

Abstract
Liver fibrosis is a clinically significant finding that has major impacts on patient morbidity and mortality. The mechanism of fibrosis involves many different cellular pathways, but the major cell type involved appears to be hepatic stellate cells. Many liver diseases, including Hepatitis B, C and fatty liver disease cause ongoing hepatocellular damage leading to liver fibrosis. No matter the cause of liver disease, liver related mortality increases exponentially with increasing fibrosis. The progression to cirrhosis brings more dramatic mortality and higher incidence of hepatocellular carcinoma. Fibrosis can also affect outcomes following liver transplantation in adult and pediatric patients and require retransplantation. Drugs exist to treat Hepatitis B and Hepatitis C that reverse fibrosis in patients with those viral diseases, but there are currently no therapies to directly treat liver fibrosis. Several mouse models of chronic liver diseases have been successfully reversed using novel drug targets with current therapies focusing mostly on prevention of myofibroblast activation. Further research in these areas could lead to development of drugs to treat fibrosis, which will have invaluable impact on patient survival.
Graphical Abstract
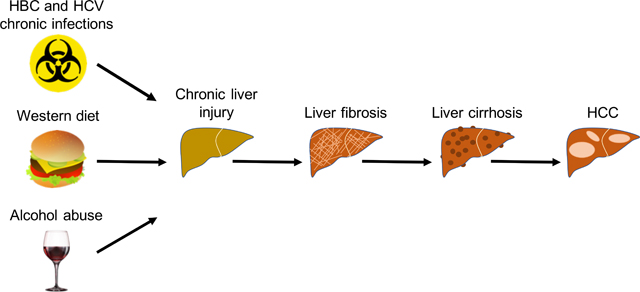
1. Introduction:
Liver fibrosis is a clinically significant finding that has major impact on patients’ morbidity and mortality. Many liver diseases, including Hepatitis B, C and fatty liver diseases, cause ongoing hepatocellular damage leading to liver fibrosis. Fibrosis is the single histopathologic feature with the greatest impact on mortality (Stal, 2015). It eventually leads to cirrhosis, which is plagued by other complications including hepatocellular carcinoma and liver failure, leaving liver transplant as the only therapy. New medications to treat hepatitis C may halt the progression of fibrosis, but currently no definitive treatment for fibrosis exists.
Liver fibrosis involves multiple cellular mechanisms. Current models for liver fibrosis show an injury driven response from the hepatic cells which release C-C motif chemokine ligand 2 (CCL2), also known as monocyte chemoattractant protein 1 (MCP1), and TGF-ß1 promoting inflammation and activation of hepatic stellate cells (HSCs) into collagen producing myofibroblasts. Myofibroblasts have been shown as a key mechanism in the development of liver fibrosis and are the primary target of antifibrotic therapy (Kisseleva & Brenner, 2011). Research has been mainly focused on developing strategies to understand the molecular mechanisms behind liver fibrosis so that effective treatments for prevention and progression can be developed. Our review emphasizes the pathophysiology, cellular mechanisms clinical significance of fibrosis and its effect on healthcare, and explores opportunities for the development of treatments.
2. Pathophysiology of liver fibrosis
Liver fibrosis is caused by chronic liver injury of two different etiologies: hepatotoxic and cholestatic injuries. Hepatotoxic injury is triggered by cellular injury from outside factors including : hepatitis B and C (HBV and HCV) viral infections, alcoholic (ASH) and non-alcoholic (NASH) steatohepatitis (Bataller & Brenner, 2005). Cholestatic injury, which is characterized by reduced or obstructed bile flow in the liver, is caused by primary (and secondary) disease including: primary biliary cholangitis (PBC), primary sclerosing cholangitis (PSC) and biliary atresia (Bataller & Brenner, 2005). Liver fibrosis results in accumulation of extracellular matrix (ECM) proteins, mostly collagens Type I and Type III, followed by formation of fibrous scar, which can ultimately compromise normal liver function (Figure 1). Regardless of the etiology, liver fibrosis is characterized by common molecular mechanisms such as hepatocyte death, chronic inflammation with cytokine release, activation of HSCs and disruption of the epithelial or endothelial barrier (Dhar, Baglieri, Kisseleva, & Brenner, 2020). Therefore, hepatic fibrogenesis is a complex process requiring cellular and extracellular signaling. In the next section we describe the different cell types and pathways involved in the pathophysiology of liver fibrosis.
Figure. 1. Pathophysiology of liver fibrosis.

2.1. Cell types in liver fibrosis
Hepatic Stellate Cells
HSCs are the main cell type involved in liver fibrosis. In normal livers HSCs are quiescent, reside in the space of Disse, store vitamin A in lipid droplets and they serve as liver pericytes (Kisseleva et al., 2012; Troeger et al., 2012). However, in response to continuous liver injury HSCs reduce the expression of genes such as glial fibrillar acidic protein (GFAP) and peroxisome proliferator-activated receptor gamma (PPARγ), lose lipid droplets and activate into myofibroblasts. Myofibroblasts start expressing fibrogenic genes such alpha-smooth muscle actin (α-SMA) and Collagen Type I. They proliferate and migrate to the liver injury site where they secrete ECM (Kisseleva & Brenner, 2008). Myofibroblasts also release vascular endothelial growth factor (VEGF) which directly promotes HSC proliferation (Kukla, 2013). Interestingly, studies performed in patients and experimental fibrosis models have shown that fibrosis can be reversed when the injury is removed (Iredale et al., 1998; Issa et al., 2003). In particular myofibroblasts can undergo apoptosis or inactivation once the causative etiology is cleared, leading to fibrosis regression (Kisseleva et al., 2012). Several mechanisms have been suggested to trigger HSC apoptosis. Following reduction of fibrogenic signals, HSCs increase expression of death-receptor mediated genes like Fas receptor or TNF receptor 1 (TNFR1), they upregulate pro-apoptotic proteins (p53, Bax, caspase 9) and downregulate anti-apoptotic factors (Bcl-2) (Iredale, 2001). Alternatively, interferon-γ (IFN-γ)-activated natural killer (NK) cells have also been involved in resolution of liver fibrosis by eliminating HSCs (Gao, Radaeva, & Park, 2009). Moreover, recent studies showed that besides senescence and apoptosis, myofibroblasts can also revert to an inactive phenotype during liver fibrosis regression (Kisseleva et al., 2012; Troeger et al., 2012). Interestingly these inactivated HSCs are more responsive to fibrogenic stimuli than quiescent HSCs
Several studies have intensively investigated the cellular origin of myofibroblasts. Following carbon tetrachloride (CCl4)-induced liver fibrosis, HSCs have been seen as the main source of myofibroblasts. However, in early cholestatic injury, myofibroblast origin is from portal fibroblasts (Iwaisako et al., 2014). Portal fibroblasts localize underneath the bile duct epithelium and, similar to HSCs, they activate into α-SMA- and Collagen Type I-expressing myofibroblasts in response to biliary injury (Desmoulière et al., 1997; Dranoff & Wells, 2010). Bone marrow-derived cells such as fibrocytes and mesenchymal stem cells (MSCs) have also been suggested to originate myofibroblasts (Kisseleva et al., 2006; Scholten et al., 2011). There is no evidence however that parenchymal cells like hepatocytes can contribute to the myofibroblast population (Taura et al., 2010).
Hepatocytes
Following liver injury, hepatocytes start producing several fibrogenic factors including osteopontin, NADPH oxidase 4 (NOX4), TAZ, Indian Hedgehog and Notch (Lan, Kisseleva, & Brenner, 2015; Wang et al., 2016; Xie et al., 2013; Zhu et al., 2018). Moreover, exosomes containing micro RNAs (miRNAs) that activate HSCs can also be released by injured hepatocytes (Lee et al., 2017). Nonetheless, hepatocyte-derived fibrogenic factors cannot lead to liver fibrosis in absence of chronic inflammation.
Inflammatory cells and cytokines
Inflammation caused by acute liver injury is considered to be beneficial in supporting hepatic regeneration. However, chronic inflammation is detrimental and has a critical role in the pathogenesis of liver fibrosis. In vitro and in vivo studies have demonstrated that inflammatory cells like neutrophils, Kupffer cells (the resident macrophages of the liver) bone marrow-derived monocytes and Th17 cells can promote HSC activation by secreting cytokines and growth factors (Seki & Schwabe, 2015). Liver macrophages, including Kupffer cells, are the main source of transforming growth factor-β (TGF-β), which plays a key role in liver fibrogenesis (Dooley & ten Dijke, 2012). TGF-β binds to its receptor in HSCs, promoting their activation into myofibroblasts and the synthesis of Collagen Type I and III. Therefore, blocking TGF-β or its genetic deletion has been shown to reduce liver fibrosis (de Gouville et al., 2005; Hellerbrand, Stefanovic, Giordano, Burchardt, & Brenner, 1999). Th17 cells secrete IL-17, which is also a profibrogenic cytokine, and the disruption of IL-17 signaling attenuates development of liver fibrosis (Meng et al., 2012). Th17 cells also produce also IL-22. Interestingly, some studies have shown that IL-22 protects from liver injury and reduces liver fibrosis (Feng et al., 2012; Ki et al., 2010; Kong et al., 2012; Radaeva, Sun, Pan, Hong, & Gao, 2004), however other reports have suggested that IL-22 is profibrogenic and its pharmacological inhibition ameliorates liver fibrosis (Fabre et al., 2018). Another fibrogenic cytokine is CCL2 which is secreted by macrophages in response to liver injury (Marra et al., 1998). CCL2 promotes activation of HSCs by recruiting monocyte-derived macrophages in the liver (Karlmark et al., 2009). Platelet-derived growth factor (PDGF) is released by macrophages and is widely considered a powerful mitogen for HSCs. The PDGF signaling pathway has been shown to be crucial to HSC activation and the development of liver fibrosis (Borkham-Kamphorst et al., 2015; Czochra et al., 2006). During liver injury neutrophils and activated Kupffer cells also release also reactive oxygen species (ROS), which promote HSC activation (Luangmonkong et al., 2018). Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) is responsible for ROS production. Mice lacking the NOX1 and NOX4 isoforms show reduced liver inflammation, reduced HSC activation and fibrosis (Lan et al., 2015). Similar results were obtained by pharmacological inhibition of NOX1 and NOX4 in mouse models of liver injury (Aoyama et al., 2012; Bettaieb et al., 2015; Jiang et al., 2012). Interestingly, macrophages not only promote liver fibrosis, but can also support fibrosis regression. Macrophages promote myofibroblast apoptosis (Ramachandran et al., 2012) and phagocytize the apoptotic cells. Furthermore, they secrete matrix metalloproteinases like MMP9, MMP12 and MMP13 which degrade ECM, a crucial process in fibrosis resolution (Popov et al., 2010; Uchinami, Seki, Brenner, & D’Armiento, 2006). Increased activity of MMPs downregulate the expression of tissue inhibitor of metalloproteinases (TIMP1). TIMP1 is a promoter of HSC activation and survival (Parsons et al., 2004).
Liver sinusoidal endothelial cells
In healthy livers liver sinusoidal endothelial cells (LSECs) are crucial for transport of nutrients, recruitment of lymphocytes from the blood stream and secretion of cytokines and growth factors (Asahara et al., 1999). Uninjured LSECs are differentiated, they have fenestrations and have been shown to maintain HSCs in a quiescent state (Deleve, Wang, & Guo, 2008; Xie et al., 2012). Production of nitric oxide (NO) by endothelial NO synthase (eNOS) is important in maintaining a physiological LSECs phenotype, in preventing HSCs activation and in promoting reversal of activated HSCs to quiescence (DeLeve, Wang, Hu, McCuskey, & McCuskey, 2004). However, in chronic liver injury LSECs undergo capillarization, which consists in loss of fenestration and reduction in eNOS activity and NO synthesis (Iwakiri, Grisham, & Shah, 2008). Therefore, in the injured liver LSECs lack of the gatekeeper function that promotes HSCs quiescence. Moreover, following disruption of the endothelium, LSECs will secrete profibrogenic cytokines like TGF-β1, PDGF, interleukins, tumor necrosis factor α (TNFα), and VEGF, which will recruit inflammatory cells to the site of injury and activate HSCs (DeLeve, 2015). Interestingly, VEGF has a biphasic nature; it induces fibrosis-associated angiogenesis (Baeck et al., 2012) and also promotes fibrosis resolution by regulating vascular permeability, monocyte migration and scar-associated macrophages function (Deleve et al., 2008; L. Yang et al., 2014). As described above, NO produced by LSECs is important in preventing HSCs activation. Furthermore, it has also been shown that NO promotes apoptosis of VEGF-activated HSCs during fibrosis reversal (Langer et al., 2008). NO-dependent apoptosis occurs through a mechanism which involves mitochondria, generation of ROS and is independent of caspase activation.
Chronic liver damage of different etiologies causes injured hepatocytes and LSEC to release proinflammatory and profibrogenic factors, which lead to chronic liver inflammation followed by activation of quiescent HSCs into Collagen Type I and III-producing myofibroblasts. As a result, fibrous scar accumulates into the liver impairing its normal functions. (See text for details).
2.2. Animal models of fibrotic liver diseases
The use of animal models is of key importance to understand the fibrotic process and to develop novel antifibrotic therapies. In the following section we describe the main features of current rodent models of liver injuries.
Toxic models of liver fibrosis
Administration of carbon tetrachloride (CCl4) in mice or rats is the most commonly used model of toxin-induced experimental liver fibrosis. In mice generally 0.5 to 2 mL/Kg body weight of CCl4 is administered either intraperitoneally or by oral gavage two to three times per week (Liedtke et al., 2013). CCl4 treatment causes activation of HSCs followed by deposition of ECM and development of highly reproducible liver fibrosis after 4 to 6 weeks from the first CCl4 injection. Cytochrome P450 2E1 (CYP2E1) in centrilobular hepatocytes metabolizes CCl4 to generate toxic trichloromethyl (CCl3) radicals, which promote liver necrosis (Slater, Cheeseman, & Ingold, 1985). Interestingly, withdrawal of CCl4 treatment results in full regression of liver fibrosis (Iredale et al., 1998; Kisseleva et al., 2012)
Thioacetamide (TAA) is another model of experimental liver fibrosis in rodents. TAA can be either administered either intraperitoneally (150–200 mg/Kg body weight) three times a week (Ding & Zhuo, 2013) or orally by adding TAA (200 mg/L) to the drinking water. In rats TAA treatment causes fibrosis and cirrhosis between 12–16 weeks, in mice between 16–24 weeks (Reif et al., 2004; Salguero Palacios et al., 2008). TAA affects both zone 1 and zone 3 hepatocytes causing portal-portal and portal-central fibrosis, which eventually recapitulates a state similar to human cirrhosis (Kang et al., 2008; Li, Benjamin, & Alexander, 2002). Moreover, unlike CCl4-induced fibrosis which regresses rapidly, fibrosis lasts for more than 2 months after cessation of TAA treatment (Reif et al., 2004). Similarly to CCl4, TAA is also bioactivated by CYP2E1 in the liver, giving rise to S,S-dioxide, which is most likely the agent causing hepatotoxicity (Hajovsky et al., 2012).
Administration of dimethylnitrosamine (DMN) is less commonly used as fibrosis model (Jenkins et al., 1985). 10 mg/Kg body weight of DMN is administered intraperitoneally twice a week. DMN induces activation of HSCs and Kupffer cells with liver fibrosis development within 4 weeks (Kitamura et al., 2002). A disadvantage of using DMN is its carcinogenic properties, which may complicate interpretation of the results.
Models of biliary fibrosis
Cholestatic liver diseases like PBC and PSC are characterized by injury of the biliary epithelium and bile duct, which can cause liver fibrosis, cirrhosis and end-stage liver disease. Several animal models are available to mimic cholestatic liver injuries (Delire, Stärkel, & Leclercq, 2015).
The most common model of obstructive cholestatic injury in rodents is surgical bile duct ligation (BDL) in which the common extrahepatic bile duct is ligated. Fibrosis is generally observed after 21–28 days (Scholten et al., 2010).
Another model is phospholipid transporter multi-drug resistant protein 2 (MDR2) deficient mice. MDR2 is encoded by the Abcb4 gene. Abcd4 knock out mice (Abcb4−/−) are not able to secrete phospholipid into bile, resulting in increased concentration of free bile acids. This damages hepatocytes and cholangiocytes, resulting in inflammatory cholangitis, portal inflammation and periductal fibrosis(Mauad et al., 1994). In this model biliary fibrosis appears shortly after birth and progresses to end-stage disease in 3–6 months.
Cholestatic liver injury can also be induced by modified diets in mice. A diet supplemented with 0.1% 3,5-diethoxycarbonyl-1,4 dihydrocollidine (DDC) for 8 weeks causes biliary liver fibrosis similar to human PSC (Fickert et al., 2007).
A diet supplemented with low doses (0.025%) of α-naphthylisothiocynate (ANIT) is another xenobiotic model of biliary injury (ELIAKIM, EISNER, & UNGAR, 1959). ANIT is toxic to hepatocytes and bile duct cells. Mice subjected to this diet will develop periportal inflammation and fibrosis with mild hepatocellular injury (Sullivan, Weinreb, Violette, & Luyendyk, 2010).
Models of alcohol-induced fibrosis
Although currently there are no animal models able to mimic all features of alcoholic liver disease (ALD), several animal models for ALD have been generated (Mathews, Xu, Wang, Bertola, & Gao, 2014). In the Lieber-De Carli model (Iseri, Lieber, & Gottlieb, 1966), animals are fed solely with an alcohol-containing isocalorically controlled liquid diet in which 36% of calories comes from alcohol. After 4–12 weeks of feeding, animals develop mild liver steatosis and inflammation, which recapitulates chronic drinking patterns seen in humans. However, no fibrosis is observed in this model (Bala et al., 2012). Another drawback is the natural aversion of animals to alcohol consumption. The Tsukamoto-French model (Tsukamoto et al., 1985) overcomes these issues and obtains prolonged high blood alcohol levels (Rouach et al., 1997). In this model animals are infused with alcohol by an intragastric canula and develop steatosis, inflammation and necrosis in 2–4 weeks, and fibrosis in 6–8 weeks (Iimuro, Ikejima, Rose, Bradford, & Thurman, 1996). However, the surgical procedure of insertion and maintenance of the intragastric canula requires intensive medical care limiting the use of this model (Ueno et al., 2012). Additional models were developed combining liquid diets containing different concentrations of alcohol (1–5%) together with intraperitoneal injections of CCl4 (Chiang et al., 2013; Jeong, Park, & Gao, 2008). Activation of HSCs and severe fibrosis was observed in these models after 5–8 weeks of feeding. Interestingly, these studies demonstrated that chronic administration of alcohol exacerbates CCl4-induced liver fibrosis.
Models of NASH-induced liver fibrosis
Human NASH is characterized by fatty liver, ballooning hepatocytes, inflammation and fibrosis (Rinella, 2015). To date no animal models can fully recapitulate the histology and pathophysiology of human NASH. Nonetheless, several dietary and genetic animal models have been described (Kucera & Cervinkova, 2014).
The methionine- and choline-deficient diet (MCDD) lack 2 key factors in the production and secretion of very low density protein (VLDL) from the liver (Ghoshal, 1995). Shortly after the start of the feeding, animals develop steatohepatitis and by 7–10 weeks perisinusoidal fibrosis (Ip, Farrell, Hall, Robertson, & Leclercq, 2004). However, unlike human NASH, MCDD diet causes weight loss and insulin hypersensitivity, which need to be taken into account in interpretation of the results (Rinella & Green, 2004).
Unlike MCDD, the choline-deficient, L-amino acid-defined diet (CDAA) contains low levels of methionine. The CDAA diet mechanism of action is similar to MCDD diet, however it causes moderate pericellular fibrosis. Animals chronically fed with CDAA may also develop HCC (Kodama et al., 2009).
In high fat diets (HFD), animals receive 30–60% of calories from fat. As a consequence they will gain weight, acquire insulin resistance and develop hepatic steatosis (Riordan & Nadeau, 2014). Diets that contain high glucose and fructose levels have also been developed, as well as diets enriched in both lipids and carbohydrates (cafeteria diet, atherogenic diet, western diet) (Kucera & Cervinkova, 2014).
Several genetic animal models of NASH have also been generated (Larter & Yeh, 2008). Among these, one of the most used is the ob/ob (ob=obese) mouse (Lindström, 2007). These mice are deficient of leptin, which is a hormone produced by the adipose tissue. The mice are hyperphagic, become obese and develop hyperglycemia, hyperinsulemia and hepatic steatosis. However, these mice require additional stimulation, such as MCDD or HFD diets to develop steatohepatitis (Brix et al., 2002).
Another genetic mouse model that recapitulates the progression of NASH in patients is the Mup-uPA mouse (Nakagawa et al., 2014). This mouse expresses urokinase-type plasminogen activator (uPA) under the control of major urinary protein (MUP) promoter. Overexpression of the transgene makes hepatocytes sensitive to injury. When these mice are fed with HFD for 8 months they will develop steatosis, steatohepatitis and eventually HCC.
3. Clinical aspects of liver fibrosis.
3.1. Stages and classification of liver fibrosis
Although several noninvasive methods exist to diagnose hepatic fibrosis, such as MRI fibroscans, they are not precise enough for the staging of fibrosis. These can be used as a screening process to determine the presence of hepatic fibrosis, but the gold standard for diagnosis and staging remains the liver biopsy. The actual staging of fibrosis has several classification systems and the most common clinically used scores are illustrated in Table 1. The Knodell score was proposed in 1981 and names three stages of fibrosis (Knodell et al., 1981). The favored Batts-Ludwig and Scheuer Stages divide fibrosis into 4 stages (Locke, Therneau, Ludwig, Dickson, & Lindor, 1996; Scheuer, 1991). In the Ishak scale instead fibrosis stages range from 0–6 (Everhart et al., 2010; Ishak et al., 1995; Pinzani & Luong, 2018).
Table 1.
Basic liver fibrosis classifications
| GENERAL CLASS | KNODELL | BATTS-LUDWIG | SCHEUER | ISHAK |
|---|---|---|---|---|
| NO FIBROSIS | 0 - NO FIBROSIS | 0 - NORMAL CONNECTIVE TISSUE | 0 - NO FIBROSIS | 0 - NO FIBROSIS |
|
| ||||
| FIBROUS PORTAL EXPANSION | 1 - FIBROUS PORTAL EXPANSION | 1 - ENLARGED, FIBROTIC PORTAL TRACTS | 1 - FIBROUS EXPANSION OF SOME PORTAL AREAS WITH OR WITHOUT SHORT FIBROUS SEPTA |
|
| EARLY FIBROSIS | 2 - FIBROUS EXPANSION OF MOST PORTAL AREAS WITH OR WITHOUT SHORT FIBROUS SEPTA | |||
|
|
||||
| PERIPORTAL FIBROSIS | BRIDGING FIBROSIS (PORTAL-PORTAL OR PORTAL-CENTRAL LINKAGE) | 2 - PERIPORTAL FIBROSIS +/− PORTAL-PORTAL SEPTA | 2 - PERIPORTAL FIBROSIS OR PORTAL-PORTAL SEPTA, INTACT ARCHITECTURE | 3 - FIBROUS EXPANSION OF MOST PORTAL AREAS WITH OCCASIONAL PORTAL TO PORTAL BRIDGING |
| 4 - FIBROUS EXPANSION OF PORTAL AREAS WITH MARKED PORTAL-PORTAL AND PORTAL-CENTRAL BRIDGING | ||||
|
|
||||
| LATE BRIDGING FIBROSIS | 3 - BRIDGING FIBROSIS BUT NO OBVIOUS CIRRHOSIS | 3 - FIBROSIS WITH ARCHITECTURAL DISTORTION | 5 - MARKED BRIDGING WITH OCCASIONAL NODULES (INCOMPLETE CIRRHOSIS) | |
|
| ||||
| CIRRHOSIS | CIRRHOSIS | 4 - REGENERATIVE NODULES ENCIRCLED BY FIBROUS SEPTA | 4 - PROBABLY OR DEFINITE CIRRHOSIS | 6 - CIRRHOSIS, PROBABLY OR DEFINITE |
3.2. Significance of Fibrosis Stage
3.2.1. Clinical Classification of Liver Disease
The severity of clinical liver disease is classified by several different systems, including the Child Pugh Score and the Model for End Stage Liver Disease (MELD) score. Each of these predicts mortality with cirrhosis (Brown et al., 2002; Kamath et al., 2001). The Childs Pugh Score was initially developed to measure operative mortality in cirrhotic patients. It is calculated with a mix of laboratory and clinical factors including ascites and encephalopathy. Patients have a score from 5–15 and a classification (A, B, or C). The most compensated cirrhotic patient would be 5A, and the most decompensated 15C. MELD score is calculated using a patient’s laboratory values (creatinine, bilirubin, INR, and sodium) and correlates strongly with 3-months mortality. MELD score is used for the liver transplant waitlist to prioritize transplanting the patients with highest mortality.
3.2.2. Mortality in fibrosis
Epidemiological studies have demonstrated that staging of fibrosis has significant clinical implications. Increased fibrosis stage is associated with higher all-cause mortality when compared to patients without fibrosis, and even stage 1 fibrosis. Different disease etiologies of liver fibrosis may affect the degree of mortality as well. However, no matter the cause of liver disease, liver related mortality increases exponentially with increasing fibrosis (Dulai et al., 2017; Ekstedt et al., 2015). In alcoholic liver disease severe fibrosis, prior to developing cirrhosis, has a major impact on 10-year mortality (Lackner et al., 2017). In non-alcoholic fatty liver disease (NAFLD) higher stages of fibrosis are proven to be the most important predictor of mortality (Angulo et al., 2015; Ekstedt et al., 2015; Lackner et al., 2017; Lackner & Tiniakos, 2019) In one NAFLD study, the ten-year transplant free survival rate with stage 4 fibrosis (and Childs Pugh A6) was shown to be only 17%. In contrast, ten-year transplant free survival in stage 4 fibrosis with Child Pugh A5 was 74%, and in stage 3 fibrosis was 94% (Vilar-Gomez et al., 2018).
3.3. Cirrhosis
With the progression of fibrosis to cirrhosis comes more dramatic liver related mortality (Vilar-Gomez et al., 2018). In 2017 chronic liver disease and cirrhosis were the 11th leading cause of death in the United States (Melonie Heron, June 24, 2019).
An Italian study evaluating the effects of the degree of decompensation in 494 patients over 3 years showed higher mortality with increasing decompensation. Patients with compensated cirrhosis had a 5-year mortality of 1.5% while patients with two decompensating events had an 88% 5-year mortality. These results were shown to be independent from Child Pugh score or MELD score (D’Amico et al., 2014). Examining the natural history of cirrhosis in Sweden in 1,317 patients over a 10-year period, the 10-year incidence of decompensation was found to be 89%. 75% of those patients died over the study period, most commonly due to liver failure and complications of cirrhosis (Nilsson, Anderson, Sargenti, Lindgren, & Prytz, 2019).
Rates of decompensation may vary by etiology of disease, with 10-year decompensation rates of 89% in alcoholic cirrhosis, 58% in hepatitis C and 75% in cryptogenic cirrhosis. One study that followed patients from four countries (Australia, USA, UK, and Italy) found a higher rate of liver related complications, including HCC, in HCV cirrhosis than in NAFLD cirrhosis (Bhala et al., 2011). Alcoholic liver disease may also progress to cirrhosis, and complications, including HCC, are faster than in non-alcoholic fatty liver disease (Shoreibah et al., 2016). Patients with cirrhosis, especially decompensated patients, have high rates or hospital readmissions and contribute to higher healthcare costs (Sempokuya et al., 2019). In a study involving Veterans Health Administration patients, annual costs of care increased as patients progressed from non-advanced fibrosis to advanced fibrosis, HCC, and liver transplant (Gidwani-Marszowski et al., 2019).
3.4. Hepatocellular Carcinoma Development
Fibrosis and cirrhosis are also associated with the development of hepatocellular carcinoma (HCC) (White, Thrift, Kanwal, Davila, & El-Serag, 2017). HCC can be treated with transplant if the tumors are confined to the liver and meet size criteria. The most common criteria are called the Milan criteria (Mazzaferro et al., 1996). The Milan criteria were developed after studying the size of HCC at transplant that would result in acceptable survival and low recurrence rates (Table 2). HCC is a common cause of death in cirrhotic patients, and the major cause of liver related death in compensated cirrhotic patients (Fattovich, Stroffolini, Zagni, & Donato, 2004). In the Swedish cohort outlined above, 15% of deaths in cirrhotic patients over the 10-year period were due to HCC (Nilsson et al., 2019). Although, patients with cirrhosis are more likely to develop HCC than non-cirrhotic, or patients with stage 1 or 2 fibrosis, the incidence increases as the stage of fibrosis increases (Danielsson Borssen et al., 2015; Fattovich et al., 2004; Gronbaek, Vilstrup, & Jepsen, 2014). The rate of HCC development varies in different studies and by etiology of liver disease. A study evaluating 634 Swedish patients with autoimmune hepatitis (AIH) found 4% of cirrhotic patients developed HCC with an incidence rate of 0.3% (Danielsson Borssen et al., 2015). Another study in Danish patients with AIH showed a 10-year cumulative risk of 0.7% of HCC (Gronbaek et al., 2014). The highest rates of HCC development appear to be with hereditary hemochromatosis (5-year incidence of 21%), hepatitis B infection (5-year risk of 10–15%), and HCV cirrhosis. In HCV cirrhosis the 5-year risk is as high as 17% in Western countries and even up to 30% in Japan (Fattovich et al., 2004). When evaluating the effects of treating hepatitis C virus with new direct acting antiviral medications on HCC incidence, patients with cirrhosis and treatment failure had higher rates of HCC than patients without cirrhosis and those who obtained sustained virologic response (SVR) (Ioannou, Green, & Berry, 2017). Other studies have also shown significantly decreased rates of HCC in patients with fibrosis and cirrhosis who have obtained SVR (Beste et al., 2017; Pinero et al., 2019).
Table 2.
Milan criteria for liver transplantation
| MILAN CRITERIA FOR LIVER TRANSPLANTATION and HEPATOCELLULAR CARCINOMA |
|---|
|
|
| - One single lesion less than 5 cm |
| - 3 lesions or less with none exceeding 3 centimeters |
| - No extrahepatic involvement |
| - No evidence of vascular invasion |
| - HCC diagnosed radiologically |
| - Biopsy only obtained if radiologic diagnosis can’t be made |
3.5. Liver Transplant and Fibrosis
3.5.1. Liver Transplantation in Adults
Recurrent fibrosis after liver transplant has shown to be a significant indicator of chronic failure of the graft. The most common causes of late graft failure after transplant are recurrence of primary disease, de novo disease (autoimmune hepatitis or NAFLD) and chronic rejection, all which lead to fibrosis and cirrhosis (Berumen & Hemming, 2017; Kitchens, Yeh, & Markmann, 2014). In HCV disease recurrence post-transplant, fibrosis has been found to progress faster than pre-transplant (Berenguer et al., 2000). Nonalcoholic steatohepatitis also has a high rate of recurrence post-transplant, but a low incidence of fibrosis development, with only 5% and 10% of patients developing bridging fibrosis or cirrhosis after 5 and 10 years, respectively (Yalamanchili, Saadeh, Klintmalm, Jennings, & Davis, 2010).
3.5.2. Pediatric Liver Transplantation
Pediatric liver disease has also seen an increase in the last several years, mainly with a higher incidence of nonalcoholic fatty liver disease. This is linked to an increase in metabolic syndrome and obesity seen in children. NAFLD in these children is linked with a higher mortality rate than age matched children and can lead to the need for transplant at a young age (Feldstein et al., 2009; Hadzic, Baumann, McKiernan, McLin, & Nobili, 2017). Biliary atresia remains the most common indication for liver transplant in children (“Scientific Registry of Transplant Recipients. 2018 Annual Data Report,”).
In pediatric allograft recipients, there is an association with acute cellular rejection and donor specific antibody (DSA) formation with the development of fibrosis in the transplanted liver. Patients who developed antibodies demonstrated significantly higher stages of fibrosis when compared to patients who had episodes of rejection without DSA formation, regardless of other factors, including initial primary disease (Tokodai et al., 2018). Some pediatric liver transplant patients with chronic hepatitis had progression over several years to fibrosis, and some subsequently to cirrhosis. HSCs also appear to be involved in the process, with higher levels of α-SMA secreted by HSCs correlating with higher stages of fibrosis in pediatric liver transplant recipient biopsies (Varma et al., 2017). Fibrosis can occur in a periportal pattern, perisinusoidal pattern, or perivenular pattern. The mechanism of each is not completely understood, but immunologic factors seem to have a significant role(Kelly et al., 2016; Markiewicz-Kijewska et al., 2015). One study looking at 47 liver biopsies of children post-transplant at 1 and 5 years showed some specific associations with the pathologic fibrosis patterns. Portal fibrosis was linked to biliary complications post-transplant, sinusoidal fibrosis was associated with prior rejection episodes, and centrilobular fibrosis was associated with immunologic mismatch (related to DSA) (Baas et al., 2017) .
3.6. Treatment and management of fibrosis
3.6.1. Existing therapies
Removal of the underlying etiological agent in liver disease may halt or improve liver fibrosis. This likely results from decreasing hepatocyte injury, which in turn decreases inflammation, and subsequent activation of hepatic stellate cells to become scar-producing myofibroblasts (Bataller & Brenner, 2005; Kisseleva & Brenner, 2011; Manka, Zeller, & Syn, 2019). Phlebotomy and iron chelation with subsequent depletion of iron overload may cause the regression of liver fibrosis in hereditary hemochromatosis (Falize et al., 2006). Copper chelating agents can improve or stabilize liver disease in patients with liver disease and cirrhosis from Wilson’s disease (Merle, Schaefer, Ferenci, & Stremmel, 2007). Specific therapies exist for specific liver diseases, including direct acting antivirals for hepatitis C virus, which decrease progression to cirrhosis and rates of HCC in HCV (Ioannou et al., 2017; Pinero et al., 2019) Specific drugs treating chronic hepatitis B have resulted in the regression of fibrosis and cirrhosis (Marcellin et al., 2013). Ceasing alcohol consumption in patients with alcoholic liver disease can improve outcomes and prevent progression to cirrhosis. Weight reduction either by bariatric surgery or by diet has been shown to improve inflammation and fibrosis in NASH (Promrat et al., 2010).
3.6.2. Future therapies
Although there are currently no therapies to directly treat liver fibrosis, such therapies are under development. Fibrosis in several mouse models of chronic liver disease has been successfully reversed using novel drug targets (Ellis & Mann, 2012). A common theme of anti-fibrotic drugs is blocking the activation of hepatic stellate cells to become myofibroblasts. Such targets include the transforming growth factor ß (TGF ß) pathway, which is the main fibrogenic pathway. Other targets include the platelet-derived growth factor (PDGF) pathway, which is the major mitogen for activated HSCs and the CB1 cannabinoid receptor, which functions as a co-activator of HSCs. Drugs are now being developed particularly in NASH for use in humans and several Phase III trials are underway (Alkhouri, Lawitz, & Noureddin, 2019; Bataller & Gao, 2015). Interestingly, a recent work has shown that inhibiting the synthesis of hyaluronan (HA), which is a component of ECM and a marker of cirrhosis, by using 4-methylumbelliferone reduced HSC activation and progression of liver fibrosis in mice (Y. M. Yang et al., 2019). This suggests that HA inhibition may be a novel anti-fibrotic therapeutic target.
Another antifibrotic therapeutic approach consists of reducing inflammation to avoid HSCs activation. Anti-inflammatory agents like corticosteroids have been used for treating autoimmune hepatitis, however they have several adverse effects in long-term treatment (Bansal, Nagórniewicz, & Prakash, 2016). Pentoxifylline (PTX) inhibits the production of TNFα, therefore blocking therefore the release of inflammatory cytokines (Vircheva et al., 2010). NASH patients treated with PTX show reduction in liver fibrosis (Zein et al., 2011). Although this study demonstrated that PTX lacks adverse effects, correlation between PTX and TNFα downregulation was not proved.
Cenicriviroc (CVC) blocks the inflammatory cytokine receptors CCR2 and CCR5 which could potentially ameliorate liver fibrosis (Seki, De Minicis, et al., 2009; Seki, de Minicis, et al., 2009). CVC has been shown to be safe (Lefebvre et al., 2016) and it is currently under evaluation for the treatment of NASH.
Galectin-3 (Gal-3) is a β-galactosidase binding lectin and a powerful activator of macrophages. Gal-3 deficiency has been shown to reduce susceptibility to CCl4-induced fibrosis in mice (Henderson et al., 2006). Gal-3 inhibition by using the inhibitor GR-MD-02 (galactoarabino-rhamnogalacturonate) was demonstrated to be successful in reducing liver fibrosis in rats (Traber et al., 2013). Clinical trials demonstrated that GR-MD-02 was safe in NASH patients and ameliorated liver fibrosis and inflammation(Harrison et al., 2016).
Inflammation can also be reduced by inhibition of ROS generation and oxidative stress. Therefore, several antioxidants like vitamin E, phosphatidylcholine, silymarin and N-acetylcysteine (NAC) have been tested with encouraging results as antifibrotic drugs (Sanyal, 2010).
A different category of antifibrotic therapies focus on inhibiting the formation of scar tissue. A key component of the scar tissue deposition process is lysyl oxidase-like-2 (LOXL2), which is involved in Collagen Type I cross-linking(Barry-Hamilton et al., 2010). Since LOXL2 is a copper-dependent enzyme, copper-binding ligands like β-aminopropionitrile and D-penicillamine have been studied as LOXL2 inhibitors (Jung, Kim, Seo, Kim, & Kim, 2003; Rodriguez et al., 2010; Vadasz et al., 2005). However, more successful strategies are the use of monoclonal anti-LOXL2 antibodies. AB0023 is a murine monoclonal antibody that has been shown to reduce cytokines release and HSCs activation (Barry-Hamilton et al., 2010). Simtuzumab (GS-6624) which is in clinical trials for liver fibrosis, is a monoclonal antibody targeting human LOXL2. GS-6624 is well tolerated in NASH patients and appears to be a promising antifibrotic drug alone or in combination with selonsertib (GS-4997) (Schuppan & Kim, 2013).
4. Conclusions
Considering the significant impact on survival with the progression of fibrosis to cirrhosis, early detection of disease at the fibrosis stage is important in order to enact interventions to block the progression of fibrosis. There is an increasing incidence of non-alcoholic steatohepatitis in the population with an estimated 25% of the world’s population with NAFLD, and with over 4 million people assumed to have NAFLD associated fibrosis in the US in 2010 (Kabbany et al., 2017; Younossi et al., 2016). The increase in NAFLD is also likely contributing to a trend in increased diagnosis of HCC in the United States. Alcoholic liver disease related fibrosis (stage 2 and 3) has been increasing over the last several years, now with an increase in alcoholic cirrhosis related deaths, particularly in younger patients aged 25 to 34 years (Tapper & Parikh, 2018; Wong, Dang, Ladhani, Singal, & Wong, 2019). The continued growing number of patients worldwide with liver disease will mean that therapies for fibrosis and liver disease will become even more necessary.
No specific fibrosis drug treatment currently exists in practice, but the ability to stop and potentially reverse fibrosis progression in other interventions (weight loss, alcohol cessation, HCV treatment, etc) give hope that fibrosis can be easily treated in the future. With the success of direct acting antivirals in HCV, and antivirals in hepatitis B there is proof of success in reversal and treatment of fibrosis with removal of the insult. There are also diseases, like alpha-1-antitrypsin deficiency, that cause liver disease and have no interventions to reduce fibrosis, ultimately leading to cirrhosis and the need for liver transplant. HSCs are activated by a chronic cycle of cell death and regeneration in alpha-1-antitrysin deficiency (Teckman & Blomenkamp, 2017). Current antifibrotic research is focusing on specific pathways, including blocking the activation of HSCs into myofibroblasts. Targeted drug therapies are currently being developed with many focusing on NASH diseases. If we are able to prevent progression and development of fibrosis, the impact on healthcare resources and patients’ survival would be invaluable. The need for liver transplant could be significantly reduced and countless lives could be saved.
5. Acknowledgements
This work is funded by the National Institutes of Health R01 DK101737-01A1, U01 AA022614-01A1, R01 DK099205-01A1. The authors want to thank Karin Diggle for proofreading the manuscript.
Grant support: Supported by the National Institutes of Health R01 DK101737-01A1, U01 AA022614-01A1, R01 DK099205-01A1
Abbreviations:
- NAFLD
non-alcoholic fatty liver disease
- NASH
non-alcoholic steatohepatitis
- HCC
hepatocellular carcinoma
- MELD
Model for End Stage Liver Disease
- HCV
Hepatitis C virus
REFERENCES:
- Alkhouri N, Lawitz E, & Noureddin M (2019). Looking Into the Crystal Ball: Predicting the Future Challenges of Fibrotic NASH Treatment. Hepatol Commun, 3(5), 605–613. doi: 10.1002/hep4.1342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angulo P, Kleiner DE, Dam-Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P, . . . Bendtsen F (2015). Liver Fibrosis, but No Other Histologic Features, Is Associated With Long-term Outcomes of Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology, 149(2), 389–397 e310. doi: 10.1053/j.gastro.2015.04.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoyama T, Paik YH, Watanabe S, Laleu B, Gaggini F, Fioraso-Cartier L, . . . Brenner DA (2012). Nicotinamide adenine dinucleotide phosphate oxidase in experimental liver fibrosis: GKT137831 as a novel potential therapeutic agent. Hepatology, 56(6), 2316–2327. doi: 10.1002/hep.25938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asahara T, Takahashi T, Masuda H, Kalka C, Chen D, Iwaguro H, . . . Isner JM (1999). VEGF contributes to postnatal neovascularization by mobilizing bone marrow-derived endothelial progenitor cells. EMBO J, 18(14), 3964–3972. doi: 10.1093/emboj/18.14.3964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baas M, Gouw ASH, van den Heuvel MC, Hepkema BG, Peeters P, Verkade H, & Scheenstra R (2017). Unique clinical conditions associated with different acinar regions of fibrosis in long-term surviving pediatric liver grafts. Pediatr Transplant, 21(7). doi: 10.1111/petr.12988 [DOI] [PubMed] [Google Scholar]
- Baeck C, Wehr A, Karlmark KR, Heymann F, Vucur M, Gassler N, . . . Tacke F (2012). Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut, 61(3), 416–426. doi: 10.1136/gutjnl-2011-300304 [DOI] [PubMed] [Google Scholar]
- Bala S, Petrasek J, Mundkur S, Catalano D, Levin I, Ward J, . . . Szabo G (2012). Circulating microRNAs in exosomes indicate hepatocyte injury and inflammation in alcoholic, drug-induced, and inflammatory liver diseases. Hepatology, 56(5), 1946–1957. doi: 10.1002/hep.25873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal R, Nagórniewicz B, & Prakash J (2016). Clinical Advancements in the Targeted Therapies against Liver Fibrosis. Mediators Inflamm, 2016, 7629724. doi: 10.1155/2016/7629724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry-Hamilton V, Spangler R, Marshall D, McCauley S, Rodriguez HM, Oyasu M, . . . Smith V (2010). Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nat Med, 16(9), 1009–1017. doi: 10.1038/nm.2208 [DOI] [PubMed] [Google Scholar]
- Bataller R, & Brenner DA (2005). Liver fibrosis. J Clin Invest, 115(2), 209–218. doi: 10.1172/JCI24282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bataller R, & Gao B (2015). Liver fibrosis in alcoholic liver disease. Semin Liver Dis, 35(2), 146–156. doi: 10.1055/s-0035-1550054 [DOI] [PubMed] [Google Scholar]
- Berenguer M, Prieto M, Rayon JM, Mora J, Pastor M, Ortiz V, . . . Berenguer J (2000). Natural history of clinically compensated hepatitis C virus-related graft cirrhosis after liver transplantation. Hepatology, 32(4 Pt 1), 852–858. doi: 10.1053/jhep.2000.17924 [DOI] [PubMed] [Google Scholar]
- Berumen J, & Hemming A (2017). Liver Retransplantation: How Much Is Too Much? Clin Liver Dis, 21(2), 435–447. doi: 10.1016/j.cld.2016.12.013 [DOI] [PubMed] [Google Scholar]
- Beste LA, Green PK, Berry K, Kogut MJ, Allison SK, & Ioannou GN (2017). Effectiveness of hepatitis C antiviral treatment in a USA cohort of veteran patients with hepatocellular carcinoma. J Hepatol, 67(1), 32–39. doi: 10.1016/j.jhep.2017.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettaieb A, Jiang JX, Sasaki Y, Chao TI, Kiss Z, Chen X, . . . Török NJ (2015). Hepatocyte Nicotinamide Adenine Dinucleotide Phosphate Reduced Oxidase 4 Regulates Stress Signaling, Fibrosis, and Insulin Sensitivity During Development of Steatohepatitis in Mice. Gastroenterology, 149(2), 468–480.e410. doi: 10.1053/j.gastro.2015.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhala N, Angulo P, van der Poorten D, Lee E, Hui JM, Saracco G, . . . George J (2011). The natural history of nonalcoholic fatty liver disease with advanced fibrosis or cirrhosis: an international collaborative study. Hepatology, 54(4), 1208–1216. doi: 10.1002/hep.24491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borkham-Kamphorst E, Meurer SK, Van de Leur E, Haas U, Tihaa L, & Weiskirchen R (2015). PDGF-D signaling in portal myofibroblasts and hepatic stellate cells proves identical to PDGF-B via both PDGF receptor type α and β. Cell Signal, 27(7), 1305–1314. doi: 10.1016/j.cellsig.2015.03.012 [DOI] [PubMed] [Google Scholar]
- Brix AE, Elgavish A, Nagy TR, Gower BA, Rhead WJ, & Wood PA (2002). Evaluation of liver fatty acid oxidation in the leptin-deficient obese mouse. Mol Genet Metab, 75(3), 219–226. doi: 10.1006/mgme.2002.3298 [DOI] [PubMed] [Google Scholar]
- Brown RS Jr., Kumar KS, Russo MW, Kinkhabwala M, Rudow DL, Harren P, . . . Emond JC (2002). Model for end-stage liver disease and Child-Turcotte-Pugh score as predictors of pretransplantation disease severity, posttransplantation outcome, and resource utilization in United Network for Organ Sharing status 2A patients. Liver Transpl, 8(3), 278–284. doi: 10.1053/jlts.2002.31340 [DOI] [PubMed] [Google Scholar]
- Chiang DJ, Roychowdhury S, Bush K, McMullen MR, Pisano S, Niese K, . . . Nagy LE (2013). Adenosine 2A receptor antagonist prevented and reversed liver fibrosis in a mouse model of ethanol-exacerbated liver fibrosis. PLoS One, 8(7), e69114. doi: 10.1371/journal.pone.0069114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czochra P, Klopcic B, Meyer E, Herkel J, Garcia-Lazaro JF, Thieringer F, . . . Kanzler S (2006). Liver fibrosis induced by hepatic overexpression of PDGF-B in transgenic mice. J Hepatol, 45(3), 419–428. doi: 10.1016/j.jhep.2006.04.010 [DOI] [PubMed] [Google Scholar]
- D’Amico G, Pasta L, Morabito A, D’Amico M, Caltagirone M, Malizia G, . . . Pagliaro L (2014). Competing risks and prognostic stages of cirrhosis: a 25-year inception cohort study of 494 patients. Aliment Pharmacol Ther, 39(10), 1180–1193. doi: 10.1111/apt.12721 [DOI] [PubMed] [Google Scholar]
- Danielsson Borssen A, Almer S, Prytz H, Wallerstedt S, Friis-Liby IL, Bergquist A, . . . Werner M (2015). Hepatocellular and extrahepatic cancer in patients with autoimmune hepatitis--a long-term follow-up study in 634 Swedish patients. Scand J Gastroenterol, 50(2), 217–223. doi: 10.3109/00365521.2014.983154 [DOI] [PubMed] [Google Scholar]
- de Gouville AC, Boullay V, Krysa G, Pilot J, Brusq JM, Loriolle F, . . . Huet S (2005). Inhibition of TGF-beta signaling by an ALK5 inhibitor protects rats from dimethylnitrosamine-induced liver fibrosis. Br J Pharmacol, 145(2), 166–177. doi: 10.1038/sj.bjp.0706172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLeve LD (2015). Liver sinusoidal endothelial cells in hepatic fibrosis. Hepatology, 61(5), 1740–1746. doi: 10.1002/hep.27376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleve LD, Wang X, & Guo Y (2008). Sinusoidal endothelial cells prevent rat stellate cell activation and promote reversion to quiescence. Hepatology, 48(3), 920–930. doi: 10.1002/hep.22351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLeve LD, Wang X, Hu L, McCuskey MK, & McCuskey RS (2004). Rat liver sinusoidal endothelial cell phenotype is maintained by paracrine and autocrine regulation. Am J Physiol Gastrointest Liver Physiol, 287(4), G757–763. doi: 10.1152/ajpgi.00017.2004 [DOI] [PubMed] [Google Scholar]
- Delire B, Stärkel P, & Leclercq I (2015). Animal Models for Fibrotic Liver Diseases: What We Have, What We Need, and What Is under Development. J Clin Transl Hepatol, 3(1), 53–66. doi: 10.14218/JCTH.2014.00035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmoulière A, Darby I, Costa AM, Raccurt M, Tuchweber B, Sommer P, & Gabbiani G (1997). Extracellular matrix deposition, lysyl oxidase expression, and myofibroblastic differentiation during the initial stages of cholestatic fibrosis in the rat. Lab Invest, 76(6), 765–778. [PubMed] [Google Scholar]
- Dhar D, Baglieri J, Kisseleva T, & Brenner DA (2020). Mechanisms of liver fibrosis and its role in liver cancer. Exp Biol Med (Maywood), 245(2), 96–108. doi: 10.1177/1535370219898141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Z, & Zhuo L (2013). Attenuation of hepatic fibrosis by an imidazolium salt in thioacetamide-induced mouse model. J Gastroenterol Hepatol, 28(1), 188–201. doi: 10.1111/j.1440-1746.2012.07265.x [DOI] [PubMed] [Google Scholar]
- Dooley S, & ten Dijke P (2012). TGF-β in progression of liver disease. Cell Tissue Res, 347(1), 245–256. doi: 10.1007/s00441-011-1246-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dranoff JA, & Wells RG (2010). Portal fibroblasts: Underappreciated mediators of biliary fibrosis. Hepatology, 51(4), 1438–1444. doi: 10.1002/hep.23405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulai PS, Singh S, Patel J, Soni M, Prokop LJ, Younossi Z, . . . Loomba R (2017). Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: Systematic review and meta-analysis. Hepatology, 65(5), 1557–1565. doi: 10.1002/hep.29085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekstedt M, Hagstrom H, Nasr P, Fredrikson M, Stal P, Kechagias S, & Hultcrantz R (2015). Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology, 61(5), 1547–1554. doi: 10.1002/hep.27368 [DOI] [PubMed] [Google Scholar]
- ELIAKIM M, EISNER M, & UNGAR H (1959). Experimental intrahepatic obstructive jaundice following ingestion of alphanaphthyl-iso-thiocyanate. Bull Res Counc Isr Sect E Exp Med, 8E, 7–17. [PubMed] [Google Scholar]
- Ellis EL, & Mann DA (2012). Clinical evidence for the regression of liver fibrosis. J Hepatol, 56(5), 1171–1180. doi: 10.1016/j.jhep.2011.09.024 [DOI] [PubMed] [Google Scholar]
- Everhart JE, Wright EC, Goodman ZD, Dienstag JL, Hoefs JC, Kleiner DE, . . . Group, H.-C. T. (2010). Prognostic value of Ishak fibrosis stage: findings from the hepatitis C antiviral long-term treatment against cirrhosis trial. Hepatology, 51(2), 585–594. doi: 10.1002/hep.23315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabre T, Molina MF, Soucy G, Goulet JP, Willems B, Villeneuve JP, . . . Shoukry NH (2018). Type 3 cytokines IL-17A and IL-22 drive TGF-β-dependent liver fibrosis. Sci Immunol, 3(28). doi: 10.1126/sciimmunol.aar7754 [DOI] [PubMed] [Google Scholar]
- Falize L, Guillygomarc’h A, Perrin M, Laine F, Guyader D, Brissot P, . . . Deugnier Y (2006). Reversibility of hepatic fibrosis in treated genetic hemochromatosis: a study of 36 cases. Hepatology, 44(2), 472–477. doi: 10.1002/hep.21260 [DOI] [PubMed] [Google Scholar]
- Fattovich G, Stroffolini T, Zagni I, & Donato F (2004). Hepatocellular carcinoma in cirrhosis: incidence and risk factors. Gastroenterology, 127(5 Suppl 1), S35–50. doi: 10.1053/j.gastro.2004.09.014 [DOI] [PubMed] [Google Scholar]
- Feldstein AE, Charatcharoenwitthaya P, Treeprasertsuk S, Benson JT, Enders FB, & Angulo P (2009). The natural history of non-alcoholic fatty liver disease in children: a follow-up study for up to 20 years. Gut, 58(11), 1538–1544. doi: 10.1136/gut.2008.171280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng D, Kong X, Weng H, Park O, Wang H, Dooley S, . . . Gao B (2012). Interleukin-22 promotes proliferation of liver stem/progenitor cells in mice and patients with chronic hepatitis B virus infection. Gastroenterology, 143(1), 188–198.e187. doi: 10.1053/j.gastro.2012.03.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fickert P, Stöger U, Fuchsbichler A, Moustafa T, Marschall HU, Weiglein AH, . . . Trauner M(2007). A new xenobiotic-induced mouse model of sclerosing cholangitis and biliary fibrosis. Am J Pathol, 171(2), 525–536. doi: 10.2353/ajpath.2007.061133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao B, Radaeva S, & Park O (2009). Liver natural killer and natural killer T cells: immunobiology and emerging roles in liver diseases. J Leukoc Biol, 86(3), 513–528. doi: 10.1189/JLB.0309135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoshal AK (1995). New insight into the biochemical pathology of liver in choline deficiency. Crit Rev Biochem Mol Biol, 30(4), 263–273. doi: 10.3109/10409239509083487 [DOI] [PubMed] [Google Scholar]
- Gidwani-Marszowski R, Owens DK, Lo J, Goldhaber-Fiebert JD, Asch SM, & Barnett PG (2019). The Costs of Hepatitis C by Liver Disease Stage: Estimates from the Veterans Health Administration. Appl Health Econ Health Policy, 17(4), 513–521. doi: 10.1007/s40258-019-00468-5 [DOI] [PubMed] [Google Scholar]
- Gronbaek L, Vilstrup H, & Jepsen P (2014). Autoimmune hepatitis in Denmark: incidence, prevalence, prognosis, and causes of death. A nationwide registry-based cohort study. J Hepatol, 60(3), 612–617. doi: 10.1016/j.jhep.2013.10.020 [DOI] [PubMed] [Google Scholar]
- Hadzic N, Baumann U, McKiernan P, McLin V, & Nobili V (2017). Long-term challenges and perspectives of pre-adolescent liver disease. Lancet Gastroenterol Hepatol, 2(6), 435–445. doi: 10.1016/S2468-1253(16)30160-1 [DOI] [PubMed] [Google Scholar]
- Hajovsky H, Hu G, Koen Y, Sarma D, Cui W, Moore DS, . . . Hanzlik RP (2012). Metabolism and toxicity of thioacetamide and thioacetamide S-oxide in rat hepatocytes. Chem Res Toxicol, 25(9), 1955–1963. doi: 10.1021/tx3002719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison SA, Marri SR, Chalasani N, Kohli R, Aronstein W, Thompson GA, . . . Traber PG (2016). Randomised clinical study: GR-MD-02, a galectin-3 inhibitor, vs. placebo in patients having non-alcoholic steatohepatitis with advanced fibrosis. Aliment Pharmacol Ther, 44(11–12), 1183–1198. doi: 10.1111/apt.13816 [DOI] [PubMed] [Google Scholar]
- Hellerbrand C, Stefanovic B, Giordano F, Burchardt ER, & Brenner DA (1999). The role of TGFbeta1 in initiating hepatic stellate cell activation in vivo. J Hepatol, 30(1), 77–87. doi: 10.1016/s0168-8278(99)80010-5 [DOI] [PubMed] [Google Scholar]
- Henderson NC, Mackinnon AC, Farnworth SL, Poirier F, Russo FP, Iredale JP, . . . Sethi T (2006). Galectin-3 regulates myofibroblast activation and hepatic fibrosis. Proc Natl Acad Sci U S A, 103(13), 5060–5065. doi: 10.1073/pnas.0511167103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iimuro Y, Ikejima K, Rose ML, Bradford BU, & Thurman RG (1996). Nimodipine, a dihydropyridine-type calcium channel blocker, prevents alcoholic hepatitis caused by chronic intragastric ethanol exposure in the rat. Hepatology, 24(2), 391–397. doi: 10.1002/hep.510240217 [DOI] [PubMed] [Google Scholar]
- Ioannou GN, Green PK, & Berry K (2017). HCV eradication induced by direct-acting antiviral agents reduces the risk of hepatocellular carcinoma. J Hepatol. doi: 10.1016/j.jhep.2017.08.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip E, Farrell G, Hall P, Robertson G, & Leclercq I (2004). Administration of the potent PPARalpha agonist, Wy-14,643, reverses nutritional fibrosis and steatohepatitis in mice. Hepatology, 39(5), 1286–1296. doi: 10.1002/hep.20170 [DOI] [PubMed] [Google Scholar]
- Iredale JP (2001). Hepatic stellate cell behavior during resolution of liver injury. Semin Liver Dis, 21(3), 427–436. doi: 10.1055/s-2001-17557 [DOI] [PubMed] [Google Scholar]
- Iredale JP, Benyon RC, Pickering J, McCullen M, Northrop M, Pawley S, . . . Arthur MJ (1998). Mechanisms of spontaneous resolution of rat liver fibrosis. Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J Clin Invest, 102(3), 538–549. doi: 10.1172/JCI1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iseri OA, Lieber CS, & Gottlieb LS (1966). The ultrastructure of fatty liver induced by prolonged ethanol ingestion. Am J Pathol, 48(4), 535–555. [PMC free article] [PubMed] [Google Scholar]
- Ishak K, Baptista A, Bianchi L, Callea F, De Groote J, Gudat F, . . . et al. (1995). Histological grading and staging of chronic hepatitis. J Hepatol, 22(6), 696–699. doi: 10.1016/0168-8278(95)80226-6 [DOI] [PubMed] [Google Scholar]
- Issa R, Zhou X, Trim N, Millward-Sadler H, Krane S, Benyon C, & Iredale J (2003). Mutation in collagen-1 that confers resistance to the action of collagenase results in failure of recovery from CCl4-induced liver fibrosis, persistence of activated hepatic stellate cells, and diminished hepatocyte regeneration. FASEB J, 17(1), 47–49. doi: 10.1096/fj.02-0494fje [DOI] [PubMed] [Google Scholar]
- Iwaisako K, Jiang C, Zhang M, Cong M, Moore-Morris TJ, Park TJ, . . . Kisseleva T (2014). Origin of myofibroblasts in the fibrotic liver in mice. Proc Natl Acad Sci U S A, 111(32), E3297–3305. doi: 10.1073/pnas.1400062111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwakiri Y, Grisham M, & Shah V (2008). Vascular biology and pathobiology of the liver: Report of a single-topic symposium. Hepatology, 47(5), 1754–1763. doi: 10.1002/hep.22203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins SA, Grandison A, Baxter JN, Day DW, Taylor I, & Shields R (1985). A dimethylnitrosamine-induced model of cirrhosis and portal hypertension in the rat. J Hepatol, 1(5), 489–499. doi: 10.1016/s0168-8278(85)80747-9 [DOI] [PubMed] [Google Scholar]
- Jeong WI, Park O, & Gao B (2008). Abrogation of the antifibrotic effects of natural killer cells/interferon-gamma contributes to alcohol acceleration of liver fibrosis. Gastroenterology, 134(1), 248–258. doi: 10.1053/j.gastro.2007.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang JX, Chen X, Serizawa N, Szyndralewiez C, Page P, Schröder K, . . . Török NJ (2012). Liver fibrosis and hepatocyte apoptosis are attenuated by GKT137831, a novel NOX4/NOX1 inhibitor in vivo. Free Radic Biol Med, 53(2), 289–296. doi: 10.1016/j.freeradbiomed.2012.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung ST, Kim MS, Seo JY, Kim HC, & Kim Y (2003). Purification of enzymatically active human lysyl oxidase and lysyl oxidase-like protein from Escherichia coli inclusion bodies. Protein Expr Purif, 31(2), 240–246. doi: 10.1016/s1046-5928(03)00217-1 [DOI] [PubMed] [Google Scholar]
- Kabbany MN, Conjeevaram Selvakumar PK, Watt K, Lopez R, Akras Z, Zein N, . . . Alkhouri N (2017). Prevalence of Nonalcoholic Steatohepatitis-Associated Cirrhosis in the United States: An Analysis of National Health and Nutrition Examination Survey Data. Am J Gastroenterol, 112(4), 581–587. doi: 10.1038/ajg.2017.5 [DOI] [PubMed] [Google Scholar]
- Kamath PS, Wiesner RH, Malinchoc M, Kremers W, Therneau TM, Kosberg CL, . . . Kim WR (2001). A model to predict survival in patients with end-stage liver disease. Hepatology, 33(2), 464–470. doi: 10.1053/jhep.2001.22172 [DOI] [PubMed] [Google Scholar]
- Kang JS, Wanibuchi H, Morimura K, Wongpoomchai R, Chusiri Y, Gonzalez FJ, & Fukushima S (2008). Role of CYP2E1 in thioacetamide-induced mouse hepatotoxicity. Toxicol Appl Pharmacol, 228(3), 295–300. doi: 10.1016/j.taap.2007.11.010 [DOI] [PubMed] [Google Scholar]
- Karlmark KR, Weiskirchen R, Zimmermann HW, Gassler N, Ginhoux F, Weber C, . . . Tacke F (2009). Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology, 50(1), 261–274. doi: 10.1002/hep.22950 [DOI] [PubMed] [Google Scholar]
- Kelly D, Verkade HJ, Rajanayagam J, McKiernan P, Mazariegos G, & Hubscher S (2016). Late graft hepatitis and fibrosis in pediatric liver allograft recipients: Current concepts and future developments. Liver Transpl, 22(11), 1593–1602. doi: 10.1002/lt.24616 [DOI] [PubMed] [Google Scholar]
- Ki SH, Park O, Zheng M, Morales-Ibanez O, Kolls JK, Bataller R, & Gao B (2010). Interleukin-22 treatment ameliorates alcoholic liver injury in a murine model of chronic-binge ethanol feeding: role of signal transducer and activator of transcription 3. Hepatology, 52(4), 1291–1300. doi: 10.1002/hep.23837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisseleva T, & Brenner DA (2008). Mechanisms of fibrogenesis. Exp Biol Med (Maywood), 233(2), 109–122. doi: 10.3181/0707-MR-190 [DOI] [PubMed] [Google Scholar]
- Kisseleva T, & Brenner DA (2011). Anti-fibrogenic strategies and the regression of fibrosis. Best Pract Res Clin Gastroenterol, 25(2), 305–317. doi: 10.1016/j.bpg.2011.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisseleva T, Cong M, Paik Y, Scholten D, Jiang C, Benner C, . . . Brenner DA (2012). Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc Natl Acad Sci U S A, 109(24), 9448–9453. doi: 10.1073/pnas.1201840109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisseleva T, Uchinami H, Feirt N, Quintana-Bustamante O, Segovia JC, Schwabe RF, & Brenner DA (2006). Bone marrow-derived fibrocytes participate in pathogenesis of liver fibrosis. J Hepatol, 45(3), 429–438. doi: 10.1016/j.jhep.2006.04.014 [DOI] [PubMed] [Google Scholar]
- Kitamura K, Nakamoto Y, Akiyama M, Fujii C, Kondo T, Kobayashi K, . . . Mukaida N (2002). Pathogenic roles of tumor necrosis factor receptor p55-mediated signals in dimethylnitrosamine-induced murine liver fibrosis. Lab Invest, 82(5), 571–583. doi: 10.1038/labinvest.3780452 [DOI] [PubMed] [Google Scholar]
- Kitchens WH, Yeh H, & Markmann JF (2014). Hepatic retransplant: what have we learned? Clin Liver Dis, 18(3), 731–751. doi: 10.1016/j.cld.2014.05.010 [DOI] [PubMed] [Google Scholar]
- Knodell RG, Ishak KG, Black WC, Chen TS, Craig R, Kaplowitz N, . . . Wollman J (1981). Formulation and application of a numerical scoring system for assessing histological activity in asymptomatic chronic active hepatitis. Hepatology, 1(5), 431–435. doi: 10.1002/hep.1840010511 [DOI] [PubMed] [Google Scholar]
- Kodama Y, Kisseleva T, Iwaisako K, Miura K, Taura K, De Minicis S, . . . Brenner DA (2009). c-Jun N-terminal kinase-1 from hematopoietic cells mediates progression from hepatic steatosis to steatohepatitis and fibrosis in mice. Gastroenterology, 137(4), 1467–1477.e1465. doi: 10.1053/j.gastro.2009.06.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong X, Feng D, Wang H, Hong F, Bertola A, Wang FS, & Gao B (2012). Interleukin-22 induces hepatic stellate cell senescence and restricts liver fibrosis in mice. Hepatology, 56(3), 1150–1159. doi: 10.1002/hep.25744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucera O, & Cervinkova Z (2014). Experimental models of non-alcoholic fatty liver disease in rats. World J Gastroenterol, 20(26), 8364–8376. doi: 10.3748/wjg.v20.i26.8364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukla M (2013). Angiogenesis: a phenomenon which aggravates chronic liver disease progression. Hepatol Int, 7(1), 4–12. doi: 10.1007/s12072-012-9391-2 [DOI] [PubMed] [Google Scholar]
- Lackner C, Spindelboeck W, Haybaeck J, Douschan P, Rainer F, Terracciano L, . . . Stauber RE (2017). Histological parameters and alcohol abstinence determine long-term prognosis in patients with alcoholic liver disease. J Hepatol, 66(3), 610–618. doi: 10.1016/j.jhep.2016.11.011 [DOI] [PubMed] [Google Scholar]
- Lackner C, & Tiniakos D (2019). Fibrosis and alcohol-related liver disease. J Hepatol, 70(2), 294–304. doi: 10.1016/j.jhep.2018.12.003 [DOI] [PubMed] [Google Scholar]
- Lan T, Kisseleva T, & Brenner DA (2015). Deficiency of NOX1 or NOX4 Prevents Liver Inflammation and Fibrosis in Mice through Inhibition of Hepatic Stellate Cell Activation. PLoS One, 10(7), e0129743. doi: 10.1371/journal.pone.0129743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer DA, Das A, Semela D, Kang-Decker N, Hendrickson H, Bronk SF, . . . Shah VH (2008). Nitric oxide promotes caspase-independent hepatic stellate cell apoptosis through the generation of reactive oxygen species. Hepatology, 47(6), 1983–1993. doi: 10.1002/hep.22285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larter CZ, & Yeh MM (2008). Animal models of NASH: getting both pathology and metabolic context right. J Gastroenterol Hepatol, 23(11), 1635–1648. doi: 10.1111/j.1440-1746.2008.05543.x [DOI] [PubMed] [Google Scholar]
- Lee YS, Kim SY, Ko E, Lee JH, Yi HS, Yoo YJ, . . . Byun KS (2017). Exosomes derived from palmitic acid-treated hepatocytes induce fibrotic activation of hepatic stellate cells. Sci Rep, 7(1), 3710. doi: 10.1038/s41598-017-03389-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre E, Gottwald M, Lasseter K, Chang W, Willett M, Smith PF, . . . Utay NS (2016). Pharmacokinetics, Safety, and CCR2/CCR5 Antagonist Activity of Cenicriviroc in Participants With Mild or Moderate Hepatic Impairment. Clin Transl Sci, 9(3), 139–148. doi: 10.1111/cts.12397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Benjamin IS, & Alexander B (2002). Reproducible production of thioacetamide-induced macronodular cirrhosis in the rat with no mortality. J Hepatol, 36(4), 488–493. doi: 10.1016/s0168-8278(02)00011-9 [DOI] [PubMed] [Google Scholar]
- Liedtke C, Luedde T, Sauerbruch T, Scholten D, Streetz K, Tacke F, . . . Weiskirchen R (2013). Experimental liver fibrosis research: update on animal models, legal issues and translational aspects. Fibrogenesis Tissue Repair, 6(1), 19. doi: 10.1186/1755-1536-6-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindström P (2007). The physiology of obese-hyperglycemic mice [ob/ob mice]. ScientificWorldJournal, 7, 666–685. doi: 10.1100/tsw.2007.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locke GR 3rd, Therneau TM, Ludwig J, Dickson ER, & Lindor KD (1996). Time course of histological progression in primary biliary cirrhosis. Hepatology, 23(1), 52–56. doi: 10.1002/hep.510230108 [DOI] [PubMed] [Google Scholar]
- Luangmonkong T, Suriguga S, Mutsaers HAM, Groothuis GMM, Olinga P, & Boersema M (2018). Targeting Oxidative Stress for the Treatment of Liver Fibrosis. Rev Physiol Biochem Pharmacol. doi: 10.1007/112_2018_10 [DOI] [PubMed] [Google Scholar]
- Manka P, Zeller A, & Syn WK (2019). Fibrosis in Chronic Liver Disease: An Update on Diagnostic and Treatment Modalities. Drugs, 79(9), 903–927. doi: 10.1007/s40265-019-01126-9 [DOI] [PubMed] [Google Scholar]
- Marcellin P, Gane E, Buti M, Afdhal N, Sievert W, Jacobson IM, . . . Heathcote EJ (2013). Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: a 5-year open-label follow-up study. Lancet, 381(9865), 468–475. doi: 10.1016/S0140-6736(12)61425-1 [DOI] [PubMed] [Google Scholar]
- Markiewicz-Kijewska M, Kalicinski P, Kluge P, Piatosa B, Jankowska I, Rekawek A, . . . Kurowski PN (2015). Immunological factors and liver fibrosis in pediatric liver transplant recipients. Ann Transplant, 20, 279–284. doi: 10.12659/AOT.892544 [DOI] [PubMed] [Google Scholar]
- Marra F, DeFranco R, Grappone C, Milani S, Pastacaldi S, Pinzani M, . . . Gentilini P (1998). Increased expression of monocyte chemotactic protein-1 during active hepatic fibrogenesis: correlation with monocyte infiltration. Am J Pathol, 152(2), 423–430. [PMC free article] [PubMed] [Google Scholar]
- Mathews S, Xu M, Wang H, Bertola A, & Gao B (2014). Animals models of gastrointestinal and liver diseases. Animal models of alcohol-induced liver disease: pathophysiology, translational relevance, and challenges. Am J Physiol Gastrointest Liver Physiol, 306(10), G819–823. doi: 10.1152/ajpgi.00041.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauad TH, van Nieuwkerk CM, Dingemans KP, Smit JJ, Schinkel AH, Notenboom RG, . . . Oude Elferink RP (1994). Mice with homozygous disruption of the mdr2 P-glycoprotein gene. A novel animal model for studies of nonsuppurative inflammatory cholangitis and hepatocarcinogenesis. Am J Pathol, 145(5), 1237–1245. [PMC free article] [PubMed] [Google Scholar]
- Mazzaferro V, Regalia E, Doci R, Andreola S, Pulvirenti A, Bozzetti F, . . . Gennari L (1996). Liver transplantation for the treatment of small hepatocellular carcinomas in patients with cirrhosis. N Engl J Med, 334(11), 693–699. doi: 10.1056/NEJM199603143341104 [DOI] [PubMed] [Google Scholar]
- Melonie Heron P (June 24, 2019). National Vital Statistics Report: Deaths: Leading Causes for 2017. Retrieved from https://www.cdc.gov/nchs/data/nvsr/nvsr68/nvsr68_06-508.pdf [PubMed] [Google Scholar]
- Meng F, Wang K, Aoyama T, Grivennikov SI, Paik Y, Scholten D, . . . Kisseleva T (2012). Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology, 143(3), 765–776.e763. doi: 10.1053/j.gastro.2012.05.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merle U, Schaefer M, Ferenci P, & Stremmel W (2007). Clinical presentation, diagnosis and long-term outcome of Wilson’s disease: a cohort study. Gut, 56(1), 115–120. doi: 10.1136/gut.2005.087262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa H, Umemura A, Taniguchi K, Font-Burgada J, Dhar D, Ogata H, . . . Karin M (2014). ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell, 26(3), 331–343. doi: 10.1016/j.ccr.2014.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson E, Anderson H, Sargenti K, Lindgren S, & Prytz H (2019). Clinical course and mortality by etiology of liver cirrhosis in Sweden: a population based, long-term follow-up study of 1317 patients. Aliment Pharmacol Ther, 49(11), 1421–1430. doi: 10.1111/apt.15255 [DOI] [PubMed] [Google Scholar]
- Parsons CJ, Bradford BU, Pan CQ, Cheung E, Schauer M, Knorr A, . . . Brenner DA (2004). Antifibrotic effects of a tissue inhibitor of metalloproteinase-1 antibody on established liver fibrosis in rats. Hepatology, 40(5), 1106–1115. doi: 10.1002/hep.20425 [DOI] [PubMed] [Google Scholar]
- Pinero F, Mendizabal M, Ridruejo E, Herz Wolff F, Ameigeiras B, Anders M, . . . Lalrean. (2019). Treatment with direct-acting antivirals for HCV decreases but does not eliminate the risk of hepatocellular carcinoma. Liver Int, 39(6), 1033–1043. doi: 10.1111/liv.14041 [DOI] [PubMed] [Google Scholar]
- Pinzani M, & Luong TV (2018). Pathogenesis of biliary fibrosis. Biochim Biophys Acta Mol Basis Dis, 1864(4 Pt B), 1279–1283. doi: 10.1016/j.bbadis.2017.07.026 [DOI] [PubMed] [Google Scholar]
- Popov Y, Sverdlov DY, Bhaskar KR, Sharma AK, Millonig G, Patsenker E, . . . Schuppan D (2010). Macrophage-mediated phagocytosis of apoptotic cholangiocytes contributes to reversal of experimental biliary fibrosis. Am J Physiol Gastrointest Liver Physiol, 298(3), G323–334. doi: 10.1152/ajpgi.00394.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Promrat K, Kleiner DE, Niemeier HM, Jackvony E, Kearns M, Wands JR, . . . Wing RR (2010). Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology, 51(1), 121–129. doi: 10.1002/hep.23276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radaeva S, Sun R, Pan HN, Hong F, & Gao B (2004). Interleukin 22 (IL-22) plays a protective role in T cell-mediated murine hepatitis: IL-22 is a survival factor for hepatocytes via STAT3 activation. Hepatology, 39(5), 1332–1342. doi: 10.1002/hep.20184 [DOI] [PubMed] [Google Scholar]
- Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A, . . . Iredale JP (2012). Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc Natl Acad Sci U S A, 109(46), E3186–3195. doi: 10.1073/pnas.1119964109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reif S, Aeed H, Shilo Y, Reich R, Kloog Y, Kweon YO, & Bruck R (2004). Treatment of thioacetamide-induced liver cirrhosis by the Ras antagonist, farnesylthiosalicylic acid. J Hepatol, 41(2), 235–241. doi: 10.1016/j.jhep.2004.04.010 [DOI] [PubMed] [Google Scholar]
- Rinella ME (2015). Nonalcoholic fatty liver disease: a systematic review. JAMA, 313(22), 2263–2273. doi: 10.1001/jama.2015.5370 [DOI] [PubMed] [Google Scholar]
- Rinella ME, & Green RM (2004). The methionine-choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. J Hepatol, 40(1), 47–51. doi: 10.1016/j.jhep.2003.09.020 [DOI] [PubMed] [Google Scholar]
- Riordan JD, & Nadeau JH (2014). Modeling progressive non-alcoholic fatty liver disease in the laboratory mouse. Mamm Genome, 25(9–10), 473–486. doi: 10.1007/s00335-014-9521-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez HM, Vaysberg M, Mikels A, McCauley S, Velayo AC, Garcia C, & Smith V (2010). Modulation of lysyl oxidase-like 2 enzymatic activity by an allosteric antibody inhibitor. J Biol Chem, 285(27), 20964–20974. doi: 10.1074/jbc.M109.094136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouach H, Fataccioli V, Gentil M, French SW, Morimoto M, & Nordmann R (1997). Effect of chronic ethanol feeding on lipid peroxidation and protein oxidation in relation to liver pathology. Hepatology, 25(2), 351–355. doi: 10.1002/hep.510250216 [DOI] [PubMed] [Google Scholar]
- Salguero Palacios R, Roderfeld M, Hemmann S, Rath T, Atanasova S, Tschuschner A, . . . Roeb E (2008). Activation of hepatic stellate cells is associated with cytokine expression in thioacetamide-induced hepatic fibrosis in mice. Lab Invest, 88(11), 1192–1203. doi: 10.1038/labinvest.2008.91 [DOI] [PubMed] [Google Scholar]
- Sanyal AJ (2010). ACP Journal Club: vitamin E, but not pioglitazone, improved nonalcoholic steatohepatitis in nondiabetic patients. Ann Intern Med, 153(6), JC3–12. doi: 10.7326/0003-4819-153-6-201009210-02012 [DOI] [PubMed] [Google Scholar]
- Scheuer PJ (1991). Classification of chronic viral hepatitis: a need for reassessment. J Hepatol, 13(3), 372–374. doi: 10.1016/0168-8278(91)90084-o [DOI] [PubMed] [Google Scholar]
- Scholten D, Osterreicher CH, Scholten A, Iwaisako K, Gu G, Brenner DA, & Kisseleva T (2010). Genetic labeling does not detect epithelial-to-mesenchymal transition of cholangiocytes in liver fibrosis in mice. Gastroenterology, 139(3), 987–998. doi: 10.1053/j.gastro.2010.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholten D, Reichart D, Paik YH, Lindert J, Bhattacharya J, Glass CK, . . . Kisseleva T (2011). Migration of fibrocytes in fibrogenic liver injury. Am J Pathol, 179(1), 189–198. doi: 10.1016/j.ajpath.2011.03.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuppan D, & Kim YO (2013). Evolving therapies for liver fibrosis. J Clin Invest, 123(5), 1887–1901. doi: 10.1172/JCI66028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scientific Registry of Transplant Recipients. 2018. Annual Data Report.
- Seki E, De Minicis S, Gwak GY, Kluwe J, Inokuchi S, Bursill CA, . . . Schwabe RF (2009). CCR1 and CCR5 promote hepatic fibrosis in mice. J Clin Invest, 119(7), 1858–1870. doi: 10.1172/jci37444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki E, de Minicis S, Inokuchi S, Taura K, Miyai K, van Rooijen N, . . . Brenner DA (2009). CCR2 promotes hepatic fibrosis in mice. Hepatology, 50(1), 185–197. doi: 10.1002/hep.22952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki E, & Schwabe RF (2015). Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology, 61(3), 1066–1079. doi: 10.1002/hep.27332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sempokuya T, Zhang G, & Nakagawa K (2019). Temporal trends of cirrhosis associated conditions. World J Hepatol, 11(1), 74–85. doi: 10.4254/wjh.v11.i1.74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoreibah M, Raff E, Bloomer J, Kakati D, Rasheed K, Kuo YF, & Singal AK (2016). Alcoholic liver disease presents at advanced stage and progresses faster compared to non-alcoholic fatty liver diseas. Ann Hepatol, 15(2), 183–189. doi: 10.5604/16652681.1193707 [DOI] [PubMed] [Google Scholar]
- Slater TF, Cheeseman KH, & Ingold KU (1985). Carbon tetrachloride toxicity as a model for studying free-radical mediated liver injury. Philos Trans R Soc Lond B Biol Sci, 311(1152), 633–645. doi: 10.1098/rstb.1985.0169 [DOI] [PubMed] [Google Scholar]
- Stal P (2015). Liver fibrosis in non-alcoholic fatty liver disease - diagnostic challenge with prognostic significance. World J Gastroenterol, 21(39), 11077–11087. doi: 10.3748/wjg.v21.i39.11077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan BP, Weinreb PH, Violette SM, & Luyendyk JP (2010). The coagulation system contributes to alphaVbeta6 integrin expression and liver fibrosis induced by cholestasis. Am J Pathol, 177(6), 2837–2849. doi: 10.2353/ajpath.2010.100425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapper EB, & Parikh ND (2018). Mortality due to cirrhosis and liver cancer in the United States, 1999–2016: observational study. BMJ, 362, k2817. doi: 10.1136/bmj.k2817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taura K, Miura K, Iwaisako K, Osterreicher CH, Kodama Y, Penz-Osterreicher M, & Brenner DA (2010). Hepatocytes do not undergo epithelial-mesenchymal transition in liver fibrosis in mice. Hepatology, 51(3), 1027–1036. doi: 10.1002/hep.23368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teckman JH, & Blomenkamp KS (2017). Pathophysiology of Alpha-1 Antitrypsin Deficiency Liver Disease. Methods Mol Biol, 1639, 1–8. doi: 10.1007/978-1-4939-7163-3_1 [DOI] [PubMed] [Google Scholar]
- Tokodai K, Miyagi S, Nakanishi C, Hara Y, Nakanishi W, Miyazawa K, . . . Kamei T (2018). Association of post-transplant donor-specific HLA antibody with liver graft fibrosis during long-term follow-up after pediatric liver transplantation. Pediatr Transplant, 22(3), e13169. doi: 10.1111/petr.13169 [DOI] [PubMed] [Google Scholar]
- Traber PG, Chou H, Zomer E, Hong F, Klyosov A, Fiel MI, & Friedman SL (2013). Regression of fibrosis and reversal of cirrhosis in rats by galectin inhibitors in thioacetamide-induced liver disease. PLoS One, 8(10), e75361. doi: 10.1371/journal.pone.0075361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troeger JS, Mederacke I, Gwak GY, Dapito DH, Mu X, Hsu CC, . . . Schwabe RF (2012). Deactivation of hepatic stellate cells during liver fibrosis resolution in mice. Gastroenterology, 143(4), 1073–1083.e1022. doi: 10.1053/j.gastro.2012.06.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto H, French SW, Benson N, Delgado G, Rao GA, Larkin EC, & Largman C (1985). Severe and progressive steatosis and focal necrosis in rat liver induced by continuous intragastric infusion of ethanol and low fat diet. Hepatology, 5(2), 224–232. doi: 10.1002/hep.1840050212 [DOI] [PubMed] [Google Scholar]
- Uchinami H, Seki E, Brenner DA, & D’Armiento J (2006). Loss of MMP 13 attenuates murine hepatic injury and fibrosis during cholestasis. Hepatology, 44(2), 420–429. doi: 10.1002/hep.21268 [DOI] [PubMed] [Google Scholar]
- Ueno A, Lazaro R, Wang PY, Higashiyama R, Machida K, & Tsukamoto H (2012). Mouse intragastric infusion (iG) model. Nat Protoc, 7(4), 771–781. doi: 10.1038/nprot.2012.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vadasz Z, Kessler O, Akiri G, Gengrinovitch S, Kagan HM, Baruch Y, . . . Neufeld G (2005). Abnormal deposition of collagen around hepatocytes in Wilson’s disease is associated with hepatocyte specific expression of lysyl oxidase and lysyl oxidase like protein-2. J Hepatol, 43(3), 499–507. doi: 10.1016/j.jhep.2005.02.052 [DOI] [PubMed] [Google Scholar]
- Varma S, Stephenne X, Komuta M, Bouzin C, Ambroise J, Smets F, . . . Sokal EM (2017). The histological quantification of alpha-smooth muscle actin predicts future graft fibrosis in pediatric liver transplant recipients. Pediatr Transplant, 21(1). doi: 10.1111/petr.12834 [DOI] [PubMed] [Google Scholar]
- Vilar-Gomez E, Calzadilla-Bertot L, Wai-Sun Wong V, Castellanos M, Aller-de la Fuente R, Metwally M, . . . Romero-Gomez M (2018). Fibrosis Severity as a Determinant of Cause-Specific Mortality in Patients With Advanced Nonalcoholic Fatty Liver Disease: A Multi-National Cohort Study. Gastroenterology, 155(2), 443–457 e417. doi: 10.1053/j.gastro.2018.04.034 [DOI] [PubMed] [Google Scholar]
- Vircheva S, Alexandrova A, Georgieva A, Mateeva P, Zamfirova R, Kubera M, & Kirkova M (2010). In vivo effects of pentoxifylline on enzyme and non-enzyme antioxidant levels in rat liver after carrageenan-induced paw inflammation. Cell Biochem Funct, 28(8), 668–672. doi: 10.1002/cbf.1705 [DOI] [PubMed] [Google Scholar]
- Wang X, Zheng Z, Caviglia JM, Corey KE, Herfel TM, Cai B, . . . Tabas I (2016). Hepatocyte TAZ/WWTR1 Promotes Inflammation and Fibrosis in Nonalcoholic Steatohepatitis. Cell Metab, 24(6), 848–862. doi: 10.1016/j.cmet.2016.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White DL, Thrift AP, Kanwal F, Davila J, & El-Serag HB (2017). Incidence of Hepatocellular Carcinoma in All 50 United States, From 2000 Through 2012. Gastroenterology, 152(4), 812–820 e815. doi: 10.1053/j.gastro.2016.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong T, Dang K, Ladhani S, Singal AK, & Wong RJ (2019). Prevalence of Alcoholic Fatty Liver Disease Among Adults in the United States, 2001–2016. JAMA, 321(17), 1723–1725. doi: 10.1001/jama.2019.2276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie G, Karaca G, Swiderska-Syn M, Michelotti GA, Krüger L, Chen Y, . . . Diehl AM (2013). Cross-talk between Notch and Hedgehog regulates hepatic stellate cell fate in mice. Hepatology, 58(5), 1801–1813. doi: 10.1002/hep.26511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie G, Wang X, Wang L, Atkinson RD, Kanel GC, Gaarde WA, & Deleve LD (2012). Role of differentiation of liver sinusoidal endothelial cells in progression and regression of hepatic fibrosis in rats. Gastroenterology, 142(4), 918–927.e916. doi: 10.1053/j.gastro.2011.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yalamanchili K, Saadeh S, Klintmalm GB, Jennings LW, & Davis GL (2010). Nonalcoholic fatty liver disease after liver transplantation for cryptogenic cirrhosis or nonalcoholic fatty liver disease. Liver Transpl, 16(4), 431–439. doi: 10.1002/lt.22004 [DOI] [PubMed] [Google Scholar]
- Yang L, Kwon J, Popov Y, Gajdos GB, Ordog T, Brekken RA, . . . Shah VH (2014). Vascular endothelial growth factor promotes fibrosis resolution and repair in mice. Gastroenterology, 146(5), 1339–1350.e1331. doi: 10.1053/j.gastro.2014.01.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YM, Noureddin M, Liu C, Ohashi K, Kim SY, Ramnath D, . . . Seki E (2019). Hyaluronan synthase 2-mediated hyaluronan production mediates Notch1 activation and liver fibrosis. Sci Transl Med, 11(496). doi: 10.1126/scitranslmed.aat9284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, & Wymer M (2016). Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology, 64(1), 73–84. doi: 10.1002/hep.28431 [DOI] [PubMed] [Google Scholar]
- Zein CO, Yerian LM, Gogate P, Lopez R, Kirwan JP, Feldstein AE, & McCullough AJ (2011). Pentoxifylline improves nonalcoholic steatohepatitis: a randomized placebo-controlled trial. Hepatology, 54(5), 1610–1619. doi: 10.1002/hep.24544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Kim K, Wang X, Bartolome A, Salomao M, Dongiovanni P, . . . Pajvani UB (2018). Hepatocyte Notch activation induces liver fibrosis in nonalcoholic steatohepatitis. Sci Transl Med, 10(468). doi: 10.1126/scitranslmed.aat0344 [DOI] [PMC free article] [PubMed] [Google Scholar]