

Abstract
The use of carbon dioxide as a C1 chemical feedstock remains an active field of research. Here we showcase the use of milled dry ice as a method to promote the availability of CO2 in a reaction solution, permitting practical synthesis of arylcarboxylic acids. Notably, the use of milled dry ice produces marked increases in yields relative to those obtained with gaseous CO2, as previously reported in the literature.
Keywords: carboxylation, carbon dioxide, C1 source, dry ice, arylcarboxylic acids
Graphical Abstract

Carboxylic acid functional groups play a major role in chemistry as biologically relevant substituents, medicinally relevant pharmacophores, or synthetically relevant handles that can be used to deliver more complex moieties.1–3 Whereas the synthesis of carboxylic acids has been well studied, the impetus provided by green chemistry has driven a demand for new methods that use renewable chemical feedstocks.4 To meet this demand, carbon dioxide (CO2) has been widely employed as a cheap, renewable, and atom-economical C1 building block.5–7 The use of solid CO2 in the synthesis of carboxylic acids from aryl lithiates or Grignard reagents has been known for some time.8,9 Green chemistry examples have also been reported in which catalytic methods are used to deliver the desired carboxylic acids through the use of CO2 gas. Recently, Zhang and co-workers reported a visible-light-mediated carboxylation of aryl halides with gaseous CO2.10 Interestingly, this method employed a phosphine ligand whose twofold role involved first impeding the formation of the undesired protodehalogentated product through use of a basic moiety and secondly encouraging emulsion formation in the biphasic mixture of water and toluene. This surfactant-promoted emulsion technique exploits the high availability of CO2 present in solution to deliver the desired carboxylic acids in good yields. Alternative methods have reported the use of ultrahigh pressures and temperatures as forcing conditions in carboxylation reactions.11 We recently reported a convenient synthetic route to deliver xanthylium and acridinium photocatalysts, in which ortho-lithiated biaryl ethers were condensed onto methyl benzoate derivatives in high yield.12 While the scope of this reaction proved to be robust, its scalability was limited by difficulties in obtaining the benzoic acid starting materials. Notably, lithium–halogen exchange conditions of the corresponding aryl bromide followed by subsequent addition of gaseous CO2 at ambient pressures gave 4-fluoro-2,6-dimethylbenzoic acid (1; see Scheme 1 below) in only a modest 17% yield.
Scheme 1.
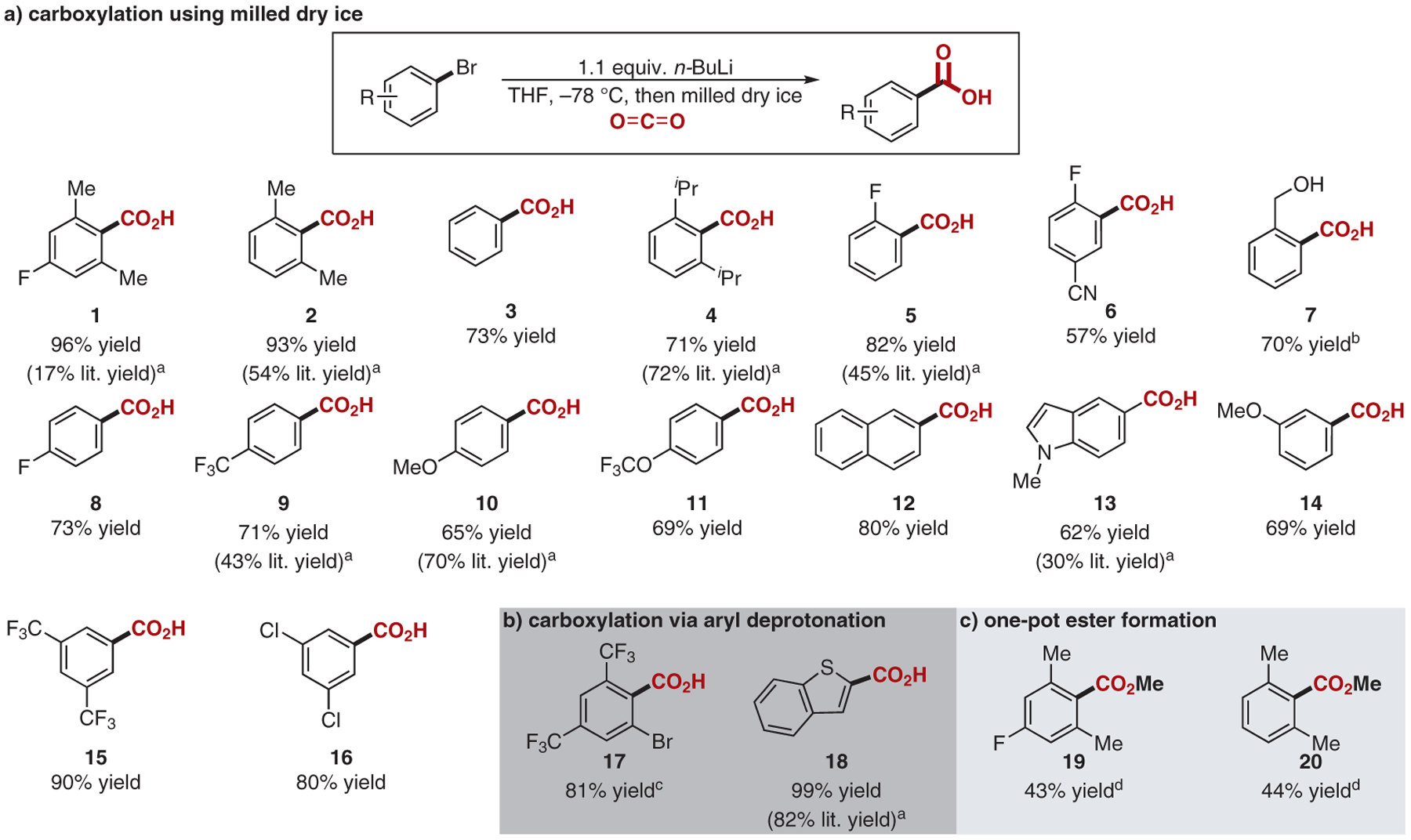
Substrate scope. Yields reported as the average isolated yield of two separate trials run on 1.0 mmol scale. a Highest yield reported in the literature through aryllithium or arylmagnesium addition to gaseous CO2 at atmospheric pressure.12,18–25 b n-BuLi (2.1 equiv). cLiTMP (1.0 equiv). d After the standard reaction conditions were employed, K2CO3 (1.5 equiv), MeI (1.2 equiv), and DMF were added to the reaction flask.
These syntheses entail many of the drawbacks associated with the use of CO2 as a reagent, chiefly that of delivering readily available dry CO2 in solution. This task is often cumbersome, requiring the gas to be passed through a drying agent such as calcium sulfate, molecular sieves, or neat sulfuric acid, thereby greatly limiting the practicality of the method.12,13 Furthermore, the solubility of CO2 varies greatly in organic solvents, leading to solvent–reaction incompatibilities.14,15 To address these shortcomings, we envisioned the deployment of dry ice, milled in a pestle and mortar, as a desirable CO2 source that has a markedly enhanced surface area and, in turn, delivers a greatly increased availability of CO2 in solution. The solid CO2 provides a marked practical improvement upon CO2 gas in terms of both its ease of use and in permitting larger scale reactions.
To deliver acid 1 in a more synthetically useful yield than the 17% yield previously reported, we sought to develop reaction conditions that implement principles discussed in the literature, and that also overcome the undesired introduction of water through condensation on milled dry ice, a common problem associated with its use. These goals were accomplished by washing the milled dry ice with THF under nitrogen.16 With milled dry ice as the CO2 source, the use of THF as a solvent instead of diethyl ether gave the desired acid 1 in an improved 43% yield. Decreasing the temperature of the lithium–halogen exchange from 0 to −78 °C compounded this improvement, resulting in a yield of 96% (Scheme 1a). This significant increase in yield is probably due to suppression of the decomposition of the aryl–lithium intermediate, which occurs at higher temperatures.17 Fortunately, the low reagent loading and the high efficiency of the reaction permitted the delivery of the desired product after a simple acid–base workup, circumventing the need for column chromatography or solvent recrystallization. This practicality was supportive of a multigram-scale reaction (3.04 g from 14.8 mmol of the starting aryl bromide), permitting the delivery of 1 in a consistent 96% yield.
With these improved reaction conditions in hand, we applied our method to a range of other substrates. The general reaction procedure gave 2,6-dimethylbenzoic acid (2) in a comparable 93% yield. 2,6-Diisopropylbenzoic acid (4), with bulkier isopropyl substituents at the two ortho-positions, was also obtained in good yield (71%). Changing the electronics of the para-substituent had little impact on reaction yield: fluoro (8, 73%), trifluoromethyl (9, 71%), methoxy (10, 65%), and trifluoromethoxy (11, 69%) substituents were all well tolerated. Notably, these reaction conditions are chemoselective, delivering the desired carboxylic acid 16 in 80% yield in the presence of two chloro substituents.
We then sought to elaborate this method to the lithiation of aryl moieties through proton removal instead of lithium–halogen exchange (Scheme 1b). Notably, removal of an aryl proton was achieved in the presence of a bromo substituent through the use of LiTMP instead of n-BuLi, delivering the bromobenzoic acid (17) in a good 81% yield. Moreover, lithiation and subsequent carboxylation of 1-benzothiophene to give 18 proceeded almost quantitatively (99% yield).
We also sought to elaborate our carboxylation method to install additional substrate functionality in one-pot fashion via the carboxylate intermediate. After formation of 2,6-dimethylbenzoic acid (2) under the reaction conditions previously described, 1.2 equivalents of iodomethane were added to the reaction mixture in an attempt to deliver the corresponding methyl ester 19. However, after 24 hours, none of the desired product was obtained. A subsequent acidic quench, workup, and trituration, delivered acid 2 in a slightly reduced yield (80%). Simple subsequent addition of DMF, potassium carbonate, and methyl iodide to the general reaction conditions then gave the desired methyl ester 19 in a decent 43% yield. Again, the corresponding methyl 2,6-dimethylbenzoate (20) was delivered in a comparable yield (44%). The apparent decrease in yield relative to the two-step method reported in the literature is probably a result of the strong coordination of the lithium ion to the carboxylate intermediate. Consequently, a stepwise synthetic route to these products is probably more desirable.12
In summary, the direct carboxylation of aryl bromides by using milled dry ice as a C1 source is demonstrated. The use of milled dry ice produced a significant increase in yields compared with methods previously reported in the literature that employ gaseous CO2. Aryl and hetaryl substrates with ether, halogen, nitrile, or alcohol functional groups underwent conversions in yields of 57–99%. The reaction provides rapid access to carboxylic acid derivatives with a low reagent loading and permits the production of these synthetically useful products without the need for column chromatography or solvent recrystallization. The reactive carboxylate intermediates were also elaborated to provide ester functionalities in a one-pot synthesis.
Supplementary Material
Funding Information
National Institute of General Medical Sciences (R35 GM136330)
Footnotes
Supporting Information
Supporting information for this article is available online at https://doi.org/10.1055/a-1384-0159.
References and Notes
- (1).Ruzi R; Liu K; Zhu C; Xie J Nat. Commun 2020, 11, 3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Lassalas P; Gay B; Lasfargeas C; James MJ; Tran V; Vijayendran KG; Brunden KR; Kozlowski MC; Thomas CJ; Smith AB III.; Huryn DM; Ballatore C J. Med. Chem 2016, 59, 3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Ballatore C; Huryn DM; Smith AB III. ChemMedChem 2013, 8, 385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Sheldon RA Green Chem. 2014, 16, 950. [Google Scholar]
- (5).Vechorkin O; Hirt N; Hu X Org. Lett 2010, 12, 3567. [DOI] [PubMed] [Google Scholar]
- (6).Fujihara T; Tsuji Y Front. Chem 2019, 7, 430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Liu Q; Wu L; Jackstell R; Beller M Nat. Commun 2015, 6, 5933. [DOI] [PubMed] [Google Scholar]
- (8).Gilman H; Beel JA J. Am. Chem. Soc 1949, 71, 2328. [Google Scholar]
- (9).Hussey AS J. Am. Chem. Soc 1951, 73, 1364. [Google Scholar]
- (10).Zhang Z; Roisnel T; Dixneuf PH; Soulé JF Angew. Chem. Int. Ed 2019, 58, 14110. [DOI] [PubMed] [Google Scholar]
- (11).Luo J; Larrosa I ChemSusChem 2017, 10, 3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).White AR; Wang L; Nicewicz DA Synlett 2019, 30, 827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Wiberg E; Holleman AF; Wiberg N Inorganic Chemistry; Academic Press; 2001. [Google Scholar]
- (14).Miller MB; Chen D-L; Luebke DR; Johnson JK; Enick RM J. Chem. Eng. Data 2011, 56, 1565. [Google Scholar]
- (15).Kunerth W Phys. Rev 1922, 19, 512. [Google Scholar]
- (16).4-Fluoro-2,6-dimethylbenzoic Acid (1); Typical Procedure Under a positive pressure of N2, 2-bromo-5-fluoro-1,3-dimethylbenzene (3.040 g, 1.0 equiv, 14.97 mmol) was added to a flame-dried 100 mL round-bottomed flask and then diluted with anhyd THF (20 mL). The solution was cooled to −78 °C and a 1.6 M solution of n-BuLi (1.055 g, 1.1 equiv, 16.47 mmol) in hexanes (10.29 mL) was added dropwise. The resulting solution was then stirred at −78 °C for 1 h.Separately, under a constant stream of N2, anhyd THF (~50 mL) was added to a flame-dried, 250 mL Erlenmeyer flask and cooled to −78 °C. Freshly milled dry ice (~75 g) was slowly added to the flask from a powder funnel. The flask was vigorously swirled, ensuring that the milled dry ice remained completely submerged in the solvent. The resulting slurry was filtered by vacuum filtration under a constant stream of N2. After complete removal of the solvent, the resulting milled dry ice was quickly transferred to the original round-bottomed reaction flask by using a powder funnel (see the Supporting Information for additional details).The resulting reaction mixture was removed from the cooling bath and stirred at r.t. until complete sublimation of the dry ice was observed. (Safety Note: Sublimation of dry ice produces a large amount of gas. To prevent a dangerous buildup of pressure, the reaction flask was left open to the atmosphere after dry ice addition.) The solution was then concentrated in vacuo, and the residue was dissolved in H2O (25 mL) and CH2Cl2 (25 mL). The resulting bilayer mixture was transferred to a separatory funnel, and the organic layer was removed. The aqueous layer was washed with CH2Cl2 (3 × 25 mL) and acidified to pH 2–3 with 1 M aq HCl. The resulting aqueous solution was then extracted with CH2Cl2 (4 × 25 mL), and the organic layers were collected, dried (MgSO4), filtered, and concentrated in vacuo to give a white solid; yield: 2.409 g (14.32 mmol, 95.68%, n = 4). 1H NMR (400 MHz, CDCl3): δ = 6.81 (d, J = 9.3 Hz, 2 H), 2.47 (s, 6 H). The spectral data agreed with those previously reported.
- (17).Wu J; Yang X; He Z; Mao X; Hatton TA; Jamison TF Angew. Chem. Int. Ed 2014, 53, 8416. [DOI] [PubMed] [Google Scholar]
- (18).Shiina I; Miyao R Heterocycles 2008, 76, 1313. [Google Scholar]
- (19).Zhang Q; Zhang FM; Zhang CS; Liu SZ; Tian JM; Wang SH; Zhang XM; Tu YQ Nat. Commun 2019, 10, 2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Paridala K; Lu S-M; Wang M-M; Li C Chem. Commun 2018, 54, 11574. [DOI] [PubMed] [Google Scholar]
- (21).Schlosser M; Cottet F; Heiss C; Lefebvre O; Marull M; Masson E; Scopelliti R Eur. J. Org. Chem 2006, 729. [Google Scholar]
- (22).Correa A; Martín RJ Am. Chem. Soc 2009, 131, 15974. [DOI] [PubMed] [Google Scholar]
- (23).Screttas CG; Steele BR J. Org. Chem 1989, 54, 1013. [Google Scholar]
- (24).Nagaraj HKM; Bandodkar BS; Ravilla L; Yellapu S; Rudresha AS; Singh JK; G V WO 2016027285, 2016.
- (25).Gschwend HW; Rodriguez HR Org. React 1979, 26, 1 DOI: 10.1002/0471264180.or026.01. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.