

Abstract
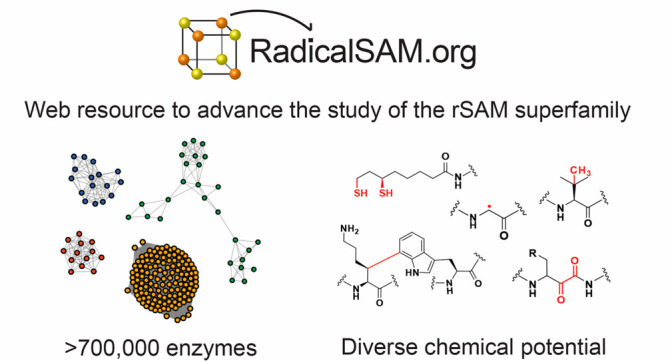
The radical SAM superfamily (RSS), arguably the most functionally diverse enzyme superfamily, is also one of the largest with ∼700 K members currently in the UniProt database. The vast majority of the members have uncharacterized enzymatic activities and metabolic functions. In this Perspective, we describe RadicalSAM.org, a new web-based resource that enables a user-friendly genomic enzymology strategy to explore sequence-function space in the RSS. The resource attempts to enable identification of isofunctional groups of radical SAM enzymes using sequence similarity networks (SSNs) and the genome context of the bacterial, archaeal, and fungal members provided by genome neighborhood diagrams (GNDs). Enzymatic activities and in vivo functions frequently can be inferred from genome context given the tendency for genes of related function to be clustered. We invite the scientific community to use RadicalSAM.org to (i) guide their experimental studies to discover new enzymatic activities and metabolic functions, (ii) contribute experimentally verified annotations to RadicalSAM.org to enhance the ability to predict novel activities and functions, and (iii) provide suggestions for improving this resource.
Keywords: Radical SAM superfamily, genomic enzymology, web tools, functional assignment, isofunctional families, protein sequence similarity networks, genome neighborhood diagrams
This issue celebrates the 20th anniversary of the discovery of the radical SAM superfamily (RSS).1 The seminal bioinformatic study of 645 proteins by Sofia et al. in 2001 revealed a conserved CX3CX2C motif located near the N-terminus of a (β/α)6-barrel domain that coordinates a [4Fe–4S] center that binds S-adenosylmethionine (SAM). As of mid-2021, genome projects had identified ∼700 K additional members that include orthologues of characterized members as well as many uncharacterized members with potentially new enzyme activities and metabolic functions. However, the sheer size and accelerating growth of the RSS create a classic “big data” problem: exploration and interpretation of the sequence-function space has become untenable for non-bioinformaticians. In this Perspective, we describe RadicalSAM.org (https://radicalsam.org/), an open-access “genomic enzymology” web resource designed for experimental biochemists that leverages the UniProt2 (protein) and European Nucleotide Archive3 (ENA; nucleotide) databases (Figure 1). This Perspective provides a general textual and graphical overview of the resource; we also provide videos on the RadicalSAM.org tutorial page (https://radicalsam.org/tutorials.php) that describe the features of RadicalSAM.org.
Figure 1.
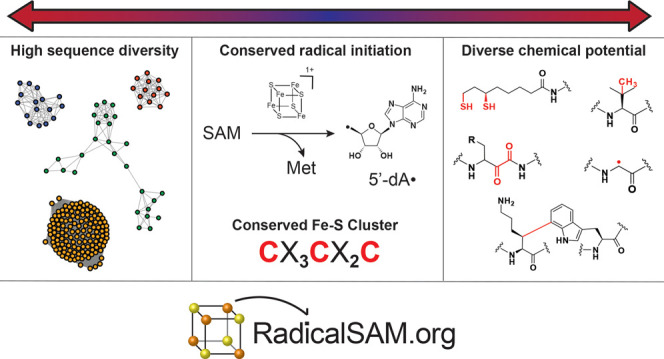
Despite high diversity in sequence and reaction outcome, all members of the RSS generate 5′-deoxyadenosyl (5′-dA) radical using a conserved [4Fe–4S]-forming motif that binds and reductively liberates Met from SAM.4−7 RadicalSAM.org provides easy access to a genomic enzymology strategy to catalog known enzymatic activities and discover new ones within the RSS.
RSS in the Structure–Function Linkage Database (SFLD)
The now archival Structure–Functional Linkage Database (SFLD) linked sequence-structure features to different chemical capabilities in several functionally diverse superfamilies,8 including the RSS (http://sfld.rbvi.ucsf.edu/archive/django/superfamily/29/index.html). The SFLD segregated the sequence similarity network (SSN) of the RSS into 20 subgroups with functionally characterized members and 22 subgroups with uncharacterized members (designated by numbers and colors in Figure 2; see expanded image in the Supporting Information); the 20 subgroups and their names are provided in Table 1. In the last update (2017),9 the SSN was generated using ∼114 K sequences then available in Pfam10 family PF04055 and InterPro11 family IPR007197 collected at 50% sequence identity into 10,741 representative nodes (PF04055 and additional families defined by SFLD and PROSITE12 are incorporated into IPR007197). The subgroups were identified by segregating the nodes in the SSN using a maximum e-value threshold of 1e-20 to draw edges.
Figure 2.

SSN generated with a maximum e-value edge threshold of 1e-20 used by the SFLD to identify its 20 functionally characterized subgroups (colored/numbered clusters) and 22 uncharacterized subgroups. Large nodes represent experimentally characterized proteins: downward arrows indicate a structurally characterized protein; diamonds indicate no structural characterization. Reproduced with permission from ref (9). Copyright 2018 Elsevier.
Table 1. SFLD Subgroups, Names, and (Mega)clusters in RadicalSAM.org.
| subgroup | subgroup name | (Mega)cluster |
|---|---|---|
| 1 | 7-carboxy-7-deazaguanine synthase-like | Megacluster-3-1 |
| 2 | coproporphyrinogen III oxidase-like | Megacluster-2-2 |
| 3 | antiviral proteins (viperin) | Megacluster-1-5 |
| 4 | avilamycin synthase | Megacluster-1 |
| 5 | B12-binding domain containing | Megacluster-2-1 |
| 6 | biotin and thiazole synthase domain containing | Megacluster-4 |
| 7 | DesII-like | Megacluster-1-8 |
| 8 | ELP3/YhcC | Megacluster-2-4, -2-5 |
| 9 | F420, menaquinone cofactor biosynthesis | Megacluster-4-2 |
| 10 | FeMo-cofactor biosynthesis protein | Megacluster-1-4 |
| 11 | lipoyl synthase like | Cluster-8 |
| 12 | methylthiotransferase | Megacluster-2-3 |
| 13 | methyltransferase class A | Cluster-6 |
| 14 | methyltransferase class D | Megacluster-1-3 |
| 15 | organic radical activating enzymes | Megacluster-3 |
| 16 | PLP-dependent | Megacluster-7 |
| 17 | SPASM/twitch domain containing | Megacluster-1-1 |
| 18 | spectinomycin biosynthesis | Megacluster-1-1 |
| 19 | spore photoproduct lyase | Megacluster-5-3 |
| 20 | tRNA wybutosine-synthesizing | Cluster-10 |
| protein MJ0683-like | Megacluster-5-1 | |
| uncharacterized protein family UPF0313 | Cluster-10 | |
| DUF5131 | Megacluster-5-2 | |
| 3′,8-cyclase/Mo cofactor synthesis | Megacluster-1-2 |
As the protein databases continue to grow (doubling time of ∼2 years), the archival SFLD provides an increasingly outdated description of the RSS sequence-function space. Additionally, not all RSS subgroups are curated as Pfam and/or InterPro families, so current membership in these cases is unavailable. Furthermore, many of the hidden Markov models (HMMs) that were used to define the membership for the previously curated subgroups are no longer sufficient to reliably classify individual RSS proteins.
RadicalSAM.org: A Resource to Accelerate the Discovery of New Enzyme Chemistry
We anticipate that many readers want to assign in vitro enzymatic activities and in vivo metabolic functions to uncharacterized members of the RSS. We propose that this can be facilitated using a “genomic enzymology” strategy.13 With this strategy, the members of a functionally diverse superfamily are segregated into potential isofunctional families using SSNs (separate SSN clusters). Then, the genomic contexts of the bacterial, archaeal, and fungal members of the clusters are retrieved and profiled. When the genomic neighborhood is reminiscent of a known pathway, the local context can be leveraged to rapidly formulate high quality hypotheses and aid in the design of experiments to confirm or refute the predicted enzymatic activity. When the genomic neighborhood is not similar to that for a known pathway, the user can use contextual clues and other information to inform decisions on further experimental characterization.
The identification of isofunctional families cannot be accomplished using sequence identity alone—the sequence boundaries between homologues (proteins derived from a common ancestor) and orthologues (homologues separated by speciation that likely exhibit the same enzymatic activities and metabolic functions) are not easily determined. As a further complication, a single pairwise minimum sequence identity threshold (or minimum edge alignment score threshold in SSNs) likely will not separate orthologous groups (clusters) across any superfamily, especially those that are functionally diverse like the RSS.
Genome context is a powerful approach for inferring isofunctionality for bacterial, archaeal, and fungal enzymes: shared genome context (encoding the same metabolic pathway) can be used as evidence for shared function. However, genome context is not necessarily preserved across diverse taxa, so the ability to readily survey the genome contexts for all members of isofunctional families is essential for inferring metabolic functions and enzymatic activities.
To “democratize” the genome enzymology strategy, we developed and provide a web-based resource14−17 (https://efi.igb.illinois.edu/) with tools for (i) generating SSNs for protein families and separating these into clusters based on pairwise sequence identity thresholds (EFI-EST; https://efi.igb.illinois.edu/efi-est/); and (ii) collecting, visualizing, and analyzing genome context of proteins in the SSN clusters using genome neighborhood networks (GNNs) and genome neighborhood diagrams (GNDs; EFI-GNT; https://efi.igb.illinois.edu/efi-gnt/). The resource also provides a tool for prioritizing uncharacterized isofunctional SSN clusters for functional discovery based on metagenome abundance using chemically guided functional profiling (CGFP; EFI-CGFP; https://efi.igb.illinois.edu/efi-cgfp/).
Although the tools have accelerated the biochemical characterization of many proteins,15,18 the size of the RSS prevents the experimental community from fully utilizing our resource, i.e., the necessary computational resources for visualizing the SSNs far exceed what is common for personal computers. Therefore, we developed RadicalSAM.org (https://radicalsam.org/), an open-access, web-based resource, for exploring sequence-function space for radical SAM enzymes that share the conserved CX3CX2C motif identified by Sofia et al. (Figure 3).
Figure 3.

Home page of RadicalSAM.org.
Following the approach used by the SFLD,9 the SSN for the RSS is segregated into functionally curated as well as uncharacterized subgroups (vide infra). RadicalSAM.org provides lists of the UniProt IDs for the sequences in the various subgroups as well as precalculated SSNs so that users can explore their regions of interest within the larger sequence-function space. RadicalSAM.org also enables straightforward access to the genome context (GNDs) for members of the subgroups as well as for individual proteins in UniProt.
The remainder of this Perspective describes general features and salient details of RadicalSAM.org. Readers interested in using RadicalSAM.org to guide their experimental work are encouraged to view the videos on the RadicalSAM.org tutorial page (https://radicalsam.org/tutorials.php) that provide an overview of RadicalSAM.org as well as descriptions of some of its tools. The first video (Index of Tutorial Videos at the end of the paper) provides an overview of RadicalSAM.org.
Sequences in RadicalSAM.org
RadicalSAM.org includes sequences for radical SAM enzymes that share the conserved CX3CX2C motif identified by Sofia et al.1 Three known radical SAM families that do not share this motif are not included: (i) diphthamide synthase19 (PF01866), (ii) phosphomethylpyrimidine synthase20 (ThiC; PF01964), and (iii) α-d-ribose 1-methylphosphonate C-P lyase21 (PhnJ; PF06007).
The UniProt 2020_05 (October 7, 2020) and InterPro 82 (October 8, 2020) databases were used to develop the release of RadicalSAM.org described in this Perspective. We used Option B of EFI-EST to specify one Pfam family and 172 InterPro families/domains (including PF04055 and IPR007197 used by the SFLD) for collecting sequences to include in the SSN, with the goal of providing a more comprehensive inventory of the membership than was provided by the SFLD. These families/domains are listed on the “Sequence Families” subtab of the “Current Release” tab on Home page of RadicalSAM.org.
We identified 664,196 UniProt IDs representing 579,102 unique sequences in 66,428 UniRef50 clusters. RadicalSAM.org uses UniRef50 and UniRef90 sequence clusters (sequences that share ≥50% and ≥90% sequence identity, respectively; https://www.uniprot.org/help/uniref) to decrease the computational requirements for generating SSNs and enable visualization of the SSNs with Cytoscape.22 A representative UniProt accession within each UniRef cluster is assigned by UniProt as its identifier. The sequence of the identifier is used to generate SSNs, multiple sequence alignments (MSAs), HMMs, and length histograms.
Not every retrieved sequence is “full length” (vide infra). We removed truncated sequences from RadicalSAM.org to (1) improve the quality and reliability of the multiple sequence alignments used to generate WebLogos23 and HMMs and (2) reduce the number of isolated nodes (singletons) in SSNs generated with alignment score edge thresholds that collect sequences into putative isofunctional clusters.
We used two procedures to remove truncated sequences:
(1) UniProt designates a “Sequence Status” for each accession: “Complete” if the encoding DNA includes both a start and stop codon; “Fragment” if either a start or stop codon is absent. After excluding fragments with the “Fragment Option” of Option B of EFI-EST, the sequence set contained 620,386 UniProt IDs represented by 535,892 unique sequences in 52,886 UniRef50 clusters.
(2) A “Complete” sequence may be truncated because of sequencing errors. The shortest “Complete” sequence in PF04055 (UniProt ID A0A351TBI7) contains 39 residues and includes the CX3CX2C motif; another “Complete” sequence (UniProt ID A0A376TI31) contains 58 residues without the CX3CX2C motif. We inspected the UniProt ID length histograms for all clusters in a prototype version of RadicalSAM.org and identified anaerobic ribonucleotide-triphosphate reductase activating enzymes as the “shortest” family with ≥140 residues. Therefore, we used 140 residues for the minimum length filter for generating the SSN used by RadicalSAM.org. The final sequence set contained 616,009 UniProt IDs represented by 531,705 unique sequences (in 50,232 UniRef50 clusters).
In unpublished SSNs generated with more recent Pfam/InterPro releases, we used three additional Pfam families and 28 additional InterPro families/domains to collect sequences missing in the sequence set used for the current release of RadicalSAM.org (Table S1). As an example, PoyD,24,25 a radical SAM enzyme involved in polytheonamide biosynthesis that epimerizes l-amino acids within a peptide substrate to the d-configuration (UniProt ID J9ZW29), is missing in RadicalSAM.org; it is included in IPR030950 that was added. If/when users recognize the absence of bona fide members of the RSS, they should contact us so that these can be included in future updates of RadicalSAM.org. These omissions could occur if additional Pfam/InterPro families and domains are needed to identify sequences; alternatively, sequences will be missing if they had not been deposited in the UniProt database used to identify sequences.
The NCBI nr protein database is much larger than the UniProt database (∼424 M sequences on October 8, 2021 vs ∼220 M sequences in UniProt Release 2021_03), so it includes more members of the RSS. As a result, radical SAM proteins that have been described/characterized in the literature may not be deposited in the UniProt database and, therefore, are not in RadicalSAM.org. As an example, NxxcB, a radical SAM enzyme from Streptococcus orisratti that installs a β-thioether bond in a ribosomally synthesized and post-translationally modified peptide (RiPP),26 is present in the NCBI database (accession identifier WP_018375754.1) but not UniProt or RadicalSAM.org. Orthologues from other Streptococcus sp. can be located in RadicalSAM.org with the “Search by Sequence” feature (vide infra) and are present in Megacluster-1-1, SFLD subgroup 17, SPASM/twitch domain-containing.
When this paper was in preparation (October 2021), the UniProt 2021_03 and InterPro 86 databases were the most current. Using the current list of Pfam families and InterPro families/domains, we identified 754,719 UniProt IDs representing 664,851 unique sequences in 78,535 UniRef50 clusters. After excluding fragments, the sequence set contained 700,346 UniProt IDs represented by 611,187 unique sequences in 62,118 UniRef50 clusters. After applying the 140 residue minimum length filter, the sequence set contained 694,831 UniProt IDs represented by 605,897 unique sequences in 58,733 UniRef50 clusters.
RSS SSN
The SSN generated with the full length UniRef50 cluster identifiers can be visualized and edited with Cytoscape 3.8.222 installed on a Mac Pro computer with 768 GB RAM. By visual inspection of SSNs with nodes colored by SFLD subgroup (with curated InterPro families/domains), a minimum edge alignment score threshold of 11 groups UniRef50 clusters into SFLD subgroups and also allows segregation of the SFLD subgroups (Figure 4). The resulting large cluster contained 615,705 UniProt IDs represented by 531,425 unique sequences in 50,084 UniRef50 clusters. All further analysis and subgroup identification used these sequences.
Figure 4.
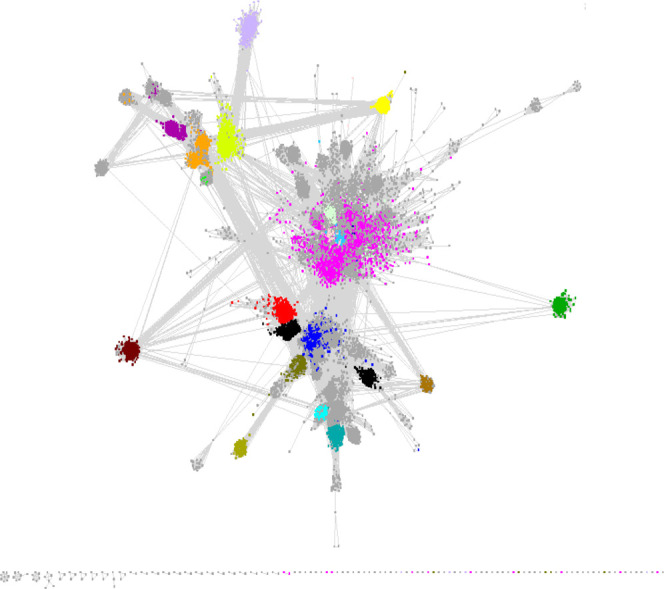
SSN of the starting point for RSS subgroup identification. The network is visualized with 11 as the minimum edge alignment score threshold and colored based on previously assigned SFLD subgroups (Figure 2 and Table 1).
Identification of Subgroups
In contrast to the SFLD’s SSN, none of the functionally characterized SFLD subgroups are separated into distinct clusters in this SSN, the result of the larger number of UniProt IDs/UniRef50 clusters and the choice of a smaller minimum edge alignment score threshold to prevent separation of the SFLD subgroups into multiple clusters (the SFLD’s SSN used a maximum edge e-value threshold of 1e-20).
The SSN clusters containing the SFLD subgroups were segregated by manual deletion of edges using Cytoscape as described in the SUBGROUPS/SUBGROUP IDENTIFICATION tab on the RadicalSAM.org home page. The resulting SSN contained 10 clusters: 5 have been designated “megaclusters” because they contain multiple SFLD subgroups (and uncharacterized subgroups), and 5 have been designated as “clusters” that contain only a single SFLD subgroup (Clusters 6–10). The (mega)clusters are numbered in order of decreasing number of UniRef50 IDs/nodes; Megacluster-1 through Megacluster-5 then Cluster-6 through Cluster-10 (Figure 5). The megaclusters were also segregated by manual deletion of edges into SFLD-curated and uncharacterized subgroups as described in the SUBGROUPS tab on the RadicalSAM.org home page.
Figure 5.

Five megaclusters (containing multiple SFLD subgroups) and five standard clusters (containing single SFLD subgroups) after manual deletion of “long” edges that correspond to larger alignment scores and connect functionally divergent nodes/subgroups in the large cluster in Figure 4. The nodes are colored based on previously assigned SFLD subgroups (Figure 2 and Table 1).
The segregation resulted in the 56 clusters/subgroups included in RadicalSAM.org (Figure 6). As described on their Explore pages (next section), some of the subclusters in Megaclusters-3 and -4 were further manually segregated (using increased minimum edge alignment score thresholds) to separate UniProtKB/SwissProt functions.
Figure 6.

Segregated subgroups in RadicalSAM.org. The nodes are colored based on previously assigned SFLD subgroups (Figure 2 and Table 1). (A) UniRef50 SSNs for Megaclusters-1-2 through -1-8 were removed from Megacluster-1 using an alignment score edge threshold of 30 and displayed with 16 as the minimum edge alignment score threshold. The remaining Megacluster-1-1 is displayed using 11 as the minimum edge alignment score threshold. (B) UniRef50 SSNs for Megaclusters-2-1 through -2-7 were segregated using 12 as the minimum edge alignment score threshold. (C) UniRef90 SSNs for Megaclusters-3-1 through -3-12 segregated using 18 as the minimum edge alignment score threshold. (D) UniRef90 SSNs for Megaclusters-4-1 through -4-11 segregated using 22 as the minimum edge alignment score threshold. (E) UniRef50 SSNs for Megaclusters-5-1 through -5-3 were segregated using 12 as the minimum edge alignment score threshold. (F) UniRef50 SSNs for Clusters 6-10 from the segregated SSN in Figure 5.
Explore Pages
RadicalSAM.org provides bioinformatic and genome context information (GNDs) for each of the clusters/subgroups on cluster-specific Explore pages. A representative Explore page, for Megacluster-3-1, 7-carboxy-7-deazaguanine synthase-like, SFLD subgroup 1, is shown in Figure 7 (see expanded image in the Supporting Information).
Figure 7.
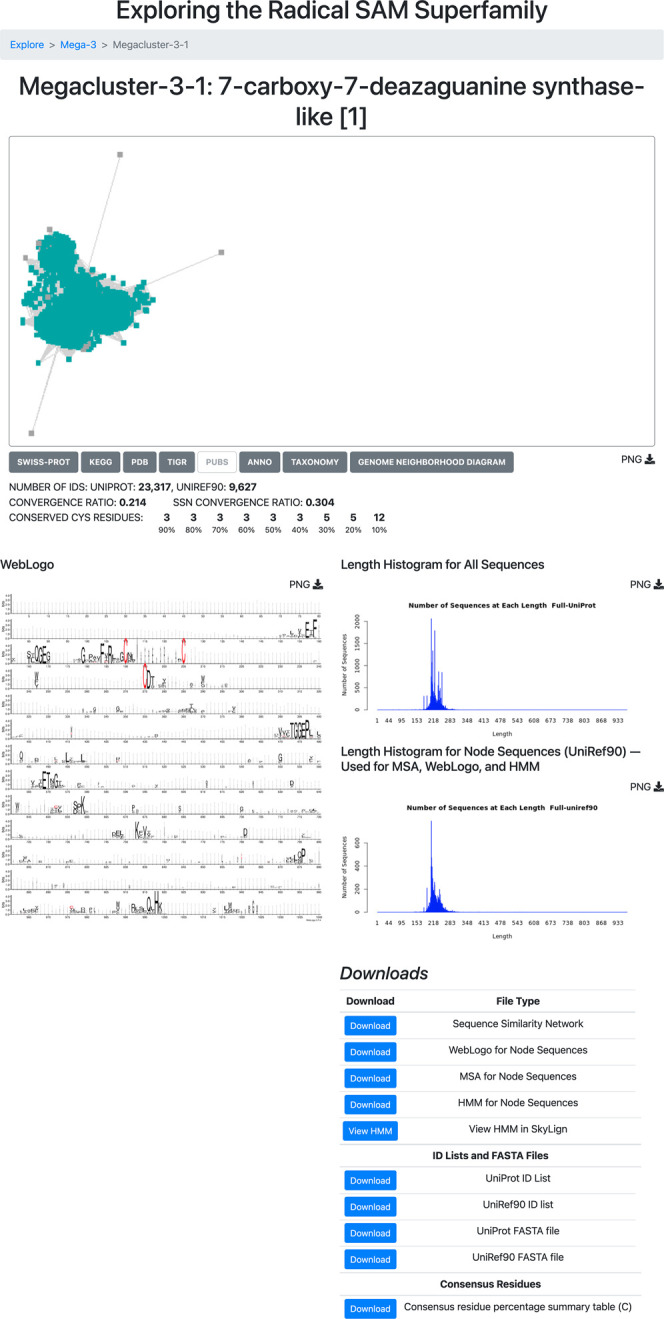
Example of an Explore page using Megacluster-3-1, the 7-carboxy-7-deazaguanine synthase-like radical SAM proteins (SFLD subgroup 1).
This section highlights several types of useful information provided by Explore pages. The second video (Index of Tutorial Videos at the end of the paper) provides a description of the contents of an Explore page.
(1) The convergence ratios (CRs) calculated using the UniRef 50 or UniRef90 cluster identifiers and the UniProt IDs in the SSN cluster using the minimum edge alignment score threshold (e-value) used to generate the cluster. The CR is a measure of sequence similarity and is the ratio of the number of edges at the specified alignment score relative to the total number of sequence pairs (maximum number of edges). The value decreases from 1.0 for sequences that are highly similar (not necessarily identical, depending on the value of the alignment score) to a value approaching 0 for very divergent sequences.
The CR is particularly useful for analyzing the “diced” clusters (next section) for which RadicalSAM.org provides a series of SSNs generated as a function of increasing minimum edge alignment score thresholds (increasing pairwise percent identity); as the clusters become isofunctional and the sequences become orthologous, the value of CR approaches 1.0. Exceptions occur when an isofunctional cluster contains orthologues from diverse taxa; i.e., the value of CR in an isofunctional cluster may decrease as the minimum edge alignment score threshold increases as taxonomic divergence in sequence dominates the CR.
(2) The numbers of conserved Cys residues in the multiple sequence alignment (MSA) calculated from 90 to 10% conservation in steps of 10%. This number is influenced by both sequence and length heterogeneity within the cluster (vide infra). By definition, all members of the RSS contain three conserved Cys residues in the CX3CX2C motif that participates in the [4Fe–4S] center than binds SAM; however, many subgroups contain additional [Fe–S] centers coordinated to additional conserved Cys residues. Examples are provided in a later section.
(3) A list of community-provided annotations (ANNO button). The UniProtKB/SwissProt database is not a comprehensive list of experimentally verified functions; annotations provided by the community using the form provided on the SUBMIT tab at the top of each page inform inference of possible uncharacterized functions. We compiled the annotations currently provided; moving forward, we ask users to contribute annotations to add to the resource (vide infra).
(4) The taxonomy sunburst (Figure 8) provides a graphical display of the taxonomic distribution of sequences in the cluster (TAXONOMY button; https://github.com/vasturiano/sunburst-chart). Clicking on a wedge expands the view to include the sequences represented in that wedge. Lists of accession IDs and FASTA files are available for download at any selected taxonomic level.
Figure 8.
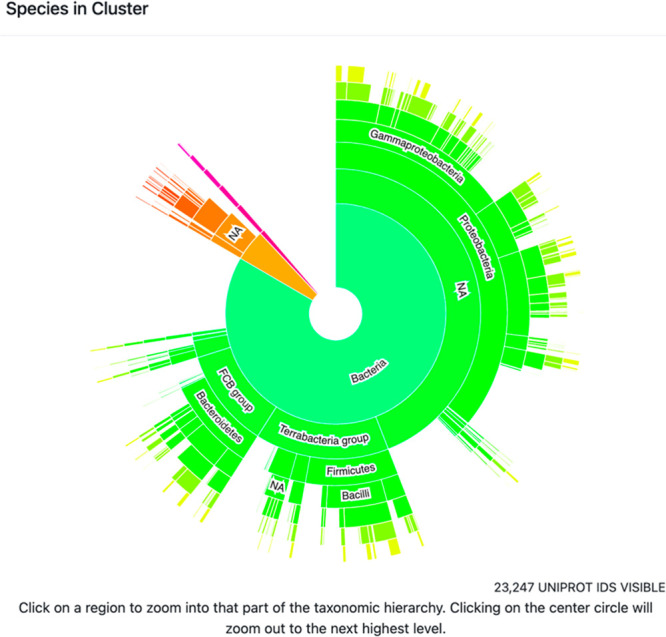
Example of a taxonomy sunburst display. Shown is Megacluster-3-1, 7-carboxy-7-deazaguanine synthase-like (SFLD subgroup 1). Green, bacteria; orange, archaea; magenta, eukaryota.
(5) GNDs (GENOME NEIGHBORHOOD DIAGRAM button; Figure 9; see expanded image in the Supporting Information) can be viewed for the UniRef50 cluster identifiers, UniRef90 cluster identifiers, and UniProt IDs for UniRef50 clusters and for the UniRef90 cluster identifiers and UniProt IDs for UniRef90 SSN clusters. The GNDs are used in the identification of isofunctional clusters as well as to provide genome context (metabolic pathway context) for discovering novel enzymatic activities and metabolic functions. The third video (Index of Tutorial Videos at the end of the paper) explains the use of the GND viewer.
Figure 9.

Representative GND Viewer page: Cluster-6 in the “diced” SSN for Megacluster-1-1 generated with a minimum edge alignment score threshold of 60.
(6) The multiple sequence alignment (MSA) for the UniRef cluster identifiers in an SSN cluster is generated with MUSCLE27,28 and can be opened with Jalview29,30 (https://www.jalview.org/). The MSA is used to assess function/sequence heterogeneity.
(7) The WebLogo23 (http://weblogo.threeplusone.com) for the SSN cluster generated from the MSA. Given their importance in RSS structure and function, Cys residues are highlighted in red to allow their easy identification.
(8) The HMM for the SSN cluster displayed using Skylign31 (https://skylign.org/; Figure 10). This HMM viewer allows quick visualization of the consensus sequence for the cluster. HMMs provide the probability of a residue at any position in the sequence, not the percent conservation.
Figure 10.

Portion of the Skylign display of the HMM for Megacluster-3-1, 7-carboxy-7-deazaguanine synthase-like, SFLD subgroup 1, showing the conserved CX3CX2C motif that binds SAM.
“Dicing” of Functionally Diverse SFLD Subgroups
SFLD subgroups 17, 5, 2, and 16, are particularly large and functionally diverse. In RadicalSAM.org, these are Megacluster-1-1 (SPASM/twitch domain-containing), Megacluster-2-1 (B12-binding domain-containing), Megacluster-2-2 (anaerobic coproporphyrinogen III oxidase-like), and Cluster-7 (PLP-dependent), respectively. As noted previously, the SSNs for functionally diverse superfamilies do not segregate into isofunctional families/SSN clusters using a single minimum edge alignment score threshold, making identification of orthologues a challenge. To aid in determining isofunctionality, RadicalSAM.org provides a series of SSNs for these subgroups with an increasing minimum edge alignment score threshold that we designate as SSN “dicing”. RadicalSAM.org also provides GNDs for each cluster in the diced SSNs to permit genome context to be used to infer isofunctionality and possible enzymatic activities and metabolic pathways.
The diced SSNs are generated with UniRef90 cluster identifiers instead of the UniRef50 cluster identifiers used to generate the SSN for the RSS so the sequences within each node are more likely to be isofunctional (sequences with ≥90% sequence identity). The sequences in the lower resolution UniRef50 clusters may be heterofunctional so the GNDs for the cluster identifiers as well as the UniProt IDs in the cluster may provide misleading information about metabolic context.
Using Megacluster-1-1 (SPASM32/twitch33 domain-containing) as an example, a series of 33 SSNs was generated as a function of minimum edge alignment score threshold, ranging from 25 in which the SSN is dominated by a large, complex cluster to 300 in which the SSN contains only a few small clusters with large CR values and likely isofunctional nodes (Figure 11). In the diced SSNs, only clusters with ≥3 nodes are shown (an Explore page is provided for each cluster).
Figure 11.

Representative diced SSNs for Megacluster-1-1 (SPASM/twitch domain-containing).
As the minimum edge alignment score threshold increases, the SSN clusters that emerge are more similar in sequence and, therefore, are more likely to carry out similar reactions. However, since a single alignment score edge threshold for generating isofunctional clusters does not apply across the entire SSN, each diced SSN will be a mixture of heterofunctional and isofunctional clusters. The GND that can be accessed for each cluster in each SSN can be used to assess functional homogeneity in the cluster, realizing that genome context for orthologues often is not conserved across taxonomic divisions, as well as potentially provide metabolic pathway context for inferring functions of uncharacterized enzymes.
The Explore page for each cluster in each diced SSN provides the ability (Figure 12; see expanded image in the Supporting Information) to (i) step forward/backward through the clusters in each SSN (left panel), (ii) select any cluster in the current SSN (center panel), or (iii) select any SSN in the diced series (right panel).
Figure 12.

Navigation through diced megaclusters. (A) “Previous” and “Next” navigation buttons (green arrows) direct the cluster selection backward and forward, respectively, in the selected diced SSN. (B) “Cluster” drop down menu allows the user to select any cluster in the currently viewed diced SSN. (C) Alignment score drop down menu allows the user to view the cluster in the selected diced SSN.
The alignment score (AS) “AS Walk-Through” button is located above the image for each cluster in the “diced” SSNs (Figure 13; see expanded image in the Supporting Information). This button opens a pop-up window that identifies the progeny clusters at the next alignment score or the progenitor cluster at the previous alignment score; these can be accessed by clicking the name of the cluster, thereby allowing the identification of functionally diverse homologues, i.e., homologues that share a common mechanistic attributes such regiochemistry/stereochemistry of hydrogen abstraction. The walk-through window provides the UniProtKB/SwissProt function(s), if any, and user-supplied annotation(s), if any, in each progenitor and progeny cluster. The fourth video (Index of Tutorial Videos at the end of the paper) describes navigation through a series of diced SSNs.
Figure 13.

AS Walk-Through pop-up window showing the identity of the progenitor cluster (“Previous Cluster) and progeny clusters (“Next Clusters”).
Alternatively, using the Search tab in the menu bar at the top of the page (Figure 14; see expanded image in the Supporting Information), the diced SSNs can be searched with either a UniProt ID (“Find by UniProt ID”) or a sequence (“Find by Sequence”). “Find by UniProt ID” identifies the cluster containing the specified UniProt ID in each diced SSN and provides its CR; clicking on the cluster number opens the Explore page for the cluster. “Find by Sequence” uses hmmscan34 to scan the HMMs for the clusters in each SSN in the series and provides a list of matching clusters, their e-values, and CRs; clicking on the cluster number opens the Explore window for the cluster. The fifth video (Index of Tutorial Videos at the end of the paper) provides a description of the Search tab.
Figure 14.
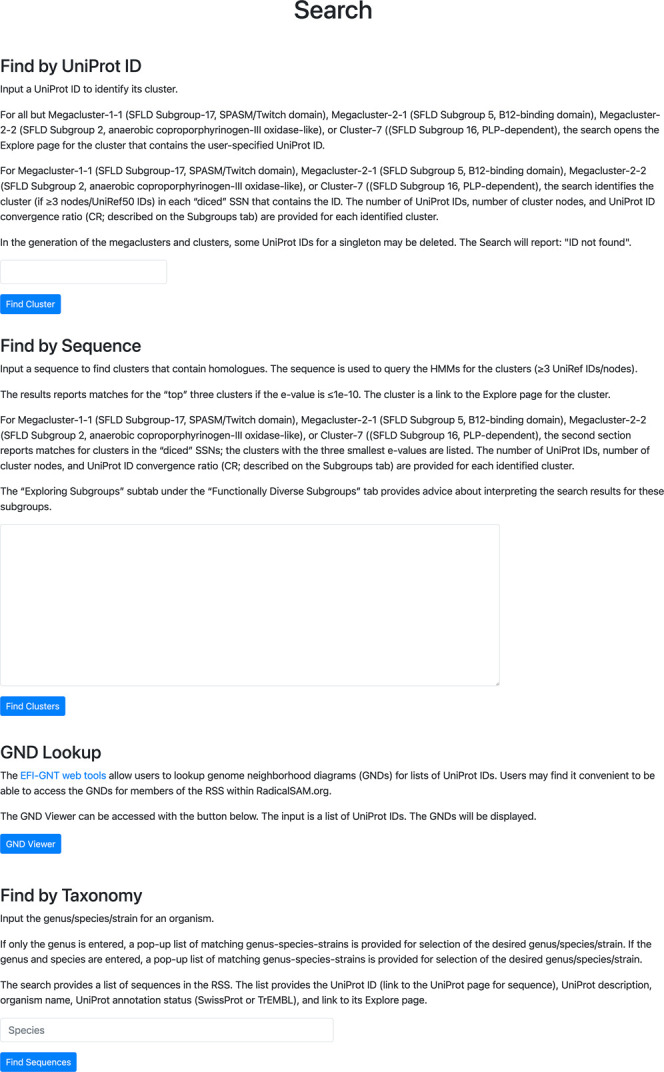
Search functions: “Find by UniProt ID” identifies the cluster(s) containing the user-specified accession ID; “Find by Sequence” identifies the cluster(s) with the best HMM match to the user-specified sequence; “GND Lookup” provides the GND(s) for the user-specified UniProt ID(s); “Find by Taxonomy” provides a list of the UniProt accession IDs and their clusters for the user-specified genus/species.
If the diced SSNs are searched with a UniProt ID, the AS Walk-Through window identifies the progeny cluster that contains the UniProt ID. If the diced SSNs are searched with a sequence, the walk-through window identifies the progeny cluster with the lowest E-value to the HMM for the cluster (best match to the cluster HMM). These features facilitate the selection of progeny clusters for more detailed investigation. The sixth video (Index of Tutorial Videos at the end of the paper ) illustrates the use of dicing to aid in the identification of isofunctional families/clusters using PqqE, the PqqA peptide cyclase in pyrroloquinoline quinone (PQQ) biosynthesis, as a case study.
Conserved Cys Residues
The chemistry of [Fe–S] centers is central to understanding reaction mechanisms in the RSS. RadicalSAM.org provides easy access to the number and positions of conserved Cys residues in isofunctional clusters, e.g., allowing identification of Cys motifs that form [Fe–S] centers in addition to the CX3CX2C motif that binds SAM as well other conserved Cys residues not involved in forming [Fe–S] centers but may be involved in the reaction mechanism.
Each Explore page provides a list of number of Conserved Cys Residues at 90, 80, 70, 60, 50, 40, 30, 20, and 10% sequence conservation (from the downloadable Consensus Residue table). The number almost always is a function of percent conservation, with the number increasing as percent conservation decreases. This occurs for two reasons: (1) length heterogeneity resulting from the presence of “truncated” sequences, although we have tried to remove as many of these as possible; and (2) sequence heterogeneity resulting from functional heterogeneity (the cluster is not isofunctional).
Inspection of the WebLogo, MSA, and/or Skylign display of the HMM (“View HMM in Skylign”) for a cluster allows visual identification of conserved Cys motifs and their locations in the sequence. These motifs are most quickly identified by inspection of the Skylign HMM display (“View HMM in Skylign”) since the HMM display “removes” insertions/deletions.
By definition, a member of the RSS contains three conserved Cys residues in the conserved CX3CX2C motif that form the SAM-binding [4Fe–4S] center. However, variations of this motif can be identified using RadicalSAM.org, e.g., Megacluster-2-4-1 with a CX11CX2C motif, Megacluster-2-5 with a CX7CX2C motif, Megaclusters-4-6-1, -4-6-3, -4-6-5, and -4-6-6 with CX8CX2C motifs, Megacluster-4-6-4 with a CX9CX2C motif, Megacluster-4-10 with a CX5CX2C motif, Megacluster-4-11 with a CX4CX2C motif, and Megacluster-5-2 with a CX6CX2C motif.
Many subgroups/clusters contain additional conserved Cys motifs. Although widely distributed across the RSS, many are found in Megacluster-1-1, which contains SPASM/twitch domains. SPASM domains located C-terminal to the (β/α)6-barrel domain contain seven or eight conserved Cys residues that bind two additional [4Fe–4S] centers;32 similarly positioned twitch domains contain three or four conserved Cys residues that bind one additional [4Fe–4S] center.33 Many isofunctional clusters that contain SPASM or twitch domains can be identified in the diced SSNs for Megacluster-1-1 generated with alignment scores ≥ 60 (as inferred from conserved genome context in the GNDs); interestingly, the conserved Cys residues do not share common sequence motifs; i.e., they occur with different inter-Cys residue spacings.
The diced SSNs for Megacluster-1-1 also contain many clusters with other numbers of conserved Cys residues, ranging from 1 to >28 (e.g., Megacluster-1-1-11 at alignment score 300). Although most of the additional conserved Cys residues are located C-terminal to the (β/α)6-barrel domain, some are located N-terminal to the (β/α)6-barrel domain; in some members of Megacluster-1-1, additional conserved Cys residues are found both N- and C-terminal to the (β/α)6-barrel domain. Although structures are not available for these proteins, perusal of models generated by trRosetta35/RoseTTAfold36/AlphaFold37 suggests that these Cys residues usually are located in additional domains. Users of RadicalSAM.org can submit sequences to trRosetta (https://yanglab.nankai.edu.cn/trRosetta/) or RoseTTAfold (https://robetta.bakerlab.org/) and then highlight the positions of Cys residues when viewing the predicted structures to determine whether the additional conserved Cys residues are proximal in space and, therefore, may participate in binding of additional [Fe–S] centers.
Additional conserved Cys motifs are also found in other subgroups/clusters. Notable examples include SFLD subgroup 14, methyltransferase Class D (Megacluster-1-3), with six conserved Cys residues N-terminal to the (β/α)6-barrel domain; SFLD subgroup 12, methylthiotransferase (Megacluster-2-3), with three conserved Cys residues N-terminal to the (β/α)6-barrel domain; SFLD subgroup 15, organic radical activating enzymes (Megacluster-3-2-1-1), with nine additional conserved Cys residues in the (β/α)6-barrel domain; SFLD subgroup 15, organic radical activating enzymes (Megacluster-3-2-1-3) with two conserved Cys residues N-terminal to the (β/α)6-barrel domain and six additional conserved Cys residues in the RSS (β/α)6-barrel domain.
A Community Resource
We invite members of the community to enhance the capabilities of this resource by contributing their experimentally determined enzymatic activities and metabolic functions for previously uncharacterized members of the RSS. The SUBMIT tab on the Home page provides a form for providing this information (Figure 15; see expanded image in the Supporting Information).
Figure 15.
Submit page for community submission of enzymatic activities and metabolic functions.
Informed inference of function in sequence-function space requires as many experimentally confirmed landmarks as possible. Although the SSNs generated by EFI-EST include a SwissProt Description node attribute (from UniProt), the SwissProt database is not a comprehensive (and accurate/reliable) list of characterized functions. Mining the literature for experimentally verified functions is tedious, and unfortunately, many publications fail to include an accession identifier (UniProt or NCBI) for experimentally investigated proteins. Therefore, it is challenging (sometimes impossible) to associate a published experimentally established function with a specific protein. Nonetheless, we have provided a large number of literature citations for experimentally characterized members that are accessible using the ANNO(TATION) buttons on the Explore pages and AS Walk-Through pop-up windows in the diced SSNs. The “Submit” tab provides users with the ability to submit annotations and publication DOI’s for specific accession IDs. With each update of RadicalSAM.org, we will make these functions and publications available.
We ask that members of the community include the UniProt and/or NCBI accession IDs for proteins that are functionally characterized in their publications. The IDs make it easier not only for UniProtKB/SwissProt to target new annotations for curation but also for members of the community to associate enzymatic activities and metabolic functions with specific proteins in the databases. BIOCHEMISTRY requires that authors include accession identifiers;38 we hope that additional journals will do the same in the future. In the meantime, please help future scientific data curation efforts by including accession identifiers in all of your publications!
Finally, we encourage suggestions for improving RadicalSAM.org. Our guiding principle has been to provide information in a format that is friendly to experimentalists. Indeed, as RadicalSAM.org has been developed, we realized the need for new useful features that have been incorporated, e.g., “dicing” and the AS Walk-Through pop-up windows. The CONTACT tab on the Home page can be used to submit suggestions (Figure 16).
Figure 16.

Contact page form for submitting feedback or questions.
Future/Planned Enhancements
RadicalSAM.org will be most useful when it is updated regularly, ideally with each update of the UniProt database (every 8 weeks). We plan to implement a pipeline for updates in which the subgroups and their HMMs are defined annually (using the first annual release of UniProt) by manual dissection of the SSN; then, for subsequent releases in the calendar year, the members of the subgroups will be retrieved using the HMMs.
The current release of RadicalSAM.org uses dicing to facilitate identification of isofunctional clusters/families in four of the largest, functionally diverse SFLD subgroups, i.e., the ability to provide genome context/GNDs for inference of function is the central concept of genomic enzymology. We are pleased with this feature and would like to extend it to all other subgroups. In principle, this can be automated, i.e., generating SSNs as a function of alignment score followed by using the EFI-EST Cluster Analysis and Convergence Ratio utilities to obtain the information provided on the Explore pages. In practice, the automated generation of images of individual clusters for diced SSNs with thousands of clusters is computationally demanding but required for the user to assess divergence of sequence (and function) as the clusters segregate into smaller clusters as the minimum edge alignment score threshold increases.
We plan to provide predicted three-dimensional structures for the UniRef90 cluster identifiers in the various SSN clusters/subgroups generated by AlphaFold/DeepMind.37 We will implement this feature when UniProt/InterPro makes this comprehensive set of AlphaFold predictions available; currently predicted structures are available for the proteins encoded by 22 prototype organisms (https://www.alphafold.ebi.ac.uk/). These structures will enable the visualization of domain organization/topology in multidomain RSS members as well as the locations of conserved Cys motifs that bind [Fe–S] centers. Perhaps some of these structures will be useful for the prediction of substrates, products, and mechanisms using virtual ligand/intermediate docking.39,40
We would like to adapt our tools to use the NCBI and JGI-IMG databases, both of which are larger than UniProt; we have received requests from the community to do this. The sequences in the NCBI and JGI-IMG databases are not as well curated as those in UniProt, so any SSNs using these databases would contain fewer node attributes. Also, the NCBI database does not always assign membership in Pfam families and InterPro families/domains to its sequences so identification of the members of the RSS is associated with certain challenges. As time and resources permit, we will investigate whether future versions of RadicalSAM.org can be developed to be compatible with NCBI and JGI-IMG.
Acknowledgments
The conceptualization and computational infrastructure that enabled the development of RadicalSAM.org was supported by two grants from NIH/NIGMS (U54GM093342 and P01GM118303). We thank Daniel Davidson and David Slater for their assistance with programming and cluster support. We also thank members of the Mitchell group for locating many UniProt IDs for previously characterized radical SAM enzymes by literature and database mining.
Index of Tutorial Videos
-
1.
RadicalSAM.org empowers researchers in the field of radical SAM enzymology to discover novel functions of uncharacterized members of the RSS by democratizing the genomic enzymology tools. This video provides a tour of the site and encourages the user to explore the tools using their own radical SAM enzymes.
-
2.
The Explore tab gives the user the ability to maneuver through the enormous protein data set in RadicalSAM.org. This video provides a tour of the Explore tab and details the various tools in the tab.
-
3.
GNDs are powerful tools in determining the potential function of a radical SAM enzyme. This video describes their use and how they have been incorporated into RadicalSAM.org.
-
4.
RadicalSAM.org uses SSNs generated at a series of increasing alignment scores, through a process known as dicing, to assist the user in determining points of isofunctionality in the radical SAM superfamily. This video provides a brief description of that process.
-
5.
The Search tab enables the user to search for a specific radical SAM in RadicalSAM.org. This video provides a tour of the Search tab and describes how a user can find useful information about their radical SAM enzyme.
-
6.
This video provides a tutorial on determining an isofunctional alignment score for a radical SAM of interest using PqqE, the PqqA peptide cyclase in pyrroloquinoline quinone (PQQ) biosynthesis, as a case study.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsbiomedchemau.1c00048.
List of additional Pfam families and InterPro families/domains used to identify members of the RSS; expanded image of Figure 2 in the text to provide visual clarity; expanded image of Figure 7 in the text to provide visual clarity; expanded image of Figure 9 in the text to provide visual clarity; expanded image of Figure 12A in the text to provide visual clarity; expanded image of Figure 12B in the text to provide visual clarity; expanded image of Figure 12C in the text to provide visual clarity; expanded image of Figure 13 in the text to provide visual clarity; expanded image of Figure 14 in the text to provide visual clarity; expanded image of Figure 15 in the text to provide visual clarity; expanded image of Figure 16 in the text to provide visual clarity (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Sofia H. J.; Chen G.; Hetzler B. G.; Reyes-Spindola J. F.; Miller N. E. Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional characterization using new analysis and information visualization methods. Nucleic Acids Res. 2001, 29, 1097–1106. 10.1093/nar/29.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. 10.1093/nar/gkaa1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison P. W.; Ahamed A.; Aslam R.; Alako B. T. F.; Burgin J.; Buso N.; Courtot M.; Fan J.; Gupta D.; Haseeb M.; Holt S.; Ibrahim T.; Ivanov E.; Jayathilaka S.; Balavenkataraman Kadhirvelu V.; Kumar M.; Lopez R.; Kay S.; Leinonen R.; Liu X.; O’Cathail C.; Pakseresht A.; Park Y.; Pesant S.; Rahman N.; Rajan J.; Sokolov A.; Vijayaraja S.; Waheed Z.; Zyoud A.; Burdett T.; Cochrane G. The European Nucleotide Archive in 2020. Nucleic Acids Res. 2021, 49, D82–D85. 10.1093/nar/gkaa1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey P. A.; Hegeman A. D.; Ruzicka F. J. The Radical SAM Superfamily. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 63–88. 10.1080/10409230701829169. [DOI] [PubMed] [Google Scholar]
- Vey J. L.; Drennan C. L. Structural insights into radical generation by the radical SAM superfamily. Chem. Rev. 2011, 111, 2487–2506. 10.1021/cr9002616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booker S. J. Radical SAM enzymes and radical enzymology. Biochim. Biophys. Acta, Proteins Proteomics 2012, 1824, 1151–1153. 10.1016/j.bbapap.2012.07.006. [DOI] [PubMed] [Google Scholar]
- Broderick W. E.; Hoffman B. M.; Broderick J. B. Mechanism of Radical Initiation in the Radical S-Adenosyl-l-methionine Superfamily. Acc. Chem. Res. 2018, 51, 2611–2619. 10.1021/acs.accounts.8b00356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiva E.; Brown S.; Almonacid D. E.; Barber A. E. 2nd; Custer A. F.; Hicks M. A.; Huang C. C.; Lauck F.; Mashiyama S. T.; Meng E. C.; Mischel D.; Morris J. H.; Ojha S.; Schnoes A. M.; Stryke D.; Yunes J. M.; Ferrin T. E.; Holliday G. L.; Babbitt P. C. The Structure-Function Linkage Database. Nucleic Acids Res. 2014, 42, D521–530. 10.1093/nar/gkt1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holliday G. L.; Akiva E.; Meng E. C.; Brown S. D.; Calhoun S.; Pieper U.; Sali A.; Booker S. J.; Babbitt P. C. Atlas of the Radical SAM Superfamily: Divergent Evolution of Function Using a ″Plug and Play″ Domain. Methods Enzymol. 2018, 606, 1–71. 10.1016/bs.mie.2018.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry J.; Chuguransky S.; Williams L.; Qureshi M.; Salazar G. A.; Sonnhammer E. L. L.; Tosatto S. C. E.; Paladin L.; Raj S.; Richardson L. J.; Finn R. D.; Bateman A. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. 10.1093/nar/gkaa913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum M.; Chang H. Y.; Chuguransky S.; Grego T.; Kandasaamy S.; Mitchell A.; Nuka G.; Paysan-Lafosse T.; Qureshi M.; Raj S.; Richardson L.; Salazar G. A.; Williams L.; Bork P.; Bridge A.; Gough J.; Haft D. H.; Letunic I.; Marchler-Bauer A.; Mi H.; Natale D. A.; Necci M.; Orengo C. A.; Pandurangan A. P.; Rivoire C.; Sigrist C. J. A.; Sillitoe I.; Thanki N.; Thomas P. D.; Tosatto S. C. E.; Wu C. H.; Bateman A.; Finn R. D. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 2021, 49, D344–D354. 10.1093/nar/gkaa977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigrist C. J.; de Castro E.; Cerutti L.; Cuche B. A.; Hulo N.; Bridge A.; Bougueleret L.; Xenarios I. New and continuing developments at PROSITE. Nucleic Acids Res. 2012, 41, D344–347. 10.1093/nar/gks1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlt J. A.; Babbitt P. C. Divergent evolution of enzymatic function: mechanistically diverse superfamilies and functionally distinct suprafamilies. Annu. Rev. Biochem. 2001, 70, 209–246. 10.1146/annurev.biochem.70.1.209. [DOI] [PubMed] [Google Scholar]
- Gerlt J. A.; Bouvier J. T.; Davidson D. B.; Imker H. J.; Sadkhin B.; Slater D. R.; Whalen K. L. Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST): A web tool for generating protein sequence similarity networks. Biochim. Biophys. Acta, Proteins Proteomics 2015, 1854, 1019–1037. 10.1016/j.bbapap.2015.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlt J. A. Genomic Enzymology: Web Tools for Leveraging Protein Family Sequence-Function Space and Genome Context to Discover Novel Functions. Biochemistry 2017, 56, 4293–4308. 10.1021/acs.biochem.7b00614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zallot R.; Oberg N. O.; Gerlt J. A. ’Democratized’ genomic enzymology web tools for functional assignment. Curr. Opin. Chem. Biol. 2018, 47, 77–85. 10.1016/j.cbpa.2018.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zallot R.; Oberg N.; Gerlt J. A. The EFI Web Resource for Genomic Enzymology Tools: Leveraging Protein, Genome, and Metagenome Databases to Discover Novel Enzymes and Metabolic Pathways. Biochemistry 2019, 58, 4169–4182. 10.1021/acs.biochem.9b00735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zallot R.; Oberg N.; Gerlt J. A. Discovery of new enzymatic functions and metabolic pathways using genomic enzymology web tools. Curr. Opin. Biotechnol. 2021, 69, 77–90. 10.1016/j.copbio.2020.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Zhu X.; Torelli A. T.; Lee M.; Dzikovski B.; Koralewski R. M.; Wang E.; Freed J.; Krebs C.; Ealick S. E.; Lin H. Diphthamide biosynthesis requires an organic radical generated by an iron-sulphur enzyme. Nature 2010, 465, 891–896. 10.1038/nature09138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee A.; Li Y.; Zhang Y.; Grove T. L.; Lee M.; Krebs C.; Booker S. J.; Begley T. P.; Ealick S. E. Reconstitution of ThiC in thiamine pyrimidine biosynthesis expands the radical SAM superfamily. Nat. Chem. Biol. 2008, 4, 758–765. 10.1038/nchembio.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamat S. S.; Williams H. J.; Dangott L. J.; Chakrabarti M.; Raushel F. M. The catalytic mechanism for aerobic formation of methane by bacteria. Nature 2013, 497, 132–136. 10.1038/nature12061. [DOI] [PubMed] [Google Scholar]
- Shannon P.; Markiel A.; Ozier O.; Baliga N. S.; Wang J. T.; Ramage D.; Amin N.; Schwikowski B.; Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks G. E.; Hon G.; Chandonia J. M.; Brenner S. E. WebLogo: a sequence logo generator. Genome Res. 2004, 14, 1188–1190. 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent A.; Benjdia A.; Guillot A.; Kubiak X.; Balty C.; Lefranc B.; Leprince J.; Berteau O. Mechanistic Investigations of PoyD, a Radical S-Adenosyl-l-methionine Enzyme Catalyzing Iterative and Directional Epimerizations in Polytheonamide A Biosynthesis. J. Am. Chem. Soc. 2018, 140, 2469–2477. 10.1021/jacs.7b08402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morinaka B. I.; Vagstad A. L.; Helf M. J.; Gugger M.; Kegler C.; Freeman M. F.; Bode H. B.; Piel J. Radical S-adenosyl methionine epimerases: regioselective introduction of diverse D-amino acid patterns into peptide natural products. Angew. Chem., Int. Ed. 2014, 53, 8503–8507. 10.1002/anie.201400478. [DOI] [PubMed] [Google Scholar]
- Caruso A.; Bushin L. B.; Clark K. A.; Martinie R. J.; Seyedsayamdost M. R. Radical Approach to Enzymatic beta-Thioether Bond Formation. J. Am. Chem. Soc. 2019, 141, 990–997. 10.1021/jacs.8b11060. [DOI] [PubMed] [Google Scholar]
- Edgar R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. C. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinf. 2004, 5, 113. 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse A. M.; Procter J. B.; Martin D. M.; Clamp M.; Barton G. J. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Procter J. B.; Carstairs G. M.; Soares B.; Mourao K.; Ofoegbu T. C.; Barton D.; Lui L.; Menard A.; Sherstnev N.; Roldan-Martinez D.; Duce S.; Martin D. M. A.; Barton G. J. Alignment of Biological Sequences with Jalview. Methods Mol. Biol. 2021, 2231, 203–224. 10.1007/978-1-0716-1036-7_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler T. J.; Clements J.; Finn R. D. Skylign: a tool for creating informative, interactive logos representing sequence alignments and profile hidden Markov models. BMC Bioinf. 2014, 15, 7. 10.1186/1471-2105-15-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haft D. H.; Basu M. K. Biological systems discovery in silico: radical S-adenosylmethionine protein families and their target peptides for posttranslational modification. J. Bacteriol. 2011, 193, 2745–2755. 10.1128/JB.00040-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grell T. A.; Goldman P. J.; Drennan C. L. SPASM and twitch domains in S-adenosylmethionine (SAM) radical enzymes. J. Biol. Chem. 2015, 290, 3964–3971. 10.1074/jbc.R114.581249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy S. R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Anishchenko I.; Park H.; Peng Z.; Ovchinnikov S.; Baker D. Improved protein structure prediction using predicted interresidue orientations. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 1496–1503. 10.1073/pnas.1914677117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek M.; DiMaio F.; Anishchenko I.; Dauparas J.; Ovchinnikov S.; Lee G. R.; Wang J.; Cong Q.; Kinch L. N.; Schaeffer R. D.; Millan C.; Park H.; Adams C.; Glassman C. R.; DeGiovanni A.; Pereira J. H.; Rodrigues A. V.; van Dijk A. A.; Ebrecht A. C.; Opperman D. J.; Sagmeister T.; Buhlheller C.; Pavkov-Keller T.; Rathinaswamy M. K.; Dalwadi U.; Yip C. K.; Burke J. E.; Garcia K. C.; Grishin N. V.; Adams P. D.; Read R. J.; Baker D. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. 10.1126/science.abj8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J.; Evans R.; Pritzel A.; Green T.; Figurnov M.; Ronneberger O.; Tunyasuvunakool K.; Bates R.; Zidek A.; Potapenko A.; Bridgland A.; Meyer C.; Kohl S. A. A.; Ballard A. J.; Cowie A.; Romera-Paredes B.; Nikolov S.; Jain R.; Adler J.; Back T.; Petersen S.; Reiman D.; Clancy E.; Zielinski M.; Steinegger M.; Pacholska M.; Berghammer T.; Bodenstein S.; Silver D.; Vinyals O.; Senior A. W.; Kavukcuoglu K.; Kohli P.; Hassabis D. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlt J. A. The Need for Manuscripts to Include Database Identifiers for Proteins. Biochemistry 2018, 57, 4239–4230. 10.1021/acs.biochem.8b00705. [DOI] [PubMed] [Google Scholar]
- Hermann J. C.; Marti-Arbona R.; Fedorov A. A.; Fedorov E.; Almo S. C.; Shoichet B. K.; Raushel F. M. Structure-based activity prediction for an enzyme of unknown function. Nature 2007, 448, 775–779. 10.1038/nature05981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L.; Kalyanaraman C.; Fedorov A. A.; Fedorov E. V.; Glasner M. E.; Brown S.; Imker H. J.; Babbitt P. C.; Almo S. C.; Jacobson M. P.; Gerlt J. A. Prediction and assignment of function for a divergent N-succinyl amino acid racemase. Nat. Chem. Biol. 2007, 3, 486–491. 10.1038/nchembio.2007.11. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.