

Abstract
Lysophosphatidic acid receptor 1 (LPA1) is one of the six G protein-coupled receptors activated by the bioactive lipid, lysophosphatidic acid (LPA). LPA1 is a drug target for various diseases, including cancer, inflammation, and neuropathic pain. Notably, LPA1 agonists have potential therapeutic value for obesity and urinary incontinence. Here, we report a cryo-electron microscopy structure of the active human LPA1-Gi complex bound to ONO-0740556, an LPA analog with more potent activity against LPA1. Our structure elucidated the details of the agonist binding mode and receptor activation mechanism mediated by rearrangements of transmembrane segment 7 and the central hydrophobic core. A structural comparison of LPA1 and other phylogenetically-related lipid-sensing GPCRs identified the structural determinants for lipid preference of LPA1. Moreover, we characterized the structural polymorphisms at the receptor-G-protein interface, which potentially reflect the G-protein dissociation process. Our study provides insights into the detailed mechanism of LPA1 binding to agonists and paves the way toward the design of drug-like agonists targeting LPA1.
Subject terms: Cryoelectron microscopy, Structure-based drug design
LPA1 is one of the GPCRs that are drug targets for various diseases. Here the authors report a cryo-EM structure of the active human LPA1-Gi complex bound to an LPA analog with more potent activity against LPA1 and clarified the ligand recognition mechanism.
Introduction
Lysophospholipids are simple phospholipids that activates GPCRs to evoke signals involved in a broad range of biological processes1. They are characterized by a single hydrocarbon chain and a polar head group, which can be divided into two subgroups; molecules containing the glycerol backbone (lysoglycerophospholipids) and the sphingoid base backbone (lysosphingolipids). The representative lysophospholipids in each of the two groups are lysophosphatidic acid (LPA) and sphingosine-1-phosphate (S1P), which activate the LPA receptors (LPA1–6) and the S1P receptors (S1P1–5), respectively2. LPA1–3 and S1P1–5 belong to the endothelial differentiation gene (EDG) family, based on the amino acid sequence identity, and exhibit conserved structural features in the ligand-binding pocket. By contrast, LPA4–6 belong to the non-EDG family, which is more closely related to the purinergic P2Y receptor family2,3. Furthermore, phosphate-modified derivatives of LPA exist in vivo to mediate signaling through different GPCRs. For example, lysophosphatidylserine and lysophosphatidylinositol activate the lysophosphatidylserine receptors (LPS1–3) and GPR55, respectively4–6. A dephosphorylated LPA derivative, 2-arachidonyl glycerol (2-AG), activates cannabinoid receptors (CB1, 2), which are most related to the EDG family at the phylogenetic level7,8. These diverse lipid-sensing GPCRs precisely discriminate between the chemical structures of lipid ligands9.
In 1996, LPA1 was the first identified LPA receptor10, and thus LPA-LPA1 signaling is the best-studied among the LPA receptors11. LPA1 couples to the G proteins such as Gαi, Gαq, and Gα12/13, and transduces various intracellular signals, e.g., increased Ca2+ concentration and actin reorganization by the Rho/ROCK pathway. LPA1 is widely expressed in several organs to control cell proliferation and survival, cell–cell contact, cell migration, and cytoskeletal morphological changes. The essential physiological functions of LPA1 are nervous-system tissue development and chondrocyte differentiation. LPA1 is associated with various diseases such as cancer, inflammation, and neuropathic pain, and thus is a pathologically important receptor that is an essential drug target. Because LPA1 signaling promotes cancer progression in many tissues12, LPA1 antagonists have been well studied as anti-cancer drugs. Moreover, some preclinical studies suggested the potential therapeutic value of selective LPA1 agonists for obesity13,14 and urinary incontinence14,15. However, the metabolic instability of LPA and its resultant short half-life have complicated the functional characterization of supplemented LPA16. Nonlipid LPA1–3 agonists are poorly reported, and thus identifying new potent and more stable agonists would be useful to explore and consolidate the potential therapeutic benefits of LPA receptors agonistic drugs. To date, the agonist-bound structures of the S1P receptors and CB receptors have been reported, revealing their lipid-ligand recognition mechanisms relevant for LPA13,17–24. While the antagonist-bound LPA1 inactive structure was also reported24, little is known about how LPA selectively activates the LPA receptors among the lipid-sensing GPCRs, limiting the design of drug-like LPA receptor agonists.
Here we report the 3.5 Å-resolution cryo-electron microscopy (cryo-EM) structure of the human LPA1-Gi signaling complex bound to an LPA analog with more potent activity against LPA1. Close examination of the LPA1 structure reveals the mechanisms of ligand-lipid binding, receptor activation, and G-protein coupling.
Results
Overall structure
For the structural study, we developed a chemically stable analog of LPA (Supplementary Method). The glycerol backbone of sn−2 LPA was partially replaced by an amide bond, and the cis-9 double bond in the acyl chain was replaced by an aromatic moiety (Fig. 1a). In a NanoBiT-G-protein dissociation assay25, the resulting compound ONO-0740556 showed agonist activity with an EC50 value of 0.26 nM for the human LPA1, which is 30-fold higher than that of LPA (Fig. 1b, Supplementary Fig. 1, Supplementary Table 1, and Supplementary Methods). This result indicates that ONO-0740556 is more suitable for the structural study toward the design of a drug-like LPA receptor agonist.
Fig. 1. Overall structure of the LPA1 -Gαi1β1γ2-ScFv16 complex.

a Chemical structures of LPA and ONO-0740556. b Ligand-induced Gi activation by LPA1-Gi activation was measured by the NanoBiT-G-protein dissociation assay. Concentration–response curves are shown as means ± s.e.m. (standard error of the mean) from three independent experiments. Source data are provided as a Source Data file. c Sharpened cryo-EM maps and refined structures. d Densities around the agonist at different density levels. We observed three strong densities, and assigned the phosphate group, glycerol backbone, and aromatic group of ONO-0740556 to them, given the surrounding environment. Furthermore, we also observed a density above W2105.43, so we extended the acyl chain to it.
We independently expressed and purified LPA1, Gi trimer, and scFv16 in insect cells and mixed them, and then purified the complex by anti-Flag affinity chromatography and size exclusion chromatography. The structure of the purified complex was determined by single-particle cryo-EM analysis with an overall resolution of 3.5 Å (PDB 7YU3) (Fig. 1c, Supplementary Fig. 2, Supplementary Table 2, and “Methods”). In this analysis, we subtracted the minimal apparent density for the micelle and the α-helical domain of the Gαi1 subunit to consider their flexibilities. The local resolution analysis demonstrated that the interaction site of Gαi1, β1, scFv16 and the interface between the Gαi1 subunit and the intracellular side of the receptor have higher resolutions. In contrast, the extracellular part of the receptor has a lower resolution (Supplementary Fig. 3). Thus, we performed a refinement with a mask on the receptor, and obtained the receptor structure with a nominal resolution of 3.7 Å (PDB 7YU4) (Supplementary Fig. 3 and Supplementary Table 2). Moreover, in this procedure, the density of ONO-0740556 ligand became more clearly observed within the orthosteric site (Fig. 1d). Based on this structure, we analyzed the modes of agonist binding and receptor activation.
ONO-0740556 binding site
ONO-0740556 provides an extensive interaction network with N-term, ECL1, 2, and TMs 2, 3, 5, 6, and 7 of the receptor (Fig. 2a–d). The binding site consists of a polar recognition region on the extracellular side and a hydrophobic pocket within the transmembrane region (Fig. 2b–d). This binding manner configuration is also found in S1P receptors17,21–23. The head phosphate and glycerol moieties of ONO-0740556 are located in the polar recognition site (Fig. 2a–d). Two oxygen atoms of the head phosphate form salt bridges with K39N-term and R1243.28 (superscripts indicate Ballesteros–Weinstein numbers) (Fig. 2a–d). The phosphate group also forms a hydrogen bond with Y34N-term, and is tightly recognized by the positively charged resides K2947.36. Moreover, the nitrogen atom in the amide bond forms a hydrogen bond with E2937.35 (Fig. 2a–d). The agonist binding mode at the polar recognition region is consistent with the previous molecular dynamics simulation24,26, which revealed that Y34N-term and K39N-term bind the head group, and with the mutational analysis in which the mutations of Y34N-term, K39N-term and R1243.28 to alanine reduced the responses elicited by ONO-0740556 (Fig. 2e).
Fig. 2. ONO-0740556 binding site.

Binding pocket for ONO-0740556, viewed from the extracellular side (a) and membrane plane (b, c). ONO-0740556 and receptor residues involved in agonist binding are shown as orange and blue sticks, respectively. The dashed lines indicate hydrogen bonds. d Schematic representation of the interactions between ONO-0740556 and the receptor within 4.5 Å. e NanoBiT-G-protein dissociation assays for LPA1 and its mutants. Concentration–response curves for ONO-074055-dependent G-protein dissociation signals for LPA1 are shown as means ± s.e.m. from three independent experiments. To match the expression of LPA1-WT to those of mutants with lower expression, 1:5 volume [WT (1:5)] plasmid was used. Source data are provided as a Source Data file. It should be noted that mutations of these head phosphate group-recognizing residues enhanced the G-protein dissociation in high concentration of the agonist, while the reason is uncertain. f Conservation of the ONO-0740556 binding site in LPA1–6.
By contrast, the long acyl chain fits into the transmembrane pocket in a bent conformation and forms extensive hydrophobic interactions with the receptor (Fig. 2b–d). Notably, the aromatic moiety in the middle of the acyl chain is sandwiched between two leucines, L2786.55 and L2977.39. Among them, L2977.39 forms a CH–π interaction with the moiety. Consistently, both the L2786.55A and L2977.39A mutations reduced the affinity, and L2977.39A had a more pronounced reduction (Fig. 2e). These data indicates that L2977.39 plays a critical role in ONO-0740556 binding. Furthermore, in the acyl chain, the C14 carbon forms a CH–π interaction with W2105.43 (Fig. 2b–d). The W2105.43A mutant completely lost the response for ONO-0740556, although its expression level was similar to that of wild type, indicating the functional importance of W2105.43 for ONO-0740556 binding and receptor activation (Fig. 2e, Supplementary Fig. 4, and Supplementary Table 3). The residues involved in the agonist binding are highly conserved among the EDG family LPA receptors (LPA1–3), suggesting a similar mechanism for LPA recognition. However, K39N-term in LPA1 is replaced by T19N-term in LPA3, suggesting that the head phosphate recognition is different between LPA1 and LPA3 (Fig. 2f).
Structural insight into LPA selectivity
The lysophospholipids LPA and S1P and the dephosphorylated LPA derivative 2-AG can selectively activate the evolutionarily related LPA receptors, the S1P receptors, and CB receptors, respectively7,8. To elucidate the mechanism of their lipid preference, we compared the agonist-bound structures of LPA1, S1P327, and CB128. Their transmembrane regions superimposed well (Fig. 3a), but the N-terminus of CB1 is different, with only partial structures observed (Fig. 3b). Focusing on the extracellular side of CB1, F1772.64 is present at the position occupied by the head phosphates of the agonists in LPA1 and S1P3 (Fig. 3c–e). Thus, lipid ligands lacking phosphate groups selectively activate CB1. A comparison of the phosphate recognition sites in LPA1 and S1P3 revealed that the head phosphate group is in almost identical positions and forms a salt bridge with lysine (K39N-term and K27N-term in LPA1 and S1P3, respectively) and arginine (R1243.28 and R1143.28 in LPA1 and S1P3, respectively). Moreover, the phosphate group also forms a hydrogen bond with the tyrosine (Y34N-term and Y22N-term in LPA1 and S1P3, respectively) (Fig. 3f and Supplementary Table 4). Thus, S1P3 and LPA1 similarly recognize phosphate groups. Overall, the salt bridges near the lysine and arginine residues enhance the recognition of the head phosphate group in LPA1 and create selectivity for LPA over other lysophospholipid mediators that have modified phosphate groups with weaker negative charges.
Fig. 3. Comparison of lipid binding modes.

a, b Superimposition of the agonist-bound LPA1, S1P3 (PDB 7C4S), and CB1 (PDB 5XRA) structures, viewed from the membrane plane (a) and the extracellular side (b). Cross sections of the ligand binding pockets in LPA1 (c), S1P3 (d), and CB1 (e). Superimposition of the LPA1 and S1P3 structures, focused on the phosphate recognition site (f) and the bottom of the hydrophobic pocket (g).
We next focused on the hydrophobic pockets accommodating the acyl chain. At the position 5.43, a tryptophan residue creates the bottom of the pocket in LPA1 and CB1 (Fig. 3c, e). The presence of tryptophan in this position only occurs in 1% of all class A GPCRs and is unique to the LPA and CB receptors24, and it is involved in the agonist binding in both receptors (Fig. 3c, e). The corresponding residue in S1P3 is C2005.43, with a smaller side chain (Fig. 3d). This amino-acid difference allows to create a deeper pocket in S1P3 as compared to LPA1 and CB1 (Fig. 3c–e, g). Furthermore, F1193.33, L189ECL2, L2596.51, and F2636.55 in S1P3 are replaced by D1293.33, A199ECL2, G2746.51, and L2786.55 in LPA1, respectively. As a result, they create a bulge of the hydrophobic pocket toward TM 5–7 in LPA1 (Fig. 3g). These amino-acid replacements allow the hydrophobic pockets of LPA1 and CB1 to be spherical (Fig. 3c–e, g) and thus they can accommodate long and bent unsaturated acyl chains (Fig. 3c), accounting for the fact that LPA1 prefers unsaturated LPA species with a cis-9 double bond in bent shapes (oleic (18:1), linoleic (18:2), and linolenic (18:3))29. By contrast, the S1P in the human body has only 18:1, with the trans-4 double bond in a linear configuration. Thus, linear S1P can activate S1P receptors with a deep, linear pocket, in contrast to LPA1 with a shallow, wide pocket. Together, the polar recognition site, which strongly recognizes phosphate groups, and the hydrophobic pocket region, which recognizes an unsaturated acyl chain, contribute to the LPA selectivity by LPA1.
Receptor activation
To examine the activation mechanism of LPA1, we compared the LPA1 structures in the present agonist-bound active state and the previously-reported antagonist-bound inactive states24. On the intracellular side, TM6 is displaced outward by about 8.2 Å, and TM7 is shifted inward by about 4.1 Å. Such structural changes are typical of class A GPCRs and allow G-protein coupling and activation30 (Fig. 4a, b). At the ligand-binding site, the positively charged residues K39N-term and R1243.28 similarly recognize the negative charges in both antagonists and agonists (Supplementary Fig. 5a, b). The antagonist is close to TM7, and the methoxycarbonyl group sterically prevents K2947.36 from accessing the polar head (Fig. 4c and Supplementary Fig. 5a, b). By contrast, the agonist is closer to TM3 than the antagonist. Since the agonist closely interacts with TM7, the extracellular side of TM7 is shifted inwardly by 1.3 Å (Fig. 4c–e). Accompanied by the shift of TM7, A3007.42, and N3037.45 move toward TM6 and push the W2716.48 rotamers inwardly (Fig. 4d, f). W2716.48 is a part of the C6.47W6.48xP6.50 motif, an essential mechanical activation switch conserved in class A GPCRs30. These observations suggest that the agonist interaction with TM7 affects the essential residue W2716.48, leading to the receptor activation on the intracellular side, as discussed later.
Fig. 4. Comparison of the agonist- and antagonist-bound LPA1 structures.

Superimposition of the agonist- and antagonist-bound LPA1 structures, colored cyan and gray (PDB 4Z34), respectively, viewed from the membrane plane (a), the intracellular side (b), and the extracellular side (c). D251 in TM6 and Y311 in TM7 are shifted by about 8.2 and 4.1 Å, respectively. d–f Structural changes of the intramolecular interactions induced by agonist binding (d). Panels (e) and (f) are focused on the extracellular side of TM7 and the receptor core, respectively. Rearrangement of the PIF (g), NPxxY (h), and DRY (I) motifs upon receptor activation. Hydrogen bonds are indicated by black dashed lines.
The bottom of the pocket also affects the rearrangement of the C6.47W6.48xP6.50 motif. In the antagonist-bound structure24, L1323.36, W2105.43, and W2716.48 constitute the bottom of the pocket, forming extensive hydrophobic interactions with the antagonist. Notably, L1323.36 forms a CH–π interaction with W2716.48, stabilizing the inactive conformation, while in the agonist-bound structure, C14 in the acyl chain of the agonist forms CH–π interactions with W2105.43 and induces its side chain flipping. The rotamer change of W2105.43 leads L1323.36 to point towards the ligand. These structural changes weaken the interaction between L1323.36 and W2716.48 and allow their synergistic conformational changes (Fig. 4f and Supplementary Fig. 5c). A similar structural rearrangement is observed in CB1, in which the homologous residues F2003.36 and W3566.48 are flipped upon agonist binding (referred to as a twin toggle switch)18,19,28 (Supplementary Fig. 5d). The density corresponding to C14 in the agonist is relatively well-observed (Fig. 1d), and the W2105.43A mutant showed no Gi dissociation signal (Fig. 2e), indicating the strength and importance of the interaction with W2105.43. These observations suggest that the inward movement of TM7 and the acyl chain interaction with W2105.43 cooperatively induce the toggle switch activation of W2716.48 (Fig. 4f).
The movement of the C6.47W6.48xP6.50 motif upon agonist binding causes a structural rearrangement in the P5.50I3.36F6.44 motif, which is also essential for receptor activation30–32. The inward rotations of the W2716.48 rotamer and N3037.45 allow the F2676.44 flipping toward TM5 (Fig. 4f), followed by the significant displacement of F2185.51 proximal to the motif (Fig. 4g). The movement of the P5.50I3.40F6.44 motif is responsible for the large outward movement of the intracellular portion of TM6. Accompanying the movement, structural rearrangements are observed in the N7.49P7.50xxY7.53 and D3.49R3.50Y3.51 motifs conserved in most class A GPCRs30. In the N7.49P7.50xxY7.53 motif, Y3117.53 shows a significant displacement toward the intracellular core and contacts L1393.43, I1423.46, and R1463.50, leading to the inward movement of TM7 (Fig. 4h). In the D3.49R3.50Y3.51 motif, R1463.50 forms a hydrogen bond with Y2255.58 and enables interactions with the C-terminal residues of the α5-helix of Gi (Fig. 4I).
These conformation changes create an intracellular cavity for G-protein recognition (Fig. 5a). The cavity closely contacts with the C-terminal α5-helix of Gi, which is the primary determinant for the G-protein coupling25. Specifically, R1463.50 forms a hydrogen bond with the backbone carbonyl of C351G.H5.23 (superscript indicates the common Gα numbering [CGN] system33), which is typically observed in other GPCR-Gi complexes34,35. Additional hydrogen-bonding interactions are observed between the α5-helix and ICL2 (Fig. 5a). In addition to these polar contacts, there are extensive hydrophobic contacts between the receptor and Gi (Supplementary Fig. 6). These interactions allow the receptor to couple with Gi.
Fig. 5. Binding modes of Gi.

a Main hydrogen-bonding interactions between the receptor and the α5 helix of Gαi1. b, c Structural comparisons of LPA1-Gi with other GPCR-G-protein complexes at the interface, viewed from the cytoplasmic side (b) and membrane plane (c). Structural comparisons of the interactions between ICL2 and Gi in the NTSR1 C state (d), NTSR1 NC state (e), LPA1 (f), and S1P1 (g). Residues are shown as stick models. Hydrogen bonds are indicated by black dashed lines.
Structural polymorphism at the receptor-Gi interface
Previous structural studies showed that the Gαi binding manner is variable, with different Gαi rotations relative to the receptor35. Moreover, canonical (C) and non-canonical (NC) states were observed in the NTSR1-Gi complex, with a 45° rotation of the G-protein relative to the receptor36. Compared with the C and NC states, the Gi protein in the LPA1 structure resides in their intermediate positions (Fig. 5b, c). This difference seems to be derived from the receptor-Gi interaction at ICL2. In most class A GPCRs, ICL2 adopts a short α-helix in the active state18,19,37–39. PositionICL2/34.50 (F174 in the NTSR1 C state) binds within the hydrophobic pocket formed by L194G.S3.01, F336G.H5.08, T340G.H5.12, and I343G.H5.15 in Gαi (Fig. 5d) and plays an essential role in G-protein activation. In the NTSR1 NC state, F174ICL2/34.51 is located away from the hydrophobic pocket of Gi (Fig. 5e), closely correlating with the Gi position (Fig. 5b, c). By contrast, the ICL2 of LPA1 adopts a disordered conformation (Fig. 5f). The corresponding residue at position ICL2/34.51 is glutamine in LPA1, which does not participate in a hydrophobic interaction. Instead, M153ICL2/34.50 binds within the hydrophobic pocket. Moreover, L15534.52 forms hydrophobic interactions with L343G.H5.15 and Q346G.H5.18. Above these hydrophobic interactions, R1523.56 forms a salt bridge with D350G.H5.22. Taken together, the disordered ICL2 tightly interacts with the α5-helix, shifting it away from ICL2 and TM3 as compared with the NTSR1 C state (Fig. 5c). These structural features are responsible for the different positions of the Gi protein in the NTSR1 C and NC states. The disordered ICL2 and the G-protein position in the LPA1-Gi complex are similar to those in other S1P-Gi complexes21–23 (Fig. 5b, c, g), illuminating the conserved structural feature for Gi coupling in EDG family members.
To determine whether the conformational transition of the Gi coupling is observed, as in NTSR1, we performed 3D classifications focusing on the alignment of LPA1 and G protein. Accordingly, we obtained cryo-EM maps for four classes (S1–S4) with nominal resolutions of 3.7, 3.9, 4.5, and 5.6 Å (PDB 7YU5, 7YU6, 7YU7, and 7YU8) (Fig. 6a and Supplementary Table 2). The maps of S1 and S2 enabled model building and refinement. Moreover, those of S3 and S4 enabled them with accuracy of the Cα atoms (Supplementary Fig. 7). Thus, we discuss the conformational changes in the main chains.
Fig. 6. Structural polymorphism of the LPA1-Gi interface.
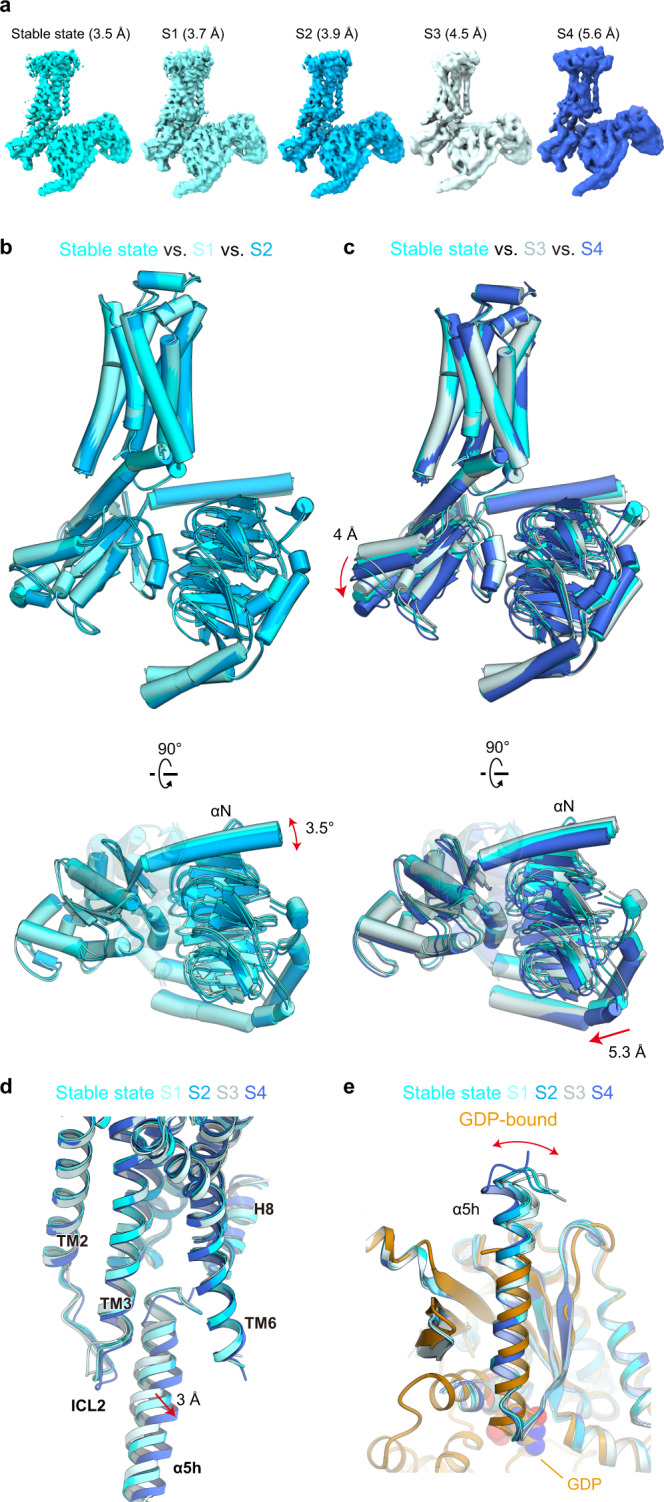
a Density maps of the 3.5 Å resolution stable state and S1–4. b Comparison of the stable state, S1, and S2. c Comparison of the stable state, S3, and S4. d Superimposition of the stable state and S1–4, aligned at the receptor. TM5 is omitted. e Superimposition of the Gαi1 subunits in the stable state, S1–4, and the GDP-bound inactive Gi heterotrimer (PDB 1GG2).
To visualize the G-protein movement, we superimposed the 3.5 Å-resolution structure described above (stable state) and S1–4 at the receptor. S1 and S2 superimposed well on the stable state, with limited in-plane rotations within 3–4° of the G protein (Fig. 6b), suggesting that this is the most stable position of the G-protein in the nucleotide-free state. By contrast, S3 and S4 both moved from the stable state in opposite directions from each other (Fig. 6c). As compared to S3, the entire Gαi1 in S4 is shifted downward by about 4 Å, followed by the lateral movement of Gβ1 by 5.3 Å. Focusing on the α5-helix, it moves 3 Å away from the receptor with the structural changes in the C-terminal residues (Fig. 6d). ICL2 follows the movement of the α5-helix to maintain the interaction with it. When aligned the S1–S4 and the stable state at the Gαi1 protein, the orientations of the C-terminal 2-turn helix are variable (Fig. 6e). This region does not adopt the α-helix in the GDP-bound inactive Gi heterotrimer40, and receptor interaction induces its helix formation. This notion suggests the innate structural flexibility in the C-terminal residues of the α5-helix, which is responsible for the structural polymorphism observed in this study, reflecting the dynamic equilibrium of the receptor-Gi interface. These movements are totally distinct from the rotational movements observed in NTSR1 (Fig. 5b), which reflect the activation pathway of G protein36. Since the downward movement of Gαi1 weakens the receptor–Gαi1 interactions (Fig. 6d), S4 might represent the dissociation process of the receptor and G protein upon GTP binding.
Discussion
We determined the structure of the LPA1-Gi complex bound to the LPA analog ONO-0740556, which revealed the tight recognition of the head phosphate and the accommodation of the bent acyl chain in the spherical pocket. Close examination of the active and inactive LPA1 structures elucidated that two factors cooperatively play key roles in receptor activation. One is the recognition of the phosphate groups and glycerol backbone by TM7, and the other is the hydrophobic interactions with a long acyl chain by the residues at the bottom of the pocket. The ligand recognition by TM7 agrees with the properties of the binding module, in which the ligand is closer to TM7 and the hydrophobic pocket is more expanded to TM7 in LPA1 than in S1P3 (Fig. 3f, g). This is unique to LPA1, among the currently reported structures of lysophospholipid receptors. At the binding site, the antagonist impedes receptor activation by its methoxycarbonyl group and indan, which prevent the inward movement of TM7. Moreover, at the bottom of the pocket, the position of the dimethoxyphenyl clashes with L1323.36 and L2977.39, which are essential for receptor activation (Supplementary Fig. 5a, e). This observation indicates that the antagonist inhibits L1323.36 and L2977.39 from moving towards activation.
After the submission of this manuscript, the structure of LPA1 bound to the endogenous agonist LPA was reported (PDB 7TD0)41. We performed a structural comparison of the LPA1 bound to LPA with our structure bound to ONO-0740556. The two structures of LPA1 superimposed well with a root mean square deviation of Cα atoms of 0.583 Å (Fig. 7a), and there are no significant differences in the recognition of polar regions on the extracellular side, and in the interaction of the ligand with W2105.43 and L2977.39 in the hydrophobic pocket (Fig. 7b, c). Given that these interactions mediate the receptor activation, LPA and ONO-0740556 activate the receptor in similar manners. However, interestingly, the route of the acyl chain is different between our new compound and LPA. The acyl chain of LPA folds on the TM5 side and extends toward TM7, but ONO-0740556 goes from TM7 to TM5 (Fig. 7c). This fact suggests that LPA1 permits the acceptance of various forms of acyl chains within the spherical hydrophobic pocket. The interaction of L2977.39 with the hydrocarbon chain of LPA is weaker than the CH–π interaction with the aromatic moiety of ONO-0740556. This difference would be one of the factors causing the distinct affinities of the agonists (Fig. 1b and Supplementary Table 1). Our study clarifies the detailed structure-activity relationship of LPA1 and will facilitate the design of novel LPA-mimetic agonists to explore the therapeutic potential of LPA1.
Fig. 7. Structural comparison of LPA1 bound to LPA and ONO-0740556.

a Superimposition of the LPA- and ONO-0740556-bound LPA1 structures, colored green (PDB 7TD0) and cyan, respectively. b, c Superimposition of the binding pocket for LPA and ONO-0740556 in polar regions on the extracellular side (b), and in the hydrophobic pocket (c). d Superimposition of LPA-LPA1-Gi states a (PDB 7TD1) and a’ (PDB 7TD2) aligned at the receptor. e Superimposition of S3 and S4 aligned at the receptor.
Gi movement was observed in the LPA-bound LPA1-Gi complex, as in our study. The 3D variability analysis (3DVA) of the LPA-bound complex identified two states (Fig. 7d) distinguished by the relative rotation of Gαi1 about LPA1 in the plane of the membrane, ~5° in both directions away from the consensus structure41. By contrast, in the ONO-0740556 bound complex, the entire Gαi1 in S4 is shifted downward by about 4 Å (Fig. 7e), indicating a weakening of the receptor–Gαi1 interactions. However, there are significant differences in the experimental conditions between our study and previous studies (e.g., ligands, detergents, analysis methods, etc.), and thus we cannot ignore their influence on the Gi movements. Moreover, the 3DVA analysis of LPA1 and S1P1 elucidated the rocking, twisting, and flexing motions of the receptor about the G protein41. These structural polymorphisms indicated the flexible coupling between GPCR and G protein, which may be observed in other GPCR-G-protein complexes by more careful analysis. The G-protein movement upon dissociation is also observed in the recently reported PTH1R-Gs complex42. Future studies will shed light on whether the observed structural polymorphism reflects the structural flexibility in the purified condition, or the process of G-protein activation and dissociation by GPCRs.
Methods
NanoBiT-G-protein dissociation assay
LPA1-induced Gi activation was measured by a NanoBiT-G-protein dissociation assay25, in which the LPA1-induced dissociation of a Gα subunit from a Gβγ subunit was monitored by a NanoBiT system (Promega). Specifically, a NanoBiT-Gi1 protein consisting of a large fragment (LgBiT)-containing Gαi1 subunit and a small fragment (SmBiT)-fused Gγ2 subunit with the C68S mutation, along with the untagged Gβ1 subunit, was expressed with a test LPA1 construct, and the ligand-induced change in the luminescent signal was measured. We used the N-terminal FLAG (DYKDDDDK)-tagged constructs of human LPA1. HEK293T cells were seeded in a six-well culture plate at a concentration of 2 × 105 cells ml−1 (2 ml per well in DMEM supplemented with 10% fetal bovine serum), 1 d before transfection. The transfection solution was prepared by combining 2.5 µl (per well hereafter) of Lipofectamine 2000 (ThermoFisher Scientific) and a plasmid mixture consisting of 100 ng LgBiT-containing Gαi1 subunit, 500 ng Gβ1, 500 ng SmBiT-fused Gγ2 with the C68S mutation, and 200 ng LPA1 in 500 µl of Opti-MEM (ThermoFisher Scientific). After an incubation for 1 d, the transfected cells were harvested with 0.5 mM EDTA-containing PBS, centrifuged, and suspended in 2 ml of HBSS containing 0.01% bovine serum albumin (BSA fatty acid-free grade, SERVA) (assay buffer). The cell suspension was dispensed into a white 96-well plate at a volume of 80 µl per well, and loaded with 20 μl of 50 μM coelenterazine diluted in the assay buffer. After 2 h of incubation, the plate was measured for baseline luminescence and then 20 µl of 6× test compound, diluted in the assay buffer, was manually added. After an incubation for 8–10 min at room temperature, the plate was read for the second measurement. The second luminescence counts were normalized to the initial counts, and the fold changes in the signals were plotted for the G-protein dissociation response. Using the Prism 7 software (GraphPad Prism), the G-protein dissociation signals were fitted to a four-parameter sigmoidal concentration–response curve, from which the pEC50 values (negative logarithmic values of half-maximum effective concentration (EC50) values) and Emax were used to calculate the mean and s.e.m.
Measurement of receptor cell-surface expression by ELISA
To measure the cell surface expression level of wild-type LPA1 and its mutants, HEK293T cells were transiently transfected in 12-well plates and incubated for 1 d. Transfection was performed by following the same procedure as described in the NanoBiT-G-protein dissociation assay section, with a downscaled volume (250 µl transfection solution). The transfected cells were harvested with 0.5 mM EDTA-containing PBS, centrifuged, and blocked with 5% (w/v) BSA at room temperature for 1 h. Anti-Flag HRP conjugate (Sigma) was then added to a dilution of 1:20,000 and incubated for 1 h at room temperature. After washing with PBS, the cells were suspended in 100 µL of PBS and 10 µl portions were dispensed into the tubes. A 50 µl aliquot of TMB, HRP Microwell Substrate (SurModics, Inc.) was added. The reactions were quenched by adding an equal volume of 450 nm Liquid Stop Solution for TMB Microwell Substrate (SurModics, Inc.) and the optical density at 450 nm was measured using Nanodrop One (Thermo Fischer Scientific).
Expression and purification of the human LPA1
The human LPA1 gene (UniProtKB, Q92633) was subcloned into a modified pFastBac vector43, with an N-terminal haemagglutinin signal peptide followed by the Flag-tag epitope (DYKDDDD) and a C-terminal tobacco etch virus (TEV) protease recognition site followed by an EGFP-His10 tag. The recombinant baculovirus was prepared using the Bac-to-Bac baculovirus expression system (Thermo Fischer Scientific). Spodoptera frugiperda Sf9 insect cells (Thermo Fischer Scientific) were infected with the virus at a cell density of 4.0 × 106 cells per milliliter in Sf900 II medium (Gibco), and grown for 48 h at 27 °C. The harvested cells were disrupted by sonication, in buffer containing 20 mM Tris-HCl, pH8.0, 200 mM NaCl, and 10% glycerol. The crude membrane fraction was collected by ultracentrifugation at 180,000 × g for 2 h. The membrane fraction was solubilized in buffer, containing 20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% n-dodecyl-beta-D-maltopyranoside (DDM) (Calbiochem), 0.2 % CHS, 10% glycerol, and 2 μM ONO-0740556 for 1 h at 4 °C. The supernatant was separated from the insoluble material by ultracentrifugation at 180,000 × g for 30 min, and incubated with TALON resin (Clontech) for 30 min. The resin was washed with ten column volumes of buffer, containing 20 mM Tris-HCl, pH 8.0 500 mM NaCl, 0.05% glyco-diosgenin (GDN) (Anatrace), 0.1 μM ONO-0740556, and 15 mM imidazole. The receptor was eluted in buffer, containing 20 mM Tris-HCl, pH 8.0, 500 mM NaCl, 0.01% GDN, 0.1 μM ONO-0740556, and 200 mM imidazole. The receptor was concentrated and loaded onto a Superdex200 10/300 Increase size-exclusion column, equilibrated in buffer containing 20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.01% GDN, and 0.1 μM ONO-0740556. Peak fractions were pooled and frozen in liquid nitrogen.
Expression and purification of the Gi heterotrimer
The Gi heterotrimer was expressed and purified using the Bac-to-Bac baculovirus expression system, according to the method reported previously35. Sf9 insect cells were infected at a density of 3–4 × 106 cells ml−1 with a one 100th volume of two viruses, one encoding the WT human Gαi1 subunit and the other encoding the WT bovine Gγ2 subunit and the WT rat Gβ1 subunit containing a His8 tag followed by an N-terminal TEV protease cleavage site. The infected Sf9 cells were incubated in Sf900II medium at 27 °C for 48 h. The Sf9 cells were collected by centrifugation at 6200 × g for 10 min. The collected cells were lysed in buffer containing 20 mM Tris, pH 8.0, 150 mM NaCl, and 10% glycerol. The Gαi1β1γ2 heterotrimer was solubilized at 4 °C for 1 h, in buffer containing 20 mM Tris (pH 8.0), 150 mM NaCl, 10% glycerol, 1% (w/v) n-dodecyl-beta-D-maltopyranoside (DDM) (Anatrace), 50 μM GDP (Roche), and 10 mM imidazole. The soluble fraction containing Gi heterotrimers was isolated by ultracentrifugation (186,000 × g for 20 min) and the supernatant was mixed with Ni-NTA Superflow resin (Qiagen) and stirred at 4 °C for 1 h. The resin was washed with 10 column volumes of buffer, containing 20 mM Tris, pH 8.0, 150 mM NaCl, 0.02% DDM, 10% glycerol, 10 μM GDP, and 30 mM imidazole. Next, the Gi heterotrimers were eluted with two column volumes of buffer, containing 20 mM Tris, pH 8.0, 150 mM NaCl, 0.02% (w/v) DDM, 10% (v/v) glycerol, 10 μM GDP and 300 mM imidazole. The eluted fraction was dialyzed overnight at 4 °C against 20 mM Tris, pH 8.0, 50 mM NaCl, 0.02% DDM, 10% glycerol, and 10 μM GDP. To cleave the histidine tag, TEV protease was added during the dialysis. The dialyzed fraction was incubated with Ni-NTA Superflow resin at 4 °C for 1 h. The flow-through was collected and purified by ion-exchange chromatography on a HiTrapQ HP column (GE), using buffer I1 (20 mM Tris, pH 8.0, 50 mM NaCl, 0.02% DDM, 10% glycerol, and 1 μM GDP) and buffer I2 (20 mM Tris, pH 8.0, 1 M NaCl, 0.02% DDM, 10% glycerol, and 1 μM GDP).
Expression and purification of scFv16
The gene encoding scFv16 was synthesized (GeneArt) and subcloned into a modified pFastBac vector, with the resulting construct encoding the GP67 secretion signal sequence at the N terminus, and a His8 tag followed by a TEV cleavage site at the C terminus35. The His8-tagged scFv16 was expressed and secreted by Sf9 insect cells, as previously reported35 The Sf9 cells were removed by centrifugation at 5000 × g for 10 min, and the secreta-containing supernatant was combined with 5 mM CaCl2, 1 mM NiCl2, 20 mM HEPES (pH 8.0), and 150 mM NaCl. The supernatant was mixed with Ni Superflow resin (GE Healthcare Life Sciences) and stirred for 1 h at 4 °C. The collected resin was washed with buffer containing 20 mM HEPES (pH 8.0), 500 mM NaCl and 20 mM imidazole, and further washed with 10 column volumes of buffer containing 20 mM HEPES (pH 8.0), 500 mM NaCl and 20 mM imidazole. Next, the protein was eluted with 20 mM Tris (pH 8.0), 500 mM NaCl and 400 mM imidazole. The eluted fraction was concentrated and loaded onto a Superdex200 10/300 Increase size-exclusion column, equilibrated in buffer containing 20 mM Tris (pH 8.0) and 150 mM NaCl. Peak fractions were pooled, concentrated to 5 mg ml−1 using a centrifugal filter device (Millipore 10 kDa MW cutoff), and frozen in liquid nitrogen.
Formation and purification of the LPA1-Gi complex
Purified LPA1-GFP was mixed with a 1.2 molar excess of Gi heterotrimer, ScFv16, and TEV protease. After the addition of apyrase to catalyze hydrolysis of unbound GDP, and ONO-0740556 (final 10 µM) the coupling reaction was performed at 4 °C for overnight. To remove excess G protein, the complexing mixture was purified by M1 anti-Flag affinity chromatography. Bound complex was washed in buffer, containing 20 mM Tris-HCl, pH8.0, 150 mM NaCl, 0.01% GDN, 10 μM ONO-0740556, 10% Glycerol, and 5 mM CaCl2. The complex was then eluted in 20 mM Tris-HCl, pH8.0, 150 mM NaCl, 0.01% GDN, 10 μM ONO-0740556, 10% Glycerol, 5 mM EDTA, and Flag peptide. The LPA1-Gi-scFv16 complex was purified by size exclusion chromatography on a Superdex 200 10/300 column in 20 mM Tris-HCl, pH8.0, 150 mM NaCl, 0.01% GDN, and 1 μM ONO-0740556. Peak fractions were concentrated to ~12 mg/ml for electron microscopy studies.
Sample vitrification and cryo-EM data acquisition
The purified complex was applied onto a freshly glow-discharged Quantifoil holey carbon grid (R1.2/1.3, Au, 300 mesh), and plunge-frozen in liquid ethane by using a Vitrobot Mark IV. Data collections were performed on a 300 kV Titan Krios G3i microscope (Thermo Fisher Scientific) and equipped with a BioQuantum K3 imaging filter and a K3 direct electron detector (Gatan). In total, 6,227 movies were acquired with a calibrated pixel size of 0.83 Å pix−1 and with a defocus range of −0.8 to −1.6 μm, using the SerialEM software44. Each movie was acquired for 2.57 s and split into 48 frames, resulting in an accumulated exposure of about 49.530 e− Å−2 at the grid.
Image processing
All acquired movies were dose-fractionated and subjected to beam-induced motion correction implemented in RELION 3.145. The contrast transfer function (CTF) parameters were estimated using CTFFIND 4.046 (Rohou & Grigorieff, 2015). A total of 3,021,676 particles were extracted. The initial model was generated in RELION 3.147,48. The particles were subjected to several rounds of 2D and 3D classifications, resulting in the optimal classes of particles, which contained 363,784 particles. Next, the particles were subjected to 3D refinement, CTF refinement, and Bayesian polishing49 (Zivanov et al., 2018).
The GDN detergent micelles and the α-helical domain of the Gαi subunit of the 363,784 particles were subtracted to obtain a higher signal-to-noise ratio. The subtracted particles were subjected to 3D classifications. The best class of particles was subjected to 3D refinement and then subjected to No-alignment classifications. The best class of particles were subjected to 3D refinement, postprocessing yielded a map with a nominal overall resolution of 3.5 Å, with the gold standard Fourier Shell Correlation (FSC = 0.143) criteria50.
Moreover, the 3D model was refined with a mask on the receptor. As a result, the receptor has a higher resolution with a nominal resolution of 3.7 Å. The local resolution was estimated by RELION 3.1. The processing strategy is described in Fig. S2.
Apart from that, the 3D model of the 363,784 particles was refined with a mask on the Gi and ScFv16, and then No-alignment classification with a mask on receptor obtained multiple conformations with different Gi-couplings. Each class of particles was subjected to 3D refinement, micelles and α-helical domains were subtracted and then was subjected to 3D refinement.
Model building and refinement
The quality of the micelle-subtracted density map was sufficient to build a model manually in COOT51,52. The model building was facilitated by the predicted LPA1 model in AlphaFold Protein Structure Database (https://alphafold.ebi.ac.uk/entry/Q92633) and the cryo-EM structure of the LPA1-Gi and μOR-Gi complex (PDB 7TD0 and 6DDE, respectively)34,41. We manually modeled LPA1, the Gi heterotrimer and scFv16 into the map by jiggle fit using COOT. We then manually readjusted the model into the density map using COOT and refined it using phenix.real_space_refine53,54 (v.1.19) with the secondary-structure restraints using phenix secondary_structure_restraints. Finally, we refined the model using servalcat55.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Acknowledgements
We thank K. Ogomori and C. Harada for technical assistance, Inoue, A for the advice on the assay experiment and Ono Pharmaceutical Co., Ltd. for the synthesis and characterization of ONO-0740556. This work was supported by grants from the Platform for Drug Discovery, Informatics and Structural Life Science by the Ministry of Education, Culture, Sports, Science and Technology (MEXT), and JSPS KAKENHI grants 21H05037 (O.N.), 22K19371 and 22H02751 (W.S.), and 21J20692 (T.T.); ONO Medical Research Foundation (W.S.); The Kao Foundation for Arts and Sciences (W.S.); The Takeda Science Foundation (W.S.); The Uehara Memorial Foundation (W.S.); the Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED, under grant numbers JP19am01011115 (support no. 1109, O.N.).
Source data
Author contributions
H.A. performed all of the experiments. T.T. assisted with the grid preparation, the cryo-EM data collection, and the single particle analysis. F.S. assisted with the single particle analysis. Y.M. assisted with the NanoBiT-G-protein dissociation assay. W.S. performed the initial screening of the LPA1 expression. The manuscript was mainly prepared by H.A. and W.S., with assistance from O.N.
Peer review
Peer review information
Nature Communications thanks Arun Shukla, Abby Parill-Baker, and Haitao Zhang for their contribution to the peer review of this work. Peer review reports are available.
Data availability
The data that support this study are available from the corresponding authors upon reasonable request. The cryo-EM density map and atomic coordinates for the LPA1-Gi complex have been deposited in the Electron Microscopy Data Bank and the PDB, under accession codes: EMD-34097 (LPA1–Gi stable state), EMD-34098 (focused on LPA1), EMD-34099 (LPA1–Gi state 1), EMD-34100 (LPA1–Gi state 2), EMD-34101 (LPA1–Gi state 3), EMD-34102 (LPA1–Gi state 4), and PDB 7YU3 (LPA1–Gi stable state), 7YU4 (focused on LPA1), 7YU5 (LPA1–Gi state 1), 7YU6 (LPA1–Gi state 2), 7YU7 (LPA1–Gi state 3), 7YU8 (LPA1–Gi state 4). Source data are provided with this paper.
Competing interests
O.N. is a co-founder and scientific advisor for Curreio. All other authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Wataru Shihoya, Email: wtrshh9@gmail.com.
Osamu Nureki, Email: nureki@bs.s.u-tokyo.ac.jp.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-022-33121-2.
References
- 1.Tan ST, Ramesh T, Toh XR, Nguyen LN. Emerging roles of lysophospholipids in health and disease. Prog. Lipid Res. 2020;80:101068. doi: 10.1016/j.plipres.2020.101068. [DOI] [PubMed] [Google Scholar]
- 2.Kihara Y, Maceyka M, Spiegel S, Chun J. Lysophospholipid receptor nomenclature review: IUPHAR Review 8. Br. J. Pharm. 2014;171:3575–3594. doi: 10.1111/bph.12678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taniguchi R, et al. Structural insights into ligand recognition by the lysophosphatidic acid receptor LPA6. Nature. 2017;548:356–360. doi: 10.1038/nature23448. [DOI] [PubMed] [Google Scholar]
- 4.Makide K, et al. Novel lysophosphoplipid receptors: Their structure and function. J. Lipid Res. 2014;55:1986–1995. doi: 10.1194/jlr.R046920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Omi J, Kano K, Aoki J. Current knowledge on the biology of lysophosphatidylserine as an emerging bioactive lipid. Cell Biochem. Biophys. 2021;79:497–508. doi: 10.1007/s12013-021-00988-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tudurí E, et al. GPR55: A new promising target for metabolism? J. Mol. Endocrinol. 2017;58:R191–R202. doi: 10.1530/JME-16-0253. [DOI] [PubMed] [Google Scholar]
- 7.Hurst DP, Schmeisser M, Reggio PH. Endogenous lipid activated G protein-coupled receptors: emerging structural features from crystallography and molecular dynamics simulations. Chem. Phys. Lipids. 2013;169:46–56. doi: 10.1016/j.chemphyslip.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yung YC, Stoddard NC, Chun J. LPA receptor signaling: Pharmacology, physiology, and pathophysiology. J. Lipid Res. 2014;55:1192–1214. doi: 10.1194/jlr.R046458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blaho VA, Chun J. ‘Crystal’ clear? Lysophospholipid receptor structure insights and controversies. Trends Pharmacol. Sci. 2018;39:953–966. doi: 10.1016/j.tips.2018.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hecht JH, Weiner JA, Post SR, Chun J. Ventricular zone gene-1 (vzg-1) encodes a lysophosphatidic acid receptor expressed in neurogenic regions of the developing cerebral cortex. J. Cell Biol. 1996;135:1071–1083. doi: 10.1083/jcb.135.4.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi JW, et al. LPA receptors: Subtypes and biological actions. Annu. Rev. Pharmacol. Toxicol. 2010;50:157–186. doi: 10.1146/annurev.pharmtox.010909.105753. [DOI] [PubMed] [Google Scholar]
- 12.Houben AJS, Moolenaar WH. Autotaxin and LPA receptor signaling in cancer. Cancer Metastasis Rev. 2011;30:557–565. doi: 10.1007/s10555-011-9319-7. [DOI] [PubMed] [Google Scholar]
- 13.Rancoule C, et al. Involvement of autotaxin/lysophosphatidic acid signaling in obesity and impaired glucose homeostasis. Biochimie. 2014;96:140–143. doi: 10.1016/j.biochi.2013.04.010. [DOI] [PubMed] [Google Scholar]
- 14.Guillot E, et al. Lysophosphatidic acid receptor agonism: Discovery of potent nonlipid benzofuran ethanolamine structures. J. Pharmacol. Exp. Ther. 2020;374:283–294. doi: 10.1124/jpet.120.265454. [DOI] [PubMed] [Google Scholar]
- 15.Terakado M, et al. Discovery of ONO-7300243 from a novel class of lysophosphatidic acid receptor 1 antagonists: From hit to lead. ACS Med. Chem. Lett. 2016;7:913–918. doi: 10.1021/acsmedchemlett.6b00225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Salous AK, et al. Mechanism of rapid elimination of lysophosphatidic acid and related lipids from the circulation of mice. J. Lipid Res. 2013;54:2775–2784. doi: 10.1194/jlr.M039685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanson MA, et al. Crystal structure of a lipid G protein-coupled receptor. Science. 2012;335:851–855. doi: 10.1126/science.1215904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krishna Kumar K, et al. Structure of a signaling cannabinoid receptor 1-G protein complex. Cell. 2019;176:448–458.e12. doi: 10.1016/j.cell.2018.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hua T, et al. Activation and signaling mechanism revealed by cannabinoid receptor-Gi complex structures. Cell. 2020;180:655–665.e18. doi: 10.1016/j.cell.2020.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xing C, et al. Cryo-EM structure of the human cannabinoid receptor CB2-Gi signaling complex. Cell. 2020;180:645–654.e13. doi: 10.1016/j.cell.2020.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu, Z. et al. Structural basis of sphingosine-1-phosphate receptor 1 activation and biased agonism. Nat. Chem. Biol. 10.1038/s41589-021-00930-3 (2021). [DOI] [PubMed]
- 22.Zhao, C. et al. Structural insights into sphingosine-1-phosphate recognition and ligand selectivity of S1PR3-Gi signaling complexes. Cell Res. 10.1038/s41422-021-00567-w (2021). [DOI] [PMC free article] [PubMed]
- 23.Yuan Y, et al. Structures of signaling complexes of lipid receptors S1PR1 and S1PR5 reveal mechanisms of activation and drug recognition. Cell Res. 2021;31:1263–1274. doi: 10.1038/s41422-021-00566-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chrencik JE, et al. Crystal structure of antagonist bound human lysophosphatidic acid receptor 1. Cell. 2015;161:1633–1643. doi: 10.1016/j.cell.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Inoue A, et al. Illuminating G-protein-coupling selectivity of GPCRs. Cell. 2019;177:1933–1947.e25. doi: 10.1016/j.cell.2019.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Omotuyi OI, Nagai J, Ueda H. Lys39-Lysophosphatidate carbonyl oxygen interaction locks LPA1 N-terminal cap to the orthosteric site and partners Arg124 during receptor activation. Sci. Rep. 2015;5:13343. doi: 10.1038/srep13343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maeda S, et al. Endogenous agonist-bound S1PR3 structure reveals determinants of G protein-subtype bias. Sci. Adv. 2021;7:eabf5325. doi: 10.1126/sciadv.abf5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hua T, et al. Crystal structures of agonist-bound human cannabinoid receptor CB1. Nature. 2017;547:468–471. doi: 10.1038/nature23272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tigyi G. Aiming drug discovery at lysophosphatidic acid targets. Br. J. Pharm. 2010;161:241–270. doi: 10.1111/j.1476-5381.2010.00815.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Venkatakrishnan AJ, et al. Molecular signatures of G-protein-coupled receptors. Nature. 2013;494:185–194. doi: 10.1038/nature11896. [DOI] [PubMed] [Google Scholar]
- 31.Shihoya W, et al. Activation mechanism of endothelin ETB receptor by endothelin-1. Nature. 2016;537:363–368. doi: 10.1038/nature19319. [DOI] [PubMed] [Google Scholar]
- 32.Shihoya W, et al. Crystal structures of human ETB receptor provide mechanistic insight into receptor activation and partial activation. Nat. Commun. 2018;9:4711. doi: 10.1038/s41467-018-07094-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Flock T, et al. Universal allosteric mechanism for Gα activation by GPCRs. Nature. 2015;524:173–179. doi: 10.1038/nature14663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koehl A, et al. Structure of the µ-opioid receptor-Gi protein complex. Nature. 2018;558:547–552. doi: 10.1038/s41586-018-0219-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Okamoto HH, et al. Cryo-EM structure of the human MT1-Gi signaling complex. Nat. Struct. Mol. Biol. 2021;28:694–701. doi: 10.1038/s41594-021-00634-1. [DOI] [PubMed] [Google Scholar]
- 36.Kato HE, et al. Conformational transitions of a neurotensin receptor 1-Gi1 complex. Nature. 2019;572:80–85. doi: 10.1038/s41586-019-1337-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Y, et al. Single-particle cryo-EM structural studies of the β2AR-Gs complex bound with a full agonist formoterol. Cell Discov. 2020;6:45. doi: 10.1038/s41421-020-0176-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kobayashi K, et al. Cryo-EM structure of the human PAC1 receptor coupled to an engineered heterotrimeric G protein. Nat. Struct. Mol. Biol. 2020;27:274–280. doi: 10.1038/s41594-020-0386-8. [DOI] [PubMed] [Google Scholar]
- 39.Nagiri C, et al. Cryo-EM structure of the β3-adrenergic receptor reveals the molecular basis of subtype selectivity. Mol. Cell. 2021;81:3205–3215.e5. doi: 10.1016/j.molcel.2021.06.024. [DOI] [PubMed] [Google Scholar]
- 40.Wall MA, et al. The structure of the G protein heterotrimer Gi alpha 1 beta 1 gamma 2. Cell. 1995;83:1047–1058. doi: 10.1016/0092-8674(95)90220-1. [DOI] [PubMed] [Google Scholar]
- 41.Liu S, et al. Differential activation mechanisms of lipid GPCRs by lysophosphatidic acid and sphingosine 1-phosphate. Nat. Commun. 2022;13:731. doi: 10.1038/s41467-022-28417-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kobayashi, K. et al. Endogenous ligand recognition and structural transition of a human PTH receptor. Mol. Cell10.1016/j.molcel.2022.07.003 (2022). [DOI] [PubMed]
- 43.Fukuhara S, et al. Structure of the human secretin receptor coupled to an engineered heterotrimeric G protein. Biochem. Biophys. Res. Commun. 2020;533:861–866. doi: 10.1016/j.bbrc.2020.08.042. [DOI] [PubMed] [Google Scholar]
- 44.Mastronarde DN. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 2005;152:36–51. doi: 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 45.Zheng SQ, et al. MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods. 2017;14:331–332. doi: 10.1038/nmeth.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rohou A, Grigorieff N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 2015;192:216–221. doi: 10.1016/j.jsb.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Scheres SHW. Processing of structurally heterogeneous Cryo-EM data in RELION. Methods Enzymol. 2016;579:125–157. doi: 10.1016/bs.mie.2016.04.012. [DOI] [PubMed] [Google Scholar]
- 48.Zivanov J, et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife. 2018;7:e42166. doi: 10.7554/eLife.42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zivanov J, Nakane T, Scheres SHW. A Bayesian approach to beam-induced motion correction in cryo-EM single-particle analysis. IUCrJ. 2019;6:5–17. doi: 10.1107/S205225251801463X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rosenthal PB, Henderson R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol. 2003;333:721–745. doi: 10.1016/j.jmb.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 51.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 52.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adams PD, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D. Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Afonine PV, et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. Sect. Struct. Biol. 2018;74:531–544. doi: 10.1107/S2059798318006551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yamashita K, Palmer CM, Burnley T, Murshudov GN. Cryo-EM single-particle structure refinement and map calculation using Servalcat. Acta Crystallogr. Sect. Struct. Biol. 2021;77:1282–1291. doi: 10.1107/S2059798321009475. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support this study are available from the corresponding authors upon reasonable request. The cryo-EM density map and atomic coordinates for the LPA1-Gi complex have been deposited in the Electron Microscopy Data Bank and the PDB, under accession codes: EMD-34097 (LPA1–Gi stable state), EMD-34098 (focused on LPA1), EMD-34099 (LPA1–Gi state 1), EMD-34100 (LPA1–Gi state 2), EMD-34101 (LPA1–Gi state 3), EMD-34102 (LPA1–Gi state 4), and PDB 7YU3 (LPA1–Gi stable state), 7YU4 (focused on LPA1), 7YU5 (LPA1–Gi state 1), 7YU6 (LPA1–Gi state 2), 7YU7 (LPA1–Gi state 3), 7YU8 (LPA1–Gi state 4). Source data are provided with this paper.