMR-mediated cytosolic DNA sensing by cGAS-STING pathway in tumor cells contributes to such clinical benefits, while impaired expression of cGAS-STING pathway associates with drug resistance.
Introduction
The DNA mismatch repair (MMR) pathway maintains DNA-replication fidelity, and deficiency of MMR (dMMR) is the initiating event in a wide range of cancer types (Modrich and Lahue, 1996). The characteristics of dMMR cancers include high tumor mutational burden (TMB), favorable prognosis (Boland and Goel, 2010) and high sensitivity to immune checkpoint blockade (ICB) therapy (Le et al., 2017; Le et al., 2015). The current dogma is that hypermutation-generated neoantigens are responsible for favorable clinical responses to various therapies, especially ICB (Germano et al., 2017; Le et al., 2017; Le et al., 2015; Mandal et al., 2019). However, despite the correlation of TMB and clinical benefit, the studies using anti-CTLA4 and anti-PDL1 have shown that some patients with a high TMB yield no benefit (Chan et al., 2015) (Rosenberg et al., 2016), implying TMB alone is insufficient. Moreover, a retrospective analysis shows that TMB is not significantly associated with the efficacy of anti-PD-1 plus chemotherapy (Paz-Ares et al., 2019). Specifically, in dMMR cancers treated with ICB, the TMB is not significantly different between responders and non-responders (Le et al., 2017). Meanwhile, the objective response rate (ORR) to anti-PD-1 therapy is quite variable, ranging from 28%−53% (Diaz et al., 2017; Le et al., 2017; Marabelle et al., 2020; Overman et al., 2017). These observations imply that in addition to TMB, other functional changes mediated by dMMR may play roles. Antigen-presentation is indispensable for T-cell mediated anti-tumor effects, and mutations in its associated pathways could attenuate cytotoxic attack. Mutations in B2M are frequent in dMMR cancers, whereas its loss correlates with a favorable clinical course and absence of metastatic disease (Koelzer et al., 2012). In contrast, recent studies have demonstrated that mutations in B2M only exist in the metastatic sites while the primary dMMR tumors are resistant to ICB (Le et al., 2017). Interferons are known to potently upregulate antigen-presentation genes. Loss-of-function mutations in JAK1/2, key genes for interferon signal transduction, have been found in ICB-resistant dMMR cancers (Shin et al., 2017). However, the frequency is very limited, and not all cases with such JAK1/2 mutation are ICB-resistant (Kopetz et al., 2018). Overall, evidence for explaining ICB-resistance is still limited (Ganesh et al., 2019).
Preclinical and clinical studies have shown that pre-existing CD8+ T cells in the tumor microenvironment are required for effective ICB therapy (Heemskerk et al., 2013; Maleki Vareki, 2018; Tang et al., 2016; Tumeh et al., 2014). However, immunogenic antigens do not surely correlate with the T cell–inflamed tumor microenvironment (Spranger et al., 2016). It has been reported that in patients with Lynch syndrome arising from dMMR, the immune profile is independent of mutational rate in colorectal tumor tissue (Chang et al., 2018). Increasing evidence suggests that tumors hamper T-cell infiltration through various mechanisms (Mariathasan et al., 2018; Peng et al., 2015; Peng et al., 2016; Spranger et al., 2015). Specifically, about 50% of colorectal cancer samples with both microsatellite-instability-high (MSI-H) and TMB-high displays a low level of T-cell infiltration (Cristescu et al., 2018). Recently, a negative correlation is identified by proteogenomic analysis between glycolytic activity and CD8+ T cell-infiltration in MSI-H colon cancers (Vasaikar et al., 2019), suggesting an underlying mechanism of rendering TMB-high tumor progressive by impairing tumor-infiltration of T cells. However, additional molecular insights are needed to figure out the major factors that account for the high or low level of T-cell infiltration in dMMR cancers (Llosa et al., 2015).
Here, we show that deficiency of MLH1 and subsequent accumulation of cytosolic DNA activate the cGAS-STING pathway, contributing to increased immunity. Conversely, diminishing this DNA sensing by MLH1 rescue or STING/cGAS knockout in tumor cells leads to progressive tumor growth and ICB resistance. Our findings reveal additional mechanisms of responsiveness versus unresponsiveness to ICB therapy in dMMR cancer hosts, and provide biomarkers for future clinical practice.
Results
Deficiency of Sting in tumor cells accelerates dMLH1 tumor progression
To explore why some hosts of dMMR cancers respond to immunotherapies while others fail, we developed dMMR murine tumor models by knocking out the key MMR gene Mlh1, whose inactivation contributes to the majority of cancers with MSI (Boland and Goel, 2010). Starting with commonly used mouse mammary carcinoma cell line 4T1 and melanoma cell line B16-OVA, we generated Mlh1 null variants through CRISPR-Cas9, permitting the isolation of clones with MLH1 protein deficiency confirmed by Western blot. The Mlh1 deficient (dMLH1) clones were passaged extensively in vitro to permit accumulation of mutations. The growth patterns in vitro among different passages were similar. Then engraftment into immunocompetent syngeneic mice was evaluated by subcutaneous injection of different passages of these dMLH1 cells. Tumors derived from cells cultured extensively at the day-170 (D170) grew more slowly than those from cells cultured for 35 or 57 days (Figures 1A and 1B), suggesting that a hypermutated phenotype indeed suppresses tumor progression. However, the in vitro growth rate was not affected (Figure S1A), despite that amounts of mutations were identified in these tumors by whole exome sequencing (Table S1). The growth kinetics both in vivo and in vitro were consistent with recent reports (Germano et al., 2017; Mandal et al., 2019), in which increased TMB or neoantigens were confirmed by sequencing and were closely associated with decreased tumorigenesis and increased immunity.
Figure 1. dMLH1 tumor cell-intrinsic STING pathway regulates tumor progression.
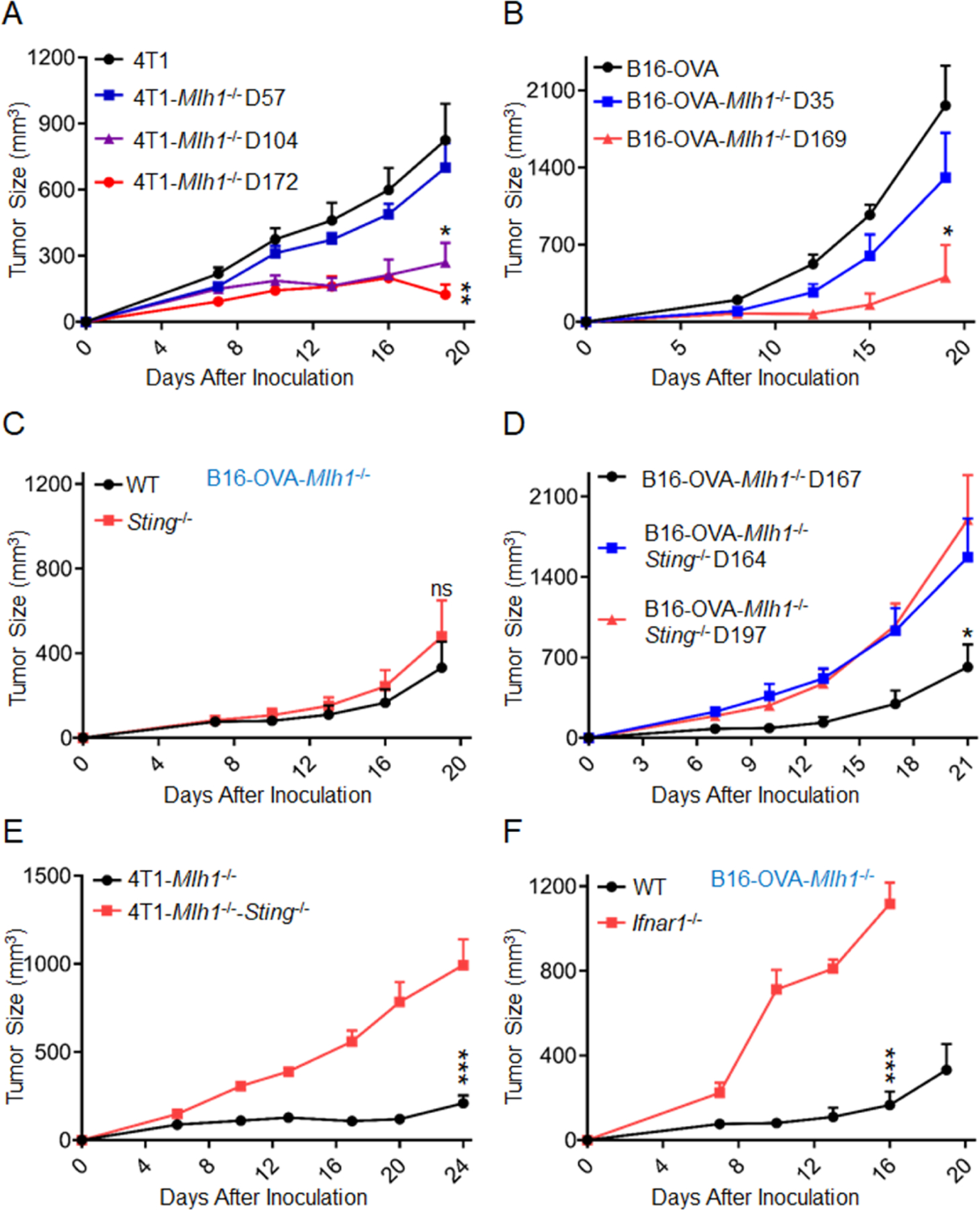
(A) Different generations of 4T1 cells with dMLH1 were inoculated into WT BALB/C mice (n=5). *P = F.18 between 4T1-Mlh1−/− D104 vs. 4T1; **P=0.0034 between 4T1-Mlh1−/− D172 vs. 4T1.
(B) Different generations of B16-OVA cells with dMLH1 were inoculated into WT C57BL/6 mice (n=4). *P = 0.0116 at end point between B16-OVA- Mlh1−/− D169 vs. B16-OVA.
(C) B16-OVA cells with dMLH1 (D170) were inoculated into WT and Sting−/− mice (n=5). n.s, P = 0.5024 at end point between the two groups.
(D) B16-OVA cells with dMLH1 (D167), and double deficiency cells of Mlh1 plus Sting (D164 and D197) were inoculated into WT C57BL/6 mice (n=4). *P=0.0253 at end point between B16-OVA-Mlh1−/− D167 vs. B16-OVA-Mlh1−/−-Sting−/− D197.
(E) 4T1 cells with dMLH1 (D166), and double deficiency cells of Mlh1 plus Sting (D169) were inoculated into WT BALB/C mice (n=5). ***P=0.0004 at end point between the two groups.
(F) B16-OVA cells with dMLH1 (D170) were inoculated into WT and IFNAR1−/− mice (n=5). ***P<0.0001 at indicated point between the two groups.
Data are represented as mean ± SEM. Tumor size was measured twice weekly. Experiments were repeated at least 2 times. Unpaired t test was used to determine significance.
MMR is a highly conserved DNA repair pathway, and we proposed that its defect might increase the exposure of tumor DNA to trigger innate sensing. MyD88 is one of the major adaptors through which host cells sense exogenous damage signaling. However, MyD88-deficient mice and wild type (WT) mice showed similar tumor-growth suppression (Figure S1B). Engraftment rate and subsequent tumor growth could also depend on the host cGAS-STING pathway (Woo et al., 2014). To explore its role in tumor progression by inoculating late passaged cells (D170) into Sting-deficient and WT mice, we surprisingly observed no significant differences between them with respect to engraftment or subsequent tumor growth (Figure 1C). To address whether the tumor intrinsic DNA sensing pathway is required, we knocked out Sting in dMLH1 cells and isolated clones at different passages by counting cultured days from the day of Mlh1 knockout as before (Figure S1C). Intriguingly, Sting-deficient dMLH1 tumors grew much faster than dMLH1 tumors (Figures 1D and 1E), suggesting that STING pathway inside tumor cells rather than host cells is essential.
The type I IFN signal is a key effector of the STING pathway for promoting antigen presentation and dendritic cells (DCs) activation (Sceneay et al., 2019). We assessed its role in type I IFN receptor (IFNAR1) deficient mice, and found that highly immunogenic B16-OVA tumors were more aggressive in these mice than in IFNAR1-proficient mice (Figure 1F). To avoid the potential interference of developmental defects in the immune system due to IFNAR1 deficiency, we repeated this experiment in 4T1 model by blocking type I IFN signal with an anti-IFNAR1 monoclonal antibody (mAb) in WT mice and found similar results (Figure S1D). DCs are the key cells that respond to IFN for cross-priming functions (Joffre et al., 2012). To study whether type I IFN signal on DCs was required, dMLH1 cells were inoculated into CD11c-Cre; IFNAR1f/f mice, where IFNAR1 is conditionally depleted in DCs. Lack of IFNAR1 in DCs allowed rapid growth of immunogenic and MLH1-deficient B16-OVA tumors (Figure S1E). CD103+ DCs, a subpopulation of DCs, plays a key role in cross-presenting tumor antigens to CD8+ T cells. We therefore inoculated dMLH1 tumors in Batf3-deficient mice lacking CD103+ and CD8α+ DCs. Batf3-deficient mice completely failed to control dMLH1 tumors (Figures S1B and S1F). Further, the results of depleting CD8+ T cells indicated that CD8+ T cells are essential for limiting dMMR tumors (Figures S1F and S1G). Taken together, the data presented here indicate that dMLH1 tumors trigger tumor cell-intrinsic DNA sensing to enhance cross-priming by dendritic cells upon CD8+ T cells.
MLH deficiency induces activation of type I IFN pathway
Next, we investigated whether and how the STING-IFN pathway in dMLH1 cells were activated. First, we detected the expression changes of interferon-induced genes (ISGs), and found that they were significantly elevated in cultured dMLH1 4T1 cells and B16-OVA cells. We reproduced this result in mouse lung cancer cell line TC-1 and colon cancer cell line MC38-OVA (Figures 2A and S2A), as well as in B16-OVA cells with Mlh1 knockout by another set of gRNA (Figure S2B). Meanwhile, activation of IFN signal was confirmed by the enhanced phosphorylation of STAT1 (Figure 2B). To directly determine IFN production, we quantified IFN-β in the supernatant of cultured dMLH1 cells by ELISA and readily detected IFN-β (Figure 2C). However, the detected levels of IFN-β did not induce apoptosis (Figure S2C) or impair growth rate (Figure S1A). The aggressor phenotype of dMLH1 tumor with Sting-deficiency implies that STING is involved in the upregulation of type I IFN. Concordantly, these type I IFN signals disappeared in Sting-deficient or cGas-deficient cells (Figures 2D, 2E and S2D). We performed the same tests in human lung cancer cell line H460 and breast cancer cell line MCF7 and obtained similar results (Figures 2F, S2E and S2F). To determine whether type I IFN signal was activated in human dMMR cancers, we generated a list of type I ISGs differentiable from type II IFN-induced ISGs, and analyzed their expression in MSI-H versus microsatellite-stable (MSS) colorectal tumors from The Cancer Genome Atlas (TCGA). About two-thirds of type I ISGs (44/68) were significantly up-regulated in MSI-H tumors (Figure 2G). For the increased ISGs, tumor cells and non-tumor cells may be the main cellular source that encounters tumor-cell derived type I IFN; Meanwhile immune cells that are able to engulf dMMR tumor cells may also contribute. Taken together, these data reveal that dMLH1 activates the cGAS-STING-IFN pathway in various mouse tumor models, and possibly in human tumors. More clinical studies are needed to further confirm this hypothesis,
Figure 2. Type I IFN signal pathway is activated in dMLH1 tumor cells.
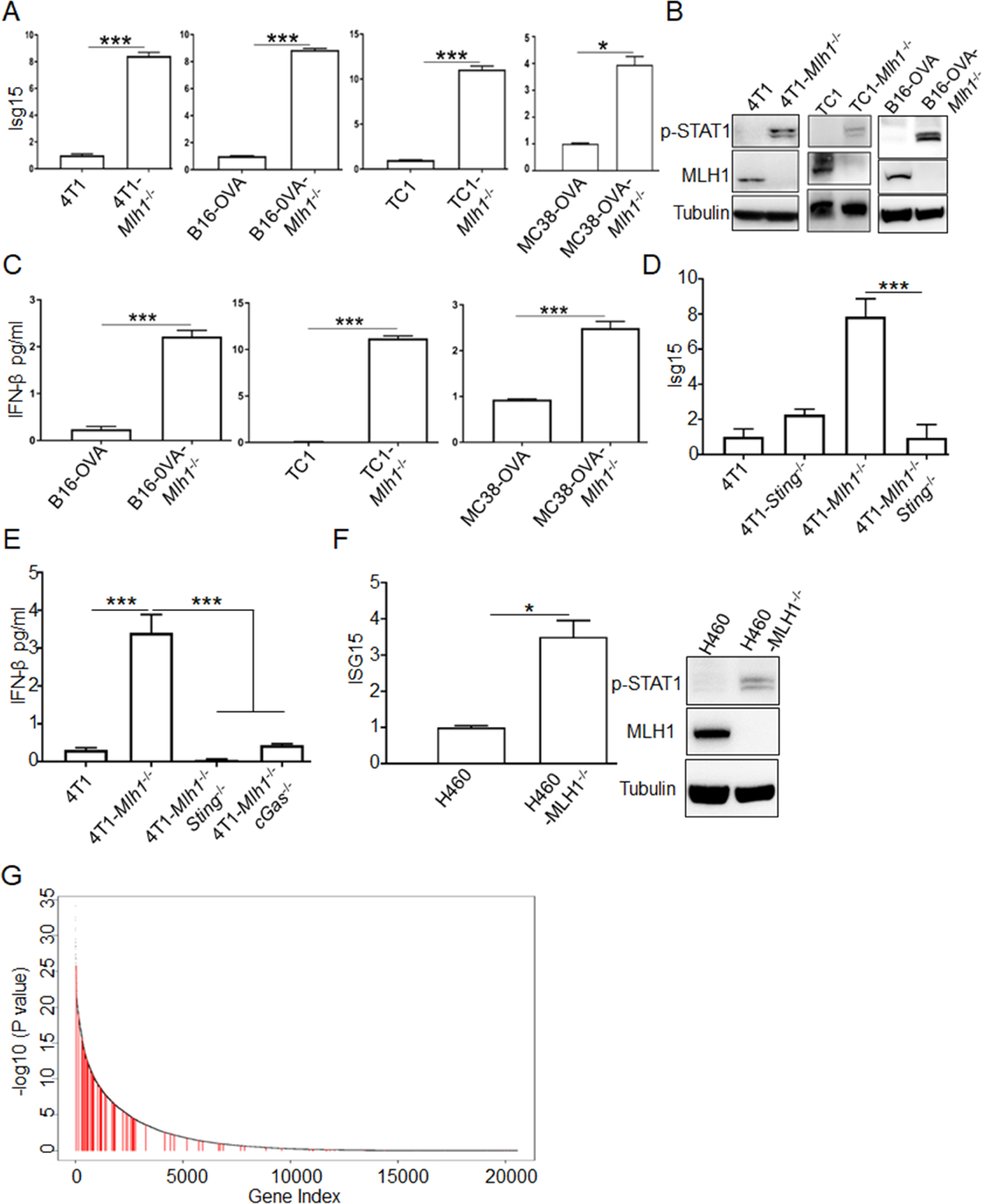
(A) ISGs expression at the mRNA level in cultured cells (n=3) was determined by qPCR. Relative expression fold change of representative ISG (Isg15) was shown. (also see Figure S2A).
(B) Phosphorylation of STAT1 at Y701 was shown by WB.
(C) IFN-β was quantified by ELISA in the supernatant of indicated cell lines (n=3).
(D and E) Isg15 and IFN-β were quantified by qPCR and ELISA, respectively, in Sting or cGAS-deficient cells lines (n=3).
(F) Isg15 and phosphorylation of STAT1 at Y701 were determined in H460 cells (n=3).
(G) The plot shows the ranked p values (-log10 scaled) estimated from Wilcoxon rank test comparing gene expression levels in MSI-H versus MSS colorectal tumors from the TCGA database. The test was one-sided, to test if genes in MSI-H are expressed at higher levels than in MSS tumors. Red vertical lines marked the type I IFN induced ISGs (see Table S1 for ISGs list and expression).
Data are represented as mean ± SEM. Representative data of over 2 independent experiments are shown. Unpaired t test was used to determine significance in A-F. Wilcoxon rank sum test was used to determine significance in G.
MLH1-deficient cells accumulate cytosolic DNA
We next investigated the mechanism of STING activation. Given that mutation is an ongoing event in dMLH1 cells, one possibility is that dMLH1-mediated mutations occur in certain genes upstream of STING, thus enhancing the STING pathway; another possibility is that MLH1 itself negatively regulates the STING pathway. MLH1 rescue should distinguish between these two possibilities, because mutations that were generated previously would not be reversed by MLH1 rescue. As shown in Figures 3A, 3B, S3A and S3C, all elevated IFN signals disappeared when MLH1 was restored to dMLH1 4T1 cells or TC1 cells, which supported the second hypothesis. Our previous findings have shown that MutLα (a heterodimer of MLH1 and PMS2), promptly terminates exonuclease 1 (Exo1)-catalyzed DNA excision upon mismatch removal; In the absence of MutLα, Exo1 conducts unnecessary hyper-excision (Zhang et al., 2005). We have recently shown that MutLα also regulates Exo1 activity in DNA end resection, which is required for homologous recombination-mediated double strand break repair (Guan et al., 2021). Excess DNA excision by Exo1 in the absence of MLH1 could result in severe DNA damage triggering the release of genomic DNA from nucleus to cytoplasm, activating the cGAS-STING pathway to produce IFN. We stained cytosolic dsDNA with PicoGreen and found that a significantly higher percentage of dMLH1 cells than WT cells showed cytosolic DNA positive, which diminished to the level of WT cells after rescue of MLH1 (Figures 3C and S3B). This result was confirmed by qPCR analysis (Figure 3D) and PicoGreen dsDNA quantification assay (Figure S3D) using cytosolic DNA fragments isolated from cultured cells. Similar results were obtained in B16-OVA cells (Figure S3E) and human cell lines (Figures 3E and 3F). To confirm this phenotype in spontaneous human dMMR cancers, we transfected MLH1 into an MLH1-deficient human colon cancer cell line HCT116 (Figure S3F), and observed decreased cytosolic DNA after rescue (Figure 3G). Consistent with the in vitro results, dMLH1 tumors showed regressor phenotype, but rescue of MLH1 reversed this to a progressor phenotype (Figures 3H and S3G). We conclude that MLH1 negatively regulates cytosolic DNA and restrains cGAS-STING-mediated DNA sensing.
Figure 3. MLH1 regulates accumulation of cytosolic DNA in tumor cells and tumor progression in vivo.
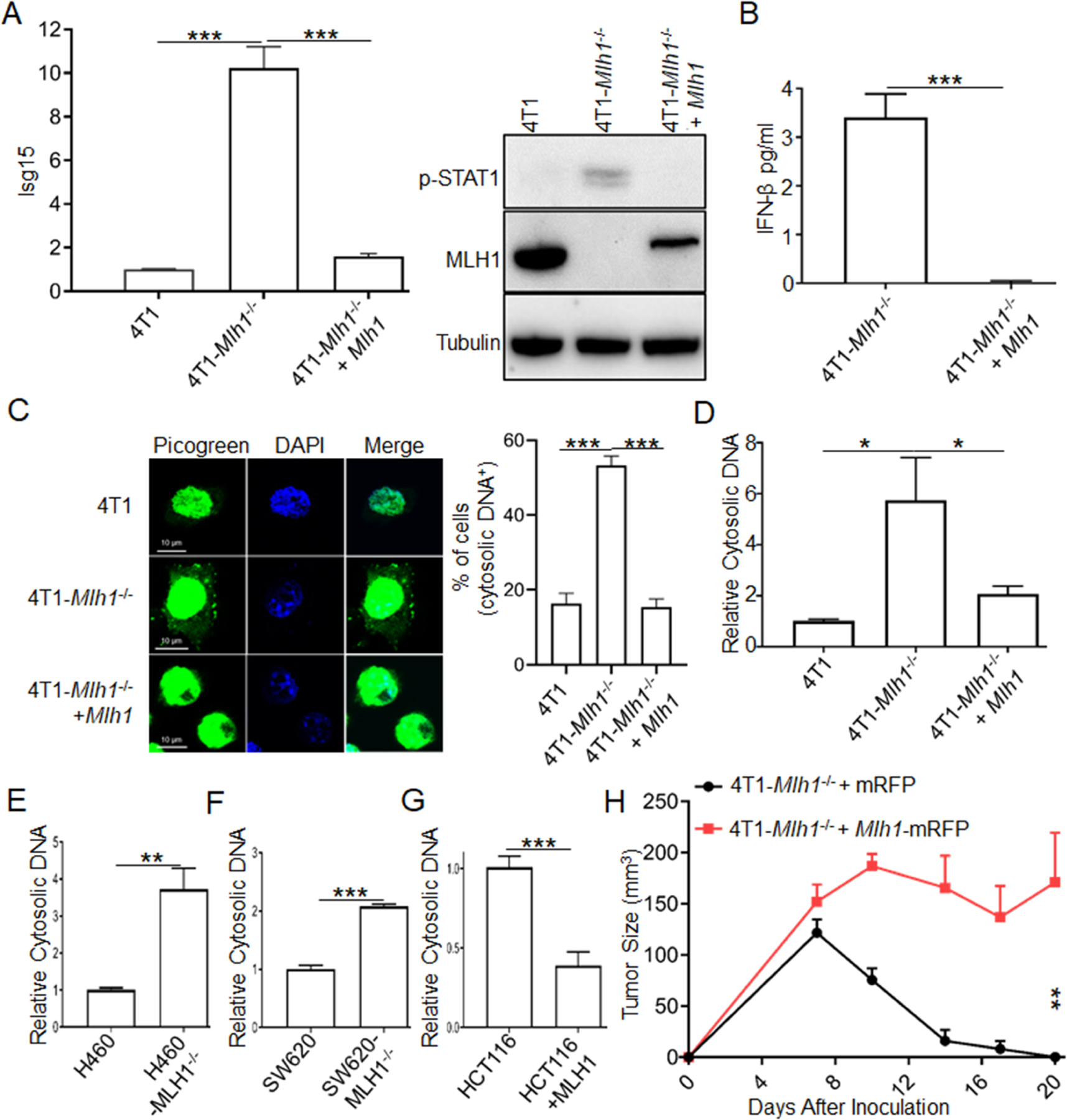
(A) Isg15 expression and phosphorylation of STAT1 at Y701 were determined in MLH1-rescued cells.
(B) IFN-β was quantified by ELISA in the supernatant of MLH1-rescued cells.
(C) dsDNA was determined by PicoGreen dsDNA quantitation assay, and extra-nuclear dsDNA was counted. Statistical data are shown (also see Figure S3B).
(D) Cytosolic DNA was isolated by a commercial kit and quantified by qPCR with genomic DNA specific primers.
(E-G) Cytosolic DNA was isolated and quantified in human cancer cell lines H460, SW620 and HCT116.
(H) 2×106 4T1-Mlh1−/− cells with either rescued MLH1 or control vector were inoculated into WT BALB/C mice (n=5). Tumor size was measured twice weekly.
Data are represented as mean ± SEM. Unpaired t test was used to determine significance.
dMLH1-mediated DNA sensing promotes epitope-specific T cell activation
To specifically elucidate the essential role of DNA sensing in eliciting anti-tumor CD8+ T cell responses independent of neoantigens, we designed experiments to exclude mutation-derived neoantigens by utilizing neoantigens-fixed models in vitro and in vivo. First, to determine whether the increased STING-IFN signal in dMLH1 cells provides an activated signal for single epitope-specific CD8+ T cell priming by antigen-presenting cells (APCs), we co-cultured bone marrow-derived dendritic cells (BMDCs) with OVA-expressing tumor cells, then used purified BMDCs to cross-prime OT-I T cells for 2 days, and finally detected T cell proliferation and function. The results showed that BMDCs pre-educated by dMLH1 tumor cells provided a potent activated signal for optimal single epitope-specific T cell proliferation and cytokine production (Figures 4A, 4B, S4A and S4B), while Sting deficiency diminished such effects. In addition, when pre-cultured Ifnar-deficient BMDCs with dMLH1 tumor cells, we found that the proliferation levels of T cells were dramatically reduced in both MMR-proficient and -deficient groups (Figure S4C), which was consistent with in vivo results that anti-dMMR tumor immunity is dependent on type I IFN signal in dendritic cells (Figures 1F and S1E).
Figure 4. dMLH-mediated DNA sensing promotes T-cell priming independent of TMB.
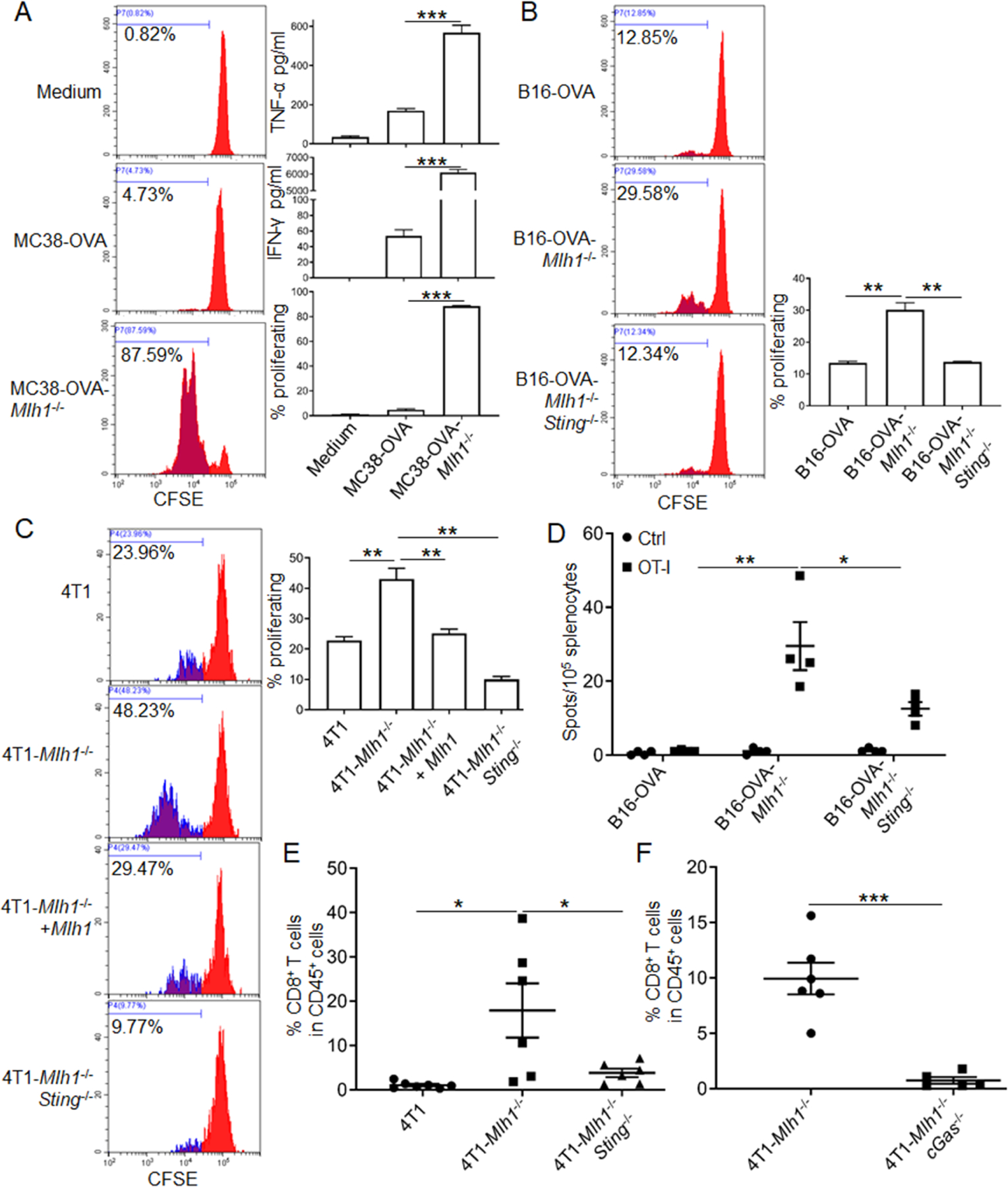
(A and B) BMDCs pre-educated with MC38-OVA and B16-OVA cells were co-cultured with OT-I T cells, then T-cell proliferation was determined. IFN-γ and TNF-α were quantified. Representative FACS histograms and statistic data are shown (also see Figures S4A and S4B).
(C) Supernatants were added into co-culture system of BMDCs and OT-I T cells, then T-cell proliferation was determined. Representative FACS histograms and statistical data are shown.
(D) B16-OVA cells were inoculated into WT C57BL/6 mice (n=4). One week later, cells from the spleen were isolated and re-stimulated by OT-I peptide or control SIY peptide in vitro. T-cell responses were determined by IFN-γ ELISPOT assay.
(E and F) Fragmented tumor tissues derived from 4T1 cells with d Mlh1 (D129), double deficiency cells of Mlh1 plus Sting (D136), and double deficiency cells of Mlh1 plus cGAS (D144) were implanted into WT BALB/C mice (n=6–7). Eleven days later, tumor-infiltrating T cells were detected by FACS.
Data are represented as mean ± SEM. Unpaired t test was used to determine significance.
To better understand the role of type I IFN in BMDC-mediated cross-priming, we added the supernatant from cultured tumor cells into the co-culture system of BMDCs and OT-I cells in the presence of OVA protein instead of tumor cells, and the results showed that rescue of MLH1 or deficiency of Sting in tumor cells impaired T cell proliferation (Figure 4C). All of these effects were independent of mutation-derived neoantigens, as the proliferation of OT-I T cells was only induced by mutation-independent OT-I epitope from tumor cells or exogenously added OVA protein at equal level. For the T cell responses in vivo, we inoculated OVA protein-expressing tumor cells into immunocompetent mice and detected anti-tumor immune responses by IFN-γ ELISPOT. The results showed that dMLH1 tumors triggered a stronger OT-I epitope-specific T cell response, while knockout of Sting significantly impaired such effects (Figures 4D and S4D). Consistent with the change in tumor-specific T cell responses, the overall tumor-infiltrating CD8+ T cells increased significantly in dMLH1 tumors, while deficiency of Sting damaged CD8+ T cell infiltration (Figure 4E). These data reveal that dMLH1 tumors activate anti-tumor CD8+ T cells responses in a manner dependent on tumor-cell intrinsic cGAS-STING pathway, but not necessarily on mutation-mediated neoantigens, and that deficiency of the cGAS-STING pathway in dMMR tumor cells dramatically diminishes tumor-infiltration of CD8+ T cells.
dMLH1-mediated DNA sensing enhances immune checkpoint blockade
To evaluate the role of DNA sensing in the therapeutic setting of ICB, we implanted fragmented tumor tissues into immunocompetent mice, followed by ICB treatment. As shown in Figures 5A and S5A, dMLH1 4T1 tumors had a complete response to ICB treatment, while MLH1-rescued tumors had no such effects. Similarly, knockout of Sting also impaired the therapeutic effects (Figure 5B). To eliminate the contribution of neoantigens generated due to Mlh1-deficiency in ICB therapy, we inoculated MC38-OVA cells into TCR transgenic 2C mice, then adoptively transferred OVA-specific OT-I T cells and administered ICB. The utilized TCR transgenic 2C mice here have two advantages: 1, the endogenous T cells only recognize the SIY epitope (SIYRYYGL), which is not present in tumor cells; 2, the endogenous T cells could inhibit homeostasis-driven proliferation of adoptive transferred T cells (Cho et al., 2000). As shown in Figure 5C, MC38-OVA tumors showed no response to ICB therapy, while dMLH1 tumor initially regressed within one week after T-cell transfer in non-ICB group, and relapsed later, but ICB treatment persistently controlled tumor progression. Similar therapeutic effects were observed with the 4T1-HA model (Figures 5D and S5B–S5E). All data above suggest that dMLH1-induced DNA sensing elicits potent anti-tumor T-cell responses independent of neoantigens.
Figure 5. dMLH1-mediated DNA sensing enhances ICB therapy.
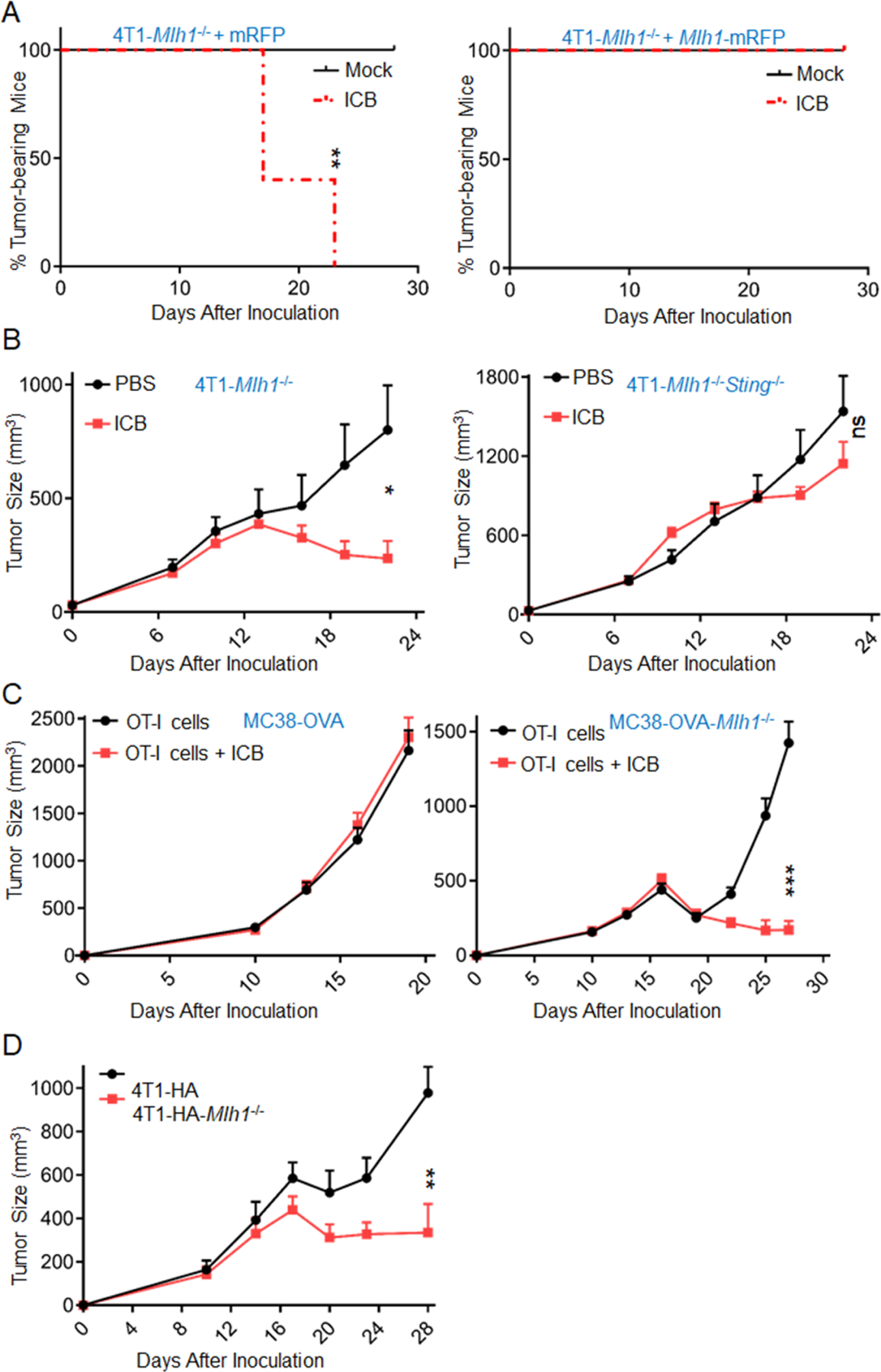
(A) Fragmented tumor tissues derived from 4T1-Mlh1−/− and 4T1-Mlh1−/−+Mlh1 cells in Rag1 mice were implanted into WT mice (n=4–5), and treated with ICB drugs at day 11, 14 and 17. Tumor-bearing mice percentages are shown. (also see Figures S5A)
(B) Fragmented tumor tissues derived from 4T1-Mlh1−/− (D173) and 4T1-Mlh1−/−Sting−/− (D170) cells in Rag1 mice were implanted into WT mice (n=6–8), and treated with ICB drugs at day 7, 10, 13 and 16. *P=0.0156 between the two groups in 4T1-Mlh1−/− model; ns, P=0.2467 between the two groups in 4T1-Mlh1−/−Sting−/− model.
(C) MC38-OVA and MC38-OVA-dMLH1 cells were inoculated into Rag1/2C mice (n=9–10), followed by OT-I T-cell transfer at day 10, then ICB treatment was administrated at day 11, 14 and 17.
(D) 4T1-HA and 4T1-HA-dMLH1 cells were inoculated into F1 mice (n=5) of Rag2× Rag2/OT-I mice, followed by CL4 T-cell transfer at day 10 and ICB treatment at day 11, 14, 17 and 20. (also see Figure S5D).
Data are represented as mean ± SEM. The log rank test (Mantel–Cox) was used to assess the significance of differences in A. Unpaired t test was used to determine significance in B-D.
Impaired cGAS expression contributes to ICB resistance
To explore clinical relevance of our findings, we analyzed dozens of human dMLH1 cancer cell lines in the Cancer Cell Line Encyclopedia (CCLE) of the Broad Institute of Massachusetts Institute of Technology and Harvard (Ghandi et al., 2019), and found 60% (18/30) of them were deficient in cGAS expression (Figure S6A). To confirm this correlation of dMMR and cGAS expression in clinics, we analyzed their expression in uterine corpus endometrial carcinoma (UCEC), which has the most MSI-H samples (TCGA). Results showed that cGAS expression was significantly lower in MSI-H group than in the MSS group (Figure 6A). To explore the consequences of impaired cGAS expression in dMMR cancers, we implanted cGAS-rescued HCT116 (Figure S6B) cells into NSG-SGM3 mice chimerized with human immune cells (Li et al., 2019), and administered ICB therapy. Compared to cGAS-rescued HCT116, cGAS-deficient HCT116 tumors had less CD8+ T cell infiltration (Figures 6B and S6C), and were more resistant to ICB therapy (Figure 6C). Consistent with our mouse data, we found that cGAS expression level positively correlated with CD8+ T cell–inflamed-gene expression (Figure S6D). Moreover, higher cGAS expression predicted better survival of patients with MSI-H cancers (Figure 6D). To confirm it in immunotherapy setting, we identified 7 samples with dMLH1 in a clinical trial in which patients received pembrolizumab until progression or unacceptable toxicity (NCT01876511), and found that higher cGAS or STING expression translated into better survival (Figure 6E). Strikingly, the responders had higher cGAS and STING expression than non-responders (Figure 6F). Additionally, we analyzed the correlation between cGAS expression in tumor tissue and survival from melanoma patients treated with anti-CTLA4 (Van Allen et al., 2015). The results showed that higher cGAS expression associated with better survival (Figure S6E). As there were very few samples with dMMR in melanoma, the non-significantly correlative trends imply that cGAS expression may predict prognosis in MMR-proficient cancers, in which we speculate that to a less extent, the basal level of cytosolic DNA may still activate cGAS-STING pathway, or there are other ways to trigger DNA sensing. Collectively, our data from human tumors in humanized mouse models and the samples collected from clinical trial reveal that down-regulation of cGAS/STING expression in MSI-H/dMLH1 cancers diminish tumor-infiltration of CD8+ T cells and associate with impaired ICB therapy.
Figure 6. impaired cGAS expression renders resistant to ICB therapy.
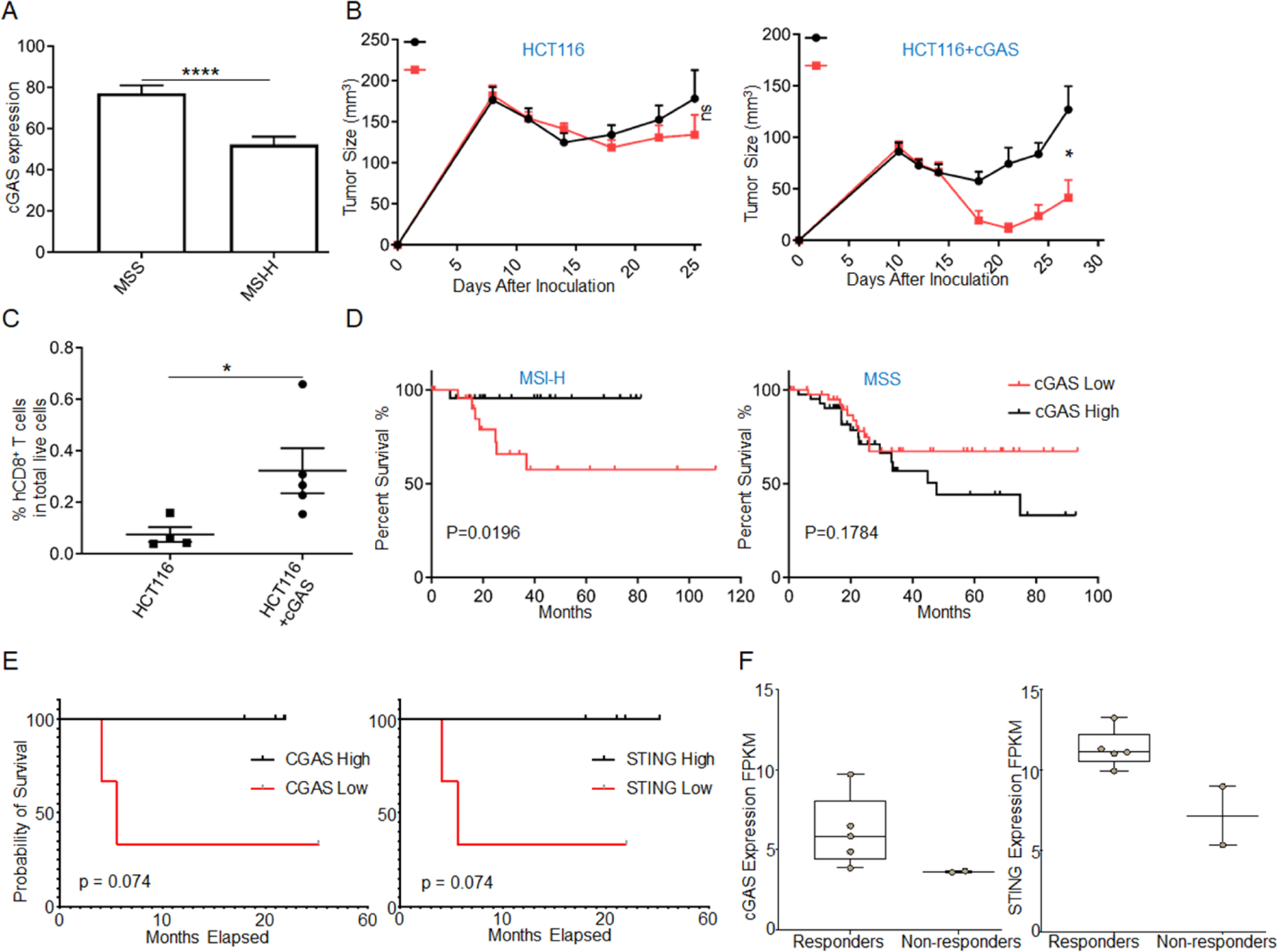
(A) cGAS expression between MSS (n=321) and MSI-H (n=170) UCEC samples, ****p<0.0001 (see Table S2 for patient information).
(B) CD8+ T cells in HCT116 tumor are detected by FACS (n=4–5).
(C) The growth curves of HCT116 and HCT116+cGAS cells-derived tumors (n=5–7) are shown. *P=0.011 between the two groups in HCT116+cGAS model; ns, P=0.3140 between the two groups in HCT116 model.
(D) Curves for disease-free survival are shown between the high and low expression of cGAS in UCEC samples with MSS (n=44) and MSI-H (n=24) (see Table S2 for patient information).
(E) Curves for overall survival are shown between the high and low expression of cGAS and STING in pan-dMLH1 tumors (n=3–4).
(F) cGAS and STING expression levels are shown between responders and non-responders treated with pembrolizumab.
Data are represented as mean ± SEM. Unpaired t test was used to determine significant differences in A-C. The log rank test (Mantel–Cox) for D. The gehan wilcoxon test for E.
Discussion
Although hypermutation-mediated neoantigens are believed to be the major reason for the high ORR in dMMR cancers, here we show that lack of T-cell infiltration still makes dMMR tumor insensitive to ICB treatment. Specifically, with the same level of neoantigens, compared to MMR-proficient tumors, MMR-deficient tumors are more potent to elicit DCs-mediated cross-activation of CD8+ T cells and more responsive to ICB treatment, revealing the indispensable role of tumor-infiltrating T cells recruited independent of neoantigens, and implying a programmed ability of dMMR tumor cells in activating T cells. Interestingly, a recent study reported that higher T cell-infiltration is found in dMMR gastroesophageal adenocarcinomas, which harbor loss-of-function mutations in HLA and B2M genes (von Loga et al., 2020). The subsequently reduced antigen-presentation obviously does not account for increased T-cell infiltration, but may imply that highly-mutated tumors may also have mutations in certain genes that negatively regulate T-cell infiltration.
Fifty percent of cancers with both MSI-H and TMB-H are lack of T-cell infiltration (Cristescu et al., 2018), but the reasons are not clearly defined (Vasaikar et al., 2019). Here, we present new evidence suggesting increased DNA sensing in dMMR tumor cells is essential for increased T-cell immunity. Notably, in a variety of therapeutic settings, such as chemotherapy, radiotherapy, DNA repair pathway-targeted therapy and so on, damaged DNA enhances T-cell activation and anti-tumor effects by triggering the Toll-like receptors or the cGAS-STING dependent type I IFN signal (Deng et al., 2014; Pantelidou et al., 2019; Shen et al., 2019; Sistigu et al., 2014). However, we find that in the absence of treatment, the cGAS-STING pathway has already been activated in dMMR tumor cells, triggering anti-tumor immune responses. Such effects are stimulated by accumulated cytosolic DNA. The disappearance of cytosolic DNA and IFN signal after MLH1 rescue demonstrates that MLH1 plays dual roles as a tumor suppressor: 1) as a key MMR component, MLH1 suppresses tumor development by correcting biosynthetic errors generated during DNA replication (Li, 2008); 2) MLH1 deficiency limits tumor progression via triggering DNA sensing-mediated anti-tumor immune surveillance. In the latter case, loss of MLH1 deprives tumor cells of Exo1’s regulator MutLα, such that Exo1 conducts uncontrolled DNA excision during DNA repair, including MMR and homologous recombination-mediated DNA double strand break repair. This uncontrolled excision causes RPA exhaustion, DNA breaks and chromosomal breaks, which triggers cells to release of damaged DNA to the cytoplasm to activate the cGAS-STING pathway (Guan et al., 2021)
With DNA-damage-inducing therapies, both tumor cell-derived and host-derived cGAS-STING pathway have been reported to contribute to tumor control (Deng et al., 2014; Pantelidou et al., 2019; Sen et al., 2019; Wang et al., 2017). However, in non-treatment setting, increasing evidence suggests that tumor cell-intrinsic cGAS/STING activation plays a role of promoting tumor initiation, development and metastasis (Bakhoum et al., 2018; Kwon and Bakhoum, 2020; Lemos et al., 2016; Liu et al., 2018). Here, we show that in a dMMR background, the STING-cGAS pathway within tumor cells, but not host cells, is essential for suppressing tumor progression. Mechanistically, the STING-cGAS pathway regulates tumor-infiltration of CD8+ T cells, as the increased CD8+ T cells disappear in STING or cGAS knockout tumors. To explain the conflicting resource of the cGAS-STING pathway, we propose two potential possibilities. On one hand, in therapeutic settings, ionizing radiation, for example, induces an acute death of tumor cells, which can readily release tumor DNA or cGAMP into tumor microenvironment, subsequently activating the cGAS-STING pathway in immune cells (Mardjuki et al., 2020); While without treatment, dMMR tumors should have no such acute death. On the other hand, although immune cells are professional in producing type I IFN via the cGAS-STING pathway, they are still replaceable if tumor cells could produce significant type I IFN on their own, and in dMLH1 tumors, this is just the case.
With both DNA sensing and neoantigens being increased in dMMR tumor cells, it is hard to evaluate the role of DNA sensing in tumor progression. As neoantigens may vary between Mlh1 single knockout cells and Mlh1 plus Sting double knockout cells even at the same passage, and each neoantigen-specific T cells may influence others, which makes it unconvincing to argue that STING but not neoantigens in dMLH1 tumor cells mainly affects tumor progression and ICB therapy in WT mice. Therefore, we fixed the level of neoantigens using OT-I or HA epitope-expressing tumor cells, and evaluated the role of dMLH1-mediated DNA sensing in anti-tumor T-cell responses by importing these epitopes-specific T cells in vitro and in vivo. Our results clearly demonstrate that the increased DNA sensing itself in dMMR tumor cells promotes T-cell priming and anti-tumor efficiency. Meanwhile, we noticed that MC38-OVA tumor, a widely used high-immunogenicity model, progressed very aggressively in such TCR transgenic mice. Several reasons may explain this: 1) the high-immunogenicity is only true in WT mice, but in our model, only the transferred number-limited OT-I T cells recognize the OVA antigen in tumor cells, so the immunogenicity of MC38-OVA model is actually rather low in our system; 2) in other studies, adoptively transferred anti-tumor T cells are usually pre-stimulated by anti-CD3 plus anti-CD28 mAbs in vitro to be fully activated, resulting in better capacity to regress tumor; 3) since we transferred un-activated T cells after tumor establishment, so there is limited basal level of innate sensing for T-cell priming while dMMR provides strong DNA sensing that helps T-cell activation; 4) when T cells are transferred into Rag mice, they would be activated because of homeostasis-driven proliferation. However, we transferred un-stimulated OT-I T cells after tumor inoculation into 2C-TCR transgenic mice, where endogenous T cells could effectively prevent homeostasis-driven proliferation.
MSI-H/dMMR has been approved as the only pan-tumor biomarkers for ICB therapy. However, the responses vary. By exploring the capacity of dMLH1 tumor cells for T-cell responses, we observed that dMLH1 simultaneously and unexpectedly created the two signals, mutation-derived neoantigen signal and DNA sensing signal, which well enabled T-cell priming. This should account for the high tumor-infiltration of T cells and the high ORR to ICB therapy in dMMR patients. However, to better explain why about 50% of dMMR cancers do not have ORR to ICB therapy, we found that the downregulation of cGAS is widespread in common human dMMR cancer cell lines. The loss of cGAS in 60% of human dMMR cancer cell lines raises the possibility that loss of cGAS promotes aggressive outcome of the parental cancers, as human cancer cell lines are usually developed from advanced tumors. Consistently, the expression level of cGAS is significantly lower in MSI-H human tumor samples than in MSS ones, implying an unknown mechanism of negative regulation. Moreover, using mouse and human dMMR tumor models, we reveal that the impaired cGAS-STING pathway in tumor cells confers resistance to ICB therapy. Accordingly, the cGAS/STING expression predicts survival of patients with MMR-deficient cancers. Loss or impaired cGAS-STING-IFN pathway might allow dMMR tumors to evade immune rejection, which, besides to neoantigens, might be another way of immunoediting (Dunn et al., 2006). Therefore, our findings suggest that the cGAS-STING-IFN could be an independent biomarker for immunotherapy in patients with dMMR cancers. This study might provide better rationales to use various immunotherapies that could bypass such deficiencies and supple downstream signals to re-activate DCs and T cells inside tumor microenvironment.
STAR★Methods
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Yang-Xin Fu (Yang-Xin.Fu@UTSouthwestern.edu).
Material Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement
Data and Code Availability
Original TCGA data in the paper is available here https://gdac.broadinstitute.org/.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse strains
C57BL/6J and BALB/c mice were purchased from the University of Texas Southwestern Medical Center Breeding Core or the Jackson Laboratory. 2C TCR transgenic mice (2C) were kept in house. Rag1−/− and Batf3−/− in both C57BL/6J and BALB/c background (Rag1 and Batf3−/−), B6[Cg]-Tmem173tm1.2Camb/J [Sting−/−] C57BL/6J-Tg[TcraTcrb]1100Mjb/J [OT-I], B6.Cg-Tg[Itgax-cre]1–1Reiz/J [CD11c-Cre], B6[Cg]-Ifnar1tm1.1Ees/J [IFNAR1f/f],B6[Cg]-Ifnar1tm1.2Ees/J [Ifnar1−/−], B6.129P2[SJL]-Myd88tm1.1Defr/J [MyD88−/−], C.129S6[B6]-Rag2tm1Fwa N12 [Rag2], B6.129S6-Rag2tm1Fwa Tg[TcraTcrb]1100Mjb [Rag2/OT-I] mice were purchased from Jackson Laboratory or Taconic Farms. Clone4-Tg (CL4) mice were provided by Dr. David Farrar from the University of Texas Southwestern Medical Center. IFNAR1f/f mice were crossed with CD11c-Cre mice to generate the conditionally deficient mice. Rag2 mice (BALB/C) were crossed with Rag2/OT-I (C57BL/6) mice to generate F1 mice. Rag1 mice (C57BL/6) were crossed with 2C mice to generate the Rag1/2C mice. All mice were maintained in a specific-pathogen-free animal facility, and all experiments were conducted according to regulations of the Institutional Animal Care and Use Committee of the University of Texas Southwestern Medical Center.
Cell lines
4T1 is a mouse mammary carcinoma cell line and TC1 is a mouse lung cancer cell line. 4T1-HA and TC1-OTI were sub-cloned from parental cells infected by a lentivirus expressing the HA-IRES-GFP cassette and OT-I epitope (SIINFEKL, OVA 257–64), respectively. B16-OVA and MC38-OVA are OVA-transfected clones derived from murine melanoma and colorectal tumor cell lines B16 and MC38, respectively. SW620, H460 and MCF-7 are human colon cancer, lung cancer and breast cancer cell lines, respectively. For cell lines with genes knocked out, guide RNA sequences targeting mouse Mlh1, Sting and human MLH1, Sting were inserted into CRISPR-Cas9 system plasmid PX458 and transfected into the above cell lines with Lipofectamine™ 2000, GFP positive cells were sorted by FACS and cultured in vitro for 5–7 days, then GFP negative cells were sorted and diluted into 96-well plates at one cell per well. Clones were isolated and confirmed by Western Blotting. To rescue MLH1, 4T1-Mlh1−/− and TC1-Mlh1−/− cells were infected by lentivirus harboring MLH1-IRES-mRFP cassette or control cassette with IRES-mRFP. Then, mRFP positive cells with similar expression levels were sorted by FACS using a BD FACSAria II cell sorter. The passage of Mlh1 knockout cells or Mlh1&Sting double knockout cells were determined by all in vitro cultured days counted from the day of transfection of plasmid pX458-gMlh1. Knockout of Tmem173 was performed on 2 months’ passage of dMLH1 cells. All cell lines were cultured in 5% CO2 at 37°C and maintained in vitro in Dulbecco’s modified Eagle medium supplemented with 10% heat-inactivated fetal bovine serum (Sigma-Aldrich), 100 U/mL penicillin, and 100 mg/mL streptomycin. All cells lines were tested and confirmed to be free of Mycoplasma. The whole-exome sequencing in 4T1-Mlh1−/− cells (day170) was performed at Genewiz with Illumina HiSeq 2×150 bp sequencing, and variant calling and data analyzed were performed by Genewiz.
METHOD DETAILS
RNA Extraction and Quantitative Real-Time PCR
After 2-day culturing in a T25 flask, tumor cell lines were resolved into TRIzol and processed to RNA extraction. Real-time PCR was performed with SsoAdvanced™ Universal SYBR Green Supermix according to the manufacturer’s instructions. The levels of gene expression were normalized to β-actin in mouse cell lines and GAPDH in human cell lines.
Western Blotting
For Western Blotting, cells were washed twice with cold PBS, and whole cell proteins were extracted in lysis buffer (40 mM Tris pH8.0, 150 mM NaCl, 1% NP40, 0.5% sodium deoxycholate, 0.1% SDS, 2 mM sodium orthovanadate) containing phosphatase inhibitor cocktail. The protein concentration was normalized using a protein assay dye reagent. Samples were boiled at 95 °C for 5 minutes and loaded to SDS-PAGE. After transferring, membranes were blocked with 5% milk in TBST buffer (0.1% Tween 20) for 1 hour. Primary antibodies were diluted and incubated at 4°C overnight. After washing three times with TBST buffer, goat anti-rabbit or anti-mouse secondary antibody and HRP conjugates were incubated with membranes for 1 hour. After washing three times, membranes were covered with the Amersham™ ECL Select™ reagent and imaged by the ChemiDocTM MP imaging system.
Quantification of Cytosolic DNA
Cultured cells (2×107) were digested to obtain nuclear, cytosolic, and mitochondrial fractions using the mitochondrial isolation kit as reported (Bakhoum et al., 2018). Mitochondria were removed by centrifugation at 17,000g for 20 minutes to minimize their contamination in the cytosolic fraction. dsDNA in the cytosolic fraction was quantified by real-time PCR with genomic DNA primers. For the immunofluorescence microscope assay, cells were cultured in a 6-well plate with a cover-glass. Twenty-four hours later, cells were washed twice with cold PBS and fixed with cold methanol at −20°C for 10 minutes. After being washed three times with PBS, cells were blocked with 1% BSA in PBS for 1 hour and stained with Pico488 dsDNA quantification reagent for 1 hour. After being washed three times with PBS, the cover-glass was mounted on white microscope slides using the ProlongTM Diamond Antifade Mountant with DAPI and imaged on a Leica TCS SP8 confocal microscope. For the cytosolic DNA quantification assay, cells in a 10-cm dish were washed two times with PBS and scraped. Half of the cells were lysed as whole cell extracts, and protein concentration was quantified for normalization. The other half of the cells were centrifuged at 720 g for 5 minutes and suspended in 500 µL hypotonic buffer (20 mM HEPEs pH7.4, 250 mM sucrose, 10 mM KCL, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, and 1 mM DTT, phosphatase inhibitor cocktail) for 30 minutes on ice and then centrifuged again at 800g for 5 minutes. Supernatants were transferred into a new EP tube and centrifuged at 20,000 g for 30 minutes. The supernatants were considered cytosolic fractionation, and 1 µl cytosolic fractionation was diluted with 49 µl TE buffer and incubated with 50 µl Pico488 dsDNA quantification reagent (1:200). The mixture was added into a costar white flat 96-well plate and measured by the Picogreen dsDNA quantification fluorescence method using a Tecan SPARK–multimode Microplate Reader.
In vitro cross-priming of T cells by BMDCs
Bone marrow cells were collected from mouse tibias and femurs, and cultured in a 6-well plate with complete RPMI 1640 medium containing recombinant mouse FLT3L (200 ng/ml). Fresh medium with 200 ng/ml FLT3L was added into the wells on day 3. On day 6, immature BMDCs were collected for co-culturing directly with T cells, or were added into a 6-well plate harboring attached tumor cells plated one day earlier. After overnight incubation, suspended BMDCs were collected and positively isolated using a mouse CD11c positive selection kit. CD8+ T cells were isolated from lymph nodes and spleens of OT-I or CL4 mice with a CD8+ T cell isolation kit. Purified T cells were labelled with 2 µM CFSE by incubating at room temperature for 10 minutes and washing three times. BMDCs were cultured with CFSE-labelled T cells at ratios of 1:2 and 1:20 in U-bottomed 96-well plates and incubated at 37°C with 5% CO2. Two days later, the supernatant was collected for cytokine quantitation by Cytometric Bead Array. T-cell proliferation was analyzed by FACS in terms of CFSE dilution. In some experiments, 50 µl of supernatant from 2-day-cultured tumor cell lines were added into a 200 µl co-culture system of BMDCs and purified OT-I T cells (CFSE labelled) in the presence of 100 µg/ml OVA protein. Three days later, T-cell proliferation was evaluated by CFSE dilution.
Cytokines measurement
IFN-γ and TNF-α in the supernatant were quantified using a BD Cytometric Bead Array, according to the manufacturer’s protocol. After staining, samples were run on a CytoFLEX flow cytometer and data were analyzed with CytExpert or FlowJo software. IFN-β in the medium of 2-day cultured tumor cells was quantified by a PBL mouse IFN-β ELISA kit.
IFN-γ Enzyme-Linked Immunosorbent Spot Assay
B16-OVA and B16-OVA-Mlh1−/−, TC1-OTI and TC1-OTI-Mlh1−/− cells were injected subcutaneously into the left flanks of WT C57BL/6 mice at 2 × 106 cells per mouse. Seven days later, single cell suspensions were prepared from tumor draining lymph nodes and spleens, then 1×105 cells were re-stimulated with 10 µg/ml OT-I peptide or control SIY peptide. After culturing for two days, ELISPOT assay was performed using the IFN-γ ELISPOT kit, according to the manufacturer’s instructions. Spots were calculated using an ImmunoSpot Analyzer.
FACS detection of tumor-infiltrating T cells
4T1-Mlh1−/− and its derivative cells were injected subcutaneously into Rag1 (BALB/C) mice, 2 weeks later, tumors were collected and cut into fragments of 2–5 mm in diameter. Tumor pieces were implanted subcutaneously beneath the incision on the left flanks of WT BALB/C mice. Eleven days later, tumors were collected and digested into single cell suspensions, which were incubated with anti-CD16/32 (anti-FcγIII/II receptor, clone 2.4G2) for 20 minutes to block nonspecific binding and then stained with antibodies. The fixable viability dye eFluor 506 was used to exclude dead cells. Data were collected on the CytoFLEX (Beckman Coulter Inc.) and analyzed with the CytExpert (Beckman Coulter Inc.) or FlowJo (Tree StarInc., Ashland, OR) software.
Human immune cells chimeric mice
The mice were developed as previously described (Li et al., 2019). Briefly, four-week-old NSG-SGM3 female mice were irradiated with 100 cGy. One day later, human CD34+ cells were purified from umbilical cord blood (UT Southwestern Parkland Hospital) by density gradient centrifugation, followed by positive immunomagnetic selection with anti-human CD34 microbeads. About 1 × 105 CD34+ cells were intravenously injected into each recipient mouse. Eight weeks after engraftment, humanized mice with over 50% human CD45+ cells reconstitution were inoculated with 1 × 106 HCT116 cells subcutaneously on the right flank. At day 7–10, ICB treatment (200 µg/mouse anti-CTLA4 ipilimumab and 200 µg/mouse anti-PDL1 atezolizumab) were injected every 3 days for a total of 3 times. Tumor volumes were measured twice weekly.
Tumor growth and treatments
B16-OVA, 4T1, TC1, and their derivative cell lines, were injected subcutaneously into the left flanks of WT C57BL/6 and BALB/c mice or the indicated genetically engineered mice at 1 × 106 cells per mouse, unless otherwise specified. Tumor size was measured twice weekly and calculated by the following formula: Length × Width × Width / 2. For CD8+ T cell depletion and IFNAR1 blocking experiments, anti-CD8b mAb and anti-IFNAR1 mAb were injected intraperitoneally at 200 µg/mouse on the day of inoculation and continued every 3 days for a total of 3 times. To transfer CL4 or 2C T cells, inguinal lymph nodes and spleens were collected and processed to T cell isolation using a mouse CD8+ T cell isolation kit. Then 1 × 105 (CL4) or 2 × 104 (OT-I) purified T cells were injected intravenously into each mouse 10–14 days after tumor inoculation. One to four days later, ICB treatment (200 µg/mouse anti-CTLA4 and 100 µg/mouse anti-PDL1 atezolizumab) were injected intraperitoneally every 3 days for a total of 3–4 times. To transplant fragmented tumor tissue, 4T1-Mlh1−/− and its derivative cells were injected subcutaneously into Rag1 (BALB/C) mice, 2 weeks later, tumors were collected and cut into fragments of 2–5 mm in diameter. Fragmented tumor tissues were implanted subcutaneously beneath the incision on the left flanks of WT BALB/C mice, assuming the initial tumor size was 30 mm3. Seven to eleven days later, ICB (200 µg/mouse anti-CTLA4 and 100 µg/mouse anti-PDL1) was administered every 3 days for a total of 3–4 times. All experiments were performed in compliance with UTSW Human Investigation Committee protocol and UTSW Institutional Animal Care and Use.
NCT01876511 phase II clinical trial
FFPE patient samples from a pan-tumor dMMR cohort treated with pembrolizumab, an anti-PD-1 therapy, (ClinicalTrials.gov number NCT01876511) were obtained for gene expression studies. Standard RNASeq with rRNA depletion was performed on RNA extracted from 7 samples that were annotated for the tumor region. For gene expression normalization, FPKM values were calculated. To account for variations in tumor purity between individual samples, FPKM values were adjusted by tumor purity. The gene expression levels of cGAS and STING were compared among responders (complete and partial responses) and non-responders (Stable Disease and Progressive Disease) using RECIST v1.1 as well as their effects on overall survival. MLH1 loss of expression was determined by immunohistochemistry. Responses and Overall Survival according to CGAS, STING mRNA expression were studied within that MLH1 deficient patients. Gehan Wilcoxon Test was used to assess the significance of differences. The protocol was approved by the institutional review board at each site. All patients provided written informed consent before study entry.
Quantification and statistical analysis
Data are represented as means ± SEM. The method of statistical analysis was indicated in figure legend. Specifically, tumor volumes are compared between two groups at the indicated time points using unpaired Student’s t-tests. The log rank test (Mantel–Cox) is used to assess the significance of differences of survival time in TCGA data. All statistical tests are two-sided, except for Figure 2G, which was one-sided and performed using R software. Gene set enrichment analysis was performed in MSI-H versus MSS colorectal tumors from the TCGA database. To profile the genes that are higher in MSI-H patients than in MSS patients, a one-sided Wilcoxon rank test was used to compare gene expression at the mRNA level. Type I-ISGs was defined as genes that were 5 times more strongly induced by type I IFN than by type II IFN, as previously reported (Der et al., 1998; Hartman et al., 2005; Indraccolo et al., 2007; Liu et al., 2012; Sanda et al., 2006). A value of p < 0.05 was considered statistically significant (ns, no significance, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Supplementary Material
Table S1 Distinct Type I ISGs list, Related to Figure 2
Table S3 Melanoma samples of clinical outcome, Related to STAR Methods and Figure S6
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| InVivoMAb anti-mouse CD8β (Lyt 3.2) | BioXcell | Cat# BE0223 |
| InVivoMAb anti-mouse PDL1 (10F.9G2) | BioXcell | Cat# BE0101 |
| InVivoMAb anti-mouse CTLA4 (9D9) | BioXcell | Cat# BP0164 |
| InVivo Plus anti-mouse IFNAR1 (MAR1-5A3) | BioXcell | Cat# BP0241 |
| APC anti-mouse CD8a Antibody | BioLegend | Cat# 100712 |
| PE anti-mouse CD3 Antibody | BioLegend | Cat# 100206 |
| Anti-FcgIII/II receptor (clone 2.4G2) | BD Biosciences | Cat# 553141 |
| PE anti-mouse H-2Kb bound to SIINFEKL Antibody | BD Biosciences | Cat# 141604 |
| STING (D2P2F) Rabbit mAb | Cell Signaling Technology | Cat# 13647S |
| cGAS (D3O8O) Rabbit mA | Cell Signaling Technology | Cat# 31659S |
| Purified Mouse Anti-Human MLH-1 | BD Biosciences | Cat# 550838 |
| Phospho-Stat1 (Tyr701) (58D6) Rabbit mAb | Cell signaling technology | Cat# 9167 |
| Alexa Fluor® 647 Annexin V | Biolegend | Cat# 640911 |
| FITC anti-human CD8 Antibody | Biolegend | Cat# 344703 |
| Brilliant Violet 421™ anti-human CD45 Antibody | Biolegend | Cat# 368521 |
| Propidium Iodide Solution | Biolegend | Cat# 421301 |
| PerCP/Cyanine5.5 anti-human CD4 Antibody | Biolegend | Cat# 317427 |
| APC/Cyanine7 anti-mouse CD45 Antibody | Biolegend | Cat# 103116 |
| atezolizumab | pharmacy | N/A |
| ipilimumab | pharmacy | N/A |
| Tubulin Antibody (B-5-1-2) | Santa Cruz Biotechnology | Cat# sc-23948 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant Mouse FLT3L | BioLegend | Cat# 576306 |
| Ovalbumin | Sigma- Aldrich | Cat# A2512 |
| OVA257-264 (SIINFEKL) | Invivogen | Cat# vac-sin |
| SIYRYYGL (SIY) peptide | Sigma- Aldrich | N/A |
| Critical Commercial Assays | ||
| CFSE Cell Division Tracker Kit | BioLegend | Cat# 423801 |
| Mouse IFN Beta ELISA Kit | PBL Assay Science | Cat# 42410-2 |
| BD Cytometric Bead Array (CBA) Mouse Inflammation Kit | BD Biosciences | Cat# 552364 |
| BD Mouse IFN-g ELISPOT Sets | BD Biosciences | Cat# 551083 |
| SsoAdvanced Uni SYBR Grn Supmix | Bio-Rad | Cat# 1725272 |
| EasySep™ Mouse CD11c Positive Selection Kit II | STEMCELL | Cat# 18780 |
| EasySep Mouse CD8+ T Cell Isolation Kit | STEMCELL | Cat# 19853 |
| Pico488 dsDNA quantification reagent | Lumiprobe life science solutions | Cat# 42010 |
| ProlongTM Diamond Antifade Mountant with DAPI | Thermo Fisher Scientific | Cat# 36962 |
| Cell Counting Kit-8 | Fisher | Cat# 50-190-5565 |
| Experimental Models: Cell Lines | ||
| 4T1 | ATCC | Cat# HTB-177™ |
| 4T1-HA | THIS PAPER | N/A |
| B16-OVA | THIS PAPER | N/A |
| TC-1 | ATCC | Cat# CRL-2785™ |
| TC1-OTI | this manuscript | N/A |
| MC38-OVA | THIS PAPER | N/A |
| H460 | ATCC | Cat# HTB-177™ |
| SW620 | ATCC | Cat# CCL-227™ |
| MCF7 | ATCC | Cat# HTB-22™ |
| HCT116 | ATCC | ATCC® CCL-247™ |
| 4T1-Mlh1−/− | this manuscript | N/A |
| B16-OVA-Mlh1−/− | this manuscript | N/A |
| MC38-OVA-Mlh1−/− | this manuscript | N/A |
| TC1-Mlh1−/− | this manuscript | N/A |
| 4T1-Mlh1−/−-Sting−/− | this manuscript | N/A |
| 4T1-Mlh1−/−-cGAS−/− | this manuscript | N/A |
| B16-OVA-Mlh1−/−-Sting−/− | this manuscript | N/A |
| SW620-MLH1−/− | this manuscript | N/A |
| H460-MLH1−/− | this manuscript | N/A |
| MCF7-MLH1−/− | this manuscript | N/A |
| 4T1-HA | this manuscript | N/A |
| HCT116+cGAS | this manuscript | N/A |
| HCT116+MLH1 | this manuscript | N/A |
| 4T1-Mlh1−/−+MLH1-mRFP | this manuscript | N/A |
| TC1-Mlh1−/−+MLH1 | this manuscript | N/A |
| 4T1-Mlh1−/−-HA | this manuscript | N/A |
| 4T1-Mlh1−/−+mRFP | this manuscript | N/A |
| Experimental Models: Organisms/Strains | ||
| BALB/c | UTSW breeding Core | N/A |
| C57BL/6 | UTSW breeding Core | N/A |
| C.129S7(B6)-Rag1tm1Mom/J | Jackson Laboratory | Cat# 003145 |
| B6(Cg)-Ifnar1tm1.2Ees/J | Jackson Laboratory | Cat# 028256 |
| B6(Cg)-Tmem173tm1.2Camb/J | Jackson Laboratory | Cat# 025805 |
| B6.129S(C)-Batf3tm1Kmm/J | Jackson Laboratory | Cat# 013755 |
| C.129S-Batf3tm1Kmm/J | Jackson Laboratory | Cat# 013756 |
| C57BL/6-Tg(TcraTcrb)1100Mjb/J | Jackson Laboratory | Cat# 003831 |
| B6.129P2(SJL)-Myd88tm1.1Defr/J | Jackson Laboratory | Cat# 009088 |
| B6.129S6-Rag2tm1Fwa Tg(TcraTcrb)1100Mjb | Jackson Laboratory | Cat# 2334 |
| B6.129S6-Rag2tm1Fwa Tg(TcraTcrb)1100Mjb | Taconic Farms | Cat# 2334 |
| C.129S6(B6)-Rag2tm1Fwa N12 | Taconic Farms | Cat# 601 |
| Clone4-Tg | a gift from David Farrar | n/a |
| Oligonucleotides | ||
| Actb (cytosolic DNA in mouse cells) | Forward CCAGGTAAGTGACCTGTTAC Reverse GGGGTACTTCAGGGTCAGGA |
N/A |
| Gapdh (cytosolic DNA in mouse cells) | Forward CAACTGCTTAGCCCCCCTGG Reverse GCAGGGTAAGATAAGAAATG |
N/A |
| Actb (qPCR in mouse cells) | Forward AGATCAAGATCATTGCTCCTCCT Reverse ACGCAGCTCAGTAACAGTCC |
N/A |
| Mx2 (qPCR in mouse cells) | Forward ACCAGGCTCCGAAAAGAGTTC Reverse AGCTCGTACAATTTCAGTGACC |
N/A |
| Isg15 (qPCR in mouse cells) | Forward GAGCTAGAGCCTGCAGCAAT Reverse TCACGGACACCAGGAAATCG |
N/A |
| Irf7 (qPCR in mouse cells) | Forward TTGGGCAAGACTTGTCAGCA Reverse ATACCCATGGCTCCAGCTTC |
N/A |
| Pdl1 (qPCR in mouse cells) | Forward TGATCATCCCAGAACTGCCTG Reverse AGAAGAGGAGGACCGTGGAC |
N/A |
| Mlh1 (gRNA-1 for mouse gene) | Forward CACCGCAACCAGGGCACCCTGATCA Reverse AAACTGATCAGGGTGCCCTGGTTGC |
N/A |
| Mlh1 (gRNA-2 for mouse gene) | Forward CACCGCTAATTCAGATCCAAGACAA Reverse AAACTTGTCTTGGATCTGAATTAGC |
N/A |
| Sting (gRNA for mouse gene) | Forward CACCGTGCCCAGGGCGTCTCCTTG Reverse AAACCAAGGAGACGCCCTGGGCAC |
N/A |
| cGAS (gRNA for mouse gene) | Forward CACCGCGCAAAGGGGGGCTCGATCG Reverse AAACCGATCGAGCCCCCCTTTGCGC |
N/A |
| MLH1 (gRNA for human gene) | Forward CACCGTGATAGCATTAGCTGGCCGC Reverse AAACCGCGGCCAGCTAATGCTATCAC |
N/A |
| ACTB (cytosolic DNA in human cells) | Forward TACAATGAGCTGCGTGTGGC Reverse GCGGCCACCAGAAGAGGTAG |
N/A |
| GAPDH (cytosolic DNA in human cells) | Forward AGCCACATCGCTCAGACAC Reverse GCCCAATACGACCAAATCC |
N/A |
| GAPDH (qPCR in human cells) | Forward AGCCACATCGCTCAGACAC Reverse GCCCAATACGACCAAATCC |
N/A |
| ISG15 (qPCR in human cells) | Forward GCGAACTCATCTTTGCCAGTA Reverse CCAGCATCTTCACCGTCAG |
N/A |
| MX1 (qPCR in human cells) | Forward TTCAGCACCTGATGGCCTA Reverse AAAGGGATGTGGCTGGAGAT |
N/A |
| Recombinant DNA | ||
| Mlh1 (NM_026810) Mouse Tagged ORF Clone | Origene | Cat# MR210511 |
| human cGAS cDNA | THIS PAPER | N/A |
| human MLH1 cDNA | THIS PAPER | N/A |
| Software and Algorithms | ||
| GraphPad Prism 8 | GraphPad Software, Inc. | https://www.graphpad.com/scientific-software/prism/ |
| Image Lab Software | Bio-Rad | https://www.bio-rad.com/en-us/product/image-lab-software?ID=KRE6P5E8Z |
| CytExpert 2.3 | Beckman Coulter, Inc | https://www.beckman.com/flow-cytometry/instruments/cytoflex/software |
| FlowJo | Tree Star Inc. | https://www.flowjo.com/solutions/flowjo/downloads |
| Other | ||
| TRIZOL REAGENT | Thermo-Invitrogen | Cat# 15596018 |
| ProtoScript® II Reverse Transcriptase | NEB | Cat# M0368L |
| Dulbecco’s Modified Eagle’s Medium | Sigma- Aldrich | Cat# D6429 |
Highlight:
dMLH1 tumor cells accumulate cytosolic DNA and produce IFN-β
Knockout of cGAS or STING in dMLH tumor cells renders resistance to checkpoint blockade
Downregulating cGAS-STING in human dMLH1 cancers impairs checkpoint blockade therapy
Acknowledgements
We thank the UT Southwestern Flow Cytometry Facility, Institutional Animal Care and Use Committee Animal Resources Center, and Animal Research Center. Y.-X.F. holds the Mary Nell and Ralph B. Rogers Professorship in Immunology, and G.-M.L. holds the Reece A. Overcash Jr. Distinguished Chair for Research on Colon Cancer. We thank Professor David Farrar for providing the CL4 mice, and Drs. Damiana Chiavolini and Jonathan Feinberg for language polishing on the manuscript. This work was supported in part by Cancer Prevention & Research Institute of Texas (United States) grants RR150072 and RP180725 to Y.-X.F., and RR160101 to G.-M.L. Both Y.-X.F. and G.-M.L. are CPRIT Scholors in Cancer Research. Y.-X.F. holds Mary Nell and Ralph B. Rogers Prefessorship, G.-M.L. holds the Reece A. Overcash, Jr, Distinguished Chair for Research on Colon Cancer. B.R. has support from The Swim Across America Foundation Postdoctoral Fellowship. Research was partially supported by the Stand Up to Cancer Colorectal Cancer Dream Team Translational Research Grant (SU2C-AACR-DT22-17). Stand Up to Cancer is a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
B.R. has served in a consulting/advisory role for Bayer, Roche, Novartis, Gilead, and Servier, and has received travel, accommodations, and expenses from Bayer, Servier, and Astellas. L.A.D. is a member of the board of directors of Personal Genome Diagnostics (PGDx) and Jounce Therapeutics. He is a paid consultant to PGDx, 4Paws (PetDx), Innovatus CP, Se’er, Kinnate and Neophore. He is an uncompensated consultant for Merck but has received research support for clinical trials from Merck. L.A.D. is an inventor of multiple licensed patents related to technology for circulating tumor DNA analyses and mismatch repair deficiency for diagnosis and therapy from Johns Hopkins University. Some of these licenses and relationships are associated with equity or royalty payments directly to Johns Hopkins and L.A.D.. He holds equity in PGDx, Jounce Therapeutics, Thrive Earlier Detection, Se’er, Kinnate and Neophore. His spouse holds equity in Amgen. The terms of all these arrangements are being managed by Johns Hopkins and Memorial Sloan Kettering in accordance with their conflict of interest policies.
References
- Bakhoum SF, Ngo B, Laughney AM, Cavallo JA, Murphy CJ, Ly P, Shah P, Sriram RK, Watkins TBK, Taunk NK, et al. (2018). Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553, 467-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland CR, and Goel A (2010). Microsatellite instability in colorectal cancer. Gastroenterology 138, 2073–2087 e2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan TA, Wolchok JD, and Snyder A (2015). Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. N Engl J Med 373, 1984. [DOI] [PubMed] [Google Scholar]
- Chang K, Taggart MW, Reyes-Uribe L, Borras E, Riquelme E, Barnett RM, Leoni G, San Lucas FA, Catanese MT, Mori F, et al. (2018). Immune Profiling of Premalignant Lesions in Patients With Lynch Syndrome. JAMA Oncol 4, 1085–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho BK, Rao VP, Ge Q, Eisen HN, and Chen J (2000). Homeostasis-stimulated proliferation drives naive T cells to differentiate directly into memory T cells. J Exp Med 192, 549–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, Sher X, Liu XQ, Lu H, Nebozhyn M, et al. (2018). Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, Li XD, Mauceri H, Beckett M, Darga T, et al. (2014). STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 41, 843–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Der SD, Zhou A, Williams BR, and Silverman RH (1998). Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc Natl Acad Sci U S A 95, 15623–15628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz L, Marabelle A, Kim T, Geva R, Van Cutsem E, André T, Ascierto P, Maio M, Delord J, and Gottfried M.J.A.o.O. (2017). 386PEfficacy of pembrolizumab in phase 2 KEYNOTE-164 and KEYNOTE-158 studies of microsatellite instability high cancers 28. [Google Scholar]
- Dunn GP, Koebel CM, and Schreiber RD (2006). Interferons, immunity and cancer immunoediting. Nat Rev Immunol 6, 836–848. [DOI] [PubMed] [Google Scholar]
- Ganesh K, Stadler ZK, Cercek A, Mendelsohn RB, Shia J, Segal NH, and Diaz LA Jr. (2019). Immunotherapy in colorectal cancer: rationale, challenges and potential. Nat Rev Gastroenterol Hepatol 16, 361–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germano G, Lamba S, Rospo G, Barault L, Magri A, Maione F, Russo M, Crisafulli G, Bartolini A, Lerda G, et al. (2017). Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 552, 116–120. [DOI] [PubMed] [Google Scholar]
- Ghandi M, Huang FW, Jane-Valbuena J, Kryukov GV, Lo CC, McDonald ER 3rd, Barretina J, Gelfand ET, Bielski CM, Li H, et al. (2019). Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan J, Lu C, Jin Q, Lu H, Chen X, Tian L, Zhang Y, Ortega J, Zhang J, Siteni S, Chen M, et al. (2021). MLH1 Deficiency-Triggered DNA Hyper-Excision by Exonuclease 1 Activates the cGAS-STING Pathway. Cancer Cell in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman SE, Bertone P, Nath AK, Royce TE, Gerstein M, Weissman S, and Snyder M (2005). Global changes in STAT target selection and transcription regulation upon interferon treatments. Genes Dev 19, 2953–2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heemskerk B, Kvistborg P, and Schumacher TN (2013). The cancer antigenome. EMBO J 32, 194–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indraccolo S, Pfeffer U, Minuzzo S, Esposito G, Roni V, Mandruzzato S, Ferrari N, Anfosso L, Dell’Eva R, Noonan DM, et al. (2007). Identification of genes selectively regulated by IFNs in endothelial cells. J Immunol 178, 1122–1135. [DOI] [PubMed] [Google Scholar]
- Joffre OP, Segura E, Savina A, and Amigorena S (2012). Cross-presentation by dendritic cells. Nat Rev Immunol 12, 557–569. [DOI] [PubMed] [Google Scholar]
- Koelzer VH, Baker K, Kassahn D, Baumhoer D, and Zlobec I (2012). Prognostic impact of beta-2-microglobulin expression in colorectal cancers stratified by mismatch repair status. J Clin Pathol 65, 996–1002. [DOI] [PubMed] [Google Scholar]
- Kopetz S, Andre T, Overman MJ, Zagonel V, Lonardi S, Aglietta M, Gelsomino F, McDermott R, Wong KYM, Hendlisz A, et al. (2018). Exploratory analysis of Janus kinase 1 (JAK1) loss-of-function (LoF) mutations in patients with DNA mismatch repair-deficient/microsatellite instability-high (dMMR/MSI-H) metastatic colorectal cancer (mCRC) treated with nivolumab plus ipilimumab in CheckMate-142. Cancer Research 78. [Google Scholar]
- Kwon J, and Bakhoum SF (2020). The Cytosolic DNA-Sensing cGAS-STING Pathway in Cancer. Cancer Discov 10, 26–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, et al. (2017). Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et al. (2015). PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med 372, 2509–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos H, Mohamed E, Huang L, Ou R, Pacholczyk G, Arbab AS, Munn D, and Mellor AL (2016). STING Promotes the Growth of Tumors Characterized by Low Antigenicity via IDO Activation. Cancer Research 76, 2076–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GM (2008). Mechanisms and functions of DNA mismatch repair. Cell Res 18, 85–98. [DOI] [PubMed] [Google Scholar]
- Li X, Liu Z, Zhang A, Han C, Shen A, Jiang L, Boothman DA, Qiao J, Wang Y, Huang X, et al. (2019). NQO1 targeting prodrug triggers innate sensing to overcome checkpoint blockade resistance. Nat Commun 10, 3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Zhang H, Wu X, Ma D, Wu J, Wang L, Jiang Y, Fei Y, Zhu C, Tan R, et al. (2018). Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature 563, 131–136. [DOI] [PubMed] [Google Scholar]
- Liu SY, Sanchez DJ, Aliyari R, Lu S, and Cheng G (2012). Systematic identification of type I and type II interferon-induced antiviral factors. Proc Natl Acad Sci U S A 109, 4239–4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM, Blosser RL, Fan H, Wang H, Luber BS, et al. (2015). The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov 5, 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maleki Vareki S (2018). High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J Immunother Cancer 6, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal R, Samstein RM, Lee KW, Havel JJ, Wang H, Krishna C, Sabio EY, Makarov V, Kuo F, Blecua P, et al. (2019). Genetic diversity of tumors with mismatch repair deficiency influences anti-PD-1 immunotherapy response. Science 364, 485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marabelle A, Le DT, Ascierto PA, Di Giacomo AM, De Jesus-Acosta A, Delord JP, Geva R, Gottfried M, Penel N, Hansen AR, et al. (2020). Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J Clin Oncol 38, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardjuki RE, Carozza JA, and Li L (2020). Development of cGAMP-Luc, a sensitive and precise coupled enzyme assay to measure cGAMP in complex biological samples. J Biol Chem 295, 4881–4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, Kadel EE III, Koeppen H, Astarita JL, Cubas R, et al. (2018). TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modrich P, and Lahue R (1996). Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu Rev Biochem 65, 101–133. [DOI] [PubMed] [Google Scholar]
- Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, Desai J, Hill A, Axelson M, Moss RA, et al. (2017). Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol 18, 1182–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantelidou C, Sonzogni O, De Oliveria Taveira M, Mehta AK, Kothari A, Wang D, Visal T, Li MK, Pinto J, Castrillon JA, et al. (2019). PARP Inhibitor Efficacy Depends on CD8(+) T-cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov 9, 722–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz-Ares L, Langer CJ, Novello S, Halmos B, Cheng Y, Gadgeel SM, Hui R, Sugawara S, Borghaei H, Cristescu R, et al. (2019). LBA80Pembrolizumab (pembro) plus platinum-based chemotherapy (chemo) for metastatic NSCLC: Tissue TMB (tTMB) and outcomes in KEYNOTE-021, 189, and 407. Annals of Oncology 30. [Google Scholar]
- Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, Sun Y, Zhao E, Vatan L, Szeliga W, et al. (2015). Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 527, 249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, Xu C, McKenzie JA, Zhang C, Liang X, et al. (2016). Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov 6, 202–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, Dawson N, O’Donnell PH, Balmanoukian A, Loriot Y, et al. (2016). Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet 387, 1909–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanda C, Weitzel P, Tsukahara T, Schaley J, Edenberg HJ, Stephens MA, McClintick JN, Blatt LM, Li L, Brodsky L, et al. (2006). Differential gene induction by type I and type II interferons and their combination. J Interferon Cytokine Res 26, 462–472. [DOI] [PubMed] [Google Scholar]
- Sceneay J, Goreczny GJ, Wilson K, Morrow S, DeCristo MJ, Ubellacker JM, Qin Y, Laszewski T, Stover DG, Barrera V, et al. (2019). Interferon Signaling Is Diminished with Age and Is Associated with Immune Checkpoint Blockade Efficacy in Triple-Negative Breast Cancer. Cancer Discov 9, 1208–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen T, Rodriguez BL, Chen LM, Della Corte CM, Morikawa N, Fujimoto J, Cristea S, Nguyen T, Diao LX, Li LR, et al. (2019). Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer Discovery 9, 646–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Zhao W, Ju Z, Wang L, Peng Y, Labrie M, Yap TA, Mills GB, and Peng G (2019). PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res 79, 311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, Grasso CS, Hugo W, Sandoval S, Torrejon DY, et al. (2017). Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov 7, 188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, Vitale I, Goubar A, Baracco EE, Remedios C, et al. (2014). Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med 20, 1301–1309. [DOI] [PubMed] [Google Scholar]
- Spranger S, Bao R, and Gajewski TF (2015). Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 523, 231–235. [DOI] [PubMed] [Google Scholar]
- Spranger S, Luke JJ, Bao R, Zha Y, Hernandez KM, Li Y, Gajewski AP, Andrade J, and Gajewski TF (2016). Density of immunogenic antigens does not explain the presence or absence of the T-cell-inflamed tumor microenvironment in melanoma. Proc Natl Acad Sci U S A 113, E7759–E7768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Wang Y, Chlewicki LK, Zhang Y, Guo J, Liang W, Wang J, Wang X, and Fu YX (2016). Facilitating T Cell Infiltration in Tumor Microenvironment Overcomes Resistance to PD-L1 Blockade. Cancer Cell 30, 500. [DOI] [PubMed] [Google Scholar]
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. (2014). PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Foppen MHG, Goldinger SM, et al. (2015). Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350, 207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasaikar S, Huang C, Wang X, Petyuk VA, Savage SR, Wen B, Dou Y, Zhang Y, Shi Z, Arshad OA, et al. (2019). Proteogenomic Analysis of Human Colon Cancer Reveals New Therapeutic Opportunities. Cell 177, 1035–1049 e1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Loga K, Woolston A, Punta M, Barber LJ, Griffiths B, Semiannikova M, Spain G, Challoner B, Fenwick K, Simon R, et al. (2020). Extreme intratumour heterogeneity and driver evolution in mismatch repair deficient gastro-oesophageal cancer. Nat Commun 11, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Hu SQ, Chen X, Shi HP, Chen C, Sun LJ, and Chen ZJJ (2017). cGAS is essential for the antitumor effect of immune checkpoint blockade. P Natl Acad Sci USA 114, 1637–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, Duggan R, Wang Y, Barber GN, Fitzgerald KA, et al. (2014). STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41, 830–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Yuan F, Presnell SR, Tian K, Gao Y, Tomkinson AE, Gu L, and Li GM (2005). Reconstitution of 5’-directed human mismatch repair in a purified system. Cell 122, 693–705. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Distinct Type I ISGs list, Related to Figure 2
Table S3 Melanoma samples of clinical outcome, Related to STAR Methods and Figure S6
Data Availability Statement
Original TCGA data in the paper is available here https://gdac.broadinstitute.org/.