

Abstract
Chimeric antigen receptor (CAR)-T cells have emerged as a promising treatment modality for various hematologic and solid malignancies over the past decade. Animal models remain the cornerstone of pre-clinical evaluation of human CAR-T cell products and are generally required by regulatory agencies prior to clinical translation. However, pharmacokinetics and pharmacodynamics of adoptively transferred T cells are dependent on various recipient factors, posing challenges for accurately predicting human engineered T cell behavior in non-human animal models. For example, murine xenograft models did not forecast now well-established cytokine-driven systemic toxicities of CAR-T cells seen in humans, highlighting the limitations of animal models that do not perfectly recapitulate complex human immune systems. Understanding the concordance as well as discrepancies between existing pre-clinical animal data and human clinical experiences, along with established advantages and limitations of each model, will facilitate investigators’ ability to appropriately select and design animal models for optimal evaluation of future CAR-T cell products. We summarize the current state of animal models in this field, and the advantages and disadvantages of each approach depending on the pre-clinical questions being asked.
Keywords: animal models, pre-clinical development, CAR-T cells, efficacy, toxicity, trafficking
Graphical abstract
Animal models are crucial for the development and improvement of adoptive cellular therapies, such as CAR-T cells. Murine xenografts, transgenic mouse models, and non-human primate models have proven essential. Every model system has inherent advantages and disadvantages that must be weighed when considering a given question.
Introduction
The field of chimeric antigen receptor (CAR)-T cell therapy has grown exponentially since the first CAR product, tisagenlecleucel (tisa-cel) was approved by the FDA in 2017,1,2 paving the way for the approval of five additional CAR-T cell products for lymphoid malignancies as of June 2022, targeting CD19 for lymphomas or acute lymphoblastic leukemia or B cell maturation antigen (BCMA) for multiple myeloma. Clinical testing has begun for multiple new CAR-T cell approaches targeting additional antigens, including CD22 for lymphoid malignancies, CD33 and CD123 for myeloid malignancies, and GD2 and mesothelin for solid tumors, giving hope that CAR-T cell therapies can be extended to a broader range of diseases.
As with any developmental therapy, in vivo animal studies establishing biological plausibility and pre-clinical safety are generally required before moving CAR-T cell therapies to the clinic.3,4 Very few products identified as promising in the laboratory ultimately make it to clinical trials, let alone gain FDA approval. Successful clinical translation of new cell therapy products is even more challenging than for new drug therapies, partly due to difficulty generating clinically relevant pharmacology, toxicology, and safety data from non-human animal models.5 In contrast to regular pharmacological agents, CAR-T cells are “living drugs” that proliferate, migrate, and persist in patients to varying degrees depending on in vivo environmental signals that they receive, making design of animal models particularly challenging. As clinical data accumulates for pioneering CAR-T cell therapies, it has become increasingly apparent that individual pre-clinical animal models do not fully predict clinical behaviors of CAR-T cells. Indeed, a comparative study that assessed the divergence of marketing authorization applications from the expected regulatory data requirements revealed that the applications for cell therapies reflected difficulties of using animal models to address toxicology and mechanisms of action more so than those for other biological agents.6 In this review, we summarize available information generated in pre-clinical CAR-T cell animal models and assess their utility and limitations, in light of results in humans. We then discuss animal studies on select timely topics, namely CAR-T cell trafficking and CAR-T cell differentiation, and we assess their implications for future engineered immune cell therapies.
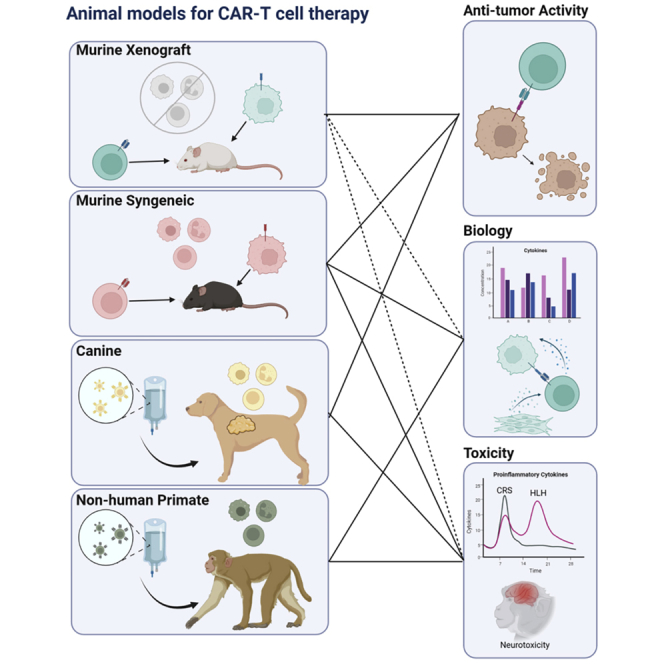
General classifications of animal models used in CAR-T cell research
Xenograft mouse models
Human tumor cell lines and some types of primary human cells can be transplanted as xenografts into immunodeficient mice that lack fully functional immune systems, permitting sustained engraftment of human cells. Less immunodeficient strains, such as athymic nude mice that lack T cells and CB17-scid mice that lack both T and B cells, continue to produce natural killer (NK) cells, which can potentially mediate anti-tumor activity and confound data. Mouse strains created on the non-obese diabetic (NOD)-severe combined immunodeficiency (SCID) background are severely immunodeficient, with complete absence of an adaptive immune system and varying degrees of impairments in innate immunity. NOD/SCID/IL2Rγc-KO (NSG) mice lack B, T, and NK cells and have become one of the most commonly utilized mouse strains for studying human CAR-T cell therapies. A broad range of human tumor cells robustly engraft in these mice, providing a model for development of adoptive immune cell therapies directed at a wide variety of human cancers.7
Patient-derived xenograft (PDX) models, in which primary patient-derived tumors are implanted into immunodeficient mice, enable evaluation of CAR-T cell activity against cells with more physiologic characteristics, including unique clonal dynamics that arise from patient-inherent tumor heterogeneity and thus variable susceptibility to CAR-T cells.8 In general, primary tumors able to engraft in PDX animals have a tendency to closely mimic characteristics of metastatic rather than primary disease.9,10
Human xenografts can also be established in mice that are reconstituted with a human immune and hematopoietic system. These “humanized” models may better capture interactions of adoptively transferred CAR-T cells in the context of tissue microenvironments and other human immune cells, potentially recapitulating human physiology more closely compared with standard xenografted mice lacking these components of the human immune system. Additional genetic modifications to improve the engraftment and support of human cells include strains such as the NSG-SGM3 mouse, engineered to express the human cytokines stem cell factor, GM-CSF, and IL3 that facilitate engraftment of human hematopoietic cells.11 A comparison of commonly utilized mouse models is depicted in Figure 1.
Figure 1.

Murine models for CAR-T therapies
Human cells are depicted in aqua and mouse cells in pink throughout the figure.
While xenograft mouse models have many advantages for studying human T cell therapies, notable limitations include xenogeneic graft-versus host disease (GVHD) and still imperfect human immunity due to incomplete cross-species cellular and soluble factor interactions. Lack of human stromal cells and other supportive elements may also interfere with maintenance and trafficking of human CAR-T as well as preclude the study of therapies specifically targeting the stromal elements of solid tumors since tumor-associated stroma supporting injected human tumor cells develop from murine fibroblasts.
Syngeneic mouse models
Syngeneic murine models allow analyses of tumor and CAR-T cell interactions in the context of an intact host immune system, including species-specific cytokines, stromal and other microenvironmental elements, inhibitory cells, and other potentially influential factors. These models are particularly beneficial for testing hypotheses based on complex interactions between adoptively transferred T cells and host elements. Pitfalls include the rapid growth of tumor cell lines in these models, rapid enough to preclude generation of an immunosuppressive tumor microenvironment playing a major role in tampering down immune responses to human solid tumors. Alternatively, mouse models genetically engineered to develop spontaneous tumors may permit more physiologic tumor stromal development and better model characteristics of primary tumor cells, as opposed to immortalized tumor cell lines.12,13 Historically, transgenic mouse strain generation is expensive and time consuming, with transgene integration variability sometimes creating undesirable phenotypic differences. In recent years, CRISPR-Cas9 targeted genome editing has greatly reduced the time and cost required to create these models. Knocking in human target antigens into murine tissues also allows evaluation of human antigen-directed T cells in otherwise syngeneic systems.14 Foreign proteins encountered in tumor cell lines, such as luciferase and GFP, are known to be highly immunogenic when given to immunocompetent mice. Transgenic mouse models have been developed allowing tolerance to these immunogenic reporter molecules, enabling the use of imaging to track tumor burden in syngeneic mouse models over time.15, 16, 17
Non-murine species
Though mice are the most frequently used model species for adoptive cell therapies, based on availability, cost, and familiarity, non-murine species have also been used for in vivo studies of CAR-T cells. Approximately four million dogs in the United States per year spontaneously develop cancer and therefore may serve as natural models for the spontaneous process of oncogenesis. This stands in contrast with the artificial introduction of cancer cell lines or germline genetic manipulation of mice to induce “spontaneous” tumor development. Across breeds, dogs harbor a much greater degree of genetic diversity than mice, which are inbred for genetic homogeneity. Breed predilections for certain tumor types exist, with large-breed dogs at greater risk for osteosarcoma, golden retrievers at risk for mast cell tumors, and smaller terrier breeds at greater risk for transitional cell carcinomas, for example.18 Humans also have diverse genetic backgrounds, making canine heterogeneity an advantage of this model system. Environmental factors leading to cancer in humans often similarly impact their canine companions, and these vary widely across different parts of the world.19 Due to dogs’ domestic evolution alongside humans, they have been exposed to many of the same pathogens and have developed immune systems that function similarly to those of their human counterparts. Dogs are immunologically competent at birth, but like humans, postnatal maturation of the immune system develops with pathogenic exposures and antigen priming.20 Pet owners and veterinarians can be highly motivated to contribute to the clinical development of new therapies, both to extend the life and health of their companions, as well as contribute to medical advances relevant to humans.
Non-human primates (NHPs) most closely resemble humans, regarding lifespan, size, telomere length, immune system characteristics, and genetic heterogeneity, conferring high predictive value for the potency and safety of cell therapies. Taraseviciute et al. established an NHP model of anti-CD20 CAR-T cell therapy, recapitulating human CAR-T cell phenotype, expansion, persistence, and toxicities.21 This achievement underscores the importance and unique strengths of NHP models. Drawbacks include limited animal availability and high costs of both animal purchase and husbandry, limiting experiments to small cohorts and thus limited statistical power, as well as lack of sufficient animals with spontaneous tumors. Induction of tumors is not feasible to date in NHP models. For these reasons, NHPs have limited utility in anti-tumor efficacy studies, but they remain the gold standard for evaluating toxicity. Each model has distinct advantages and disadvantages, which have been outlined in Table 1.
Table 1.
A comparison of animal model systems for pre-clinical CAR-T cell testing
| Species | Advantages | Disadvantages | Efficacy | Toxicity | |
|---|---|---|---|---|---|
| Zebrafish |
|
|
In vivo efficacy reflects similar results as in vitro cytotoxicity assays | Unclear utility for toxicity modeling | |
| Mouse | Xenograft |
|
|
Most CAR-T undergo proof-of-concept efficacy studies in xenografts.
|
Limited reports of toxicity, but some exist |
| Patient-derived xenograft (PDX) |
|
|
Advantageous for testing efficacy of dual-target and gated CAR-T cells | Limited toxicity data | |
| Genetically engineered |
|
|
No CAR-T studies published to date | ||
| Humanized |
|
|
Important for modeling efficacy in the face of other human immune cells and tissues Improved B and T cell maturation in NSG-SGM3 BLT mice |
Allows study of the role other cell types play in toxicity | |
| Syngeneic |
|
|
Critical for understanding role of lymphodepletion on CAR-T efficacy | On-target off-tumor toxicity may not directly apply to humans HLH, MAS modeled in perforin-deficient CD19 CAR-T cells |
|
| Transgenic |
|
|
Evaluation of human CAR construct in otherwise all murine system | Ideal for modeling on-target off-tumor toxicity Evaluation of host immune system involvement |
|
| Dog |
|
|
Preliminary efficacy with similar escape mechanisms in humans | No published CAR-T studies of toxicity | |
| Non-human primate |
|
|
Efficacy in infectious disease models and anti-B cell antigen CAR-T (B cell aplasia) Limited utility in anti-tumor efficacy |
Ideal for modeling cytokine-mediated cytotoxicity (CRS, ICANS) Useful for on-target off-tumor toxicity modeling in antigens with cross-species reactivity |
|
Studying efficacy of CAR-T cells in animal models
Murine models
Estimation of a cell product’s ability to induce complete remission
Xenograft murine models are often the most accessible and commonly utilized to test proof-of-concept activity of a new CAR-T cell product. Immunodeficient mice are first engrafted with human cancer cell lines or PDX tumors, followed by treatment with human T cells transduced with CARs, which allows direct observation of the effects of the therapeutic human T cell product in vivo. All CAR-T cell products currently in clinic were tested in xenograft models and most have been described in publications reporting in vivo anti-tumor activity, including CARs targeting hematologic tumor antigens,22,23 dual antigen targeting (e.g., CD20 and CD19,24 CD19 and CD2225,26) and solid tumor antigens (e.g., EGFRvIII,27 mesothelin28).
Estimation of clinically effective and safe cell dose
Predicting biologically active dose levels and identifying a safe starting dose are considered central objectives of pre-clinical studies by regulatory bodies such as the FDA.3 Unfortunately, xenograft mouse models have been of limited utility in defining appropriate doses of CAR-T cells for first-in-human trials. Cell doses in xenograft studies are often chosen arbitrarily or start very high in order to quickly assess an anti-tumor response. For example, anti-CD19 and CD22 human CAR-T cell doses employed in many early pre-clinical studies were often as high as 1 × 106 to 1 × 107 cells per mouse,29, 30, 31 which extrapolates to 5 × 107 to 5 × 108 cells per kilogram, based on an average mouse weighing 20 grams. In contrast, clinically safe and effective cell doses of anti-CD19, CD22, and BCMA CAR-T cells in humans are far lower, only approximately 2 × 105 to 5 × 106 cells/kg.32, 33, 34, 35, 36, 37, 38, 39, 40 These stark differences in cell doses reflect the fact that mice tend to be much less sensitive to CAR-T cell-mediated toxicities than humans (discussed further later in the context of toxicity models). Lower CD19 CAR-T cell doses in NSG xenograft models (5 × 104 to 2 × 105 cells/mouse) do have some in vivo anti-leukemia activity,41 more in alignment with clinically feasible doses. In humans, duration of clinical response and long-term survival are the critical relevant outcomes. However, the lifespan of xenografted mice is limited by both rapid progression of engrafted tumor cells and xeno-GVHD-related morbidity and mortality. Thus, dose-finding in these models is not feasible, and investigators are more often forced to rely on dosing data from previous human clinical trials to set starting parameters for new therapies, sometimes with unanticipated intolerable toxicities at doses required for efficacy.
Evaluation of lymphodepletion and long-term therapeutic effects
The syngeneic murine system models physiologic species-specific cytokine and immune cell interactions without the complication of xenogeneic GVHD, overcoming some of the major limitations of xenograft models. While direct evaluation of actual human CAR-T cells is not possible in a syngeneic murine system, studies of equivalent murine CAR-T cells can provide translationally valuable information. One of the earliest syngeneic mouse CAR-T cell studies was conducted by Cheadle et al. using a retroviral vector containing a first-generation CAR-T cell targeting murine CD19. This work helped establish that efficacy of CAR-T cells is enhanced by lymphodepletion with irradiation or cyclophosphamide prior to CAR-T infusion and also uncovered the limited persistence of first-generation CAR-T cells.42,43 Pre-clinical syngeneic studies of a second-generation CAR-T construct targeting CD19 and containing co-stimulatory domains demonstrated the ability of these CAR-T cells to persist long term and induce B cell aplasia for up to 3–12 months following treatment, enabling the evaluation of long-term therapeutic outcomes.43, 44, 45, 46, 47, 48 Work by Davila et al. used syngeneic tumor-prone Eμ-myc C57BL/6 transgenic mice to demonstrate anti-leukemia CD19 second-generation CAR-T cell efficacy, as well as showing that intensive preconditioning with cyclophosphamide was more central to efficacy than CAR-T cell dose escalation.49,50 Clinical studies have confirmed the importance of achieving the appropriate degree of lymphodepletion, with higher overall response rates and progression-free survival in patients receiving higher doses of lymphodepletion treatment prior to CAR-T cell infusion.51 The similarity of the results from syngeneic mouse models and human trials highlights that the presence of an intact immune system and ability to follow animals long term are valuable for pre-clinical optimization of cell therapies.
One of the most important outcomes that can be modeled most easily in a syngeneic model is post-CAR-T cell relapse. Despite the remarkable ability of CAR-T cells to induce complete remissions, sustained disease control is achieved in no more than half of patients.52,53 Disease relapse occurs in part due to the emergence of cell-surface antigen-low or negative tumor subclones that evade CAR-T cell targeting. Interestingly, lineage switch of B cell malignancies to non-B cell lineages under CAR-T cell pressure has been shown to contribute to post CAR-T cell therapy relapses as well.44,54,55 Jacoby et al. demonstrated leukemia lineage reprogramming in a syngeneic model as an escape mechanism from CD19 CAR-T cell pressure, characterized by epigenetic suppression of B cell lineage transcription factors such as Pax5 and Ebf1, with concurrent upregulation of myeloid-lineage transcription factors such as Cebpa.44 In clinic, KMT2A-rearranged leukemias, most often seen in infants, are particularly vulnerable to CD19 negative relapses after CD19-directed therapies.54,56 Extensive efforts are underway to model KMT2A-rearranged leukemia (as reviewed in Liao et al.57), which will further elucidate the mechanisms of leukemia lineage switch provoked by CAR-T cell pressure.
Comparing the potency of CARs with different structural designs
There are substantial ongoing efforts to improve CAR-T cell efficacy by fine-tuning CAR designs, including co-stimulatory domains, linker lengths, and signaling strength. Animal models, especially xenograft mouse models, have been essential to evaluate the function of various parts of human antigen-directed CARs. As summarized by Cappell and Kochenderfer in a recent review,58 various murine studies demonstrated heterogeneous results regarding the efficacy between varying co-stimulatory domains. Mouse xenograft models and immunocompetent syngeneic models have been used to evaluate the effects of modulation of CAR signaling by partially inactivating CD3z immunoreceptor tyrosine-based activation motifs (ITAMs)59 or by incorporating mutations in subdomains of a co-stimulatory molecule.60 In addition to different combinations of hinge and transmembrane domains,61,62 the length of linker connecting the variable heavy (VH) and light chain (VL) of the scFv were also found to influence anti-tumor efficacy of CAR-T cells in murine xenografts,63 informing the design of subsequent clinical trials (NCT02650414 and NCT03620058).
Evaluating CAR-T cell efficacy in non-murine species
Zebrafish
One group has created a xenografted CAR-T cell model in embryonic zebrafish, arguing that this system is fast, inexpensive, and offers the ability to perform live imaging for the study of CAR-T cell migration and killing.64 They used human anti-CD19 CAR-T cells labeled with the DiI membrane dye in fish engrafted with human GFP-expressing human leukemia cells, and they measured in vivo cytotoxicity over 24 hours via luminescence imaging. Mouse studies, on the other hand, typically require 2–6 weeks at a minimum to evaluate in vivo anti-tumor activity. The related downside of this short-term zebrafish model is that it does not allow analysis of CAR-T cell persistence, exhaustion, or tumor escape, all requiring much longer periods of in vivo observation. Furthermore, application to solid tumor models, in which orthotopic tumor inoculation is desired, may be difficult due to the size of zebrafish embryos. Lastly, the zebrafish embryonic immune system varies significantly from humans in that it consists of only innate immune cells with unclear cross-species cytokine reactivity. The lack of adaptive endogenous immunity prevents immediate rejection of human CAR-T cells, but it does not accurately recapitulate the immune milieu of humans.
Canine models
Dogs with spontaneous hematologic malignancies have been treated with canine CAR-T cells targeting CD20.65, 66, 67 Dogs bearing sarcomas have also been treated with B7-H3 CAR-T cells, which are also in human clinical trials (NCT046670068), demonstrating feasibility and safety.68 Many of the observations made in human CAR-T cells trials have been recapitulated in dogs, including the development of cytokine release syndrome, the emergence of antigen-negative tumor relapse, and the development of anti-mouse antibodies generated against the murine portion of the scFv used in many human and dog CAR constructs (Figure 2).69 While thus far use of the canine CAR-T cell models and clinical utilization by veterinarians is lagging behind, canine models can serve as a promising translational bridge, and activity in this area is increasing.
Figure 2.

Non-murine models for CAR-T cell therapies
Non-human primate models
The primary limitation for efficacy testing in NHP models is the lack of high incidence spontaneous malignancies in these animals, in part because cancer is often a disease of aging, and housing enormous (and thus expensive) colonies of aging NHPs for many decades would be required to obtain any animals with relevant primary tumors. Use of tumor cell lines is not feasible in non-inbred and immunocompetent NHPs, and any tumor cell artificially introduced from another animal would be immediately rejected. However, B cell aplasia can be used as a surrogate marker for modeling persistence of CAR-T cell therapies directed at B lymphoid target antigens.21
NHPs, specifically various old world macaques, have been central platforms for the pre-clinical development of immune therapies, including vaccines, cell therapies, and monoclonal antibodies against infectious pathogens, given the many macaque viruses that are highly homologous to human pathogens such as HIV, CMV, EBV, Ebola, Zika, and SARS-CoV-2.70, 71, 72, 73 The first CAR-T cells given to humans were actually targeting HIV-infected cells via CARs with a CD4/CD3zeta first-generation chimeric receptor binding HIV envelope expressed on the surface of HIV-infected T cells. Pre-clinical testing of CAR-transduced human hematopoietic stem and progenitor cells (HSPCs) resulted in efficacy against an HIV-infected leukemia cell line in a xenograft model.74 However, pioneering clinical trials using first-generation CAR-T cells in HIV-infected patients were ineffective, with limited in vivo expansion in the absence of lymphodepletion.75,76 More recently, Zhen et al. developed a lentiviral vector expressing a CD4/CD3z CAR along with a C46 fusion inhibitor to prevent viral infection of CAR-expressing T cells.77 These investigators also engineered HSPCs rather than peripheral T cells to express CARs, with successful differentiation into CAR-expressing T cells in vivo following autologous transplantation. CAR-T cells then persisted for over 2 years and provided sustained anti-viral immunity. The NHP SIV model also enabled testing of the combination therapy of CD4 CAR-T cells plus immune checkpoint blockade or antigen boosting.78
Modeling CAR-T cell toxicities
Cytokine-driven toxicities
CAR-T cell therapies are associated with unique toxicities associated with systemic inflammation, linked to various cytokines secreted by both CAR-T cells and the recipients’ endogenous immune cells. The spectrum of toxic manifestations has been categorized into cytokine release syndrome (CRS), immune effector cell-associated neurotoxicity syndrome (ICANS), hemophagocytic lymphohistiocytosis (HLH), and macrophage activation syndrome (MAS).79,80 Prior to the advent and clinical translation of CAR-T cells, the potential for cytokine-driven toxicities resulting from adoptive cell transfer was not anticipated, in large part due to lack of similar events in standard murine models. The unexpected nature of the toxicities may be reflected in the study design of the first-in-human clinical trial of CD19 CAR-T cells administered together with high-dose IL-2.81 Exogenous IL-2 continues to be frequently co-administered with polyclonal or TCR-based T cell products, but it is not now administered with CAR-T cells, given the well-recognized risks of cytokine-related toxicities that occur even without exogenous cytokine administration. Over the past decade, significant progress has been made in establishing animal models for CAR-T-associated toxicities. This section will highlight representative animal models recapitulating the spectra of inflammatory toxicities seen in humans (Figure 3), with a particular focus on B cell antigen-directed CAR-T cells.
Figure 3.

Modeling cytokine-mediated CAR-T cell toxicities
The mechanism of CAR-T-mediated cytokine-related toxicities involves a surge of T cell activation, resulting in release of cytokines, which in turn results in myeloid cell release of other pro-inflammatory cytokines. CAR-T-mediated lysis and induction of tumor cell apoptosis further feeds the cycle of pro-inflammatory cytokine production. Limitations of the xenograft mice for modeling toxicity stem from lack of interspecies cytokine cross-reactivity and inadequate host immune functions, although CRS and neurotoxicity have been successfully modeled in humanized mice. Both CRS and HLH have been successfully modeled in syngeneic or transgenic immunocompetent mouse models. Similarly, neurotoxicity and CRS modeling in NHPs has mimicked human toxicities and provided valuable insight into toxicity mechanisms.
Difficulties predicting toxicities using animal models
Novel human CAR-T cell products intended for clinical translation are often tested in murine immunodeficient xenograft models to demonstrate pre-clinical anti-tumor activity, as discussed above. However, NSG mice receiving CAR-T cells targeting CD19, CD22, or other human B cell antigens generally do not manifest obvious CRS or other CAR-T toxicities, likely due to incomplete cross-species reactivity of cytokines, lack of human immune effector cells other than CAR-T cells, and lack of additional cell targets such as normal B cells. Xenogeneic GVHD can further complicate interpretation of possible CAR-T toxicities in immunodeficient mice. Syngeneic,46,82 transgenic,83 and humanized murine models84 along with non-human primate models21 have been more valuable tools for understanding the pathophysiology of CAR-T cell-mediated toxicities. Despite this, animal models are somewhat limited in pre-clinical ability to predict all possible untoward effects of a given human CAR-T cell product, since no model can fully represent the complex immune system and diversity of cell types and antigen expression within a patient with cancer. These models have been more robust in advancing our understanding of the pathophysiology of these side effects and initial exploration of the efficacy of potential interventions.
Xenograft models for studying pathophysiology of cytokine-driven toxicities
Multiple models suggest the important role that recipient-derived myeloid cells, such as monocytes and macrophages, play in cytokine-mediated toxicities. In a xenograft mouse model using SCID-beige mice, which lack B and T cells but have a relatively normal myeloid compartment, severe CRS developed in the context of high intraperitoneal disease burden in mice treated with human CD19-CAR-T cells.85 Taking advantage of the xenograft model, the cellular source of cytokines could be distinguished by specifically measuring levels of human versus murine cytokines generated from either adoptively transferred human CAR-T cells or murine immune cells, respectively. Mice with severe CRS had elevated mouse-derived IL-6, CCL2, and CXCL9, and murine myeloid cells with high levels of pro-inflammatory gene expression were recruited to the sites of tumor in the peritoneum. Notably, human CAR-T cells engineered to co-express mouse CD40L resulted in more severe CRS, indicating that physical CD40/CD40L-mediated interactions between CAR-T and recipients’ myeloid cells expressing mouse CD40 receptor contributed to toxicity. IL-6 and genes involved in IL-1 signaling were upregulated in recipient mice myeloid cells, consistent with observations in humans,32,86,87 and blockade of IL-6, IL-1, and inducible nitric oxide synthase improved CRS without negatively impacting anti-leukemia efficacy of CAR-T cells.
The critical roles played by recipients’ myeloid cells and IL-6/IL-1 cytokine axes were further emphasized in a humanized NSG-SGM3 murine model expressing human SCF, GM-CSF, and IL-3. Newborn NSG-SGM3 mice engrafted with human cord blood HSPCs develop human T cells xenotolerant to murine antigens (termed nHuSGM3 T cells). Use of nHuSGM3 T cells for CAR-T generation mitigated the confounding issue of xeno-GVHD.84 Compared with SGM3 recipient mice without human hematopoietic reconstitution, humanized SGM3 recipient mice with human myeloid cells experienced more severe and sometimes fatal CRS within a week of CAR-T cell infusion. Depletion of monocytes, determined to be the major source of IL-6 and IL-1, mitigated CRS and further underscored the central role of recipient myeloid cells. Neurotoxicity was also observed in humanized SGM3 recipients, characterized by multifocal meningeal thickening and human macrophage infiltration into the subarachnoid space. Importantly, blockade of IL-1 with anakinra effectively treated both CRS and neurotoxicity, but IL-6 blockade with tocilizumab only addressed CRS, mirroring the efficacies of each drug in human CRS versus ICANS.88,89
In addition to IL-6 and IL-1, other myeloid cell-derived cytokines such as GM-CSF have also been implicated in CRS and neurotoxicity in murine xenograft models.90 Mice that suffered neurotoxicity had CD11b+ myeloid cell infiltration into the brain accompanied by inflammation. Pharmacologic GM-CSF blockade or genetic deletion of GM-CSF from CAR-T cells improved toxicities while enhancing the anti-tumor activity of CAR-T cells. These data provided the basis for an ongoing clinical trial prospectively evaluating the use of lenzilumab, a human GM-CSF-neutralizing antibody, with axicabtagene ciloleucel (NCT04314843).
The role of catecholamines as an upstream myeloid factor influencing cytokine release has also been studied in the context of xenograft NSG-SGM3 models.91 The myeloid-specific deletion of tyrosine hydroxylase, which blocks conversion of tyrosine to L-DOPA, led to the reduction of catecholamine production following bacterial infection or other inflammatory stimuli. CD19 CAR-T cell-driven CRS in NSG-SGM3 mice was improved by pharmacologic blockade of tyrosine hydroxylase without impacting anti-leukemia efficacy, demonstrating an alternative avenue to address cytokine-mediated toxicities.
Syngeneic immunocompetent murine models for toxicities
Syngeneic and transgenic murine models allow for the study of CAR-T cell biology in the presence of a fully competent host immune system. It is important to note, however, that mice maintained in a standard animal facility have an antigen-inexperienced immune system due to facility cleanliness standards, not accurately resembling humans exposed to a multitude of exogenous antigens.92,93 This may partially explain why symptomatic CRS or neurotoxicity typically does not occur in standard syngeneic murine models with CD19 CAR-T cells on a C57BL/6 background.45,46 One of the most faithful recapitulations of clinical CRS and neurotoxicity was achieved in a transgenic mouse system in which mice with B cell-restricted human CD19 expression were engrafted with a mouse lymphoma cell line (TBL12) engineered to co-express human CD19 and given syngeneic murine T cells expressing a human CD19-directed CAR.83 CAR-T cell dose-dependent acute and lethal cytokine-mediated toxicities consistent with CRS and neurotoxicity occurred, accompanied by depletion of brain microglial cells. Blockade of IL-6 and IFNγ ameliorated toxicities. Another syngeneic murine model utilized perforin-deficient T cells to capture late-onset cytokine-mediated toxicities resembling the poorly understood clinical CAR-T toxicities of HLH and MAS.46 Perforin-deficient T cells expressing anti-CD19 CAR had initial in vivo expansion concurrent with leukemia clearance, and then re-expanded in the absence of detectable antigens several weeks after CAR-T cell infusion. This secondary expansion was accompanied by concomitant expansion and activation of recipient-derived lymphocytes and myeloid cells, splenomegaly, and pro-inflammatory cytokine upregulation, resembling HLH. The model mirrored clinical observations of patients treated with anti-CD22 CAR-T cells who experienced biphasic inflammation that was chronologically and phenotypically distinguishable, manifesting as CRS in the first phase and HLH/MAS in the second.32,94 IL-1 family cytokines were among many pro-inflammatory cytokines significantly elevated in the murine model as well as in patients who experienced HLH/MAS compared with patients with CRS alone. Anakinra, an IL-1 receptor antagonist antibody, is increasingly utilized in clinic as an adjunctive treatment to manage CAR-T-related toxicities.95 Murine models recapitulating different aspects of human clinical manifestations may aid in elucidating the pathophysiology of each toxicity, which may be distinct from one another.
NHP model of CRS and neurotoxicity
Lastly, CRS and neurotoxicity have been modeled in NHPs.21 Feasible study sizes for NHP studies are small but offer the unparalleled advantage of studying clinical toxicities in animals with immune systems closely related to humans and body size permitting longitudinal sampling of CSF and other relevant tissues. CD20-specific CAR-T cells were administered to four rhesus macaques preconditioned with cyclophosphamide.21 All experienced CRS followed by overt neurotoxicity. Various cytokines, including IL-6, GM-CSF, IL-1β, and MCP-1, were significantly elevated in CSF compared with concurrent blood samples from each animal. Scheduled autopsies at the onset of neurotoxicity, corresponding to maximal in vivo CAR-T expansion, demonstrated diffuse infiltration of brain parenchyma with CAR-positive T cells as well as CAR-negative T cells. These findings suggested that CAR-T-associated neurotoxicity is not target antigen dependent.
Antigen-specific toxicities
CAR-T cells also have the potential for inducing antigen-specific toxicity via on-target/off-tumor and off-target reactivities. In contrast to CRS, ICANS, and HLH/MAS, in which clinical manifestations are driven by systemic inflammatory responses, antigen-specific toxicity is a direct result of CAR-T cell-mediated damage to otherwise healthy tissues. Off-target toxicity occurs when the CAR cross-reacts with an unintended target expressed on healthy tissues. Off-target toxicities and clinical trial fatalities have occurred with TCR-engineered T cell infusions,96, 97, 98 but none have been reported with CAR-T cells to date. Off-target toxicities of human engineered T cells cannot be studied in animal models because they do not express the complete repertoire of human antigens required to study these side effects. In the absence of suitable animal models, off-target screening of CAR-T cells is generally achieved via in vitro evaluations using cell lines and tissue arrays, in addition to extrapolating experiences from antibody therapies, from which the antigen-recognition domains of CARs (i.e., scFv) are often derived.
On-target off-tumor toxicities occur when a known target antigen is shared by both tumor and normal tissues. B cell aplasia after B cell lineage antigen-directed CAR-T cell therapy is the classic example of on-target off-tumor toxicity. B cells are uniquely “dispensable” normal cells, since B cell aplasia and resultant hypogammaglobulinemia can be managed with intravenous immunoglobulin (IVIG) supplementation, but the vast majority of other potential target antigens are found on cells and organs indispensable for survival. On-target toxicities can be avoided if antigen expression is restricted to cancerous cells alone (i.e., tumor-specific antigens); however, many, if not all, cell-surface proteins on tumors are shared by normal cells. Target antigen vetting and in vitro methods of antigen expression evaluation have been previously reviewed.99 The following section will discuss examples of animal models employed to define and avoid adverse effects of on-target off-tumor toxicities.
HER2 and ERBB family antigens
HER2 (ERBB2) is an example of tumor-associated antigen with known variable protein-level expression by various normal tissues.100 The first clinical trial of trastuzumab-based CAR-T cells targeting HER2 led to acute respiratory compromise and a fatality,101 hypothesized to be due to on-target off-tumor targeting of lung epithelial cells encountered upon first pass circulation. However, subsequent clinical trials of different anti-HER2 constructs based on the alternative FRP5 antibody given at lower cell doses demonstrated improved safety profiles without on-target off-tumor toxicities.102, 103, 104 These examples imply that antigen expression on normal tissue is not the only determinant of on-target off-tumor toxicity, underscoring the importance of pre-clinical models to predict the therapeutic window of CAR-T cells directed against tumor-associated antigens. NSG mice gene-transferred to express human HER2 in liver cells experienced on-target off-tumor hepatic toxicities when given human HER2 CAR-T cells.105 Interestingly, when low- and high-affinity CAR-T cells were compared in these mice, the low-affinity CAR-T cells mediated superior anti-tumor activity compared with high-affinity CAR-T cells due to on-target off-tumor sequestration of the high-affinity CAR-T cells in the liver. The study demonstrated the importance of evaluating on-target off-tumor effects not only to achieve a better understanding of toxicity but also to identify potential mechanisms for improving CAR-T cell efficacy. CAR-T cells that target several ErbB dimers, named T1E28z CAR-T cells,106 have been evaluated in SCID-beige mice107 and cross-react with both human and mouse ErbB, making this murine model suitable for on-target off-tumor toxicity evaluation. Mice treated with intravenous or intratumoral injection of T1E28z CAR-T cells did not experience toxicities, but intraperitoneal delivery led to toxicities reminiscent of CRS. Toxicities were considered on-target off-tumor effects since mice developed toxicity regardless of the presence of tumor in the peritoneum. The model highlights the relevance of T cell administration route.
GD2
GD2 is a tumor target antigen also known to be expressed by normal brain cells. A recent pioneering clinical study suggested that GD2 CAR-T cells, given either intravenously or intracerebroventricularly, are surprisingly safe and potentially effective, without apparent on-target off-tumor toxicities.108 There is strong homology of the GD2 antigen between human and rodents, and certain scFvs may bind GD2 expressed by both species,109 enabling the modeling of on-target off-tumor toxicities in a murine system. Pre-clinical murine models have shown different degrees of toxicity depending on the CAR construct, ranging from complete absence to lethality.109, 110, 111 Murine GD2 CAR-T cell models have been valuable for testing the effectiveness of various toxicity mitigation strategies, such as a ligand-inducible system to degrade CAR on the cell surface110 and SynNotch CAR designs.112 In the case of SynNotch-gated GD2-B7H3 CAR-T cells interrogated in murine xenograft models of neuroblastoma, GD2 and anti-GD2 interactions were required to induce B7H3 CAR expression, ensuring that CAR-T cells are reactive only with cells that co-express both GD2 and B7H3, reducing the likelihood of tumor non-specific targeting by CAR.112
CD33 and CD123
Animal models have been particularly important for pre-clinical evaluation of predicted serious on-target off-tumor toxicities of CAR-T cells directed against myeloid leukemia target antigens such as CD33 and CD123, also expressed by normal HSPCs and mature myeloid cells. When NSG mice engrafted with human HSPCs were treated with anti-CD123 CAR-T cells, human hematopoiesis was depleted,113 contrasting with in vitro hematopoietic colony assays suggesting CD123 CAR-T could mediate specific cytotoxicity against leukemia cells while sparing normal HSPCs,114,115 emphasizing the importance of in vivo models. CD33 CAR-T cell on-target off-tumor toxicity to human hematopoiesis was mitigated by engrafting mice with CD33-knockout (KO) human HSPCs generated utilizing CRISPR-Cas9 technology.116 Importantly, the authors also demonstrated that CD33-KO HSPCs were fully functional and capable of establishing long-term multilineage hematopoiesis in rhesus macaques transplanted with autologous CD33-KO HSPCs, setting the stage for the approach to genetically engineer the recipients’ cells to mitigate on-target, off-tumor toxicities.
Other on-target toxicities studied in NHP models
NHP models have been particularly useful in assessing the impact of potential on-target off-tumor toxicities for antigens that are homologous between humans and macaques. For example, ROR1 is a tumor-associated antigen expressed by B cell malignancies and some epithelial cancers. Berger and colleagues treated macaques with autologous ROR1 CAR-T cells after demonstrating that ROR1 is highly homologous between the two species, with similar tissue expression patterns.117 ROR1 CAR-T cells did not induce toxicities against normal tissues expressing ROR1 at low levels. Another example is a study of EPHB4-CAR-T cells118 that exploited the fact that the human EPHB4 ligand Ephrin B2-based CAR binds to both human and NHP EPHB4. Macaques given lymphodepleting conditioning followed by human T cells expressing the EPHB4-CAR experienced no acute on-target off-tumor toxicity. However, given expected xenogeneic T cell rejection, this model could not be used to study later-onset toxicities. Nonetheless, the study demonstrated that lymphodepleted NHPs may serve as a model to study acute on-target toxicities of human CAR-T cells.
CAR-T cell biology and animal models
Trafficking
An obvious prerequisite for effective CAR-T cell therapy involves the cells’ ability to reach the tumor site, which is a concept that cannot be adequately studied in in vitro and necessitates use of animal models. T cell migration relies on a variety of factors, including cytokines, chemokines, adhesion molecules, and their respective receptors. Though many of these factors are species specific, some adhesion molecules and chemokine receptors can engage with murine receptors/ligands.119 Evaluation of the impact of different routes of CAR-T cells administration also requires animal studies. Murine xenograft studies by Parente-Pereira et al. demonstrated that intravenous administration mimicked patterns seen with either human or murine T cells in recipients of the same species.120 Locoregional administration of CAR-T cells via either intraperitoneal or subcutaneous routes showed no systemic trafficking of CAR-T cells, which may reduce systemic exposure and on-target off-tumor toxicity.121 This work led to a clinical trial (NCT01818323) of intratumoral administration of CAR-T cells for head and neck cancers. Trials in murine xenografts have also shown improved results when CAR-T cells for brain tumors were administered directly into the CNS compared with systemic administration.122,123 Safety and feasibility of implanted CNS reservoirs for delivery of CAR-T cells were demonstrated in NHPs,124 paving the way for clinical trials of locoregional administration of CAR-T cells for glioblastoma.125,126
Non-invasive tracking systems using reporter molecules and radiotracers have enabled researchers to visualize CAR-T cells in animal models using PET/SPECT imaging. Although immunodeficient mice lack chemokines and cytokines that might otherwise influence cellular trafficking patterns, murine xenografts have allowed proof-of-concept studies to demonstrate that CAR-T cells can be effectively labeled and imaged, which is necessary prior to conducting large mammal studies to ensure tracking methods are tolerated. The sodium iodide symporter (NIS)127,128 and somatostatin receptor 2 (SSTR2) 129 are reporter molecules successfully employed for non-invasive imaging of CAR-T cells in murine xenografts.
CAR-T cell stemness, differentiation, and exhaustion
T cell terminal differentiation is an undesirable state. Less differentiated T cells mediate superior anti-tumor responses in in vivo models characterized by enhanced persistence and sustained anti-tumor activities (excellent reviews on the concept are available130,131). The hypothesis that CD8+ effector memory (TEM) and central memory (TCM) T cells may variably survive and persist following adoptive transfer has been investigated in both non-human primates132,133 and murine xenografts,134 influencing later phase I clinical trial development evaluating the safety and feasibility of CD19 CAR TCM therapy post-autologous hematopoietic stem cell transplantation (HSCT) in relapsed/refractory diffuse large B cell lymphoma (DLBCL).135 T memory stem cells (TSCM) are even less differentiated and retain stem cell-like properties such as high self-renewal capacity and multipotency,136,137 and CAR-TSCM cells demonstrate superior anti-tumor responses in murine xenografts compared with conventional CAR-T cells.138 Similarly, CAR-T cells generated from pre-selected naive and stem cell memory T cells demonstrated superior and sustained anti-leukemia activity in a humanized mouse model compared with bulk CAR-T cells.139 CAR-TSCM cells can be reliably generated at clinical scale and were tested in a clinical trial (NCT01087294).
The concept of combining human CAR-T cells with various agents that prevent T cell terminal differentiation, either during the CAR-T manufacturing process or as combination systemic therapy, has been tested in murine xenograft models.140, 141, 142 Some CAR constructs are prone to tonic signaling that induces CAR-T cell exhaustion, leading to suboptimal anti-tumor efficacy in murine xenograft models.111 A drug-regulatable platform allowing control of CAR surface expression with dasatinib, which is known to suppress CAR-T cell activation, mitigated exhaustion of anti-GD2 CAR-T cells in murine xenografts,143 which provided a basis for an ongoing clinical trial (NCT04539366).
Conclusion
It is important to understand the benefits and limitations of available animal models for pre-clinical evaluation of CAR-T cells. Retrospective interpretation of animal model-derived data under the light of amassing human clinical observations helps us better delineate the appropriate application of each model. Establishment of safety may be the most important objective of pre-clinical animal studies, but animal models remain an imperfect system for evaluating the toxicology of human CAR-T cell products. Nonetheless, animal models have been indispensable to advancing the cell therapy field, and increasingly refined models will continue to aid us in better understanding the biology of CAR-T cells and provide critical information for the development of novel next-generation cell therapy products.
Acknowledgments
This work was funded by the NIH Intramural Research Program. Figures were created with BioRender.com.
Author contributions
B.B.D., K.I., and C.E.D. wrote the manuscript.
Declaration of interests
No conflict of interest.
References
- 1.Maude S.L., Frey N., Shaw P.A., Aplenc R., Barrett D.M., Bunin N.J., Chew A., Gonzalez V.E., Zheng Z., Lacey S.F., et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maude S.L., Laetsch T.W., Buechner J., Rives S., Boyer M., Bittencourt H., Bader P., Verneris M.R., Stefanski H.E., Myers G.D., et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 2018;378:439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.FDA . 2013. Preclinical Assessment of Investigational Cellular and Gene Therapy Products. Report Number: FDA-2012-D-1038. [Google Scholar]
- 4.FDA . 2022. Considerations for the Development of Chimeric Antigen Receptor (CAR) T Cell Products, Draft Guidance for Industry. Report Number: FDA-2021-D-0404. [Google Scholar]
- 5.Abou-El-Enein M., Angelis A., Appelbaum F.R., Andrews N.C., Bates S.E., Bierman A.S., Brenner M.K., Cavazzana M., Caligiuri M.A., Clevers H., et al. Evidence generation and reproducibility in cell and gene therapy research: a call to action. Mol. Ther. Methods Clin. Dev. 2021;22:11–14. doi: 10.1016/j.omtm.2021.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elsallab M., Bravery C.A., Kurtz A., Abou-El-Enein M. Mitigating deficiencies in evidence during regulatory assessments of advanced therapies: a comparative study with other biologicals. Mol. Ther. Methods Clin. Dev. 2020;18:269–279. doi: 10.1016/j.omtm.2020.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hudson W.A., Li Q., Le C., Kersey J.H. Xenotransplantation of human lymphoid malignancies is optimized in mice with multiple immunologic defects. Leukemia. 1998;12:2029–2033. doi: 10.1038/sj.leu.2401236. [DOI] [PubMed] [Google Scholar]
- 8.Cassidy J.W., Caldas C., Bruna A. Maintaining tumor heterogeneity in patient-derived tumor xenografts. Cancer Res. 2015;75:2963–2968. doi: 10.1158/0008-5472.Can-15-0727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ding L., Ellis M.J., Li S., Larson D.E., Chen K., Wallis J.W., Harris C.C., McLellan M.D., Fulton R.S., Fulton L.L., et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature. 2010;464:999–1005. doi: 10.1038/nature08989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garrido-Laguna I., Uson M., Rajeshkumar N.V., Tan A.C., de Oliveira E., Karikari C., Villaroel M.C., Salomon A., Taylor G., Sharma R., et al. Tumor engraftment in nude mice and enrichment in stroma- related gene pathways predict poor survival and resistance to gemcitabine in patients with pancreatic cancer. Clin. Cancer Res. 2011;17:5793–5800. doi: 10.1158/1078-0432.Ccr-11-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jangalwe S., Shultz L.D., Mathew A., Brehm M.A. Improved B cell development in humanized NOD-scid IL2Rγ(null) mice transgenically expressing human stem cell factor, granulocyte-macrophage colony-stimulating factor and interleukin-3. Immun. Inflamm. Dis. 2016;4:427–440. doi: 10.1002/iid3.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen Z., Trotman L.C., Shaffer D., Lin H.K., Dotan Z.A., Niki M., Koutcher J.A., Scher H.I., Ludwig T., Gerald W., et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–730. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hooijkaas A.I., Gadiot J., van der Valk M., Mooi W.J., Blank C.U. Targeting BRAFV600E in an inducible murine model of melanoma. Am. J. Pathol. 2012;181:785–794. doi: 10.1016/j.ajpath.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 14.Sanmamed M.F., Chester C., Melero I., Kohrt H. Defining the optimal murine models to investigate immune checkpoint blockers and their combination with other immunotherapies. Ann. Oncol. 2016;27:1190–1198. doi: 10.1093/annonc/mdw041. [DOI] [PubMed] [Google Scholar]
- 15.Bresser K., Dijkgraaf F.E., Pritchard C.E.J., Huijbers I.J., Song J.Y., Rohr J.C., Scheeren F.A., Schumacher T.N. A mouse model that is immunologically tolerant to reporter and modifier proteins. Commun. Biol. 2020;3:273. doi: 10.1038/s42003-020-0979-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aoyama N., Miyoshi H., Miyachi H., Sonoshita M., Okabe M., Taketo M.M. Transgenic mice that accept Luciferase- or GFP-expressing syngeneic tumor cells at high efficiencies. Gene Cell. 2018;23:580–589. doi: 10.1111/gtc.12592. [DOI] [PubMed] [Google Scholar]
- 17.Day C.P., Carter J., Weaver Ohler Z., Bonomi C., El Meskini R., Martin P., Graff-Cherry C., Feigenbaum L., Tüting T., Van Dyke T., et al. “Glowing head” mice: a genetic tool enabling reliable preclinical image-based evaluation of cancers in immunocompetent allografts. PLoS One. 2014;9:e109956. doi: 10.1371/journal.pone.0109956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gardner H.L., Fenger J.M., London C.A. Dogs as a model for cancer. Annu. Rev. Anim. Biosci. 2016;4:199–222. doi: 10.1146/annurev-animal-022114-110911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ranieri G., Gadaleta C.D., Patruno R., Zizzo N., Daidone M.G., Hansson M.G., Paradiso A., Ribatti D. A model of study for human cancer: spontaneous occurring tumors in dogs. Biological features and translation for new anticancer therapies. Crit. Rev. Oncol. Hematol. 2013;88:187–197. doi: 10.1016/j.critrevonc.2013.03.005. [DOI] [PubMed] [Google Scholar]
- 20.Felsburg P.J. Overview of immune system development in the dog: comparison with humans. Hum. Exp. Toxicol. 2002;21:487–492. doi: 10.1191/0960327102ht286oa. [DOI] [PubMed] [Google Scholar]
- 21.Taraseviciute A., Tkachev V., Ponce R., Turtle C.J., Snyder J.M., Liggitt H.D., Myerson D., Gonzalez-Cuyar L., Baldessari A., English C., et al. Chimeric antigen receptor T cell-mediated neurotoxicity in nonhuman primates. Cancer Discov. 2018;8:750–763. doi: 10.1158/2159-8290.CD-17-1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kochenderfer J.N., Feldman S.A., Zhao Y., Xu H., Black M.A., Morgan R.A., Wilson W.H., Rosenberg S.A. Construction and preclinical evaluation of an anti-CD19 chimeric antigen receptor. J. Immunother. 2009;32:689–702. doi: 10.1097/CJI.0b013e3181ac6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Friedman K.M., Garrett T.E., Evans J.W., Horton H.M., Latimer H.J., Seidel S.L., Horvath C.J., Morgan R.A. Effective targeting of multiple B-cell maturation antigen-expressing hematological malignances by anti-B-cell maturation antigen chimeric antigen receptor T cells. Hum. Gene Ther. 2018;29:585–601. doi: 10.1089/hum.2018.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schneider D., Xiong Y., Wu D., Nӧlle V., Schmitz S., Haso W., Kaiser A., Dropulic B., Orentas R.J. A tandem CD19/CD20 CAR lentiviral vector drives on-target and off-target antigen modulation in leukemia cell lines. J. Immunother. Cancer. 2017;5:42. doi: 10.1186/s40425-017-0246-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fry T.J., Shah N.N., Orentas R.J., Stetler-Stevenson M., Yuan C.M., Ramakrishna S., Wolters P., Martin S., Delbrook C., Yates B., et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 2018;24:20–28. doi: 10.1038/nm.4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qin H., Ramakrishna S., Nguyen S., Fountaine T.J., Ponduri A., Stetler-Stevenson M., Yuan C.M., Haso W., Shern J.F., Shah N.N., Fry T.J. Preclinical development of bivalent chimeric antigen receptors targeting both CD19 and CD22. Mol. Ther. Oncolytics. 2018;11:127–137. doi: 10.1016/j.omto.2018.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson L.A., Scholler J., Ohkuri T., Kosaka A., Patel P.R., McGettigan S.E., Nace A.K., Dentchev T., Thekkat P., Loew A., et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci. Transl. Med. 2015;7:275ra22. doi: 10.1126/scitranslmed.aaa4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carpenito C., Milone M.C., Hassan R., Simonet J.C., Lakhal M., Suhoski M.M., Varela-Rohena A., Haines K.M., Heitjan D.F., Albelda S.M., et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc. Natl. Acad. Sci. USA. 2009;106:3360–3365. doi: 10.1073/pnas.0813101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haso W., Lee D.W., Shah N.N., Stetler-Stevenson M., Yuan C.M., Pastan I.H., Dimitrov D.S., Morgan R.A., FitzGerald D.J., Barrett D.M., et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. 2013;121:1165–1174. doi: 10.1182/blood-2012-06-438002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sommermeyer D., Hudecek M., Kosasih P.L., Gogishvili T., Maloney D.G., Turtle C.J., Riddell S.R. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia. Leukemia. 2016;30:492–500. doi: 10.1038/leu.2015.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brentjens R.J., Santos E., Nikhamin Y., Yeh R., Matsushita M., La Perle K., Quintás-Cardama A., Larson S.M., Sadelain M. Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin. Cancer Res. 2007;13(18 Pt 1):5426–5435. doi: 10.1158/1078-0432.CCR-07-0674. [DOI] [PubMed] [Google Scholar]
- 32.Shah N.N., Highfill S.L., Shalabi H., Yates B., Jin J., Wolters P.L., Ombrello A., Steinberg S.M., Martin S., Delbrook C., et al. CD4/CD8 T-Cell selection affects chimeric antigen receptor (CAR) T-cell potency and toxicity: updated results from a phase I anti-CD22 CAR T-cell trial. J. Clin. Oncol. 2020;38:1938–1950. doi: 10.1200/JCO.19.03279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang M., Munoz J., Goy A., Locke F.L., Jacobson C.A., Hill B.T., Timmerman J.M., Holmes H., Jaglowski S., Flinn I.W., et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N. Engl. J. Med. 2020;382:1331–1342. doi: 10.1056/NEJMoa1914347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Munshi N.C., Anderson L.D., Jr., Shah N., Madduri D., Berdeja J., Lonial S., Raje N., Lin Y., Siegel D., Oriol A., et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N. Engl. J. Med. 2021;384:705–716. doi: 10.1056/NEJMoa2024850. [DOI] [PubMed] [Google Scholar]
- 35.Schuster S.J., Tam C.S., Borchmann P., Worel N., McGuirk J.P., Holte H., Waller E.K., Jaglowski S., Bishop M.R., Damon L.E., et al. Long-term clinical outcomes of tisagenlecleucel in patients with relapsed or refractory aggressive B-cell lymphomas (JULIET): a multicentre, open-label, single-arm, phase 2 study. Lancet Oncol. 2021;22:1403–1415. doi: 10.1016/S1470-2045(21)00375-2. [DOI] [PubMed] [Google Scholar]
- 36.Schuster S.J., Bishop M.R., Tam C.S., Waller E.K., Borchmann P., McGuirk J.P., Jäger U., Jaglowski S., Andreadis C., Westin J.R., et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N. Engl. J. Med. 2019;380:45–56. doi: 10.1056/NEJMoa1804980. [DOI] [PubMed] [Google Scholar]
- 37.Raje N., Berdeja J., Lin Y., Siegel D., Jagannath S., Madduri D., Liedtke M., Rosenblatt J., Maus M.V., Turka A., et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N. Engl. J. Med. 2019;380:1726–1737. doi: 10.1056/NEJMoa1817226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shah B.D., Ghobadi A., Oluwole O.O., Logan A.C., Boissel N., Cassaday R.D., Leguay T., Bishop M.R., Topp M.S., Tzachanis D., et al. KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. Lancet. 2021;398:491–502. doi: 10.1016/S0140-6736(21)01222-8. [DOI] [PubMed] [Google Scholar]
- 39.Turtle C.J., Hanafi L.A., Berger C., Gooley T.A., Cherian S., Hudecek M., Sommermeyer D., Melville K., Pender B., Budiarto T.M., et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Invest. 2016;126:2123–2138. doi: 10.1172/JCI85309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brudno J.N., Lam N., Vanasse D., Shen Y.W., Rose J.J., Rossi J., Xue A., Bot A., Scholler N., Mikkilineni L., et al. Safety and feasibility of anti-CD19 CAR T cells with fully human binding domains in patients with B-cell lymphoma. Nat. Med. 2020;26:270–280. doi: 10.1038/s41591-019-0737-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao Z., Condomines M., van der Stegen S.J.C., Perna F., Kloss C.C., Gunset G., Plotkin J., Sadelain M. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell. 2015;28:415–428. doi: 10.1016/j.ccell.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheadle E.J., Hawkins R.E., Batha H., O'Neill A.L., Dovedi S.J., Gilham D.E. Natural expression of the CD19 antigen impacts the long-term engraftment but not antitumor activity of CD19-specific engineered T cells. J. Immunol. 2010;184:1885–1896. doi: 10.4049/jimmunol.0901440. [DOI] [PubMed] [Google Scholar]
- 43.Kochenderfer J.N., Yu Z., Frasheri D., Restifo N.P., Rosenberg S.A. Adoptive transfer of syngeneic T cells transduced with a chimeric antigen receptor that recognizes murine CD19 can eradicate lymphoma and normal B cells. Blood. 2010;116:3875–3886. doi: 10.1182/blood-2010-01-265041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jacoby E., Nguyen S.M., Fountaine T.J., Welp K., Gryder B., Qin H., Yang Y., Chien C.D., Seif A.E., Lei H., et al. CD19 CAR immune pressure induces B-precursor acute lymphoblastic leukaemia lineage switch exposing inherent leukaemic plasticity. Nat. Commun. 2016;7:12320. doi: 10.1038/ncomms12320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jacoby E., Yang Y., Qin H., Chien C.D., Kochenderfer J.N., Fry T.J. Murine allogeneic CD19 CAR T cells harbor potent antileukemic activity but have the potential to mediate lethal GVHD. Blood. 2016;127:1361–1370. doi: 10.1182/blood-2015-08-664250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ishii K., Pouzolles M., Chien C.D., Erwin-Cohen R.A., Kohler M.E., Qin H., Lei H., Kuhn S., Ombrello A.K., Dulau-Florea A., et al. Perforin-deficient CAR T cells recapitulate late-onset inflammatory toxicities observed in patients. J. Clin. Invest. 2020;130:5425–5443. doi: 10.1172/JCI130059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ghosh A., Smith M., James S.E., Davila M.L., Velardi E., Argyropoulos K.V., Gunset G., Perna F., Kreines F.M., Levy E.R., et al. Donor CD19 CAR T cells exert potent graft-versus-lymphoma activity with diminished graft-versus-host activity. Nat. Med. 2017;23:242–249. doi: 10.1038/nm.4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang Y., Kohler M.E., Chien C.D., Sauter C.T., Jacoby E., Yan C., Hu Y., Wanhainen K., Qin H., Fry T.J. TCR engagement negatively affects CD8 but not CD4 CAR T cell expansion and leukemic clearance. Sci. Transl. Med. 2017;22:eaag1209. doi: 10.1126/scitranslmed.aag1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Davila M.L., Kloss C.C., Gunset G., Sadelain M. CD19 CAR-targeted T cells induce long-term remission and B Cell Aplasia in an immunocompetent mouse model of B cell acute lymphoblastic leukemia. PLoS One. 2013;8:e61338. doi: 10.1371/journal.pone.0061338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brentjens R.J., Rivière I., Park J.H., Davila M.L., Wang X., Stefanski J., Taylor C., Yeh R., Bartido S., Borquez-Ojeda O., et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817–4828. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gauthier J., Bezerra E.D., Hirayama A.V., Fiorenza S., Sheih A., Chou C.K., Kimble E.L., Pender B.S., Hawkins R.M., Vakil A., et al. Factors associated with outcomes after a second CD19-targeted CAR T-cell infusion for refractory B-cell malignancies. Blood. Jan 21 2021;137:323–335. doi: 10.1182/blood.2020006770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Majzner R.G., Mackall C.L. Clinical lessons learned from the first leg of the CAR T cell journey. Nat. Med. 2019;25:1341–1355. doi: 10.1038/s41591-019-0564-6. [DOI] [PubMed] [Google Scholar]
- 53.Shah N.N., Fry T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019;16:372–385. doi: 10.1038/s41571-019-0184-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gardner R., Wu D., Cherian S., Fang M., Hanafi L.A., Finney O., Smithers H., Jensen M.C., Riddell S.R., Maloney D.G., Turtle C.J. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood. 2016;127:2406–2410. doi: 10.1182/blood-2015-08-665547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nagel I., Bartels M., Duell J., Oberg H.H., Ussat S., Bruckmueller H., Ottmann O., Pfeifer H., Trautmann H., Gökbuget N., et al. Hematopoietic stem cell involvement in BCR-ABL1-positive ALL as a potential mechanism of resistance to blinatumomab therapy. Blood. 2017;130:2027–2031. doi: 10.1182/blood-2017-05-782888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lamble A., Myers R.M., Taraseviciute A., John S., Yates B., Steinberg S.M., Sheppard J.D., Kovach A.E., Wood B.L., Borowitz M.J., et al. Preinfusion factors impacting relapse immunophenotype following CD19 CAR T cells. Blood Adv. 2022 doi: 10.1182/bloodadvances.2022007423. bloodadvances.2022007423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liao W., Kohler M.E., Fry T., Ernst P. Does lineage plasticity enable escape from CAR-T cell therapy? Lessons from MLL-r leukemia. Exp. Hematol. 2021;100:1–11. doi: 10.1016/j.exphem.2021.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cappell K.M., Kochenderfer J.N. A comparison of chimeric antigen receptors containing CD28 versus 4-1BB costimulatory domains. Nat. Rev. Clin. Oncol. 2021;18:715–727. doi: 10.1038/s41571-021-00530-z. [DOI] [PubMed] [Google Scholar]
- 59.Feucht J., Sun J., Eyquem J., Ho Y.J., Zhao Z., Leibold J., Dobrin A., Cabriolu A., Hamieh M., Sadelain M. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat. Med. 2019;25:82–88. doi: 10.1038/s41591-018-0290-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Boucher J.C., Li G., Kotani H., Cabral M.L., Morrissey D., Lee S.B., Spitler K., Beatty N.J., Cervantes E.V., Shrestha B., et al. CD28 costimulatory domain-targeted mutations enhance chimeric antigen receptor T-cell function. Cancer Immunol. Res. 2021;9:62–74. doi: 10.1158/2326-6066.Cir-20-0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Majzner R.G., Rietberg S.P., Sotillo E., Dong R., Vachharajani V.T., Labanieh L., Myklebust J.H., Kadapakkam M., Weber E.W., Tousley A.M., et al. Tuning the antigen density requirement for CAR T-cell activity. Cancer Discov. 2020;10:702–723. doi: 10.1158/2159-8290.Cd-19-0945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hudecek M., Sommermeyer D., Kosasih P.L., Silva-Benedict A., Liu L., Rader C., Jensen M.C., Riddell S.R. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol. Res. 2015;3:125–135. doi: 10.1158/2326-6066.Cir-14-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Singh N., Frey N.V., Engels B., Barrett D.M., Shestova O., Ravikumar P., Cummins K.D., Lee Y.G., Pajarillo R., Chun I., et al. Antigen-independent activation enhances the efficacy of 4-1BB-costimulated CD22 CAR T cells. Nat. Med. 2021;27:842–850. doi: 10.1038/s41591-021-01326-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pascoal S., Salzer B., Scheuringer E., Wenninger-Weinzierl A., Sturtzel C., Holter W., Taschner-Mandl S., Lehner M., Distel M. A preclinical embryonic zebrafish xenograft model to investigate CAR T cells in vivo. Cancers. 2020;12:E567. doi: 10.3390/cancers12030567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Panjwani M.K., Atherton M.J., MaloneyHuss M.A., Haran K.P., Xiong A., Gupta M., Kulikovsaya I., Lacey S.F., Mason N.J. Establishing a model system for evaluating CAR T cell therapy using dogs with spontaneous diffuse large B cell lymphoma. OncoImmunology. 2020;9:1676615. doi: 10.1080/2162402x.2019.1676615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Panjwani M.K., Smith J.B., Schutsky K., Gnanandarajah J., O'Connor C.M., Powell D.J., Mason N.J. Feasibility and safety of RNA-transfected CD20-specific chimeric antigen receptor T cells in dogs with spontaneous B cell lymphoma. Mol. Ther. 2016;24:1602–1614. doi: 10.1038/mt.2016.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rotolo A., Atherton M.J., Kasper B.T., Haran K.P., Mason N.J. Genetic re-direction of canine primary T cells for clinical trial use in pet dogs with spontaneous cancer. STAR Protoc. 2021;2:100905. doi: 10.1016/j.xpro.2021.100905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang S., Black R.G., Kohli K., Hayes B.J., Miller C., Koehne A., Schroeder B.A., Abrams K., Schulte B.C., Alexiev B.A., et al. B7-H3 specific CAR T cells for the naturally occurring, spontaneous canine sarcoma model. Mol. Cancer Ther. 2022;21:999–1009. doi: 10.1158/1535-7163.Mct-21-0726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Migliorini D., Mason N.J., Posey A.D. Keeping the engine running: the relevance and predictive value of preclinical models for CAR-T cell development. ILAR J. 2018;59:276–285. doi: 10.1093/ilar/ilz009. [DOI] [PubMed] [Google Scholar]
- 70.Mercado N.B., Zahn R., Wegmann F., Loos C., Chandrashekar A., Yu J., Liu J., Peter L., McMahan K., Tostanoski L.H., et al. Single-shot Ad26 vaccine protects against SARS-CoV-2 in rhesus macaques. Nature. 2020;586:583–588. doi: 10.1038/s41586-020-2607-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McMahan K., Yu J., Mercado N.B., Loos C., Tostanoski L.H., Chandrashekar A., Liu J., Peter L., Atyeo C., Zhu A., et al. Correlates of protection against SARS-CoV-2 in rhesus macaques. Nature. 2021;590:630–634. doi: 10.1038/s41586-020-03041-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Colonna L., Peterson C.W., Schell J.B., Carlson J.M., Tkachev V., Brown M., Yu A., Reddy S., Obenza W.M., Nelson V., et al. Evidence for persistence of the SHIV reservoir early after MHC haploidentical hematopoietic stem cell transplantation. Nat. Commun. 2018;9:4438. doi: 10.1038/s41467-018-06736-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Barouch D.H., Alter G., Broge T., Linde C., Ackerman M.E., Brown E.P., Borducchi E.N., Smith K.M., Nkolola J.P., Liu J., et al. Protective efficacy of adenovirus/protein vaccines against SIV challenges in rhesus monkeys. Science. 2015;349:320–324. doi: 10.1126/science.aab3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hege K.M., Cooke K.S., Finer M.H., Zsebo K.M., Roberts M.R. Systemic T cell-independent tumor immunity after transplantation of universal receptor-modified bone marrow into SCID mice. J. Exp. Med. 1996;184:2261–2269. doi: 10.1084/jem.184.6.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Deeks S.G., Wagner B., Anton P.A., Mitsuyasu R.T., Scadden D.T., Huang C., Macken C., Richman D.D., Christopherson C., June C.H., et al. A phase II randomized study of HIV-specific T-cell gene therapy in subjects with undetectable plasma viremia on combination antiretroviral therapy. Mol. Ther. 2002;5:788–797. doi: 10.1006/mthe.2002.0611. [DOI] [PubMed] [Google Scholar]
- 76.Walker R.E., Bechtel C.M., Natarajan V., Baseler M., Hege K.M., Metcalf J.A., Stevens R., Hazen A., Blaese R.M., Chen C.C., et al. Long-term in vivo survival of receptor-modified syngeneic T cells in patients with human immunodeficiency virus infection. Blood. 2000;96:467–474. [PubMed] [Google Scholar]
- 77.Zhen A., Peterson C.W., Carrillo M.A., Reddy S.S., Youn C.S., Lam B.B., Chang N.Y., Martin H.A., Rick J.W., Kim J., et al. Long-term persistence and function of hematopoietic stem cell-derived chimeric antigen receptor T cells in a nonhuman primate model of HIV/AIDS. PLoS Pathog. 2017;13:e1006753. doi: 10.1371/journal.ppat.1006753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rust B.J., Kean L.S., Colonna L., Brandenstein K.E., Poole N.H., Obenza W., Enstrom M.R., Maldini C.R., Ellis G.I., Fennessey C.M., et al. Robust expansion of HIV CAR T cells following antigen boosting in ART-suppressed nonhuman primates. Blood. Oct 8 2020;136:1722–1734. doi: 10.1182/blood.2020006372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Morris E.C., Neelapu S.S., Giavridis T., Sadelain M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat. Rev. Immunol. 2022;22:85–96. doi: 10.1038/s41577-021-00547-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Neelapu S.S., Tummala S., Kebriaei P., Wierda W., Gutierrez C., Locke F.L., Komanduri K.V., Lin Y., Jain N., Daver N., et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018;15:47–62. doi: 10.1038/nrclinonc.2017.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kochenderfer J.N., Wilson W.H., Janik J.E., Dudley M.E., Stetler-Stevenson M., Feldman S.A., Maric I., Raffeld M., Nathan D.A.N., Lanier B.J., et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116:4099–4102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cheadle E.J., Sheard V., Rothwell D.G., Bridgeman J.S., Ashton G., Hanson V., Mansoor A.W., Hawkins R.E., Gilham D.E. Differential role of Th1 and Th2 cytokines in autotoxicity driven by CD19-specific second-generation chimeric antigen receptor T cells in a mouse model. J. Immunol. 2014;192:3654–3665. doi: 10.4049/jimmunol.1302148. [DOI] [PubMed] [Google Scholar]
- 83.Pennell C.A., Barnum J.L., McDonald-Hyman C.S., Panoskaltsis-Mortari A., Riddle M.J., Xiong Z., Loschi M., Thangavelu G., Campbell H.M., Storlie M.D., et al. Human CD19-targeted mouse T cells induce B cell aplasia and toxicity in human CD19 transgenic mice. Mol. Ther. 2018;26:1423–1434. doi: 10.1016/j.ymthe.2018.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Norelli M., Camisa B., Barbiera G., Falcone L., Purevdorj A., Genua M., Sanvito F., Ponzoni M., Doglioni C., Cristofori P., et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat. Med. 2018;24:739–748. doi: 10.1038/s41591-018-0036-4. [DOI] [PubMed] [Google Scholar]
- 85.Giavridis T., van der Stegen S.J.C., Eyquem J., Hamieh M., Piersigilli A., Sadelain M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat. Med. 2018;24:731–738. doi: 10.1038/s41591-018-0041-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Teachey D.T., Lacey S.F., Shaw P.A., Melenhorst J.J., Maude S.L., Frey N., Pequignot E., Gonzalez V.E., Chen F., Finklestein J., et al. Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Cancer Discov. 2016;6:664–679. doi: 10.1158/2159-8290.CD-16-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hay K.A., Hanafi L.A., Li D., Gust J., Liles W.C., Wurfel M.M., López J.A., Chen J., Chung D., Harju-Baker S., et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood. 2017;130:2295–2306. doi: 10.1182/blood-2017-06-793141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gust J., Hay K.A., Hanafi L.A., Li D., Myerson D., Gonzalez-Cuyar L.F., Yeung C., Liles W.C., Wurfel M., Lopez J.A., et al. Endothelial activation and blood-brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discov. 2017;7:1404–1419. doi: 10.1158/2159-8290.CD-17-0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wehrli M., Gallagher K., Chen Y.B., Leick M.B., McAfee S.L., El-Jawahri A.R., DeFilipp Z., Horick N., O'Donnell P., Spitzer T., et al. Single-center experience using anakinra for steroid-refractory immune effector cell-associated neurotoxicity syndrome (ICANS) J. Immunother. Cancer. 2022;10:e003847. doi: 10.1136/jitc-2021-003847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sterner R.M., Sakemura R., Cox M.J., Yang N., Khadka R.H., Forsman C.L., Hansen M.J., Jin F., Ayasoufi K., Hefazi M., et al. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood. 2019;133:697–709. doi: 10.1182/blood-2018-10-881722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Staedtke V., Bai R.Y., Kim K., Darvas M., Davila M.L., Riggins G.J., Rothman P.B., Papadopoulos N., Kinzler K.W., Vogelstein B., Zhou S. Disruption of a self-amplifying catecholamine loop reduces cytokine release syndrome. Nature. 2018;564:273–277. doi: 10.1038/s41586-018-0774-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Beura L.K., Hamilton S.E., Bi K., Schenkel J.M., Odumade O.A., Casey K.A., Thompson E.A., Fraser K.A., Rosato P.C., Filali-Mouhim A., et al. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature. 2016;532:512–516. doi: 10.1038/nature17655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Abolins S., King E.C., Lazarou L., Weldon L., Hughes L., Drescher P., Raynes J.G., Hafalla J.C.R., Viney M.E., Riley E.M. The comparative immunology of wild and laboratory mice, Mus musculus domesticus. Nat. Commun. 2017;8:14811. doi: 10.1038/ncomms14811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lichtenstein D.A., Schischlik F., Shao L., Steinberg S.M., Yates B., Wang H.W., Wang Y., Inglefield J., Dulau-Florea A., Ceppi F., et al. Characterization of HLH-like manifestations as a CRS variant in patients receiving CD22 CAR T cells. Blood. 2021;138:2469–2484. doi: 10.1182/blood.2021011898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Diorio C., Vatsayan A., Talleur A.C., Annesley C., Jaroscak J.J., Shalabi H., Ombrello A.K., Hudspeth M., Maude S.L., Gardner R.A., Shah N.N. Anakinra utilization in refractory pediatric CAR T-cell associated toxicities. Blood Adv. 2022;6:3398–3403. doi: 10.1182/bloodadvances.2022006983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cameron B.J., Gerry A.B., Dukes J., Harper J.V., Kannan V., Bianchi F.C., Grand F., Brewer J.E., Gupta M., Plesa G., et al. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci. Transl. Med. 2013;5:197ra103. doi: 10.1126/scitranslmed.3006034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Linette G.P., Stadtmauer E.A., Maus M.V., Rapoport A.P., Levine B.L., Emery L., Litzky L., Bagg A., Carreno B.M., Cimino P.J., et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. 2013;122:863–871. doi: 10.1182/blood-2013-03-490565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Morgan R.A., Chinnasamy N., Abate-Daga D., Gros A., Robbins P.F., Zheng Z., Dudley M.E., Feldman S.A., Yang J.C., Sherry R.M., et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother. 2013;36:133–151. doi: 10.1097/CJI.0b013e3182829903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hinrichs C.S., Restifo N.P. Reassessing target antigens for adoptive T-cell therapy. Nat. Biotechnol. 2013;31:999–1008. doi: 10.1038/nbt.2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Uhlén M., Fagerberg L., Hallström B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A., et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 101.Morgan R.A., Yang J.C., Kitano M., Dudley M.E., Laurencot C.M., Rosenberg S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ahmed N., Brawley V., Hegde M., Bielamowicz K., Kalra M., Landi D., Robertson C., Gray T.L., Diouf O., Wakefield A., et al. HER2-Specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol. 2017;3:1094–1101. doi: 10.1001/jamaoncol.2017.0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ahmed N., Brawley V.S., Hegde M., Robertson C., Ghazi A., Gerken C., Liu E., Dakhova O., Ashoori A., Corder A., et al. Human epidermal growth factor receptor 2 (HER2) -specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J. Clin. Oncol. 2015;33:1688–1696. doi: 10.1200/JCO.2014.58.0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hegde M., Joseph S.K., Pashankar F., DeRenzo C., Sanber K., Navai S., Byrd T.T., Hicks J., Xu M.L., Gerken C., et al. Tumor response and endogenous immune reactivity after administration of HER2 CAR T cells in a child with metastatic rhabdomyosarcoma. Nat. Commun. 2020;11:3549. doi: 10.1038/s41467-020-17175-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Castellarin M., Sands C., Da T., Scholler J., Graham K., Buza E., Fraietta J.A., Zhao Y., June C.H. A rational mouse model to detect on-target, off-tumor CAR T cell toxicity. JCI Insight. 2020:136012. doi: 10.1172/jci.insight.136012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Davies D.M., Foster J., Van Der Stegen S.J.C., Parente-Pereira A.C., Chiapero-Stanke L., Delinassios G.J., Burbridge S.E., Kao V., Liu Z., Bosshard-Carter L., et al. Flexible targeting of ErbB dimers that drive tumorigenesis by using genetically engineered T cells. Mol. Med. 2012;18:565–576. doi: 10.2119/molmed.2011.00493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.van der Stegen S.J.C., Davies D.M., Wilkie S., Foster J., Sosabowski J.K., Burnet J., Whilding L.M., Petrovic R.M., Ghaem-Maghami S., Mather S., et al. Preclinical in vivo modeling of cytokine release syndrome induced by ErbB-retargeted human T cells: identifying a window of therapeutic opportunity? J. Immunol. 2013;191:4589–4598. doi: 10.4049/jimmunol.1301523. [DOI] [PubMed] [Google Scholar]
- 108.Majzner R.G., Ramakrishna S., Yeom K.W., Patel S., Chinnasamy H., Schultz L.M., Richards R.M., Jiang L., Barsan V., Mancusi R., et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature. 2022;603:934–941. doi: 10.1038/s41586-022-04489-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Richman S.A., Nunez-Cruz S., Moghimi B., Li L.Z., Gershenson Z.T., Mourelatos Z., Barrett D.M., Grupp S.A., Milone M.C. High-affinity GD2-specific CAR T cells induce fatal encephalitis in a preclinical neuroblastoma model. Cancer Immunol. Res. 2018;6:36–46. doi: 10.1158/2326-6066.CIR-17-0211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Richman S.A., Wang L.C., Moon E.K., Khire U.R., Albelda S.M., Milone M.C. Ligand-Induced degradation of a CAR permits reversible remote control of CAR T cell activity in vitro and in vivo. Mol. Ther. 2020;28:1600–1613. doi: 10.1016/j.ymthe.2020.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Long A.H., Haso W.M., Shern J.F., Wanhainen K.M., Murgai M., Ingaramo M., Smith J.P., Walker A.J., Kohler M.E., Venkateshwara V.R., et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015;21:581–590. doi: 10.1038/nm.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Moghimi B., Muthugounder S., Jambon S., Tibbetts R., Hung L., Bassiri H., Hogarty M.D., Barrett D.M., Shimada H., Asgharzadeh S. Preclinical assessment of the efficacy and specificity of GD2-B7H3 SynNotch CAR-T in metastatic neuroblastoma. Nat. Commun. 2021;12:511. doi: 10.1038/s41467-020-20785-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gill S., Tasian S.K., Ruella M., Shestova O., Li Y., Porter D.L., Carroll M., Danet-Desnoyers G., Scholler J., Grupp S.A., et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood. 2014;123:2343–2354. doi: 10.1182/blood-2013-09-529537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mardiros A., Dos Santos C., McDonald T., Brown C.E., Wang X., Budde L.E., Hoffman L., Aguilar B., Chang W.C., Bretzlaff W., et al. T cells expressing CD123-specific chimeric antigen receptors exhibit specific cytolytic effector functions and antitumor effects against human acute myeloid leukemia. Blood. 2013;122:3138–3148. doi: 10.1182/blood-2012-12-474056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tettamanti S., Marin V., Pizzitola I., Magnani C.F., Giordano Attianese G.M.P., Cribioli E., Maltese F., Galimberti S., Lopez A.F., Biondi A., et al. Targeting of acute myeloid leukaemia by cytokine-induced killer cells redirected with a novel CD123-specific chimeric antigen receptor. Br. J. Haematol. 2013;161:389–401. doi: 10.1111/bjh.12282. [DOI] [PubMed] [Google Scholar]
- 116.Kim M.Y., Yu K.R., Kenderian S.S., Ruella M., Chen S., Shin T.H., Aljanahi A.A., Schreeder D., Klichinsky M., Shestova O., et al. Genetic inactivation of CD33 in hematopoietic stem cells to enable CAR T cell immunotherapy for acute myeloid leukemia. Cell. 2018;173:1439–1453.e19. doi: 10.1016/j.cell.2018.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Berger C., Sommermeyer D., Hudecek M., Berger M., Balakrishnan A., Paszkiewicz P.J., Kosasih P.L., Rader C., Riddell S.R. Safety of targeting ROR1 in primates with chimeric antigen receptor-modified T cells. Cancer Immunol. Res. 2015;3:206–216. doi: 10.1158/2326-6066.CIR-14-0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yagyu S., Mochizuki H., Yamashima K., Kubo H., Saito S., Tanaka M., Sakamoto K., Shimoi A., Nakazawa Y. A lymphodepleted non-human primate model for the assessment of acute on-target and off-tumor toxicity of human chimeric antigen receptor-T cells. Clin. Transl. Immunology. 2021;10:e1291. doi: 10.1002/cti2.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Taub D.D., Tsarfaty G., Lloyd A.R., Durum S.K., Longo D.L., Murphy W.J. Growth hormone promotes human T cell adhesion and migration to both human and murine matrix proteins in vitro and directly promotes xenogeneic engraftment. J. Clin. Invest. 1994;94:293–300. doi: 10.1172/jci117320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Parente-Pereira A.C., Burnet J., Ellison D., Foster J., Davies D.M., van der Stegen S., Burbridge S., Chiapero-Stanke L., Wilkie S., Mather S., Maher J. Trafficking of CAR-engineered human T cells following regional or systemic adoptive transfer in SCID beige mice. J. Clin. Immunol. 2011;31:710–718. doi: 10.1007/s10875-011-9532-8. [DOI] [PubMed] [Google Scholar]
- 121.Papa S., Adami A., Metoudi M., Achkova D., van Schalkwyk M., Parente Pereira A., Bosshard-Carter L., Whilding L., van der Stegen S., Davies D., et al. A phase I trial of T4 CAR T-cell immunotherapy in head and neck squamous cancer (HNSCC) J. Clin. Oncol. 2018;36(suppl):3046. doi: 10.1200/JCO.2018.36.15_suppl.3046. [DOI] [Google Scholar]
- 122.Theruvath J., Sotillo E., Mount C.W., Graef C.M., Delaidelli A., Heitzeneder S., Labanieh L., Dhingra S., Leruste A., Majzner R.G., et al. Locoregionally administered B7-H3-targeted CAR T cells for treatment of atypical teratoid/rhabdoid tumors. Nat. Med. 2020;26:712–719. doi: 10.1038/s41591-020-0821-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Donovan L.K., Delaidelli A., Joseph S.K., Bielamowicz K., Fousek K., Holgado B.L., Manno A., Srikanthan D., Gad A.Z., Van Ommeren R., et al. Locoregional delivery of CAR T cells to the cerebrospinal fluid for treatment of metastatic medulloblastoma and ependymoma. Nat. Med. 2020;26:720–731. doi: 10.1038/s41591-020-0827-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Nellan A., Rota C., Majzner R., Lester-McCully C.M., Griesinger A.M., Mulcahy Levy J.M., Foreman N.K., Warren K.E., Lee D.W. Durable regression of Medulloblastoma after regional and intravenous delivery of anti-HER2 chimeric antigen receptor T cells. J. Immunother. Cancer. 2018;6:30. doi: 10.1186/s40425-018-0340-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Brown C.E., Badie B., Barish M.E., Weng L., Ostberg J.R., Chang W.C., Naranjo A., Starr R., Wagner J., Wright C., et al. Bioactivity and safety of IL13Rα2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin. Cancer Res. 2015;21:4062–4072. doi: 10.1158/1078-0432.Ccr-15-0428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Keu K.V., Witney T.H., Yaghoubi S., Rosenberg J., Kurien A., Magnusson R., Williams J., Habte F., Wagner J.R., Forman S., et al. Reporter gene imaging of targeted T cell immunotherapy in recurrent glioma. Sci. Transl. Med. 2017;9:eaag2196. doi: 10.1126/scitranslmed.aag2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sakemura R., Bansal A., Siegler E.L., Hefazi M., Yang N., Khadka R.H., Newsom A.N., Hansen M.J., Cox M.J., Manriquez Roman C., et al. Development of a clinically relevant reporter for chimeric antigen receptor T-cell expansion, trafficking, and toxicity. Cancer Immunol. Res. 2021;9:1035–1046. doi: 10.1158/2326-6066.CIR-20-0901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Volpe A., Lang C., Lim L., Man F., Kurtys E., Ashmore-Harris C., Johnson P., Skourti E., de Rosales R.T.M., Fruhwirth G.O. Spatiotemporal PET imaging reveals differences in CAR-T tumor retention in triple-negative breast cancer models. Mol. Ther. 2020;28:2271–2285. doi: 10.1016/j.ymthe.2020.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Vedvyas Y., Shevlin E., Zaman M., Min I.M., Amor-Coarasa A., Park S., Park S., Kwon K.W., Smith T., Luo Y., et al. Longitudinal PET imaging demonstrates biphasic CAR T cell responses in survivors. JCI Insight. 2016;17:e90064. doi: 10.1172/jci.insight.90064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gattinoni L., Klebanoff C.A., Restifo N.P. Paths to stemness: building the ultimate antitumour T cell. Nat. Rev. Cancer. 2012;12:671–684. doi: 10.1038/nrc3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gattinoni L., Speiser D.E., Lichterfeld M., Bonini C. T memory stem cells in health and disease. Nat. Med. 2017;23:18–27. doi: 10.1038/nm.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Berger C., Jensen M.C., Lansdorp P.M., Gough M., Elliott C., Riddell S.R. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J. Clin. Invest. 2008;118:294–305. doi: 10.1172/jci32103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Minang J.T., Trivett M.T., Bolton D.L., Trubey C.M., Estes J.D., Li Y., Smedley J., Pung R., Rosati M., Jalah R., et al. Distribution, persistence, and efficacy of adoptively transferred central and effector memory-derived autologous simian immunodeficiency virus-specific CD8+ T cell clones in rhesus macaques during acute infection. J. Immunol. 2010;184:315–326. doi: 10.4049/jimmunol.0902410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wang X., Berger C., Wong C.W., Forman S.J., Riddell S.R., Jensen M.C. Engraftment of human central memory-derived effector CD8+ T cells in immunodeficient mice. Blood. 2011;117:1888–1898. doi: 10.1182/blood-2010-10-310599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wang X., Popplewell L.L., Wagner J.R., Naranjo A., Blanchard M.S., Mott M.R., Norris A.P., Wong C.W., Urak R.Z., Chang W.C., et al. Phase 1 studies of central memory-derived CD19 CAR T-cell therapy following autologous HSCT in patients with B-cell NHL. Blood. 2016;127:2980–2990. doi: 10.1182/blood-2015-12-686725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gattinoni L., Lugli E., Ji Y., Pos Z., Paulos C.M., Quigley M.F., Almeida J.R., Gostick E., Yu Z., Carpenito C., et al. A human memory T cell subset with stem cell-like properties. Nat. Med. 2011;17:1290–1297. doi: 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lugli E., Dominguez M.H., Gattinoni L., Chattopadhyay P.K., Bolton D.L., Song K., Klatt N.R., Brenchley J.M., Vaccari M., Gostick E., et al. Superior T memory stem cell persistence supports long-lived T cell memory. J. Clin. Invest. 2013;123:594–599. doi: 10.1172/JCI66327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sabatino M., Hu J., Sommariva M., Gautam S., Fellowes V., Hocker J.D., Dougherty S., Qin H., Klebanoff C.A., Fry T.J., et al. Generation of clinical-grade CD19-specific CAR-modified CD8+ memory stem cells for the treatment of human B-cell malignancies. Blood. 2016;128:519–528. doi: 10.1182/blood-2015-11-683847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Arcangeli S., Bove C., Mezzanotte C., Camisa B., Falcone L., Manfredi F., Bezzecchi E., El Khoury R., Norata R., Sanvito F., et al. CAR T-cell manufacturing from naive/stem memory T-lymphocytes enhances antitumor responses while curtailing cytokine release syndrome. J. Clin. Invest. 2022;132:e150807. doi: 10.1172/jci150807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hebbar N., Epperly R., Vaidya A., Thanekar U., Moore S.E., Umeda M., Ma J., Patil S.L., Langfitt D., Huang S., et al. CAR T cells redirected to cell surface GRP78 display robust anti-acute myeloid leukemia activity and do not target hematopoietic progenitor cells. Nat. Commun. 2022;13:587. doi: 10.1038/s41467-022-28243-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Klebanoff C.A., Crompton J.G., Leonardi A.J., Yamamoto T.N., Chandran S.S., Eil R.L., Sukumar M., Vodnala S.K., Hu J., Ji Y., et al. Inhibition of AKT signaling uncouples T cell differentiation from expansion for receptor-engineered adoptive immunotherapy. JCI Insight. 2017;2:95103. doi: 10.1172/jci.insight.95103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mestermann K., Giavridis T., Weber J., Rydzek J., Frenz S., Nerreter T., Mades A., Sadelain M., Einsele H., Hudecek M. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci. Transl. Med. 2019:eaau5907. doi: 10.1126/scitranslmed.aau5907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Weber E.W., Parker K.R., Sotillo E., Lynn R.C., Anbunathan H., Lattin J., Good Z., Belk J.A., Daniel B., Klysz D., et al. Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science. 2021:eaba1786. doi: 10.1126/science.aba1786. [DOI] [PMC free article] [PubMed] [Google Scholar]