

This study shows that spatial characterization can address the challenge of finding efficient biomarkers, revealing that localization of macrophages and T cells in melanoma predicts patient response to ICI.
Abstract
While immune checkpoint–based immunotherapy (ICI) shows promising clinical results in patients with cancer, only a subset of patients responds favorably. Response to ICI is dictated by complex networks of cellular interactions between malignant and nonmalignant cells. Although insights into the mechanisms that modulate the pivotal antitumoral activity of cytotoxic T cells (Tcy) have recently been gained, much of what has been learned is based on single-cell analyses of dissociated tumor samples, resulting in a lack of critical information about the spatial distribution of relevant cell types. Here, we used multiplexed IHC to spatially characterize the immune landscape of metastatic melanoma from responders and nonresponders to ICI. Such high-dimensional pathology maps showed that Tcy gradually evolve toward an exhausted phenotype as they approach and infiltrate the tumor. Moreover, a key cellular interaction network functionally linked Tcy and PD-L1+ macrophages. Mapping the respective spatial distributions of these two cell populations predicted response to anti-PD-1 immunotherapy with high confidence. These results suggest that baseline measurements of the spatial context should be integrated in the design of predictive biomarkers to identify patients likely to benefit from ICI.
Significance:
This study shows that spatial characterization can address the challenge of finding efficient biomarkers, revealing that localization of macrophages and T cells in melanoma predicts patient response to ICI.
Introduction
Treatment of malignant melanoma has been revolutionized by the introduction of anti-PD-1–based immune checkpoint inhibitors (ICI; refs. 1, 2). Despite having improved overall survival and providing durable response, the outcome of immune checkpoint blockade is very variable, with most stage IV patients still succumbing to the disease (1, 2). Nonetheless, ICI therapy has become a standard of care in melanoma and many other solid cancers.
The widespread use of immunotherapy, the variable results obtained in different tumor types, the considerable and sometimes irreversible toxicity in roughly 20% of the patients and the treatment-associated costs have made the search for predictive biomarkers a pressing issue more than ever before in oncology. Consequently, researchers were prompted to investigate the tumor microenvironment (TME) more thoroughly. As a result, a plethora of different factors correlating with immunotherapy outcome have emerged, including both tumor intrinsic [e.g., tumor mutational burden (3), DNA mismatch repair deficiencies, and microsatellite instability (4), (epi)genetic aberrations leading to defects in antigen presentation (5, 6), signaling defects in JAK/STAT pathway (7), extracellular vesicles (8), and tumoral MHC-II expression (9)] features and TME-related features (e.g., tumor-infiltrating lymphocytes (TIL; refs. 10, 11), specific cytotoxic T cell (Tcy) subtypes such as progenitor-exhausted Tcy (12), tumor-infiltrating B cells and tertiary lymphoid structures (13, 14), intratumoral and gut microbiota (15, 16), myeloid-derived suppressor cells (17), cancer-associated fibroblasts (18), and tumor-associated high-endothelial venules (19)]. However, none of these factors have thus far been broadly implemented in the clinic, partly due to a low predictive value when used as a single marker. In addition, we are still lacking a profound understanding of the mechanism(s) that make anti-PD-1–based therapy successful in 1 patient and unsuccessful in another. Similarly to how next-generation sequencing has made it possible to match mutational profiles to specific targeted therapies (i.e., BRAF and MEK inhibitors; ref. 20), unraveling and improving response to immunotherapy will require an accurate spatial description of the inflammatory landscape within the tumor area (TA) and the specific understanding of the contribution of each factor of the complex immune response machinery.
Recently, the preexistent CD8+ T-cell effector response resulting from chronic tumor antigen exposure and the dynamics following checkpoint blockade has been elucidated more thoroughly. A subset of Tcy that display stem-like properties and that promote tumor control in response to checkpoint blockade immunotherapy has been identified (12, 21, 22). These so-called progenitor exhausted T cells are responsible for the proliferative T-cell burst after administration of anti PD-1 (12, 23). There is substantial evidence that response to checkpoint blockade is not due to reversal of a T-cell exhaustion phenotype but rather depends on proliferation of this stem-like subset of T cells. After eliciting a cytotoxic effect these cells can further differentiate into “terminally exhausted” Tcy. Data supporting this model are mostly generated by methods based on tissue dissociation followed by single-cell analysis, such as single-cell RNA sequencing (scRNA-seq) or mass cytometry. Obviously, these techniques exclude spatial information, thereby precluding the analysis of crucial cell–cell interactions within the context of the original tissue. To partially overcome this, algorithms for analyzing scRNA-seq data have been developed to screen for ligand-receptor interactions across all cell types present in a TME [CellPhoneDB (24)/Nichenet (25)]. These predictive methods are based on prior knowledge of signaling and gene regulatory networks, and while very interesting at producing novel hypotheses, they still require validation at the tissue level. In addition, computational methods to analyze dissociated cells are devoid of visual control and thus bear the risk for inaccurate results. Finally, RNA-based findings may also not translate to the proteomic level, thereby leaving the final executors of most biological interactions unidentified. Considering that immune reactions rely on multiple cell–cell interactions, a study of the mechanism of an immune response upon anti-PD-1 treatment should also include spatial information. Knowing the spatial distribution of the different inflammatory cell subtypes could further fuel our understanding on how immunotherapy works while revealing novel potential therapeutic mechanisms or novel targets with translational clinical utility.
In this study, we used high-dimensional multiplexed IHC (mIHC) according to the Multiple Iterative Labeling by Antibody Neodeposition (MILAN; ref. 26) method through which we visualized 77 immune and tumor-related markers at single-cell resolution. As such, we characterized the cellular composition, architecture, and sociology of the immune landscape in metastatic melanoma, which allowed us to create a detailed, high-dimensional pathology map of Tcy, in which the detailed phenotype of the different Tcy subsets and their exact location in clinical biopsies is identified. This analysis did not only confirm previous findings at the proteomic level, but it also allowed us to visualize and study the interactions between the cellular subsets within their native tissue context. By subsequently applying novel spatial analysis approaches, we were able to construct a spatial trajectory within the tumor in parallel to and corresponding with a pseudotime differentiation trajectory of Tcy, showing that Tcy gradually evolved toward more exhausted phenotypes as they approached and infiltrated the tumor. Finally, we studied the interaction between Tcy and their local immune microenvironment, thereby revealing a role for interactions between Tcy and PD-L1+ M1-like macrophages in the distinction between responders (RESP) and nonresponders (NRESP) to anti-PD-1 immunotherapy.
Materials and Methods
Clinical data
For the discovery cohort A, a set of 16 pretreatment, frozen melanoma metastasis lesions from 14 different patients was selected for NanoString gene expression analysis. All patients were treated with anti-PD-1 monotherapy (nivolumab or pembrolizumab) after the biopsy was taken. Only biopsies taken < 365 days before the start of checkpoint inhibition therapy were included. Furthermore, only patients with measurable disease were selected, hence enabling tumor response assessment according to RECIST 1.1 (27). Patients were classified according to the best objective response to immunotherapy during their time of follow-up, as defined by RECIST 1.1 (27). Complete response and partial response were classified as RESP, progressive disease or stable disease as NRESP. According to these criteria, 7 patients could be classified as RESP (eight samples) and 7 patients as NRESP (eight samples). In addition, 24 pretreatment, formalin-fixed, paraffin-embedded (FFPE) melanoma metastasis samples from 21 patients from the University Hospital of Leuven were collected. Of these samples, 12 were also included in the NanoString analysis. For validation cohort B, 19 pretreatment FFPE melanoma metastasis samples from 16 patients from the University Hospital of Leuven were collected. For both patient cohorts, only metastatic samples were eligible for inclusion. Pathologists selected the most representative areas of the tumors for tissue microarray (TMA) construction. For each metastasis, one to five representative cores/regions of interest were sampled having at least a size of 1 mm in diameter. The number of samples taken was determined by the specimen and the morphologic heterogeneity of both the melanoma and the inflammatory infiltrate. Therefore, a smaller number of cores were taken from small and homogeneous samples whereas a larger number was taken from large but heterogeneous specimens. Specifically for cohort B, cores were sampled only at the tumor-stroma interface (TSI). In addition, core selection was done blinded for patients’ response to immunotherapy. In total, for cohort A 70 and for cohort B 27 cores/regions of interest were selected for analysis. The TMAs were constructed with the TMA Grand Master (3DHistech Ltd.). A subset of patients included were treated with anti-PD-1 monotherapy (nivolumab or pembrolizumab) after the biopsy was taken. Biopsies taken ≥ 365 days before the start of checkpoint inhibition therapy were excluded for response stratification. Following similar response stratification criteria used in the NanoString patient cohort, in cohort A 7 of 21 patients could be classified as RESP (seven samples) and 8 of 21 patients as NRESP (nine samples). For 6 patients (eight samples), response assessment was not possible due to several reasons (e.g., no subsequent therapy with anti-PD-1 treatment, sample too old according to our cutoff of 365 days). To exclude noise in the downstream analysis regarding factors associated with response, these 6 patients were included for the evaluation of the immune landscape but were excluded from the correlation analysis with response. For cohort B, 9 of 16 patients could be classified as RESP (11 samples) and 7 as NRESP (eight samples). An overview of the samples/patients included in this study is provided in Supplementary Tables S1 and S2. Written informed consent was obtained from each patient. The studies were conducted in accordance with recognized ethical guidelines (Declaration of Helsinki). This project was approved by the Ethical Commission of the University Hospital of Leuven and approved by the review board.
NanoString gene expression analysis
The frozen material was analyzed using the PanCancer Immune Profiling Panel of the nCounter technology from NanoString. Transcriptomic counts were log2 transformed. Differential gene expression of RESP patients versus NRESP patients was calculated using the limma R package (28). Enriched pathways were identified using the piano R package (29). Differentially expressed genes were network mapped using stringDB (30). From the different gene set analysis methods included in the Piano pipeline, we selected the following 10 using the gene-level statistic defined in parenthesis: Fisher (P value), stouffer (P value), reporter (P value), page (t value), tailStrength (P value), gsea (t value), mean (logFC), median (logFC), sum (logFC), and maxmean (t value). P values from the different gsa methods were summarized by calculating the median ranking of the individual methods and then mapping the corresponding P value for the obtained rank. For the gene sets, the Molecular Signatures Database (MSigDB; ref. 31), curated pathways (c2), canonical pathways (cp), version 7.2 was used. The enrichment of selected pathways was visualized using fGSEA plots generated with the fgsea R package (32). Insights in general sample cytometry were gained using CibersortX (33) with the LM22 signature matrix. Insights in Tcy specific cell populations cytometry were obtained by first creating a custom signature matrix using the data and defined cell states by Sade-Feldman and colleagues (21) and CibersortX's “Create Signature Matrix” module. Tcy proportions were then estimated using the “Impute Cell Fractions” module. The NanoString results were further validated using the publicly available data reported in Chen and colleagues (11). The expression data were directly downloaded from the supplementary data reported in the publication. Only pretreatment samples were included for analysis (TimePoint = “Pre aPD1”). For this dataset, the same abovementioned differential gene expression and pathway analysis were performed.
MILAN multiplex staining and image acquisition
Multiplex immunofluorescent staining was performed according to the previously published MILAN protocol (26). The antibody panel for MILAN was designed to allow a phenotypic identification of the most abundant cell types based on the results from the NanoString analysis and the scRNA-seq data from others (21), including a more in-depth functional characterization of T cells based on literature review. An overview of the panel with the 77 markers included for the discovery cohort and the specifications about the primary and secondary antibodies can be found in Supplementary Table S3. A second mIHC staining was performed using the validation cohort B with a limited panel of seven markers plus DAPI (DAPI, CD68, CD163, PDL1, CD8, MLANA, SOX10, and S100A1). For cohort A, immunofluorescence images were scanned using the NanoZoomer S60 Digital slide scanner (Hamamatsu) at 20× objective with resolution of 0.45 μm/pixel. The hematoxylin and eosin slides were digitized using the Axio scan.Z1 slidescanner (Zeiss) in brightfield modus using a 20× objective with resolution of 0.22 μm/pixel. For cohort B, immunofluorescence images were scanned using the Axio scan.Z1 slidescanner (Zeiss) at 10× objective with resolution of 0.65 μm/pixel. All downstream analyses took into consideration the differences in pixel size between cohorts A and B to ensure that the same metric distances were evaluated.
Image quality control and analysis
The stainings were visually evaluated for quality by digital image experts and experienced pathologists (F.M. Bosisio, G. Cattoretti, and Y. Van Herck, triple blinded). Multiple approaches were taken to ensure the quality of the single-cell data. On the image level, the cross-cycle image registration and tissue integrity were reviewed; regions that were poorly registered or contained severely deformed tissues and artifacts were identified, and cells inside those regions were excluded. Antibodies that gave low confidence staining patterns by visual evaluation were excluded from the analyses. Image analysis was performed in Fiji/ImageJ following a procedure described previously (34). Briefly, DAPI images from consecutive rounds were aligned (registered) using the Turboreg and MultiStackReg plugins from Fiji/ImageJ (version 1.51 u). The coordinates of the registration were saved as Landmarks and applied to the rest of the channels. Tissue autofluorescence was subtracted from an acquired image in a dedicated channel, for FITC, TRITC, and Pacific Orange. The TMA was segmented into tissue cores using a custom macro. Core segmentation was followed by watershed cell segmentation and single-cell measurements, which were performed using the EBImage R package (35). For every cell, the extracted features included: X/Y coordinates, nuclear size, and mean fluorescence intensity (MFI) for all the measured markers.
Phenotypic identification
MFI values were normalized within each core to Z-scores as recommended in Caicedo and colleagues (36). Z-scores were trimmed in the [0, 5] range to avoid a strong influence of any possible outliers in downstream analyses. Single cells were mapped to known cell phenotypes using three different clustering methods: PhenoGraph (37), FlowSom (38), and KMeans as implemented in the Rphenograph, FlowSOM, and stats R packages. While FlowSom and KMeans require the number of clusters as input, PhenoGraph can be executed by defining exclusively the number of nearest neighbors to calculate the Jaccard coefficient (37), which was set to 12. PhenoGraph groups the input cells into a number of numeric clusters (1,2,…,n) with similar expression profiles. The number of clusters identified by PhenoGraph was then passed as an argument for FlowSom and KMeans.
Clustering was performed exclusively in a subset of the identified cells (50, 000) selected by stratified proportional random sampling and using only the markers defined as phenotypic (Supplementary Table S3). The stratification was performed by selecting a number of cells in each sample equal to the relative proportion of the number of cells in that sample in the entire dataset. That is:
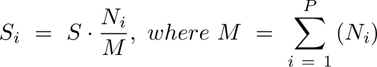 |
where Si is the number of cells to be sampled for the ith sample, S is the total number of cells to be sampled (here 50,000), Ni is the number of cells in the ith sample, and M is the total number of cells in the dataset (sum of all samples, P).
For each clustering method, clusters were mapped to known cell phenotypes following manual annotation from domain experts (F.M. Bosisio, Y. Van Herck, double blinded). If two or more clustering methods agreed on the assigned phenotype, the cell was annotated as such. If all three clustering methods disagreed on the assigned phenotype, the cell was annotated as “not otherwise specified, NOS.” For each phenotype, a fingerprint summarizing the average expression of each marker for all the cells of the given phenotype was constructed. These fingerprints were used to predict the phenotype of all the cells included in the dataset (minimum of Euclidean distance).
In silico tissue microdissection
We further dissected the analyzed samples into TAs, TSI, and nontumor areas (NTA; Supplementary Fig. S1). To that end, tissues were fragmented into 50×50 pixel tiles (22.5 sq. micrometers). Tiles with at least one cell identified as melanoma were initially defined as TA. To reduce the impact of potential outliers, a median filter was applied to the obtained tumor masks. The tumor edge was defined as the overlap between the dilated tumor mask (box kernel of 5px of diameter), a dilated nontumor mask (complementary of the tumor mask, box kernel of 5px of diameter), and the complement of a dilated mask positive for the areas without tissue (i.e., outside mask, box kernel of 5 tiles, 251px of diameter). The complement of the outside mask was used to remove the tumor edge in areas close to the contour of the tissue as predictions around the contour are often unreliable (39). The TSI was defined at 150 pixels (67.5 μm) symmetrically toward and away from the tumor edge (i.e., the border between the tumor and the stroma). This was calculated by dilating the tumor edge with a box kernel of 301 pixels in diameter. The rest of the tissue was defined as NTA. We evaluated the immune populations present in each microdissected area by comparing the proportion of each identified immune phenotype relative to the total immune population (pairwise, Wilcoxon test). Adjustment for multiple comparisons was performed using the FDR method.
High-dimensional proteomic profiling of Tcy
Tcy were further profiled on the basis of the expression of the following 10 markers: CD137/4-1BB, CD69, CD7, CD74, CXCR5, granzyme B (GrB), PD-1, TCF7, TIM-3, and VISTA (“Tcy panel”; Supplementary Table S3). A subset of all the identified Tcy (stratified sampling, 6,000 cells) was clustered using KMeans and manually annotated by domain experts (F.M. Bosissio, Y. Van Herck). Fingerprints were used to calculate the status of all the Tcy included in the dataset (minimum of Euclidean distance). For all the identified subtypes, we looked for enrichment in the different in silico microdissected areas by checking the proportion relative to all the immune cells of each identified subtype between the areas (pairwise, Wilcoxon test). Adjustment for multiple comparisons was performed using the FDR method.
In addition, we assigned an activation score to each Tcy based on a previously published model that integrates the expression of TIM-3 and LAG3 as exhaustion markers and CD69 and OX40 as activation markers into a single score in the [−1, 1] range (34). We looked for differences in Tcy activation in the different in-silico microdissected areas (generally for all Tcy and specifically for each identified subtype) by applying t tests (two-tails, FDR corrected) on the average activation of each core. We also investigated how the distance to the tumor edge affected Tcy activation. To that end, we calculated the Euclidean distance of each Tcy to the closest point in the tumor edge, grouped the Tcy in bins of 10 μm, and calculated the average activation score of all the Tcy in each bin. Distances toward the tumor were defined negative while distances away from the tumor were defined positive. To avoid patient-to-patient variability, we previously normalized the activation of the Tcy by taking z-scores in each core. We fitted a cubic smoothing spline function to the generated curve, calculated its first-order derivative, and looked for the point in the X-axis (distance to the tumor edge) where the Y-axis (derivative of Tcy activation) was maximum, that is, the point where the Tcy activation score changes the most.
Spatial dynamics of Tcy differentiation
Next, we assessed the spatial evolution of Tcy subtypes based on their distance to the tumor edge. To that end, first, we calculated the probability (Pi,j) of each Tcy (i) belonging to any of the categories manually annotated (j) as follows:
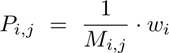 |
where Mi,j is the euclidean distance of the expression of Tcyi to the fingerprint of the subtype j, and wi is the sum of the inverse distances of Tcyi for all the subtypes, that is:
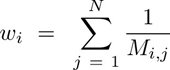 |
where N is the total number of identified subtypes. Essentially, wi guarantees that:
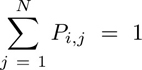 |
Then, we calculated the distance of each Tcy to the tumor edge. Distances toward the tumor were defined negative while distances away from the tumor were defined positive. Continuous distance values were grouped in bins of 10 μm. For each bin, we calculated the probability of belonging to each subtype by averaging the probabilities of all the Tcy included in the bin.
Spatial lineage analysis
We correlated the spatial distance of Tcy to the tumor edge with a pseudotime value obtained after applying Slingshot (40). To that end, PD-1, GrB, TIM-3, CD69, TCF7, and Ki67 expression values were normalized (z-scores) in a number of preselected Tcy subtypes. After normalization, a subset of the data (500 cells per Tcy subtype) was randomly sampled. Then, diffusion maps as implemented in the destiny R package (41) were used to dimensionally reduce the data to three components. The rest of the dataset was projected on the reduced space using the dm_predict function. Lineages and trajectories were calculated using the getLineages and getCurves functions from the slingshot R package (40). The obtained pseudotime score was normalized in the [0–1] range. We correlated the density of Tcy subtypes, their spatial location, and the expression of each of the included markers in this analysis with the obtained normalized pseudotime score.
Survival analysis
For each candidate biomarker, we evaluated their prognostic value by evaluating all the potential cutoffs between the 10% and 90% quantiles. In each potential threshold, we calculated the P value associated with a log-rank test. To increase the robustness of our cutoff, we applied a running average on the continuous distribution of P values and selected the threshold that returned the lowest P value after applying the running average. This P value was then used to classify each patient as high or low for each given biomarker. This classification of patients was finally used to create the Kaplan–Meier curves and estimate the log-rank P value associated with each potential biomarker.
Data reporting
All samples within each patient cohort were stained simultaneously. Image acquisition order was distributed spatially and independently of patient or tumor replicates. Image acquisition, single-cell quantification, and clustering were blinded to patient identifiers and clinical metadata. No statistical methods were used to predetermine sample size.
Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
Results
NanoString Gene expression analysis
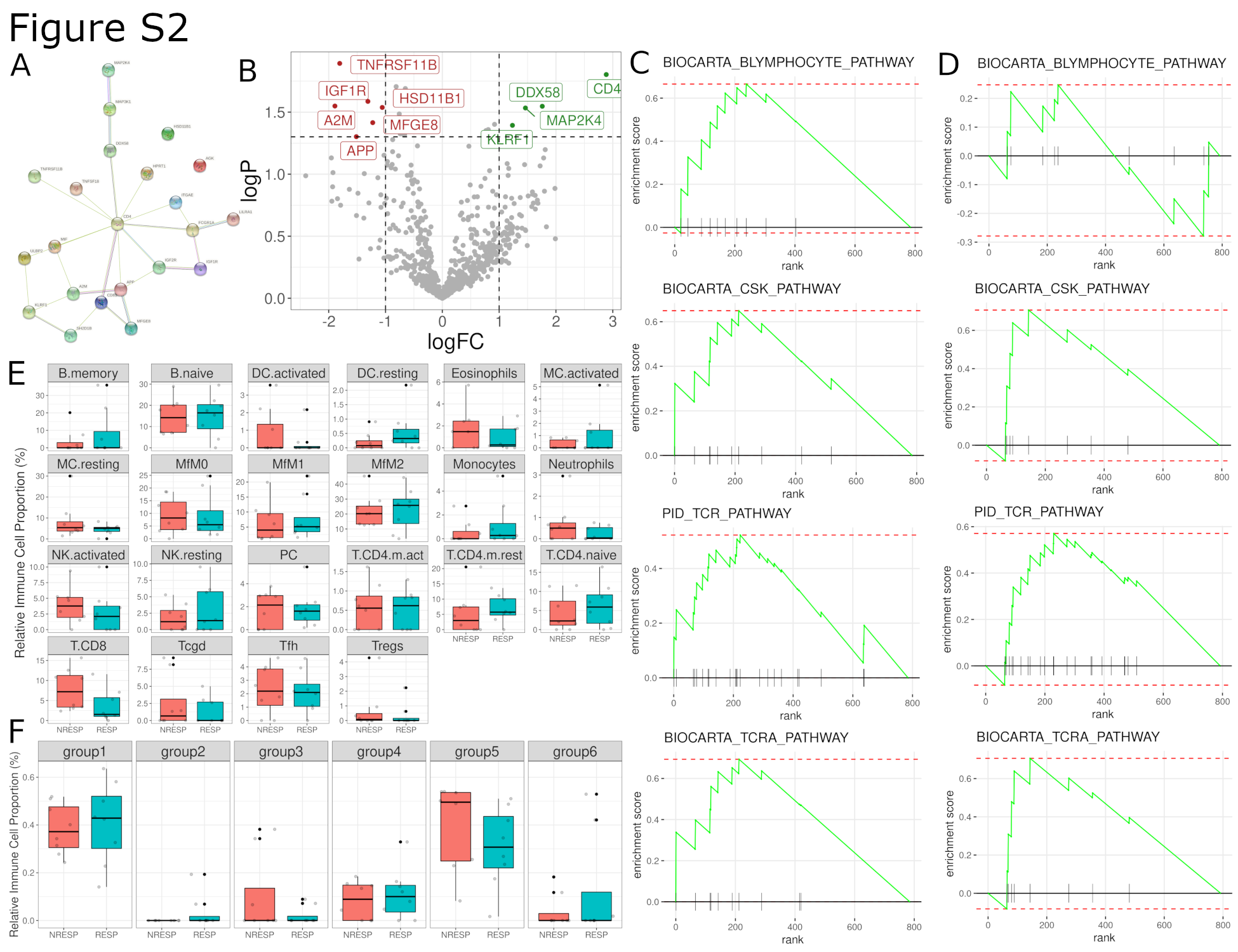
We first performed bulk transcriptomic analysis using the PanCancer Immune Profiling Panel of the NanoString nCounter technology on the frozen material available (whole tumoral nodules) from 16 metastatic melanoma samples (8 RESP, 8 NRESP, clinical data in Supplementary Table S1). In total, 10 of 784 genes included in the gene panel were differentially expressed (Supplementary Table S4). Differentially expressed genes were network projected using String-DB (Supplementary Fig. S2A; ref. 30). CD4, DDX58, MAP2K4, and KLRF1 were overexpressed in RESP patients, while HSD11B1, MFGE8, IGF1R, APP, TNFRSF11B (OPG), and A2M were underexpressed (Supplementary Fig. S2B). DDX58 and MAP2K4 have been associated with response to anti-CTLA4 and MEK inhibition, respectively (42, 43), whereas KLRF1 is associated with natural killer cell infiltration, T-cell exhaustion, and reduced TNF/IFNγ production (44). On the other hand, several of the underexpressed genes are known to be associated with specific remodeling of the tumoral microenvironment, for instance the production of OPG or alpha-2 macroglobuline (A2M) with potential cytokine scavenger properties (45), M2 polarization of tumor-associated macrophages via IGF1 and MFG-E8 (46, 47), or the inhibition of proliferation among tumor-specific CD8+ T-cells by means of increased local cortisol with the induction of HSD11B1 (48). Performing gene set enrichment and pathway analysis (Supplementary Table S5), we observed in RESP an upregulation of B cell–related and T cell–related pathways and of pathways involved in the adaptive immune system like B lymphocyte pathway, CSK pathway, T-cell receptor signaling pathway, TCRA pathway, among others (Supplementary Fig. S2C). The importance of these pathways was validated in an independent dataset that shows an upregulation of all reported pathways, with the exception of B lymphocyte pathway, that could be influenced by sample to sample variability in B-cell fraction (Supplementary Fig. S2D; ref. 11). When evaluating the cell fractions obtained after using CibersortX, we did not find significant cell differences in general cell composition (LM22) or in Tcy subtypes (Supplementary Fig. S2E and S2F). We used these results to assemble a specific antibody panel for the subsequent in situ single-cell analysis, focusing on the lymphocytic, macrophagic, and antigen-presenting cells compartment.
Immune landscape and in silico microdissection
Using MILAN in the discovery cohort (see Materials and Methods), we were able to identify 1,426,617 cells (average of 67,934.14 cells per patient, SD of 52,588.52; range of 7,316–247,806) containing information on nuclear size, X/Y coordinates, and expression (MFI) for each of the 77 markers included in the analysis (Supplementary Table S3). Applying unsupervised consensus clustering (see Materials and Methods, phenotypic identification) by using a panel of 37 selected phenotypic markers, we identified 67 clusters that were manually annotated to 18 cell phenotypes, each corresponding to a specific protein signature and/or fingerprint, characterized by the following main markers (Fig. 1A and B; Supplementary Fig. S3A–S3D): B cells (“BC”; CD20, CD79a, and PAX5), plasma cells (“PC”; CD138 and PRDM1), classical dendritic cells type 1 (“cDC1”; CD141 and IRF8), classical dendritic cells type 2 (“cDC2”; CD1c), follicular dendritic cells (“fDC”; CD23 and CD21), plasmacytoid dendritic cells (“pDC”; CD303), cytotoxic T-cells (“Tcy”; CD3 and CD8), Th cells (“Th”; CD3 and CD4), regulatory T cells (“Treg”; CD3, CD4 and FOXP3), blood vessels (“BV”; CD31 and CD34), high endothelial venules (“HEV”; PNAd), lymphatic vessels (“LV”; Podoplanin), epithelial cells (“EC”; CK), M1-like macrophages (“M1Mf”, CD68, CD64, and LYZ), M2-like Macrophages (“M2Mf”, CD68, CD64, and CD163), and melanoma (“Mel”, S100B and Melan-A). Clusters with the expression of several markers without an obvious phenotypic profile were annotated as “not otherwise specified, NOS.” Clusters with no expression of any phenotypic marker were annotated as “blank.” A Uniform Manifold Approximation and Projection (UMAP) colored by patient and metastatic site showed a homogeneous distribution indicating the absence of batch effects regarding these two parameters (Supplementary Fig. S3E and S3F). From the 49,998 cells included in the clustering (two cells less than the 50,000 due to rounding effects on the stratification), 24,767 (49.54%) showed agreement in the assigned phenotype between all three included clustering methods, 21,685 (43.37%) showed agreement between two clustering methods, and 3,546 (7.09%) showed an inconsistent phenotype assignation. Cells with an inconsistent labeling between the different clustering methods were also labeled as “NOS” (Supplementary Fig. S3G–S3I). On the basis of the predicted phenotypes and the X/Y coordinates of each individual cell, the tissue was digitally reconstructed, resembling the morphology of the corresponding hematoxylin and eosin staining of a previous section, but showing the phenotypic identity of each cell type (Fig. 1C–E). General sample composition is summarized in Supplementary Table S6. We did not find significant differences in general cell composition between RESP and NRESP (Wilcoxon rank-sum test, two tails).
Figure 1.

Phenotypic identification and in silico microdissection. A, UMAP showing the 18 different cell phenotypes identified during phenotypic clustering. The colors in the UMAP represent the populations after manual annotation of the 67 clusters obtained with the preselected 37 protein markers. B, Heatmap showing the protein signatures of the different cell phenotypes. Rows represent identified cell types; columns represent protein markers. The score inside each cell of the matrix indicates the average expression of the marker in the identified population. C, Representative core from sample MEL9 from patient PT7 with hematoxylin and eosin staining. D–F, Composite fluorescent image of four markers (+DAPI) after image processing (D), digital reconstruction of the core highlighting the phenotypic identify of each individual cell (E), and in silico microdissection into three areas and the tumor edge (solid black line) within the TSI (F). G, Cell composition analysis of selected cell phenotypes comparing the relative proportion of immune cells in the different in silico microdissected areas (NTA, nontumor area; TSI, tumor-stroma interface; TA, tumor area), using pairwise Wilcoxon test. BC, B cells; PC, plasma cells; cDC1, classical dendritic cells type I; cDC2, classical dendritic cells type II; fDC, follicular dendritic cells; pDC, plasmacytoid dendritic cells; Th, T helper cells; BV, blood vessels; HEV, high endothelial venules; LV, lymphatic vessels; M1Mf, M1-like macrophages; M2Mf, M2-like macrophages; Mel, melanoma cells; NOS, not otherwise specified. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. P values > 0.05 are not reported.
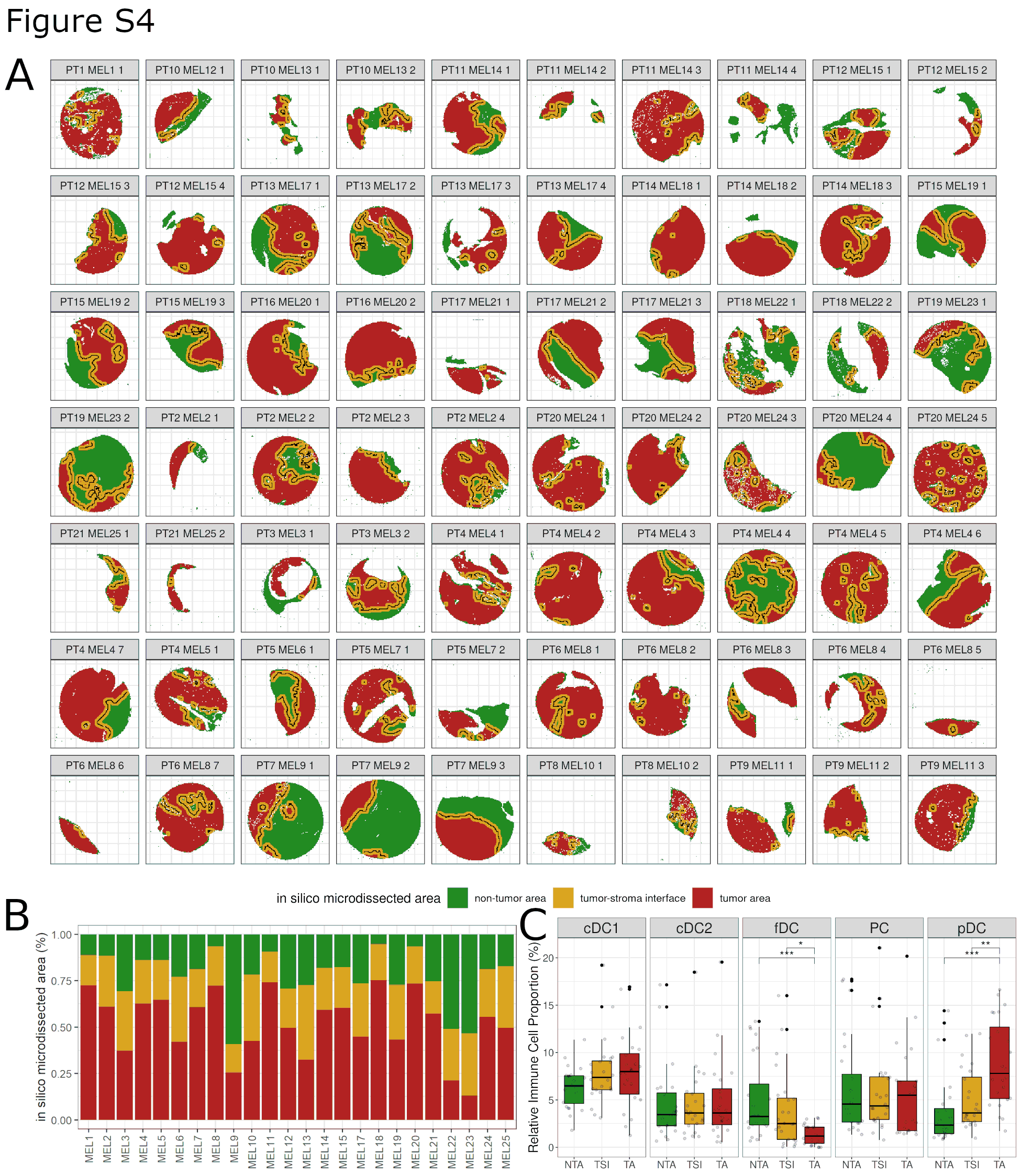
Next, using the phenotypical identity of each cell within the digitally reconstructed tissue, we further in silico microdissected the tissue, separating TA, TSI, and NTA (Fig. 1F; Supplementary Fig. S4A). Overall, 51% of the areas were labeled as tumor (interpatient SD = 17.44%; range, 13.24%–75.28%), 24.6% as TSI (SD = 6.97%; range, 15.07%–39.88%), and 24.4% as nontumor (SD = 14.4%; range, 5.29%–59.32%), with a large interpatient variability (Supplementary Fig. S4B). On the basis of the different areas, the global cell composition was redefined into an area-specific cell composition. In the TSI 29.54% of the cells were melanoma cells (compared with 76.39% in the TA). On the contrary, in the TA, approximately 23.61% of the cells were identified as nonmelanoma cells, mainly infiltrating immune cells (12.77%; Supplementary Table S6). Subsequently, the relative proportion of the various immune cells was compared between the different areas (Fig. 1G; Supplementary Fig. S4C). BC, Th, and Treg were significantly enriched in the NTA compared with the TA with an intermediate level in the TSI. Tcy on the contrary peaked within the TSI with a significant enrichment compared with the TA but no significant enrichment compared to the NTA. Both M1-like and M2-like macrophages were significantly enriched in the TA compared with the TSI and NTA. In fact, macrophages represented the most abundant immune cell type within the TA (3.13% M1-like and 2.56% M2-like) in our patient cohort. No significant differences could be found regarding the immune composition of RESP versus NRESP, both overall (Supplementary Fig. S5A) as well as in the different microdissected areas (Supplementary Fig. S5B).
High-dimensional proteomic profiling of Tcys
To gain deeper insights into the different Tcy subsets present in situ at the proteomic level, we performed a second level of clustering using the expression of 10 markers that were not included in the first level clustering, now exclusively focusing on the previously identified Tcy. KMeans identified 34 clusters that after manual annotation were mapped to nine Tcy subtypes, each with a specific protein signature and/or fingerprint (Fig. 2A–C; Supplementary Fig. S6A and S6B, biologically explained in Supplementary Table S7).
Figure 2.

Profiling of Tcys: cytometry and activation. A, UMAP showing the nine different Tcy subtypes identified during Tcy clustering. The colors represent the populations after manual annotation of the 34 clusters identified by KMeans with the selected 10 markers. B, Heatmap showing the protein signatures of the different Tcy subtypes. Rows represent identified Tcy subtypes; columns represent protein markers. The score inside each cell of the matrix represents the average expression of the marker in the subtype. C, Relative immune cell proportion of different Tcy subtypes. D, Cell composition analysis of Tcy subtypes comparing the relative proportion within Tcy in the different in silico microdissected areas (NTA, nontumor area; TSI, tumor-stroma interface; TA, tumor area), using pairwise Wilcoxon test. E, Average activation of Tcy in the different in-silico microdissected areas, using pairwise t test. F, Boxplots representing the activation of different selected Tcy subtypes. G, Boxplots representing the activation of the different subtypes within the different in silico microdissected areas using pairwise t test. H, Activation gradients around the tumor edge (all patients). Green-shaded area, NTA; gold-shaded area, tumor-stroma interface; red-shaded area, tumor area. Each black dot represents the average activation level of the Tcy at a given discretized distance from the tumor edge. The blue line represents the curve fitting for the population of black dots. The red dashed line represents the first-order derivative of the blue line. I, Activation gradients around the tumor edge (RESP, responders vs. NRESP, nonresponders). **, P < 0.01; ***, P < 0.001. P values > 0.05 are not reported.
In addition, using the in silico microdissection and the previously described proteomic Tcy signatures, we analyzed the possible enrichment of specific subtypes in the different areas of the tumor. No significant differences could be found for most of the different Tcy, although an expected trend toward enrichment of the most exhausted subtype (Tcy09) within the TA seemed to emerge (Fig. 2D). On the contrary, the TCF7-expressing subtypes, that is, Tcy05 and Tcy06, were blocked at the TSI and were not detected in the tumor bed, confirming the finding described in a previous report (Fig. 2D; ref. 12). Finally, comparing RESP versus NRESP, no significant differences were found in bulk (Supplementary Fig. S6C) and in silico microdissected areas (Supplementary Fig. S6D).
Spatial dynamics of Tcy differentiation
As Tcy infiltrate the tumor and are chronically exposed to tumor-specific antigens (tumor–immune interaction) and to the immune microenvironment (immune–immune interaction), a spatial differentiation trajectory is expected. We hypothesized that we did not completely capture the spatial differences in Tcy subtypes with our previous analysis because of the use of microdissected areas (TA, TSI, and NTA) as opposed to a continuous spatial description from the NTA toward the TA. First, based on our observation of a trend toward enrichment of the exhausted phenotypes within the tumor, we evaluated the spatial dynamics of Tcy activation, applying a simplified model that integrates the simultaneous expression of four markers (CD69, OX40, LAG3, and TIM-3) as published previously (34). By comparing the average level of activation in the three different microdissected areas, not surprisingly Tcy are significantly more active in the NTA compared with Tcy in the TA (Fig. 2E and F). Furthermore, the same spatial dynamics is observed when analyzing Tcy activation for each Tcy subtype in the different microdissected areas although only that of Tcy06 is significant (Fig. 2G).
Next, to profile how this level of activation dynamically evolves even in a higher resolution, the average level of activation of Tcy at specific distances from the tumor edge was analyzed. This confirmed the largest change in activation status as happening in the TSI at 60 μm of the tumor edge toward the NTA (Fig. 2H). Interestingly, at 180 μm from the tumor edge toward the NTA the normalized Tcy activation starts decreasing as indicated by the negative value of the first derivative of the fitted curve (see Materials and Methods; Fig. 2H). This suggests that active Tcy accumulate in the vicinity of the tumor edge still within the NTA. When comparing RESP versus NRESP, the gradient in activation is more pronounced for RESP patients (Fig. 2I).
We expected to see a similar spatial transition in Tcy differentiation. Indeed, when plotting the probability of each Tcy belonging to any of the proteomic Tcy signatures at different distances to the tumor edge (see Materials and Methods), it was apparent that Tcy outside the tumor, far from the edge, are likely to be part of cluster Tcy05. On the contrary, when closer to the tumor edge, this likelihood decreases, and Tcy are more likely to be part of cluster Tcy06 and Tcy07 (Fig. 3A). Tcy infiltrating the tumor are more likely to be part of Tcy08 and eventually Tcy09 as they infiltrate deeper in the tumor, very much in line with the results obtained from our Tcy activation analysis (Fig. 2E–G). This spatial trajectory of Tcy subtype signature from outside the tumor to inside the tumor (Tcy05–Tcy09), is also notable by the mean distance to the tumor edge for each of the Tcy subtypes (Supplementary Fig. S7). Moreover, when comparing the expression values of selected markers of activation and exhaustion in Tcy05–09, we observed a gradient in expression of these markers in these subtypes (Fig. 3B). On the basis of these results, we hypothesized these spatial changes in Tcy subcluster signature correlates with a spatial differentiation trajectory within the tissue and this trajectory in T-cell phenotype and function is caused by the dynamic process of T cells infiltrating the tumor from the peritumoral niche. To further corroborate this spatial behavior, we correlated the spatial distance of Tcy to the tumor edge with a pseudotime value obtained after cell lineage and pseudotime inference using Slingshot (40) to a selected number of Tcy subtypes (Fig. 3C–F). Indeed, we could observe a nonrandom behavior between the spatial location of Tcy and the pseudotime values, confirming that Tcys that are deeply infiltrative in the tumor are indeed the more terminally differentiated/exhausted cells and Tcy outside the tumor are generally more functional/less exhausted and hence are at the beginning of their differentiation trajectory. Our results show a peak in cytotoxicity (as determined by GrB expression) and proliferation (as determined by Ki67 expression) combined with a lower TCF7 and/or higher PD-1 expression at the inner border of the TSI compared with the outer rim of the tumor stroma interface or nontumoral areas (Fig. 3F).
Figure 3.

Profiling of Tcys: lineage. A, Probability gradients around the TSI. Green-shaded area, NTA; gold-shaded area, TSI; red-shaded area, tumor area. Black dots represent the estimated probability of a Tcy in that region to belong to any of the subtypes. The blue curve represents the curve fitting for that cloud of dots. B, Barplots showing the average expression of six preselected markers (CD69, GrB, Ki67, PD-1, TCF7, and TIM3) for a subset of the identified Tcy subtypes (Tcy05:Tcy09). C, Diffusion map representing the projection of the trajectory described by the pseudotime analysis (black line). The scatter plot is colored by the different preselected Tcy subtypes (Tcy05:Tcy09). D, Density plot of the Tcy subtypes within the trajectory defined by the normalized pseudotime. E, Scatter plot showing the correlation between the distance to the tumor edge and the inferred pseudotime trajectory. Black dots represent the average normalized pseudotime for the set of Tcy located at a given distance from the tumor edge. The dashed red line represents the first-order derivative of the fitted curve. F, Histogram plots showing the average expression value of the markers used to define the pseudotime along the trajectory. The normalized distance to the tumor edge (DTE) is also represented.
Spatial Tcy macrophage interaction
Other groups have shown by using ex vivo and/or functional assays that the myeloid compartment and Tcy closely interact and influence each other in multiple ways, such as, myeloid cell triggered suppression of Tcy effector function and/or Tcy exhaustion or Tcy-induced macrophage polarization (49–51). Nonetheless, in situ confirmation of these findings by spatial analysis is rather limited. We were therefore specifically interested in further deciphering this Tcy–macrophage interaction.
Spatial effect of neighboring Tcy on PD-L1 expression in macrophages
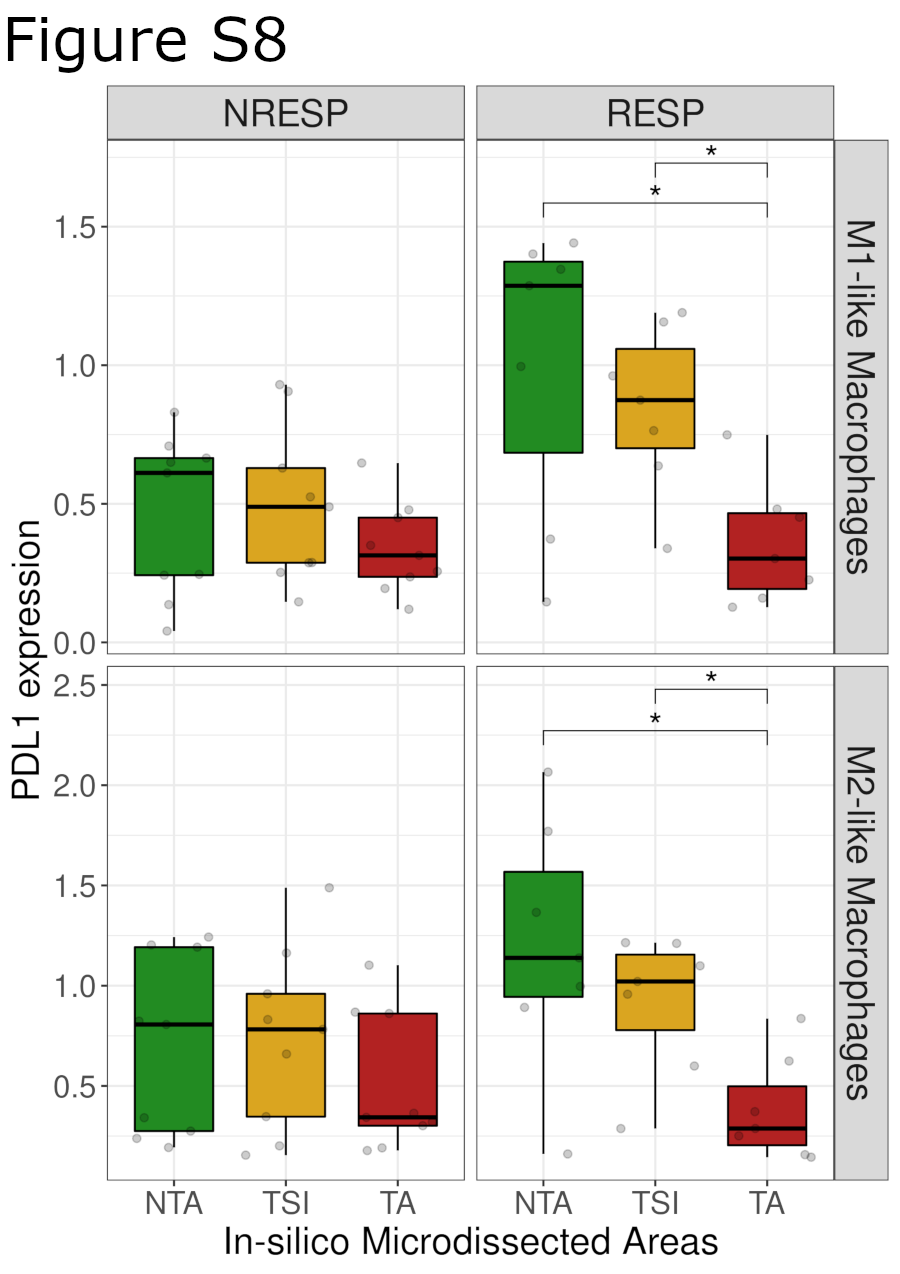
We first analyzed the correlation between neighboring Tcy and the expression of PD-L1 in macrophages. PD-L1 expression in macrophages has been described to have predictive value in patients treated with anti-PD-1 antibodies in melanoma, ovarian cancer, sarcoma, and non–small cell lung cancer, suggesting that the PD-L1 status of macrophages is important in addition to their mere presence in the tumor stroma (52, 53). Similar to Tcy activation, we analyzed how the expression of PD-L1 in both M1-like and M2-like macrophages differs depending on their localization relative to the tumor. For both types of macrophages, the PD-L1 expression is significantly lower in the TA when compared with the NTA and the TSI (Fig. 4A). Next, by increasing the spatial resolution around the tumor edge, we observed a peak in PD-L1 expression close to the tumor edge outside of the tumor for M1-like and M2-like macrophages (peak at 40 and 70 μm, respectively, from the tumor edge), with the highest expression observed in M2-like macrophages (Fig. 4B). On the basis of the predictive value of PD-L1 expression in macrophages for checkpoint inhibitors described by others (52, 53), we repeated the analysis only for the anti-PD-1–treated patients (7 RESP, 8 NRESP). Remarkably, for both types of macrophages, these differences are only preserved for the subset of RESP patients whereas for the NRESP patients the differences are not significant (Fig. 4C; Supplementary Fig. S8). Next, we hypothesized that the expression of PD-L1 in macrophages is also influenced by the local microenvironment, specifically by Tcy, for example via IFNγ secretion (54). For this analysis, we calculated the shortest distance to the closest Tcy for all macrophages irrespective of their location and defined a cut-off value of 100 μm to discriminate between macrophages that could be considered close to at least one Tcy (“Tcy-close”) and macrophages that could be considered far from any Tcy (“Tcy-far”). Comparing PD-L1 expression in these two groups of macrophages, PD-L1 was found significantly higher in “Tcy-close” macrophages (Fig. 4D). Coherently, when analyzing the expression of PD-L1 relative to a gradual increase in distance to the closest Tcy, we observed a gradual decrease in the level of expression with the distance away from a Tcy (Fig. 4E). Interestingly, when comparing RESP and NRESP, an inverse correlation between PD-L1 expression and distance to closest Tcy is only present in the RESP patients (Fig. 4F).
Figure 4.

PD-L1 expression in macrophages. A, Boxplots showing PD-L1 expression in M1-like and M2-like macrophages in the different in silico microdissected areas (NTA, nontumor area; TSI, tumor-stroma interface; TA, tumor area), using pairwise t test test. B, PD-L1 gradient based on distance to the tumor edge. Green-shaded area, NTA; gold-shaded area, TSI; red-shaded area, tumor area. Dots represent the average PD-L1 expression in M1-like and M2-like macrophages at a certain distance of the tumor edge. Lines represent the fitted curves for those clouds of points. C, PD-L1 gradient based on distance to the tumor edge stratified by patient response. D, Boxplots showing PD-L1 expression based on discretized distance to closest Tcy (cut-off distance value, 100 μm) using pairwise t test (FDR corrected). E, Scatter plot showing PD-L1 expression in M1-like and M2-like macrophages based on their distance to the closest Tcy. Each dot represents the average expression of the macrophages located at a certain distance from the closest Tcy (bins of 10 μm). Yellow-shaded area, “close” region; light-blue-shaded area, “far” region. F, Scatter plot showing the PD-L1 expression in M1-like and M2-like macrophages based on their distance to closest Tcy stratified by patient response (RESP, responders; NRESP, nonresponders). G, Boxplots showing PD-L1 expression based on discretized distance to the edge and to closest Tcy using pairwise t test (FDR corrected). CC, Close-Close; CF, Close-Far; FC, Far-Close; FF, Far-Far. H, Boxplots showing PD-L1 expression based on discretized distance to the edge and to closest Tcy stratified by patient response using pairwise t test (FDR corrected). *, P < 0.05; **, P < 0.01; ***, P < 0.001. P values > 0.05 are not reported.
On the basis of the previous analysis, two spatial components influence the expression of PD-L1 in M1-like and M2-like macrophages: (i) The localization of the macrophage relative to the tumor, and (ii) distance to Tcy, specifically for RESP patients. To evaluate whether both spatial components have a synergistic effect on PD-L1 expression, we compared the level of PD-L1 in the following four groups, both for M1-like and M2-like macrophages: (i) macrophages close to the tumor edge and close to Tcy (“Close-Close”; CC), (ii) macrophages close to the tumor edge but far from Tcy (“Close-Far”; CF), (iii) macrophages far from the tumor edge but close to Tcy (“Far-Close”; FC), and (iv) macrophages far from the tumor edge and far from Tcy (“Far-Far”; FF). Confirming the previous analysis, both spatial features (distance to tumor edge and distance to Tcy) were associated with increased expression of PD-L1 (comparing both “Close-Far” and “Far-Close” with “Far-Far”). As expected, the highest expression is observed in the “Close-Close” subgroup, suggesting a rather additive effect of both spatial components (Fig. 4G). When comparing RESP and NRESP, the significant difference between “Close-Close” and “Far-Far” is only preserved in the RESP patients both in M1-like and M2-like macrophages (Fig. 4H).
Spatial effect of neighboring macrophages on Tcy activation
Next, we investigated the effect of neighboring macrophages on the activation status of Tcy. We previously showed a gradual onset of exhaustion as Tcy infiltrate the tumor (Fig. 2E). Similar to the influence of neighboring Tcy on the expression of PD-L1 in macrophages, we explored how this spatial behavior of Tcy activation is affected by the presence of macrophages close to Tcy. To start, using the shortest distance to the closest macrophage, we separated Tcy into “M1 close”/“M2 close” Tcy and “M1 far”/“M2 far” Tcy using 30 μm as threshold to define “close” versus “far” (Fig. 5A and B). We found that Tcy close to M1-like macrophages are significantly more exhausted than Tcy that are far away from M1-like macrophages, which was not significant for M2-like macrophages (Fig. 5C). When comparing RESP with NRESP, remarkable differences emerge. Whereas Tcy close to M1-like macrophages are significantly more exhausted compared with Tcy far from M1-like macrophages in the RESP patients, no significant difference in level of activation of Tcy in relation to macrophages is observed in the NRESP patients (Fig. 5D).
Figure 5.

Tcy activation levels based on distance to closest macrophage. A, Scatter plot showing average activation value of Tcy based on distance to closest macrophage. Yellow-shaded area, “close” region; light-blue-shaded area, “far” region. B, Scatter plot showing average activation value of Tcy based on distance to closest macrophage stratified by patient response. C, Boxplots showing the activation level of Tcy based on discretized distance to closest macrophage using pairwise t test (FDR corrected). D, Boxplots showing the activation level of Tcy based on discretized distance to closest macrophage stratified by patient response using pairwise t test (FDR corrected). E, Boxplots showing the activation level of Tcy based on discretized distance to closest macrophage and distance to the edge using pairwise t test (FDR corrected). CC, Close-Close; CF, Close-Far; FC, Far-Close; FF, Far-Far. F, Boxplots showing the activation level of Tcy based on discretized distance to closest macrophage and distance to the edge stratified by patient response using pairwise t test (FDR corrected). *, P < 0.05; **, P < 0.01; ***, P < 0.001. P values > 0.05 are not reported.
Finally, we combined both spatial features (location of Tcy relative to the tumor edge and distance to closest M1/M2-like macrophage) comparing the level of Tcy activation in the following four groups: (i) Tcy close to the tumor edge and close to macrophage (“Close-Close”), (ii) Tcy close to the tumor edge but far from macrophage (“Close-Far”), (iii) Tcy far from the tumor edge but close to macrophage (“Far-Close”), and (iv) Tcy far from the tumor edge and far from macrophage (“Far-Far”). Both for M1- and M2-like macrophages, the two spatial features work synergistically in exhausting Tcy with the highest exhaustion present in Tcy close to the tumor edge and close to macrophages (Fig. 5E). Similarly, when comparing the level of exhaustion in these four different spatial groups of Tcy within RESP and NRESP, the significant difference is only preserved in the RESP for M1-like macrophages, showing again the highest exhaustion in “Close-Close” Tcy (Fig. 5F).
Spatial biomarkers predicting response to anti-PD-1 therapy
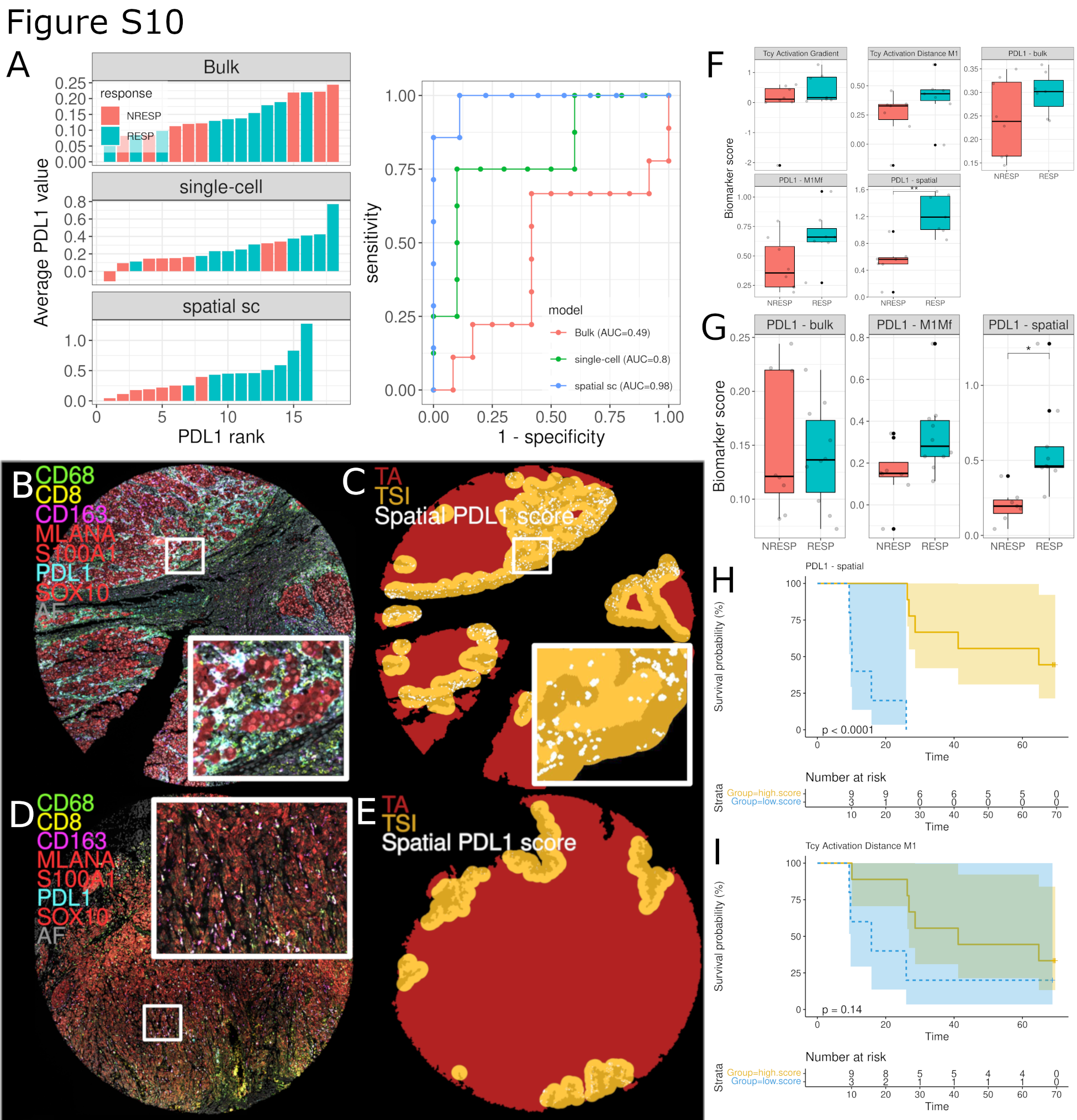
The ultimate goal of mIHC is to serve as a predictive tool for patients undergoing ICI therapy. We therefore explored to which extent spatially extracted features can be used to predict response to anti-PD-1 treatment. On one hand, we observed significant spatial differences in the expression of PD-L1 in macrophages between RESP and NRESP (Fig. 4). On the other hand, the predictive value of bulk PD-L1 expression in melanoma has not yet been shown to have predictive ability in serving as a clinical biomarker. This is in contrast to other immune checkpoint inhibitor sensitive diseases where the “bulk analysis” of PD-L1 is used as a stratifying factor for anti-PD-1 therapy (58, 59). We were therefore wondering whether adding spatial information would further improve the predictive value of PD-L1 expression. First, considering the average PD-L1 expression in the entire sample (“bulk analysis”) carries, as expected, a rather low predictive value (AUC = 0.68; Fig. 6A, top). When comparing the PD-L1 expression in M1-like and M2-like macrophages between RESP and NRESP (“single-cell, nonspatial”), we observed an increase in AUC only for the expression in M1-like macrophages (AUC = 0.80; Fig. 6A, center). By adding spatial information and focusing on the expression of PD-L1 exclusively in macrophages that are close to the tumor edge and close to Tcy, we observed a further increase in the predictive value in M1-like macrophages reaching an AUC of 0.98 (Fig. 6A, bottom). Next, we explored the predictive performance of this spatially resolved biomarker in an independent validation cohort of patients with anti-PD-1–treated melanoma collected retrospectively (11 samples from 9 RESP and eight samples from 7 NRESP). For this, MILAN was executed using a limited panel of markers designed to specifically apply only this spatial biomarker (Supplementary Fig. S9). Indeed, in this independent patient cohort, predictive performance was comparable, reaching an AUC of 0.98 (Supplementary Fig. S10A). An illustrative composite immunofluorescence image and digitization of the spatial PD-L1 score from both a RESP and NRESP can be found in Supplementary Fig. S10B–S10E.
Figure 6.

High order biomarkers. A, Left, barplots showing the average PD-L1 expression in all cells (top; “bulk”), M1-like macrophages (middle; “single-cell”), and spatially selected (distance to tumor edge less than 30 μm and distance to the closest Tcy less than 10 μm) M1-like macrophages (bottom; “spatial single-cell”) for each patient and colored by patient response. Responders, RESP; nonresponders, NRESP. Right, ROC curves corresponding to the score rankings for the different candidate biomarkers. B, Left, barplot showing the difference in Tcy activation between tumor and NTAs for RESP and NRESP patients. Each bar represents a patient. The module of the bar is calculated as the difference between the average Tcy activation in the NTA and the average Tcy activation in the TA. Activation values were first normalized (z-scores) for each core. Right, ROC curve corresponding to the score ranking. C, Left, barplot showing the average Tcy activation level at less than 10 μm from the closest M1-like macrophage. Right, ROC curve corresponding to the score ranking.
Likewise, an improvement in the predictive value of Tcy activation could be obtained by adding spatial information. Considering the differences in spatial dynamics in Tcy activation between RESP and NRESP (Fig. 2I), and by comparing the difference in Tcy activation between TA and NTA, it was apparent that RESP patients were poorly separated from NRESP patients (AUC 0.71; Fig. 6B). However, when considering only those Tcy that are close to M1-like macrophages and comparing the level of Tcy activation, the predictive value increased, reaching an AUC of 0.82 (Fig. 6C). Notably, across all candidate biomarkers, only “PD-L1 expression in spatially selected M1-like macrophages” provided statistical significance in both the discovery and validation cohort (Supplementary Fig. S10F and S10G). This highlights the potential value of this candidate biomarker not only showing good classification performance in two independent datasets, but also significantly different value distributions.
Finally, to investigate the prognostic value of both spatial biomarkers (Tcy activation close to M1-like macrophages and PD-L1 expression in spatially selected M1-like macrophages), patients were classified in a “high” and “low” score group, independent of response to immunotherapy. Indeed, for both features a high score indicates better survival, yet only reaching significance when using PD-L1 expression in spatially selected M1-like macrophages (Supplementary Fig. S10H and S10I).
Discussion
The race to identify biomarkers to predict a response to checkpoint inhibition therapy in patients with malignant melanoma has produced a plethora of potential candidates over the past few years. Nevertheless, for melanoma, none of these therapies has been implemented in clinical practice as they have not elicited sufficient predictive power thus far. In contrast to targeted therapy, where the presence of a single driver mutation poses a simple and effective marker for selecting patients most likely to respond to the corresponding therapy, immunotherapy-treated patients’ responses are vastly more complex. Obviously, the immune microenvironment poses a multicellular system, which amongst others is constituted of multiple types of inflammatory cells, each of them present in many different functional states and locations. It has become obvious that to adequately interrogate the tumor immune ecosystem, the use of single-cell technologies that include as many parameters as possible is required. Importantly, these technologies should also enable them to capture the spatial distribution of the different cell types and shed light on their spatial relationships. In this study, we have employed mIHC to create a detailed immune landscape of untreated metastatic melanoma, with a focus on the role of Tcy and their spatial interactions with other components of the TME.
Previous reports based on low-plex IHC have found preliminary evidence that the location, and not just the composition of the inflammatory cells is associated with functional differences that are fundamental for an antitumoral immune response (55, 56). With our high-plex spatial technique, we were able to precisely localize each inflammatory cell in the tissue. This allowed us to study the evolution of some specific functional states in relation to the spatial distribution of these cells. In particular, we identified Tcy subtypes based on a marker profile described in previous literature (21) and identified their location in the tissue. The approach we used could be applied on much larger numbers of cells compared with other single-cell techniques, for example, scRNA-seq. Moreover, by applying novel spatial analysis tools in combination with our previously described Tcy activation score (34), we visualized an increase of Tcy exhaustion along a gradient that ran perpendicular from outside the tumor, across the TSI toward the inside of the tumor.
In addition, by studying cell–cell interactions, we have demonstrated that PD-L1 expression is a discriminator for response in our melanoma cohort when analyzed in the spatial context. Our findings suggest that, at least in metastatic melanoma, PD-L1 expression should not be analyzed on a bulk level but using technologies with much higher resolution. Specifically, when integrating spatial information and cell–cell interaction between Tcy and macrophages, the technique we employed allowed us to further decipher differences between RESP and NRESP. It is known that PD-L1 expression in macrophages is induced by an IFNγ-rich environment secreted by lymphocytes (54), and that PD-L1 expression by macrophages is typically present at the TSI (57). Our data demonstrate that in RESP patients specifically, PD-L1 expression levels are very high in M1-like and M2-like macrophages at the TSI followed by a rapid decrease in expression inside the TA, with even higher PD-L1 expression when close to Tcy. Others observed an enhanced frequency of IFNγ-producing T cells correlating with a numerical expansion and higher PD-L1 expression in the myeloid compartment only in responding patients using mass cytometry analysis of peripheral blood mononuclear cell (58). Although lacking spatial information, these data suggest that even in the periphery PD-L1 expression in myeloid-derived cells harbors predictive information, which is in line with the data we present. Recently, an in-depth spatial characterization of primary melanoma has been performed by others to investigate immune evasion mechanisms (59). Interestingly, the authors could observe a spatially restricted suppressive microenvironment along the tumor-stromal boundary, partially driven by PD-1/PD-L1–mediated cell contacts involving macrophages and T cells. This suggests the TSI-restricted TME involving close macrophage–Tcy interactions with a pivotal role for the PD-L1 axis described in our work, is already present at early stages of the disease. When investigating the level of Tcy activation, the opposite trend is evident. The closer Tcy are located to M1-like and M2-like macrophages at the TSI, the larger the decrease in activation markers, hereby suggesting that in RESP patients, PD-L1 expression and Tcy activation follow a more orderly spatial regulation while in NRESP patients these are more arbitrary.
Our study has several limitations. First of all, the number of patients included in the discovery cohort is rather limited, which could hamper the broad applicability of our findings to the general melanoma population. As a direct result, some generally accepted but less specific predictive biomarkers, such as TIL infiltration, cannot be reproduced within this study. Nonetheless, the integration of spatial information on top of high-dimensional proteomic data of >1.4 million single cells allows the discovery of novel biological insights with clinical potential, when analyzing cell–cell interactions combined with marker expression. The validation of our spatially resolved predictive biomarker in an independent patient cohort confirms the latter. Next, the mIHC method itself harbors the risk for selection bias with the a priori selection of antibody markers to include in the experiment. For example, confirming the importance of macrophage–Tcy interaction, our current panel does not allow a higher level of macrophage subtyping. The stratification of macrophages into M1-like and M2-like macrophages is an oversimplification of macrophage biology by excluding all intermediate states. To investigate this interaction even more in depth, an expanded panel of macrophage markers would need to be included. Finally, mIHC is not a dynamic functional assay and all Tcy activity (e.g., Tcy activation status) is inferred from the static quantitative and qualitative immunofluorescence staining of the included markers. Therefore, it is not clear how the spatial dynamics of our Tcy activation model would translate into in vivo functional Tcy activity.
In summary, our data show the feasibility of high-dimensional mIHC as a valuable technique in the armamentarium of single-cell analysis in the quest for biomarkers associated with response to anti-PD-1 therapy in melanoma. We demonstrate that marker expression on cellular components of the TME is a very dynamic process, and that spatial information is crucial in identifying relevant cell–cell interactions and dynamics of marker expression. Given the complexity of the mechanisms of an immune response we believe that integrating multiple techniques will be necessary to gain a deeper understanding of what makes immunotherapy successful.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
A. Antoranz was partially funded by the KU Leuven funding (C3/19/053), Opening the Future Campaign of the KU Leuven University Fund, and by Kom op tegen Kanker (Stand Up To Cancer), the Flemish cancer society. F.M. Bosisio was funded by KUL INTERNE FONDSEN MIDDEL-Zware infrastructuren EMH-D8191-AKUL/19/30 I005920N, FWO Fundamenteel Klinisch Mandaat EMH-D8972-FKM/20, Impulsbudget UZ Leuven 2018. W.M. Gallagher, S.M. Lynch, and A. Rahman were funded by the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement #642295 (MEL-PLEX), by the European Union's FP7 Marie Skłodowska-Curie Industry-Academia Partnership and Pathways research programme under the grant agreement #611107 (SYS-MEL), and by the Science Foundation Ireland Investigator Programme (OPTi-PREDICT; 15/IA/3104) and the Science Foundation Ireland Strategic Partnership Programme (Precision Oncology Ireland; 18/SPP/3522). M.M. Bolognesi and G. Cattoretti were funded by the Regione Lombardia POR FESR 2014–2020, Call HUB Ricerca ed Innovazione: ImmunHUB.
The authors would like to acknowledge the technical support of Dr. John A. Browne, UCD School of Agriculture and Food Science.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Authors' Disclosures
A. Antoranz reports grants from Kom op teger kanker during the conduct of the study; in addition, A. Antoranz has a patent for European patent application EP22182632.4 filed on July 1, 2022 pending. W.M. Gallagher reports grants from European Union, European Union, Science Foundation Ireland, and Science Foundation Ireland during the conduct of the study and other support from OncoMark and OncoAssure outside the submitted work. F. De Smet reports grants from Kom op tegen kanker, Flanders Research Foundation (FWO), KU Leuven, and Leuven Cancer Institute during the conduct of the study. F.M. Bosisio reports grants from Kom op tegen kanger, FWO, and UZ Leuven during the conduct of the study; in addition, F.M. Bosisio has a patent for European patent application EP22182632.4 filed on July 1, 2022 pending. No disclosures were reported by the other authors.
Authors' Contributions
A. Antoranz: Conceptualization, data curation, formal analysis, investigation, visualization, methodology, writing–original draft, project administration. Y. Van Herck: Conceptualization, formal analysis, visualization, writing–original draft. M.M. Bolognesi: Resources, data curation, formal analysis, writing–review and editing. S.M. Lynch: Resources, writing–review and editing. A. Rahman: Resources, writing–review and editing. W.M. Gallagher: Resources, writing–review and editing. V. Boecxstaens: Resources, data curation, writing–review and editing. J.-C. Marine: Supervision, writing–review and editing. G. Cattoretti: Resources, data curation, writing–review and editing. J.J. van den Oord: Funding acquisition, writing–review and editing. F. De Smet: Supervision, writing–original draft, writing–review and editing. O. Bechter: Conceptualization, writing–original draft. F.M. Bosisio: Conceptualization, resources, data curation, formal analysis, supervision, funding acquisition, writing–original draft, writing–review and editing.
References
- 1. Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015;372:320–30. [DOI] [PubMed] [Google Scholar]
- 2. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015;373:1270–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Forschner A, Battke F, Hadaschik D, Schulze M, Weißgraeber S, Han CT, et al. Tumor mutation burden and circulating tumor DNA in combined CTLA-4 and PD-1 antibody therapy in metastatic melanoma - results of a prospective biomarker study. J Immunother Cancer 2019;7:180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 2015;372:2509–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 Blockade in melanoma. N Engl J Med 2016;375:819–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sucker A, Zhao F, Real B, Heeke C, Bielefeld N, Maßen S, et al. Genetic evolution of T-cell resistance in the course of melanoma progression. Clin Cancer Res 2014;20:6593–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell 2016;167:397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Turiello R, Capone M, Morretta E, Monti MC, Madonna G, Azzaro R, et al. Exosomal CD73 from serum of patients with melanoma suppresses lymphocyte functions and is associated with therapy resistance to anti-PD-1 agents. J Immunother Cancer 2022;10:e004043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Johnson DB, Estrada MV, Salgado R, Sanchez V, Doxie DB, Opalenik SR, et al. Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat Commun 2016;7:10582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515:568–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen PL, Roh W, Reuben A, Cooper ZA, Spencer CN, Prieto PA, et al. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov 2016;6:827–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Siddiqui I, Schaeuble K, Chennupati V, Marraco SAF, Calderon-Copete S, Ferreira DP, et al. Intratumoral Tcf1 + PD-1 + CD8 + T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity 2019;50;195–211. [DOI] [PubMed] [Google Scholar]
- 13. Cabrita R, Lauss M, Sanna A, Donia M, Larsen MS, Mitra S, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020;577:561–5. [DOI] [PubMed] [Google Scholar]
- 14. Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020;577:549–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nejman D, Livyatan I, Fuks G, Gavert N, Zwang Y, Geller LT, et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 2020;368:973–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018;359:97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Holtzhausen A, Harris W, Ubil E, Hunter DM, Zhao J, Zhang Y, et al. TAM family receptor kinase inhibition reverses MDSC-mediated suppression and augments anti–PD-1 therapy in melanoma. Cancer Immunol Res 2019;7:1672–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wong PF, Wei W, Gupta S, Smithy JW, Zelterman D, Kluger HM, et al. Multiplex quantitative analysis of cancer-associated fibroblasts and immunotherapy outcome in metastatic melanoma. J Immunother Cancer 2019;7:194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Asrir A, Tardiveau C, Coudert J, Laffont R, Blanchard L, Bellard E, et al. Tumor-associated high endothelial venules mediate lymphocyte entry into tumors and predict response to PD-1 plus CTLA-4 combination immunotherapy. Cancer Cell 2022;40:318–34. [DOI] [PubMed] [Google Scholar]
- 20. Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 2014;371:1877–88. [DOI] [PubMed] [Google Scholar]
- 21. Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell 2018;175:998–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kallies A, Zehn D, Utzschneider DT. Precursor exhausted T cells: key to successful immunotherapy? Nat Rev Immunol 2020;20:128–36. [DOI] [PubMed] [Google Scholar]
- 23. Kurtulus S, Madi A, Escobar G, Klapholz M, Nyman J, Christian E, et al. Checkpoint blockade immunotherapy induces dynamic changes in PD-1-CD8 + tumor-infiltrating T cells. Immunity 2019;50:181–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Efremova M, Vento-Tormo M, Teichmann SA, Vento-Tormo R. CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nat Protoc 2020;15:1484–506. [DOI] [PubMed] [Google Scholar]
- 25. Browaeys R, Saelens W, Saeys Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat Methods 2020;17:159–62. [DOI] [PubMed] [Google Scholar]
- 26. Bolognesi MM, Manzoni M, Scalia CR, Zannella S, Bosisio FM, Faretta M, et al. Multiplex staining by sequential immunostaining and antibody removal on routine tissue sections. J Histochem Cytochem 2017;65:431–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228–47. [DOI] [PubMed] [Google Scholar]
- 28. Smyth GK. limma: Linear models for microarray data. Bioinformatics and Computational Biology Solutions Using R Bioconductor. New York, NY: Springer; 2005. [Google Scholar]
- 29. Väremo L, Nielsen J, Nookaew I. Enriching the gene set analysis of genome-wide data by incorporating directionality of gene expression and combining statistical hypotheses and methods. Nucleic Acids Res 2013;41:4378–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 2019;47:D607–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The molecular signatures database hallmark gene set collection. Cell Syst 2015;1:417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Korotkevich G, Sukhov V, Budin N, Shpak B, Artyomov MN, Sergushichev A. Fast gene set enrichment analysis. bioRxiv; 2021. Available from:http://biorxiv.org/content/early/2021/02/01/060012.abstract.
- 33. Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol 2019;37:773–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bosisio FM, Antoranz A, Van Herck Y, Bolognesi MM, Marcelis L, Chinello C, et al. Functional heterogeneity of lymphocytic patterns in primary melanoma dissected through single-cell multiplexing. Elife 2020;9:e53008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pau G, Fuchs F, Sklyar O, Boutros M, Huber W. EBImage-an R package for image processing with applications to cellular phenotypes. Bioinformatics 2010;26:979–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Caicedo JC, Cooper S, Heigwer F, Warchal S, Qiu P, Molnar C, et al. Data-analysis strategies for image-based cell profiling. Nat Methods 2017;14:849–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Levine JH, Simonds EF, Bendall SC, Davis KL, Amir EAD, Tadmor MD, et al. Data-driven phenotypic dissection of AML reveals Progenitor-like cells that correlate with prognosis. Cell 2015;162:184–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Van Gassen S, Callebaut B, Van Helden MJ, Lambrecht BN, Demeester P, Dhaene T, et al. FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytometry A 2015;87:636–45. [DOI] [PubMed] [Google Scholar]
- 39. Parra ER. Methods to determine and analyze the cellular spatial distribution extracted from multiplex immunofluorescence data to understand the tumor microenvironment. Front Mol Biosci 2021;8:668340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Street K, Risso D, Fletcher RB, Das D, Ngai J, Yosef N, et al. Slingshot: Cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics 2018;19:477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Haghverdi L, Buettner F, Theis FJ. Diffusion maps for high-dimensional single-cell analysis of differentiation data. Bioinformatics 2015;31:2989–98. [DOI] [PubMed] [Google Scholar]
- 42. Such L, Zhao F, Liu D, Thier B, Le-Trilling VTK, Sucker A, et al. Targeting the innate immunoreceptor RIG-I overcomes melanoma-intrinsic resistance to T cell immunotherapy. J Clin Invest 2020;140:4266–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xue Z, Vis DJ, Bruna A, Sustic T, Van Wageningen S, Batra AS, et al. MAP3K1 and MAP2K4 mutations are associated with sensitivity to MEK inhibitors in multiple cancer models. Cell Res 2018;28:719–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Truong KL, Schlickeiser S, Vogt K, Boës D, Stanko K, Appelt C, et al. Killer-like receptors and GPR56 progressive expression defines cytokine production of human CD4+ memory T cells. Nat Commun 2019;10:2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Oliver JL, Alexander MP, Norrod AG, Mullins IM, Mullins DW. Differential expression and tumor necrosis factor-mediated regulation of TNFRSF11b/osteoprotegerin production by human melanomas. Pigment Cell Melanoma Res 2013;26:571–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Simpson A, Petnga W, Macaulay VM, Weyer-Czernilofsky U, Bogenrieder T. Insulin-like growth factor (IGF) pathway targeting in cancer: role of the IGF axis and opportunities for future combination studies. Target Oncol 2017;12:571–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yamada K, Uchiyama A, Uehara A, Perera B, Ogino S, Yokoyama Y, et al. MFG-E8 drives melanoma growth by stimulating mesenchymal stromal cell-induced angiogenesis and M2 polarization of tumor-associated macrophages. Cancer Res 2016;76:4283–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cirillo N, Morgan DJ, Pedicillo MC, Celentano A, Muzio LL, McCullough MJ, et al. Characterisation of the cancer-associated glucocorticoid system: Key role of 11β-hydroxysteroid dehydrogenase type 2. Br J Cancer 2017;117:984–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Awad RM, De Vlaeminck Y, Maebe J, Goyvaerts C, Breckpot K. Turn back the TIMe: Targeting tumor infiltrating myeloid cells to revert cancer progression. Front Immunol 2018;31:1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhang W, Zhang C, Li W, Deng J, Herrmann A, Priceman SJ, et al. CD8+ T-cell immunosurveillance constrains lymphoid premetastatic myeloid cell accumulation. Eur J Immunol 2015;45:71–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Peranzoni E, Lemoine J, Vimeux L, Feuillet V, Barrin S, Kantari-Mimoun C, et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti–PD-1 treatment. Proc Natl Acad Sci U S A 2018;115:E4041–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lin H, Wei S, Hurt EM, Green MD, Zhao L, Vatan L, et al. Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade–mediated tumor regression. J Clin Invest 2018;128:805–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Liu Y, Zugazagoitia J, Ahmed FS, Henick BS, Gettinger SN, Herbst RS, et al. Immune cell PD-L1 colocalizes with macrophages and is associated with outcome in PD-1 pathway blockade therapy. Clin Cancer Res 2020;26:970–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Qian J, Wang C, Wang B, Yang J, Wang Y, Luo F, et al. The IFN-γ/PD-L1 axis between T cells and tumor microenvironment: hints for glioma anti-PD-1/PD-L1 therapy. J Neuroinflammation 2018;15:290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Egelston CA, Avalos C, Tu TY, Rosario A, Wang R, Solomon S, et al. Resident memory CD8+ T cells within cancer islands mediate survival in breast cancer patients. JCI Insight 2019;4:e130000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rasmusson A, Zilenaite D, Nestarenkaite A, Augulis R, Laurinaviciene A, Ostapenko V, et al. Immunogradient indicators for antitumor response assessment by automated tumor-stroma interface zone detection. Am J Pathol 2020;190:1309–22. [DOI] [PubMed] [Google Scholar]
- 57. Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 2013;39:782–95. [DOI] [PubMed] [Google Scholar]
- 58. Krieg C, Nowicka M, Guglietta S, Schindler S, Hartmann FJ, Weber LM, et al. High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat Med 2018;24:144–53. [DOI] [PubMed] [Google Scholar]
- 59. Nirmal AJ, Maliga Z, Vallius T, Quattrochi B, Chen AA, Jacobson CA, et al. The spatial landscape of progression and immunoediting in primary melanoma at single cell resolution. Cancer Discov 2022;12:1518–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors upon reasonable request.