

Abstract

Electron crystallography has a storied history which rivals that of its more established X-ray-enabled counterpart. Recent advances in data collection and analysis have sparked a renaissance in the field, opening a new chapter for this venerable technique. Burgeoning interest in electron crystallography has spawned innovative methods described by various interchangeable labels (3D ED, MicroED, cRED, etc.). This Review covers concepts and findings relevant to the practicing crystallographer, with an emphasis on experiments aimed at using electron diffraction to elucidate the atomic structure of three-dimensional molecular crystals.
1. Introduction and Historical Background
In 1927, Davisson and Germer conducted one of
the most consequential
experiments of the twentieth century.1−3 Using a heated tungsten
filament as a thermionic gun, they fired a collimated beam of slow-moving
electrons (accelerated by a potential of ∼60 V) at a polished
chunk of crystalline nickel. As a makeshift detector, they installed
a galvanometer enclosed within a Faraday box capable of rotating along
a 135° arc. To their astonishment, Davisson and Germer observed
that the reflected electrons displayed a discrete distribution of
scattering angles, precisely analogous to diffraction of X-ray photons.
Invoking Bragg’s law, Davisson and Germer then found very good
agreement between their putative electron wavelength and the theoretical
value predicted by the de Broglie relation 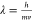 , which de Broglie had proposed only three
years earlier.4 Their discovery, widely
recognized as the first demonstration of electron diffraction (ED),
provided powerful experimental evidence that electrons conformed to
wave-particle duality, an idea still nascent at the time. Several
months later, Davisson and Germer’s results were echoed by
Thomson and Reid, who bombarded a thin film of polycrystalline celluloid
using a beam of higher-energy electrons propagated through a greater
potential drop (∼13 kilovolts).5 On a photographic plate, Thomson and Reid observed a series of concentric
rings evocative of X-ray powder diffraction. In subsequent studies,
Thomson went on to disclose similar ring-like patterns formed upon
irradiation of metallic films composed of polycrystalline platinum,
aluminum, and gold.6,7 Thomson’s calculations,
just like Davisson and Germer’s, showed excellent agreement
between the theoretical de Broglie wavelength and the experimental
electron wavelength back-calculated from Bragg’s law. Naturally,
he concluded that such diffraction patterns could only have originated
if the scattered electrons had behaved as waves. Merely a decade after
the publication of these seminal papers, Davisson and Thomson received
the 1937 Nobel Prize in Physics for “their experimental discovery
of the diffraction of electrons by crystals.” Their pioneering
work created the field of electron crystallography.
, which de Broglie had proposed only three
years earlier.4 Their discovery, widely
recognized as the first demonstration of electron diffraction (ED),
provided powerful experimental evidence that electrons conformed to
wave-particle duality, an idea still nascent at the time. Several
months later, Davisson and Germer’s results were echoed by
Thomson and Reid, who bombarded a thin film of polycrystalline celluloid
using a beam of higher-energy electrons propagated through a greater
potential drop (∼13 kilovolts).5 On a photographic plate, Thomson and Reid observed a series of concentric
rings evocative of X-ray powder diffraction. In subsequent studies,
Thomson went on to disclose similar ring-like patterns formed upon
irradiation of metallic films composed of polycrystalline platinum,
aluminum, and gold.6,7 Thomson’s calculations,
just like Davisson and Germer’s, showed excellent agreement
between the theoretical de Broglie wavelength and the experimental
electron wavelength back-calculated from Bragg’s law. Naturally,
he concluded that such diffraction patterns could only have originated
if the scattered electrons had behaved as waves. Merely a decade after
the publication of these seminal papers, Davisson and Thomson received
the 1937 Nobel Prize in Physics for “their experimental discovery
of the diffraction of electrons by crystals.” Their pioneering
work created the field of electron crystallography.
Davisson and Thomson’s results prompted a flurry of activity during the interwar period. In 1933, Laschkarew and Usyskin disclosed a painstaking electron-diffraction analysis of Debye lines generated by polycrystalline ammonium chloride (NH4Cl), through which they managed to estimate the N—H covalent bond length with remarkable accuracy (0.95 ± 0.07 Å).8 Although very sporadically cited, Laschkarew and Usyskin’s work represents the first (albeit indirect) detection and localization of hydrogen atoms by electron diffraction, a feat which was subsequently reinvestigated many times in later decades.9−11 This early report explicitly underscored a key distinction between ED and conventional X-ray diffraction, where observation of H atoms is comparatively more difficult. Three years later, Rigamonti conducted an ED study of several straight-chain n-alkane crystals.12 Intriguingly, Rigamonti’s work paired quantitative experimental intensities alongside theoretical structure-factor amplitudes, foreshadowing later attempts at reconstruction by Fourier synthesis. Subsequently, Charlesby et al. carried out a detailed single-crystal investigation of anthracene, complete with photocopied electron diffraction patterns meticulously indexed by hand.13 Their results largely confirmed unit cell vectors and angles previously measured by X-ray diffraction, providing a compelling validation of ED as a capable standalone method for crystallographic analysis. Taken in tandem, these three reports paint a portrait of early electron crystallography as a vibrant field of study already producing impactful discoveries only a few short years after its birth in 1927.
In the postwar years, however, progress in the field began to decelerate considerably. ED never quite came into its own as a widely used means of structure determination. Instead, it was rapidly eclipsed by single-crystal X-ray diffraction, which by the mid-twentieth century had become well-established as the gold standard for crystallographic analysis. This remarkable shift in trajectory, which initially appears perplexing given the impressive heights scaled by ED in the 1930s, was spurred by increasingly strident fears over multiple scattering, a physical phenomenon intrinsic to ED.14 These concerns were buttressed by historical constraints (such as low operating voltages), which amplified the probability of observing multiple scattering artifacts, ultimately inhibiting ED’s development as an independent experimental technique. For decades, ED was relegated to a niche method championed mostly by Vainshtein and co-workers, who developed a specialized electron diffractometer capable of collecting so-called texture patterns from 3D crystallites. An excellent summary of their work is available in Vainshtein’s 1964 monograph Structure Analysis by Electron Diffraction, which details >30 3D structures, ranging from inorganic salts to organic small molecules, methodically solved by electron diffraction.15 Nevertheless, it was not until Dorset’s retroactive validation of Vainshtein’s work in the 1990s that the stigma surrounding multiple scattering began to dissipate.16 ED then experienced a belated resurgence in activity in the mid-2010s, driven by methodological and hardware-based advances which enabled collection of diffraction patterns minimizing the deleterious influence of multiple scattering.17 Nearly a century after Davisson and Germer, ED now appears poised to reclaim its mantle as one of the most promising techniques for structure elucidation of 3D molecular crystals.
2. Theoretical Foundations
2.1. Differences Between X-ray and Electron Scattering
In real space, X-ray photons scatter solely off the periodic charge density distribution ρ(r⃗), which emanates from the electron clouds encapsulating atoms within the crystal lattice. In chemistry parlance, ρ(r⃗) is often referred to simply as “electron density.” Following Fourier synthesis, X-ray diffraction (XRD) ultimately recapitulates a real-space map of ρ(r⃗). As uncharged, massless quanta, however, incident X-rays interact with matter quite weakly. Practically, in a routine XRD experiment conducted on an in-house diffractometer, a macroscopic crystal at least ∼105 μm3 in volume is desired to generate enough signal for structure determination. High-flux microfocus beamlines at third-generation synchrotron facilities can push this lower-size threshold down to ∼103 μm3; these highly brilliant X-ray sources have enabled viable diffraction from crystals with dimensions as small as 1–10 μm on one side.18 Below this 1 μm limit, crystals quickly become smaller than the wavelength of visible light, rendering them invisible to optical microscopy. At this submicrometric scale, only the exceptionally intense pulses produced by X-ray free-electron lasers (XFELs) can extract diffraction from slurries of submicrometer-sized crystals. Nevertheless, XFELs currently do not present a widely accessible or convenient means for routine structure elucidation.
In this context, electron diffraction, typically conducted in a transmission electron microscope (TEM), provides a powerful alternative which empowers us to interrogate nanocrystals inaccessible to conventional XRD. Disparities in intrinsic physical properties cause X-rays and electrons to interact with atoms differently. Because of their nonzero mass and inherent negative charge, incident electrons experience electrostatic attraction toward protons in atomic nuclei in addition to repulsion from ρ(r⃗). As a direct consequence of this remarkably strong Coulombic interaction, incident electrons can produce tractable diffraction from minuscule crystals many orders of magnitude smaller in volume (∼10–2 μm3) than those needed for conventional XRD. Unlike X-ray scattering, elastic electron scattering is dictated by electrostatic potential (ESP), or V(r⃗). V(r⃗) amalgamates contributions from both ρ(r⃗) and nuclear charge density δ(r⃗), which is usually expressed as a point charge weighted by atomic number. The key relation between atomic charge density and ESP is given by Poisson’s equation:
where ∇2 is the Laplace operator, Z is the atomic number, δ(r⃗) is a Dirac delta function representing nuclear charge density, e is the elementary charge, and ϵ0 is the permittivity of free space.19
A central pillar of crystallography is the notion that every
diffraction
pattern encodes critical information about the Fourier transform of
the periodic real-space density distribution which produced it. In
Fourier space, the atomic scattering factor or form factor f(s) describes the scattering amplitude
of an isolated, stationary atom by an incident wave, where 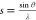 . Formally, f(s) is defined as
the probability amplitude of the exit spherical wave
relative to the incoming plane wave. Informally, f(s) simply provides us with a way to quantify the
scattering power of different atoms in reciprocal space. It follows
that f(s) is highly dependent on
the identity of the impinging quanta. Mathematically, f(s) is derived via Fourier transform
of its corresponding real-space counterpart: ρ(r⃗) for X-rays and V(r⃗) for
electrons. To convert between X-ray and electron scattering factors,
we invoke the Mott–Bethe formula, which functionally provides
a reciprocal-space equivalent to Poisson’s equation:
. Formally, f(s) is defined as
the probability amplitude of the exit spherical wave
relative to the incoming plane wave. Informally, f(s) simply provides us with a way to quantify the
scattering power of different atoms in reciprocal space. It follows
that f(s) is highly dependent on
the identity of the impinging quanta. Mathematically, f(s) is derived via Fourier transform
of its corresponding real-space counterpart: ρ(r⃗) for X-rays and V(r⃗) for
electrons. To convert between X-ray and electron scattering factors,
we invoke the Mott–Bethe formula, which functionally provides
a reciprocal-space equivalent to Poisson’s equation:
where m0 is the electron rest mass, ℏ is the reduced Planck constant, and we denote X-ray scattering factors as fx(s) and electron scattering factors as fe(s).20
Inspection of these equations unveils several key distinctions between X-ray and electron scattering factors. First, the Mott–Bethe formula indicates no simple, monotonic relationship between fx(s) and fe(s). Instead, we observe a nonlinear scaling factor of s–2. Second, unlike their X-ray counterparts, electron scattering amplitudes do not always scale linearly with Z. fe(s) is directly proportional to atomic number only at high spatial frequencies, where electron scattering is dominated by Z-weighted nuclear charge density. At low spatial frequencies, electron scattering is influenced by repulsion from outer-shell valence electrons, which causes fx(s) and fe(s) to exhibit disparate behavior in the limit as s → 0. For instance, following a shared inflection point at ∼0.16 Å–1 (∼3 Å), boron becomes a stronger electron scatterer than carbon, nitrogen, and oxygen at low resolution despite its smaller atomic mass. This order is reversed at scattering angles corresponding to high resolution (Figure 1A). Conversely, all X-ray scattering factors obey the constraint
where Z0 is the number of electrons associated with each atom. A straightforward consequence of this limit is that heavier atoms always scatter X-rays more strongly than lighter atoms, regardless of resolution (Figure 1B). Furthermore, X-ray scattering amplitudes for adjacent neutral elements never converge to a shared point (apart from collectively dwindling to zero as s → ∞), whereas this behavior is permissible for electron scattering amplitudes. As a result, in ED, certain elements become physically indistinguishable at specific scattering angles (Figure 1C). Broadly, relative differences between elements shrink in ED; at 0.2 Å–1 (2.5 Å), for example, iron scatters electrons merely 2.4× more strongly than carbon. This ratio grows to approximately 6× for X-rays, which is much more commensurate with the discrepancy in atomic mass between C and Fe. By the same token, however, lighter elements contribute a greater fraction of scattering signal in ED relative to XRD. This property empowers ED to detect and localize atoms such as hydrogen, which typically scatter X-rays very weakly. Finally, arguably the most drastic disparity between electron and X-ray scattering factors lies in electrons’ ability to visualize charged states.21V(r⃗) contains an explicit contribution from nuclear charge density δ(r⃗), which renders its Fourier transform fe(s) innately sensitive to the excess nuclear charge intrinsic to ionized atoms. Consequently, electron scattering amplitudes for neutral atoms diverge strikingly from those of their ionic counterparts, especially at low spatial frequencies. As s → 0, fe(s) skyrockets toward ∞ for cations and plummets toward −∞ for anions (Figure 1E). These differences materialize much more subtly in X-ray scattering, which remains comparatively uninfluenced by nuclear charge density (Figure 1F).
Figure 1.

(A) Neutral electron scattering factors for
seven representative
elements. All neutral scattering factors were parametrized into five
Gaussians and plotted within the range [0 < 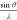 < 0.6 Å–1], equivalent
to [∞ < d < 0.83 Å]. (To convert
between
< 0.6 Å–1], equivalent
to [∞ < d < 0.83 Å]. (To convert
between 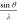 and d, recall Bragg’s
law:
and d, recall Bragg’s
law:  .) (B) Neutral X-ray
scattering factors.
(C) Neutral electron scattering relative to carbon. Relative scattering
amplitudes were calculated by dividing each scattering factor by f(s) for neutral carbon. (D) Neutral X-ray
scattering relative to carbon. (E) Ionic vs neutral
electron scattering factors for O and Fe. To avoid physically unrealistic
values in the limit as
.) (B) Neutral X-ray
scattering factors.
(C) Neutral electron scattering relative to carbon. Relative scattering
amplitudes were calculated by dividing each scattering factor by f(s) for neutral carbon. (D) Neutral X-ray
scattering relative to carbon. (E) Ionic vs neutral
electron scattering factors for O and Fe. To avoid physically unrealistic
values in the limit as 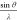 tends to zero, O1– was
truncated at 0.02 Å–1 before parametrization
into five Gaussians, while Fe2+ and Fe3+ were
truncated at 0.05 Å–1. (F) Ionic vs neutral X-ray scattering factors.
tends to zero, O1– was
truncated at 0.02 Å–1 before parametrization
into five Gaussians, while Fe2+ and Fe3+ were
truncated at 0.05 Å–1. (F) Ionic vs neutral X-ray scattering factors.
2.2. Differences Between X-ray and Electron Wavelengths
Conventional TEMs accelerate electrons to a significant fraction of the speed of light, exploiting voltage differences to produce a high-energy beam (i.e., 100–300 keV) in which each constituent electron is forcibly propagated through a potential drop. At these energies, an accurate calculation of the de Broglie electron wavelength must incorporate relativistic contraction, as follows:
where h is Planck’s
constant, c is the speed of light in a vacuum, m0 is the rest mass of the electron, and E is the kinetic energy (in keV) imparted by the accelerating
voltage (in kV). Discrepancies between the nonrelativistic (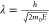 ) and relativistic
calculations widen significantly
as E rises (Figure 2). At 300 keV, for instance, the error grows to approximately
13.7%; these two wavelengths would generate distinct Ewald spheres
with markedly different radii, underscoring the importance of using
the relativistically corrected value.
) and relativistic
calculations widen significantly
as E rises (Figure 2). At 300 keV, for instance, the error grows to approximately
13.7%; these two wavelengths would generate distinct Ewald spheres
with markedly different radii, underscoring the importance of using
the relativistically corrected value.
Figure 2.
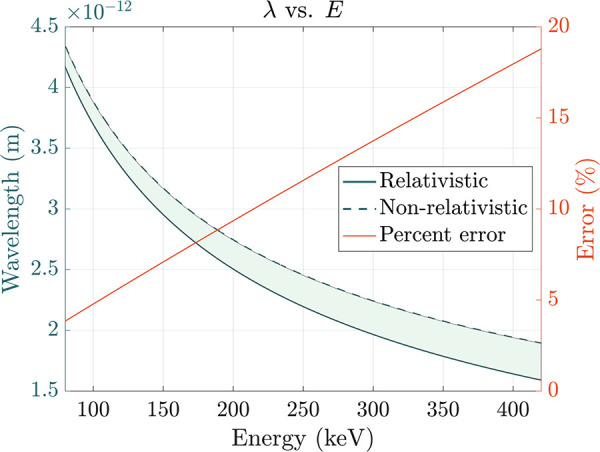
Relativistic (solid blue line) and nonrelativistic (dashed blue line) electron wavelengths plotted as a function of incident energy (E) at a range of accelerating voltages accessible to TEM. Percent error between the two calculations is plotted in orange; characteristic values include ∼4.7% at 100 keV, ∼9.3% at 200 keV, ∼13.7% at 300 keV, and ∼17.9% at 400 keV.
For additional perspective, a systematic comparison of typical X-ray and electron wavelengths is given in Table 1. These numbers indicate that 100–300 keV electrons exhibit relativistic wavelengths roughly 50–100× shorter than their X-ray counterparts, which leads to an array of experimental consequences. Because the radius λ–1 of the Ewald sphere scales inversely with the wavelength of the impinging quanta, electrons at these energies divulge expansive Ewald spheres which intercept the reciprocal lattice along gently sloping arcs (Figure 3A). This geometry stands in stark contrast to X-ray diffraction, where inherently longer wavelengths produce smaller Ewald spheres featuring distinct surface curvature (Figure 3B). As a result, the cascades of circular lunes seen in X-ray diffraction patterns give way to nearly planar slices in electron diffraction patterns, which resemble canonical precession photographs (Figure 3C).
Table 1. Systematic Comparison of X-ray and Electron Wavelengths at a Range of Relevant Energies.
| energy (keV) | quanta | β (v/c) | wavelength (Å) | radius of Ewald sphere (Å–1) |
|---|---|---|---|---|
| 8.042 | X-rays (Cu Kα) | 1.0 | 1.5418 | 0.6485 |
| 12.65 | X-rays (Se K) | 1.0 | 0.9795 | 1.0209 |
| 17.44 | X-rays (Mo Kα) | 1.0 | 0.7107 | 1.4070 |
| 100 | electrons | 0.548 | 0.0370 | 27.027 |
| 200 | electrons | 0.695 | 0.0251 | 39.840 |
| 300 | electrons | 0.776 | 0.0197 | 50.761 |
Figure 3.

X-ray vs electron Ewald spheres and experimental diffraction patterns. Superimposed X-ray (rendered in blood orange, E = 8.042 keV, λ = 1.541 Å, radius = 0.6485 Å–1, volume = 1.142 Å–3) and electron (rendered in blue, E = 300 keV, λ = 0.0197 Å, radius = 50.76 Å–1, volume = 5.478 × 105 Å–3) Ewald spheres are drawn intersecting a cubic reciprocal lattice. The X-ray Ewald sphere is comfortably dwarfed by its much more voluminous electron counterpart. (A) 2D orthographic projection viewed normal to an arbitrary reciprocal lattice vector. (B) Alternate view revealing the three-dimensionality of the reciprocal lattice. (C) Electron diffraction pattern acquired using an accelerating voltage of 300 kV. Inset shows a close-up view and somewhat noisy 3D peak profile of a 0.95 Å resolution Bragg reflection. (D) X-ray diffraction pattern acquired on an in-house diffractometer equipped with a Cu Kα anode (8.042 keV). Inset shows a close-up view and strong 3D peak profile of a 1.56 Å Bragg reflection.
Each sampled Bragg peak represents an intersection between a reciprocal lattice vector and the surface of the Ewald sphere. A wider, flattened Ewald sphere causes ED patterns to accommodate different groups of reflections per scattering angle relative to XRD. For instance, observation of several Friedel mates within a singular diffraction pattern is commonplace in ED, whereas the curvature of the X-ray Ewald sphere curtails this in XRD. Furthermore, because of planarity, a singular ED pattern generally only permits deduction of two unit cell vectors at once (exceptions include strongly diffracting samples in materials science, where higher-order Laue zone reflections can reveal three-dimensionality in the reciprocal lattice22). Conversely, a lone XRD pattern typically samples all three dimensions of the reciprocal lattice simultaneously. In practice, to reliably determine all three unit cell parameters, indexing requires comparatively more consecutive frames in ED (often covering a ∼15–25° angular wedge of reciprocal space) than it does in XRD, where one or two can theoretically suffice. Finally, the set of permissible scattering angles in ED (i.e., values of θ which satisfy the Bragg condition) encompasses a much smaller numerical range versus XRD, a direct consequence of substituting shorter wavelengths into Bragg’s law. To compensate for this, ED requires a significantly longer detector distance than XRD to discriminate between Bragg peaks, often in the vicinity of ∼1 m. Another key distinction is that adjustments to detector distance in XRD involve physically moving a piece of hardware. Conversely, in a transmission electron microscope, the physical distance between the sample and the detector is fixed. ED performed in TEMs utilizes a system of postspecimen electromagnetic lenses to generate virtual camera lengths, effectively either magnifying or demagnifying the reciprocal lattice projected onto the detector.
2.3. Multiple Scattering
As another consequence of their augmented cross-sections relative to X-rays, incident electrons have a higher relative likelihood of undergoing multiple scattering events while traversing an illuminated crystal.23−27 This phenomenon, frequently referred to as “dynamical” scattering, a term which specifically encompasses multiple elastic events, was for decades considered a daunting bulwark against accurate structure determination by electron crystallography. Broadly, the probability of detecting multiple scattering is chiefly influenced by three factors: (a) the incident electron energy, (b) the irradiated crystal’s density and thickness, and (c) its geometric orientation relative to the impinging beam. Within an energy range germane to TEM (i.e., accelerating voltages between 80–300 kV), electron cross-sections for all neutral elements vary as a monotonic function of kinetic energy E (Figure 4B). As E becomes progressively larger (i.e., as the relativistic electron velocity asymptotically approaches c), the likelihood of any singular scattering event, and, by extension, the likelihood of multiple scattering, becomes progressively lower. For instance, the elastic cross-section of carbon at 300 keV is ∼4× smaller than its counterpart at 80 keV (Figure 4A). In principle, the probability of multiple scattering is therefore diminished at higher incident energies and maximized at lower incident energies.
Figure 4.

(A) Elastic cross-sections for neutral carbon at 80 keV (green) and 300 keV (yellow); cross-sectional areas expressed as concentric circles. (B) Elastic cross-section for neutral carbon decreasing as a monotonic function of incident energy, plotted at a range of accelerating voltages relevant to TEM.
Furthermore, substrate-specific attributes such as crystal density determine the incident electrons’ elastic and inelastic mean free paths (MFPs). MFPs provide a statistical estimate of the average distance traveled between each respective scattering event. Assuming a randomly distributed set of point scatterers, the MFP is defined as
where N is the number of atoms per unit cell volume and σ is a weighted mean cross-section which represents an “average atom” within the unit cell. Clearly, MFPs scale inversely with N and σ, indicating that the probability of multiple scattering is amplified if the incident electrons must penetrate (a) dense, tightly packed lattices or (b) unit cells containing strong scatterers such as heavy metals, whose cross-sections eclipse those of lighter elements. These scenarios lead to shorter MFPs. Theoretically, if the crystal under interrogation is several MFPs thick, multiple scattering becomes a statistical inevitability.
Finally, geometric orientations where the incident beam illuminates major zone axes can cause excitation of many Bragg reflections all at once. If an incident electron undergoes exclusively multiple elastic scattering, its ultimate fate likely lies within a Bragg peak regardless of how many scattering events it experiences. Thus, because zone-axis diffraction patterns feature simultaneous excitation of a wide range of Bragg peaks, they effectively open many more avenues through which multibeam interference could potentially occur. This effect is intensified by low mosaicity. In sum, ED studies which report severe multiple scattering typically feature some combination of low accelerating voltages, near-perfect or minimally mosaic crystals, alignment at major zone-axis orientations, or thick and dense samples. All these experimental conditions maximize the occurrence of dynamical effects.
If singular elastic or “kinematical” scattering holds, the integrated intensity of each Bragg peak is proportional to the squared modulus of its corresponding structure factor:
In conventional X-ray crystallography, this relationship is almost universally observed. In ED, however, multiple elastic scattering stochastically redistributes some fraction of the diffracted intensities, a process mathematically described by self-convolution of Ihkl.28 Such self-convolution breaks a key tenet of kinematical scattering, where the intensity of any random Bragg reflection is decoupled from that of its neighbor. Conversely, dynamical scattering imbues the intensities of compromised reflections with some degree of dependence on the intensities of their simultaneously excited counterparts. Two diagnostic markers of this effect include (a) violation of Friedel’s law29−31 and (b) appearance of symmetry-forbidden Bragg peaks at reciprocal lattice points where glide planes, screw axes, or nonprimitive lattices would normally mandate systematic extinctions.14,32−39 In space groups which contain these symmetry operators, a useful metric to quantify the extent of multiple scattering is the ratio between average intensities of symmetry-forbidden versus symmetry-allowed reflections within a particular zone axis.35,37,38 If the recorded diffraction pattern is sufficiently marred by these artifacts, the fundamental link between Ihkl and |Fhkl|2 becomes increasingly tenuous, undermining the validity of the measured intensities. In milder cases, multiple elastic scattering would simply intensify weaker reflections and attenuate stronger reflections. In severe cases, multiple scattering would theoretically sever this link altogether, producing a pseudouniform distribution of intensities which ablates distinctions between ideally independent reflections.39 This homogenization of relative differences between Bragg peaks would render any structure-factor amplitudes derived from such intensities meaningless.
For many years, these concerns led to a self-imposed moratorium on structure elucidation by electron diffraction, as ED intensities were considered too corrupted to yield reliable atomic coordinates.40 Such sentiments were succinctly expressed in The Determination of Crystal Structures, the classic 1966 textbook by Lipson and Cochran.41 Following a perfunctory summary of Vainshtein’s work, the authors concluded that electron diffraction was “inferior to the other two diffraction techniques [X-ray and neutron] because of the many difficulties which stand in the way of making accurate intensity measurements.” In some laboratories, this belief rapidly ossified into dogma, and the steady stream of small-molecule ED structures solved by Vainshtein and co-workers in the Soviet Union was treated with suspicion. In 1968, Cowley14 felt compelled to write that it was “perhaps significant that the first work on structure analysis by electron diffraction, and most of the subsequent work, was done in the USSR and Australia, countries well removed [emphasis added] from the leading pre-war experimental electron diffraction groups in England and the groups in Japan which had the most complete knowledge of dynamical theory.”
A key breakthrough was provided by Hauptman and Karle’s development of direct methods, which supplied an objective means of phase retrieval from integrated intensities.42−45 Because direct methods leverage statistical relationships between accurately sampled structure-factor amplitudes, untethering Ihkl and |Fhkl|2 should have nullified any possibility of ab initio phasing. Dynamically corrupted intensities would have led direct methods to formulate incorrect phase relationships between structure-factor amplitudes, ultimately generating a nonsense structure. However, in a seminal 1976 study, Dorset and Hauptman deployed ab initio phasing to successfully decipher the subcell structures of two organic compounds, n-hexatriacontane and racemic 1,2-dipalmitoyl-glycerophosphoethanolamine, via electron diffraction.46 This work provided robust experimental evidence that structure elucidation using the kinematical approximation was plausible despite the countervailing influence of multiple scattering. Specifically, Dorset and Hauptman found that the utility of the triplet and quartet phase invariants (as well as the centrosymmetric phase restriction ϕhkl = 0 or π) emerged unscathed, notwithstanding usage of amplitudes presumably distorted by multiple scattering. Dorset and Hauptman’s results were especially compelling given their relatively low operating voltages of 80–100 kV (i.e., energies at which the probability of multiple scattering was already amplified). In a steadily increasing number of counterexamples, ominous predictions about multiple scattering have generally failed to hold true outside specific extenuating circumstances, and dynamical effects have not impeded structure solution by direct methods (Table 3). In sum, multiple elastic scattering rarely distorts intensities with enough severity to generate an experimental Patterson map out of sync with the autocorrelation function of the genuine structure.47
Table 3. List of Small-Molecule 3D ED Structures Deposited in the CSD as of October 2021, Excluding Duplicatesa,b.
| compound name | empirical formula | CSD accession code | year | resolution (Å) | space group | R1 | ref |
|---|---|---|---|---|---|---|---|
| biotin | C10H16N2O3S | BIOTIN13 | 2018 | 0.9 | P212121 | 17.81 | (200) |
| carbamazepine | C15H12N2O | CBMZPN28 | 2016 | 0.8 | P21/n | 25.45 | (225) |
| epicorazine A | C18H16N2O6S2 | BISGAO | 2019 | 0.83 | P212121 | 15.43 | (144) |
| dehydrocurvularin | C16H18O5 | IRELOH01 | 2019 | 0.82 | P212121 | 14.95 | (144) |
| (+)-limaspermidine | C19H26N2O | CAHKUU01 | 2018 | 0.77 | P212121 | 18.22 | (200) |
| cimetidine | C10H16N6S | CIMETD06 | 2019 | 1.0 | C2/c | 19.69 | (227) |
| cinchonine | C19H22N2O | CINCHO11 | 2018 | 1.0 | P21 | 17.80 | (200) |
| paracetamol (monoclinic polymorph) | C8H9NO2 | COTZAN07 | 2018 | 0.86 | P21/n | 26.46 | (199) |
| Schwartz’s reagent | C24H34Cl2Zr2 | DIZZUK | 2019 | 1.15 | Pnnm | 14.95 | (163) |
| Pd(II) ethylene insertion product | C36H40B18O2P2Cl18Pd2 | DOBBEE | 2019 | 0.9 | P1̅ | 18.22 | (163) |
| Pd(dba)(PHOX) | C43H43NO2PPd | DOBCAB | 2019 | 1.0 | P212121 | 14.32 | (163) |
| polyamylose–propanol complex | (C42H70O35)n, (C3H8O)4n, (H2O)6n | GUTGAF | 2015 | 3.03 | P212121 | 34.19 | (275) |
| paracetamol (orthorhombic polymorph) | C8H9NO2 | HXACAN41 | 2018 | 0.8 | Pcab | 8.89a | (143) |
| ibuprofen | C13H18O2 | IBPRAC20 | 2018 | 0.9 | P21/c | 25.41 | (200) |
| Ni(dppf)Cl2 | C34H28Cl2FeNiP2 | KADXES02 | 2019 | 1.0 | Pna21 | 11.25 | (163) |
| methylene blue derivative (MBBF4) | (C30H31N7S) 2+,2(C30H30N7S)+,4(BF4)− | LIMZAL01 | 2018 | 0.9 | C2/c | 25.83 | (199) |
| brucine | C23N2O4 | MAJRIZ02 | 2018 | 0.9 | P21 | 18.29 | (200) |
| [Co(ddpd)2](BF4)2 | (C34H34N10Co)2+,2(BF4)− | MOTNUG | 2015 | 1.2 | P21/c | 28.81 | (276) |
| nicotinic acid | C6H5NO2 | NICOAC05 | 2016 | 0.75 | P21/c | 30.26 | (225) |
| ethisterone | C21H28O2 | POSJAI01 | 2018 | 0.9 | P21 | 22.21 | (200) |
| Grubbs’ catalyst (1st generation) | C43H72Cl2P2Ru | IKORIK03 | 2019 | 0.85 | P21/n | 15.95 | (163) |
| progesterone | C21H30O2 | PROGST15 | 2018 | 0.9 | P212121 | 17.65 | (200) |
| HKL-I-029 | C19H17NO5 | QILJUT | 2018 | 1.0 | P21/n | 22.23 | (200) |
| n-tritriacontane | C33H68 | QQQFVD03 | 1999 | N/Ab | A21am | 21.00 | (277) |
| HRh(CO) (PPh3)3 | C55H46OP3Rh | RCOHPH04 | 2019 | 1.0 | P21/n | 13.24 | (163) |
| Fe(acac)3 | C15H21FeO6 | XAQVIX01 | 2019 | 0.9 | Pbca | 16.07 | (163) |
| C68-warped nanographene | C68H28 | AQETUO | 2021 | 0.85 | P42 | 16.47 | (218) |
| loratadine | C22H23N2O2Cl | BEQGIN08 | 2020 | 1.2 | C2/c | 57.58 | (125) |
| (−)-lomaiviticin C | C68H82N4O24 | ERUHEH | 2021 | 1.05 | P21 | 12.06 | (207) |
| sofosbuvir/l-proline cocrystal | C22H29FN3 O9P, C5H9N O2 | EYIQEL | 2019 | 1.0 | P212121 | 9.62a | (175) |
| polycyclic indole-derived ester | C19H14N4O2 | FABTIP | 2020 | 0.83 | R3̅ | 15.77 | (217) |
| remdesivir | C27H35N6O8P | IQIMAZ02 | 2021 | 0.9 | P21 | 16.09 | (232) |
| glycine (α-polymorph) | C2H5NO2 | KUFDIB | 2020 | 0.703 | P21/n | 21.88 | (278) |
| glycine (β-polymorph) | C2H5NO2 | KUFDOH | 2020 | 0.751 | P21 | 12.76 | (278) |
| glycine (γ-polymorph) | C2H5NO2 | KUFDUN | 2020 | 0.7 | P31 | 30.64 | (278) |
| dipyrrolidine perylene diimide | C32H24N4O4 | LACPAJ01 | 2020 | 0.6 | Cc | 19.91 | (54) |
| dicyano naphthalene diimide | C16H4N4O4 | TUKVON | 2020 | 0.57 | P21/c | 13.76 | (54) |
| diketopyrrolopyrrole | C54H70N8O6S2 | TUKVUT | 2020 | 0.9 | P21/n | 23.5 | (54) |
| l-histidine | C6H9N3O2 | LHISTD15 | 2019 | 0.88 | P212121 | 19.81 | (227) |
| nickel carbene complex | C27H31N3O2Ni | LUZZUE | 2020 | 0.85 | Pca21 | 24.63 | (279) |
| [Fe(bpy)3](PF6)2 | (C30H24FeN6)2+, 2(PF6)− | NUZKOI13 | 2020 | N/A | P3̅c1 | N/Ab | (279) |
| [11]helicene | C88H92O10 | QADMUH | 2020 | 1.0 | P212121 | 11.73 | (216) |
| [11]helicene monoquinone | C86H86O10 | QADNAO | 2020 | 1.1 | Iba2 | 17.04 | (216) |
| [11]helicene diquinone | C84H80O10 | QADNES | 2020 | 1.0 | P1̅ | 17.16 | (216) |
| [11]helicene diquinoxaline | C96H88N4O6 | QADNIW | 2020 | 1.0 | I4̅c2 | 15.41 | (216) |
| [11]helicene monoquinoxaline | C92H90N2O8 | QADNOC | 2020 | 1.0 | Iba2 | 18.80 | (216) |
| B/N-doped p-arylenevinylene chromophore | C102H114B2N2 | SADGEN | 2020 | 0.95 | P1̅ | 24.29 | (281) |
| spiroconjugated carbon-bridged p-phenylenevinylene | C42H26O | SUVJOL | 2020 | 0.95 | P1̅ | 24.29 | (282) |
| copper(II) perchlorophthalocyanine | C32N8Cl16Cu | UZEMIY | 2021 | 0.8 | C2/m | 27.85 | (205) |
| olanzapine/ phenol cocrystal | C17H20N4S, C6H6O | WACDEN | 2020 | 1.0 | P1̅ | 31.40 | (229) |
| tryptophan-derived oxindole | C12H14N2O3 | YOYXAO | 2019 | 0.9 | P21/c | 17.77 | (213) |
| tryptophan-derived indanone | C13H15NO3 | YOYYOD | 2019 | 0.9 | P212121 | 17.07 | (213) |
| glucopyranosyl uric acid derivative | C11H14N4O8 | YURNIL | 2020 | 1.0 | P1 | 14.01 | (215) |
| metaxalone | C12H15NO3 | ZUQXIV | 2020 | 0.78 | P212121 | 38.95 | (230) |
| orthocetamol | C8H9NO2 | WOFXEX | 2019 | 0.9 | C2/c | 32.70 | (204) |
| bismuth subgallate | C7H5BiO6 | JAXSUZ | 2017 | 0.7 | Pmna | 11.80 | (226) |
| teniposide | C32H32O13S | KUXJUL | 2021 | 0.9 | P212121 | 9.76 | (231) |
| thiophene-fused cyclooctatetraene | C36H36O12NS4 | AQECOR | 2021 | 0.8 | P1̅ | 23.96 | (223) |
Cases applying dynamical refinement.
CIF files for entries NUZKOI03 and QQQFVD03 do not contain any structure-factor amplitudes or phases, simply atomic coordinates. Abbreviations used: acac = acetylacetonate, bpy = 2,2′-bipyridine, dppf = 1,1′-bis(diphenylphosphino)ferrocene, dba = dibenzylideneacetone, ddpd = N,N′-dimethyl-N,N′-dipyridine-2-yl-pyridine-2,6-diamine. If any discrepancies were found between the R-factors reported in the CSD vs the R-factors quoted in the associated publications, we cited those listed in the CSD.
An impactful portion of this dogma-busting work was conducted by Dorset, who embarked on a quest to apply direct methods to electron-diffraction amplitudes originally recorded at ∼50 kV by Vainshtein, Zvyagin, and other pioneering electron crystallographers in the 1950s.48−52 Because these ED data were collected prior to the advent of ab initio phasing, Vainshtein and co-workers usually relied on pairing experimental ED amplitudes with phases borrowed from corresponding X-ray structures. Naturally, this approach invited concerns regarding phase bias. Nevertheless, armed with the objectivity of direct methods, Dorset was able to replicate Vainshtein’s structures of diketopiperazine, urea, and thiourea, all using a simple kinematical approximation. This resounding vindication of Vainshtein’s early work, nearly three-and-a-half decades after it was first published, dispelled much of the stigma projected by dynamical scattering. In 2010, Dorset concluded the diketopiperazine saga with another reevaluation of Vainshtein’s results, this time equipped with contemporary crystallographic software.53 A full-matrix least-squares refinement of 60-year-old data in SHELXL proved remarkably successful, yielding an R1 residual comparable to recent ED structures obtained using modern instrumentation.
3. Experimental Setup
3.1. Sample Preparation
Sample preparation for 3D electron crystallography involves dispersing a micro- or nanocrystalline powder onto an EM grid 3.05 mm in diameter. For a wide range of small molecules, this procedure is quite simple; it merely entails inserting an EM grid into a scintillation vial containing a few milligrams of substrate and vigorously shaking for ∼10 s (Figure 5B). If this “shake-n-bake” method produces an unduly sparse distribution of crystals, an alternative strategy involves immersing a small quantity of powder in a volatile solvent (ideally one in which the substrate is completely insoluble), drop-casting 2–3 μL of the resultant slurry directly onto the grid using a micropipette, and allowing it to air-dry at RT. Alternatively, crystals suitable for ED can be grown or annealed directly on EM grids by drop-casting a dilute solution of analyte and letting it evaporate, prompting in situ nucleation and crystallization.54,55 Optionally, excess solvent can be wicked away using filter paper or drained under reduced pressure by a vacuum pump.56 Because the amorphous carbon surface of many grids is somewhat hydrophobic, it generally interferes with adherence of aqueous solvents. This mismatch can prevent the drop-casted suspension from spreading uniformly across the film. A highly uneven distribution of crystallites can lead to a few overly congested grid squares, prohibiting isolation of a single crystal within the selected area aperture. To combat this, the surface of the grid can be rendered hydrophilic by glow-discharging before use.
Figure 5.

(A–C) Optical microscopy of several crystalline compounds suitable for 3D electron crystallography. (A,C) Formally recrystallized material (an organic small molecule suspended in glycerol in A, an oligopeptide suspended in a hanging drop in C) requiring additional pulverization before ED due to their macroscopic size. (B) An inherently microcrystalline powder amenable to a direct “shake-n-bake” approach with a standard 3.05 mm lacey carbon EM grid, encircled in blue. (D–F) Transmission electron microscopy reveals micro- and nanocrystalline specimens with a range of morphologies, all suitable for ED analysis.
An added layer of complexity is presented by crystals which contain disordered channels of volatile solvent, such as proteins.57−59 These species can undergo swift lattice collapse when subjected to the high vacuum (typically <10–4 Pa) of the TEM. Therefore, as a prophylactic measure, electron diffraction of solvated crystals is generally recorded under cryogenic (−175 °C) conditions facilitated by liquid nitrogen. Common practice involves implementing well-established cryo-preservation techniques borrowed from single-particle cryo-EM.60−63 Encasing susceptible crystals within a thin layer of vitreous ice shields them from the TEM vacuum and preserves the lattice in a frozen-hydrated state. Cryogenic temperatures also delay the onset and progression of radiation damage, which is frequently quite severe for macromolecular crystals at RT. For proteins and oligopeptides, several step-by-step protocols detailing cryo-preservation procedures have been published.64,65 A glow-discharged EM grid is first loaded with 2–3 μL of an aqueous suspension of protein crystals (usually immersed in mother liquor from a successful crystallization trial, such as the hanging drop in Figure 5C). Subsequently, the grid is blotted and rapidly plunged into a small reservoir (∼4 mL) of liquid ethane. Ethane’s high specific heat capacity allows it to function as a ruthlessly efficient cryogen, ensuring complete vitrification of residual water without cocrystallization of adventitious ice. Because pure ethane solidifies upon prolonged exposure to liquid nitrogen, eutectic mixtures of ethane and propane have also been proposed as alternatives with depressed freezing points.66 This step is typically carried out at high speed by automated vitrification robots, although manual plunge-freezing is also an option. Frozen grids can then be immediately cryo-transferred to the TEM or indefinitely stored in liquid nitrogen for future use. Finally, for substrates such as beam-sensitive, unsolvated small molecules, vitrification is generally unnecessary. Nonetheless, these crystals may still benefit considerably from the reduced radiation damage engendered by cryogenic conditions. A typical tactic therefore involves skipping vitrification and simply slow-cooling the sample within a cryo-holder following insertion into the TEM.
Atomic-resolution ED data have been routinely recorded from crystals hundreds of nanometers thick. Nevertheless, as crystal thicknesses approach the 1 μm mark, data quality rapidly deteriorates, largely due to prohibitive amounts of inelastic scattering overwhelming productive signal from Bragg peaks.67 Thus, sonicating the microcrystalline slurry (or vortexing with acid-washed glass beads) is often necessary to shatter crystals into smaller, thinner shards amenable to ED. In cases where a suitably inert drop-casting solvent is unavailable, simply grinding dry powder between two glass coverslips can achieve an analogous effect via shear force. Alternatively, focused ion-beam (FIB) milling can shave excessively thick crystals down to thin electron-transparent lamellae with precision.68−71 Although quite powerful, FIB milling requires usage of specialized ancillary equipment (a scanning electron microscope), as well as multiple cumbersome cryo-transfer steps if dealing with vitrified samples.
3.2. 3D ED Data Collection Procedures
Historically, electron diffraction patterns were recorded after tilting the crystal to a low-index zone-axis orientation.13 In principle, in-zone diffraction patterns near-perfectly coincide with sets of parallel Bragg planes within the reciprocal lattice (Figure 6A). As a result, these slices of the Ewald sphere contain an especially high density of simultaneously excited reflections. Such circumstances present a double-edged sword. On one hand, a well-defined zone-axis geometry facilitates indexing and simplifies determination of unit cell parameters. By the same token, however, this method is blind to reflections located between zone axes, leaving interstitial corridors of reciprocal space undersampled. Furthermore, zone-axis orientations maximize the probability of observing multiple elastic scattering, impeding accurate integration of quasi-kinematical intensities needed for structure solution. To compound matters, merging intensities recorded solely from disparate still-frame in-zone patterns is often quite difficult. Excitation error can cause even small angular deviations to produce prohibitive variations in intensities recorded slightly outside their exact Bragg condition. As the relatively small handful of successful examples attests,72−74ab initio structure determination from oriented zone-axis patterns was never widely adopted as a robust means of solving 3D structures.
Figure 6.
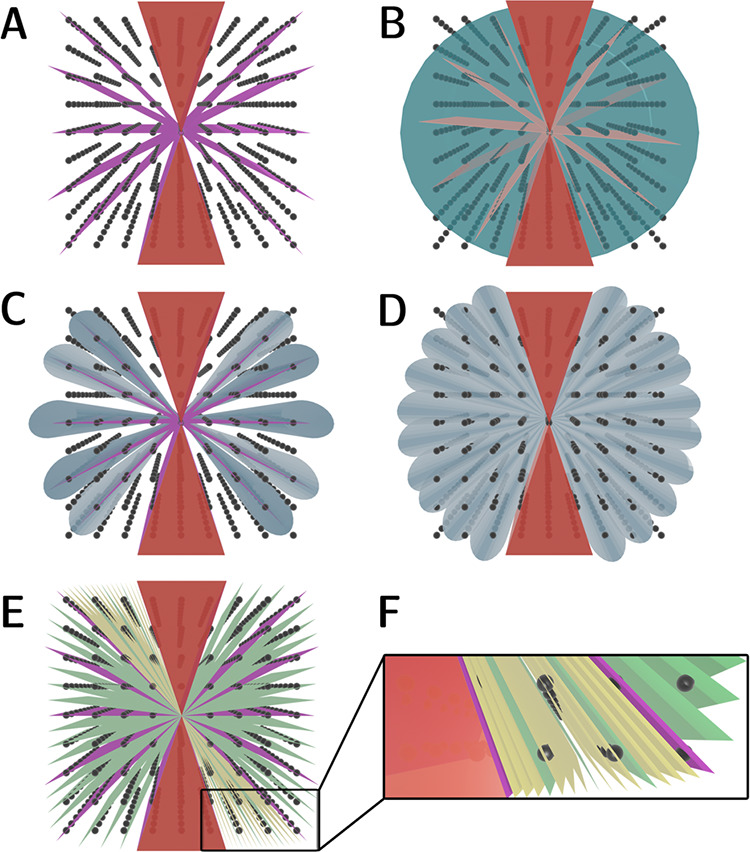
Different modalities of 3D ED data collection. In all schematics, the hourglass-shaped missing wedge intrinsic to the TEM goniometer is depicted in red. (A) Zone-axis orientations (purple planes) accessed via stepwise angular tilts. This approach maximizes the density of Bragg reflections per diffraction pattern, streamlining deduction of unit cell parameters. It also leaves several corridors of reciprocal space between zone axes (white) unsampled, hampering completeness. (B) Continuous-rotation electron diffraction. Blue wedges correspond to regions of reciprocal space sampled during the exposure time, whereas red planes represent gaps left unsampled while the TEM stage continues to rotate during the detector readout time; these become negligibly small with modern active-pixel sensors. (C) Zone-axis precession electron diffraction (PED). Thanks to the gyrating motion of the incident beam (blue cones), this method intercepts several off-zone reflections neglected in (A). (D) Precession-assisted electron diffraction tomography (PEDT). This technique combines beam precession with rotation about the goniometer axis, further enhancing coverage of reciprocal space. (E) Automated diffraction tomography (ADT). Stepwise tilts about the goniometer axis ensure that most diffraction patterns (green planes) represent off-zone orientations. (F) Rotation electron diffraction (RED). Exploitation of electron beam tilt enables finer sampling of reciprocal space (closely spaced yellow planes) than relying on the mechanical precision of the TEM goniometer alone (green planes).
In 1994, Vincent and Midgley pioneered precession electron diffraction (PED), a novel means of data collection that mitigated some of these issues.75 In their method, the incident electron beam is effectively precessed within a fixed, hollow cone whose vertex is coincident with the plane of the illuminated crystal.76 The resultant diffraction patterns contain signal averaged over elongated conical sections of the Ewald sphere rather than planar slices through zone axes (Figure 6C). These cones encompass both in-zone reflections and several previously neglected off-zone reflections. Critically, because the gyrating beam captures most off-zone reflections sequentially and not all at once, they generally do not undergo simultaneous excitation. Consequently, PED reduces the number of plausible multibeam pathways for dynamical scattering. Furthermore, accurate measurement of PED intensities is facilitated by integration over a more complete snapshot of the Bragg condition for each observed reflection. As an ensemble, these intensities largely behave quasi-kinematically.77−79 A straightforward tactic to further minimize dynamical effects involves widening the angle of precession,77 which has been shown to systematically diminish the intensities of symmetry-forbidden reflections.35 PED also expands coverage of reciprocal space relative to sampling exclusively in-zone reflections. Nevertheless, this technique still favors locating zone-axis orientations and adds only a subset of off-zone reflections (i.e., those proximal to their in-zone counterparts). As a result, ab initio structures solved by zone-axis PED often relied on high-symmetry centrosymmetric space groups to simplify phasing and bolster completeness.80,81
A crucial step forward was taken by Kolb et al. in 2007; these researchers proposed collecting a tomographic series of diffraction patterns, using the TEM goniometer to tilt the substrate in a sequence of discrete angular steps (Figure 6E).82,83 Because the axis of the TEM goniometer is geometrically arbitrary with respect to the orientation of the crystal, ED data collected in this way represent slices of the Ewald sphere which overlap “only accidentally” with crystallographic zone axes.82 Therefore, this approach, originally termed automated diffraction tomography (ADT), banished the persistent specter of zone-axis orientations amplifying multiple scattering. Indeed, ADT deliberately ensured that most diffraction patterns were collected off-zone, providing ideal conditions for observing quasi-kinematical scattering.84,85
ADT’s most salient limitation was its tendency to leave unsampled gaps in reciprocal space between angular tilts: essentially a less severe version of the large swaths overlooked by zone-axis diffraction. Several subsequent strategies were developed to address this. ADT was swiftly combined with beam precession by Mugnaioli et al., who developed a hybrid technique coined precession-assisted electron diffraction tomography (PEDT; Figure 6D).86 PEDT represented the first ED technique to gain some level of traction as a generally applicable method for structure elucidation despite the necessity of specialized external hardware to implement beam precession.87 Alternatively, Hovmöller, Zou, and co-workers devised a means of slicing reciprocal space more finely by supplementing coarse mechanical tilts with electron beam tilts (Figure 6F).88 This approach was dubbed rotation electron diffraction (RED); it utilized custom software to enable data collection in very granular angular steps (Δη < 0.1°), which eclipsed the precision of the TEM goniometer.
These developments paved the way for arguably the most impactful methodological advance in 3D electron crystallography: continuous rotation, which was formulated nearly in parallel by Nederlof et al. and Nannenga et al. in 2013 and 2014, respectively.89,90 Unlike PEDT or RED, no ancillary hardware or software is strictly required to implement continuous-rotation ED; most commercially available TEMs can collect continuous-rotation data with little to no reconfiguration. In this technique, reciprocal space is regularly sampled in periodic intervals, while the irradiated crystal is unidirectionally rotated about the TEM goniometer axis (Figure 6B). Each diffraction pattern thus represents signal averaged over an oscillation range whose thickness in reciprocal space is given by
where ωrot is the rotational velocity of the goniometer (typically expressed in degrees per second) and τexp is the exposure time (for instance, 2–3 s).91 In practice, because no detector operates instantaneously, Δη is modified by adding a hardware-specific parameter τdead, which represents the readout time needed to store the data collected during τexp:
Especially in older systems containing slow-scan charge-coupled device (CCD) detectors, τdead can become significant. In such cases, each consecutive diffraction pattern is separated by a missing wedge (Δηdead = ωrotτdead) corresponding to the angular range left unsampled during the readout period. A viable tactic to minimize dead time entails spatial subsampling or binning each recorded frame, although this may ultimately compromise maximum achievable resolution. Nannenga et al. circumvented this issue by using a complementary metal–oxide–semiconductor (CMOS) detector in rolling-shutter mode, which provided a readout speed sufficiently high that τdead was rendered negligible relative to τexp. This breakthrough allowed continuous-rotation ED to fully sample all regions of reciprocal space accessible to the TEM goniometer. By integrating signal over an angular wedge, continuous-rotation ED also evaded all the canonical problems associated with multiple elastic scattering and partially recorded reflections. Nannenga et al. demonstrated this by using molecular replacement to solve a 2.5 Å ED structure of hen egg white lysozyme (HEWL), a protein frequently used as a standard in X-ray crystallography. HEWL crystallizes in the primitive tetragonal space group P43212, which features several sets of systematic absences orchestrated by the 43 and 21 screw axes. Critically, Nannenga et al. hunted for symmetry-forbidden reflections and found that their intensities were quite weak, contributing only ∼2.5% of observed signal relative to their symmetry-allowed counterparts (vs ∼5% for a previous ED investigation37 of HEWL using still frames collected at discrete tilts).
These results sparked a renaissance in the field. In recent years, continuous rotation has clearly emerged as the method of choice for ED data collection. This has been accompanied by a variety of acronyms, including microcrystal electron diffraction (MicroED),90 integrated electron diffraction tomography (IEDT),91 and continuous-rotation electron diffraction (cRED).92 Ultimately, these all describe the same technique. We find Gemmi et al.’s adoption of the umbrella term 3D electron diffraction (3D ED)17 a useful construct and follow this convention throughout.
3.3. Serial Electron Diffraction
To maximize sampling of reciprocal space, diffraction experiments have often relied on merging data sets collected from multiple crystals. Serial X-ray crystallography stretches this idea to its limit, exploiting X-ray free-electron lasers to collect and combine one-shot diffraction patterns extricated from hundreds of thousands of randomly oriented specimens.93 Almost instantly after producing diffraction, these exceptionally brilliant lasers leave a bleak obliteration zone in their wake, vaporizing every crystal they touch. Ironically, XFELs come closest to generating diffraction patterns undistorted by radiation damage because each successive crystal is exposed to a femtosecond-scale X-ray pulse only once before it is annihilated (as encapsulated in the mantra “diffraction before destruction”).94 Ever-faster detectors at synchrotron facilities have driven serial X-ray crystallography’s proliferation to many beamlines. Likewise, growing digitization and improved hardware have also enabled more ambitious, automated data collection strategies in ED.95−97 Recent studies have exploited the automation capabilities of modern TEMs to collect data from thousands of crystals per hour. This approach, termed serial electron diffraction (serial ED),98 generally relies on merging snapshots recorded from disparate crystals at distinct orientations, foregoing conventional sampling of a lone crystal at multiple angles. As with serial XRD, this technique exploits single exposures in an attempt to outrun radiation damage. With plenty of real estate on a typical EM grid, an experimentalist (or algorithm) can easily find dozens or possibly thousands of well-diffracting crystals during a routine search. Although many publications only report the number of crystals merged to produce a structure solution, hundreds more are typically probed and then belatedly abandoned.
Serial ED has successfully determined a small handful of structures, including HEWL, granulovirus occlusion bodies, and several highly symmetric zeolites.98,99 Elucidation of entirely novel structures remains a challenge, as it would require ab initio indexing, merging, and phasing. Nevertheless, serial ED has rapidly emerged as a potent microscopic alternative to the much larger-scale experiments conducted at synchrotrons or X-ray free-electron laser facilities. In addition to greater accessibility, the TEM unlocks another crucial advantage over conventional XFEL experiments: the power of real-space imaging. Indeed, the ability to visualize target crystals greatly streamlines the hunt for well-diffracting specimens, which for nanocrystals can be a blind and comparatively inefficient process in serial X-ray crystallography.
In this context, 4D scanning transmission electron microscopy (4DSTEM) also merits discussion because it too harmoniously combines real-space screening with reciprocal-space sampling.100 This method leverages a scanning nanobeam to record ED patterns at an array of real-space points defined by a 2D raster scan across a user-selected region of a crystalline specimen.101,102 For instance, within an illuminated area of 500 nm2, individual diffraction patterns can be collected every 20 nanometers. Conceptually, therefore, 4DSTEM provides an inherently serial approach to diffraction, simply localized with nanoscale precision onto the canvas of a single crystal. In principle, 4DSTEM’s ability to digitally pinpoint a specific nanoscale volume for data collection is quite powerful; for instance, it could allow facile deconvolution of signal from twinned, metamict, or otherwise imperfect regions present within an already submicrometer-sized crystal. 4DSTEM analysis can reveal complex mosaic substructures even in crystals anticipated to contain monolithic lattices.101 A conventional selected-area aperture is far too large to permit such granular spatial subsampling. Thanks to cryogenic conditions, 4DSTEM has also proved compatible with a range of beam-sensitive materials,103 and stepwise rotation of the TEM stage has allowed for tomographic data collection amenable to 3D structure determination. In sum, this approach permits ex post facto extraction and summation of diffraction signal from arbitrary regions of a 4DSTEM scan. These slices can subsequently be assembled into a more conventional tilt series comprehensible to standard data processing pipelines.
4. Data Processing
4.1. Data Reduction
Prior to the widespread adoption of continuous rotation, 3D ED data processing was nontrivial and somewhat opaque to the nonspecialist; it was typically handled by a suite of dedicated programs104−107 developed by a coterie of seasoned electron crystallographers. Continuous-rotation 3D ED, however, is directly analogous to rotation of a mounted crystal on an X-ray diffractometer equipped with a single-axis goniometer. As a result, 3D ED data collected in this way can undergo indexing, integration, merging, and scaling routines implemented in several software packages originally written for X-ray crystallography. With minimal modification, well-established programs such as iMosflm,108 DIALS,109 and XDS110 have all been successfully applied to continuous-rotation 3D ED data reduction. Detailed tutorials (such as for DIALS111 and XDS65) easily comprehensible to any practicing X-ray crystallographer have subsequently appeared in the literature. Likewise, current processing pipelines for serial diffraction (such as crystFEL112) have also been ported to ED data.113 Indexed lists of integrated intensities generated by these programs can directly serve as input for phasing algorithms.
4.2. Phasing by Direct Methods
It is a truth universally acknowledged that any diffraction experiment must overcome the phase problem, and ED is no exception. Since its initial demonstration by Dorset and Hauptman in 1976,46ab initio phasing has been successfully deployed on virtually all small-molecule substrates solved by 3D ED. If Sheldrick’s criterion114,115 is met (i.e., if the illuminated crystal diffracts to at least ∼1.2 Å resolution and completeness in the outermost 1.2–1.1 Å shell exceeds 50%) or exceeded, direct methods (DM) has proved a robust and reliable means of phasing ED data. As in X-ray diffraction, the presence of (a) centrosymmetry, (b) sparsely populated unit cells, and (c) heavy atoms often permits some relaxation of Sheldrick’s criterion (which is simply a conservative empirical estimate). Prior to the advent of automated software, venerable statistical approaches such as the Sayre equation and the tangent formula were applied manually, phase-by-phase. Today, widely used programs such as SHELXT116 and SHELXD117 have also found routine utility in ED data processing.
As currently implemented, DM algorithms generally hinge on two key constraints: atomicity and positivity. Because X-ray scattering amplitudes for all atoms remain non-negative regardless of resolution, positivity is a clearly justified postulate in X-ray crystallography. Indeed, the periodic electron density function recapitulated from X-ray diffraction is universally positive. An intriguing phenomenon intrinsic to ED, however, is that electron scattering amplitudes for negatively charged ions dip well below zero at low resolution, analogous to the negative scattering lengths exhibited by elements like H or Li in neutron diffraction. Consequently, in ED, anionic species can legally contribute negative density to electrostatic potential maps, a nuance to which ab initio phasing intended for X-ray diffraction is currently blind. As discussed in detail by Altomare et al., violation of the positivity postulate is expected to alter the triplet phase invariant relationships traditionally exploited by direct methods.118 Evidence from difference Fourier maps indicates that this limitation may have contributed to erroneous assignment of charged moieties (such as deprotonated carboxylates) as neutral atoms.119−121
Nevertheless, ab initio phasing by DM remains the gold standard in 3D ED, and the diverse array of structures determined by this approach has played a pivotal role in dispelling doomsday predictions about multiple scattering. Ab initio phasing has proved remarkably successful even on 3D ED data recorded from crystals hundreds of nanometers thick, despite multislice simulations122 suggesting a much lower thickness threshold for purportedly irreversible dynamical corruption. This yawning chasm between theory and experiment is fueled by many factors, such as complex mosaicity at the nanoscale,101 unmodeled inelastic scattering,123 and the now-widespread usage of off-zone data collection. DM continues to face a stiff, often insurmountable challenge from macromolecular crystals containing >50% disordered solvent, which generally fail to diffract to atomic resolution. For lower-quality diffraction data extracted from small molecules, phasing by simulated annealing124 has also proved a useful approach in 3D ED, often in conjunction with DM.125,126
4.3. Phasing by Molecular Replacement
Intrinsic disorder often prevents macromolecular crystals from diffracting to a resolution sufficiently high for direct methods. In such cases, if a search model with adequate sequence homology (generally at least 25%) is available, molecular replacement (MR) is a tried-and-tested means of phasing 3D ED data. Programs such as Phaser127 and MOLREP128 have been applied relatively seamlessly to ED; almost all protein structures solved by 3D ED have been phased via MR using an existing X-ray structure as a template. A substantial fraction originates from studies demonstrating new methodological approaches to 3D ED; this has resulted in well-studied proteins typically used as standards in X-ray crystallography (especially proteinase K, lysozyme, and catalase), accounting for over 40% of macromolecular ED structures deposited in the PDB. Comparatively few de novo structures have been determined by MR; currently, these remain limited to a handful of oligopeptides (with a maximum sequence length of 11 residues; see Table 2) and a single novel protein, R2lox129 (which was later supplanted by a higher-quality X-ray structure130).
Table 2. List of Amyloid or Amyloid-adjacent 3D ED Structures Deposited in the PDB as of October 2021, Excluding Duplicatesa.
| parent protein | amino acid sequence | PDBaccession code | phasing method | resolution (Å) | space group | Rwork/Rfree | ref |
|---|---|---|---|---|---|---|---|
| α-synuclein (68–78) | GAVVTGVTAVA | 4RIL | MR | 1.4 | C2 | 0.248/0.275 | (186) |
| α-synuclein (47–56) | GVVHGVTTVA | 4ZNN | MR | 1.4 | P21 | 0.235/0.282 | (186) |
| human islet amyloid polypeptide (19–29, S20G) | SGNNFGAILSS | 5KNZ | MR | 1.9 | P212121 | 0.228/0.275 | (188) |
| human islet amyloid polypeptide (15–25) | FLVHSSNNFGA | 5KO0 | MR | 1.4 | P1 | 0.225/0.259 | (188) |
| Sup35 (8–13) | Zn-NNQQNY | 5K2E | DM | 1.0 | P21 | 0.152/0.194 | (187) |
| Sup35 (8–13) | Cd-NNQQNY | 5K2F | DM | 1.0 | P21 | 0.220/0.241 | (187) |
| Sup35 (7–13) | GNNQQNY | 5K2G | DM | 1.1 | P21 | 0.187/0.224 | (187) |
| Sup35 (7–13) | GNNQQNY | 5K2H | DM | 1.05 | P212121 | 0.177/0.186 | (187) |
| Tau (306–311) | VQIVYK | 5K7N | DM | 1.1 | C2 | 0.210/0.223 | (64) |
| Tau (591–600) | KVQIINKKLD | 5V5B | MR | 1.5 | P21 | 0.190/0.213 | (189) |
| Tau (592–597) | VQIINK | 5V5C | MR | 1.25 | P21212 | 0.219/0.266 | (189) |
| Tau (305–310) | SVQIVY | 6ODG | DM | 1.0 | P21 | 0.245/0.266 | (190) |
| TDP-43 (333–343) | SWGMMGMLASQ | 6CFH | MR | 1.5 | P1 | 0.280/0.313 | (192) |
| TDP-43 (312–317, A315pT) | NFGpTFS | 6CF4 | DM | 0.75 | P212121 | 0.232/0.251 | (192) |
| TDP-43 (312–317, A315E) | NFGEFS | 5WKB | DM | 1.0 | P21212 | 0.220/0.270 | (192) |
| TDP-43 (247–257) | DLIIKGISVHI | 5W52 | MR | 1.4 | P1 | 0.262/0.306 | (191) |
| bank vole prion protein (168–176) | QYNNQNNFV | 6AXZ | DM | 0.75 | P1 | 0.242/0.246 | (165) |
| human prion protein (169–175) | GSNQNNF | 6CLC | DM | 1.01 | P1 | 0.159/0.178 | (261) |
| InaZ (707–712) | rac-GSTSTA | 6M9J | DM | 0.9 | P21/c | 0.233/0.252 | (193) |
| InaZ (707–712) | GSTSTA | 6M9I | DM | 0.9 | P212121 | 0.217/0.232 | (193) |
| Nup98 (116–123) | GFGNFGTS | 6BZM | DM | 0.9 | P1 | 0.226/0.264 | (183) |
| amyloid-β (20–34, D23iD) | FAEiDVGSNKGAIIGL | 6NB9 | DM | 1.05 | P21 | 0.198/0.246 | (195) |
| amyloid-β (20–34) | FAEDVGSNKGAIIGL | 6OIZ | DM | 1.1 | P21 | 0.194/0.213 | (195) |
| amyloid-β (24–34) | VGSNKGAIIGL | 5VOS | MR | 1.42 | P21 | 0.234/0.292 | (273) |
| heterogeneous nuclear ribonucleoprotein A1 (209–217) | GFGGNDNFG | 6J60 | DM | 0.96 | P212121 | 0.233/0.248 | (196) |
| fused in sarcoma (77–82) | STGGYG | 6BZP | DM | 1.1 | P212121 | 0.219/0.255 | (183) |
| fused in sarcoma (37–42) | SYSGYS | 5XSG | DM | 0.73 | P21 | 0.261/0.289 | (197) |
| fused in sarcoma (37–42) | SYSGYS | 6KJ1 | DM | 0.65 | P21 | 0.229/0.240 | (198) |
| fused in sarcoma (37–42) | SYSGYS | 6KJ3 | DM | 0.6 | P21 | 0.307/0.326 | (198) |
| Tau (591–599) | KVQIINKKL | 6NK4 | MR | 1.99 | P61 | 0.260/0.299 | (274) |
| amyloid-β (16–26, D23N) | KLVFFAENVGS | 6O4J | MR | 1.4 | P21 | 0.237/0.283 | (194) |
| OsPYL/RCAR5 (24–29) | AVAAGA | 6UOR | DM | 0.9 | P212121 | 0.206/0.240 | (102) |
Abbreviations used: MR = molecular replacement, DM = direct methods, pT = phosphorylated l-threonine, rac = racemic, iD = l-isoaspartic acid.
If existing models prove insufficient for MR and >1.2 Å resolution nullifies DM, fragment-based phasing (FBP) has emerged as a potential alternative enabling structure determination. As implemented in the ARCIMBOLDO suite of programs, this approach mines focused fragment libraries derived from distant homologues or idealized elements of secondary structure (such as polyalanine α-helices). Iterative omission or placement of these fragments into a nascent structure solution allows for assessment of their respective phasing power. Ultimately, structures phased by this method fall conceptually closer to MR than to DM, although not quite as phase-biased as MR from a unitary model. Originally demonstrated on a variety of X-ray data sets by Usón and co-workers,131,132 FBP has recently been extended to a few ED cases where MR and DM had collectively proved ineffective,102,133 in addition to a proof-of-concept FBP structure of proteinase K.134 Interestingly, FBP appears uniquely suited to probe smaller species with less predictable folds, including polymorphic amyloid oligopeptides.133 Like MR, FBP is also theoretically compatible with fragments harvested from computationally generated models (i.e., AlphaFold135 or RoseTTAFold136 for proteins, or DFT for small molecules), removing the need for an experimentally determined template.
5. Structure Refinement
5.1. Theoretical Background
3D ED recapitulates a three-dimensional map of electrostatic potential derived from interaction between the incident electron beam and the substrate under interrogation. Refinement of ESP maps is carried out by programs such as Phenix,137 REFMAC,138 and SHELXL,139 which attempt to iteratively minimize the discrepancy between theoretically calculated (Fcalc) and experimentally observed (Fobs) structure factors in reciprocal space. Ultimately, the agreement between Fcalc and Fobs is encapsulated in a residual or R-factor, which is defined as
and is generally reported as a universal validation metric to assess map quality. Computation of Fcalc hinges on approximations of constituent atoms in terms of their parametrized electron scattering factors:
where fj(s) is the individual electron scattering factor for the jth atom, h, k, and l correspond to the Miller indices, and x, y, and z give the fractional coordinates of the jth atom in real space. Just like XRD, each atomic scattering factor is treated as a sum of Gaussians, given the computational tractability of calculating Fourier transforms on Gaussian functions. These take the general form
where j = 4 or 5 and aj and bj represent arbitrary fitting coefficients. Some approximations also add a scalar constant
which can augment the accuracy of the Gaussian fit. Specifically for ionic electron scattering factors, a divergent charge-correction term is historically used
where ΔZ = Z – Z0 and therefore represents excess nuclear charge.20 The above equation yields a very accurate fit for ionic electron scattering factors. Unfortunately, because of the resultant singularity at s = 0, inclusion of a divergent charge-correction term is incompatible with widely used refinement programs, rendering such parametrizations unusable for routine analysis of continuous-rotation 3D ED data. This dearth has forced groups interested in the process of refining charged species to compute their own parametrizations.140 As a resource for the community, we have developed a publicly accessible web server, factors of atomic electron scattering (FAES, https://srv.mbi.ucla.edu/faes), which returns refinement-friendly parametrizations of all electron scattering factors currently tabulated in the International Tables for Crystallography, as well as fractionally charged scattering factors computed via linearly weighted combinations of integer parents. We also harness FAES’ 5 Gaussian parametrization to derive elastic and estimated inelastic cross-sections for all neutral elements.
A survey of published 3D ED structures, encompassing oligopeptides (Table 2), small molecules (Table 3), and proteins (Table 4) reveals average refinement residuals in the ∼20–30% range, markedly greater than values typically observed in XRD (Figure 7). To a certain extent, however, this gap is cosmetic. In many cases, structures generated by 3D ED have yielded stubbornly inflated refinement R-factors despite featuring no errors in atomic assignment or placement. Additional validation of these ESP maps is provided by all-atom RMSD analyses relative to known X-ray structures, which often compare very favorably. Especially if initial data reduction statistics (such as Rmeas, ⟨I/σ(I)⟩, and CC1/2) appear well-behaved, elevated refinement R-factors may partially reflect systematic inaccuracies in computation of Fcalc rather than deficiencies in the atomic model itself. For instance, although 3D ED modalities such as continuous rotation and precession minimize the effects of multiple elastic scattering, dynamical diffraction can still distort structure-factor amplitudes. Conventional refinement procedures (in programs originally written for X-ray diffraction) neglect this and simply assume singular elastic scattering. To rectify this oversight, a series of studies by Palatinus and co-workers has formulated a refinement approach which incorporates dynamical diffraction theory into calculation of model structure factors.107,141,142 As implemented in Jana2006, this procedure has diminished refinement R-factors for 3D ED data and seemingly enhanced the ability to detect granular details such as H atoms in Fourier difference maps.143 Nevertheless, dynamical refinement is not yet a routine procedure, partially because its computational expense renders it currently unsuitable for larger systems like macromolecules. Alternative approaches involve application of various correction factors to measured intensities,140,144 including off-label use of a primary extinction parameter originally intended for X-ray diffraction.145 These methods may help compensate for lingering dynamical effects.
Table 4. List of Macromolecular 3D ED Structures Deposited in the PDB as of October 2021a.
| protein | sequence length | PDB accession code | phasing method | resolution (Å) | space group | Rwork/Rfree | ref |
|---|---|---|---|---|---|---|---|
| HEWL (tetragonal polymorph) | 129 | 3J4G | MR | 2.9 | P43212 | 0.255/0.278 | (37) |
| HEWL (tetragonal polymorph) | 129 | 3J6K | MR | 2.5 | P43212 | 0.220/0.255 | (38) |
| catalase | 527 | 3J7B | MR | 3.2 | P212121 | 0.262/0.308 | (90) |
| calcium ATPase | 994 | 3J7T | MR | 3.4 | C2 | 0.277/0.315 | (157) |
| HEWL (orthorhombic polymorph) | 129 | 5A3E | MR | 2.5 | P212121 | 0.213/0.253 | (38) |
| catalase | 527 | 5GKN | MR | 3.2 | P212121 | 0.251/0.304 | (158) |
| proteinase K | 279 | 5I9S | MR | 1.75 | P43212 | 0.217/0.266 | (283) |
| HEWL (tetragonal polymorph) | 129 | 5K7O | MR | 1.8 | P43212 | 0.239/0.284 | (64) |
| xylanase | 190 | 5K7P | MR | 2.3 | P212121 | 0.230/0.267 | (64) |
| thaumatin | 207 | 5K7Q | MR | 2.5 | P41212 | 0.251/0.294 | (64) |
| trypsin | 223 | 5K7R | MR | 1.7 | P212121 | 0.248/0.281 | (64) |
| proteinase K | 279 | 5K7S | MR | 1.6 | P43212 | 0.224/0.255 | (64) |
| thermolysin | 316 | 5K7T | MR | 2.5 | P6122 | 0.290/0.310 | (64) |
| HEWL (orthorhombic polymorph) | 129 | 5O4W | MR | 2.11 | P21212 | 0.335/0.350 | (284) |
| HEWL (orthorhombic polymorph) | 129 | 5OCV | MR | 2.2 | P21212 | 0.236/0.270 | (285) |
| TGF-β/TGF-β receptor 2 complex | 103/97 | 5TY4 | MR | 2.9 | P212121 | 0.292/0.328 | (64) |
| proteinase K | 279 | 6CL7 | MR | 1.71 | P43212 | 0.221/0.253 | (261) |
| NaK ion channel | 96 | 6CPV | MR | 2.5 | I4 | 0.218/0.263 | (286) |
| HEWL (tetragonal polymorph) | 129 | 6H3V | MR | 1.9 | P43212 | 0.291/0.283 | (68) |
| HEWL (monoclinic polymorph) | 129 | 6HU5 | MR | 2.8 | P21 | 0.297/0.339 | (287) |
| catalase (energy-filtered) | 527 | 6JNT | MR | 3.0 | P212121 | 0.251/0.283 | (170) |
| catalase (energy-filtered) | 527 | 6JNU | MR | 3.0 | P212121 | 0.207/0.251 | (170) |
| thiostrepton | 19 | 6MXF | MR | 1.91 | P43212 | 0.190/0.218 | (200) |
| CTD-SP1 fragment of HIV-1 Gag | 110 | 6N3J | MR | 3.0 | C2 | 0.254/0.292 | (250) |
| proteinase K (FIB-milled) | 279 | 6N4U | MR | 2.75 | P43212 | 0.238/0.263 | (70) |
| R2-like ligand-binding oxidase (R2lox) | 328 | 6QRZ | MR | 3.0 | P21212 | 0.318/0.335 | (129) |
| proteinase K | 279 | 6V8R | FBP | 1.6 | P43212 | 0.195/0.232 | (134) |
| acetazolamide-bound human carbonic anhydrase II | 260 | 6YMA | MR | 2.5 | P21 | 0.224/0.255 | (249) |
| human carbonic anhydrase II | 260 | 6YMB | MR | 2.5 | P21 | 0.249/0.276 | (249) |
| granulovirus occlusion body | 248 | 6S2O | MR | 1.55 | I23 | 0.171/0.197 | (99) |
| HEWL (tetragonal polymorph) | 129 | 6S2N | MR | 1.8 | P43212 | 0.272/0.316 | (99) |
| catalase | 527 | 7DI8 | MR | 3.2 | P212121 | 0.309/0.348 | (288) |
| thermolysin | 316 | 6ZHJ | MR | 3.26 | P6122 | 0.210/0.292 | (140) |
| thaumatin | 207 | 6ZHN | MR | 2.76 | P41212 | 0.280/0.321 | (140) |
| voltage-dependent anion-selective channel protein 1 | 295 | 7KUH | MR | 3.12 | C2 | 0.257/0.287 | (289) |
| bovine insulin | 21/30 | 6ZHB | MR | 3.25 | H3 | 0.181/0.319 | (140) |
| myeloid differentiation primary response 88 | 151 | 7BEQ | MR | 3.0 | C2 | 0.223/0.280 | (290) |
| proteinase K (LCP) | 279 | 6PQ0 | MR | 2.0 | P43212 | 0.217/0.267 | (291) |
| proteinase K (LCP) | 279 | 6PQ4 | MR | 2.0 | P43212 | 0.244/0.282 | (291) |
| CypA | 165 | 6U5G | MR | 2.5 | P212121 | 0.185/0.224 | (292) |
| human adenosine receptor | 447 | 7RM5 | MR | 2.79 | C2221 | 0.248/0.288 | (248) |
| vancomycin (triclinic polymorph) | 7 | 7C4V | MR | 1.05 | P1 | 0.232/0.268 | (293) |
| vancomycin (orthorhombic polymorph) | 7 | 7C4U | MR | 1.2 | P22121 | 0.202/0.216 | (293) |
| granulovirus occlusion body | 248 | 6YNG | MR | 2.83 | I23 | 0.184/0.226 | (99) |
| proteinase K | 279 | 6ZEV | MR | 2.4 | P43212 | 0.200/0.243 | (295) |
| proteinase K | 279 | 6ZET | MR | 2.7 | P43212 | 0.225/0.268 | (295) |
| proteinase K | 279 | 6ZEU | MR | 2.0 | P43212 | 0.199/0.234 | (295) |
Vancomycin and thiostrepton, although more akin to small molecules, are categorized here because of their presence in the PDB.
Figure 7.

Circles represent the mean resolution and refinement R-factor (R1 for small molecules, Rwork for peptides and proteins) for each category of substrate, whereas error bars signify one standard deviation in each direction. Data were taken from Tables 2, 3, and 4.
Another potential source of error lies in fe(s) itself. Inverse Fourier transforms of conventional electron scattering factors ultimately yield spherical, isotropic distributions capable of accommodating a Gaussian model. This is emblematic of electrostatic potential projected by an isolated atom. In real systems, however, ESP almost always experiences perturbations due to environmental effects. Specifically, low-angle scattering is especially sensitive to the redistribution of valence electrons which accompanies ionization or chemical bonding. Isolated scattering factors disregard these effects. Chang et al. analyzed this issue by conducting Hartree–Fock molecular orbital calculations at the 6-31G* level of theory, which they then transformed into substrate-specific molecular electron scattering factors.146 Yamashita and Kidera developed a similar treatment using the hybrid functional B3LYP, decomposing output from DFT into parametrized, atom-specific contributions.147 Both investigations concluded that ESP is represented more accurately by aspherical, anisotropic scattering factors, particularly at low spatial frequencies. However, neither of these approaches has since been applied in a generalizable or user-friendly fashion to experimental 3D ED data sets. More recent work by Dominiak and co-workers148,149 has focused on refining 3D ED data against aspherical ESP produced by applying the Mott–Bethe formula to multipolar electron density distributions tabulated in databases such as ELMAM2.150 Nonetheless, this method ultimately led only to an incremental (1–2%) improvement in refinement R-factors, suggesting that the isolated atom model (although imperfect) is fairly accurate for neutral atoms, particularly at high resolution. For charged species, however, isolated electron scattering factors’ sharp divergence to infinity is likely a significant exaggeration of ionic ESP in crystal structures, where excess charge is either balanced by the presence of proximal counterions or diluted by noncovalent interactions such as hydrogen bonding. To indirectly account for this, it is helpful to introduce fractionally charged scattering factors, which can provide a proxy for modeling effective, partial, or delocalized charge.
5.2. Charged Species
Historically, several 2D electron crystallographic studies had already demonstrated that ionic electron scattering factors’ divergent behavior as s → 0 renders ED uniquely capable of differentiating neutral atoms and ionized states. Grigorieff et al. observed effects consistent with negative charge in their 3.5 Å 2D ED structure of bacteriorhodopsin, where they visualized weakly resolved electrostatic potential enveloping the carboxylate termini of aspartate and glutamate side chains.151 Similar findings were reported by Fujiyoshi and co-workers, who recorded systematically absent ESP for several putatively deprotonated aspartate and glutamate residues in bacteriorhodopsin at 3.0 Å.152 These artifacts materialized most prominently in low-resolution shells, where ionic electron scattering amplitudes diverge strikingly from their neutral counterparts. Kimura et al. provided a compelling validation of theory by calculating experimental ESP maps omitting low-resolution reflections, which regenerated positive density around ionized carboxylates.153 Intriguingly, Fujiyoshi and co-workers also visualized negative peaks on backbone carbonyl O atoms in Fourier difference maps computed assuming neutral electron scattering factors, suggesting experimentally observable partial charge even on formally neutral moieties. Indeed, Fujiyoshi and co-workers obtained slightly diminished refinement R-factors by assigning fractional charges of +0.5 and −0.5 to carbonyl C and O atoms, respectively. Later work by Hirai et al. further validated a range of these observations via computational simulations of charged states.154 In addition to these extensive studies on proteins, ionic ESP has also been analyzed quantitatively in inorganic salts, where bonding features far less covalent character.155,156
A string of investigations by Yonekura and co-workers has propelled the study of ionized states into 3D ED territory.157−159 Their results have largely reproduced the effects previously observed by their 2D predecessors: anions contribute negative density to ESP maps, whereas cations lead to modest enhancements in scattering power. In parallel, Wang has catalogued a variety of artifacts in ESP maps which may indicate the presence of deprotonated carboxylates incorrectly modeled as neutral oxygen atoms.119 Specifically, several experimental 3D ED structures feature (a) strong negative peaks localized on carboxylate O atoms in Fourier difference maps calculated presuming neutral electron scattering factors, (b) weak or nonexistent density enveloping these O atoms in experimental ESP maps, and (c) aberrantly high, physically absurd B-factors associated with the offending atoms. Ions mistreated in this way would also increase refinement R-factors. Yonekura and co-workers managed to mitigate this via implementation of fractionally charged scattering factors.158 Collectively, these studies underscore the necessity of integrating treatment of charged states as a routine facet of 3D ED analysis. Although this has been thwarted by the nonexistence of appropriately parametrized scattering factors, we hope tools like FAES and the ScatCurve package158 developed by Yonekura and Maki-Yonekura clear a path toward refinement of ionic species in 3D ED data.
Finally, a currently underexplored strategy to unequivocally validate differences resulting from charge is joint refinement160 of 3D ED structures alongside corroborating X-ray diffraction data. Because electron density in X-ray structures is universally positive and comparatively insensitive to charge, ESP from 3D ED could potentially convey complementary information about ionized states. Furthermore, 3D ED usually suffers from relatively low completeness; this is easily rectified by addition of X-ray data, which is typically highly redundant and much more complete. More uncharted territory is also provided by the prospect of joint refinement with neutron diffraction, which could serve as a useful cross-validation metric for localization of hydrogen atoms.161,162 For instance, 3D ED has already demonstrated its potential to elucidate structures of transition-metal hydrides,163 a family of organometallic complexes which has historically relied on single-crystal neutron diffraction for solid-state detection of H atoms.164 Although the hydride ligand carries a formal negative charge, many species classified as “hydrides” nevertheless display acidic properties. 3D ED offers the tantalizing possibility of evaluating hydridic character via analysis of ionic ESP, whereas neutron diffraction can easily corroborate spatial positions of H atoms. Interestingly, however, bond lengths involving H atoms will likely prove slightly inconsistent between ED, XRD, and neutron diffraction, as these three forms of incident quanta all interact with hydrogen in appreciably different ways. Incident electrons experience perturbation due to both positively charged nuclei and atomic charge density projected by the electron cloud, placing them in between the two extremes of X-rays (which interact solely with the cloud) and neutrons (which interact solely with atomic nuclei). Such variability has already been noted in a 0.75 Å ED structure of a prion protofibril,165 as well as a 1.22 Å single-particle cryo-EM structure of apoferritin.166 In both of these cases, individual H atoms in Fourier difference maps appeared consistently different from their putative X-ray positions, indicating observable deviation from the idealized geometry of the riding model. Joint refinement would allow for a detailed analysis of such discrepancies.
5.3. Energy Filtration
Every practical aspect of crystallography is substantially influenced by the energy of the incident quanta. Thanks to the energy–time uncertainty principle, a perfectly coherent beam is forbidden by quantum mechanics, and the incident energy of the impinging electrons is properly described as a statistical distribution (with a full-width half-maximum of ΔE) in lieu of a discrete value E.167 Typical TEM instruments suitable for 3D ED employ either field-emission guns (FEGs) or thermionic cathodes (containing tungsten hairpin filaments or lanthanum hexaboride crystals) as electron sources, all of which feature their own characteristic ΔE ranges. FEGs generate an especially coherent beam, with an energy spread ΔE of < 0.7 eV; W filaments and LaB6 crystals exhibit less monochromatic ΔE values of 1.5–3 and 1–2 eV, respectively.167 Nevertheless, 3D ED structures have been routinely solved using instruments employing all three sources, which compare favorably to the monochromaticity obtained using an in-house X-ray diffractometer. In HRTEM imaging, ΔE has direct experimental repercussions; in reciprocal space, its influence on phase contrast is captured by a damped envelope function which delineates the maximum achievable resolution for an image. Such chromatic aberration (resulting from inherent fluctuations in energy within the incident beam) is also reflected in measured diffraction patterns, albeit more indirectly; it effectively causes the surface of the Ewald sphere to thicken. However, its influence on diffraction is not quite as consequential as its impact on imaging. Indeed, ΔE is quite small compared to the energy dispersion induced by the complex set of elastic and inelastic scattering events arising from the beam impinging upon an illuminated crystal.
In this context, postspecimen energy filtration (achieved, for example, via installation of a postcolumn filter) is an impactful and currently underutilized strategy in 3D ED. Energy filtration allows selective exclusion of any scattered electrons which suffered some degree of energy loss (resolved, for instance, within fixed-width windows of 10, 50, or 100 eV). Theoretically, diffraction signal is contributed largely by elastically scattered quanta residing within or very near the zero-loss peak, whereas inelastically scattered electrons mostly generate diffuse noise. Zero-loss energy filtration purges any evidence of inelastic scattering events polluting regions of reciprocal space proximal to Bragg reflections. Consequently, it significantly augments the accuracy of integrated intensities.168
This phenomenon is especially well-illustrated by filtration of diffraction recorded from thick, frozen-hydrated specimens. In these systems, a substantial fraction of scattering signal is contributed by amorphous solvent and vitreous ice. For instance, in protein crystals, unfiltered ED patterns often feature a dense halo of low-frequency noise protruding radially from the central beam. By using an in-column energy filter, Yonekura and co-workers demonstrated that much of this detrimental noise is easily eliminated; energy filtration (with a slit width of 10 eV relative to the zero-loss peak) resulted in a pronounced enhancement in signal-to-noise for all reflections.168−170 Furthermore, removal of inelastically scattered electrons disinterred a range of low-resolution reflections previously occluded by the diffuse penumbra emanating from the central beam (a dramatic illustration is provided by Figure 4 in ref (170)). Because Bragg peaks at mid-to-low spatial frequencies encipher crucial information about scattering differences between elements (as well as distinctions between neutral atoms and charged states), unveiling these reflections could have deeper consequences beyond more accurate integration of intensities. For instance, in addition to enabling proper refinement of ionized species, accentuation of scattering differences could potentially facilitate experimental phasing by multiple isomorphous replacement (MIR). Because discrepancies between elements become comparatively muted in ED versus XRD, MIR has been proposed171 but never convincingly demonstrated in ED. Presumably the likelihood of reliably detecting these discrepancies would grow if energy filtration enabled facile detection and integration of low-resolution reflections. Similar logic also applies to radiation-induced phasing.172
Intriguingly, inelastically scattered electrons which undergo relatively small energy losses can also end up within the vicinity of Bragg peaks. In fact, unfiltered Bragg reflections really represent a coalescence of signal from singular and multiple elastic scattering in tandem with a non-negligible fraction of inelastic scattering events. By removing any contributions from inelastic collisions, zero-loss energy filtration would in principle provide a more accurate measurement of multiple elastic scattering.123 Dynamical refinement would presumably profit considerably from this. Although dynamical refinement explicitly seeks to treat effects arising from multiple elastic scattering, it is currently challenged by the prospect of accurately accounting for inelastically scattered electrons also contributing to individual integrated intensities. Finally, despite yielding cleaner diffraction patterns, energy filtration’s extraction of inelastically scattered signal comes at the expense of attenuating the intensities of weak, high-resolution reflections. This tradeoff indicates that the net impact of energy filtration on 3D ED is likely to be nuanced, and future investigations would benefit from a systematic comparison of data reduction and refinement statistics against filter slit width.
5.4. Absolute Structure and Absolute Configuration
An especially impactful aspect of X-ray crystallography is its ability to routinely determine the absolute configuration of individual stereocenters in chiral molecules.173 X-ray diffraction’s sensitivity to chirality is conferred by anomalous dispersion, a resonant scattering effect which leads to enantiospecific violation of Friedel’s law.174 Analogously, in electron diffraction, a similar breakdown of Friedel symmetry is caused by multiple elastic scattering.29,30 Recent work by Brázda et al. has shown that this discrepancy is detectable using dynamical refinement, which permits discrimination between enantiomers via an R-factor comparison against the inverted structure.175 This approach derives its sensitivity to chirality from an incorporation of dynamical effects into computation of Fcalc (distinguishing it from a standard kinematical refinement, where enantiomorphic crystals would yield identical distributions of calculated structure factors and therefore identical R-values). Initially, however, this type of procedure may appear somewhat counterintuitive, as methodological developments in 3D ED have followed a trajectory specifically intended to diminish the effects of multiple scattering. For instance, electron diffraction patterns collected via continuous rotation have proven generally devoid of dramatic dynamical artifacts. Today’s status quo is a far cry from historical work, where aberrations such as violation of systematic absences were both very strong and routinely observed.14 To quote Dorset,176 “certainly the existence of higher voltage sources than used in pioneering work allows the quasi-kinematical approximation to be satisfied for samples that would have caused problems” in the past. In this context, it remains somewhat unclear exactly how much dynamical diffraction is (a) quantifiably present in 3D ED patterns and (b) strictly necessary to reliably detect disruption of Friedel symmetry and confidently assign absolute structure.
To further develop the analogy to conventional X-ray crystallography, XRD’s capacity to detect absolute chirality is directly tethered to the strength of the observed anomalous signal. As a result, X-ray methods did not always yield a reliable readout of absolute chirality in systems where resonant scattering was inherently weak, such as organic compounds composed entirely of lighter atoms. These cases necessitated the development of alternative statistical approaches with heightened sensitivity to differences in Bijvoet pair intensity, such as the Bayesian methods outlined by Hooft and co-workers.177 In 3D ED, a rigorous examination of dynamical scattering’s sensitivity to several similarly intertwined variables is currently lacking. For instance, parameters such as accelerating voltage, elemental composition, defects or imperfections in lattice structure, and variable thicknesses across data sets merged from several crystals would all systematically alter the probability of multiple elastic scattering. In cases where nanocrystals have been milled to thicknesses at or below the elastic mean free path of the material, dynamical scattering is expected to be weak or unobservable. Nevertheless, the outlook for 3D ED appears promising, as recent work by Klar et al. has extended the scope of dynamical refinement to a wide range of data sets collected using continuous rotation.178 An encouraging experiment reported by Klar et al. involves a double-blind comparison against analogous X-ray data collected on a chiral zeolite. In this case, dynamical refinement on 3D ED data returned internally consistent results with an independent assessment of absolute structure made via the Flack parameter. Remaining challenges include implementation in a realistic case where absolute chirality is genuinely unknown, such as a crystalline sample obtained from a synthetic mixture with poor enantiomeric excess. Intriguingly, simulations by Spence and Donatelli have suggested that retrieval of chirality via exploitation of dynamical effects is thwarted both by very low thicknesses and by very high thicknesses.179 A detailed experimental investigation of the conditions under which this approach is expected to falter is still required. Regardless, in the absence of appreciable dynamical signal, 3D ED remains perfectly capable of inferring stereochemistry relative to an internal chiral reference, such as another stereocenter whose absolute configuration is known a priori. One viable strategy to achieve this involves cocrystallization with enantiopure additives.180
6. Applications
6.1. Amyloids and LARKS
Continuous-rotation 3D ED has emerged as a highly useful tool for studying the atomic structure of amyloid-forming peptides. A wide range of proteins can access the amyloid state, which is marked by dense fibrillar aggregates of interdigitated β-sheets cross-linked by hydrogen bonds.181,182 Accumulation of these aggregates is implicated in several fatal diseases, such as transmissible spongiform encephalopathy, Alzheimer’s disease, Parkinson’s disease, and Huntington’s chorea. Amyloid fibrils exhibit a characteristic left-handed helical twist arising from their cross-β-sheet architecture. This makes it difficult for amyloidogenic proteins to crystallize in the fibrillar state because the translational symmetry imposed by the Bravais lattice forcibly restricts their ability to twist.182 Usually, the ensuing buildup of lattice strain prohibits the growth of X-ray-scale crystals. An analogous set of circumstances is presented by intrinsically disordered proteins (IDPs) containing low-complexity aromatic-rich kinked segments (LARKS), which often congeal into semisolid hydrogels.183 These species also exhibit amyloid-like cross-β-sheet morphology, although fibrils formed by LARKS appear more susceptible to chemically induced denaturation than their amyloid counterparts.
In 2001, Eisenberg and co-workers discovered that short oligopeptide fragments (4–7 residues) of amyloidogenic proteins do form microcrystals amenable to synchrotron X-ray diffraction at microfocus beamlines.184,185 Nevertheless, these peptides’ propensity to crystallize tended to diminish with increasing sequence length, and a number of species continued to stubbornly resist X-ray-scale crystallization. These circumstances prompted a prescient attempt at electron diffraction of nanocrystalline GNNQQNY, a seven-residue peptide from the yeast prion protein Sup35.34 Remarkably, GNNQQNY nanocrystals divulged clear Bragg peaks at comfortably subangstrom (∼0.7 Å) resolution, and the corresponding diffraction patterns permitted indexing of reasonable orthorhombic unit cell parameters. Despite their ultrahigh resolution, these 3D ED data were recorded as a discrete tilt series of still frames, which apparently thwarted reliable integration of diffracted intensities. In addition to prohibitively partial sampling of Bragg reflections, Diaz-Avalos et al. observed weak violations of 21 systematic absences, suggesting some degree of distortion by multiple scattering.
Although this initial foray into electron diffraction did not allow full structure elucidation, this study clearly foreshadowed future success, which arrived 12 years later. For nearly a decade, the 11-residue core of the amyloidogenic protein α-synuclein (termed NACore), a key component of Lewy bodies in Parkinson’s disease, had yielded only submicrometer-sized crystals invisible to optical microscopy. Despite years of extensive attempts at X-ray-scale crystallization, this species exclusively formed nanocrystals with dimensions smaller than the wavelength of visible light. In 2015, Rodriguez et al. subjected frozen-hydrated NACore nanocrystals to ED using continuous rotation, which facilitated accurate integration of intensities out to a resolution of 1.4 Å.186 These data were successfully phased via molecular replacement to yield the first novel solid-state structure solved by continuous-rotation ED. Subsequent reinvestigation of GNNQQNY by Sawaya et al. once again yielded high-resolution diffraction (∼1.0–1.1 Å), this time amenable to successful structure determination via direct methods.187 Notably, continuous rotation greatly minimized the presence of dynamical scattering artifacts, which failed to impede ab initio phasing.
Since these proof-of-concept studies, a slew of amyloidogenic peptide fragments, as well as a smaller subset of LARKS, has been investigated by continuous-rotation 3D ED (Table 2). These include segments derived from human islet amyloid polypeptide,188 several isoforms of tau,64,189,190 TAR DNA-binding protein 43,191,192 bank vole prion protein,165 the ice-nucleation protein InaZ,193 amyloid-β,194,195 heterogeneous nuclear ribonucleoprotein A1,196 fused in sarcoma (FUS),183,197,198 and nuclear pore complex protein 98.183 Several of these reports exploited the atomic-resolution information provided by 3D ED to design small-molecule or peptide inhibitors of amyloid fibril aggregation, highlighting 3D ED’s potential to elucidate key structural details relevant to drug discovery.188−190 Many of these amyloidogenic peptide structures ultimately tell a similar story: in addition to collectively displaying canonical amyloid-like features such as steric zippers, several refused to yield X-ray-scale crystals despite considerable effort. A fairly typical example is Guenther et al.’s 1.0 Å structure of NFGEFS (Figure 8A), which features face-to-back packing of parallel in-register sheets.192 Additionally, Gallagher-Jones et al.’s 0.75 Å structure of QYNNQNNFV unveiled a unique structural motif termed a polar clasp (Figure 8B),165 whereas Warmack et al.’s 1.1 Å structure of the 15-residue peptide FAEiDVGSNKGAIIGL extended the scope of direct methods to the lengthiest sequence yet (Figure 8C).195 Finally, Zhou et al.’s 0.6 Å structure of SYSGYS,198 a hexapeptide derived from the low-complexity domain of FUS, is noteworthy for its unusually granular resolution, although this species has also been solved at 1.1 Å via synchrotron X-ray diffraction.183
Figure 8.

Ab initio atomic-resolution 3D ED structures of three novel oligopeptide fragments derived from pathologically relevant proteins. Carbon atoms and the peptide backbone are rendered in blue, oxygen atoms in orange, and nitrogen atoms in purple. (A) 1.0 Å resolution structure of 312NFGEFS317 (PDB 5WKB), a hexapeptide segment from the low-complexity domain of the A315E familial mutant of TAR DNA-binding protein 43. (B) 0.75 Å resolution structure of 168QYNNQNNFV176 (PDB 6AXZ), a nonapeptide segment from the β2−α2 loop of the bank vole prion protein. (C) 1.1 Å resolution structure of 20FAEiDVGSNKGAIIGL34 (PDB 6OIZ), a 15-residue segment from wild-type amyloid-β.
6.2. Small Molecules
In 2018, the near-simultaneous release of two papers by Gruene et al.(199) and Jones et al.(200) generated an abrupt resurgence of interest in applying continuous-rotation ED techniques to small molecules.201−203 By this point, a considerable number of small-molecule structures had already been deciphered by 3D electron crystallography (by Dorset, Abrahams, Hovmöller, Kolb, and others, in addition to an extensive body of historical work by Vainshtein). Nevertheless, these two reports transformed the landscape of 3D electron crystallography by re-exposing its potential to a nonspecialist audience. Synthetic chemists, for instance, frequently produce small quantities of seemingly amorphous powders recalcitrant to X-ray-scale crystallization. In this context, ED’s ability to extract atomic-resolution diffraction from nanocrystals is potentially liberating.
For instance, Jones et al. solved a 0.77 Å structure of synthetic (+)-limaspermidine from a few milligrams of solid residue obtained after in vacuo evaporation of eluent from flash column chromatography (Figure 9A).200 Furthermore, Jones et al. went on to determine four independent structures of biotin, acetaminophen, cinchonine, and brucine from a heterogeneous mixture of powders deposited on a single grid. At the bulk scale, overlapping signals from different components in this mixture would likely have prohibited clear disambiguation via X-ray powder diffraction or NMR spectroscopy. These results demonstrated how ED could function as a powerful addition to the synthetic chemist’s toolbox. Not only does ED slot conveniently into established purification workflows, often obviating any need for formal recrystallization, it also offers elusive solid-state structural information potentially inaccessible via conventional methods.
Figure 9.

ORTEP diagrams of five ab initio small-molecule 3D ED structures, with H atoms omitted. Carbon atoms are rendered in blue, nitrogen atoms in lilac, oxygen atoms in red, chlorine atoms in sea green, and copper atoms in orange. All thermal ellipsoids are drawn at 50% probability, except for compound D, which is depicted at 15% for clarity. (A) 0.77 Å resolution structure of synthetic (+)-limaspermidine (CSD: CAHKUU01), a monoterpene indole alkaloid featuring a cis-fused azadecalin core. Suitable microcrystals were obtained directly from flash column chromatography, without any formal recrystallization. (This compound did not undergo B-factor refinement, so its thermal ellipsoids do not carry any physical meaning.) (B) 0.9 Å resolution structure of the analgesic orthocetamol (CSD: WOFXEX), refined isotropically. (C) 0.8 Å resolution structure of the viridian pigment copper(II) perchlorophthalocyanine (CSD: UZEMIY), refined anisotropically. (D) 1.05 Å resolution structure of the genotoxic natural product (−)-lomaiviticin C (CSD: ERUHEH), featuring two independent molecules in the asymmetric unit. (E) 0.57 Å resolution structure of the organic semiconductor dicyanonaphthalene diimide (CSD: TUKVON), refined anisotropically. This entry represents one of the highest-resolution small-molecule structures currently solved by 3D ED.
A handful of small-molecule studies have rapidly delivered on this promise; two illustrative examples are highlighted here. Andrusenko et al. elucidated a 0.9 Å 3D ED structure of orthocetamol, a regioisomer of the antipyretic paracetamol (Figure 9B).204 This simple compound exhibits a bizarre morphology in which assemblies of nanocrystals coalesce into flat quadrilateral platelets up to ∼300 μm in length. To further complicate matters, these tetragonal conglomerates display high susceptibility to pseudomerohedral twinning. These characteristics had thwarted structure determination of orthocetamol by X-ray crystallography for over a century. Andrusenko et al.’s ED structure supplied an unambiguous solution to this perennial problem.
In a similar vein, Gorelik et al. solved a 0.8 Å 3D ED structure of copper(II) perchlorophthalocyanine (also known as phthalo green or viridian), a widely used synthetic pigment (Figure 9C).205 Phthalo green is stubbornly insoluble in a remarkably wide range of solvents, which effectively precludes X-ray-scale recrystallization. Thanks to prior investigations by Uyeda et al.(206) and Dorset,48 ED had already established a partially complete structure of this compound. Although several subtleties remained unclear, this organometallic species had nonetheless become something of a poster child for ED, appearing on the cover of Dorset’s 1995 textbook Structural Electron Crystallography.176 Gorelik et al.’s data capped off the copper(II) perchlorophthalocyanine saga by confirming earlier results with a complete 3D structure. These cases demonstrated 3D ED’s ability to resolve two longstanding quandaries in conventional X-ray crystallography with ease.
In these examples, however, the atomic connectivity of both compounds was already well-established; 3D ED simply contributed a solid-state structure that reinforced what was previously known. In this context, Kim et al.’s 1.05 Å structure of (−)-lomaiviticin C provides a compelling case where ED data spurred reevaluation of an existing structural assignment (Figure 9D).207 (−)-Lomaiviticin C is a genotoxic bacterial metabolite which has evaded 20 years of efforts aimed at total synthesis and X-ray-scale crystallization. Intriguingly, this natural product (NP) contains an unusual monomeric aglycon moiety in which only 6 out of 19 carbon atoms feature bonds to hydrogen. This dearth of proton-attached carbons, in tandem with a high degree of unsaturation, rendered inference of connectivity quite challenging based on NMR spectroscopy alone. Ultimately, Kim et al.’s ED structure, alongside high-field (800 MHz) NMR spectroscopic studies and DFT calculations which further substantiated the ED assignment, corrected several errors originally caused by misinterpretation of fortuitously misleading HMBC coupling constants.208 This study underscores ED’s vast potential to make impactful contributions to elucidation of NPs, many of which feature some combination of forbidding structural complexity, scarcely available source material, and potentially inconclusive NMR data. In a field continually grappling with the myriad pitfalls209 associated with analysis of complex 2D NMR spectra, the clarity provided by a corroborating crystal structure seems almost cathartic. Furthermore, when applied in tandem with comparative genomics or metabolomics (to mine relevant biosynthetic gene clusters) and synthetic biology (to express those genes in model organisms), 3D ED could also significantly accelerate the rate of NP discovery.210−212 More broadly, 3D ED is rapidly finding a complementary niche within the wider context of synthetic chemistry; a growing number of reports now feature 3D ED structures of relevant synthetic targets or intermediates which proved unsuitable for single-crystal XRD.213−224 These structures include two noncanonical amino acids bearing all-carbon quaternary stereocenters,213 a trio of organic semiconductors solved at ultrahigh resolution (one of which is depicted in Figure 9E),54 a family of electron-deficient expanded helicenes,216 a pentacyclic indole-derived ester,217 and a synthetic mimic of the cuboidal subunit in the oxygen-evolving complex of photosystem II.220
Finally, 3D ED has also tackled a bevy of small-molecule active pharmaceutical ingredients (APIs), including carbamazepine,200,225 niacin (nicotinic acid),225 bismuth subgallate,226 ibuprofen,200 ethisterone,200 progesterone,200 biotin,200 paracetamol (acetaminophen),143,199,200 cimetidine,227 loratadine,125 sofosbuvir,143 ramelteon,228 tolvaptan,228 olanzapine,229 epicorazine A,144 dehydrocurvularin,144 metaxalone,230 teniposide,231 remdesivir,232 and indomethacin.126 Because many APIs exist natively as microcrystalline powders, 3D ED could potentially revolutionize solid-state structure determination in the pharmaceutical industry,231,233 where size-limited single-crystal XRD is currently the gold standard. Specifically, 3D ED’s sensitivity to variable polymorphism at the nanoscale could provide crucial insights into API stability and solubility, as different polymorphs of the same drug can often display drastically disparate pharmacokinetic profiles.234−237 For instance, in orally administered drugs, an API’s immediate bioavailability is controlled partially by its rate of dissolution in the gastrointestinal tract, which can vary considerably as a function of altered lattice packing. Ultimately, structural information supplied by 3D ED could play a pivotal role in guiding crystal engineering efforts238 aimed at designing solvates, cocrystals, or polymorphs of APIs with optimized pharmacokinetic properties.
Undoubtedly, 3D ED has plenty of potential in this area. Despite the considerable hype,201−203 however, the interested synthetic chemist is confronted with several issues that warrant caution. First, electrostatic potential maps cannot always distinguish between disparate elements as unambiguously as atomic charge density maps derived from X-ray diffraction. Unlike their X-ray counterparts, elastic electron scattering factors do not scale linearly or monotonically with Z. As a result, relative differences between elements become diminished, as discussed earlier. Therefore, electrostatic potential alone does not always provide a self-sufficient means of differentiating neutral C, N, and O, particularly if the diffraction data set samples heavily within the vicinity of 3 Å resolution. Indeed, ab initio phasing algorithms frequently assign these atoms interchangeably,199,207 particularly because they typically presume X-ray-scale scattering differences between elements. In these cases, even at the refinement stage, elemental identity can be arduous or impossible to deduce based solely on experimental difference Fourier maps. Given these limitations, if attempting to solve a challenging novel structure such as a complex natural product (generally replete with heteroatoms such as O and N) via 3D ED, rigorous corroboration with external data from NMR spectroscopy and mass spectrometry remains essential.207,212
Second, 3D ED always requires well-formed single crystals. Serendipitously, many compounds may inhabit a specific “Goldilocks zone” where they refuse to form X-ray-scale crystals yet grudgingly aggregate into crystalline assemblies at the nanoscale. ED is well-equipped to solve structures which fit this profile. Nevertheless, ED is not a panacea; it cannot salvage genuinely amorphous substrates. Species which systematically failed to form macroscopic crystals, especially if such reluctance reflects thermodynamic instability in the crystalline state, could just as easily fail at the microscopic level. Before attempting 3D ED on seemingly amorphous material, X-ray powder diffraction (XRPD) is strongly recommended as a simple, effective test to screen for the presence of microcrystalline domains. If XRPD fails to yield clear, well-resolved rings, structure determination by ED is unlikely to succeed.
Third, ceteris paribus, current 3D ED data quality is often inferior to X-ray data quality by a range of metrics (Rmeas, ⟨I/σ(I)⟩, CC1/2), although this gap is beginning to contract quickly for small molecules. An especially relevant statistic is completeness because ED’s coverage of reciprocal space is inherently limited by the restricted tilt range available to the TEM goniometer. The resultant “missing wedge” becomes particularly problematic if the space group symmetry of the crystal is low or if orientation bias is severe. X-ray diffractometers can easily collect 360° of data, unlocking regions of the reciprocal lattice potentially inaccessible by continuous-rotation ED. Moreover, some fraction of small molecules deemed “impossible” to solve by XRD may simply indicate a lack of rigorous screening. In macromolecular crystallography, screening thousands of crystallization conditions via high-throughput hanging-drop vapor diffusion is routine. Similar methods have not yet percolated widely into small-molecule work, where venerable techniques such as slow evaporation of layered solvents usually reign supreme. Thus, molecules seemingly “uncrystallizable” for XRD may benefit considerably from a broader, more systematic exploration of crystallization conditions.239 Although ED’s lower size constraint confers a distinct advantage over XRD, high-flux microfocus beamlines can now produce tractable X-ray diffraction from microcrystals with dimensions as small as 1–10 μm.18 Whenever possible, XRD remains the technique of choice for small-molecule structure determination.240 Nevertheless, if X-ray-scale crystals prove impossible to obtain despite rigorous effort, ED is a powerful alternative which can match or surpass the resolution achieved by XRD. As the technique continues to mature, the development of specialized hardware engineered exclusively for ED will undoubtedly alleviate many current issues with data quality.241
6.3. Proteins
Continuous-rotation electron diffraction was originally developed specifically for the purpose of interrogating three-dimensional macromolecular crystals.37,89 This work traces its origins to a venerable tradition of two-dimensional electron crystallography, where amplitudes derived from 2D diffraction patterns were historically paired with phases obtained via direct Fourier transform of real-space images.242−244 Key milestones in this field include Henderson et al.’s 3.5 Å structure of bacteriorhodopsin245 and Gonen et al.’s 1.9 Å structure of aquaporin,246 two intermembrane proteins whose biological roles naturally predispose formation of 2D crystals. In this context, continuous rotation emerged as a method to analyze proteins not innately suited to aggregating into 2D arrays. Shi et al.’s 2.9 Å structure of HEWL37 represented the first protein successfully solved by 3D electron crystallography; it was rapidly followed by a suite of canonical soluble proteins well-studied by conventional X-ray methods.64 Since these pioneering studies, however, 3D ED of proteins appears to have progressed more slowly than expected, especially when juxtaposed against the explosion of interest in small molecules. This is largely because sample preparation in macromolecular electron crystallography is typically much more laborious, and most major advances have therefore focused on methodological development in lieu of elucidating novel structures. For instance, a series of reports by Gonen and co-workers have demonstrated that continuous-rotation ED is procedurally compatible with focused ion-beam milling and in meso crystallogenesis within lipidic cubic phases (LCPs).70,247,248 These techniques were applied in tandem to solve a 1.9 Å structure of the human A2A adenosine receptor, marking a significant breakthrough for ED given the inherent difficulty of working with lipophilic membrane proteins.248 Another emphasis has been placed on soaking protein nanocrystals with solutions of pharmacologically relevant ligands to visualize their substrate-binding pockets. These efforts have culminated in a 2.5 Å structure of human carbonic anhydrase bound to the sulfonamide inhibitor acetazolamide,249 as well as a 3.0 Å structure of an HIV-1 Gag polyprotein fragment bound to the steroidal inhibitor bevirimat.250
Interestingly, when contrasted with analogous structures solved by single-crystal XRD, macromolecular crystals have historically diffracted to worse resolution by 3D ED, typically by a factor of 1.5–2. For instance, despite the considerable number of proteinase K ED structures currently deposited in the PDB, none have surpassed a resolution finer than 1.5 Å. Nevertheless, the PDB is replete with just over 100 sub-1.5 Å X-ray structures of proteinase K, including several determined to subangstrom resolution. No such discrepancy has been observed with small molecules, which routinely diffract to subangstrom resolution by both 3D ED and XRD. In fact, the average resolution of structures catalogued in Table 4 is only 2.5 Å (cf. 0.95 Å in Table 3); 2.5 Å is ultimately an underwhelming number, especially given the overrepresentation of well-diffracting crystallographic standards within the sample size. Relative to small molecules, protein crystals suffer from a couple of unique disadvantages in addition to innately higher disorder. Although signal-to-noise in ED is boosted by a greater number of repeating units, protein crystals’ larger unit cells provide an inherently lower bound on the maximum thickness permissible before inelastic scattering overpowers Bragg peaks. Furthermore, vitrification and frozen hydration remain experimental necessities, and insulating layers of amorphous ice will always contribute noise.
An illustrative example is provided by Xu et al.’s multipart investigation of an R2-like ligand-binding oxidase (a metalloenzyme originally isolated from Sulfolobus acidocaldarius).129,130 In 2018, Xu et al. disclosed a 3.0 Å 3D ED structure of R2lox, phased by molecular replacement using a homologous X-ray structure with 35% shared sequence identity as a template. Although novel at the time, this structure nevertheless exhibited less-than-ideal completeness (62.8%, despite merging data from 21 crystals, suggesting stark orientation bias) and unusually high Rmeas (56.1% overall) statistics. A subsequent reinvestigation of this species by synchrotron X-ray diffraction yielded a higher-quality 2.1 Å X-ray structure (featuring 99.4% completeness and 16.6% overall Rmeas), which corrected several deficiencies in the 3D ED model. Specifically, 3D ED had omitted the presence of a fatty acid ligand bound to the enzyme’s active site, as well as an 11-residue stretch between amino acids 249 and 261. While most general aspects of the 3D ED structure proved consistent with XRD, middling resolution and low completeness conspired to limit its utility in modeling biologically relevant details. Xu et al.’s commendable decision to pursue a corroborating X-ray structure in these circumstances reflects ED’s current challenge in consistently matching XRD data quality in macromolecular crystallography.
A promising step forward has recently been contributed by Gonen and co-workers’ 0.87 Å structure of triclinic HEWL.251 This report exploited the heightened sensitivity of a direct electron detector operating in counting mode to break the subangstrom resolution barrier for 3D electron crystallography of proteins, albeit on a well-diffracting standard. In addition to this study, a potential blueprint for macromolecular electron crystallography to overcome its current limitations is also provided by Yonekura and co-workers’ development of energy-filtered 3D ED,170 as well as Bücker et al.’s serial approach to data collection.99 These tactics could work in tandem to mitigate radiation damage and boost diffraction data quality, allowing 3D ED to deliver novel macromolecular structures on par with XRD.
6.4. Radioactive Minerals and Inorganic Compounds
Although slightly esoteric to chemists, mineralogy is a field replete with ideal samples for investigation by 3D ED. In fact, mineralogy has historically functioned as a key impetus behind research in 3D electron crystallography, dating back to Zvyagin’s studies of celadonite and muscovite.51 A detailed discussion on applications of 3D ED to mineralogy has been provided by Mugnaioli and Gemmi.39 Here we would like to specifically highlight radioactive metamict minerals, which comprise a fascinating and seemingly tailor-made class of substrates for 3D ED.252 Metamict systems feature an intricate lattice structure punctuated by trace impurities of radioactive elements like uranium or thorium.253 Over geologic timescales, these interstitial radionuclides undergo alpha decay, selectively destroying certain regions of the lattice from within. This process, known as metamictization, gradually results in total amorphization of crystalline order. In a compelling study, Capitani et al. used the presence of Bragg peaks in ED patterns to spatially map metamict domains in the mineral samarskite at the nanoscale.252 After targeting specific submicrometer-sized zones where crystallinity seemed best preserved, Capitani et al. collected a tomographic series of still-frame ED patterns. These 0.8 Å ED data successfully yielded a solution via direct methods, providing an elusive 3D structure of metamict samarskite. Critically, at the single-crystal X-ray scale, alpha decay had rendered the bulk sample mostly amorphous, carving a unique niche for ED. In many ways, this work also echoes the more general blueprint formulated by Baybarz and co-workers at Oak Ridge National Laboratory in the 1970s.254−256 These researchers worked primarily with inorganic salts formed by fully anthropogenic, superheavy elements like einsteinium, californium, or fermium. Synthetic Es and Fm typically decay so rapidly and destructively that formation of high-quality X-ray-scale crystals is a nonstarter; self-irradiation would likely cause lattices containing Es or Fm to collapse well before growing to X-ray size. Furthermore, synthesis of transplutonium elements is exceptionally arduous, often divulging only nanogram-scale quantities of material (which then immediately begins to decay!). Undeterred, Baybarz and co-workers exploited ED’s ability to interrogate submicrometer-sized crystals and deduced the unit cell parameters of several Es and Cf oxides from polycrystalline ED patterns recorded at 80 kV. Following a long hiatus, their torch has recently been lifted by Minor, Abergel, and co-workers.257 Given contemporary advances in data collection and analysis, ED appears uniquely poised to deliver 3D structures of inorganic systems containing either superheavy or primordial radionuclides, an exciting prospect.
6.5. Radiation Damage
As with any diffraction experiment, the maximum achievable signal-to-noise in 3D ED is ultimately constrained by radiation damage, which begins as soon as the crystal of interest is exposed to the impinging beam. ED leverages information about structure-factor amplitudes encoded in Bragg peaks, which result from elastic scattering of incident electrons. Because the low-angle elastic collisions contributing to Bragg peaks involve negligible (<1 eV) energy loss,258 they leave the crystal lattice completely intact. (At higher incident energies, elastic scattering can destructively dislocate atoms via knock-on displacement, but the likelihood of these events relative to radiolysis is negligible at TEM accelerating voltages.259) Conversely, inelastic scattering causes impinging electrons to deposit a significant fraction (10–100 eV) of their incident energy within the sample, damaging the structural integrity of the lattice. Second-row elements such as carbon, nitrogen, and oxygen possess inelastic electron cross-sections roughly 3× greater than their elastic counterparts (Figure 10). On a per electron basis, therefore, any crystal composed primarily of C, N, and O atoms is 3× more likely to undergo unproductive inelastic scattering.260 Although seemingly inauspicious, this ratio is actually superior to the corresponding fraction for X-ray diffraction; incident X-rays can inflict up to 3 orders of magnitude more collateral damage per useful elastic scattering event. (In principle, this advantage is attenuated somewhat by electrons’ propensity for multiple scattering because they interact with the substrate more frequently than X-rays.)
Figure 10.
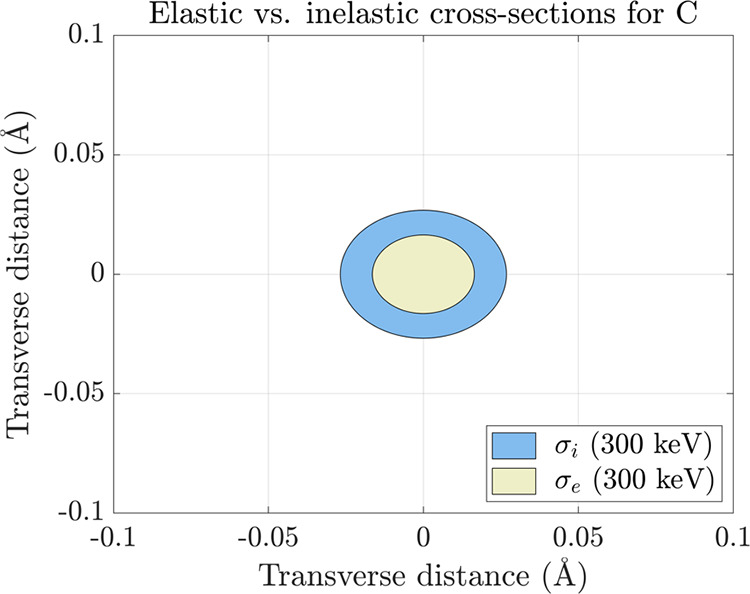
Elastic vs inelastic cross-sections for neutral carbon at 300 keV, expressed as concentric circles.
High-resolution information is especially sensitive to degradation of lattice structure. Therefore, in reciprocal space, radiation damage begins by consuming high-resolution reflections, causing Bragg peaks at the periphery of the detector to diminish in intensity until they become indistinguishable from noise. Ultimately, as crystalline order is totally destroyed, low-resolution reflections also recede into the void space of the noise floor. Statistically, this manifests as a monotonic decrease in ⟨I/σ(I)⟩ which starts in the outermost resolution shell and spreads gradually inward. In real space, radiation damage results in two major global consequences: a uniform increase in B-factors and an expansion of unit cell volume.261 Bloated B-factors represent growing uncertainty in atomic positions, whereas unit cell volume is thought to expand due to radiolytic generation of hydrogen gas within the lattice.262 A systematic study by Hattne et al. found that site-specific radiation damage inflicted upon particular functional groups largely mirrors the progression observed in X-ray diffraction.263,264 In frozen-hydrated proteinase K, Hattne et al. observed perturbation of metal cations, elongation and lysis of disulfide bonds, and radiolytic decarboxylation of aspartate and glutamate side chains, all in quick succession. Loss of near-atomic resolution (∼2 Å) information generally occurred after a total accumulated exposure of 3 e– Å–2. By collecting 3D ED data at an ultralow flux density (<0.01 e– Å–2 s–1), staying well below this threshold is quite feasible. Furthermore, this cutoff is substrate-specific. For instance, organometallic complexes, which frequently exhibit denser packing and lattices free of solvent channels, could potentially tolerate equivalent levels of fluence quite easily, even at ambient temperatures.163 Nevertheless, in virtually all cases, radiation damage is significantly abated by cryogenic temperatures, which presumably stall the thermal diffusion of destructive free radicals generated by radiolysis.265−267 Another bulwark against radical-induced decay is the presence of highly conjugated polyaromatic systems, which could facilitate delocalization of errant secondary electrons via resonance.268−270 This effectively provides a thermodynamic sink for radicals which would otherwise propagate freely throughout the crystal lattice.
Finally, some clarification on nomenclature is warranted (Table 5). Formally, dose refers to energy absorbed per unit mass271 (measured in units of MGy, or 106 J kg–1), whereas fluence corresponds to particles delivered per unit area (measured in units of e– Å–2 for electrons, or γ μm–2 for X-ray photons). Regrettably, these two terms have become thoroughly muddled in the cryo-EM literature, where they are frequently used interchangeably. For instance, a substantial fraction of ED structures deposited in the PDB currently reports a “dose” tabulated in units of e– Å–2. This value is really a misrepresentation of total accumulated fluence. Although often strongly correlated, dose and fluence do not represent fungible observables, making their conflation incorrect and potentially misleading. The key distinction is that dose is a quantity specific to the substrate under interrogation. Conversely, fluence is a property of the incident beam, which is completely decoupled from the identity of the substrate. To illustrate this point, consider two identical, isomorphous protein crystals exposed to a fixed fluence: (a) one native and (b) one derivative intercalated with heavy metal cations. Because heavy metals feature significantly higher elastic-to-inelastic cross-section ratios than lighter elements such as C, N, and O, crystal B could experience a smaller proportion of inelastic scattering events than crystal A. In that case, crystal B would experience a lower dose than crystal A, notwithstanding being illuminated with the same fluence. In other words, despite being exposed to the same number of particles per unit area, crystal A’s elevated susceptibility to inelastic scattering would cause it to absorb more energy per unit mass than crystal B. In sum, different specimens exposed to an identical fluence can accumulate variable quantities of dose based on their chemical composition. Dose is a more suitable metric for assessing radiation damage than total accumulated fluence because it situates electron exposure within the specific context of the substrate itself.272
Table 5. Definitions and Typical Units for Several Terms in Dosimetry.
| observable | unit (electrons) | unit (X-rays) | description |
|---|---|---|---|
| dose | MGy (106 J kg–1) | MGy (106 J kg–1) | energy absorbed per unit mass |
| fluence | e– Å–2 | γ μm–2 | particles delivered per unit area |
| flux | e– s–1 | γ s–1 | particles delivered per unit time |
| flux density | e– Å–2 s–1 | γ μm–2 s–1 | fluence delivered per unit time |
7. Conclusion and Outlook
As a science born in the quantum age, crystallographic analysis has been intimately shaped by our increasingly sophisticated understanding of incident quanta. Modern transmission electron microscopy is a powerful tool capable of generating highly coherent, atomically precise beams of electrons which would have been inconceivable to pioneering researchers like Davisson and Germer. Our ability to probe the atomic structure of 3D molecular nanocrystals at subangstrom resolution is a testament to electron diffraction’s burgeoning relevance and vast potential. This Review has focused on pivotal concepts and experiments which have underpinned 3D electron crystallography’s ongoing transformation from a somewhat esoteric subfield to an area of swiftly growing importance. We conclude with forward-looking recommendations organized around two central themes: increasing transparency and expanding access.
7.1. Increasing Transparency
It has become standard operating procedure to deposit fully refined structures to databases like the CSD or the PDB, although these resources have yet to flag 3D ED data in an easily identifiable or searchable way. To ensure maximum transparency, we also recommend concurrent deposition of raw, unprocessed data (i.e., a tilt series of diffraction patterns in a file format compatible with data reduction software) on public repositories such as Zenodo. Furthermore, automated validation routines, such as those embedded in checkCIF, typically raise an array of objections when presented with 3D ED structures, some of which reflect intrinsic disparities between 3D ED and XRD rather than genuine deficiencies in the deposited models. Moving forward, establishment of ED-specific validation criteria cognizant of the various differences between 3D ED and XRD would provide a more accurate record of the quality of 3D ED structures reported in the literature.
7.2. Expanding Access
Although ED is en route to becoming a more mainstream technique, its current practitioners remain limited to a relatively small (albeit growing) handful of specialists. Transmission electron microscopy presents a steeper economic barrier to entry than X-ray crystallography; a mid- to high-end TEM optimally equipped for ED carries a hefty six-digit price tag, whereas a standard X-ray diffractometer is usually up to an order of magnitude cheaper. Retrofitting used or refurbished TEMs for ED is usually a more viable option, although still expensive. Sadly, lack of widespread access to the appropriate instrumentation can thwart researchers otherwise interested in incorporating ED into their work. Furthermore, TEM maintenance is typically carried out by trained engineers or facility managers whom many institutions may not have the financial bandwidth to hire. Systemic inequities aside, however, the conceptual learning curve for ED is comparatively gentle, especially thanks to the advent of continuous rotation. Any practicing X-ray crystallographer has already attained the requisite skillset to start solving structures from continuous-rotation 3D ED data, leaving lack of access as the main bottleneck. To rectify this, investment in a subsidized infrastructure for ED data collection at dedicated user facilities, analogous to the now well-established network of synchrotron beamlines across the globe, will prove especially critical. ED will reach its considerable potential only when the technique proliferates to more users outside its current niche.
Acknowledgments
This material is based upon work supported by the National Science Foundation Graduate Research Fellowship (to A.S.) under grant no. DGE-2034835. J.A.R. is supported as a Pew Scholar, a Beckman Young Investigator, and a Packard Fellow. Work by J.A.R. and A.S. was also supported in part by DOE grant no. DE-FC02-02ER63421, NIH-NIGMS grant no. R35 GM128867, and STROBE, an NSF Science and Technology Center, through grant no. DMR-1548924. A.S. is indebted to C. A. Glynn and L. S. Richards for syntactic guidance. We thank D. S. Eisenberg, D. Cascio, M. R. Sawaya, B. C. Regan, M. H. Mecklenburg, and P. Ercius for invaluable feedback on the manuscript, as well as N. Johnson (CCDC) for graciously providing a list of ED structures deposited in the Cambridge Structural Database.
Glossary
Abbreviations
- ED
electron diffraction
- 3D ED
three-dimensional electron diffraction
- cryo-EM
cryogenic electron microscopy
- TEM
transmission electron microscopy
- XRD
X-ray diffraction
- XRPD
X-ray powder diffraction
- ESP
electrostatic potential
- MFP
mean free path
- DM
direct methods
- MR
molecular replacement
- FBP
fragment-based phasing
- FIB
focused ion beam
- PED
precession electron diffraction
- PEDT
precession-assisted electron diffraction tomography
- ADT
automated diffraction tomography
- RED
rotation electron diffraction
- CCD
charge-coupled device
- CMOS
complementary metal oxide semiconductor
- MicroED
microcrystal electron diffraction
- IEDT
integrated electron diffraction tomography
- cRED
continuous-rotation electron diffraction
- 4DSTEM
four-dimensional scanning transmission electron microscopy
- nanoEDT
nanobeam electron diffraction tomography
- HRTEM
high-resolution transmission electron microscopy
- MIR
multiple isomorphous replacement
- DFT
density functional theory
- IDP
intrinsically disordered protein
- LARKS
low-complexity aromatic-rich kinked segments
- HEWL
hen egg white lysozyme
- FUS
fused in sarcoma
- API
active pharmaceutical ingredient
- LCP
lipidic cubic phase
- MGy
megagrays
Biographies
Ambarneil Saha was born in Starkville, Mississippi, in 1996. After spending his more impressionable years growing up in Cupertino, California, he earned an A.B. in Chemistry from Cornell University in 2018, where he worked on electrochemical difunctionalization of alkenes with Professor Song Lin. He is currently a Ph.D. candidate and NSF fellow in the Rodríguez laboratory at UCLA.
Shervin S. Nia was born in Los Angeles, California, in 1999. He earned his B.S. in Biochemistry from UCLA in 2021. During his undergraduate career, he studied eye tracking and reaction times in virtual reality with Professor Katsushi Arisaka before joining the Rodríguez group. Currently, Shervin performs computational work creating reconstructions of cryoEM datasets to characterize elusive crystal structures.
José A. Rodríguez was born in Aguascalientes, Mexico, in 1985. He emigrated to and spent his formative years in Los Angeles, California, before earning a B.S. in Biophysics from UCLA in 2007. He has remained at UCLA ever since, subsequently obtaining a Ph.D. in Molecular Biology and postdoctoral training in bioimaging, crystallography, and biological chemistry. He is currently an associate professor of chemistry and biochemistry at UCLA (http://rodriguez.chem.ucla.edu/).
The authors declare the following competing financial interest(s): J.A.R. is a founder and equity stake holder of MEDSTRUC INC.
References
- Davisson C.; Germer L. H. Diffraction of Electrons by a Crystal of Nickel. Phys. Rev. 1927, 30, 705–740. 10.1103/PhysRev.30.705. [DOI] [Google Scholar]
- Davisson C.; Germer L. H. The Scattering of Electrons by a Single Crystal of Nickel. Nature 1927, 119, 558–560. 10.1038/119558a0. [DOI] [Google Scholar]
- Gehrenbeck R. K. Electron Diffraction: Fifty Years Ago. Physics Today 1978, 31, 34–41. 10.1063/1.3001830. [DOI] [Google Scholar]
- De Broglie L. XXXV. A Tentative Theory of Light Quanta. Philosophical Magazine Letters 2006, 86, 411–423. 10.1080/09500830600914721. [DOI] [Google Scholar]
- Thomson G. P.; Reid A. Diffraction of Cathode Rays by a Thin Film. Nature 1927, 119, 890–890. 10.1038/119890a0. [DOI] [Google Scholar]
- Thomson G. P. The Diffraction of Cathode Rays by Thin Films of Platinum. Nature 1927, 120, 802–802. 10.1038/120802a0. [DOI] [Google Scholar]
- Thomson G. P. Experiments on the Diffraction of Cathode Rays. Proc. R. Soc. Lond. A 1928, 117, 600–609. 10.1098/rspa.1928.0022. [DOI] [Google Scholar]
- Laschkarew W. E.; Usyskin I. D. Die Bestimmung der Lage der Wasserstoffionen im NH4Cl-Kristallgitter durch Elektronenbeugung. Z. Physik 1933, 85, 618–630. 10.1007/BF01331003. [DOI] [Google Scholar]
- Cowley J. M. Structure Analysis of Single Crystals by Electron Diffraction. II. Disordered Boric Acid Structure. Acta Crystallogr. 1953, 6, 522–529. 10.1107/S0365110X53001423. [DOI] [Google Scholar]
- Honjo G.; Shimaoka K. Determination of Hydrogen Position in Cubic Ice by Electron Diffraction. Acta Crystallogr. 1957, 10, 710–711. 10.1107/S0365110X57002479. [DOI] [Google Scholar]
- Kuwabara S. Accurate Determination of Hydrogen Positions in NH4Cl by Electron Diffraction. J. Phys. Soc. Jpn. 1959, 14, 1205–1216. 10.1143/JPSJ.14.1205. [DOI] [Google Scholar]
- Rigamonti R. La Struttura Della Catena Paraffinica Studiata Mediante i Raggi Di Elettroni. Gazz. Chim. Ital. 1936, 66, 174–182. [Google Scholar]
- Charlesby A.; Finch G. I.; Wilman H. The Diffraction of Electrons by Anthracene. Proc. Phys. Soc. 1939, 51, 479–528. 10.1088/0959-5309/51/3/312. [DOI] [Google Scholar]
- Cowley J. M. Crystal Structure Determination by Electron Diffraction. Progress in Materials Science 1968, 13, 267–321. 10.1016/0079-6425(68)90023-6. [DOI] [Google Scholar]
- Vainshtein B. K.Structure Analysis by Electron Diffraction; Pergamon Press: Oxford, 1964. [Google Scholar]
- Dorset D. L. Electron Crystallography. Acta Cryst B 1996, 52, 753–769. 10.1107/S0108768196005599. [DOI] [PubMed] [Google Scholar]
- Gemmi M.; Mugnaioli E.; Gorelik T. E.; Kolb U.; Palatinus L.; Boullay P.; Hovmöller S.; Abrahams J. P. 3D Electron Diffraction: The Nanocrystallography Revolution. ACS Cent. Sci. 2019, 5, 1315–1329. 10.1021/acscentsci.9b00394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji X.; Axford D.; Owen R.; Evans G.; Ginn H. M.; Sutton G.; Stuart D. I. Polyhedra Structures and the Evolution of the Insect Viruses. Journal of Structural Biology 2015, 192, 88–99. 10.1016/j.jsb.2015.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo J. M.; Spence J. C. H.. Advanced Transmission Electron Microscopy: Imaging and Diffraction in Nanoscience; Springer-Verlag: New York, 2017 10.1007/978-1-4939-6607-3. [DOI] [Google Scholar]
- Peng L.-M. Electron Scattering Factors of Ions and Their Parameterization. Acta Cryst A 1998, 54, 481–485. 10.1107/S0108767398001901. [DOI] [Google Scholar]
- Zheng J.-C.; Zhu Y.; Wu L.; Davenport J. W. On the Sensitivity of Electron and X-Ray Scattering Factors to Valence Charge Distributions. J. Appl. Crystallogr. 2005, 38, 648–656. 10.1107/S0021889805016109. [DOI] [Google Scholar]
- Shi H. L.; Li Z. A. UnitCell Tools, a Package to Determine Unit-Cell Parameters from a Single Electron Diffraction Pattern. IUCrJ 2021, 8, 805–813. 10.1107/S2052252521007867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bethe H. A.; Rose M. E.; Smith L. P. The Multiple Scattering of Electrons. Proc. Am. Philos. Soc. 1938, 78, 573–585. [Google Scholar]
- Blackman M.; Thomson G. P. On the Intensities of Electron Diffraction Rings. Proc. R. Soc. Lond. A 1939, 173, 68–82. 10.1098/rspa.1939.0129. [DOI] [Google Scholar]
- Cowley J. M.; Moodie A. F. The Scattering of Electrons by Atoms and Crystals. I. A New Theoretical Approach. Acta Crystallogr. 1957, 10, 609–619. 10.1107/S0365110X57002194. [DOI] [Google Scholar]
- Fujiwara K. Application of Higher Order Born Approximation to Multiple Elastic Scattering of Electrons by Crystals. J. Phys. Soc. Jpn. 1959, 14, 1513–1524. 10.1143/JPSJ.14.1513. [DOI] [Google Scholar]
- Stern R. M.; Taub H. An Introduction to the Dynamical Scattering of Electrons by Crystals. C R C Critical Reviews in Solid State Sciences 1970, 1, 221–302. 10.1080/10408437008243421. [DOI] [Google Scholar]
- Hu H.; Dorset D. L.; Moss B. Space Group Symmetry and the Location of Forbidden Reflections Due to Incoherent Multiple Scattering from Epitaxially Oriented Paraffins. Ultramicroscopy 1989, 27, 161–169. 10.1016/0304-3991(89)90084-3. [DOI] [Google Scholar]
- Miyake S.; Uyeda R. An Exception to Friedel’s Law in Electron Diffraction. Acta Crystallogr. 1950, 3, 314–314. 10.1107/S0365110X5000080X. [DOI] [Google Scholar]
- Goodman P.; Lehmpfuhl G. Observation of the Breakdown of Friedel’s Law in Electron Diffraction and Symmetry Determination from Zero-Layer Interactions. Acta Cryst A 1968, 24, 339–347. 10.1107/S0567739468000677. [DOI] [Google Scholar]
- Glaeser R. M.; Ceska T. A. High-Voltage Electron Diffraction from Bacteriorhodopsin (Purple Membrane) Is Measurably Dynamical. Acta Cryst A 1989, 45, 620–628. 10.1107/S0108767389004599. [DOI] [PubMed] [Google Scholar]
- Heidenreich R. D. Theory of the “Forbidden” (222) Electron Reflection in the Diamond Structure. Phys. Rev. 1950, 77, 271–283. 10.1103/PhysRev.77.271. [DOI] [Google Scholar]
- Cowley J. M.; Rees A. L. G.; Spink J. A. Secondary Elastic Scattering in Electron Diffraction. Proc. Phys. Soc. A 1951, 64, 609–619. 10.1088/0370-1298/64/7/302. [DOI] [Google Scholar]
- Diaz-Avalos R.; Long C.; Fontano E.; Balbirnie M.; Grothe R.; Eisenberg D.; Caspar D. L. D. Cross-Beta Order and Diversity in Nanocrystals of an Amyloid-Forming Peptide. J. Mol. Biol. 2003, 330, 1165–1175. 10.1016/S0022-2836(03)00659-4. [DOI] [PubMed] [Google Scholar]
- Ciston J.; Deng B.; Marks L. D.; Own C. S.; Sinkler W. A Quantitative Analysis of the Cone-Angle Dependence in Precession Electron Diffraction. Ultramicroscopy 2008, 108, 514–522. 10.1016/j.ultramic.2007.08.004. [DOI] [PubMed] [Google Scholar]
- Eggeman A. S.; White T. A.; Midgley P. A. Is Precession Electron Diffraction Kinematical? Part II: A Practical Method to Determine the Optimum Precession Angle. Ultramicroscopy 2010, 110, 771–777. 10.1016/j.ultramic.2009.10.012. [DOI] [PubMed] [Google Scholar]
- Shi D.; Nannenga B. L.; Iadanza M. G.; Gonen T. Three-Dimensional Electron Crystallography of Protein Microcrystals. eLife 2013, 2, e01345 10.7554/eLife.01345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nannenga B. L.; Shi D.; Leslie A. G. W.; Gonen T. High-Resolution Structure Determination by Continuous-Rotation Data Collection in MicroED. Nat Methods 2014, 11, 927–930. 10.1038/nmeth.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mugnaioli E.; Gemmi M. Single-Crystal Analysis of Nanodomains by Electron Diffraction Tomography: Mineralogy at the Order-Disorder Borderline. Zeitschrift für Kristallographie - Crystalline Materials 2018, 233, 163–178. 10.1515/zkri-2017-2130. [DOI] [Google Scholar]
- Cowley J. M. Structure Analysis of Single Crystals by Electron Diffraction. I. Techniques. Acta Crystallogr. 1953, 6, 516–521. 10.1107/S0365110X53001411. [DOI] [Google Scholar]
- Lipson H.; Cochran W.. The Determination of Crystal Structures, 3rd ed.; Cornell University Press: Ithaca, NY, 1966. [Google Scholar]
- Hauptman H.; Karle J. Structure Invariants and Seminvariants for Noncentrosymmetric Space Groups. Acta Crystallogr. 1956, 9, 45–55. 10.1107/S0365110X56000097. [DOI] [Google Scholar]
- Karle J.; Hauptman H. A Theory of Phase Determination for the Four Types of Non-Centrosymmetric Space Groups 1P222, 2P22, 3P12, 3P22. Acta Crystallogr. 1956, 9, 635–651. 10.1107/S0365110X56001741. [DOI] [Google Scholar]
- Hauptman H.; Karle J. A Unified Algebraic Approach to the Phase Problem I. Space Group P. Acta Crystallogr. 1957, 10, 267–270. 10.1107/S0365110X57000833. [DOI] [Google Scholar]
- Hauptman H.; Karle J. Phase Determination from New Joint Probability Distribution: Space Group P. Acta Crystallogr. 1958, 11, 149–157. 10.1107/S0365110X58000414. [DOI] [Google Scholar]
- Dorset D. L.; Hauptman H. A. Direct Phase Determination for Quasi-Kinematical Electron Diffraction Intensity Data from Organic Microcrystals. Ultramicroscopy 1976, 1, 195–201. 10.1016/0304-3991(76)90034-6. [DOI] [PubMed] [Google Scholar]
- Dorset D. L. Comments on the Validity of the Direct Phasing and Fourier Methods in Electron Crystallography. Acta Cryst A 1995, 51, 869–879. 10.1107/S0108767395005915. [DOI] [Google Scholar]
- Dorset D. L. Is Electron Crystallography Possible? The Direct Determination of Organic Crystal Structures. Ultramicroscopy 1991, 38, 23–40. 10.1016/0304-3991(91)90106-G. [DOI] [Google Scholar]
- Dorset D. L. Electron Diffraction Structure Analysis of Diketopiperazine - a Direct Phase Determination. Acta Crystallogr., Sect. A 1991, 47, 510–515. 10.1107/S0108767391003550. [DOI] [Google Scholar]
- Dorset D. L. Automated Phase Determination in Electron Crystallography: Thermotropic Phases of Thiourea. Ultramicroscopy 1992, 45, 357–364. 10.1016/0304-3991(92)90147-C. [DOI] [Google Scholar]
- Dorset D. L. Direct Phasing in Electron Crystallography: Determination of Layer Silicate Structures. Ultramicroscopy 1992, 45, 5–14. 10.1016/0304-3991(92)90033-G. [DOI] [Google Scholar]
- Dorset D. L.; McCourt M. P. Automated Structure Analysis in Electron Crystallography: Phase Determination with the Tangent Formula and Least-Squares Refinement. Acta Cryst A 1994, 50, 287–292. 10.1107/S010876739300916X. [DOI] [Google Scholar]
- Dorset D. L. Direct Methods and Refinement in Electron and X-Ray Crystallography - Diketopiperazine Revisited. Zeitschrift für Kristallographie 2010, 225, 86–93. 10.1524/zkri.2010.1198. [DOI] [Google Scholar]
- Levine A. M.; Bu G.; Biswas S.; Tsai E. H. R.; Braunschweig A. B.; Nannenga B. L. Crystal Structure and Orientation of Organic Semiconductor Thin Films by Microcrystal Electron Diffraction and Grazing-Incidence Wide-Angle X-Ray Scattering. Chem. Commun. 2020, 56, 4204–4207. 10.1039/D0CC00119H. [DOI] [PubMed] [Google Scholar]
- Levine A. M.; He G.; Bu G.; Ramos P.; Wu F.; Soliman A.; Serrano J.; Pietraru D.; Chan C.; Batteas J. D.; Kowalczyk M.; Jang S. J.; Nannenga B. L.; Sfeir M. Y.R.; Tsai E. H. R.; Braunschweig A. B. Efficient Free Triplet Generation Follows Singlet Fission in Diketopyrrolopyrrole Polymorphs with Goldilocks Coupling. J. Phys. Chem. C 2021, 125, 12207–12213. 10.1021/acs.jpcc.1c02737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J.; Xu H.; Lebrette H.; Carroni M.; Taberman H.; Högbom M.; Zou X. A Simple Pressure-Assisted Method for MicroED Specimen Preparation. Nat Commun 2021, 12, 5036. 10.1038/s41467-021-25335-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews B. W. Solvent Content of Protein Crystals. J. Mol. Biol. 1968, 33, 491–497. 10.1016/0022-2836(68)90205-2. [DOI] [PubMed] [Google Scholar]
- Fraser R. D. B.; MacRae T. P.; Suzuki E. An Improved Method for Calculating the Contribution of Solvent to the X-Ray Diffraction Pattern of Biological Molecules. J. Appl. Crystallogr. 1978, 11, 693–694. 10.1107/S0021889878014296. [DOI] [Google Scholar]
- Weichenberger C. X.; Afonine P. V.; Kantardjieff K.; Rupp B. The Solvent Component of Macromolecular Crystals. Acta Cryst D 2015, 71, 1023–1038. 10.1107/S1399004715006045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubochet J.; McDowall A. W. Vitrification of Pure Water for Electron Microscopy. Journal of Microscopy 1981, 124, 3–4. 10.1111/j.1365-2818.1981.tb02483.x. [DOI] [Google Scholar]
- Dubochet J.; Lepault J.; Freeman R.; Berriman J. A.; Homo J.-C. Electron Microscopy of Frozen Water and Aqueous Solutions. Journal of Microscopy 1982, 128, 219–237. 10.1111/j.1365-2818.1982.tb04625.x. [DOI] [Google Scholar]
- Dubochet J.; Adrian M.; Chang J.-J.; Homo J.-C.; Lepault J.; McDowall A. W.; Schultz P. Cryo-Electron Microscopy of Vitrified Specimens. Quarterly Reviews of Biophysics 1988, 21, 129–228. 10.1017/S0033583500004297. [DOI] [PubMed] [Google Scholar]
- Iancu C. V.; Tivol W. F.; Schooler J. B.; Dias D. P.; Henderson G. P.; Murphy G. E.; Wright E. R.; Li Z.; Yu Z.; Briegel A.; Gan L.; He Y.; Jensen G. J. Electron Cryotomography Sample Preparation Using the Vitrobot. Nat Protoc 2006, 1, 2813–2819. 10.1038/nprot.2006.432. [DOI] [PubMed] [Google Scholar]
- de la Cruz M. J.; Hattne J.; Shi D.; Seidler P.; Rodriguez J.; Reyes F. E.; Sawaya M. R.; Cascio D.; Weiss S. C.; Kim S. K.; Hinck C. S.; Hinck A. P.; Calero G.; Eisenberg D.; Gonen T. Atomic-Resolution Structures from Fragmented Protein Crystals with the CryoEM Method MicroED. Nat Methods 2017, 14, 399–402. 10.1038/nmeth.4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zee C.-T.; Saha A.; Sawaya M. R.; Rodriguez J. A.. Ab Initio Determination of Peptide Structures by MicroED. In Cryoem: Methods and Protocols; Methods in Molecular Biology; Gonen T., Nannenga B. L., Eds.; Springer: New York, 2021; pp 329–348 10.1007/978-1-0716-0966-8_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tivol W. F.; Briegel A.; Jensen G. J. An Improved Cryogen for Plunge Freezing. Microscopy and Microanalysis 2008, 14, 375–379. 10.1017/S1431927608080781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martynowycz M. W.; Clabbers M. T. B.; Unge J.; Hattne J.; Gonen T. Benchmarking the Ideal Sample Thickness in Cryo-EM. PNAS 2021, 118, 118. 10.1073/pnas.2108884118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duyvesteyn H. M. E.; Kotecha A.; Ginn H. M.; Hecksel C. W.; Beale E. V.; de Haas F.; Evans G.; Zhang P.; Chiu W.; Stuart D. I. Machining Protein Microcrystals for Structure Determination by Electron Diffraction. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 9569–9573. 10.1073/pnas.1809978115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martynowycz M. W.; Zhao W.; Hattne J.; Jensen G. J.; Gonen T. Qualitative Analyses of Polishing and Precoating FIB Milled Crystals for MicroED. Structure 2019, 27, 1594–1600. e2 10.1016/j.str.2019.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martynowycz M. W.; Zhao W.; Hattne J.; Jensen G. J.; Gonen T. Collection of Continuous Rotation MicroED Data from Ion Beam-Milled Crystals of Any Size. Structure 2019, 27, 545–548. e2 10.1016/j.str.2018.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martynowycz M. W.; Gonen T. Protocol for the Use of Focused Ion-Beam Milling to Prepare Crystalline Lamellae for Microcrystal Electron Diffraction (MicroED). STAR Protocols 2021, 2, 100686. 10.1016/j.xpro.2021.100686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolopoulos S.; Gonzalez-Calbet J. M.; Vallet-Regi M.; Corma A.; Corell C.; Guil J. M.; Perez-Pariente J. Direct Phasing in Electron Crystallography: Ab Initio Determination of a New MCM-22 Zeolite Structure. J. Am. Chem. Soc. 1995, 117, 8947–8956. 10.1021/ja00140a008. [DOI] [Google Scholar]
- Gemmi M.; Righi L.; Calestani G.; Migliori A.; Speghini A.; Santarosa M.; Bettinelli M. Structure Determination of φ-Bi8Pb5O17 by Electron and Powder X-Ray Diffraction. Ultramicroscopy 2000, 84, 133–142. 10.1016/S0304-3991(00)00007-3. [DOI] [PubMed] [Google Scholar]
- Weirich T. E.; Zou X. D.; Ramlau R.; Simon A.; Cascarano G. L.; Giacovazzo C.; Hovmöller S. Structures of Nanometre-Size Crystals Determined from Selected-Area Electron Diffraction Data. Acta Cryst A 2000, 56, 29–35. 10.1107/S0108767399009605. [DOI] [PubMed] [Google Scholar]
- Vincent R.; Midgley P. A. Double Conical Beam-Rocking System for Measurement of Integrated Electron Diffraction Intensities. Ultramicroscopy 1994, 53, 271–282. 10.1016/0304-3991(94)90039-6. [DOI] [Google Scholar]
- Eggeman A. S.; Midgley P. A.. Precession Electron Diffraction. In Advances in Imaging and Electron Physics; Advances in Imaging and Electron Physics; Hawkes P. W., Ed.; Elsevier, 2012; Chapter 1, Vol. 170, pp 1–63 10.1016/B978-0-12-394396-5.00001-4. [DOI] [Google Scholar]
- Own C. S.; Marks L. D.; Sinkler W. Precession Electron Diffraction 1: Multislice Simulation. Acta Crystallogr., Sect. A 2006, 62, 434–443. 10.1107/S0108767306032892. [DOI] [PubMed] [Google Scholar]
- Avilov A.; Kuligin K.; Nicolopoulos S.; Nickolskiy M.; Boulahya K.; Portillo J.; Lepeshov G.; Sobolev B.; Collette J. P.; Martin N.; Robins A. C.; Fischione P. Precession Technique and Electron Diffractometry as New Tools for Crystal Structure Analysis and Chemical Bonding Determination. Ultramicroscopy 2007, 107, 431–444. 10.1016/j.ultramic.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Midgley P. A.; Eggeman A. S. Precession Electron Diffraction - a Topical Review. IUCrJ 2015, 2, 126–136. 10.1107/S2052252514022283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gjønnes J.; Hansen V.; Berg B. S.; Runde P.; Cheng Y. F.; Gjønnes K.; Dorset D. L.; Gilmore C. J. Structure Model for the Phase AlmFe Derived from Three-Dimensional Electron Diffraction Intensity Data Collected by a Precession Technique. Comparison with Convergent-Beam Diffraction. Acta Cryst A 1998, 54, 306–319. 10.1107/S0108767397017030. [DOI] [Google Scholar]
- Gemmi M.; Zou X. D.; Hovmöller S.; Migliori A.; Vennström M.; Andersson Y. Structure of Ti2P Solved by Three-Dimensional Electron Diffraction Data Collected with the Precession Technique and High-Resolution Electron Microscopy. Acta Cryst A 2003, 59, 117–126. 10.1107/S0108767302022559. [DOI] [PubMed] [Google Scholar]
- Kolb U.; Gorelik T.; Kübel C.; Otten M. T.; Hubert D. Towards Automated Diffraction Tomography: Part I—Data Acquisition. Ultramicroscopy 2007, 107, 507–513. 10.1016/j.ultramic.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Kolb U.; Gorelik T.; Otten M. T. Towards Automated Diffraction Tomography. Part II—Cell Parameter Determination. Ultramicroscopy 2008, 108, 763–772. 10.1016/j.ultramic.2007.12.002. [DOI] [PubMed] [Google Scholar]
- Gorelik T. E.; van de Streek J.; Kilbinger A. F. M.; Brunklaus G.; Kolb U. Ab-Initio Crystal Structure Analysis and Refinement Approaches of Oligo p-Benzamides Based on Electron Diffraction Data. Acta Cryst B 2012, 68, 171–181. 10.1107/S0108768112003138. [DOI] [PubMed] [Google Scholar]
- Feyand M.; Mugnaioli E.; Vermoortele F.; Bueken B.; Dieterich J. M.; Reimer T.; Kolb U.; de Vos D.; Stock N. Automated Diffraction Tomography for the Structure Elucidation of Twinned, Sub-Micrometer Crystals of a Highly Porous, Catalytically Active Bismuth Metal-Organic Framework. Angewandte Chemie International Edition 2012, 51, 10373–10376. 10.1002/anie.201204963. [DOI] [PubMed] [Google Scholar]
- Mugnaioli E.; Gorelik T.; Kolb U. “Ab Initio” Structure Solution from Electron Diffraction Data Obtained by a Combination of Automated Diffraction Tomography and Precession Technique. Ultramicroscopy 2009, 109, 758–765. 10.1016/j.ultramic.2009.01.011. [DOI] [PubMed] [Google Scholar]
- Own C. S.; Subramanian A. K.; Marks L. D. Quantitative Analyses of Precession Diffraction Data for a Large Cell Oxide. Microscopy and Microanalysis 2004, 10, 96–104. 10.1017/S1431927604040322. [DOI] [PubMed] [Google Scholar]
- Zhang D.; Oleynikov P.; Hovmöller S.; Zou X. Collecting 3D Electron Diffraction Data by the Rotation Method. Zeitschrift für Kristallographie 2010, 225, 94–102. 10.1524/zkri.2010.1202. [DOI] [Google Scholar]
- Nederlof I.; van Genderen E.; Li Y.-W.; Abrahams J. P. A Medipix Quantum Area Detector Allows Rotation Electron Diffraction Data Collection from Submicrometre Three-Dimensional Protein Crystals. Acta Cryst D 2013, 69, 1223–1230. 10.1107/S0907444913009700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nannenga B. L.; Shi D.; Hattne J.; Reyes F. E.; Gonen T. Structure of Catalase Determined by MicroED. eLife 2014, 3, e03600 10.7554/eLife.03600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemmi M.; La Placa M. G. I.; Galanis A. S.; Rauch E. F.; Nicolopoulos S. Fast Electron Diffraction Tomography. J. Appl. Crystallogr. 2015, 48, 718–727. 10.1107/S1600576715004604. [DOI] [Google Scholar]
- Wang Y.; Yang T.; Xu H.; Zou X.; Wan W. On the Quality of the Continuous Rotation Electron Diffraction Data for Accurate Atomic Structure Determination of Inorganic Compounds. J. Appl. Crystallogr. 2018, 51, 1094–1101. 10.1107/S1600576718007604. [DOI] [Google Scholar]
- Schlichting I. Serial Femtosecond Crystallography: The First Five Years. IUCrJ 2015, 2, 246–255. 10.1107/S205225251402702X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman H. N.; Caleman C.; Timneanu N. Diffraction before Destruction. Philosophical Transactions of the Royal Society B: Biological Sciences 2014, 369, 20130313. 10.1098/rstb.2013.0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cichocka M. O.; Ångström J.; Wang B.; Zou X.; Smeets S. High-Throughput Continuous Rotation Electron Diffraction Data Acquisition via Software Automation. J. Appl. Crystallogr. 2018, 51, 1652–1661. 10.1107/S1600576718015145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roslova M.; Smeets S.; Wang B.; Thersleff T.; Xu H.; Zou X. InsteaDMatic: Towards Cross-Platform Automated Continuous Rotation Electron Diffraction. J. Appl. Crystallogr. 2020, 53, 1217–1224. 10.1107/S1600576720009590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaba K.; Maki-Yonekura S.; Yonekura K. Collecting Large Datasets of Rotational Electron Diffraction with ParallEM and SerialEM. Journal of Structural Biology 2020, 211, 107549. 10.1016/j.jsb.2020.107549. [DOI] [PubMed] [Google Scholar]
- Smeets S.; Zou X.; Wan W. Serial Electron Crystallography for Structure Determination and Phase Analysis of Nanocrystalline Materials. J. Appl. Crystallogr. 2018, 51, 1262–1273. 10.1107/S1600576718009500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bücker R.; Hogan-Lamarre P.; Mehrabi P.; Schulz E. C.; Bultema L. A.; Gevorkov Y.; Brehm W.; Yefanov O.; Oberthür D.; Kassier G. H.; Dwayne Miller R. J. Serial Protein Crystallography in an Electron Microscope. Nat Commun 2020, 11, 996. 10.1038/s41467-020-14793-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ophus C. Four-Dimensional Scanning Transmission Electron Microscopy (4D-STEM): From Scanning Nanodiffraction to Ptychography and Beyond. Microscopy and Microanalysis 2019, 25, 563–582. 10.1017/S1431927619000497. [DOI] [PubMed] [Google Scholar]
- Gallagher-Jones M.; Ophus C.; Bustillo K. C.; Boyer D. R.; Panova O.; Glynn C.; Zee C.-T.; Ciston J.; Mancia K. C.; Minor A. M.; Rodriguez J. A. Nanoscale Mosaicity Revealed in Peptide Microcrystals by Scanning Electron Nanodiffraction. Commun Biol 2019, 2, 1–8. 10.1038/s42003-018-0263-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher-Jones M.; Bustillo K. C.; Ophus C.; Richards L. S.; Ciston J.; Lee S.; Minor A. M.; Rodriguez J. A. Atomic Structures Determined from Digitally Defined Nanocrystalline Regions. Int. Union Crystallogr. J. 2020, 7, 490–499. 10.1107/S2052252520004030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustillo K. C.; Zeltmann S. E.; Chen M.; Donohue J.; Ciston J.; Ophus C.; Minor A. M. 4D-STEM of Beam-Sensitive Materials. Acc. Chem. Res. 2021, 54, 2543–2551. 10.1021/acs.accounts.1c00073. [DOI] [PubMed] [Google Scholar]
- Kolb U.; Mugnaioli E.; Gorelik T. E. Automated Electron Diffraction Tomography - a New Tool for Nano Crystal Structure Analysis. Crystal Research and Technology 2011, 46, 542–554. 10.1002/crat.201100036. [DOI] [Google Scholar]
- Gemmi M.; Oleynikov P. Scanning Reciprocal Space for Solving Unknown Structures: Energy Filtered Diffraction Tomography and Rotation Diffraction Tomography Methods. Zeitschrift für Kristallographie 2013, 228, 51–58. 10.1524/zkri.2013.1559. [DOI] [Google Scholar]
- Wan W.; Sun J.; Su J.; Hovmöller S.; Zou X. Three-Dimensional Rotation Electron Diffraction: Software RED for Automated Data Collection and Data Processing. J. Appl. Crystallogr. 2013, 46, 1863–1873. 10.1107/S0021889813027714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palatinus L.; Brázda P.; Jelínek M.; Hrdá J.; Steciuk G.; Klementová M. Specifics of the Data Processing of Precession Electron Diffraction Tomography Data and Their Implementation in the Program PETS2.0. Acta Cryst B 2019, 75, 512–522. 10.1107/S2052520619007534. [DOI] [PubMed] [Google Scholar]
- Battye T. G. G.; Kontogiannis L.; Johnson O.; Powell H. R.; Leslie A. G. W. IMOSFLM: A New Graphical Interface for Diffraction-Image Processing with MOSFLM. Acta Cryst D 2011, 67, 271–281. 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter G.; Beilsten-Edmands J.; Devenish N.; Gerstel M.; Gildea R. J.; McDonagh D.; Pascal E.; Waterman D. G.; Williams B. H.; Evans G. DIALS as a Toolkit. Protein Sci. 2022, 31, 232–250. 10.1002/pro.4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Cryst D 2010, 66, 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clabbers M. T. B.; Gruene T.; Parkhurst J. M.; Abrahams J. P.; Waterman D. G. Electron Diffraction Data Processing with DIALS. Acta Cryst D 2018, 74, 506–518. 10.1107/S2059798318007726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White T. A.; Mariani V.; Brehm W.; Yefanov O.; Barty A.; Beyerlein K. R.; Chervinskii F.; Galli L.; Gati C.; Nakane T.; Tolstikova A.; Yamashita K.; Yoon C. H.; Diederichs K.; Chapman H. N. Recent Developments in CrystFEL. J. Appl. Crystallogr. 2016, 49, 680–689. 10.1107/S1600576716004751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bücker R.; Hogan-Lamarre P.; Miller R. J. D. Serial Electron Diffraction Data Processing With Diffractem and CrystFEL. Frontiers in Molecular Biosciences 2021, 8, 415. 10.3389/fmolb.2021.624264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick G. M. Phase Annealing in SHELX-90: Direct Methods for Larger Structures. Acta Cryst A 1990, 46, 467–473. 10.1107/S0108767390000277. [DOI] [Google Scholar]
- Morris R. J.; Bricogne G. Sheldrick’s 1.2 Å Rule and Beyond. Acta Cryst D 2003, 59, 615–617. 10.1107/S090744490300163X. [DOI] [PubMed] [Google Scholar]
- Sheldrick G. M. SHELXT - Integrated Space-Group and Crystal-Structure Determination. Acta Cryst A 2015, 71, 3–8. 10.1107/S2053273314026370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider T. R.; Sheldrick G. M. Substructure Solution with SHELXD. Acta Cryst D 2002, 58, 1772–1779. 10.1107/S0907444902011678. [DOI] [PubMed] [Google Scholar]
- Altomare A.; Giacovazzo C.; Guagliardi A.; Siliqi D. Triplet and Quartet Relationships and the `positivity’ Postulate. Acta Cryst A 1994, 50, 311–317. 10.1107/S0108767393009857. [DOI] [Google Scholar]
- Wang J. On the Appearance of Carboxylates in Electrostatic Potential Maps. Protein Sci. 2017, 26, 396–402. 10.1002/pro.3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. Experimental Charge Density from Electron Microscopic Maps. Protein Sci. 2017, 26, 1619–1626. 10.1002/pro.3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Moore P. B. On the Interpretation of Electron Microscopic Maps of Biological Macromolecules. Protein Sci. 2017, 26, 122–129. 10.1002/pro.3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian G.; Basu S.; Liu H.; Zuo J.-M.; Spence J. C. H. Solving Protein Nanocrystals by Cryo-EM Diffraction: Multiple Scattering Artifacts. Ultramicroscopy 2015, 148, 87–93. 10.1016/j.ultramic.2014.08.013. [DOI] [PubMed] [Google Scholar]
- Latychevskaia T.; Abrahams J. P. Inelastic Scattering and Solvent Scattering Reduce Dynamical Diffraction in Biological Crystals. Acta Cryst B 2019, 75, 523–531. 10.1107/S2052520619009661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger A. T. Simulated Annealing in Crystallography. Annu. Rev. Phys. Chem. 1991, 42, 197–223. 10.1146/annurev.pc.42.100191.001213. [DOI] [Google Scholar]
- Woollam G. R.; Das P. P.; Mugnaioli E.; Andrusenko I.; Galanis A. S.; van de Streek J.; Nicolopoulos S.; Gemmi M.; Wagner T. Structural Analysis of Metastable Pharmaceutical Loratadine Form II, by 3D Electron Diffraction and DFT+D Energy Minimisation. CrystEngComm 2020, 22, 7490–7499. 10.1039/D0CE01216E. [DOI] [Google Scholar]
- Lightowler M.; Li S.; Ou X.; Zou X.; Lu M.; Xu H. Indomethacin Polymorph δ Revealed To Be Two Plastically Bendable Crystal Forms by 3D Electron Diffraction: Correcting a 47-Year-Old Misunderstanding**. Angewandte Chemie International Edition 2022, 61, e202114985 10.1002/anie.202114985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser Crystallographic Software. J. Appl. Crystallogr. 2007, 40, 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A.; Teplyakov A. Molecular Replacement with MOLREP. Acta Cryst D 2010, 66, 22–25. 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- Xu H.; Lebrette H.; Clabbers M. T. B.; Zhao J.; Griese J. J.; Zou X.; Högbom M. Solving a New R2lox Protein Structure by Microcrystal Electron Diffraction. Science Advances 2019, 5, eaax4621 10.1126/sciadv.aax4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H.; Zou X.; Högbom M.; Lebrette H.. Redetermination of the First Unknown Protein MicroED Structure by High Resolution X-Ray Diffraction. bioRxiv 2021; 438860. 10.1101/2021.04.07.438860. [DOI] [Google Scholar]
- Rodríguez D. D.; Grosse C.; Himmel S.; González C.; de Ilarduya I. M.; Becker S.; Sheldrick G. M.; Usón I. Crystallographic Ab Initio Protein Structure Solution below Atomic Resolution. Nat Methods 2009, 6, 651–653. 10.1038/nmeth.1365. [DOI] [PubMed] [Google Scholar]
- Sammito M.; Meindl K.; de Ilarduya I. M.; Millán C.; Artola-Recolons C.; Hermoso J. A.; Usón I. Structure Solution with ARCIMBOLDO Using Fragments Derived from Distant Homology Models. The FEBS Journal 2014, 281, 4029–4045. 10.1111/febs.12897. [DOI] [PubMed] [Google Scholar]
- Richards L. S.; Flores M. D.; Millán C.; Glynn C.; Zee C.-T.; Sawaya M. R.; Gallagher-Jones M.; Borges R. J.; Usón I.; Rodriguez J. A.. Fragment-Based Ab Initio Phasing of Peptidic Nanocrystals by MicroED. bioRxiv 2021; 459692. 10.1101/2021.09.13.459692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards L. S.; Millán C.; Miao J.; Martynowycz M. W.; Sawaya M. R.; Gonen T.; Borges R. J.; Usón I.; Rodriguez J. A. Fragment-Based Determination of a Proteinase K Structure from MicroED Data Using ARCIMBOLDO_SHREDDER. Acta Crystallogr D Struct Biol 2020, 76, 703–712. 10.1107/S2059798320008049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J.; Evans R.; Pritzel A.; Green T.; Figurnov M.; Ronneberger O.; Tunyasuvunakool K.; Bates R.; Žídek A.; Potapenko A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek M.; DiMaio F.; Anishchenko I.; Dauparas J.; Ovchinnikov S.; Lee G. R.; Wang J.; Cong Q.; Kinch L. N.; Schaeffer R. D.; Millán C.; Park H.; Adams C.; Glassman C. R.; DeGiovanni A.; Pereira J. H.; Rodrigues A. V.; van Dijk A. A.; Ebrecht A. C.; Opperman D. J.; Sagmeister T.; Buhlheller C.; Pavkov-Keller T.; Rathinaswamy M. K.; Dalwadi U.; Yip C. K.; Burke J. E.; Garcia K. C.; Grishin N. V.; Adams P. D.; Read R. J.; Baker D. Accurate Prediction of Protein Structures and Interactions Using a Three-Track Neural Network. Science 2021, 373, 871–876. 10.1126/science.abj8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebschner D.; Afonine P. V.; Baker M. L.; Bunkóczi G.; Chen V. B.; Croll T. I.; Hintze B.; Hung L.-W.; Jain S.; McCoy A. J.; Moriarty N. W.; Oeffner R. D.; Poon B. K.; Prisant M. G.; Read R. J.; Richardson J. S.; Richardson D. C.; Sammito M. D.; Sobolev O. V.; Stockwell D. H.; Terwilliger T. C.; Urzhumtsev A. G.; Videau L. L.; Williams C. J.; Adams P. D. Macromolecular Structure Determination Using X-Rays, Neutrons and Electrons: Recent Developments in Phenix. Acta Cryst D 2019, 75, 861–877. 10.1107/S2059798319011471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N.; Skubák P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. REFMAC5 for the Refinement of Macromolecular Crystal Structures. Acta Cryst D 2011, 67, 355–367. 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick G. M. Crystal Structure Refinement with SHELXL. Acta Cryst C 2015, 71, 3–8. 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum T. B.; Housset D.; Clabbers M. T. B.; van Genderen E.; Bacia-Verloop M.; Zander U.; McCarthy A. A.; Schoehn G.; Ling W. L.; Abrahams J. P. Statistically Correcting Dynamical Electron Scattering Improves the Refinement of Protein Nanocrystals, Including Charge Refinement of Coordinated Metals. Acta Cryst D 2021, 77, 75–85. 10.1107/S2059798320014540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palatinus L.; Petříček V.; Corrêa C. A. Structure Refinement Using Precession Electron Diffraction Tomography and Dynamical Diffraction: Theory and Implementation. Acta Cryst A 2015, 71, 235–244. 10.1107/S2053273315001266. [DOI] [PubMed] [Google Scholar]
- Palatinus L.; Corrêa C. A.; Steciuk G.; Jacob D.; Roussel P.; Boullay P.; Klementová M.; Gemmi M.; Kopeček J.; Domeneghetti M. C.; Cámara F.; Petříček V. Structure Refinement Using Precession Electron Diffraction Tomography and Dynamical Diffraction: Tests on Experimental Data. Acta Crystallographica Section B 2015, 71, 740–751. 10.1107/S2052520615017023. [DOI] [PubMed] [Google Scholar]
- Palatinus L.; Brázda P.; Boullay P.; Perez O.; Klementová M.; Petit S.; Eigner V.; Zaarour M.; Mintova S. Hydrogen Positions in Single Nanocrystals Revealed by Electron Diffraction. Science 2017, 355, 166–169. 10.1126/science.aak9652. [DOI] [PubMed] [Google Scholar]
- Clabbers M. T. B.; Gruene T.; van Genderen E.; Abrahams J. P. Reducing Dynamical Electron Scattering Reveals Hydrogen Atoms. Acta Cryst A 2019, 75, 82–93. 10.1107/S2053273318013918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T.; Xu H.; Zou X.. Improving Data Quality for 3D Electron Diffraction (3D ED) by Gatan Image Filter and a New Crystal Tracking Method. arXiv 2021; arXiv:2108.07468. 10.48550/arXiv.2108.07468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S.; Head-Gordon T.; Glaeser R. M.; Downing K. H. Chemical Bonding Effects in the Determination of Protein Structures by Electron Crystallography. Acta Cryst A 1999, 55, 305–313. 10.1107/S0108767398009726. [DOI] [PubMed] [Google Scholar]
- Yamashita H.; Kidera A. Environmental Influence on Electron Scattering from a Molecule. Acta Cryst A 2001, 57, 518–525. 10.1107/S0108767301006778. [DOI] [PubMed] [Google Scholar]
- Gruza B.; Chodkiewicz M. L.; Krzeszczakowska J.; Dominiak P. M. Refinement of Organic Crystal Structures with Multipolar Electron Scattering Factors. Acta Cryst A 2020, 76, 92–109. 10.1107/S2053273319015304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha K. K.; Gruza B.; Chodkiewicz M. L.; Jelsch C.; Dominiak P. M. Refinements on Electron Diffraction Data of β-Glycine in MoPro: A Quest for an Improved Structure Model. J. Appl. Crystallogr. 2021, 54, 1234–1243. 10.1107/S160057672100580X. [DOI] [Google Scholar]
- Domagała S.; Fournier B.; Liebschner D.; Guillot B.; Jelsch C. An Improved Experimental Databank of Transferable Multipolar Atom Models - ELMAM2. Construction Details and Applications. Acta Cryst A 2012, 68, 337–351. 10.1107/S0108767312008197. [DOI] [PubMed] [Google Scholar]
- Grigorieff N.; Ceska T. A.; Downing K. H.; Baldwin J. M.; Henderson R. Electron-Crystallographic Refinement of the Structure of Bacteriorhodopsin. J. Mol. Biol. 1996, 259, 393–421. 10.1006/jmbi.1996.0328. [DOI] [PubMed] [Google Scholar]
- Mitsuoka K.; Hirai T.; Murata K.; Miyazawa A.; Kidera A.; Kimura Y.; Fujiyoshi Y. The Structure of Bacteriorhodopsin at 3.0 Å Resolution Based on Electron Crystallography: Implication of the Charge Distribution11Edited by R. Huber. J. Mol. Biol. 1999, 286, 861–882. 10.1006/jmbi.1998.2529. [DOI] [PubMed] [Google Scholar]
- Kimura Y.; Vassylyev D. G.; Miyazawa A.; Kidera A.; Matsushima M.; Mitsuoka K.; Murata K.; Hirai T.; Fujiyoshi Y. Surface of Bacteriorhodopsin Revealed by High-Resolution Electron Crystallography. Nature 1997, 389, 206–211. 10.1038/38323. [DOI] [PubMed] [Google Scholar]
- Hirai T.; Mitsuoka K.; Kidera A.; Fujiyoshi Y. Simulation of Charge Effects on Density Maps Obtained by High-Resolution Electron Crystallography. Journal of Electron Microscopy 2007, 56, 131–140. 10.1093/jmicro/dfm019. [DOI] [PubMed] [Google Scholar]
- Anstis G. R.; Lynch D. F.; Moodie A. F.; O’Keefe M. A. N-Beam Lattice Images. III. Upper Limits of Ionicity in W4Nb26O77. Acta Cryst A 1973, 29, 138–147. 10.1107/S0567739473000379. [DOI] [Google Scholar]
- Tsirelson V. G.; Avilov A. S.; Lepeshov G. G.; Kulygin A. K.; Stahn J.; Pietsch U.; Spence J. C. H. Quantitative Analysis of the Electrostatic Potential in Rock-Salt Crystals Using Accurate Electron Diffraction Data. J. Phys. Chem. B 2001, 105, 5068–5074. 10.1021/jp0015729. [DOI] [Google Scholar]
- Yonekura K.; Kato K.; Ogasawara M.; Tomita M.; Toyoshima C. Electron Crystallography of Ultrathin 3D Protein Crystals: Atomic Model with Charges. PNAS 2015, 112, 3368–3373. 10.1073/pnas.1500724112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonekura K.; Maki-Yonekura S. Refinement of Cryo-EM Structures Using Scattering Factors of Charged Atoms. J. Appl. Crystallogr. 2016, 49, 1517–1523. 10.1107/S1600576716011274. [DOI] [Google Scholar]
- Yonekura K.; Matsuoka R.; Yamashita Y.; Yamane T.; Ikeguchi M.; Kidera A.; Maki-Yonekura S. Ionic Scattering Factors of Atoms That Compose Biological Molecules. IUCrJ 2018, 5, 348–353. 10.1107/S2052252518005237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine P. V.; Mustyakimov M.; Grosse-Kunstleve R. W.; Moriarty N. W.; Langan P.; Adams P. D. Joint X-Ray and Neutron Refinement with Phenix. Refine. Acta Cryst D 2010, 66, 1153–1163. 10.1107/S0907444910026582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardberg A. S.; Del Castillo A. R.; Weiss K. L.; Meilleur F.; Blakeley M. P.; Myles D. A. A. Unambiguous Determination of H-Atom Positions: Comparing Results from Neutron and High-Resolution X-Ray Crystallography. Acta Cryst D 2010, 66, 558–567. 10.1107/S0907444910005494. [DOI] [PubMed] [Google Scholar]
- Schmidtmann M.; Coster P.; Henry P. F.; Ting V. P.; Weller M. T.; Wilson C. C. Determining Hydrogen Positions in Crystal Engineered Organic Molecular Complexes by Joint Neutron Powder and Single Crystal X-Ray Diffraction. CrystEngComm 2014, 16, 1232–1236. 10.1039/C3CE42070A. [DOI] [Google Scholar]
- Jones C. G.; Asay M.; Kim L. J.; Kleinsasser J. F.; Saha A.; Fulton T. J.; Berkley K. R.; Cascio D.; Malyutin A. G.; Conley M. P.; Stoltz B. M.; Lavallo V.; Rodríguez J. A.; Nelson H. M. Characterization of Reactive Organometallic Species via MicroED. ACS Cent. Sci. 2019, 5, 1507–1513. 10.1021/acscentsci.9b00403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bau R.; Drabnis M. H. Structures of Transition Metal Hydrides Determined by Neutron Diffraction. Inorg. Chim. Acta 1997, 259, 27–50. 10.1016/S0020-1693(97)89125-6. [DOI] [Google Scholar]
- Gallagher-Jones M.; Glynn C.; Boyer D. R.; Martynowycz M. W.; Hernandez E.; Miao J.; Zee C.-T.; Novikova I. V.; Goldschmidt L.; McFarlane H. T.; Helguera G. F.; Evans J. E.; Sawaya M. R.; Cascio D.; Eisenberg D. S.; Gonen T.; Rodriguez J. A. Sub-Ångström Cryo-EM Structure of a Prion Protofibril Reveals a Polar Clasp. Nat Struct Mol Biol 2018, 25, 131–134. 10.1038/s41594-017-0018-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakane T.; Kotecha A.; Sente A.; McMullan G.; Masiulis S.; Brown P. M. G. E.; Grigoras I. T.; Malinauskaite L.; Malinauskas T.; Miehling J.; Uchański T.; Yu L.; Karia D.; Pechnikova E. V.; de Jong E.; Keizer J.; Bischoff M.; McCormack J.; Tiemeijer P.; Hardwick S. W.; Chirgadze D. Y.; Murshudov G.; Aricescu A. R.; Scheres S. H. W. Single-Particle Cryo-EM at Atomic Resolution. Nature 2020, 587, 152–156. 10.1038/s41586-020-2829-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimer L.; Kohl H.. Transmission Electron Microscopy: Physics of Image Formation, 5th ed.; Springer: New York, 2008. [Google Scholar]
- Yonekura K.; Maki-Yonekura S.; Namba K. Quantitative Comparison of Zero-Loss and Conventional Electron Diffraction from Two-Dimensional and Thin Three-Dimensional Protein Crystals. Biophys. J. 2002, 82, 2784–2797. 10.1016/S0006-3495(02)75619-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonekura K.; Braunfeld M. B.; Maki-Yonekura S.; Agard D. A. Electron Energy Filtering Significantly Improves Amplitude Contrast of Frozen-Hydrated Protein at 300kV. Journal of Structural Biology 2006, 156, 524–536. 10.1016/j.jsb.2006.07.016. [DOI] [PubMed] [Google Scholar]
- Yonekura K.; Ishikawa T.; Maki-Yonekura S. A New Cryo-EM System for Electron 3D Crystallography by EEFD. Journal of Structural Biology 2019, 206, 243–253. 10.1016/j.jsb.2019.03.009. [DOI] [PubMed] [Google Scholar]
- Burmester C.; Schröder R. R. Solving the Phase Problem in Protein Electron Crystallography: Multiple Isomorphous Replacement and Anomalous Dispersion as Alternatives to Imaging. Scan. Microsc. 1997, 11, 323–334. [Google Scholar]
- Martynowycz M. W.; Hattne J.; Gonen T. Experimental Phasing of MicroED Data Using Radiation Damage. Structure 2020, 28, 458–464. e2 10.1016/j.str.2020.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flack H. D.; Bernardinelli G. The Use of X-Ray Crystallography to Determine Absolute Configuration. Chirality 2008, 20, 681–690. 10.1002/chir.20473. [DOI] [PubMed] [Google Scholar]
- Bijvoet J. M.; Peerdeman A. F.; van BOMMEL A. J. Determination of the Absolute Configuration of Optically Active Compounds by Means of X-Rays. Nature 1951, 168, 271–272. 10.1038/168271a0. [DOI] [Google Scholar]
- Brázda P.; Palatinus L.; Babor M. Electron Diffraction Determines Molecular Absolute Configuration in a Pharmaceutical Nanocrystal. Science 2019, 364, 667–669. 10.1126/science.aaw2560. [DOI] [PubMed] [Google Scholar]
- Dorset D. L.Structural Electron Crystallography; Springer Science: New York, 1995. [Google Scholar]
- Hooft R. W. W.; Straver L. H.; Spek A. L. Determination of Absolute Structure Using Bayesian Statistics on Bijvoet Differences. J. Appl. Crystallogr. 2008, 41, 96–103. 10.1107/S0021889807059870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klar P.; Krysiak Y.; Xu H.; Steciuk G.; Cho J.; Zou X.; Palatinus L.. Chirality and Accurate Structure Models by Exploiting Dynamical Effects in Continuous-Rotation 3D ED Data. ChemRxiv 2021, 4jh14. 10.26434/chemrxiv-2021-4jh14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spence J. C. H.; Donatelli J. J. Inversion of Dynamical Bragg Intensities to Complex Structure Factors by Iterated Projections. For Ultramic. 2020. (“Pico” Festschrift, May 2021). Ultramicroscopy 2021, 222, 113214. 10.1016/j.ultramic.2021.113214. [DOI] [PubMed] [Google Scholar]
- Wang B.; Bruhn J.; Weldeab A.; Wilson T.; McGilvray P.; Mashore M.; Song Q.; Scapin G.; Lin Y. Absolute Configuration Determination of Pharmaceutical Crystalline Powders by MicroED via Chiral Salt Formation. Chem. Commun. 2022, 58, 4711. 10.1039/D2CC00221C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg D.; Jucker M. The Amyloid State of Proteins in Human Diseases. Cell 2012, 148, 1188–1203. 10.1016/j.cell.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg D. S.; Sawaya M. R. Structural Studies of Amyloid Proteins at the Molecular Level. Annu. Rev. Biochem. 2017, 86, 69–95. 10.1146/annurev-biochem-061516-045104. [DOI] [PubMed] [Google Scholar]
- Hughes M. P.; Sawaya M. R.; Boyer D. R.; Goldschmidt L.; Rodriguez J. A.; Cascio D.; Chong L.; Gonen T.; Eisenberg D. S. Atomic Structures of Low-Complexity Protein Segments Reveal Kinked β Sheets That Assemble Networks. Science 2018, 359, 698–701. 10.1126/science.aan6398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balbirnie M.; Grothe R.; Eisenberg D. S. An Amyloid-Forming Peptide from the Yeast Prion Sup35 Reveals a Dehydrated β-Sheet Structure for Amyloid. PNAS 2001, 98, 2375–2380. 10.1073/pnas.041617698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson R.; Sawaya M. R.; Balbirnie M.; Madsen A. Å.; Riekel C.; Grothe R.; Eisenberg D. Structure of the Cross-β Spine of Amyloid-like Fibrils. Nature 2005, 435, 773–778. 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez J. A.; Ivanova M. I.; Sawaya M. R.; Cascio D.; Reyes F. E.; Shi D.; Sangwan S.; Guenther E. L.; Johnson L. M.; Zhang M.; Jiang L.; Arbing M. A.; Nannenga B. L.; Hattne J.; Whitelegge J.; Brewster A. S.; Messerschmidt M.; Boutet S.; Sauter N. K.; Gonen T.; Eisenberg D. S. Structure of the Toxic Core of α-Synuclein from Invisible Crystals. Nature 2015, 525, 486–490. 10.1038/nature15368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaya M. R.; Rodriguez J.; Cascio D.; Collazo M. J.; Shi D.; Reyes F. E.; Hattne J.; Gonen T.; Eisenberg D. S. Ab Initio Structure Determination from Prion Nanocrystals at Atomic Resolution by MicroED. PNAS 2016, 113, 11232–11236. 10.1073/pnas.1606287113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krotee P.; Rodriguez J. A.; Sawaya M. R.; Cascio D.; Reyes F. E.; Shi D.; Hattne J.; Nannenga B. L.; Oskarsson M. E.; Philipp S.; Griner S.; Jiang L.; Glabe C. G.; Westermark G. T.; Gonen T.; Eisenberg D. S. Atomic Structures of Fibrillar Segments of HIAPP Suggest Tightly Mated β-Sheets Are Important for Cytotoxicity. eLife 2017, 6, e19273 10.7554/eLife.19273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidler P. M.; Boyer D. R.; Rodriguez J. A.; Sawaya M. R.; Cascio D.; Murray K.; Gonen T.; Eisenberg D. S. Structure-Based Inhibitors of Tau Aggregation. Nature Chem 2018, 10, 170–176. 10.1038/nchem.2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidler P. M.; Boyer D. R.; Murray K. A.; Yang T. P.; Bentzel M.; Sawaya M. R.; Rosenberg G.; Cascio D.; Williams C. K.; Newell K. L.; Ghetti B.; DeTure M. A.; Dickson D. W.; Vinters H. V.; Eisenberg D. S. Structure-Based Inhibitors Halt Prion-like Seeding by Alzheimer’s Disease-and Tauopathy-Derived Brain Tissue Samples. J. Biol. Chem. 2019, 294, 16451–16464. 10.1074/jbc.RA119.009688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther E. L.; Ge P.; Trinh H.; Sawaya M. R.; Cascio D.; Boyer D. R.; Gonen T.; Zhou Z. H.; Eisenberg D. S. Atomic-Level Evidence for Packing and Positional Amyloid Polymorphism by Segment from TDP-43 RRM2. Nat Struct Mol Biol 2018, 25, 311–319. 10.1038/s41594-018-0045-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther E. L.; Cao Q.; Trinh H.; Lu J.; Sawaya M. R.; Cascio D.; Boyer D. R.; Rodriguez J. A.; Hughes M. P.; Eisenberg D. S. Atomic Structures of TDP-43 LCD Segments and Insights into Reversible or Pathogenic Aggregation. Nat Struct Mol Biol 2018, 25, 463–471. 10.1038/s41594-018-0064-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zee C.; Glynn C.; Gallagher-Jones M.; Miao J.; Santiago C. G.; Cascio D.; Gonen T.; Sawaya M. R.; Rodriguez J. A. Homochiral and Racemic MicroED Structures of a Peptide Repeat from the Ice-Nucleation Protein InaZ. IUCrJ 2019, 6, 197–205. 10.1107/S2052252518017621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griner S. L.; Seidler P.; Bowler J.; Murray K. A.; Yang T. P.; Sahay S.; Sawaya M. R.; Cascio D.; Rodriguez J. A.; Philipp S.; Sosna J.; Glabe C. G.; Gonen T.; Eisenberg D. S. Structure-Based Inhibitors of Amyloid Beta Core Suggest a Common Interface with Tau. eLife 2019, 8, e46924 10.7554/eLife.46924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warmack R. A.; Boyer D. R.; Zee C.-T.; Richards L. S.; Sawaya M. R.; Cascio D.; Gonen T.; Eisenberg D. S.; Clarke S. G. Structure of Amyloid-β (20–34) with Alzheimer’s-Associated Isomerization at Asp23 Reveals a Distinct Protofilament Interface. Nat Commun 2019, 10, 3357. 10.1038/s41467-019-11183-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui X.; Luo F.; Li Y.; Zhou H.; Qin Z.; Liu Z.; Gu J.; Xie M.; Zhao K.; Dai B.; Shin W. S.; He J.; He L.; Jiang L.; Zhao M.; Sun B.; Li X.; Liu C.; Li D. Structural Basis for Reversible Amyloids of HnRNPA1 Elucidates Their Role in Stress Granule Assembly. Nat Commun 2019, 10, 2006. 10.1038/s41467-019-09902-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo F.; Gui X.; Zhou H.; Gu J.; Li Y.; Liu X.; Zhao M.; Li D.; Li X.; Liu C. Atomic Structures of FUS LC Domain Segments Reveal Bases for Reversible Amyloid Fibril Formation. Nat Struct Mol Biol 2018, 25, 341–346. 10.1038/s41594-018-0050-8. [DOI] [PubMed] [Google Scholar]
- Zhou H.; Luo F.; Luo Z.; Li D.; Liu C.; Li X. Programming Conventional Electron Microscopes for Solving Ultrahigh-Resolution Structures of Small and Macro-Molecules. Anal. Chem. 2019, 91, 10996–11003. 10.1021/acs.analchem.9b01162. [DOI] [PubMed] [Google Scholar]
- Gruene T.; Wennmacher J. T. C.; Zaubitzer C.; Holstein J. J.; Heidler J.; Fecteau-Lefebvre A.; De Carlo S.; Müller E.; Goldie K. N.; Regeni I.; Li T.; Santiso-Quinones G.; Steinfeld G.; Handschin S.; van Genderen E.; van Bokhoven J. A.; Clever G. H.; Pantelic R. Rapid Structure Determination of Microcrystalline Molecular Compounds Using Electron Diffraction. Angewandte Chemie International Edition 2018, 57, 16313–16317. 10.1002/anie.201811318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones C. G.; Martynowycz M. W.; Hattne J.; Fulton T. J.; Stoltz B. M.; Rodriguez J. A.; Nelson H. M.; Gonen T. The CryoEM Method MicroED as a Powerful Tool for Small Molecule Structure Determination. ACS Cent. Sci. 2018, 4, 1587–1592. 10.1021/acscentsci.8b00760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A.; Clardy J. Tiny Crystals Have Big Potential for Determining Structures of Small Molecules. Nature 2018, 564, 348–349. 10.1038/d41586-018-07756-5. [DOI] [PubMed] [Google Scholar]
- Sitsel O.; Raunser S. Big Insights from Tiny Crystals. Nature Chem 2019, 11, 106–108. 10.1038/s41557-019-0211-3. [DOI] [PubMed] [Google Scholar]
- Filer C. N. MicroED and Cannabinoid Research. Journal of Cannabis Research 2021, 3, 14. 10.1186/s42238-021-00070-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrusenko I.; Hamilton V.; Mugnaioli E.; Lanza A.; Hall C.; Potticary J.; Hall S. R.; Gemmi M. The Crystal Structure of Orthocetamol Solved by 3D Electron Diffraction. Angewandte Chemie International Edition 2019, 58, 10919–10922. 10.1002/anie.201904564. [DOI] [PubMed] [Google Scholar]
- Gorelik T. E.; Habermehl S.; Shubin A. A.; Gruene T.; Yoshida K.; Oleynikov P.; Kaiser U.; Schmidt M. U. Crystal Structure of Copper Perchlorophthalo-cyanine Analysed by 3D Electron Diffraction. Acta Cryst B 2021, 77, 662–675. 10.1107/S2052520621006806. [DOI] [Google Scholar]
- Uyeda N.; Kobayashi T.; Suito E.; Harada Y.; Watanabe M. Molecular Image Resolution in Electron Microscopy. J. Appl. Phys. 1972, 43, 5181–5189. 10.1063/1.1661094. [DOI] [Google Scholar]
- Kim L. J.; Xue M.; Li X.; Xu Z.; Paulson E.; Mercado B.; Nelson H. M.; Herzon S. B. Structure Revision of the Lomaiviticins. J. Am. Chem. Soc. 2021, 143, 6578–6585. 10.1021/jacs.1c01729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He H.; Ding W.-D.; Bernan V. S.; Richardson A. D.; Ireland C. M.; Greenstein M.; Ellestad G. A.; Carter G. T. Lomaiviticins A and B, Potent Antitumor Antibiotics from Micromonospora Lomaivitiensis. J. Am. Chem. Soc. 2001, 123, 5362–5363. 10.1021/ja010129o. [DOI] [PubMed] [Google Scholar]
- Burns D. C.; Reynolds W. F. Minimizing the Risk of Deducing Wrong Natural Product Structures from NMR Data. Magn. Reson. Chem. 2021, 59, 500–533. 10.1002/mrc.4933. [DOI] [PubMed] [Google Scholar]
- Ghosh R.; Bu G.; Nannenga B. L.; Sumner L. W. Recent Developments Toward Integrated Metabolomics Technologies (UHPLC-MS-SPE-NMR and MicroED) for Higher-Throughput Confident Metabolite Identifications. Frontiers in Molecular Biosciences 2021, 8, 844. 10.3389/fmolb.2021.720955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim L. J.; Ohashi M.; Zhang Z.; Tan D.; Asay M.; Cascio D.; Rodriguez J. A.; Tang Y.; Nelson H. M. Prospecting for Natural Products by Genome Mining and Microcrystal Electron Diffraction. Nat Chem Biol 2021, 17, 872–877. 10.1038/s41589-021-00834-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.-D.; Li Y.; Moon K.; Han E. J.; Lee S. R.; Seyedsayamdost M. R. Structural Elucidation of Cryptic Algaecides in Marine Algal-Bacterial Symbioses by NMR Spectroscopy and MicroED. Angewandte Chemie International Edition 2022, 61, e202114022 10.1002/anie.202114022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick M.; Sarai N. S.; Martynowycz M. W.; Gonen T.; Arnold F. H. Tailoring Tryptophan Synthase TrpB for Selective Quaternary Carbon Bond Formation. J. Am. Chem. Soc. 2019, 141, 19817–19822. 10.1021/jacs.9b09864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting C. P.; Funk M. A.; Halaby S. L.; Zhang Z.; Gonen T.; van der Donk W. A. Use of a Scaffold Peptide in the Biosynthesis of Amino Acid-Derived Natural Products. Science 2019, 365, 280–284. 10.1126/science.aau6232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis B. J.; Kim L. J.; Wrobel C. J. J.; Eagan J. M.; Smith R. A.; Burch J. E.; Le H. H.; Artyukhin A. B.; Nelson H. M.; Schroeder F. C. Identification of Uric Acid Gluconucleoside-Ascaroside Conjugates in Caenorhabditis Elegans by Combining Synthesis and MicroED. Org. Lett. 2020, 22, 6724–6728. 10.1021/acs.orglett.0c02038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samkian A. E.; Kiel G. R.; Jones C. G.; Bergman H. M.; Oktawiec J.; Nelson H. M.; Tilley T. D. Elucidation of Diverse Solid-State Packing in a Family of Electron-Deficient Expanded Helicenes via Microcrystal Electron Diffraction (MicroED)**. Angew. Chem. 2021, 133, 2523–2529. 10.1002/ange.202012213. [DOI] [PubMed] [Google Scholar]
- Ghashghaei O.; Pedrola M.; Seghetti F.; Martin V. V.; Zavarce R.; Babiak M.; Novacek J.; Hartung F.; Rolfes K. M.; Haarmann-Stemmann T.; Lavilla R. Extended Multicomponent Reactions with Indole Aldehydes: Access to Unprecedented Polyheterocyclic Scaffolds, Ligands of the Aryl Hydrocarbon Receptor. Angewandte Chemie International Edition 2021, 60, 2603–2608. 10.1002/anie.202011253. [DOI] [PubMed] [Google Scholar]
- Kato K.; Takaba K.; Maki-Yonekura S.; Mitoma N.; Nakanishi Y.; Nishihara T.; Hatakeyama T.; Kawada T.; Hijikata Y.; Pirillo J.; Scott L. T.; Yonekura K.; Segawa Y.; Itami K. Double-Helix Supramolecular Nanofibers Assembled from Negatively Curved Nanographenes. J. Am. Chem. Soc. 2021, 143, 5465–5469. 10.1021/jacs.1c00863. [DOI] [PubMed] [Google Scholar]
- Hall C. L.; Andrusenko I.; Potticary J.; Gao S.; Liu X.; Schmidt W.; Marom N.; Mugnaioli E.; Gemmi M.; Hall S. R. 3D Electron Diffraction Structure Determination of Terrylene, a Promising Candidate for Intermolecular Singlet Fission. ChemPhysChem 2021, 22, 1631–1637. 10.1002/cphc.202100320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H. J.; Kim K.; Kim H.; Kim T.; Malyutin A. G.; Rees D. C.; Yoo B.-K.; Song C. Microcrystal Electron Diffraction Elucidates Water-Specific Polymorphism-Induced Emission Enhancement of Bis-Arylacylhydrazone. ACS Appl. Mater. Interfaces 2021, 13, 7546–7555. 10.1021/acsami.0c21248. [DOI] [PubMed] [Google Scholar]
- Lee H. B.; Shiau A. A.; Marchiori D. A.; Oyala P. H.; Yoo B.-K.; Kaiser J. T.; Rees D. C.; Britt R. D.; Agapie T. CaMn3IVO4 Cubane Models of the Oxygen-Evolving Complex: Spin Ground States S < 9/2 and the Effect of Oxo Protonation. Angewandte Chemie International Edition 2021, 60, 17671–17679. 10.1002/anie.202105303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellen M. J.; Liepuoniute I.; Jin M.; Jones C. G.; Yang S.; Jiang X.; Nelson H. M.; Houk K. N.; Garcia-Garibay M. A. Enhanced Gearing Fidelity Achieved Through Macrocyclization of a Solvated Molecular Spur Gear. J. Am. Chem. Soc. 2021, 143, 7740–7747. 10.1021/jacs.1c01885. [DOI] [PubMed] [Google Scholar]
- Ueda M.; Aoki T.; Akiyama T.; Nakamuro T.; Yamashita K.; Yanagisawa H.; Nureki O.; Kikkawa M.; Nakamura E.; Aida T.; Itoh Y. Alternating Heterochiral Supramolecular Copolymerization. J. Am. Chem. Soc. 2021, 143, 5121–5126. 10.1021/jacs.1c00823. [DOI] [PubMed] [Google Scholar]
- Doyle L. R.; Thompson E. A.; Burnage A. L.; Whitwood A. C.; Jenkins H. T.; Macgregor S. A.; Weller A. S. MicroED Characterization of a Robust Cationic σ-Alkane Complex Stabilized by the [B(3,5-(SF5)2C6H3)4]- Anion, via on-Grid Solid/Gas Single-Crystal to Single-Crystal Reactivity. Dalton Trans. 2022, 51, 3661–3665. 10.1039/D2DT00335J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Genderen E.; Clabbers M. T. B.; Das P. P.; Stewart A.; Nederlof I.; Barentsen K. C.; Portillo Q.; Pannu N. S.; Nicolopoulos S.; Gruene T.; Abrahams J. P. Ab Initio Structure Determination of Nanocrystals of Organic Pharmaceutical Compounds by Electron Diffraction at Room Temperature Using a Timepix Quantum Area Direct Electron Detector. Acta Cryst A 2016, 72, 236–242. 10.1107/S2053273315022500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Takki S.; Cheung O.; Xu H.; Wan W.; Öhrström L.; Inge A. K. Elucidation of the Elusive Structure and Formula of the Active Pharmaceutical Ingredient Bismuth Subgallate by Continuous Rotation Electron Diffraction. Chem. Commun. 2017, 53, 7018–7021. 10.1039/C7CC03180G. [DOI] [PubMed] [Google Scholar]
- Guzmán-Afonso C.; Hong Y.; Colaux H.; Iijima H.; Saitow A.; Fukumura T.; Aoyama Y.; Motoki S.; Oikawa T.; Yamazaki T.; Yonekura K.; Nishiyama Y. Understanding Hydrogen-Bonding Structures of Molecular Crystals via Electron and NMR Nanocrystallography. Nat Commun 2019, 10, 3537. 10.1038/s41467-019-11469-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das P. P.; Mugnaioli E.; Nicolopoulos S.; Tossi C.; Gemmi M.; Galanis A.; Borodi G.; Pop M. M. Crystal Structures of Two Important Pharmaceuticals Solved by 3D Precession Electron Diffraction Tomography. Org. Process Res. Dev. 2018, 22, 1365–1372. 10.1021/acs.oprd.8b00149. [DOI] [Google Scholar]
- Andrusenko I.; Potticary J.; Hall S. R.; Gemmi M. A New Olanzapine Cocrystal Obtained from Volatile Deep Eutectic Solvents and Determined by 3D Electron Diffraction. Acta Cryst B 2020, 76, 1036–1044. 10.1107/S2052520620012779. [DOI] [PubMed] [Google Scholar]
- Hamilton V.; Andrusenko I.; Potticary J.; Hall C.; Stenner R.; Mugnaioli E.; Lanza A. E.; Gemmi M.; Hall S. R. Racemic Conglomerate Formation via Crystallization of Metaxalone from Volatile Deep Eutectic Solvents. Crystal Growth & Design 2020, 20, 4731–4739. 10.1021/acs.cgd.0c00497. [DOI] [Google Scholar]
- Bruhn J. F.; Scapin G.; Cheng A.; Mercado B. Q.; Waterman D. G.; Ganesh T.; Dallakyan S.; Read B. N.; Nieusma T.; Lucier K. W.; Mayer M. L.; Chiang N. J.; Poweleit N.; McGilvray P. T.; Wilson T. S.; Mashore M.; Hennessy C.; Thomson S.; Wang B.; Potter C. S.; Carragher B. Small Molecule Microcrystal Electron Diffraction for the Pharmaceutical Industry-Lessons Learned From Examining Over Fifty Samples. Frontiers in Molecular Biosciences 2021, 8, 354. 10.3389/fmolb.2021.648603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekharan S.; Liu X.; Yang Z.; Liu X.; Deng L.; Ruan S.; Abramov Y.; Sun G.; Li S.; Zhou T.; Shi B.; Zeng Q.; Zeng Q.; Chang C.; Jin Y.; Shi X. Selecting a Stable Solid Form of Remdesivir Using Microcrystal Electron Diffraction and Crystal Structure Prediction. RSC Adv. 2021, 11, 17408–17412. 10.1039/D1RA03100G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das P. P.; Pérez A. G.; Galanis A. S.; Nicolopoulos S. Structural Characterization of Beam Sensitive Pharmaceutical Compounds Using 3D Electron Diffraction-Micro-ED at Low Dose with Pixelated Detectors. Microscopy and Microanalysis 2020, 26, 1522–1522. 10.1017/S1431927620018395. [DOI] [Google Scholar]
- Bauer J.; Spanton S.; Henry R.; Quick J.; Dziki W.; Porter W.; Morris J. Ritonavir: An Extraordinary Example of Conformational Polymorphism. Pharm. Res. 2001, 18, 859–866. 10.1023/A:1011052932607. [DOI] [PubMed] [Google Scholar]
- Lu J.; Rohani S. Polymorphism and Crystallization of Active Pharmaceutical Ingredients (APIs). Current Medicinal Chemistry 2009, 16, 884–905. 10.2174/092986709787549299. [DOI] [PubMed] [Google Scholar]
- Censi R.; Di Martino P. Polymorph Impact on the Bioavailability and Stability of Poorly Soluble Drugs. Molecules 2015, 20, 18759–18776. 10.3390/molecules201018759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A. Y.; Erdemir D.; Myerson A. S. Crystal Polymorphism in Chemical Process Development. Annual Review of Chemical and Biomolecular Engineering 2011, 2, 259–280. 10.1146/annurev-chembioeng-061010-114224. [DOI] [PubMed] [Google Scholar]
- Blagden N.; de Matas M.; Gavan P. T.; York P. Crystal Engineering of Active Pharmaceutical Ingredients to Improve Solubility and Dissolution Rates. Adv. Drug Delivery Rev. 2007, 59, 617–630. 10.1016/j.addr.2007.05.011. [DOI] [PubMed] [Google Scholar]
- Tyler A. R.; Ragbirsingh R.; McMonagle C. J.; Waddell P. G.; Heaps S. E.; Steed J. W.; Thaw P.; Hall M. J.; Probert M. R. Encapsulated Nanodroplet Crystallization of Organic-Soluble Small Molecules. Chem 2020, 6, 1755–1765. 10.1016/j.chempr.2020.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson H. M.; Siu J. C.; Saha A.; Cascio D.; MacMillan S. N.; Wu S.-B.; Lu C.; Rodríguez J. A.; Houk K. N.; Lin S. Isolation and X-Ray Crystal Structure of an Electrogenerated TEMPO-N3 Charge-Transfer Complex. Org. Lett. 2021, 23, 454–458. 10.1021/acs.orglett.0c03966. [DOI] [PubMed] [Google Scholar]
- Heidler J.; Pantelic R.; Wennmacher J. T. C.; Zaubitzer C.; Fecteau-Lefebvre A.; Goldie K. N.; Müller E.; Holstein J. J.; van Genderen E.; De Carlo S.; Gruene T. Design Guidelines for an Electron Diffractometer for Structural Chemistry and Structural Biology. Acta Cryst D 2019, 75, 458–466. 10.1107/S2059798319003942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaeser R. M.; Thomas G. Application of Electron Diffraction to Biological Electron Microscopy. Biophys. J. 1969, 9, 1073–1099. 10.1016/S0006-3495(69)86437-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unwin P. N. T.; Henderson R. Molecular Structure Determination by Electron Microscopy of Unstained Crystalline Specimens. J. Mol. Biol. 1975, 94, 425–440. 10.1016/0022-2836(75)90212-0. [DOI] [PubMed] [Google Scholar]
- Glaeser R. M.; Downing K. H. High-Resolution Electron Crystallography of Protein Molecules. Ultramicroscopy 1993, 52, 478–486. 10.1016/0304-3991(93)90064-5. [DOI] [PubMed] [Google Scholar]
- Henderson R.; Baldwin J. M.; Ceska T. A.; Zemlin F.; Beckmann E.; Downing K. H. Model for the Structure of Bacteriorhodopsin Based on High-Resolution Electron Cryo-Microscopy. J. Mol. Biol. 1990, 213, 899–929. 10.1016/S0022-2836(05)80271-2. [DOI] [PubMed] [Google Scholar]
- Gonen T.; Sliz P.; Kistler J.; Cheng Y.; Walz T. Aquaporin-0 Membrane Junctions Reveal the Structure of a Closed Water Pore. Nature 2004, 429, 193–197. 10.1038/nature02503. [DOI] [PubMed] [Google Scholar]
- Martynowycz M. W.; Zhao W.; Hattne J.; Jensen G. J.; Gonen T. Qualitative Analyses of Polishing and Precoating FIB Milled Crystals for MicroED. Structure 2019, 27, 1594–1600. e2 10.1016/j.str.2019.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martynowycz M. W.; Shiriaeva A.; Ge X.; Hattne J.; Nannenga B. L.; Cherezov V.; Gonen T. MicroED Structure of the Human Adenosine Receptor Determined from a Single Nanocrystal in LCP. PNAS 2021, 118, 118. 10.1073/pnas.2106041118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clabbers M. T. B.; Fisher S. Z.; Coinçon M.; Zou X.; Xu H. Visualizing Drug Binding Interactions Using Microcrystal Electron Diffraction. Commun Biol 2020, 3, 1–8. 10.1038/s42003-020-01155-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purdy M. D.; Shi D.; Chrustowicz J.; Hattne J.; Gonen T.; Yeager M. MicroED Structures of HIV-1 Gag CTD-SP1 Reveal Binding Interactions with the Maturation Inhibitor Bevirimat. PNAS 2018, 115, 13258–13263. 10.1073/pnas.1806806115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martynowycz M. W.; Clabbers M. T. B.; Hattne J.; Gonen T.. Ab Initio Phasing Macromolecular Structures Using Electron-Counted MicroED Data. bioRxiv 2021, 464672. 10.1101/2021.10.16.464672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capitani G. C.; Mugnaioli E.; Guastoni A. What Is the Actual Structure of Samarskite-(Y)? A TEM Investigation of Metamict Samarskite from the Garnet Codera Dike Pegmatite (Central Italian Alps). Am. Mineral. 2016, 101, 1679–1690. 10.2138/am-2016-5605. [DOI] [Google Scholar]
- Woodhead J. A.; Rossman G. R.; Silver L. T. The Metamictization of Zircon: Radiation Dose-Dependent Structural Characteristics. Am. Mineral. 1991, 76, 74–82. [Google Scholar]
- Baybarz R. D.; Haire R. G.; Fahey J. A. On the Californium Oxide System. Journal of Inorganic and Nuclear Chemistry 1972, 34, 557–565. 10.1016/0022-1902(72)80435-4. [DOI] [Google Scholar]
- Haire R. G.; Baybarz R. D. Identification and Analysis of Einsteinium Sesquioxide by Electron Diffraction. Journal of Inorganic and Nuclear Chemistry 1973, 35, 489–496. 10.1016/0022-1902(73)80561-5. [DOI] [Google Scholar]
- Haire R. G.; Baybarz R. D. Studies of Einsteinium Metal. J. Phys. Colloques 1979, 40, C4-101. 10.1051/jphyscol:1979431. [DOI] [Google Scholar]
- Müller A.; Deblonde G. J.-P.; Ercius P.; Zeltmann S. E.; Abergel R. J.; Minor A. M. Probing Electronic Structure in Berkelium and Californium via an Electron Microscopy Nanosampling Approach. Nat Commun 2021, 12, 948. 10.1038/s41467-021-21189-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egerton R. F. Radiation Damage to Organic and Inorganic Specimens in the TEM. Micron 2019, 119, 72–87. 10.1016/j.micron.2019.01.005. [DOI] [PubMed] [Google Scholar]
- Egerton R. F. Mechanisms of Radiation Damage in Beam-Sensitive Specimens, for TEM Accelerating Voltages between 10 and 300 KV. Microscopy Research and Technique 2012, 75, 1550–1556. 10.1002/jemt.22099. [DOI] [PubMed] [Google Scholar]
- Henderson R. The Potential and Limitations of Neutrons, Electrons and X-Rays for Atomic Resolution Microscopy of Unstained Biological Molecules. Quarterly Reviews of Biophysics 1995, 28, 171–193. 10.1017/S003358350000305X. [DOI] [PubMed] [Google Scholar]
- Hattne J.; Shi D.; Glynn C.; Zee C.-T.; Gallagher-Jones M.; Martynowycz M. W.; Rodriguez J. A.; Gonen T. Analysis of Global and Site-Specific Radiation Damage in Cryo-EM. Structure 2018, 26, 759–766. e4 10.1016/j.str.2018.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meents A.; Gutmann S.; Wagner A.; Schulze-Briese C. Origin and Temperature Dependence of Radiation Damage in Biological Samples at Cryogenic Temperatures. PNAS 2010, 107, 1094–1099. 10.1073/pnas.0905481107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravelli R. B.; McSweeney S. M. The ‘Fingerprint’ That X-Rays Can Leave on Structures. Structure 2000, 8, 315–328. 10.1016/S0969-2126(00)00109-X. [DOI] [PubMed] [Google Scholar]
- Burmeister W. P. Structural Changes in a Cryo-Cooled Protein Crystal Owing to Radiation Damage. Acta Cryst D 2000, 56, 328–341. 10.1107/S0907444999016261. [DOI] [PubMed] [Google Scholar]
- Hattne J.; Shi D.; Glynn C.; Zee C.-T.; Gallagher-Jones M.; Martynowycz M. W.; Rodriguez J. A.; Gonen T. Analysis of Global and Site-Specific Radiation Damage in Cryo-EM. Structure 2018, 26, 759–766. e4 10.1016/j.str.2018.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeng T.-W.; Chiu W. Quantitative Assessment of Radiation Damage in a Thin Protein Crystal. Journal of Microscopy 1984, 136, 35–44. 10.1111/j.1365-2818.1984.tb02544.x. [DOI] [PubMed] [Google Scholar]
- Henderson R.; Clarke B. C. Cryo-Protection of Protein Crystals against Radiation Damage in Electron and X-Ray Diffraction. Proc. R. Soc. Lond. B 1990, 241, 6–8. 10.1098/rspb.1990.0057. [DOI] [Google Scholar]
- Fryer J. R. The Effect of Dose Rate on Imaging Aromatic Organic Crystals. Ultramicroscopy 1987, 23, 321–327. 10.1016/0304-3991(87)90242-7. [DOI] [Google Scholar]
- Fryer J. R.; McConnell C. H.; Zemlin F.; Dorset D. L. Effect of Temperature on Radiation Damage to Aromatic Organic Molecules. Ultramicroscopy 1992, 40, 163–169. 10.1016/0304-3991(92)90057-Q. [DOI] [Google Scholar]
- Li P.; Egerton R. F. Radiation Damage in Coronene, Rubrene and p-Terphenyl, Measured for Incident Electrons of Kinetic Energy between 100 and 200kev. Ultramicroscopy 2004, 101, 161–172. 10.1016/j.ultramic.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Dickerson J. L.; McCubbin P. T. N.; Garman E. F. RADDOSE-XFEL: Femtosecond Time-Resolved Dose Estimates for Macromolecular X-Ray Free-Electron Laser Experiments. J. Appl. Crystallogr. 2020, 53, 549–560. 10.1107/S1600576720000643. [DOI] [Google Scholar]
- Our central point here is that dose is inequivalent to fluence. Nevertheless, although dose is a better metric to assess radiation damage than fluence, this issue is quite nuanced, and our presentation is somewhat oversimplified for clarity’s sake. Different specimens exposed to the same dose can still accumulate damage in disparate ways depending on a variety of other factors, such as the presence or absence of weak or labile bonds susceptible to radiolysis. In other words, the quantity of energy absorbed per unit mass does not necessarily tell us about how that energy is dissipated within the sample, or the crystal’s ability to accommodate it without being severely damaged. A detailed discussion of these effects is beyond the scope of this review.
- Krotee P.; Griner S. L.; Sawaya M. R.; Cascio D.; Rodriguez J. A.; Shi D.; Philipp S.; Murray K.; Saelices L.; Lee Ji; Seidler P.; Glabe C. G.; Jiang L.; Gonen T.; Eisenberg D. S. Common fibrillar spines of amyloid-β and human islet amyloid polypeptide revealed by microelectron diffraction and structure-based inhibitors: common fibrillar spines of Aβ and hIAPP. J. Biol. Chem. 2018, 293, 2888–2902. 10.1074/jbc.M117.806109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipps C.; Kelly H. R.; Dahl P. J.; Yi S. M.; Vu D.; Boyer D.; Glynn C.; Sawaya M. R.; Eisenberg D.; Batista V. S.; Malvankar N. S. Intrinsic electronic conductivity of individual atomically resolved amyloid crystals reveals micrometer-long hole hopping via tyrosines. Proc. Natl. Acad. Sci. U. S. A. 2021, 118, e2014139118. 10.1073/pnas.2014139118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama Y.; Mazeau K.; Morin M.; Cardoso M. B.; Chanzy H.; Putaux J.-L.. CCDC 1427317: Experimental Crystal Structure Determination. Integro; Cambridge Crystallographic Data Centre: Cambridge, UK, 2015 10.5517/cc1jx7h5 [DOI]
- Förster C.; Gorelik T. E.; Kolb U.; Ksenofontov V.; Heinze K. Crystalline Non-Equilibrium Phase of a Cobalt(II) Complex with Tridentate Ligands. Eur. J. Inorg. Chem. 2015, 2015, 920. 10.1002/ejic.201403200. [DOI] [Google Scholar]
- Dorset D. L. Electron crystallography of the polymethylene chain. 1. The crystal structure of n-tritriacontane in the B-polymorph. Zeitschrift Fur Kristallographie 1999, 214, 223–228. [Google Scholar]
- Broadhurst E. T.; Xu H.; Clabbers M. T. B.; Lightowler M.; Nudelman F.; Zou X.; Parsons S. Polymorph evolution during crystal growth studied by 3D electron diffraction. Int. Union Crystallogr. J. 2020, 7, 5. 10.1107/S2052252519016105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cambridge Structural Database (CSD); Cambridge Structural Database: Cambridge, UK, 2022; https://dx.doi.org/10.5517/ccdc.csd.cc246dyq, https://dx.doi.org/10.5517/ccdc.csd.cc26m422.
- Lu H.; Nakamuro T.; Yamashita K.; Yanagisawa H.; Nureki O.; Kikkawa M.; Gao H.; Tian J.; Shang R.; Nakamura E. B/N-Doped p-Arylenevinylene Chromophores: Synthesis, Properties, and Microcrystal Electron Crystallographic Study. J. Am. Chem. Soc. 2020, 142, 18990. 10.1021/jacs.0c10337. [DOI] [PubMed] [Google Scholar]
- Hamada H.; Nakamuro T.; Yamashita K.; Yanagisawa H.; Nureki O.; Kikkawa M.; Harano K.; Shang R.; Nakamura E. Spiro-Conjugated Carbon/Heteroatom-Bridged p-Phenylenevinylenes: Synthesis, Properties, and Microcrystal Electron Crystallographic Analysis of Racemic Solid Solutions. Bull. Chem. Soc. Jpn. 2020, 93, 776. 10.1246/bcsj.20200065. [DOI] [Google Scholar]
- Hattne J.; Shi D.; de la Cruz M. J.; Reyes F. E.; Gonen T. Modeling truncated pixel values of faint reflections in MicroED images. J. Appl. Crystallogr. 2016, 49, 1029. 10.1107/S1600576716007196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clabbers M. T. B.; van Genderen E.; Wan W.; Wiegers E. L.; Gruene T.; Abrahams J. P. Protein structure determination by electron diffraction using a single three-dimensional nanocrystal. Acta Crystallogr. Sect. D: Struct. Biol. 2017, 73, 738. 10.1107/S2059798317010348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H.; Lebrette H.; Yang T.; Srinivas V.; Hovmöller S.; Högbom M.; Zou X. A Rare Lysozyme Crystal Form Solved Using Highly Redundant Multiple Electron Diffraction Datasets from Micron-Sized Crystals Author links open overlay panel. Structure 2018, 26, 667. 10.1016/j.str.2018.02.015. [DOI] [PubMed] [Google Scholar]
- Liu S.; Gonen T. MicroED structure of the NaK ion channel reveals a Na+ partition process into the selectivity filter. Commun. Biol. 2018, 1, 38. 10.1038/s42003-018-0040-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanza A.; Margheritis E.; Mugnaioli E.; Cappello V.; Garau G.; Gemmi M. Nanobeam precession-assisted 3D electron diffraction reveals a new polymorph of hen egg-white lysozyme. Int. Union Crystallogr. J. 2019, 6, 178. 10.1107/S2052252518017657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaba K.; Maki-Yonekura S.; Inoue S.; Hasegawa T.; Yonekura K. Protein and Organic-Molecular Crystallography With 300 kV Electrons on a Direct Electron Detector. Front. Mol. Biosci. 2021, 7, 612226. 10.3389/fmolb.2020.612226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martynowycz M. W.; Khan F.; Hattne J.; Abramson J.; Gonen T. MicroED structure of lipid-embedded mammalian mitochondrial voltage-dependent anion channel. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 32380. 10.1073/pnas.2020010117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clabbers M. T. B.; Holmes S.; Muusse T. W.; Vajjhala P. R.; Thygesen S. J.; Malde A. K.; Hunter D. J. B.; Croll T. I.; Flueckiger L.; Nanson J. D.; Rahaman Md. H.; Aquila A.; Hunter M. S.; Liang M.; Yoon C. H.; Zhao J.; Zatsepin N. A.; Abbey B.; Sierecki E.; Gambin Y.; Stacey K. J.; Darmanin C.; Kobe B.; Xu H.; Ve T. MyD88 TIR domain higher-order assembly interactions revealed by microcrystal electron diffraction and serial femtosecond crystallography. Nat. Commun. 2021, 12, 2578. 10.1038/s41467-021-22590-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L.; Bu G.; Jing L.; Shi D.; Lee M.-Y.; Gonen T.; Liu W.; Nannenga B. L. Structure Determination from Lipidic Cubic Phase Embedded Microcrystals by MicroED. Structure 2020, 28, 1149. 10.1016/j.str.2020.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff A. M.; Young I. D.; Sierra R. G.; Brewster A. S.; Martynowycz M. W.; Nango E.; Sugahara M.; Nakane T.; Ito K.; Aquila A.; Bhowmick A.; Biel J. T.; Carbajo S.; Cohen A. E.; Cortez S.; Gonzalez A.; Hino T.; Im D.; Koralek J. D.; Kubo M.; Lazarou T. S.; Nomura T.; Owada S.; Samelson A. J.; Tanaka T.; Tanaka R.; Thompson E. M.; van den Bedem H.; Woldeyes R. A.; Yumoto F.; Zhao W.; Tono K.; Boutet S.; Iwata S.; Gonen T.; Sauter N. K.; Fraser J. S.; Thompson M. C. Comparing serial X-ray crystallography and microcrystal electron diffraction (MicroED) as methods for routine structure determination from small macromolecular crystals. Int. Union Crystallogr. J. 2020, 7, 306. 10.1107/S205225252000072X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Q.; Li L.; Xue H.; Zhou H.; Zhao L.; Liu J.; Mao J.; Wu S.; Zhang S.; Wu C.; Li X.; Zhou X.; Wang J. Precise Control Over Kinetics of Molecular Assembly: Production of Particles with Tunable Sizes and Crystalline Forms. Angew. Chem., Int. Ed. Engl. 2020, 59, 15141. 10.1002/anie.202003922. [DOI] [PubMed] [Google Scholar]
- Beale E. V.; Waterman D. G.; Hecksel C.; van Rooyen J.; Gilchrist J.B.; Parkhurst J. M.; de Haas F.; Buijsse B.; Evans G.; Zhang P. A Workflow for Protein Structure Determination From Thin Crystal Lamella by Micro-Electron Diffraction.. Front. Mol. Biosci., 2020, 7, 179. 10.3389/fmolb.2020.00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Nishiyama Y.; Mazeau K.; Morin M.; Cardoso M. B.; Chanzy H.; Putaux J.-L.. CCDC 1427317: Experimental Crystal Structure Determination. Integro; Cambridge Crystallographic Data Centre: Cambridge, UK, 2015 10.5517/cc1jx7h5 [DOI]