

Supplemental Digital Content is Available in the Text.
A combination of clinical and preclinical data that support a role for the lysolipid LPC16:0 via acid-sensing ion channels 3 in chronic joint pain related to rheumatic diseases.
Keywords: Chronic pain, Joint, Osteoarthritis, Lysophosphatidylcholine, Lipid, Acid-sensing ion channels, Sodium channels
Abstract
Rheumatic diseases are often associated to debilitating chronic pain, which remains difficult to treat and requires new therapeutic strategies. We had previously identified lysophosphatidylcholine (LPC) in the synovial fluids from few patients and shown its effect as a positive modulator of acid-sensing ion channel 3 (ASIC3) able to induce acute cutaneous pain in rodents. However, the possible involvement of LPC in chronic joint pain remained completely unknown. Here, we show, from 2 independent cohorts of patients with painful rheumatic diseases, that the synovial fluid levels of LPC are significantly elevated, especially the LPC16:0 species, compared with postmortem control subjects. Moreover, LPC16:0 levels correlated with pain outcomes in a cohort of osteoarthritis patients. However, LPC16:0 do not appear to be the hallmark of a particular joint disease because similar levels are found in the synovial fluids of a second cohort of patients with various rheumatic diseases. The mechanism of action was next explored by developing a pathology-derived rodent model. Intra-articular injections of LPC16:0 is a triggering factor of chronic joint pain in both male and female mice, ultimately leading to persistent pain and anxiety-like behaviors. All these effects are dependent on ASIC3 channels, which drive sufficient peripheral inputs to generate spinal sensitization processes. This study brings evidences from mouse and human supporting a role for LPC16:0 via ASIC3 channels in chronic pain arising from joints, with potential implications for pain management in osteoarthritis and possibly across other rheumatic diseases.
1. Introduction
Chronic joint pain is the primary reason why patients seek care from rheumatologists, representing a heavy burden for modern societies.4,14 In addition to economical costs, chronic joint pain considerably impairs the patients' quality of life, leading to disabilities and psychological distress.4,36 Despite major advances in inflammatory arthritis therapies, most patients, especially those suffering from osteoarthritis (OA), continue to experience life-disabling pain. The development of new therapeutic strategies requires a better understanding of the pathophysiology of joint pain, with careful identification of the triggering factors and associated mechanisms.
In the past 2 decades, signaling lipids have been shown to contribute to the onset and maintenance of pain. Lysophosphatidylcholine (LPC) and lysophosphatidic acid (LPA), the most prominent lysoglycerophospholipids,41 are emerging as potential direct pain mediators, besides well-known thromboxanes, leukotrienes and prostaglandins.15,32 Lysophosphatidic acid has been already identified as a key promoter of nerve demyelination and certainly of neuropathic pain.22,52 Paradoxically, LPA receptor signaling has also documented physiological roles in myelinating cells during nervous system development.54–56 The role of LPC in pain remains more elusive because its effects could be partially mediated by LPA through the action of autotaxin, the LPC-to-LPA converting enzyme.50,51 Nevertheless, some studies support a direct role of LPC in pain associated to its ability to activate/potentiate pain-related ion channels.2,12,15,31,32 We already reported high levels of LPC in the synovial fluids of painful joint patients,32 and an overactivation of the conversion pathway of PC-to-LPC has been recently observed in OA patients.57 Lysophosphatidylcholine seems also important in pain originating from muscles.18
Lysophosphatidylcholine is able to activate and potentiate pain-related acid-sensing ion channel 3 (ASIC3),32 a subtype of the ASIC channel family, widely expressed in nociceptors and also gated by extracellular protons.53 Acid-sensing ion channel 3 have been involved in animal models of inflammatory and noninflammatory pain,9,43 especially in the development of chronic pain states originating from joints20,23,48 and muscles.18,44,45 Genetic deletion of ASIC3 reduced secondary pain behaviors in different models of arthritis, such as intra-articular complete Freund adjuvant (CFA) or carrageenan, and in collagen antibodies–induced arthritis model.17,20,21,46 However, its direct role in inflammation is still debated with proresolving and worsening effects reported.17,46 Blockade of ASIC3 with APETx210 attenuated both disease and pain progression of the early phase in a OA rat model.23 Finally, ASIC3-dependent pain behaviors can be induced upon injection of LPC in rodent paw or muscle.18,32 Here, we investigated the clinical relevance of LPC in chronic joint pain and explored its mechanism of action involving peripheral ASIC3 channels following intra-articular administrations of LPC16:0 in mice.
2. Methods
2.1. Patient cohorts
2.1.1. Cohort of patients with osteoarthritis (first cohort)
Thirty-five patients with knee OA (16 women and 19 men, average age, 64.8 years, range 49‐73 years) participated. This study was approved by the Regional Ethical Review Board in Stockholm, Sweden (reference number 2011/2036-31-1, 2012/2006-32) and followed the guidelines of the Declaration of Helsinki. All patients were recruited consecutively from the waiting list for total knee replacement at Ortho Center, Upplands Väsby, Sweden. The patient inclusion criteria were 25 to 75 years of age, radiologically verified knee OA, and the presence of knee pain as the dominant pain symptom and a motivation for surgical procedure. The patients were excluded if they suffered from chronic pain due to causes other than knee OA (eg, fibromyalgia, degenerative disc disease, disc herniation, inflammatory rheumatic disease, or neurologic disease) or in case of previous knee surgery planned for total knee replacement. Information regarding medication was collected from all patients. Eight patients were taking analgesics (3 codeine, 2 tramadol, 2 buprenorphin plaster, 1 ketobemidone), 12 were taking acetaminophen, and 16 had previously been taking nonsteroidal anti-inflammatory drugs at demand; however, these had been stopped 14 days before the surgical procedure. All patients received 2 g of acetaminophen (paracetamol) and 10 mg of oxycodone orally as premedication before the operation. All participants were informed about the study procedure, and written consent was obtained. Knee joint synovial fluid was collected during surgery and immediately frozen at −80°C for future analysis.
2.1.2. Cohort of patients with different joint diseases (second cohort)
Fifty patients suffering from different painful joint diseases (32 women and 18 men, average age, 68.6 years, range, 26-94 years), recruited in the Rheumatology Department of Nice University Hospital, France, participated and were distributed as follow: rheumatoid arthritis (RA, n = 6), chondrocalcinosis (CCA, n = 12), spondyloarthritis (SPA, n = 5), psoriatic arthritis (n = 4), gout (n = 5), and OA (n = 18). All subjects provided informed consent before inclusion, and the study was approved by the Nice University Institutional Review Board for Research on Human Subjects. By contrast with the first OA cohort, information about medications was not available for all patients excluding the possibility to perform correlation studies between LPC levels in synovial fluids and patient pain outcomes. The study has been conducted in accordance with the French national regulations regarding patient consent and ethical review. The study was registered in the ClinicalTrials.gov protocol registration system (NCT 01867840). All samples were obtained from patients with acute knee joint effusion requiring joint puncture for diagnosis and/or treatment. The synovial fluids remaining after biological analysis for patients' care were included in the present study and frozen at −80°C for future analysis.
2.1.3. Postmortem control subjects
Ten postmortem specimens with no history of knee or hip OA or inflammatory rheumatic diseases were included as controls. Synovial fluid from the knee joint was collected during an autopsy procedure and immediately frozen at −80°C for future analysis.
2.1.4. Questionnaires
Only OA patients from the first cohort completed questionnaires and scales within a week before the operation. Global pain intensity at the day of examination (visual analogue scale [VAS] global) and pain in the affected knee (VAS knee) were all scored using 100-mm VAS, with 0 indicating “no pain” and 100 indicating “the worst imaginable pain.” The severity of patient-reported symptoms was assessed by Knee injury and Osteoarthritis Outcome Score (KOOS), which consists of 5 subscales: (1) pain, (2) other symptoms, (3) activity in daily living, (4) function in sport and recreation, and (5) knee-related quality of life.38,39 Each KOOS subscale contains questions scored from 0 to 4, and the average score of all 5 KOOS subscales was calculated and used for the analysis.
2.1.5. Patient demographic data and lysophosphatidylcholine concentrations
The demographic data (age, sex, body mass index [BMI]) and LPC concentrations found in patients synovial fluids from the first (OA patients) and the second cohorts, as well as in postmortem control specimens, are reported in supplementary tables 1 and 3 (available at http://links.lww.com/PAIN/B579). Part of the data of the first cohort (age, sex, BMI, VAS global, VAS knee, KOOS assessment, and interleukin [IL]-6 levels in the knee synovial fluids) have also been reported elsewhere.26
2.2. Lipidomic analysis of patient samples
2.2.1. Chemical and lipid standards
Chloroform (CHCl3), methanol (CH3OH), and formic acid (HCOOH) were obtained from Sigma Aldrich (Saint Quentitn Fallavier, France). Water (H2O) used for lipid extraction was from Milli-Q quality. Lysophosphatidylcholine standards were purchased from Avanti Polar Lipids via Sigma Aldrich and then prepared at the appropriate concentration and stored at −20°C.
2.2.2. Lipid extraction
All patients' samples were processed in the same manner by the same laboratory as follow. Extraction of lipids from human synovial fluid (HSF) samples was adapted from the Folch method.13 Fifty microliters of each patient sample were diluted with water to a final volume of 1 mL. The diluted samples were then transferred into glass tubes (Pyrex Labware) and vortexed for 1 minute. Lipids were extracted using 4-mL chloroform (CHCl3)/methanol (CH3OH) (2:1, vol/vol) and shaken with an orbital shaker (IKA VX basic Vibrax, Sigma-Aldrich) at 1500 rpm for 2 hours at room temperature. After centrifugation for 10 minutes with a swingout centrifuge at 410 g, aqueous phases were eliminated, and lipid-containing organic phases were supplemented with 1 mL of a 4:1 (vol/vol) 2N KCl/CH3OH solution. Samples were shaken for 10 minutes at 1500 rpm, centrifuged for 5 minutes, and the upper aqueous phases were eliminated. The resulting organic phases were complemented with 1 mL of a 3:48:47 (vol/vol/vol) CHCl3/CH3OH/H2O solution, shaken for 10 minutes and centrifuged for 5 minutes. The aqueous phases were eliminated, and the organic phases containing whole lipids were transferred into new glass tubes. Lipid extracts were then evaporated until dryness at 60°C under a stream of nitrogen, redissolved in 500 µL CHCl3/CH3OH (1:2, vol/vol) and stored at −20°C until further analysis.
2.2.3. Mass spectrometry analysis of lysophosphatidylcholine
Lipid extracts were diluted once in CHCl3/CH3OH (1:2, vol/vol) before addition of 1% (vol/vol) formic acid. An optimized quantity of the internal standard LPC13:0 (0.1 µg of LPC13:0 for 100 µL of lipid extract) was added to each sample to quantify LPC species. Diluted lipid extracts were analyzed in the positive ion mode by direct infusion on a SYNAPT G2 High-Definition Mass Spectrometer (Waters Corporation, Milford, MA) equipped with an Electrospray Ionization Source. The flow rate was 5 µL/minute. All High Resolution full-scan mass spectrometry (MS) experiments were acquired in profile mode over 1 minute with a normal dynamic range from 300 to 1200 m/z. Ionization conditions in positive ion mode have been optimized, and all LPC species, including the LPC internal standard, were detected as protonated ions [M + H]+.
Identification of LPC species was based on their exact masses in the full-scan spectrum and on tandem MS experiments in the positive ion mode. Tandem mass spectrometry/MS fragmentation was performed by collision-induced-dissociation, which notably allowed to obtain structural information about LPC species by identifying the choline polar head group (characteristic and prominent fragment ion for all LPC species with m/z 184).
Lysophosphatidylcholine species were quantified by normalizing the intensity of the protonated ion [M + H]+ corresponding to each LPC individual species in the full-scan spectrum to the intensity of the internal standard LPC 13:0 [M + H]+ and multiplying by the amount of the internal LPC standard. In addition, corrections were applied to the data for isotopic overlap. Total LPC amount was calculated by summing the quantities of all individual LPC species. All spectra were recorded with MassLynx software© (Version 4.1, Waters). Data processing for high-resolution full-scan experiments was performed with the help of ALEX Software.19
2.3. Animals and behavioral experiments
2.3.1. Animals
Experiments were performed on adult male and/or female C57Bl6J wild‐type (WT) mice (Janvier Labs, Le Genest, France) as well as ASIC3-knockout (ASIC3−/−) and WT littermates (ASIC3+/+), breed in animal facilities of IPMC and UCA Medicine School with agreements no. C061525 and no. C63115.15, respectively. Animals were kept with a 12‐hour light/dark cycle with ad libitum access to food and water and were acclimated to housing and husbandry conditions for at least 1 week before experiments. All experiments followed guidelines of the International Association for the Study of Pain.59 All the protocols used were approved by the local ethical committees (CIEPAL-Azur and CEMEA-Auvergne) and the French government (agreements no. 02595.02, APAFIS#13499, APAFIS#17387) in accordance with European Communities Council Directive for the care of laboratory animals (86/609/EEC).
2.3.2. Joint pain models
Monoarthritis was induced by a single intra-articular injection of 12 µL of CFA (heat-killed Mycobacterium butyricum 2 mg/mL, Sigma) under isoflurane (2.5%) anesthesia and was used as a positive control. Concerning the LPC-induced joint pain model, mice received 2 consecutive intra-articular injections of saline solutions containing either LPC16:0 (10 or 20 nmol; Avanti polar lipds, Coger, France) or vehicle (EtOH 2%-4%), 5 days apart, within the ankle or knee joints (10 µL) under isoflurane (2.5%) anesthesia. The mechanical (von Frey test) and heat (thermal test) pain thresholds, as well as the weight-bearing between hind paws (static weight-bearing test) were measured before and after LPC/CFA/vehicle injections over a month period (see figures for schematic protocol of experiments).
2.3.3. Pharmacological experiments
The autotaxin inhibitor S32826 (Biotechne, France), was injected in mice ankles (10 nmol) either at the time of the first LPC16:0 injection or 5 days after the second LPC16:0 injection. When S32826 was coinjected with the first LPC16:0 administration, mechanical allodynia was assessed from 30 to 240 minutes post injection. For experiments where S32826 was administrated 5 days after the second LPC16:0 injection, the mechanical allodynia was assessed 30 and 60 minutes post injection. APETx2 (Smartox, Saint-Égrève, France), the ASIC3 pharmacological inhibitor,10 was coinjected in mice knees together with LPC16:0 (0.1 nmol APETx2 + 20 nmol LPC16:0), either at the first or the second intra-articular administration of LPC16:0.
2.3.4. Mechanical sensitivity
Mechanical sensitivity was evaluated by performing von Frey tests. Two types of von Frey tests were used, the classical up–down method of Dixon11 modified by Chaplan7 with von Frey filaments (Bioseb, Vitrolles, France) and the dynamic plantar aesthesiometer (Ugo Basile, Gemonio, Italy). For both methods, freely moving animals were placed in individual plastic boxes on a wire mesh surface, so that von Frey mechanical stimuli could be applied to the plantar surfaces of their hind paws. For up–down von Frey experiments, animals followed an habituation period before a set of calibrated von Frey filaments ranging from 0.02 to 1.4 g was applied to the plantar surface of each hind paw, alternatively or only on the ipsilateral hind paw, until withdrawal or licking of the paw to the stimulus. The 50% paw mechanical threshold was evaluated before (Baseline) and at different time points after the first and second LPC16:0 or vehicle injections, or after the CFA injection. For experiments using the dynamic plantar aesthesiometer, a ramp of force was applied through a single filament (up to 7.5 g in 10 seconds), and paw withdrawal thresholds (g) were measured in duplicate. A mean withdrawal threshold was then calculated for each animal hind paw. Three baseline measures were made prior LPC16:0 or vehicle intra-articular injections, the third baseline measure served as the reference mechanical withdrawal threshold for statistical analyses.
2.3.5. Heat sensitivity
Heat sensitivity in mice was assessed by the paw immersion test using a thermoregulated water bath maintained at 46.0 ± 0.2°C. Mice were maintained under a soft cloth except the ipsilateral hind paw, which was immersed in water until withdrawal (a cutoff of 30 seconds was used to avoid any potential tissue damage).
2.3.6. Weight-bearing experiments
Static weight-bearing apparatus (Bioseb) was used to evaluate spontaneous weight distribution between hind paws, which could be considered as a measure of ongoing pain. Following 2 training sessions to habituate animals, mice were placed in a plastic glass enclosure so that each hind paw rested on separate transducer pads. Once mice were settled in correct position, the force exerted by each hind paw was measured over a period of 5 seconds. Weight-bearing differences were recorded as an average of 4/5 trials and expressed as contralateral/ipsilateral ratio. The weight distribution was assessed before (baseline) and at different time points following vehicle, CFA or LPC injections.
2.3.7. Assessment of anxiety-like behaviors
2.3.7.1. Open field test
Mice were individually placed in the middle of an open field (50 × 50 × 50 cm) under dim light conditions (30 lux). The distance travelled and the velocity were automatically calculated during the 5 minutes of testing period as well as the time spent in the center for each mouse (Ethovision XT 13; Noldus, Wageningen, the Netherlands).
2.3.7.2. Elevated plus maze
The elevated plus maze (EPM) consists of 4 arms, 2 opposite open arms (37.5 × 5 cm) and 2 opposite closed arms (37.5 × 5 cm with 20 cm high walls), joined by a common central platform (5 × 5 cm), subjected to an equal illumination (30 lux). The maze was elevated to 60 cm above the floor. Each mouse was placed randomly into the center of the EPM facing an open arm. The time spent into the open arms, considered when head, gravity center and tail points were located within the arm, was automatically recorded for 5 minutes and calculated for each animal (Ethovision XT 13; Noldus).
2.3.7.3. Marble burying test
Briefly, mice were individually placed in the experimental cage (42.5 × 27.6 × 15.3 cm) containing 20 marbles (4 lines of 5 equidistant marbles) disposed on the top of 5-cm-thickness litter. Animals were left undisturbed during 30 minutes in a quiet room. At the end, mice were removed, and the numbers of buried marbles were quantified. A marble was considered as buried when at least 50% of its surface was covered with litter.
2.3.7.4. Hole board test
The hole board test consists of a board (39.5 × 39.5 cm) with 16 equidistant holes, 3 cm in diameter, equally distributed throughout the platform and placed 70 cm above the floor. Mice were individually placed randomly on one corner of the board facing away the experimenter and videotaped. The number of head dips in the holes was quantified for 5 minutes by a blind experimenter.
2.4. Immunohistochemistry on tibiotarsal joints and amygdala
At day 28, 4 and 5 mice of the vehicle and LPC16:0 10 nmol groups, respectively, were terminally anaesthetized using a mixture of ketamine/xylazine and quickly perfused transcardially with saline followed by 4% paraformaldehyde (PFA). Ipsilateral tibiotarsal joints were excised, post fixed in 4% PFA in phosphate buffer (0.1 M, pH 7.4) for 48 hours at 4°C and decalcified in 10% ethylenediaminetetraacetic acid (pH 7.6, Sigma) during 2 weeks at 4°C as previously described.5 Brain was also excised and post fixed in 4% PFA in phosphate buffer (0.1 M, pH 7.4) for 24 hours at 4°C. After cryoprotection (PB-Sucrose 30%) for at least 48 hours, samples were included in Tissue-Tek OCT cryo embedding compound.
Twenty-micrometer, cryostat-thick, frozen sections of joints were processed, mounted on Superfrost slides, blocked with phosphate buffered saline (PBS), bovine serum albumin (BSA) 1% and, incubated with primary antibodies in PBS + Triton 0.2% overnight at room temperature following 3 washes in PBS. Monocytes, macrophages, and osteoclasts lineage was labeled with a rat antibody against myeloid protein CD68 (1:1,000; AbD Serotec, #MCA1957, Oxford, United Kingdom) and peptide-rich sensory nerve fibers with a rabbit anti-calcitonin gene-related peptide (CGRP) (1:2,000, Calbiochem, Meudon, France, #PC205L). After washes, sections were incubated with the corresponding secondary antibody (1:1,000, AlexaFluor 488 and AlexaFluor 546 for CD68 and CGRP, respectively, Molecular Probes, Eugene, OR). After PBS washes, sections were then coverslipped with fluorescent mounting medium (Dako, Glostrup, Denmark) and observed with Nikon Eclipse Ni-E microscope. Quantitative analyses were performed with NIS-Elements software and at least 3 to 6 sections per joint per animal (n = 4-5 per group) were quantified using regions of interest (ROIs) on tibiotarsal joint. Results are presented as the mean intensity of signal per joint area in one ROI (2500 × 2500 µm) for CD68 and in 2 ROIs (570 × 570 µm) for CGRP.
Transverse sections (30 μm) of the brain containing the amygdala were cut on a cryotome (Microm HM450; Thermo Scientific, Illkirch, France). Free floating sections of the amygdala were stained for c-Fos immunohistochemistry as follows: after 3 washes in tris buffered saline (TBS) 0.05M pH 7.6, sections were incubated for 1 hour in a blocking solution (TBS 0.05M, BSA 3%, Triton 0.4%, donkey serum 1%) and then overnight at 4°C with a rabbit primary antibody anti-Fos (1:2000 in TBST; Santa Cruz, Santa Cruz, CA). After 3 washes in TBS 0.05M, sections were incubated for 2 hours with the appropriate secondary antibody (AlexaFluor 488 goat anti-rabbit IgG; 1:1000; Molecular Probes). Sections were then washed in TBS 0.05M, mounted on gelatin-coated slides and coverslipped with Dako fluorescent mounting medium. From each animal, 3 to 6 sections were randomly selected for counting c-Fos–positive cells in the ipsilateral side of the BLA (n = 4 per group) observed with Nikon Eclipse Ni-E microscope by a blinded investigator, and an average of these counts was taken.
2.5. Electronic microscopy
Potential demyelination of the saphenous nerve was assessed at day 7, after the 2 knee injections of 20 nmol of LPC16:0 or vehicle using electronic microscopy as previously described.35 Briefly, 4 animals in each group were terminally anesthetized and perfused with PAF4%/2.5% glutaraldehyde (diluted with 0.1 M sodium cacodylate buffer). A segment of the saphenous nerve was isolated proximal to the ipsilateral knee joint from vehicle and LPC16:0 groups and processed for electronic microscopy to evaluate the G ratio. This was calculated using the equation , where r is the internal axonal radius, and R is the total axonal radius.
2.6. Quantitative Polymerase Chain Reaction
At days 14 and 28, 4 to 6 mice from each of the different groups (LPC16:0 10 nmol at days 14 and 28, CFA only at day 14 or vehicle at days 14 and 28) were terminally anesthetized using a mixture of ketamine/xylazine, and ankle joints were dissected out and muscles and tendons trimmed. Samples were then flash frozen in liquid nitrogen and stored at −80°C until use. Frozen joints were pulverized with a biopulverizer (BioSpec, Bartlesville, OK) followed by RNA extraction with TRIzol using Tissue Lyser II (30 Hz frequency for 2 minutes with 2 cycles, Qiagen, Hilden, Germany). Equal amounts of RNA from each sample were reverse transcribed in 20 µL to produce complementary DNA (cDNA) using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) following the manufacturer's recommendation. SYBR Green–based detection was carried out on a real-time polymerase chain reaction instrument (Biorad CFX96 Touch, France) for Il6 (Interleukin-6; F-ACCGCTATGAAGTTCCTCTC, R-GTATCCTCTGTGAAGTCTCCTC), Tnf (tumor necrosis factor; F-GACCCTCACACTCAGATCATCTTCT, R-CCTCCACTTGGTGGTTTGCT), Apc5 (tartrate-resistant acid phosphatase; F-CACATAGCCCACACCGTTCTC, R-TGCCTACCTGTGTGGACATGA), and Tnfsf11 (receptor activator of nuclear factor kappa-B ligand; F-TCTCAGATGTCTCTTTTCGTCCAC, R- CTCAGTGTCATGGAAGAGCTG). In each experiment, samples were assayed in triplicate. Data were analyzed using the threshold cycle (Ct) relative quantification method. Relative quantities were determined using the equation: . RNA 18S (F-GCCGCTAGAGGTGAA, R-CATTCTTGGCAAATG) was used as housekeeping gene.
2.7. In vivo imaging
Inflammation and bone remodeling were assessed in vivo using an IVIS Spectrum small animal imaging system (Perkin Elmer, Waltham, MA). Each mouse received 2 nmol of MMPSense 680 (150 µL; Perkin Elmer) or Cat K 680 FAST (100 µL; Perkin Elmer) by intravenous route at day 7 and day 14, after LPC16:0 (10 nmol) or vehicle, to assess inflammation and bone remodeling, respectively (n = 5 in each group). Six hours or 24 hours later for Cat K 680 or MMP680, respectively, acquisition was performed under isoflurane anesthesia (1.5%). Imaging was performed using the ex/em = 675/720 nm band-pass filters. Quantification analysis was performed with Living Image Software (Perkin Elmer) using fixed-sized rectangular ROI focused on ipsilateral or contralateral tibiotarsal joint of the hind paws. Results are presented as the total counts of radiance efficiency (radiance photons per second per square centimeter per steradian) per incident excitation power (microwatt per square centimeter).
2.8. Cell culture and transfection
Human embryonic kidney (HEK) 293 cell line was grown in DMEM medium (Lonza BioWhittaker) supplemented with 10% of heat-inactivated fetal bovine serum (Biowest) and 1% of antibiotics (penicillin + streptomycin, Lonza BioWhittaker). One day after plating, cells were transfected with pIRES2-rASIC1a-EGFP, pIRES2-rASIC3-EGFP, or pIRES2-hASIC3a-EGFP vector using the JetPEI reagent according to the supplier's protocol (Polyplus transfection SA, Illkirch, France). Fluorescent cells were used for patch-clamp recordings 2 to 4 days after transfection.
2.9. Primary culture of dorsal root ganglia neurons
Lumbar dorsal root ganglia (DRG L1-L6) were rapidly dissected out from euthanized adult male C57Bl6J (Janvier Lab, at least 8 weeks of age) and ASIC3−/− mice, and placed in cold Ca-free/Mg-free HBSS medium (Corning) supplemented with 10 mM glucose and 5 mM 2-[4-(2-Hydroxyethyl)-1-piperazinyl]-ethanesulfonic acid (HEPES, pH 7.5 with NaOH). After removing the roots, DRGs were enzymatically dissociated at 37°C for 2 × 20 minutes in the HBSS solution supplemented with calcium (CaCl2 5 mM) and containing collagenase type II (Gibco, ThermoFisher, Les Ulis, France) and dispase (Gibco, France). Dorsal root ganglia were then mechanically triturated and washed in a complete neurobasal A (NBA) medium (Gibco Invitrogen supplemented with 2% B27, 2 mM L-glutamine and 1% antibiotics: penicillin + streptomycin, Lonza BioWhittaker), before being plated in 35-mm petri dishes. Neurons were then kept in culture at 37°C for 1 week with one-third of the culture medium renewed every 2 days (complete NBA supplemented with 10 ng/mL NT3, 2 ng/mL glial cell line-derived neurotrophic factor (GDNF), 10 ng/mL brain-derived neurotrophic factor (BDNF), 100 ng/mL nerve growth factor (NGF) and 100 nM retinoic acid) and were used for patch clamp experiments at least 2 days after plating.
2.10. Patch clamp experiments
The whole-cell configuration of the patch clamp technique was used to record either membrane currents (voltage clamp) or membrane potentials (current clamp). The patch pipettes were made by heating and pulling borosilicate glass tubes (Hilgenberg, Malsfeld, Germany) with a vertical P830 puller (Narishige, Tokyo, Japan), so that the pipette resistances were comprised between 3 and 7MΩ. Patch pipettes were filled with an intracellular solution containing either 135 mM KCl, 2.5 mM ATP-Na2, 2.1 mM CaCl2, 5 mM EGTA, 2 mM MgCl2, 10 mM HEPES, pH 7.3 with KOH for DRG neurons or 135 mM KCl, 5 mM NaCl, 5 mM EGTA, 2 mM MgCl2, 10 mM HEPES, pH 7.3 with KOH for HEK cells. Cells were bathed into an extracellular solution made of 145 mM NaCl, 5 mM KCl, 2 mM CaCl2, 2 mM MgCl2, and 10 mM HEPES for HEK cells, and this medium was supplemented with 10 mM glucose for DRG neurons. pH of extracellular solutions were adjusted to different values with NMDG-Cl, including the control pH 7.4 solution and the test pH 6.6 solution. During patch-clamp recordings, the cell under investigation was continuously superfused with extracellular solutions using a homemade 8-outlet system digitally controlled by solenoid valves (Warner Instruments, Hamden, CT), allowing rapid changes of the immediate cell environment from control to test solutions. Electrophysiological signals generated by patch-clamped cells were amplified and low-pass filtered at 2 kHz using an Axopatch 200B amplifier (Molecular Devices, Berkshire, United Kingdom), digitized with a 1550 A-D/D-A converter (Molecular Devices), sampled at 20 kHz and stored on a computer using Clampex software (V10.7, Molecular Devices). Analyses of these electrophysiological signals were then made offline using Clampfit software (V10.7, Molecular Devices). GFP+ HEK cells were considered as positively transfected when the peak amplitudes of pH6.6-evoked ASIC1a or ASIC3 currents were ≥ 200 pA. Nontransfected cells corresponded to GFP− cells with a pH6.6-evoked current < 200 pA. Dorsal root ganglia neurons were considered as ASIC+ when they exhibited a transient pH6.6-evoked current with a peak amplitude ≥ 20 pA; otherwise, they were considered as ASIC−.
2.11. In vivo electrophysiological recordings of spinal dorsal horn neurons
Single unit extracellular recordings of lumbar dorsal horn neurons were made using tungsten paralyn-coated electrodes (0.5MΩ impedance, WPI, Hertfordshire, United Kingdom). The tip of a recording electrode was initially placed at the dorsal surface of the spinal cord using a micromanipulator (M2E, France), and this initial position corresponded to 0 µm on the micromanipulator's micrometer. The electrode was then progressively moved down into the dorsal horn until the receptive field of a spinal neuron was localized on the ipsilateral plantar hind paw using mechanical stimulations including nonnoxious brushing and noxious pinching. In this study, experiments were focused on 2 types of spinal neurons that were distinguished as follow: (1) low threshold (LT) neurons only responding to non-noxious sensory stimulations and (2) high-threshold (HT) neurons that dynamically respond to noxious stimulations. The depth of spinal neurons recorded in this study was 80 ± 16 µm (range from 10 to 228 µm) and 202 ± 21 µm (range from 10 µm to 571 µm) for LT and HT neurons, respectively. Activities of neurons were sampled at 20 kHz, filtered (0.3-30 kHz band pass) and amplified using a DAM80 amplifier (WPI, Europe), digitized with a A/D-D/A converter (1401 data acquisition system, Cambridge Electronic Design, Cambridge, United Kingdom), and recorded on a computer using Spike 2 software (Cambridge Electronic Design).
2.12. Statistical analysis
Data are presented as mean ± SEM. Significant differences between the data sets are evaluated using P values, which were calculated using either parametric or nonparametric tests followed by adequate post hoc tests (see figure legends). Statistical analyses were performed using GraphPad Prism software. Association between VAS knee, VAS global, KOOS, and LPC16:0 content was assessed using analysis of variance using age, gender, BMI, and IL-6 as covariates. The analysis was performed using R 3.6.3. Differences between sets of data and correlations were considered significant when P values were less or equal to 0.05.
3. Results
3.1. Patients with painful joint diseases exhibit high levels of knee synovial LPC16:0
The knee synovial fluids of 35 OA patients from a first cohort (supplementary table 1, available at http://links.lww.com/PAIN/B579) displayed significantly higher concentrations of LPC compared with control specimens (77.4 ± 5.0 µM and 40.0 ± 3.4 µM, respectively; Fig. 1A), with LPC16:0 being the most represented species and the only one significantly increased compared with control specimens (35.2 ± 2.4 µM vs 16.3 ± 1.5 µM; Fig. 1B). Correlation studies were conducted between LPC16:0 concentrations and pain outcomes, including VAS (global and knee VAS) and KOOS, with data adjusted for patients' age, gender, BMI, and synovial fluid IL-6 level (supplementary table 2, available at http://links.lww.com/PAIN/B579). Significant correlations between LPC16:0 concentrations and pain (VAS) were observed (P = 0.016 or P = 0.031 for VAS knee and VAS global, respectively, supplementary table 2, available at http://links.lww.com/PAIN/B579), and an inverse correlation with KOOS questionnaire almost reached statistical significance (P = 0.060, supplementary table 2, available at http://links.lww.com/PAIN/B579). Including or excluding IL-6 levels in the analysis of variance did not have drastic impact on the association between LPC16:0 and pain measures (P = 0.016/0.019, P = 0.031/0.066 and P = 0.060/0.080 for VAS knee, VAS global and KOOS, respectively). In line with this, there were no significant correlations between LPC16:0 and IL-6 levels in synovial fluids (supplementary table 2, available at http://links.lww.com/PAIN/B579), indicating that LPC16:0 involvement in patient pain outcomes was not associated to an IL-6–dependent inflammation in this first OA cohort. Previously, OA symptom severity in this cohort, including pain, correlated with IL-6 levels in the synovial fluid but was inversely associated to IL-6 levels in the cerebrospinal fluid.26
Figure 1.
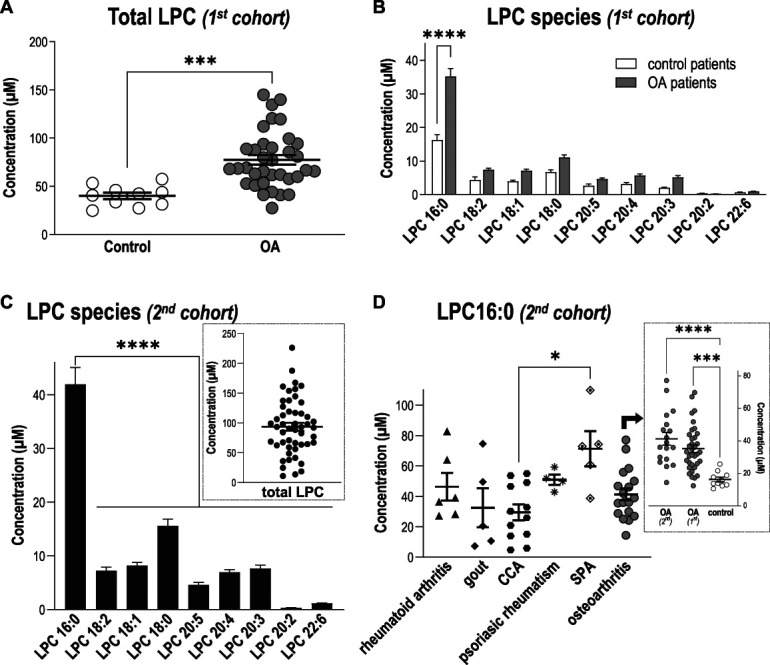
Levels of LPC16:0 are increased in synovial fluids of patients suffering from joint pain. Quantifications of LPC species were performed in knee human synovial fluid (HSF) samples. Total lipids were extracted from HSF samples and analyzed by direct infusion in mass spectrometry (MS) using an electrospray ionization source (ESI) in the positive ion mode (see “Methods” section for details). (A) Comparison of total LPC concentrations (µM) between a first cohort of patients with osteoarthritis (OA, n = 35 patients) and postmortem control subjects (n = 10), showing significantly higher levels of LPC in patients compared with control specimens (***P = 0,0003, unpaired t test). (B) Distribution of the different LPC species concentrations (µM) in the HSF of OA patients and postmortem control specimens. Although mean concentrations of most LPC species were higher in the HSF of OA patients, LPC16:0 was the most abundant and the only one to be significantly elevated compared with postmortem control subjects (***P < 0.0001, 2-way ANOVA followed by a Sidak multiple comparison test). (C) Distribution of the different LPC species concentrations (µM) in the HSF of a second cohort of patients suffering from different joint pathologies (n = 50), showing significantly higher level of LPC16:0 compared with the others species (****P < 0.0001, One-way ANOVA followed by a Dunnet multiple comparison test). Inset: Total LPC concentration in HSF of the second cohort of patients. (D) LPC16:0 concentrations in the different subgroups of patients from the second cohort: rheumatoid arthritis (RA, n = 6), gout (n = 5), CCA (n = 12), psoriatic arthritis (n = 4), SPA (n = 5), and OA (n = 18). The LPC16:0 concentrations in all the different subgroups of patients are comparable, except a difference between CCA and SPA (*, P < 0.05, Kruskal–Wallis test followed by a Dunn multiple comparison test). Inset: LPC16:0 concentrations in HSF of OA patients from the first and second cohort are similar and higher than control postmortem (***P < 0.001 and ****P < 0.0001, 1-way ANOVA followed by a Tukey multiple comparison test). ANOVA, analysis of variance; CCA, chondrocalcinosis; HSF, human synovial fluid; LPC, lysophosphatidylcholine; OA, osteoarthritis; SPA, spondyloarthritis.
We next assessed whether increase of LPC16:0 knee synovial fluid levels could be the hallmark of painful joint pathologies associated with inflammation in a second heterogeneous cohort of patients with different joint diseases (Figs. 1C and D; supplementary table 3, available at http://links.lww.com/PAIN/B579). The data from these 50 patients, irrespective of the disease, showed a distribution of LPC species (Fig. 1C) similar to the first OA cohort (Fig. 1B), with a total LPC concentration of 93.6 ± 6.7 µM (Fig. 1C, inset), significantly higher than control specimens (Fig. 1A, P = 0.0007, unpaired t test). Lysophosphatidylcholine 16:0 was also the major contributor to this elevated LPC level, with an average concentration of 41.9 ± 3.1 µM (Fig. 1C) significantly higher than control specimens (Fig. 1B, P = 0.0006, unpaired t test). When the analysis was made according to the patients' diseases (Fig. 1D and supplementary Fig. 1, available at http://links.lww.com/PAIN/B579), LPC16:0 levels in the different subgroups of patients were similar, except a difference between CCA and SPA patients. Importantly, OA patients from this second cohort displayed an elevated level of LPC16:0 similar to the first OA cohort and significantly different from control specimens (Fig. 1D, inset).
3.2. Intra-articular LPC16:0 injections induce persistent pain and anxiety-like behaviors in mice
As LPC16:0 concentration was higher in the synovial fluids of joint pain patients (Fig. 1), we investigated its potential pronociceptive and associated anxiogenic effects in mice. Two ankle intra-articular injections of LPC16:0 were made 5 days apart (Figs. 2A–E and supplementary Fig. 2, available at http://links.lww.com/PAIN/B579), and a single ankle CFA injection was used as a positive control. The dose of LPC16:0 injected (10 nmol) corresponded to the amount of lipids we had previously shown to generate acute pain-like behaviors following intraplantar injection in mice,32 and it was in the lower range of quantities assessed here in patient synovial fluids. The first LPC16:0 injection induced short-lasting ipsilateral mechanical allodynia compared with vehicle, whereas the second injection led to a long-lasting mechanical allodynia until the end of the protocol (day 28), which remained ipsilateral, similar to CFA (Fig. 2B and supplementary Fig. 2A, available at http://links.lww.com/PAIN/B579) and consistent with models based on local intra-articular injections in mice. The second injection was also associated to weight-bearing deficit and thermal hyperalgesia (Fig. 2C and supplementary Fig. 2B, available at http://links.lww.com/PAIN/B579). Importantly, the effect of intra-articular LPC16:0 was not due to its conversion to LPA by autotaxin because neither cotreeatment nor posttreatment with an autotaxin inhibitor (S32826) altered its pronociceptive effect (Figs. 2D and E). Because joint pain is often associated with comorbidities such as anxiety,36 we therefore assessed anxiety-like behaviors in our model of LPC16:0-induced persistent joint pain at days 19 to 23 (Figs. 2F and G and supplementary Fig. 2C-E, available at http://links.lww.com/PAIN/B579). LPC16:0 or CFA induced a significant decrease in the time spent in the center of an open field without any locomotor impairment (Fig. 2F and supplementary Fig. 2C), indicating increased thigmotactism. This is in good agreement with what was shown previously on locomotor impairments following CFA intraplantar injections.42 The development of anxiety-like behaviors was demonstrated in both LPC16:0 and CFA groups in the EPM, hole board, and marble burying tests (Fig. 2G, supplementary Fig. 2D–E, available at http://links.lww.com/PAIN/B579) and was associated to increased neuronal activity within the amygdala (supplementary Fig. 3, available at http://links.lww.com/PAIN/B579).
Figure 2.
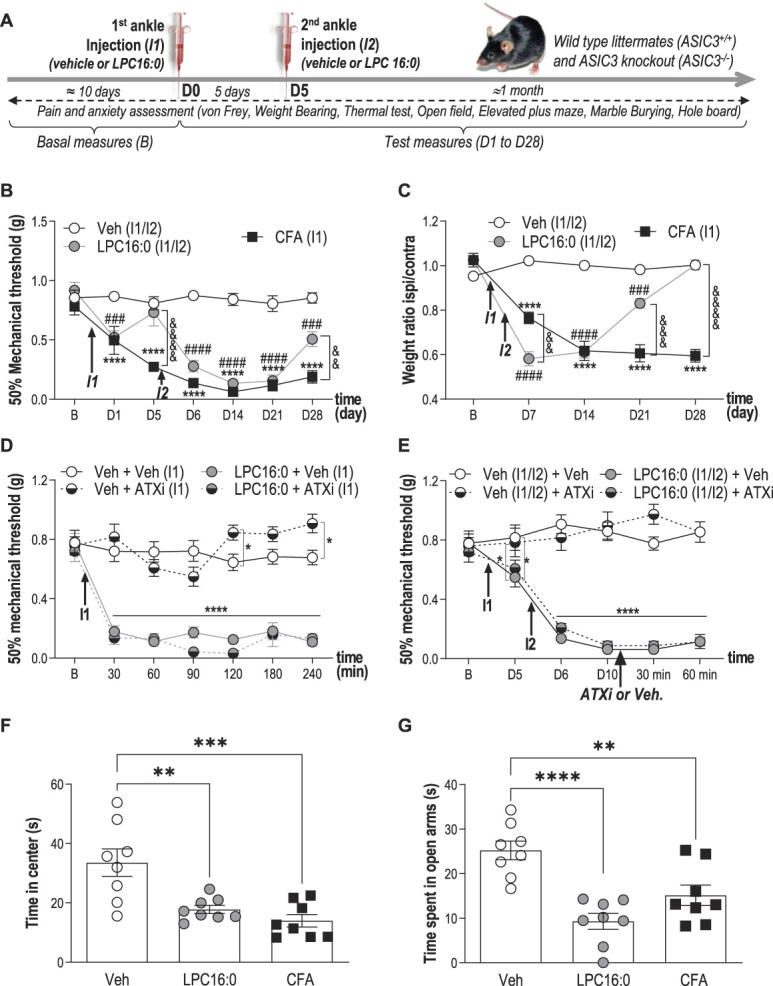
Lysophosphatidylcholine injections into mouse ankle joints produce long-lasting pain-like behaviors associated with anxiety-related behaviors. (A) Timeline of the experimental procedures used. (B) Effect of intra-articular ankle injections of LPC16:0 (10 nmol), CFA or vehicle (Veh) on mechanical allodynia in male mice. Ipsilateral mechanical paw withdrawal threshold was assessed using the up and down method with von Frey filament from D1 to D28. Results are expressed as 50% mechanical threshold (n = 8-16 mice per group; ****P < 0.0001 for Veh vs CFA; ###P < 0.001 and ####P < 0.0001 for Veh vs LPC16:0; &&P < 0.01 and &&&&P < 0.0001 for CFA vs LPC16:0; 2-way ANOVA followed by a Tukey post hoc test). (C) Effect of intra-articular injections of LPC16:0 (10 nmol), CFA or vehicle on weight-bearing in male mice. Results are expressed as the weight ratio between the ipsilateral and contralateral hind paws (n = 8 mice per group; ****P < 0.0001 for Veh vs CFA; ###P < 0.001 and ####P < 0.0001 for Veh vs LPC16:0; &&P < 0.01, &&&P < 0.001, and &&&&P < 0.0001 for CFA vs LPC16:0; 2-way ANOVA followed by a Tukey post hoc test). (D) The autotaxin inhibitor S32826 (ATXi) was co-injected at the dose of 10 nmol with LPC16:0 (10 nmol, I1) and mechanical paw withdrawal thresholds were assessed from 30 to 240 minutes post injection (n = 8 mice per group, *P < 0.05 and ****P < 0.0001, 2-way ANOVA followed by a Tukey post hoc test). (E) S32826 was injected at D10, ie, after the 2 injections of LPC16:0 (10 nmol) 5 days apart (I1/I2), in the same ankle joint, and mechanical paw withdrawal thresholds were assessed at 30 and 60 minutes post injection (n = 8 per group, *P < 0.05 and ****P < 0.0001, 2-way ANOVA followed by a Tukey post hoc test). (F–G) Effect of intra-articular injections of LPC16:0 (10 nmol), CFA or vehicle on the time spent in the center of an open field test at D20 (F) and on the time spent in the open arms in the elevated plus maze test at D23 (G). The open field and elevated plus maze test lasted 5 minutes and the time spent in the center and the open arms, respectively, was automatically calculated using Ethovision XT 13 (Noldus). One-way ANOVA followed by Tukey post hoc tests with **P < 0.01, ***P < 0.001, and ***P < 0.0001 (n = 8 mice per group). ANOVA, analysis of variance; CFA, complete Freund adjuvant; LPC, lysophosphatidylcholine.
3.3. Intra-articular LPC16:0 does not cause peripheral nerve sprouting, inflammation, or bone alterations
No peptidergic nerve fibers sprouting (CGRP staining, Fig. 3A) nor cell infiltration (CD68+ macrophage lineage cells influx, Fig. 3B) were observed in the joint of LPC16:0 mice compared with control at day 28. When potential LPC16:0-induced joint inflammation was evaluated, no fluorescence increase of the MMP680 matrix metalloprotease marker was observed at day 7 in the ipsilateral joint of LPC16:0 mice compared with the contralateral joint and control mice (Fig. 3C). A potential LPC16:0-induced bone remodeling was also investigated and no increase in the cathepsin K activity was detected at day 14 in the ipsilateral joints of LPC16:0 mice compared with the contralateral ones and control mice (Fig. 3D). In good agreement with these observations, II6 and Tnf messenger RNA levels in the ipsilateral joint of LPC16:0-injected mice were similar to controls at days 14 and 28, whereas an increase was observed in CFA mice, as expected (Figs. 3E and F). Finally, Apc5 and Tnfsf11 messenger RNA levels in the joints of LPC16:0 mice were also similar to control at days 14 and 28 (Figs. 3G and H). These results indicate no change in peripheral nerve sprouting, bone alterations, or apparent inflammation following intra-articular LPC16:0 in the subacute and chronic stages of pain behaviors.
Figure 3.
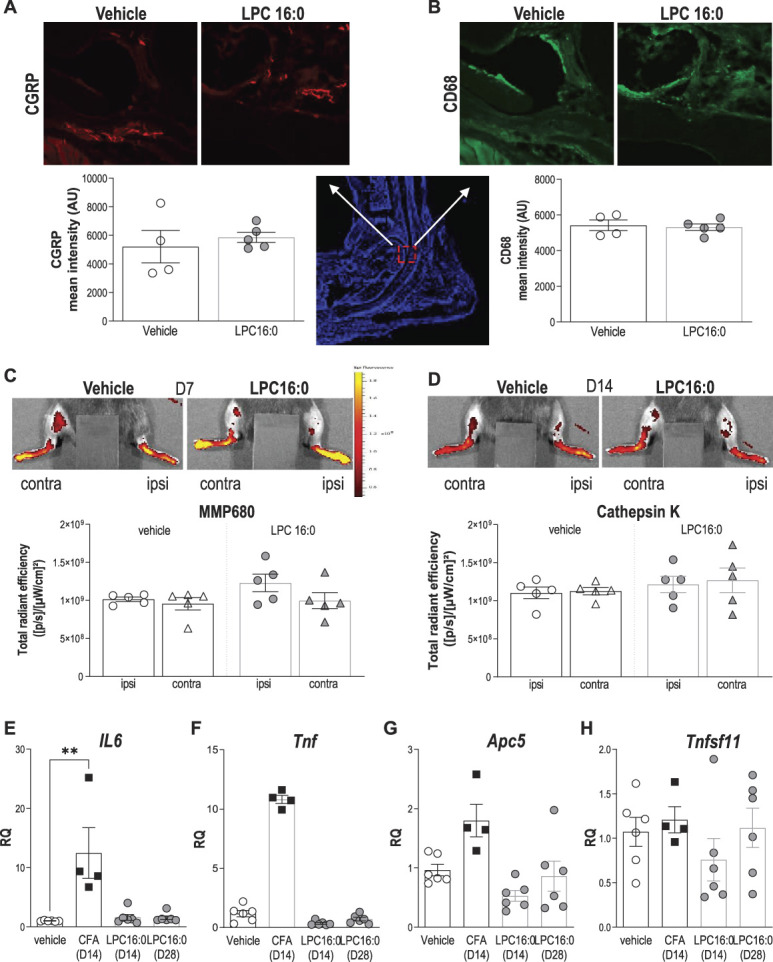
LPC16:0 injections into mouse ankle are not associated to peripheral nerve sprouting, inflammation, nor bone alterations. (A–B) Analysis of CGRP and CD68 expression in the ipsilateral tibiotarsal joint at D28, following intra-articular administrations of LPC16:0 (10 nmol) or vehicle in male mice. Top, representative photomicrographs of CGRP (A) and CD68 (B) staining in vehicle and LPC16:0-treated mice. Bottom, quantification of CGRP-positive fibers (A) in 2 ROIs (570 × 570 µm) and of CD68-positive cells (B) in one ROI (2500 × 2500 µm) from the ipsilateral tibiotarsal joint of vehicle and LPC16:0-injected mice. Results are expressed as a mean intensity (n = 4 for each group, no significant differences, Mann–Whitney tests). (C–D) Evaluation of the effect of LPC16:0 (10 nmol) or vehicle on inflammation and bone remodeling using in vivo fluorescent imaging for metalloprotease (MMP680) activity at D7 (C) and cathepsin K activity at D14 (D), respectively, in the ipsilateral and contralateral hind paws of male mice. Upper panels, illustrations; Lower panels, MMP680 ratio quantification (C) and cathepsin K activity ratio quantification (D). Data from n = 5 animals per group (no significant differences, Kruskal–Wallis tests followed by Dunn multiple comparison tests). (E–H) Relative levels of IL6 (E), Tnf (F), Apc5 (TRAP) (G), and Tnfsf11 (RANK-L) (H) messenger RNA expressed in the ipsilateral joint, determined by quantitative polymerase chain reaction after intra-articular injections of LPC16:0 (10 nmol, D14 and D28), CFA (D14), and vehicle (D14/D28; n = 4-6 for each group, **P < 0.01, Kruskal–Wallis tests followed by Dunn multiple comparison tests). CFA, complete Freund adjuvant; LPC, lysophosphatidylcholine; ROI, region of interest.
3.4. The in vivo pronociceptive effects of LPC16:0 are largely dependent on acid-sensing ion channel 3 in a sex-independent manner
LPC16:0 activated rat and human ASIC3 in transfected HEK293 cells32 (supplementary Fig. 4A–C, available at http://links.lww.com/PAIN/B579) and induced an ASIC3-dependent depolarizing current in mouse DRG neurons (Figs. 4A–D). The in vivo involvement of this channel in pain and anxiety-like behaviors following intra-articular LPC16:0 injections was thus investigated using wild-type (ASIC3+/+) and ASIC3-knockout (ASIC3−/−) mice (Fig. 5 and supplementary Fig. 5, available at http://links.lww.com/PAIN/B579). LPC16:0 injections induced long-lasting mechanical allodynia and thermal hyperalgesia in both male and female ASIC3+/+ mice, indicating no sex dimorphism (fig. 5A-B and supplementary Fig. 5A-B, available at http://links.lww.com/PAIN/B579). These pain behaviors were significantly reduced in both male and female ASIC3−/− mice. The persistent LPC16:0-induced pain state was also associated to anxiety-like behaviors assessed between days 19 and 23 in male and female ASIC3+/+ mice, which were significantly reduced in ASIC3−/− mice, as shown in the EPM and marble burying tests (Fig. 5C-F). Finally, LPC16:0 did not alter locomotor behaviors in both ASIC3+/+ and ASIC3−/− mice at day 20 (supplementary Fig. 5C, D, available at http://links.lww.com/PAIN/B579). Thus, the development of LPC16:0-induced pain and associated anxiety-like behaviors were mediated by ASIC3 in both sexes.
Figure 4.
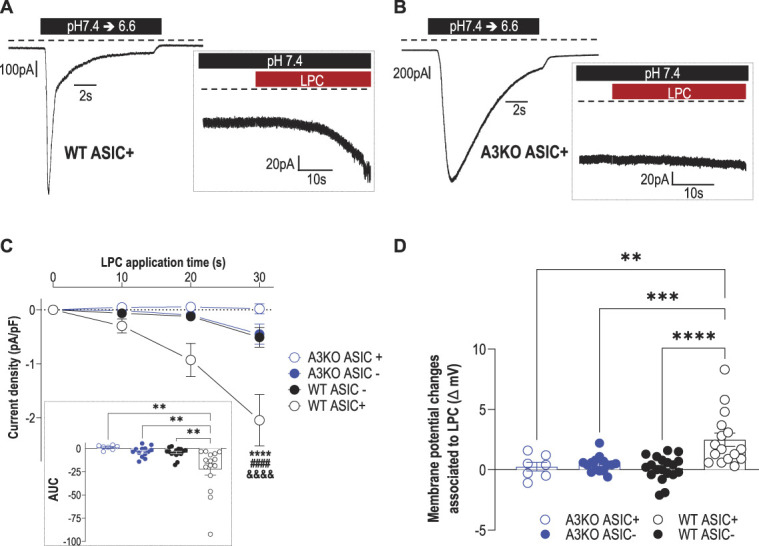
LPC16:0 induces a noninactivating, ASIC3-dependent current associated with membrane depolarization in mouse DRG neurons. Whole-cell patch clamp experiments performed on WT and ASIC3 knockout (A3KO) DRG neurons expressing (ASIC+) or not expressing (ASIC−) a pH6.6-evoked ASIC current. (A–B) Native pH6.6-evoked ASIC currents recorded at −80 mV in DRG neurons from wild-type (WT ASIC+, A) and ASIC3-knockout (A3KO ASIC+, B) mice. Insets show the effect of LPC16:0 (5 µM) applied for 30 seconds on the same neurons. (C) Analysis of the current densities measured after 10-, 20-, and 30-second applications of LPC16:0 onto WT ASIC+, WT ASIC−, A3KO ASIC+, and A3KO ASIC- neurons (see methods, n = 7-16; ****P < 0.0001 compared with WT ASIC-, ####P < 0.0001 compared with A3KO ASIC+ and &&&&P < 0.0001 compared with A3KO ASIC-; Three-way ANOVA followed by a Tukey multiple comparison test). Inset shows the AUC calculated over 30-second period from the baseline fixed at 0 (n = 7-16, **P < 0.01, 1-way ANOVA followed by a Tukey multiple comparison test). (D) Membrane potential changes associated with 30-second application of LPC16:0 on WT ASIC+, WT ASIC−, A3KO ASIC+, and A3KO ASIC- neurons (n = 7-19; **P < 0.01, ***P < 0.001, and ****P < 0.0001, 1-way ANOVA followed by a Tukey multiple comparison test). ANOVA, analysis of variance; ASIC3, acid-sensing ion channel 3; DRG, dorsal root ganglia; LPC, lysophosphatidylcholine; WT, wild‐type.
Figure 5.
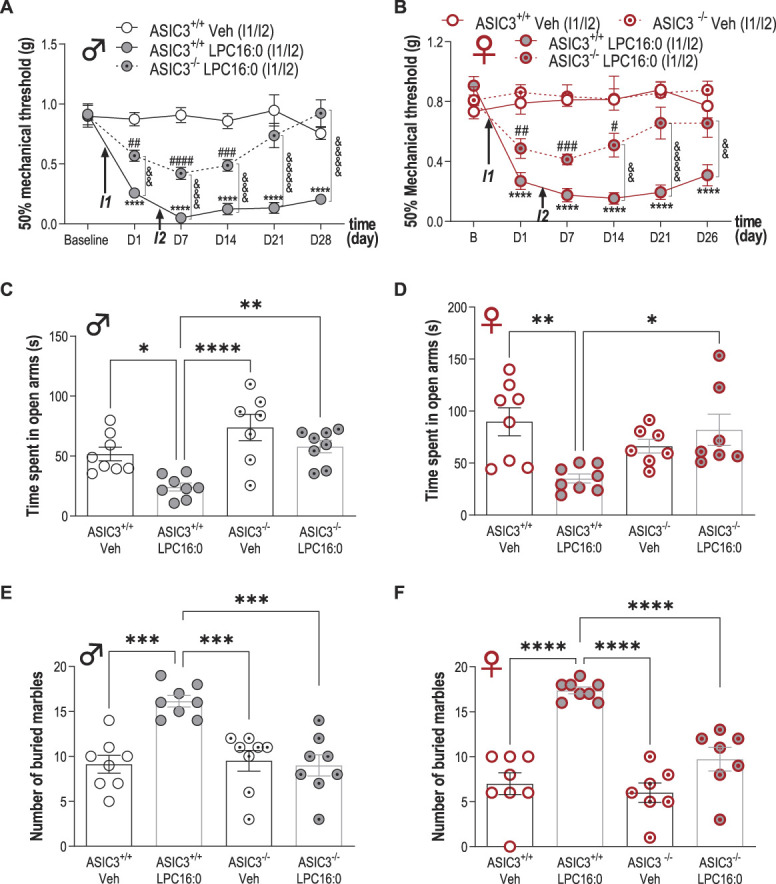
Acid-sensing ion channel 3 is crucial for the development of both pain and anxiety-like behaviors induced by LPC16:0 injections into mouse ankle joints of male and female mice. Time course effect of intra-articular ankle injections of LPC16:0 (10 nmol) or vehicle in male (A) or female (B) ASIC3+/+ and ASIC3−/− mice. Mechanical paw withdrawal threshold was assessed using the up and down method with von Frey filaments from D1 to D28 and from D1 to D26, for male and female mice, respectively (n = 7-8 mice per group, ****P < 0.0001 for ASIC3+/+ Veh vs ASIC3+/+ LPC16:0; #P < 0.05, ##P < 0.01, ###P < 0.001, and ####P < 0.0001 for ASIC3−/− LPC16:0 vs ASIC3+/+ Veh (♂) or ASIC3−/− Veh (♀); &&P < 0.01 and &&&&P < 0.0001 for ASIC3+/+ LPC16:0 vs ASIC3−/− LPC16:0; 2-way ANOVA followed by Tukey post hoc tests). In (A), both male ASIC3+/+ and male ASIC3−/− mice (Supplementary Fig. 5E, available at http://links.lww.com/PAIN/B579), did not exhibit any changes in mechanical paw withdrawal threshold following vehicle injections. (C–F) Effect of intra-articular ankle injections of LPC16:0 (10 nmol) or vehicle on the time spent in the open arm in the elevated plus maze test at D23 (C–D), on the number of buried marble in the marble burying test at D19 (E–F), in male (C–E) or female (D–F) ASIC3+/+ and ASIC3−/− mice. The elevated plus maze test lasted 5 minutes and the time spent in the open arm was automatically calculated using Ethovision XT 13 (Noldus). The marble burying test lasted 30 minutes and was analyzed by a blind experimenter (n = 7-8 mice per group, *P < 0.05, **P < 0.01, and ****P < 0.0001, 1-way ANOVA followed by Tukey post hoc tests). ANOVA, analysis of variance; LPC, lysophosphatidylcholine.
3.5. LPC16:0 injections into mouse knee joint also produce persistent pain-like behaviors, which are associated to spinal neuron sensitization
LPC16:0 was injected into mouse knee joint to determine whether the results obtained with the ankle can be extended to other joints (Fig. 6 and supplementary Fig. 6, available at http://links.lww.com/PAIN/B579). Mice injected in the knee (Fig. 6A) with LPC16:0 at 10 or 20 nmol developed dose-dependent mechanical allodynia (Fig. 6B), weight-bearing deficit, and thermal hyperalgesia (supplementary Fig. 6A and B, available at http://links.lww.com/PAIN/B579), associated with anxiety-like behaviors (supplementary Fig. 6C and D) not because of locomotor impairment (supplementary Fig. 6E, available at http://links.lww.com/PAIN/B579). We also did not observe any saphenous nerve demyelination33,47 following 20 nmol of LPC16:0 knee injections (supplementary Fig. 6F, available at http://links.lww.com/PAIN/B579). When mechanical thresholds were assessed with the dynamic plantar aesthesiometer test, LPC16:0 knees injections (20 nmol) also induced long-lasting ipsilateral allodynia in both male and female wild-type mice, which was clearly secondary to the injection site (Fig. 6C, supplementary Fig. 6G, available at http://links.lww.com/PAIN/B579). As for the ankle joint, LPC16:0 knee injection–induced pain was significantly reduced in both male and female ASIC3−/− mice compared with WT (Fig. 6D). This long-lasting pain state was also significantly reduced in male WT when APETx2, an ASIC3 blocker,10,32 was coinjected with LPC16:0 either at the first or the second administration, further supporting an effect primarily mediated through peripheral ASIC3 activation (Fig. 6E). Finally, 2 LPC16:0 intra-articular injections were necessary to induce persistent pain as for the ankle (supplementary Fig. 6H, available at http://links.lww.com/PAIN/B579). These results support a similar LPC-induced ASIC3-dependent persistent pain state upon 2 intra-articular knee or ankle injections of LPC16:0, suggesting a central sensitization process. The activity of spinal dorsal horn neurons was therefore recorded in vivo following LPC16:0 knee injections in WT mice (Figs. 6F–H). Electrophysiological recordings clearly demonstrated a sensitization of spinal HT but not LT neurons associated to LPC16:0 knee injections, which correlated with persistent secondary allodynia.
Figure 6.
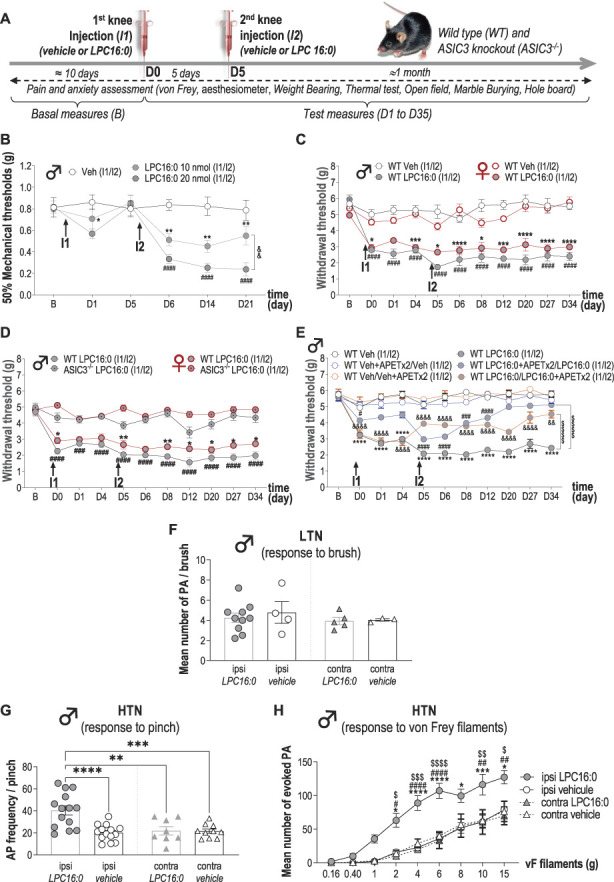
Lysophosphatidylcholine knee injections generate persistent secondary mechanical allodynia associated with sensitization of spinal dorsal horn neuron activity. (A) Timeline of the experimental procedures used. (B) Effect of intra-articular knee administrations of LPC16:0 (10 and 20 nmol), or vehicle (Veh) on mechanical allodynia in the ipsilateral paw of male mice. Paw withdrawal thresholds (PWTs) were assessed using the up and down method with von Frey filament from D1 to D21. Results are expressed as 50% mechanical thresholds (n = 8 mice per group; *P < 0.05 and **P < 0.01 for LPC16:0 10 nmol vs Veh, ####P < 0.0001 for LPC16:0 20 nmol vs Veh and &&P < 0.01 for LPC16:0 20 nmol vs LPC16:0 10 nmol; 2-way ANOVA followed by a Tukey post hoc test). (C) Ipsilateral paw PWTs of female and male WT mice injected twice in the knees with LPC16:0 (20 nmol) or vehicle (Veh) were assessed using a dynamic plantar aesthesiometer (n = 6 mice per group; *P < 0.05, ***P < 0.001, and ****P < 0.0001 for WT female Veh vs WT female LPC16:0; ####P < 0.0001 for WT male Veh vs WT male LPC16:0; 3-way ANOVA test followed by a Tukey multiple comparison test). (D) Ipsilateral PWTs of female and male, WT and ASIC3−/− mice injected twice with LPC16:0 (n = 5-18 mice per group; *P < 0.05, and **P < 0.01 for female WT LPC16:0 vs ASIC3−/− LPC16:0; ###P < 0.001, and ####P < 0.0001 for male WT LPC16:0 vs ASIC3−/− LPC16:0; 3-way ANOVA test followed by a Tukey multiple comparison test). (E) Ipsilateral PWTs of male WT mice injected twice with LPC16:0 or vehicle, with or without coinjection of APETx2 (0.1 nmol) at the first or the second injection (n = 6-12 per group; ****P < 0.0001 for WT Veh vs WT LPC16:0; #P < 0.05, ###P < 0.01, and ####P < 0.0001 for WT Veh + APETx2/Veh vs WT LPC16:0 + APETx2/LPC16:0; &&P < 0.01, and &&&&P < 0.0001 for WT Veh/Veh+APETx2 vs WT LPC16:0/LPC16:0 + APETx2; 2-way ANOVA test followed by a Tukey multiple comparison test; $$$$P < 0.0001 for main effects on curves, 2-way ANOVA test followed by a Tukey multiple comparison test). (F) Mean number of action potentials (APs) emitted by spinal low threshold neurons (LTNs) following light brushing of their receptive fields, located on the plantar surface. Spinal LTN-evoked activities were not different whether they were recorded ipsilaterally or contralaterally to the knees injected twice with LPC16:0 or vehicle (n = 3-10 LTNs, no significant differences, Kruskal–Wallis test followed by a Dunn post hoc test). (G) Frequencies of APs emitted by spinal HTNs following pinching (n = 8-14 HTNs; **P < 0.01, ***P < 0.001, and ****P < 0.0001, 1-way ANOVA followed by a Tukey multiple comparison test). (H) Mean number of APs evoked by spinal HTNs following application of von Frey filaments of increasing strength (n = 8-14 HTNs; *P < 0.05, ***P < 0.001, and ****P < 0.0001 for ipsi LPC16:0 vs ipsi vehicle; #P < 0.05, ##P < 0.01, and ####P < 0.0001 for ipsi LPC16:0 vs Contra LPC16:0; $P < 0.05, $$P < 0.01, $$$P < 0.001, and $$$$P < 0.0001 for ipsi LPC16:0 vs contra vehicle; 3-way ANOVA test followed by a Tukey multiple comparison test). ANOVA, analysis of variance; ASIC3, acid-sensing ion channel 3; LPC, lysophosphatidylcholine; WT, wild type.
4. Discussion
Here, we identify LPC16:0 as the major LPC species in HSFs and a critical trigger of chronic joint pain through peripheral ASIC3 channel activation in mice. Compared with control specimens, the knee synovial fluids from a first cohort of OA patients displayed an elevated LPC16:0 content, which correlated with their knee and global pain (VAS knee and VAS global), regardless of age, gender, BMI, and IL-6 synovial fluid levels. Correlation with knee injury and osteoarthritis outcome score (KOOS) almost reach statistical significance (P = 0.060). This absence of a statistically significant correlation can be explained by the more holistic nature of the patient's overall OA experience assessed by this questionnaire. A specific increase in LPC16:0 content was further confirmed in a second cohort of patients with different joint diseases, including OA but also other pathologies such as RA, suggesting that LPC is probably not the hallmark of a particular joint pathology. Previous works also reported an increase in plasma and/or synovial LPC contents in OA and RA patients27,28,57 but without any information on its possible contribution to chronic joint pain.
We demonstrate that ankle or knee intra-articular administrations of LPC16:0 in mice led to the development of chronic pain as well as anxiety-like behaviors, independently of the LPC-to-LPA conversion by autotaxin, excluding a role of LPA in these effects. Our data support a direct role of LPC16:0 as a triggering factor of pain-like behaviors in this new preclinical model of persistent joint pain. Because the levels of LPC16:0 correlated with pain outcomes in OA patients, these data could be also relevant in humans. The persistent pain state in mice did not seem to be associated with nerve sprouting, demyelination, or inflammatory processes because no changes in CGRP joint staining, nerve fiber G ratio, in vivo metalloprotease activity, or expression of TNF and IL-6 were observed in joints following LPC16:0 administrations. Hung et al.18 also failed to observe any inflammatory signs following intramuscular administration of LPC16:0 in mice. The elevated level of LPC, especially LPC16:0, in the synovial fluid of OA patients could therefore explain why they still suffered from chronic joint pain despite well controlled inflammation and possibly even in the absence of it, as supported here by the lack of correlation between LPC16:0 and IL-6 levels in patient synovial fluids. No change in cathepsin K activity and joint expression of TRAP and RANK-L was observed in our model, indicating that LPC16:0 also did not lead to bone alterations/remodeling, which could contribute to chronic joint pain as already suggested in rheumatic diseases such as OA34,40,58 and RA.24 This might explain the lack of clear correlation between joint damage and pain in OA.3,16
Chronic joint pain associated with rheumatic diseases is generally considered to be mainly under inflammatory condition. However, there is a frequent disconnect between disease activity such as inflammation and chronic joint pain especially in RA.29 Indeed, despite the success of conventional disease-modifying antirheumatic drugs, alone or in combination with biologic agents, on disease activity, a significant number of patients still complain from persistent pain, and more than 60% are not satisfied by their pain management.37,49 In addition, joint pain is an early marker of an emerging RA before any evident sign of inflammation.6 This discrepancy between disease activity and pain indicates that synovitis is not the only driver of joint pain at least in RA1,25 but that additional mechanisms are at play. Our LPC-induced chronic joint pain model in mice is actually not a model of rheumatic disease per se because it is not associated with any evident signs of inflammation nor joint damages, but rather a model of joint pain (arthralgia), one of the main symptoms of rheumatic diseases. We show that LPC within joints is a critical triggering factor of pain but is not sufficient by itself to mimic the whole clinical status of a particular rheumatic disease. This latest point is actually an advantage because it allows identification of a new potential noninflammatory nociceptive pathway in rheumatic diseases besides the well-described inflammatory mechanisms. Both inflammatory and noninflammatory components could therefore participate in systemic pain and its chronicity in rheumatic diseases, but their respective contributions and/or triggering roles still remain to be determined. In addition, our data indicate that intra-articular LPC16:0 administrations are not associated to joint inflammatory processes in the subacute stage of pain behaviors, but we cannot completely rule out an effect of inflammation in the hyperacute stage following intra-articular LPC injection.
The pronociceptive effects of LPC16:0 following joint administration in mice are dependent on peripheral ASIC3 channel in a sex-independent manner. Indeed, both pain and anxiety-like behaviors were significantly reduced in both male and female ASIC3 KO mice, or following local pharmacological inhibition of these channels. The contribution of ASIC3 in the development of pain-like behaviors evoked by LPC16:0 appeared to be more pronounced following injections into the knee than into the ankle of mice. Such a difference might illustrate a predominant role of ASIC3 in the development of secondary hypersensitivity, as proposed previously.20,44 Because pain measurements were taken at the plantar level, they were clearly associated to secondary pain following knee injections; however, they more likely reflected a mixture of primary and secondary pain for ankle injections. ASIC3, which was constitutively activated by LPC16:0 in cultured DRG neurons, induced a sustained membrane depolarization similar to the ASIC3-dependent depolarization we already shown to sensitize these neurons.8 It is thus very likely that intra-articular injections of LPC16:0 could drive an ASIC3-dependent nociceptive input leading to a persistent pain state originating from joints. ASIC3 has been already associated to long-lasting pain states following repeated acid injections in rodent joints48 and muscles44 and more recently following LPC injections in mouse muscle.18 The stimulation of ASIC3 channels in sensory nerve endings of deep tissues appears to be a key step for the development of chronic pain. The effect of LPC16:0 on ASIC3 expressed in joint nociceptors probably results in the sensitization of dorsal horn neurons as supported by the enhanced output of spinal HT nociceptive, but not LT, neurons. Central sensitization was also proposed to occur after repeated intramuscular injections of LPC16:0 in mice, which induced increased expression of c-fos and pERK in spinal dorsal horn neurons.18 Moreover, several clinical studies demonstrated the presence of central sensitization in OA patients.30
To conclude, LPC16:0 could be involved in joint pain from different etiologies of inflammatory and noninflammatory origins and more generally across rheumatic musculoskeletal diseases. However, it remains to be demonstrated whether an elevated level of LPC16:0 could be used as an objective biomarker of chronic joint pain across different etiologies. Our data bring both human and rodent evidences for a crucial role of this lipid in chronic joint pain and identify LPC-activated ASIC3 channels as promising targets for chronic joint pain management.
Conflict of interest statement
E. Kosek reports grants from Donation from Lundblad family, personal fees from Eli Lilly, personal fees from Sandoz, personal fees from UCB Pharma, outside the submitted work. The remaining authors have no conflicts of interest to declare.
Appendix A. Supplemental digital content
Supplemental digital content associated with this article can be found online at http://links.lww.com/PAIN/B579.
Acknowledgements
The authors thank Drs A. Baron, S. Diochot, J. Noel, and M. Salinas for helpful discussions, V. Friend and J. Salvi-Leyral for technical support, and V. Berthieux for secretarial assistance. The authors also thank Tycho Tullberg, MD, PhD, CEO of Stockholm Spine Center during the data collection period for generous support and for providing research facilities at Stockholm Spine Center. The authors thank orthopaedic surgeons Ingemar Gladh and Per Gerdin for patient recruitment and taking tissue samples during surgery at Ortho Center, Stockholm. Furthermore, the authors thank Carola Skärvinge, research nurse at Stockholm Spine Center for excellent logistic assistance and Azar Baharpoor, Department of Physiology and Pharmacology, Karolinska Insitutet for support and laboratory assistance.
This work was supported by the Centre National de la Recherche Scientifique (CNRS), the Institut National de la Santé et de la Recherche Médicale (INSERM), the Association Française contre les Myopathies (AFM grant #19618), the Agence Nationale de la Recherche (ANR-11-LABX-0015-01 and ANR-17-CE16-0018) and the NeuroMod Institute of University Côte d'Azur (UCA). This work was also supported by the Conseil Regional Auvergne-Rhone-Alpes (project Ressourcement S3, Arth-Innov) and Feder as well as the French government IDEX-ISITE initiative 16-IDEX-0001 (CAP 20-25). The authors thank the multimodal imaging platform IVIA and CICS, Clermont-Ferrand, France, for in vivo imaging and electronic microscopy, respectively. The study has also received funding from Stockholm County Council, Swedish research Council (K2013-52X-22,199-01-3), and Eli Lilly, USA. The research leading to these results has also received funding from the European Union Seventh Framework Programme (FP7/2007–2013) under grant agreement no. 602919 and from a generous donation from Leif Lundblad and family.
Author contributions: F. Jacquot, designed, performed and analyzed behavioral experiments as well as in vivo imaging; L. Delay, A. Bayle performed and analyzed behavioral experiment; J. Barbier performed and analyzed the electronic microscopy and immunohistochemistry experiments; Y. Aissouni and A. Jurczak did quantitative polymerase chain reaction experiments; D.A. Barriere helped in experimental design and critical reading of the manuscript; F. Marchand conceived, designed and analyzed behavioral experiments and wrote manuscript; C.I. Svensson helped in quantitative polymerase chain reaction experiment, experimental design and critical reading of the manuscript; A. Hugo, diagnosed patients and collected patient samples (Swedish cohort); A.S. Ahmed, performed patient data and sample collection (Swedish cohort); E. Kosek, PI for patient data and sample collection (Sweden) and critical reading of the manuscript; K. Kultima and E. Freyhult helped in data analysis related to patients and critical reading of the manuscript; S. Khoury and T. Ferreira conceived, designed and performed lipidomic experiments, and analyzed data; B. Labrum designed, performed and analyzed behavioral experiments using the dynamic aesthesiometer; K. Delanoe and L. Pidoux designed, performed and analyzed electrophysiological experiments; E. Lingueglia helped in experimental design and critical reading of the manuscript; V. Breuil coordinated the clinical aspects of the study (French cohort); E. Deval conceived, designed, performed and analyzed experiments, and wrote the manuscript. All authors participated in the critical reading of the manuscript and give their consent for this final draft.
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.painjournalonline.com).
F. Jacquot, S. Khoury, B. Labrum contributed equally to this work.
Contributor Information
Florian Jacquot, Email: fl.Jacquot@gmail.com.
Spiro Khoury, Email: spiro.khoury@univ-poitiers.fr.
Bonnie Labrum, Email: labrum@ipmc.cnrs.fr.
Kévin Delanoe, Email: delanoe@ipmc.cnrs.fr.
Ludivine Pidoux, Email: pidoux@ipmc.cnrs.fr.
Julie Barbier, Email: julie.barbier@uca.fr.
Lauriane Delay, Email: ldelay@health.ucsd.edu.
Agathe Bayle, Email: agathe.bayle1@orange.fr.
Youssef Aissouni, Email: youssef.aissouni@uca.fr.
David A. Barriere, Email: david.a.barriere@gmail.com.
Kim Kultima, Email: kim.kultima@medsci.uu.se.
Eva Freyhult, Email: eva.freyhult@medsci.uu.se.
Anders Hugo, Email: anders.hugo@orthocenter.se.
Eva Kosek, Email: eva.kosek@ki.se.
Aisha S. Ahmed, Email: aisha.ahmed@ki.se.
Alexandra Jurczak, Email: alexandra.jurczak@ki.se.
Eric Lingueglia, Email: lingueglia@ipmc.cnrs.fr.
Camilla I. Svensson, Email: camilla.svensson@ki.se.
Véronique Breuil, Email: breuil.v@chu-nice.fr.
Thierry Ferreira, Email: thierry.ferreira@univ-poitiers.fr.
Fabien Marchand, Email: fabien.marchand@uca.fr.
References
- [1].Altawil R, Saevarsdottir S, Wedren S, Alfredsson L, Klareskog L, Lampa J. Remaining pain in early rheumatoid arthritis patients treated with methotrexate. Arthritis Care Res (Hoboken) 2016;68:1061–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Andersson DA, Nash M, Bevan S. Modulation of the cold-activated channel TRPM8 by lysophospholipids and polyunsaturated fatty acids. J Neurosci 2007;27:3347–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Barr AJ, Campbell TM, Hopkinson D, Kingsbury SR, Bowes MA, Conaghan PG. A systematic review of the relationship between subchondral bone features, pain and structural pathology in peripheral joint osteoarthritis. Arthritis Res Ther 2015;17:228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Breivik H, Collett B, Ventafridda V, Cohen R, Gallacher D. Survey of chronic pain in Europe: prevalence, impact on daily life, and treatment. Eur J Pain 2006;10:287–333. [DOI] [PubMed] [Google Scholar]
- [5].Castaneda-Corral G, Jimenez-Andrade JM, Bloom AP, Taylor RN, Mantyh WG, Kaczmarska MJ, Ghilardi JR, Mantyh PW. The majority of myelinated and unmyelinated sensory nerve fibers that innervate bone express the tropomyosin receptor kinase A. Neuroscience 2011;178:196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Catrina AI, Svensson CI, Malmstrom V, Schett G, Klareskog L. Mechanisms leading from systemic autoimmunity to joint-specific disease in rheumatoid arthritis. Nat Rev Rheumatol 2017;13:79–86. [DOI] [PubMed] [Google Scholar]
- [7].Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 1994;53:55–63. [DOI] [PubMed] [Google Scholar]
- [8].Deval E, Baron A, Lingueglia E, Mazarguil H, Zajac JM, Lazdunski M. Effects of neuropeptide SF and related peptides on acid sensing ion channel 3 and sensory neuron excitability. Neuropharmacology 2003;44:662–71. [DOI] [PubMed] [Google Scholar]
- [9].Deval E, Lingueglia E. Acid-Sensing Ion Channels and nociception in the peripheral and central nervous systems. Neuropharmacology 2015;94:49–57. [DOI] [PubMed] [Google Scholar]
- [10].Diochot S, Baron A, Rash LD, Deval E, Escoubas P, Scarzello S, Salinas M, Lazdunski M. A new sea anemone peptide, APETx2, inhibits ASIC3, a major acid-sensitive channel in sensory neurons. Embo J 2004;23:1516–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Dixon WJ. The up-and-down method for small samples. J Am Stat Assoc 1965;60:967. [Google Scholar]
- [12].Flemming PK, Dedman AM, Xu SZ, Li J, Zeng F, Naylor J, Benham CD, Bateson AN, Muraki K, Beech DJ. Sensing of lysophospholipids by TRPC5 calcium channel. J Biol Chem 2006;281:4977–82. [DOI] [PubMed] [Google Scholar]
- [13].Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem 1957;226:497–509. [PubMed] [Google Scholar]
- [14].Gaskin DJ, Richard P. The economic costs of pain in the United States. J Pain 2012;13:715–24. [DOI] [PubMed] [Google Scholar]
- [15].Gentry C, Stoakley N, Andersson DA, Bevan S. The roles of iPLA2, TRPM8 and TRPA1 in chemically induced cold hypersensitivity. Mol Pain 2010;6:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hannan MT, Felson DT, Pincus T. Analysis of the discordance between radiographic changes and knee pain in osteoarthritis of the knee. J Rheumatol 2000;27:1513–17. [PubMed] [Google Scholar]
- [17].Hsieh WS, Kung CC, Huang SL, Lin SC, Sun WH. TDAG8, TRPV1, and ASIC3 involved in establishing hyperalgesic priming in experimental rheumatoid arthritis. Sci Rep 2017;7:8870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hung CH, Lee CH, Tsai MH, Chen CH, Lin HF, Hsu CY, Lai CL, Chen CC. Activation of acid-sensing ion channel 3 by lysophosphatidylcholine 16:0 mediates psychological stress-induced fibromyalgia-like pain. Ann Rheum Dis 2020;79:1644–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Husen P, Tarasov K, Katafiasz M, Sokol E, Vogt J, Baumgart J, Nitsch R, Ekroos K, Ejsing CS. Analysis of lipid experiments (ALEX): a software framework for analysis of high-resolution shotgun lipidomics data. PLoS One 2013;8:e79736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ikeuchi M, Kolker SJ, Burnes LA, Walder RY, Sluka KA. Role of ASIC3 in the primary and secondary hyperalgesia produced by joint inflammation in mice. PAIN 2008;137:662–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ikeuchi M, Kolker SJ, Sluka KA. Acid-sensing ion channel 3 expression in mouse knee joint afferents and effects of carrageenan-induced arthritis. J Pain 2009;10:336–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Inoue M, Rashid MH, Fujita R, Contos JJ, Chun J, Ueda H. Initiation of neuropathic pain requires lysophosphatidic acid receptor signaling. Nat Med 2004;10:712–18. [DOI] [PubMed] [Google Scholar]
- [23].Izumi M, Ikeuchi M, Ji Q, Tani T. Local ASIC3 modulates pain and disease progression in a rat model of osteoarthritis. J Biomed Sci 2012;19:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kleyer A, Finzel S, Rech J, Manger B, Krieter M, Faustini F, Araujo E, Hueber AJ, Harre U, Engelke K, Schett G. Bone loss before the clinical onset of rheumatoid arthritis in subjects with anticitrullinated protein antibodies. Ann Rheum Dis 2014;73:854–60. [DOI] [PubMed] [Google Scholar]
- [25].Koop SM, ten Klooster PM, Vonkeman HE, Steunebrink LM, van de Laar MA. Neuropathic-like pain features and cross-sectional associations in rheumatoid arthritis. Arthritis Res Ther 2015;17:237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kosek E, Finn A, Ultenius C, Hugo A, Svensson C, Ahmed AS. Differences in neuroimmune signalling between male and female patients suffering from knee osteoarthritis. J Neuroimmunol 2018;321:49–60. [DOI] [PubMed] [Google Scholar]
- [27].Kosinska MK, Liebisch G, Lochnit G, Wilhelm J, Klein H, Kaesser U, Lasczkowski G, Rickert M, Schmitz G, Steinmeyer J. A lipidomic study of phospholipid classes and species in human synovial fluid. Arthritis Rheum 2013;65:2323–33. [DOI] [PubMed] [Google Scholar]
- [28].Kosinska MK, Mastbergen SC, Liebisch G, Wilhelm J, Dettmeyer RB, Ishaque B, Rickert M, Schmitz G, Lafeber FP, Steinmeyer J. Comparative lipidomic analysis of synovial fluid in human and canine osteoarthritis. Osteoarthritis Cartilage 2016;24:1470–8. [DOI] [PubMed] [Google Scholar]
- [29].Lee YC, Cui J, Lu B, Frits ML, Iannaccone CK, Shadick NA, Weinblatt ME, Solomon DH. Pain persists in DAS28 rheumatoid arthritis remission but not in ACR/EULAR remission: a longitudinal observational study. Arthritis Res Ther 2011;13:R83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lluch E, Torres R, Nijs J, Van Oosterwijck J. Evidence for central sensitization in patients with osteoarthritis pain: a systematic literature review. Eur J Pain 2014;18:1367–75. [DOI] [PubMed] [Google Scholar]
- [31].Maingret F, Patel AJ, Lesage F, Lazdunski M, Honore E. Lysophospholipids open the two-pore domain mechano-gated K(+) channels TREK-1 and TRAAK. J Biol Chem 2000;275:10128–33. [DOI] [PubMed] [Google Scholar]
- [32].Marra S, Ferru-Clement R, Breuil V, Delaunay A, Christin M, Friend V, Sebille S, Cognard C, Ferreira T, Roux C, Euller-Ziegler L, Noel J, Lingueglia E, Deval E. Non-acidic activation of pain-related Acid-Sensing Ion Channel 3 by lipids. EMBO J 2016;35:414–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Meiri H, Steinberg R, Medalion B. Detection of sodium channel distribution in rat sciatic nerve following lysophosphatidylcholine-induced demyelination. J Membr Biol 1986;92:47–56. [DOI] [PubMed] [Google Scholar]
- [34].Nwosu LN, Allen M, Wyatt L, Huebner JL, Chapman V, Walsh DA, Kraus VB. Pain prediction by serum biomarkers of bone turnover in people with knee osteoarthritis: an observational study of TRAcP5b and cathepsin K in OA. Osteoarthritis Cartilage 2017;25:858–65. [DOI] [PubMed] [Google Scholar]
- [35].O'Brien MS, Philpott HTA, McDougall JJ. Targeting the Nav1.8 ion channel engenders sex-specific responses in lysophosphatidic acid-induced joint neuropathy. PAIN 2019;160:269–78. [DOI] [PubMed] [Google Scholar]
- [36].O'Brien T, Breivik H. The impact of chronic pain-European patients' perspective over 12 months. Scand J Pain 2012;3:23–9. [DOI] [PubMed] [Google Scholar]
- [37].Radawski C, Genovese MC, Hauber B, Nowell WB, Hollis K, Gaich CL, DeLozier AM, Gavigan K, Reynolds M, Cardoso A, Curtis JR. Patient perceptions of unmet medical need in rheumatoid arthritis: a cross-sectional survey in the USA. Rheumatol Ther 2019;6:461–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Roos EM, Roos HP, Lohmander LS, Ekdahl C, Beynnon BD. Knee Injury and Osteoarthritis Outcome Score (KOOS)—development of a self-administered outcome measure. J Orthop Sports Phys Ther 1998;28:88–96. [DOI] [PubMed] [Google Scholar]
- [39].Roos EM, Toksvig-Larsen S. Knee injury and Osteoarthritis Outcome Score (KOOS)—validation and comparison to the WOMAC in total knee replacement. Health Qual Life Outcomes 2003;1:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Sagar DR, Ashraf S, Xu L, Burston JJ, Menhinick MR, Poulter CL, Bennett AJ, Walsh DA, Chapman V. Osteoprotegerin reduces the development of pain behaviour and joint pathology in a model of osteoarthritis. Ann Rheum Dis 2014;73:1558–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Sevastou I, Kaffe E, Mouratis MA, Aidinis V. Lysoglycerophospholipids in chronic inflammatory disorders: the PLA(2)/LPC and ATX/LPA axes. Biochim Biophys Acta 2013;1831:42–60. [DOI] [PubMed] [Google Scholar]
- [42].Sheahan TD, Siuda ER, Bruchas MR, Shepherd AJ, Mohapatra DP, Gereau RW, IV, Golden JP. Inflammation and nerve injury minimally affect mouse voluntary behaviors proposed as indicators of pain. Neurobiol Pain 2017;2:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Sluka KA, Gregory NS. The dichotomized role for acid sensing ion channels in musculoskeletal pain and inflammation. Neuropharmacology 2015;94:58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Sluka KA, Price MP, Breese NM, Stucky CL, Wemmie JA, Welsh MJ. Chronic hyperalgesia induced by repeated acid injections in muscle is abolished by the loss of ASIC3, but not ASIC1. PAIN 2003;106:229–39. [DOI] [PubMed] [Google Scholar]
- [45].Sluka KA, Radhakrishnan R, Benson CJ, Eshcol JO, Price MP, Babinski K, Audette KM, Yeomans DC, Wilson SP. ASIC3 in muscle mediates mechanical, but not heat, hyperalgesia associated with muscle inflammation. PAIN 2007;129:102–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Sluka KA, Rasmussen LA, Edgar MM, O'Donnell JM, Walder RY, Kolker SJ, Boyle DL, Firestein GS. Acid-sensing ion channel 3 deficiency increases inflammation but decreases pain behavior in murine arthritis. Arthritis Rheum 2013;65:1194–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Smith ME, Kocsis JD, Waxman SG. Myelin protein metabolism in demyelination and remyelination in the sciatic nerve. Brain Res 1983;270:37–44. [DOI] [PubMed] [Google Scholar]
- [48].Sugimura N, Ikeuchi M, Izumi M, Kawano T, Aso K, Kato T, Ushida T, Yokoyama M, Tani T. Repeated intra-articular injections of acidic saline produce long-lasting joint pain and widespread hyperalgesia. Eur J Pain 2015;19:629–38. [DOI] [PubMed] [Google Scholar]
- [49].Taylor P, Manger B, Alvaro-Gracia J, Johnstone R, Gomez-Reino J, Eberhardt E, Wolfe F, Schwartzman S, Furfaro N, Kavanaugh A. Patient perceptions concerning pain management in the treatment of rheumatoid arthritis. J Int Med Res 2010;38:1213–24. [DOI] [PubMed] [Google Scholar]
- [50].Tokumura A, Majima E, Kariya Y, Tominaga K, Kogure K, Yasuda K, Fukuzawa K. Identification of human plasma lysophospholipase D, a lysophosphatidic acid-producing enzyme, as autotaxin, a multifunctional phosphodiesterase. J Biol Chem 2002;277:39436–42. [DOI] [PubMed] [Google Scholar]
- [51].Umezu-Goto M, Kishi Y, Taira A, Hama K, Dohmae N, Takio K, Yamori T, Mills GB, Inoue K, Aoki J, Arai H. Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. J Cell Biol 2002;158:227–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Velasco M, O'Sullivan C, Sheridan GK. Lysophosphatidic acid receptors (LPARs): potential targets for the treatment of neuropathic pain. Neuropharmacology 2017;113:608–17. [DOI] [PubMed] [Google Scholar]
- [53].Waldmann R, Bassilana F, de Weille J, Champigny G, Heurteaux C, Lazdunski M. Molecular cloning of a non-inactivating proton-gated Na+ channel specific for sensory neurons. J Biol Chem 1997;272:20975–8. [DOI] [PubMed] [Google Scholar]
- [54].Weiner JA, Chun J. Schwann cell survival mediated by the signaling phospholipid lysophosphatidic acid. Proc Natl Acad Sci U S A 1999;96:5233–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Weiner JA, Fukushima N, Contos JJ, Scherer SS, Chun J. Regulation of Schwann cell morphology and adhesion by receptor-mediated lysophosphatidic acid signaling. J Neurosci 2001;21:7069–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Weiner JA, Hecht JH, Chun J. Lysophosphatidic acid receptor gene vzg-1/lpA1/edg-2 is expressed by mature oligodendrocytes during myelination in the postnatal murine brain. J Comp Neurol 1998;398:587–98. [PubMed] [Google Scholar]
- [57].Zhai G, Pelletier JP, Liu M, Aitken D, Randell E, Rahman P, Jones G, Martel-Pelletier J. Activation of the phosphatidylcholine to lysophosphatidylcholine pathway is associated with osteoarthritis knee cartilage volume loss over time. Sci Rep 2019;9:9648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Zhu S, Zhu J, Zhen G, Hu Y, An S, Li Y, Zheng Q, Chen Z, Yang Y, Wan M, Skolasky RL, Cao Y, Wu T, Gao B, Yang M, Gao M, Kuliwaba J, Ni S, Wang L, Wu C, Findlay D, Eltzschig HK, Ouyang HW, Crane J, Zhou FQ, Guan Y, Dong X, Cao X. Subchondral bone osteoclasts induce sensory innervation and osteoarthritis pain. J Clin Invest 2019;129:1076–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. PAIN 1983;16:109–10. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental digital content associated with this article can be found online at http://links.lww.com/PAIN/B579.